 Valentin S. Bruttel
Valentin S. Bruttel Jörg Wischhusen
Jörg Wischhusen- Section for Experimental Tumor Immunology, Department of Obstetrics and Gynecology, School of Medicine, University of Würzburg, Würzburg, Germany
Cancer stem cell (CSC) biology and tumor immunology have shaped our understanding of tumorigenesis. However, we still do not fully understand why tumors can be contained but not eliminated by the immune system and whether rare CSCs are required for tumor propagation.
Long latency or recurrence periods have been described for most tumors. Conceptually, this requires a subset of malignant cells which is capable of initiating tumors, but is neither eliminated by immune cells nor able to grow straight into overt tumors. These criteria would be fulfilled by CSCs. Stem cells are pluripotent, immune-privileged, and long-living, but depend on specialized niches. Thus, latent tumors may be maintained by a niche-constrained reservoir of long-living CSCs that are exempt from immunosurveillance while niche-independent and more immunogenic daughter cells are constantly eliminated.
The small subpopulation of CSCs is often held responsible for tumor initiation, metastasis, and recurrence. Experimentally, this hypothesis was supported by the observation that only this subset can propagate tumors in non-obese diabetic/scid mice, which lack T and B cells. Yet, the concept was challenged when an unexpectedly large proportion of melanoma cells were found to be capable of seeding complex tumors in mice which further lack NK cells. Moreover, the link between stem cell-like properties and tumorigenicity was not sustained in these highly immunodeficient animals. In humans, however, tumor-propagating cells must also escape from immune-mediated destruction. The ability to persist and to initiate neoplastic growth in the presence of immunosurveillance – which would be lost in a maximally immunodeficient animal model – could hence be a decisive criterion for CSCs.
Consequently, integrating scientific insight from stem cell biology and tumor immunology to build a new concept of “CSC immunology” may help to reconcile the outlined contradictions and to improve our understanding of tumorigenesis.
Introduction to Tumor Immunosurveillance
Cancer is caused by accumulating genetic alterations in a cell. These lead to activation or multiplication of proteins promoting cell growth and survival whereas proteins promoting cell cycle arrest or cell death are lost or inactivated. Every day no less than one million molecular lesions are thought to occur in the DNA of a single cell (1). While most of these are instantly repaired, the high number implies that very effective control mechanisms must exist to prevent the formation of tumors by mutant cells. This includes both cell-intrinsic mechanisms, such as DNA repair enzymes and tumor suppressor genes [comprehensively reviewed by Hanahan and Weinberg (2)], and the cell-extrinsic control function of the immune system, as first postulated by Paul Ehrlich more than a century ago and systematically introduced by Burnet in 1953 (3). Tumor immunosurveillance relies on traits of malignant transformation that can be recognized by innate and adaptive immune cells. As oncogenic stress induces the upregulation of ligands for activating NK cell receptors (4) and other immune stimulatory surface molecules, tumor cells can be recognized and rejected by NK cells. This innate immune response can further activate a targeted adaptive immune response against antigens specifically expressed by lysed tumor cells and thereby lead to T cell dependent tumor control. Such immunogenic antigens are either highly overexpressed in tumor cells or even tumor specific (when caused by non-silent mutations that have accumulated during malignant transformation) (5). Whole genome sequencing data from lung cancer (6, 7), melanoma, and lymphoblastoid carcinoma (8) genomes indicate that tumors can harbor up to 50,000 somatic mutations of which few hundreds affect protein-coding sequences. It is estimated that this will result in about 7–10 new and unique MHC-binding peptides per HLA allele (9).
Accordingly, innate immunity may constitute a first line of defense, but long-term control is thought to depend on the subsequent adaptive immune responses and on memory cells, which also provide us with life-long protection against pathogens. The detailed roles of different immune cells in tumor immunosurveillance have been comprehensively reviewed recently (10, 11).
Key molecules for tumor immunosurveillance as identified in knockout mouse models comprise interferon-γ (IFNγ) (12), IL-12 (13) perforin (14, 15), TRAIL (tumor necrosis factor related apoptosis-inducing ligand) (16–19), and its corresponding apoptosis-inducing receptors DR4 and DR5 (20), the recombination activating genes RAG1 (21), and RAG2 (12), which are required for T cell development, the T cell receptor (22), and the activating NK cell receptor NKG2D (23). The loss of any of these molecules results in more frequent or faster spontaneous or carcinogen-induced tumorigenesis. The ability to evade immune destruction was thus recognized as an additional hallmark of cancer (24).
Tumor Latency and Immune Escape
Late recurrence indicates that tumor outgrowth can sometimes be constrained over decades. Koebel et al. (13) elegantly demonstrated that the immune system can control tumor outgrowth without actually eradicating every malignant cell. By treating immunocompetent mice with low doses of the carcinogenic compound methylcholanthrene, they induced stable masses containing only very few malignantly transformed cells. However, when CD4+ or CD8+ T cells were depleted even as late as 200 days after chemical carcinogenesis, tumors started to grow. This indicates that the immune system can control tumor growth and keep pre-malignant lesions in a dormant state rather than completely eliminating every transformed cell.
Correspondingly, immunocompromised humans such as recipients of organ transplants (25–28) or AIDS patients (29–31) have a significantly increased risk of cancer development, which may be partially attributed to the loss of immunological control over preexistent malignantly transformed cells. Advanced age remains the most important risk factor for tumor development [recently reviewed by de Magalhaes (32)], which may be due to a decline of immunosurveillance. While the stochastic increase in the number of genetic alterations over time and the deterioration of cellular repair mechanisms certainly contribute to this well-known phenomenon, age-dependent defects in the immune system (also termed “immunosenescence”) (33–35) are also likely to facilitate tumor outgrowth. This is corroborated by the finding that adoptively transferred spleen cells from young immunized mice eliminate even large tumors whereas immune cells from old mice are not protective (36). Further factors which may reduce the efficiency of anti-tumor immune responses include non-recognition of tumor antigens due to pre-established tolerance (37), evolution of poorly immunogenic subsets of tumor cells and immunosuppression via accessory cells in the tumor microenvironment. However, tumor-initiating cells need to escape from immunosurveillance even before a proper tumor microenvironment has been established.
On cellular level, continuous genetic transformation can randomly generate less-immunogenic malignant subclones. Lower levels of activatory and/or higher levels of inhibitory NK cell receptor ligands (38, 39) may enable these cells to survive in the presence of the immune system. Also antigens toward which a specific T cell immune response has been established may be lost (40, 41). Furthermore, aggressive tumors are often characterized by low levels of classical HLA class I molecules. Accordingly, those tumor antigens, which are expressed, are poorly presented which limits killing by CD8+ cytotoxic T lymphocytes (CTLs) (42). Nevertheless, a complete loss of ”self-MHC,” which would render cells most susceptible for NK cell killing (11, 43–45) does not seem to be the rule. A further important aspect is the active suppression of NK and T cells in the tumor microenvironment where immunosuppressive cytokines or hormones are abundant. Cell surface expression and/or secretion of immune-inhibitory non-classical HLA class I molecules such as HLA-G and HLA-E also contributes to local immune paralysis (42).
Physiologically, many of these factors are required for feto-maternal immune tolerance. As immune-mediated destruction of semi-allogeneic embryonic cells would preclude a successful pregnancy, tolerance toward cells expressing “non-self” antigens is essential for reproduction. This imposes certain limits to the stringent elimination of altered cells. Unfortunately, the same tolerance inducing strategies may also be used by cancer cells to overcome immunosurveillance. Nevertheless, mechanisms to prevent tumor growth still fulfill their evolutionary purpose by largely protecting individuals from cancer until reproductive age or, ideally, until their children (or even grandchildren) have become adults.
Introduction to Cancer Stem Cells
Tissue-specific stem cells have been described in many tissues prone to cancer, including breast (46, 47), lung (48), prostate (49–51), intestine (52, 53), and many others. They are exceptionally long-lived and can perpetually self-renew. They further give rise to progenitor cells, which are more restricted in their developmental potential, but capable of undergoing rapid cell divisions. The respective daughter cells then further differentiate into mature tissue-specific cells, which contribute to growth, tissue homeostasis, and wound healing (Figure 1A) [reviewed by Ref. (54, 55)].
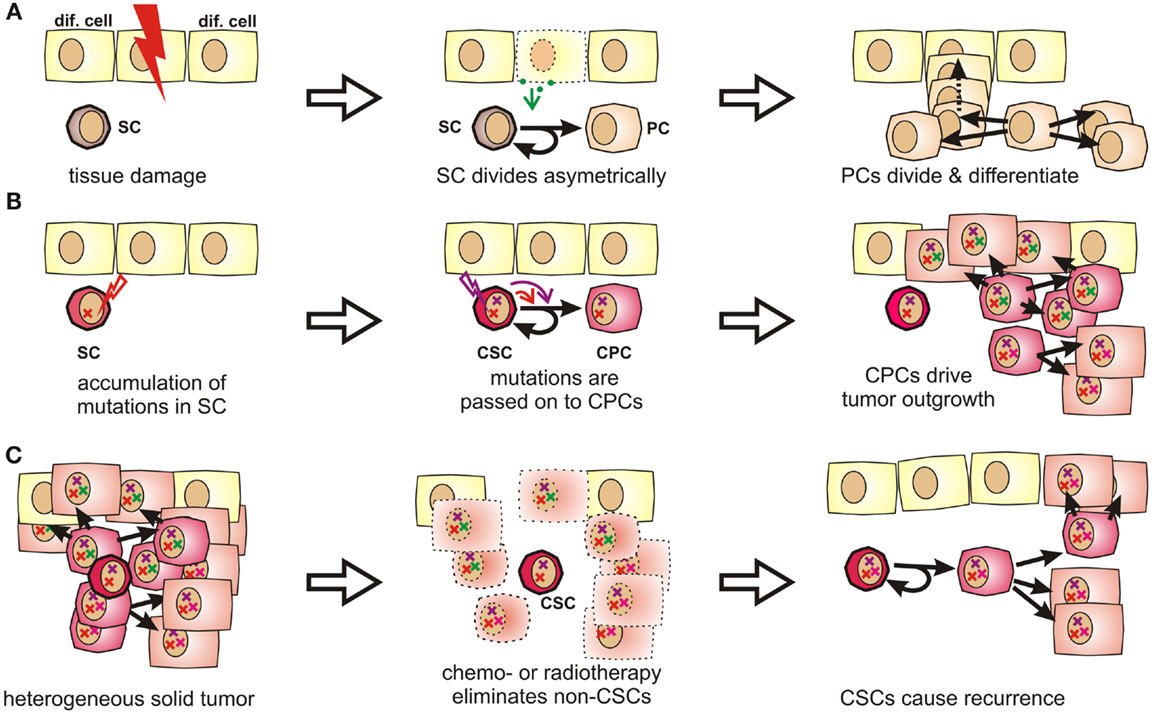
Figure 1. Stem cells and progenitor cells in tissue homeostasis, tumorigenesis, and tumor recurrence. (A) During organogenesis or in response to tissue damage, resting stem cells (SCs) are induced to undergo asymmetric divisions that produce further differentiated progenitor cells (PCs). Progenitor cells can quickly regenerate tissue by first undergoing a limited number of symmetric cell divisions. The generated daughter cells can then further differentiate to acquire organ-specific functions while generally losing the ability to reenter the cell cycle. (B) Stem cells are exceptionally long-lived, pluripotent, and can undergo an unlimited number of cell divisions. Therefore, they may accumulate more mutations than other cells and are thus stochastically more likely to initiate a tumor. These mutations are passed on to progenitor cells, which may drive tumor outgrowth as they cycle much faster by nature. (C) Stem cells and CSCs are highly resistant to chemotherapeutic agents and irradiation, which explains why these cells might selectively survive cytoreductive therapy. Therefore, these cells may cause cancer recurrence years or decades later.
This cellular hierarchy within a tissue has been studied most extensively in the hematopoietic system. Here, long term and short term stem cells, multipotent and lineage-committed progenitor cells, precursors, and fully differentiated cells have been well characterized based on their functional properties and on cell surface markers [reviewed by Orkin (56)]. Among these, pluripotent stem cells tend to cycle very slowly while the bulk of blood cells originate from various frequently dividing progenitor cell populations (57, 58). In adult mice, a population of quiescent or dormant hematopoietic stem cells has been described to undergo only about five divisions during the lifespan of a mouse (59).
Because of their longevity, pluripotency, and seemingly unlimited potential to undergo cell divisions, stem cells have long been suspected to be the culprits for tumor formation (60). It is indeed much more likely that a stem cell, which persists for decades in a given tissue, rather than a short-lived differentiated cell, can accumulate numerous mutations as required for malignant transformation (61, 62) (Figures 1B and S1 in Supplementary Material). Moreover, the presence of highly diverse cancer cell populations both in tumors and metastases indicates that their cells of origin are pluripotent. In fact, Kleinsmith and colleagues showed already in the 1960s that some teratocarcinoma cells are capable of seeding highly heterogeneous tumors even if only single cells are transplanted (63). Recently, it was shown that even some blood vessels within glioblastoma are derived from tumor cells (64). This strikingly illustrates the remaining differentiation potential of tumor-initiating cells.
Clinical observations from cancer patients teach us that tumor recurrence may still occur years or decades after a successful-appearing treatment. In this context, a recent sequencing study on acute myeloid leukemia showed that cells collected at the time of the initial diagnosis and during relapse were both closely related to stem cells. Surprisingly, cells collected after relapse had often undergone fewer cell divisions than cells collected at first diagnosis (65). This suggests that the relapsed disease originated from Cancer stem cells (CSCs), which had been largely quiescent during the initial tumor burst, but which survived tumor eradication without losing their malignant potential. Indirectly, this also confirms the existence of a long-lived and multipotent subpopulation of cancer cells, which is highly resistant to radio- and chemotherapy (Figure 1C) and too small to be detected by common screening methods.
Similar to non-malignant stem cells, CSCs are thought to cycle much more slowly than cancer progenitor cells (CPCs). This could explain their resistance toward drugs directed against dividing cells. Additionally, many stem cells can minimize intracellular drug levels via expression of ABC (ATP binding cassette) transporters (66). Accordingly, chemoresistance is expected to be an inherent trait of CSC even before the onset of therapy. Furthermore, stem cells and cancer cells share decreased p53 activity and increased telomerase activity (67).
Still, resistant CSCs are not necessarily the main drivers of rapid tumor outgrowth. While acquisition of specific mutations may alter the replicative behavior of these quiescent and niche-dependent cells (68), homeostatic, and presumably also neoplastic tissue growth depends mainly on niche-independent progenitor cells which divide much more rapidly. In this manuscript, the term CPC is used for the malignant counterparts of such rapidly dividing and lineage-committed cells. As these cells inherit genomic mutations accumulated by stem cells, malignantly transformed stem cells will give rise to malignantly transformed committed progenitor cells, which can then drive tumor growth. Luminal progenitor cells deleted for the tumor suppressor gene Brca1 were thus shown to seed basal-like breast cancer in mice (69, 70). Likewise, prostate cancer can be initiated by basal progenitor cells, which have been virally transfected with oncogenes (71). Obviously, CPCs may not only inherit mutations, but also accumulate “first genetic hits,” which likely contributes to clonal evolution and to the heterogeneity typically found in late stage tumors.
The Cancer Stem Cell Hypothesis – Ambiguities and Criticism
Cellular differentiation states are difficult to categorize. While omnipotent embryonic stem cells (ESCs) and terminally differentiated cells represent the two ends of the spectrum, numerous gradual transitions exist in between. Defining clear-cut categories is even more difficult in the tumor context where cellular functions and markers may be randomly altered due to mutations. Distinct stem cell markers have been described for CSCs, but these only help to define CSCs within a certain tissue. In an attempt to define the unifying functional characteristics of a CSC, a panel at the American Association of Cancer Research (AACR) meeting suggested in 2006: “A CSC is a cell within a tumor that possesses the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that comprise the tumor” (72). We consider this definition to be insufficient for several reasons:
1) De-differentiation. By now, a considerable body of evidence indicates that lineage-committed differentiated cells and progenitor cells can de-differentiate into multipotent stem cells. While induced pluripotent stem cells [iPS cells as first introduced by Takahashi and Yamanaka (73)] are typically generated by artificially introducing defined transcription factors into differentiated cells, de-differentiation can also occur spontaneously (74) or in response to external stimuli (75, 76). Accordingly, both tissues and tumors may not be organized in a strictly hierarchical manner with few stem cells on the apex, but rather in a more dynamic way with constantly differentiating and de-differentiating cells (77, 78). Likewise, a fairly constant percentage of CSC-like cells can be found to co-exist with basal or luminal progenitor cells in some established tumor cell lines. These populations can be isolated. However, if cultured for a few days, each of them will regenerate the depleted populations and an equilibrium containing almost identical proportions of all three original differentiation states is quickly reestablished (77).
The most prominent example of physiological transdifferentiation is the so-called epithelial to mesenchymal transition (EMT) (79–81) during which epithelial cells acquire stem cell-like properties. Interestingly, in breast cancer or melanoma EMT can be induced by CD8+ T cells (77, 80). Accordingly, there appears to be more plasticity within the cellular hierarchy than previously anticipated, especially in tumors.
2) The frequency of tumor-propagating cells depends on the respective animal model. For melanoma, CSC markers, frequencies, and their role in tumorigenesis were investigated in considerable detail. CD20 (82), CD133 (83), ABCG2 (83), MDR1 (84), or CD271 (85) have all been described as potential markers for melanoma cells with increased tumorigenic potential as assessed in non-obese diabetic (NOD)/severe combined immune-deficient (scid) mice. With about one CSC in 106 melanoma cells, these CSCs constitute only a minute subset of all tumor cells (86). However, in a highly debated (87, 88) article, Quintana et al. (89) reported that about 27% of highly aggressive melanoma cells were capable of inducing tumors in interleukin-2 receptor gamma chain deficient (Il2rg−/−) NOD/scid (NSG) mice, which do not only lack T and B, but also NK cells. Thus, the proportion of tumor cells, which can self-renew and seed heterogeneous tumors in highly immunodeficient mice may be much higher than previously anticipated. This raised the question whether the small subpopulation of CSCs is of any clinical relevance (86).
In a later study, the same authors proposed that most melanoma cells may reversibly express stem cell markers (90). They argue in favor of clonal evolution and negate any kind of hierarchical organization within a tumor cell population, thus refuting the basic idea underlying the CSC hypothesis. Accordingly, the term CSC is avoided by some authors and replaced by more descriptive alternatives such as cancer-initiating cell, tumor-propagating cell or metastasis-initiating cell. In our opinion, however, this approach has not helped to simplify matters, especially since suitable markers and physiologically correlating cell populations are ill-defined.
We believe that the ability of differentiated cancer cells to de-differentiate into CSCs may partly reconcile these conflicting findings. Moreover, it should be noted that these data were generated in mice with very different immunological properties. Thus, a unique and functionally most relevant criterion shared by stem cells and CSCs, namely their lack of immunogenicity, should be considered.
Immunological Properties of CSCs
Unfortunately, our knowledge about specific immunological properties of distinct CSC populations is still limited. It is, however, clear that high levels of anti-apoptotic proteins like bcl-2, bcl-xL, or survivin do not only protect CSCs against chemotherapeutic drugs (91), but also increase resistance toward apoptosis-inducing immune effectors like T or NK cells. In this context, the PI3K/Akt pathway seems to be of particular relevance: it is not only a known mediator of chemoresistance and CSC renewal (92), but was also found to be involved in tumor immune escape (93). Likewise, the oncogenic growth factor receptor HER2/neu (which activates PI3K/Akt signaling) does not only deliver pro-survival signals (94). HER2 does also interfere with antigen processing and presentation (95, 96) and it was reported as crucial for the maintenance of CSC in luminal breast cancer (97). Thus, mechanisms which were mainly thought to mediate chemoresistance in CSC may also confer cross protection against immune-mediated tumor eradication.
In the aggressive melanoma cell line A375, Schatton and Frank also found the immunogenic tumor-associated antigen MART-1 (melanoma antigen recognized by T cells) to be only expressed on differentiated melanoma cells, but not on malignant melanoma initiating cells (MMICs). MART-1-specific T cells thus cannot eliminate MMICs (98). Reim et al. have further shown that CD44high/CD24low breast CSCs selectively escape from NK cell mediated killing and trastuzumab-dependent ADCC (99). While these findings explain the difficulties in targeting CSCs with immune cells, stem cell-like cells are not only poor targets for immunosurveillance. They also actively suppress immune responses (100), which may be crucial during tumorigenesis (101). Breast CSCs (102) and glioblastoma stem cells (103) secrete more TGF-β as compared to normal tumor cells. Colon CSCs are further known to secrete Interleukin 4 (104), which promotes drug resistance (104) and inhibits anti-tumor immune responses (105). CSCs also express CD200 (106), a molecule that inhibits myeloid cells and could therefore play a major role in tumor immune escape (107, 108). Immunological properties of CSCs in solid tumors have also been recently reviewed by Maccalli et al. (109).
More is known about molecular immunological properties of physiological stem cells. ESCs express little classical antigen-presenting MHC class Ia molecules and no MHC class II molecules and will thus be poor antigen presenters for T cells. Additionally, they express only low levels of ligands for the activatory NK cell receptor NKp44 and no ligands for NKp30, NKp46, and CD16 (110). Among inhibitory signals, HLA-G (111) is highly expressed by both embryonic (112) and mesenchymal stem cells (MSCs) (113). Thus, studies describing higher MHC class I expression on stem cells may have failed to distinguish between immunogenic MHC class Ia (HLA-A, -B, -C) and immunosuppressive MHC class Ib (HLA-E, -F, -G) molecules. MHC class II or costimulatory molecules such as CD40, B7-1 (CD80), or B7-2 (CD86), which can also support T cell interactions with non-lymphoid cells (114) have not been found on MSCs (115–117).
T cell proliferation, macrophage activation, and Th1 responses can further be suppressed by prostaglandin E2 (PGE2), which is secreted by MSCs (118, 119). After exposure to IFN-γ, MSCs also upregulate indoleamine 2,3-dioxygenase (IDO), which metabolizes tryptophan into immunosuppressive kynurenines. The concomitant tryptophan depletion also has a profound inhibiting effect on T cells (120). Via secretion of hepatocyte growth factor (121), MSCs can further induce tolerogenic dendritic cells and regulatory T cells (122). Consequently, MSCs show beneficial effects in autoimmune disease models (123–125).
To summarize, stem cells and CSCs both express numerous and diverse membrane-bound and soluble factors (Figure 2), which enable these cells to efficiently modulate immune responses and which protect them against immune-mediated destruction in a way that is unrivaled by further or fully differentiated cells. Strikingly, mouse ESCs and human MSCs have even been shown to survive for weeks and to engraft in immunocompetent rats (126) and sheep (127, 128). The immunomodulatory potential of MSCs is now exploited clinically to counteract graft-versus-host disease (129) and to promote allograft acceptance upon solid organ transplantation (130).
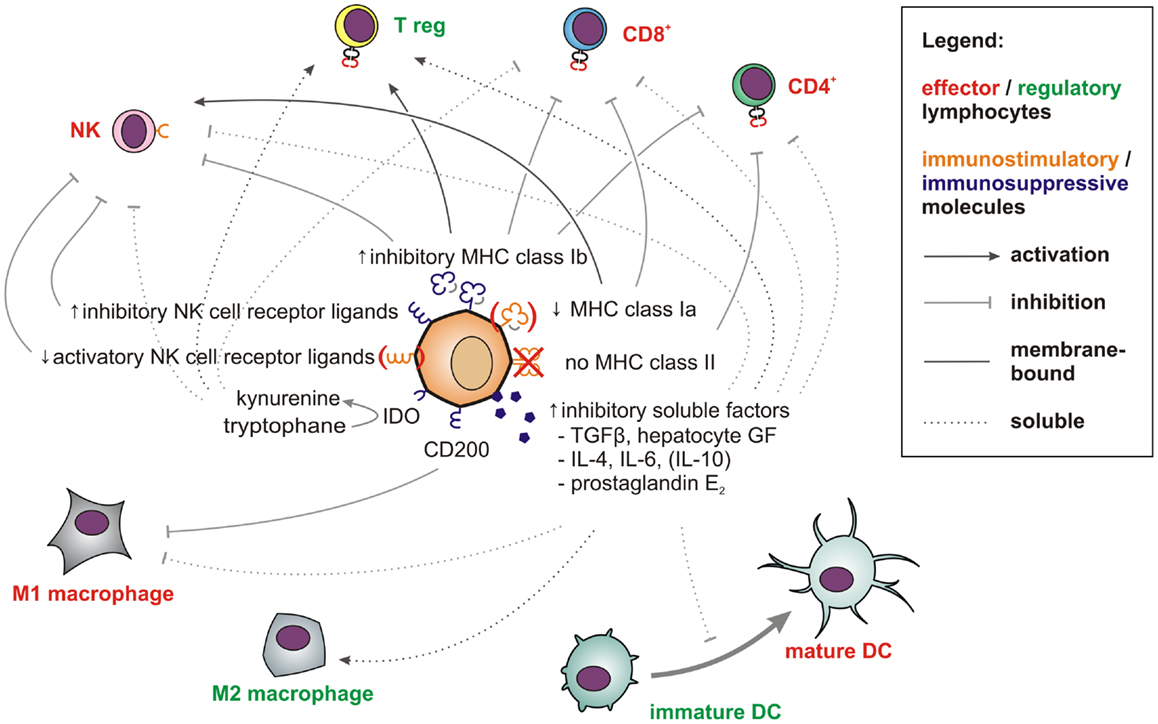
Figure 2. Factors and pathways contributing to the low immunogenicity of stem cells and CSCs. Membrane-bound (solid line) and soluble factors (dotted lines) implicated in immunotolerance toward stem cells and CSCs are schematically depicted. These include antigen-presenting and immunostimulatory molecules (orange) expressed at low levels and abundantly expressed immunosuppressive factors (blue) (see main text for references). Immune effector cells impaired by these signals are shown in red whereas tolerogenic immune cells, which may be stimulated are displayed in green.
As the immune system is incapable of eliminating allogeneic or even xenogenic stem cells, the comparatively few mutations acquired by CSC are unlikely to cause immune-mediated rejection. Thus, stem cells likely represent a unique compartment in which oncogenic mutations can accumulate without being detected by the immune system.
Immune Privilege – The Missing CSC Criterion?
The question whether or not all cancer cells are equally capable of seeding tumors obviously depends also on their ability to escape from destruction by the innate or adaptive immune system. This essential difference, however, may not be observable in a mouse model lacking functional T, B, and NK cells, such as the NSG (131) mice used by Quintana et al. (89). The conflicting data on the frequency of melanoma initiating cells can thus easily be explained by accepting that CSCs are much less sensitive toward tumor immunosurveillance than more differentiated cells. While NK cells in NOD/scid mice may clear more differentiated cancer cells (Figure 3B), they cannot eliminate immunosubversive human CSCs (Figure 3A). Thus, non-CSC-like cancer cells may replicate endlessly in vitro whereas only CSCs will seed tumors in these mice. NK cell-deficient NSG mice can, in contrast, neither eliminate CSCs (Figure 3C) nor more differentiated cancer cells, which would then also become capable of seeding tumors (Figure 3D). Their recently discovered ability to de-differentiate may then further facilitate tumor propagation.
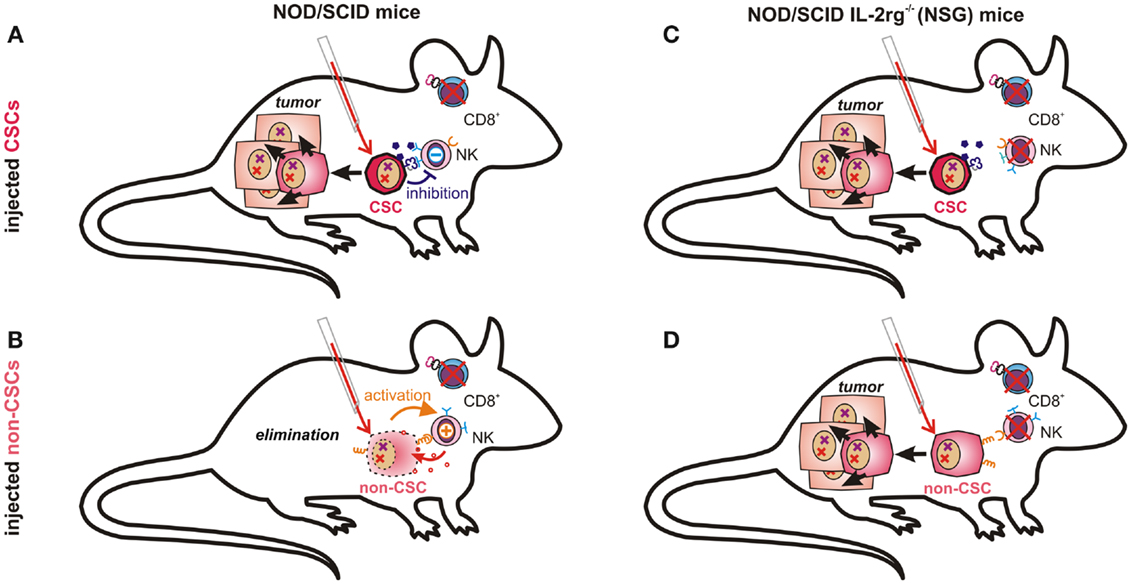
Figure 3. Tumor-propagating capacity depends on immunological properties of injected cancer cells and on the respective mouse model. NK cells in NOD/scid mice are likely incapable of eliminating CSCs due to their low immunogenicity (A). More differentiated cancer cells expressing ligands for activating NK cell receptors and fewer immunosuppressive molecules may, however, be recognized and eliminated by host NK cells (B). In NOD/scid IL2rg−/− (NSG) mice, which also lack NK cells, both poorly immunogenic CSCs (C) and further differentiated cancer cells (D) can seed tumors, as malignant progenitor cells may also possess enormous proliferative capacity.
A maximally immune-deficient mouse may therefore demonstrate the malignant potential of differentiated cancer cells in the complete absence of immunosurveillance, an aspect that may have been underestimated in the original CSC theory. We, however, wonder how relevant NSG mice can be for understanding tumor initiation (and thus tumor-initiating cells) in patients. While stem cell experts tend to favor the most completely immunodeficient animal model available, the most relevant subject of translational cancer research is the immune-competent human subject afflicted by a malignant disease. The ability to propagate tumors should therefore best be tested in models possessing a functional immune system. Furthermore, as implied by studies performed in NOD/scid mice (132–134), the ability to continuously seed tumors in presence of (residual) immunosurveillance may be a most relevant functional criterion for CSCs.
Consequently, tumor initiation may better be analyzed in congenic or syngenic animals rather than in immunodeficient xenograft models. Limitations arise from the fact that all cells contained in transplantable syngenic tumor cell lines have evidently undergone immunoediting in vivo before the cell line could be derived. Accordingly, the proportion of immune-refractory, tumor-seeding cells may be quite variable: with B cell lymphoma cells, for example, inoculation with 10 unsorted cells was sufficient to induce lethal lymphomas within a few weeks, irrespective of expression of the stem cell marker CD93 (135). In the 4T1 mammary carcinoma cell line, however, exclusion of the stem cells (Hoechst 33342 side population) by cell sorting greatly reduced both tumor take and tumor load, and most animals injected with 8 × 103 non-CSC remained tumor-free (136). Thus, the frequency of CSCs can differ widely depending on the respective tumor.
Tumor Immunoediting – The Model and Unresolved Questions
The immune privilege of CSCs may not only be relevant for the quantification of tumor-propagating cells, but could also help to elucidate ambiguities in tumor initiation and immune escape. The complex interactions between tumors and the immune system have been described by a model which differentiates between three phases of tumor immunoediting: elimination, equilibrium, and escape (137). There is now solid scientific evidence for the existence of all three phases and this model nicely correlates with clinical observations (10).
During the elimination phase, tumors may be successfully detected and destroyed by the innate and adaptive immune system, which results in the reestablishment of healthy tissue. If, however, some tumor cells escape from elimination, a dynamic equilibrium between tumor growth and immunological elimination of malignant cells was proposed to emerge. While tumor outgrowth is still constrained, elimination of tumor cells remains incomplete, which allows for mutational diversification and Darwinian selection to occur. This latency phase may persist for decades and may be functionally similar to tumor dormancy, the time between an initially successful anti-tumor therapy and tumor recurrence, which can also span decades (138). When ultimately one clone arises that is capable of escaping from a functional immune system, it may form a clinically overt tumor and initiate the escape phase.
However, for the equilibrium to be reached, this model requires “not yet immunoedited” cancer cells to survive the early elimination phase. Furthermore, it is not clear why cells, which can be immunologically constrained in equilibrium for decades, are not simply cleared by an adaptive immune response. Normally, expanded and activated CTLs efficiently eliminate cells expressing their cognate antigen whereas cells that have lost all immunologically relevant antigens could directly grow into overt tumors.
Based on these considerations, latency requires the existence of a poorly immunogenic sub-pool of cancer cells, which escape from targeted immune responses, which can give rise to new cancer cells, but which are initially incapable of growing into massive tumors. Instead, these cells apparently persist for months, years, or even decades within tissues as dormant tumor cells. In our opinion, multipotent stem cells and CSCs, which are not only long-lived, but also exceptionally well protected from immune-mediated destruction, and largely confined to stem cell niches (68, 139), would meet all these criteria. By considering the immunological properties of CSCs, a new refined concept of tumor immunoediting can thus be proposed (Figure 4 and more detailed in Figure S1 in Supplementary Material).
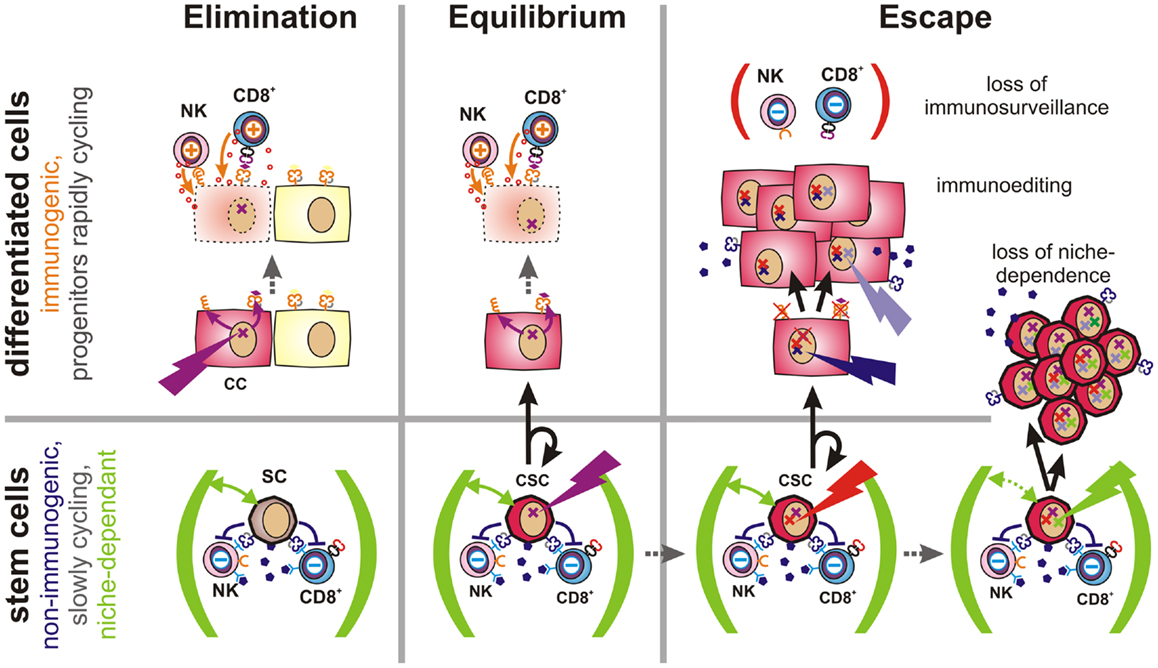
Figure 4. Proposed role of CSCs in tumor immune escape. Elimination (Left): malignant transformation requires oncogenic mutations (flash) to accumulate within an individual cell. If such mutations occur in a differentiated cell, this cell will upregulate activatory NK cell receptor ligands and present tumor-specific peptides via MHC class Ia molecules (both orange). Thus, these cells can be detected and eliminated by the immune system, leading to restoration of normal tissue. Equilibrium (Center): stem cells (SC) are long-lived and express a multitude of immunosuppressive factors (dark purple). Hence, they may accumulate oncogenic mutations without being cleared by the immune system. However, CSCs are initially confined to stem cell niches (green) and limited to asymmetric divisions. More differentiated daughter cells inherit all malignant mutations, but are more immunogenic and could thus be eliminated by the adaptive immune system. Thus, a robust equilibrium may emerge. Escape (Right): mechanisms contributing to tumor immune escape include defects in tumor immunosurveillance, immunoediting, or the expansion of CSCs. The immune system may lose its ability to constrain tumors due to aging, immunosuppressive therapies, diseases or other factors. Immunoediting describes the evolutionary adaptation of individual tumor subclones to the selection pressure exerted by the immune system. It will ultimately lead to expansion of less-immunogenic or more immunosuppressive subclones. Non-immunogenic CSCs may further acquire the ability to expand independently of their niches, which may lead to the outgrowth of poorly differentiated tumors.
Implementing CSCs in the Concept of Tumor Immunoediting
When oncogenic mutations occur in fully or partially differentiated cells, activatory NK cell receptor ligands are induced (4) and tumor antigens are presented via MHC class Ia. Thus, altered cells may be readily detected and eliminated by the immune system. Failure may be due to de-differentiation of mutated cells.
If, instead, an oncogenic mutation occurs in a poorly immunogenic stem cell, escape from immunosurveillance is much more likely. However, their dependence on stem cell niches may prevent mutated stem cells from growing into overt tumors. Upon asymmetric division, a more differentiated and more immunogenic daughter cell will be generated, which would then be subjected to immunosurveillance. As the other daughter cell will maintain stem cell characteristics, a robust equilibrium would emerge. Recent findings indicate that AML finally arises from such pre-malignant stem cells (140).
During this latency period, stochastic genetic and epigenetic changes may accumulate within the reservoir of non-immunogenic stem cells. Mutations in coding regions may generate new immunogenic peptides that would be presented on MHC molecules of malignant daughter cells. Such neoantigens may support immunosurveillance by providing further tumor antigens and, ideally, enabling tumor clearance. However, if CSCs are not eliminated by the immune system, the low frequency of niche-confined, mutated stem cells, and their low rate of cell divisions would still prevent the formation of a clinically relevant tumor as long as their daughter cells are subjected to immunosurveillance. Accordingly, the immune system could constrain a sub-clinical malignant disease in life-long equilibrium.
However, once tumors become clinically apparent immunosurveillance has failed. This can be due to a weakening of the immune system caused by aging, to therapeutic immunosuppression or to disease. In this case, also moderately immunogenic tumor cells may propagate. Alternatively, cancer cells may actively attenuate immunosurveillance (immunosubversion) by secretion or expression of immunosuppressive factors (11) or by recruitment of accessory cells, which locally suppress the immune system (141, 142). Furthermore, genetic alterations may result in reduced expression of antigens that are targeted by the immune system (immunoselection). Other factors allowing for the rapid growth of more differentiated cancer cells include the loss of the antigen processing and presentation machinery (143) or the loss or shedding of ligands for activatory NK cell receptors (144). Importantly, non-immunogenic long-lived CSCs provide an evolutionary playground for mutations to accumulate until an immune escape phenotype has emerged.
Expansion of immune-evasive CSCs represents a further possibility for immune escape. This mechanism may predominate in poorly differentiated tumors, which tend to be more aggressive and more difficult to treat (145). Alterations in cell signaling pathways are known to enable CSCs to grow outside of their niches (68). Interestingly, signals that promote stem cell self-renewal and expansion (146) are also frequently activated in cancer as evidenced by alterations in the notch, wnt, and sonic hedgehog pathways (147–149).
Oncogenic mutations may also increase the frequency of cell divisions in otherwise fairly quiescent stem cells (150) or inhibit differentiation in the respective daughter cells (151, 152). Furthermore, CSC proliferation may be promoted by conditions like chronic inflammation, which triggers physiological stem cell division. As these mechanisms are not mutually exclusive, increased cell division in CSCs will also increase the risk for emergence of less-immunogenic and more aggressive subclones.
Implications of “Cancer Stem Cell Immunology”
Stem cell research has helped to understand tumor initiation, heterogeneity, and drug resistance, while immunological knowledge has provided a framework for apprehending mechanisms that naturally limit tumor growth. Consolidating insights from these two different scientific fields may further help to explain phenomena which are still poorly understood.
As discussed in this opinion article, we propose that pluripotency, immortality, and an apparently unlimited potential to undergo cell divisions may be insufficient criteria for distinguishing CSCs from other cancer cells. In the tumor context, partly differentiated cells may also acquire these properties through mutations or de-differentiation processes. However, the unique immunological properties of stem cells and CSCs may help to further discriminate these cells from the tumor bulk (Table 1). Experimental evidence supporting this novel perspective may be drawn from model-dependent differences regarding the respective frequencies of tumor-initiating cells. This may best be explained by considering the different levels of immunogenicity coexisting within the same tumor. In addition, CSCs excellently complement current models of tumor immune escape. They can be seen as highly resistant and long-lived units of Darwinian selection, which accumulate mutations and undergo immunoediting over decades. Thus, they can either remain dormant or represent an almost invincible backbone of tumorigenesis which links “the three e’s of cancer immunoediting”: elimination, equilibrium, and escape, i.e., tumor recurrence.
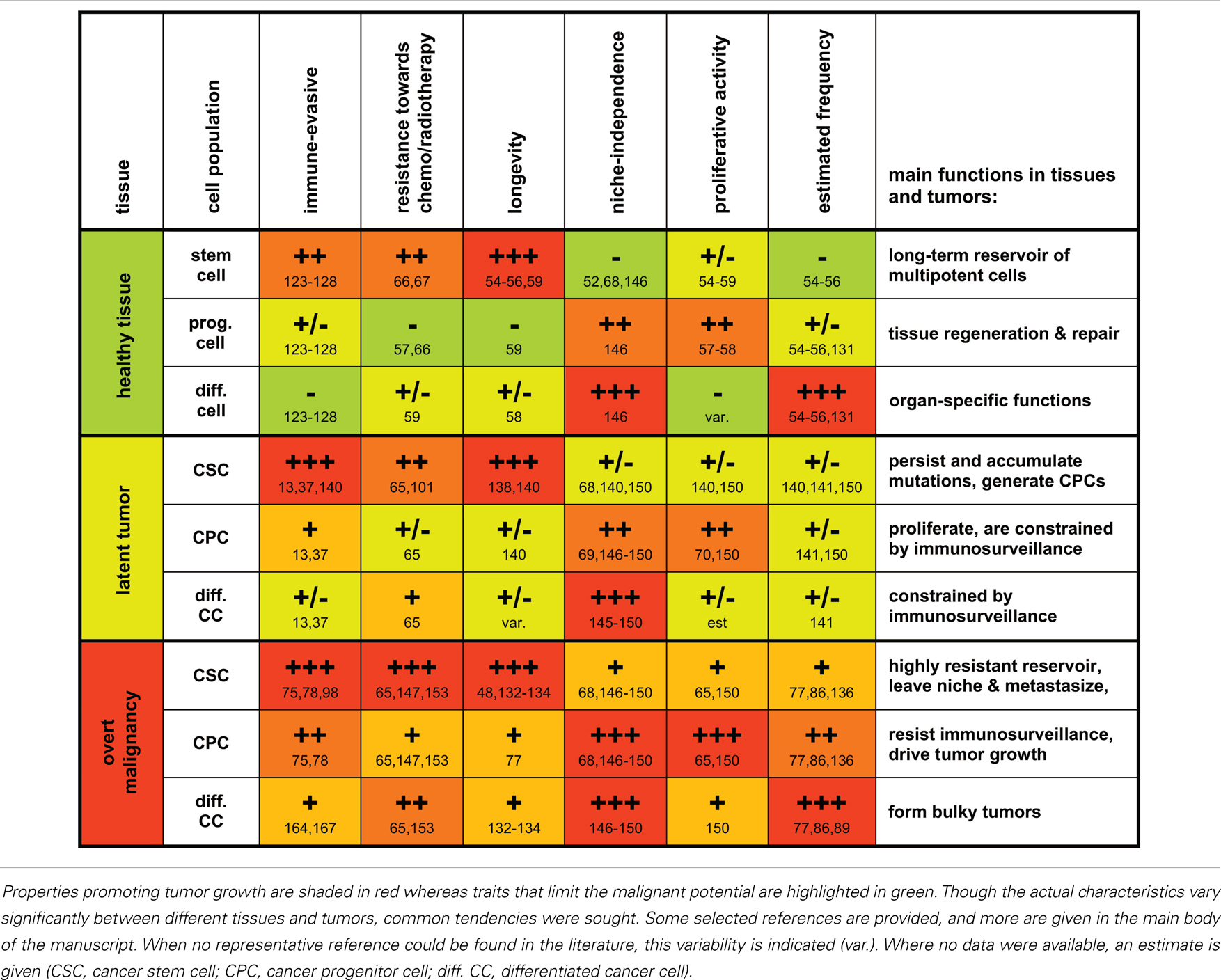
Table 1. A grossly schematic overview summarizing characteristic properties and functions of stem cells, progenitor cells, and differentiated cells and their malignant counterparts.
Yet what are the clinical implications if CSCs are not only highly resistant to chemo- and radiotherapy (153) but also to anti-tumor immune responses? Strategies to specifically target CSCs for elimination (154, 155) or to induce differentiation (156) would obviously be most desirable. However, none of the current approaches shows a high selectivity for CSCs over non-malignant stem cells. Therefore, therapies that efficiently and selectively eliminate CSCs are still far from clinical application. Still, accepting that we are currently unable to eradicate CSCs is far from accepting defeat in the war on cancer. Instead, by accepting these limitations one can rather prioritize strategies, which are more likely to work. These include, on the one hand, the indirect targeting of CSC via the tumor stroma, which represents a niche for CSCs (157–160). On the other hand, containment and control of CSCs to maintain tumors in the equilibrium phase may be a realistic clinical aim. This, however, depends on the incessant functionality of the immune system (161). Beneficial effects of radio- or chemotherapy, which can destroy some tumor cells (but presumably not the CSC) may thus largely depend on immune responses induced by provision of danger signals and by liberation of antigens from damaged tumor cells (37, 162, 163). Strengthening of cancer immunosurveillance by vaccination strategies or by directly activating the immune system (164–166) may support such a response and thus be effective in preventing tumor recurrence over prolonged periods of time (167, 168). Adjuvant immunotherapy might hence be advisable when a successful-appearing treatment has reduced overt cancer to minimal residual disease (which likely represents an immunologically sustained equilibrium). Overaggressive treatment modalities are, however, strongly immunosuppressive and may be detrimental. In fact, a comparatively mild metronomic chemotherapy may often be more effective (169, 170) than application of the maximum tolerated dose, which still fails to eliminate the seed of the evil but enables the outgrowth of more differentiated malignant cells. Assuming that CSCs represent moving targets, which need to be controlled throughout the whole life, we need to learn how the highly dynamic and adaptable forces of our immune system can best be used in the war on cancer. Fortunately, first studies are showing the potential of cancer immunotherapy in clinical reality.
Last but not least, an improved understanding of the mechanisms which enable either tumor control or escape may help to devise suitable therapeutic strategies. We thus hope that the still speculative, but hopefully coherent conceptual framework outlined in this brief article will stimulate further research in “CSC immunology.” In the best case, a mutual exchange of ideas could not only inspire both the CSC and the tumor immunology community, but eventually also lead to some benefit for patients who depend on a wisely chosen therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Valentin S. Bruttel was supported by a grant of the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fimmu.2014.00360/abstract
References
1. Lodish HBA, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, et al. Molecular Biology of the Cell. 5th ed. New York, NY: WH Freeman (2004).
2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9
4. Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol (2009) 9(8):568–80. doi:10.1038/nri2604
5. Haen SP, Rammensee HG. The repertoire of human tumor-associated epitopes – identification and selection of antigens and their application in clinical trials. Curr Opin Immunol (2013) 25(2):277–83. doi:10.1016/j.coi.2013.03.007
6. Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature (2010) 463(7278):184–90. doi:10.1038/nature08629
7. Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature (2010) 465(7297):473–7. doi:10.1038/nature09004
8. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature (2010) 463(7278):191–6. doi:10.1038/nature08658
9. Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, et al. Epitope landscape in breast and colorectal cancer. Cancer Res (2008) 68(3):889–92. doi:10.1158/0008-5472.CAN-07-3095
10. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol (2011) 29:235–71. doi:10.1146/annurev-immunol-031210-101324
11. Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol (2006) 6(10):715–27. doi:10.1038/nri1936
12. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410(6832):1107–11. doi:10.1038/35074122
13. Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature (2007) 450(7171):903–7. doi:10.1038/nature06309
14. van den Broek ME, Kagi D, Ossendorp F, Toes R, Vamvakas S, Lutz WK, et al. Decreased tumor surveillance in perforin-deficient mice. J Exp Med (1996) 184(5):1781–90. doi:10.1084/jem.184.5.1781
15. Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med (2000) 192(5):755–60. doi:10.1084/jem.192.5.755
16. Takeda K, Smyth MJ, Cretney E, Hayakawa Y, Yamaguchi N, Yagita H, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in NK cell-mediated and IFN-gamma-dependent suppression of subcutaneous tumor growth. Cell Immunol (2001) 214(2):194–200. doi:10.1006/cimm.2001.1896
17. Takeda K, Smyth MJ, Cretney E, Hayakawa Y, Kayagaki N, Yagita H, et al. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med (2002) 195(2):161–9. doi:10.1084/jem.20011171
18. Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol (2002) 168(3):1356–61. doi:10.4049/jimmunol.168.3.1356
19. Seki N, Hayakawa Y, Brooks AD, Wine J, Wiltrout RH, Yagita H, et al. Tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis is an important endogenous mechanism for resistance to liver metastases in murine renal cancer. Cancer Res (2003) 63(1):207–13.
20. Finnberg N, Klein-Szanto AJ, El-Deiry WS. TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J Clin Invest (2008) 118(1):111–23. doi:10.1172/JCI29900
21. Nepal RM, Zaheen A, Basit W, Li L, Berger SA, Martin A. AID and RAG1 do not contribute to lymphomagenesis in Emu c-myc transgenic mice. Oncogene (2008) 27(34):4752–6. doi:10.1038/onc.2008.111
22. Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, et al. Regulation of cutaneous malignancy by gammadelta T cells. Science (2001) 294(5542):605–9. doi:10.1126/science.1063916
23. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity (2008) 28(4):571–80. doi:10.1016/j.immuni.2008.02.016
24. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
25. Ulrich C, Schmook T, Sachse MM, Sterry W, Stockfleth E. Comparative epidemiology and pathogenic factors for nonmelanoma skin cancer in organ transplant patients. Dermatol Surg (2004) 30(4 Pt 2):622–7. doi:10.1097/00042728-200404020-00009
26. Tremblay F, Fernandes M, Habbab F, deB Edwardes MD, Loertscher R, Meterissian S. Malignancy after renal transplantation: incidence and role of type of immunosuppression. Ann Surg Oncol (2002) 9(8):785–8. doi:10.1007/BF02574501
27. Andres A. Cancer incidence after immunosuppressive treatment following kidney transplantation. Crit Rev Oncol Hematol (2005) 56(1):71–85. doi:10.1016/j.critrevonc.2004.11.010
28. Vial T, Descotes J. Immunosuppressive drugs and cancer. Toxicology (2003) 185(3):229–40. doi:10.1016/S0300-483X(02)00612-1
29. Dal Maso L, Serraino D, Franceschi S. Epidemiology of AIDS-related tumours in developed and developing countries. Eur J Cancer (2001) 37(10):1188–201. doi:10.1016/S0959-8049(01)00120-4
30. Goedert JJ. The epidemiology of acquired immunodeficiency syndrome malignancies. Semin Oncol (2000) 27(4):390–401.
31. Bower M, Palmieri C, Dhillon T. AIDS-related malignancies: changing epidemiology and the impact of highly active antiretroviral therapy. Curr Opin Infect Dis (2006) 19(1):14–9. doi:10.1097/01.qco.0000200295.30285.13
32. de Magalhaes JP. How ageing processes influence cancer. Nat Rev Cancer (2013) 13(5):357–65. doi:10.1038/nrc3497
33. Malaguarnera L, Cristaldi E, Malaguarnera M. The role of immunity in elderly cancer. Crit Rev Oncol Hematol (2010) 74(1):40–60. doi:10.1016/j.critrevonc.2009.06.002
34. Fulop T, Kotb R, Fortin CF, Pawelec G, de Angelis F, Larbi A. Potential role of immunosenescence in cancer development. Ann N Y Acad Sci (2010) 1197:158–65. doi:10.1111/j.1749-6632.2009.05370.x
35. Derhovanessian E, Solana R, Larbi A, Pawelec G. Immunity, ageing and cancer. Immun Ageing (2008) 5:11. doi:10.1186/1742-4933-5-11
36. Schreiber K, Arina A, Engels B, Spiotto MT, Sidney J, Sette A, et al. Spleen cells from young but not old immunized mice eradicate large established cancers. Clin Cancer Res (2012) 18(9):2526–33. doi:10.1158/1078-0432.CCR-12-0127
37. Willimsky G, Czeh M, Loddenkemper C, Gellermann J, Schmidt K, Wust P, et al. Immunogenicity of premalignant lesions is the primary cause of general cytotoxic T lymphocyte unresponsiveness. J Exp Med (2008) 205(7):1687–700. doi:10.1084/jem.20072016
38. Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, Honda S, et al. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J Exp Med (2008) 205(13):2959–64. doi:10.1084/jem.20081611
39. Elboim M, Gazit R, Gur C, Ghadially H, Betser-Cohen G, Mandelboim O. Tumor immunoediting by NKp46. J Immunol (2010) 184(10):5637–44. doi:10.4049/jimmunol.0901644
40. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature (2012) 482(7385):400–4. doi:10.1038/nature10755
41. DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature (2012) 482(7385):405–9. doi:10.1038/nature10803
42. Campoli M, Ferrone S. Tumor escape mechanisms: potential role of soluble HLA antigens and NK cells activating ligands. Tissue Antigens (2008) 72(4):321–34. doi:10.1111/j.1399-0039.2008.01106.x
43. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity (2004) 21(2):137–48. doi:10.1016/j.immuni.2004.07.017
44. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology (2007) 121(1):1–14. doi:10.1111/j.1365-2567.2007.02587.x
45. Reiman JM, Kmieciak M, Manjili MH, Knutson KL. Tumor immunoediting and immunosculpting pathways to cancer progression. Semin Cancer Biol (2007) 17(4):275–87. doi:10.1016/j.semcancer.2007.06.009
46. Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, et al. Generation of a functional mammary gland from a single stem cell. Nature (2006) 439(7072):84–8. doi:10.1038/nature04372
47. Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, et al. Purification and unique properties of mammary epithelial stem cells. Nature (2006) 439(7079):993–7. doi:10.1038/nature04496
48. Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell (2005) 121(6):823–35. doi:10.1016/j.cell.2005.03.032
49. Lawson DA, Xin L, Lukacs RU, Cheng D, Witte ON. Isolation and functional characterization of murine prostate stem cells. Proc Natl Acad Sci U S A (2007) 104(1):181–6. doi:10.1073/pnas.0609684104
50. Collins AT, Habib FK, Maitland NJ, Neal DE. Identification and isolation of human prostate epithelial stem cells based on alpha(2)beta(1)-integrin expression. J Cell Sci (2001) 114(Pt 21):3865–72.
51. Richardson GD, Robson CN, Lang SH, Neal DE, Maitland NJ, Collins AT. CD133, a novel marker for human prostatic epithelial stem cells. J Cell Sci (2004) 117(Pt 16):3539–45. doi:10.1242/jcs.01222
52. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature (2007) 449(7165):1003–7. doi:10.1038/nature06196
53. Winton DJ, Ponder BA. Stem-cell organization in mouse small intestine. Proc Biol Sci (1990) 241(1300):13–8. doi:10.1098/rspb.1990.0059
54. Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol (2009) 10(3):207–17. doi:10.1038/nrm2636
55. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature (2001) 414(6859):105–11. doi:10.1038/35102167
56. Orkin SH. Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet (2000) 1(1):57–64. doi:10.1038/35049577
57. Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med (2005) 202(11):1599–611. doi:10.1084/jem.20050967
58. Weissman IL. Stem cells: units of development, units of regeneration, and units in evolution. Cell (2000) 100(1):157–68. doi:10.1016/S0092-8674(00)81692-X
59. Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell (2008) 135(6):1118–29. doi:10.1016/j.cell.2008.10.048
60. Nowell PC. The clonal evolution of tumor cell populations. Science (1976) 194(4260):23–8. doi:10.1126/science.959840
61. Ashley DJ. The two “hit” and multiple “hit” theories of carcinogenesis. Br J Cancer (1969) 23(2):313–28. doi:10.1038/bjc.1969.41
62. Nordling CO. A new theory on cancer-inducing mechanism. Br J Cancer (1953) 7(1):68–72. doi:10.1038/bjc.1953.8
63. Kleinsmith LJ, Pierce GB Jr. Multipotentiality of single embryonal carcinoma cells. Cancer Res (1964) 24:1544–51.
64. Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med (2001) 7(9):1028–34. doi:10.1038/nm0901-1028
65. Shlush LI, Chapal-Ilani N, Adar R, Pery N, Maruvka Y, Spiro A, et al. Cell lineage analysis of acute leukemia relapse uncovers the role of replication-rate heterogeneity and miscrosatellite instability. Blood (2012) 120(3):603–12. doi:10.1182/blood-2011-10-388629
66. Chaudhary PM, Roninson IB. Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell (1991) 66(1):85–94. doi:10.1016/0092-8674(91)90141-K
67. Kooreman NG, Wu JC. Tumorigenicity of pluripotent stem cells: biological insights from molecular imaging. J R Soc Interface (2010) 7(Suppl 6):S753–63. doi:10.1098/rsif.2010.0353.focus
68. Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res (2006) 66(9):4553–7. doi:10.1158/0008-5472.CAN-05-3986
69. Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med (2009) 15(8):907–13. doi:10.1038/nm.2000
70. Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell (2010) 7(3):403–17. doi:10.1016/j.stem.2010.07.010
71. Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science (2010) 329(5991):568–71. doi:10.1126/science.1189992
72. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells – perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res (2006) 66(19):9339–44. doi:10.1158/0008-5472.CAN-06-3126
73. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126(4):663–76. doi:10.1016/j.cell.2006.07.024
74. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A (2011) 108(19):7950–5. doi:10.1073/pnas.1102454108
75. Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res (2009) 69(7):2887–95. doi:10.1158/0008-5472.CAN-08-3343
76. Sharif A, Legendre P, Prevot V, Allet C, Romao L, Studler JM, et al. Transforming growth factor alpha promotes sequential conversion of mature astrocytes into neural progenitors and stem cells. Oncogene (2007) 26(19):2695–706. doi:10.1038/sj.onc.1210071
77. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell (2011) 146(4):633–44. doi:10.1016/j.cell.2011.07.026
78. Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature (2012) 490(7420):412–6. doi:10.1038/nature11538
79. Fantozzi A, Gruber DC, Pisarsky L, Heck C, Kunita A, Yilmaz M, et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res (2014) 74(5):1566–75. doi:10.1158/0008-5472.CAN-13-1641
80. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi:10.1038/nrm3758
81. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science (2013) 342(6159):1234850. doi:10.1126/science.1234850
82. Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res (2005) 65(20):9328–37. doi:10.1158/0008-5472.CAN-05-1343
83. Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer (2007) 43(5):935–46. doi:10.1016/j.ejca.2007.01.017
84. Keshet GI, Goldstein I, Itzhaki O, Cesarkas K, Shenhav L, Yakirevitch A, et al. MDR1 expression identifies human melanoma stem cells. Biochem Biophys Res Commun (2008) 368(4):930–6. doi:10.1016/j.bbrc.2008.02.022
85. Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature (2010) 466(7302):133–7. doi:10.1038/nature09161
86. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med (2009) 15(9):1010–2. doi:10.1038/nm0909-1010
87. Eaves CJ. Cancer stem cells: here, there, everywhere? Nature (2008) 456(7222):581–2. doi:10.1038/456581a
88. Passegue E, Rafii S, Herlyn M. Cancer stem cells are everywhere. Nat Med (2009) 15(1):23. doi:10.1038/nm0109-23
89. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature (2008) 456(7222):593–8. doi:10.1038/nature07567
90. Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell (2010) 18(5):510–23. doi:10.1016/j.ccr.2010.10.012
91. Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med (2013) 2(1):3. doi:10.1186/2001-1326-2-3
92. Ni J, Cozzi P, Hao J, Duan W, Graham P, Kearsley J, et al. Cancer stem cells in prostate cancer chemoresistance. Curr Cancer Drug Targets (2014) 14(3):225–40. doi:10.2174/1568009614666140328152459
93. Hamai A, Meslin F, Benlalam H, Jalil A, Mehrpour M, Faure F, et al. ICAM-1 has a critical role in the regulation of metastatic melanoma tumor susceptibility to CTL lysis by interfering with PI3K/AKT pathway. Cancer Res (2008) 68(23):9854–64. doi:10.1158/0008-5472.CAN-08-0719
94. Knuefermann C, Lu Y, Liu B, Jin W, Liang K, Wu L, et al. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene (2003) 22(21):3205–12. doi:10.1038/sj.onc.1206394
95. Seliger B, Kiessling R. The two sides of HER2/neu: immune escape versus surveillance. Trends Mol Med (2013) 19(11):677–84. doi:10.1016/j.molmed.2013.08.003
96. Mimura K, Shiraishi K, Mueller A, Izawa S, Kua LF, So J, et al. The MAPK pathway is a predominant regulator of HLA-A expression in esophageal and gastric cancer. J Immunol (2013) 191(12):6261–72. doi:10.4049/jimmunol.1301597
97. Ithimakin S, Day KC, Malik F, Zen Q, Dawsey SJ, Bersano-Begey TF, et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: implications for efficacy of adjuvant trastuzumab. Cancer Res (2013) 73(5):1635–46. doi:10.1158/0008-5472.CAN-12-3349
98. Schatton T, Frank MH. Antitumor immunity and cancer stem cells. Ann N Y Acad Sci (2009) 1176:154–69. doi:10.1111/j.1749-6632.2009.04568.x
99. Reim F, Dombrowski Y, Ritter C, Buttmann M, Häusler S, Ossadnik M, et al. Immunoselection of breast and ovarian cancer cells with trastuzumab and natural killer cells: selective escape of CD44high/CD24low/HER2low breast cancer stem cells. Cancer Res (2009) 69(20):8058–66. doi:10.1158/0008-5472.CAN-09-0834
100. Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res (2010) 16(3):800–13. doi:10.1158/1078-0432.CCR-09-2730
101. Schatton T, Frank MH. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res (2008) 21(1):39–55. doi:10.1111/j.1755-148X.2007.00427.x
102. Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell (2007) 11(3):259–73. doi:10.1016/j.ccr.2007.01.013
103. Lottaz C, Beier D, Meyer K, Kumar P, Hermann A, Schwarz J, et al. Transcriptional profiles of CD133+ and CD133− glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res (2010) 70(5):2030–40. doi:10.1158/0008-5472.CAN-09-1707
104. Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell (2007) 1(4):389–402. doi:10.1016/j.stem.2007.08.001
105. Olver S, Groves P, Buttigieg K, Morris ES, Janas ML, Kelso A, et al. Tumor-derived interleukin-4 reduces tumor clearance and deviates the cytokine and granzyme profile of tumor-induced CD8+ T cells. Cancer Res (2006) 66(1):571–80. doi:10.1158/0008-5472.CAN-05-1362
106. Kawasaki BT, Mistree T, Hurt EM, Kalathur M, Farrar WL. Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells. Biochem Biophys Res Commun (2007) 364(4):778–82. doi:10.1016/j.bbrc.2007.10.067
107. Kawasaki BT, Farrar WL. Cancer stem cells, CD200 and immunoevasion. Trends Immunol (2008) 29(10):464–8. doi:10.1016/j.it.2008.07.005
108. Kretz-Rommel A, Qin F, Dakappagari N, Ravey EP, McWhirter J, Oltean D, et al. CD200 expression on tumor cells suppresses antitumor immunity: new approaches to cancer immunotherapy. J Immunol (2007) 178(9):5595–605. doi:10.4049/jimmunol.178.9.5595
109. Maccalli C, Volonte A, Cimminiello C, Parmiani G. Immunology of cancer stem cells in solid tumours. A review. Eur J Cancer (2014) 50(3):649–55. doi:10.1016/j.ejca.2013.11.014
110. Drukker M, Katz G, Urbach A, Schuldiner M, Markel G, Itskovitz-Eldor J, et al. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc Natl Acad Sci U S A (2002) 99(15):9864–9. doi:10.1073/pnas.142298299
111. Favier B, LeMaoult J, Carosella ED. Functions of HLA-G in the immune system. Tissue Antigens (2007) 69(Suppl 1):150–2. doi:10.1111/j.1399-0039.2006.763_6.x
112. Verloes A, Van de Velde H, LeMaoult J, Mateizel I, Cauffman G, Horn PA, et al. HLA-G expression in human embryonic stem cells and preimplantation embryos. J Immunol (2011) 186(4):2663–71. doi:10.4049/jimmunol.1001081
113. Nasef A, Mathieu N, Chapel A, Frick J, Francois S, Mazurier C, et al. Immunosuppressive effects of mesenchymal stem cells: involvement of HLA-G. Transplantation (2007) 84(2):231–7. doi:10.1097/01.tp.0000267918.07906.08
114. Kroczek RA, Mages HW, Hutloff A. Emerging paradigms of T-cell co-stimulation. Curr Opin Immunol (2004) 16(3):321–7. doi:10.1016/j.coi.2004.03.002
115. Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, Andreini A, et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells (2006) 24(2):386–98. doi:10.1634/stemcells.2005-0008
116. Le Blanc K. Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy (2003) 5(6):485–9. doi:10.1080/14653240310003611
117. Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringden O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol (2003) 31(10):890–6. doi:10.1016/S0301-472X(03)00110-3
118. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood (2005) 105(4):1815–22. doi:10.1182/blood-2004-04-1559
119. Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol (2002) 23(3):144–50. doi:10.1016/S1471-4906(01)02154-8
120. Meisel R, Zibert A, Laryea M, Gobel U, Daubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood (2004) 103(12):4619–21. doi:10.1182/blood-2003-11-3909
121. Bai L, Lennon DP, Caplan AI, Dechant A, Hecker J, Kranso J, et al. Hepatocyte growth factor mediates mesenchymal stem cell-induced recovery in multiple sclerosis models. Nat Neurosci (2012) 15(6):862–70. doi:10.1038/nn.3109
122. Benkhoucha M, Santiago-Raber ML, Schneiter G, Chofflon M, Funakoshi H, Nakamura T, et al. Hepatocyte growth factor inhibits CNS autoimmunity by inducing tolerogenic dendritic cells and CD25+Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A (2010) 107(14):6424–9. doi:10.1073/pnas.0912437107
123. Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood (2003) 101(9):3722–9. doi:10.1182/blood-2002-07-2104
124. Maitra B, Szekely E, Gjini K, Laughlin MJ, Dennis J, Haynesworth SE, et al. Human mesenchymal stem cells support unrelated donor hematopoietic stem cells and suppress T-cell activation. Bone Marrow Transplant (2004) 33(6):597–604. doi:10.1038/sj.bmt.1704400
125. Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation (2003) 75(3):389–97. doi:10.1097/01.TP.0000045055.63901.A9
126. McDonald JW, Liu XZ, Qu Y, Liu S, Mickey SK, Turetsky D, et al. Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat Med (1999) 5(12):1410–2. doi:10.1038/70986
127. Menard C, Hagege AA, Agbulut O, Barro M, Morichetti MC, Brasselet C, et al. Transplantation of cardiac-committed mouse embryonic stem cells to infarcted sheep myocardium: a preclinical study. Lancet (2005) 366(9490):1005–12. doi:10.1016/S0140-6736(05)67380-1
128. Niemeyer P, Schonberger TS, Hahn J, Kasten P, Fellenberg J, Suedkamp N, et al. Xenogenic transplantation of human mesenchymal stem cells in a critical size defect of the sheep tibia for bone regeneration. Tissue Eng Part A (2010) 16(1):33–43. doi:10.1089/ten.TEA.2009.0190
129. Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet (2008) 371(9624):1579–86. doi:10.1016/S0140-6736(08)60690-X
130. Roemeling-van Rhijn M, Weimar W, Hoogduijn MJ. Mesenchymal stem cells: application for solid-organ transplantation. Curr Opin Organ Transplant (2012) 17(1):55–62. doi:10.1097/MOT.0b013e32834ee676
131. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol (2005) 174(10):6477–89. doi:10.4049/jimmunol.174.10.6477
132. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature (1994) 367(6464):645–8. doi:10.1038/367645a0
133. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A (2003) 100(7):3983–8. doi:10.1073/pnas.0530291100
134. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature (2004) 432(7015):396–401. doi:10.1038/nature03128
135. Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science (2007) 317(5836):337. doi:10.1126/science.1142596
136. Kruger JA, Kaplan CD, Luo Y, Zhou H, Markowitz D, Xiang R, et al. Characterization of stem cell-like cancer cells in immune-competent mice. Blood (2006) 108(12):3906–12. doi:10.1182/blood-2006-05-024687
137. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3(11):991–8. doi:10.1038/ni1102-991
138. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer (2007) 7(11):834–46. doi:10.1038/nrc2256
139. Li L, Xie T. Stem cell niche: structure and function. Annu Rev Cell Dev Biol (2005) 21:605–31. doi:10.1146/annurev.cellbio.21.012704.131525
140. Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature (2014) 506(7488):328–33. doi:10.1038/nature13038
141. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature (2005) 438(7069):820–7. doi:10.1038/nature04186
142. Sceneay J, Smyth MJ, Moller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev (2013) 32(3–4):449–64. doi:10.1007/s10555-013-9420-1
143. Mehling M, Simon P, Mittelbronn M, Meyermann R, Ferrone S, Weller M, et al. WHO grade associated downregulation of MHC class I antigen-processing machinery components in human astrocytomas: does it reflect a potential immune escape mechanism? Acta Neuropathol (2007) 114(2):111–9. doi:10.1007/s00401-007-0231-8
144. Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature (2007) 447(7143):482–6. doi:10.1038/nature05768
145. Gleason DF, Mellinger GT. Prediction of prognosis for prostatic adenocarcinoma by combined histological grading and clinical staging. J Urol (1974) 111(1):58–64.
146. Campbell C, Risueno RM, Salati S, Guezguez B, Bhatia M. Signal control of hematopoietic stem cell fate: Wnt, Notch, and Hedgehog as the usual suspects. Curr Opin Hematol (2008) 15(4):319–25. doi:10.1097/MOH.0b013e328303b9df
147. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol (2011) 8(2):97–106. doi:10.1038/nrclinonc.2010.196
148. Zardawi SJ, O’Toole SA, Sutherland RL, Musgrove EA. Dysregulation of Hedgehog, Wnt and Notch signalling pathways in breast cancer. Histol Histopathol (2009) 24(3):385–98.
149. Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature (2001) 411(6835):349–54. doi:10.1038/35077219
150. Li Q, Bohin N, Wen T, Ng V, Magee J, Chen SC, et al. Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature (2013) 504(7478):143–7. doi:10.1038/nature12830
151. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (2012) 483(7390):474–8. doi:10.1038/nature10860
152. Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer (2003) 3(2):89–101. doi:10.1038/nrc989
153. Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst (2006) 98(24):1777–85. doi:10.1093/jnci/djj495
154. Beier D, Rohrl S, Pillai DR, Schwarz S, Kunz-Schughart LA, Leukel P, et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res (2008) 68(14):5706–15. doi:10.1158/0008-5472.CAN-07-6878
155. Diessner J, Bruttel V, Stein R, Horn E, Häusler S, Dietl J, et al. Targeting of pre-existing and induced breast cancer stem cells with trastuzumab and trastuzumab emtansine (T-DM1). Cell Death Dis (2014) 5:e1149. doi:10.1038/cddis.2014.115
156. Morrison BJ, Schmidt CW, Lakhani SR, Reynolds BA, Lopez JA. Breast cancer stem cells: implications for therapy of breast cancer. Breast Cancer Res (2008) 10(4):210. doi:10.1186/bcr2111
157. Yi SY, Hao YB, Nan KJ, Fan TL. Cancer stem cells niche: a target for novel cancer therapeutics. Cancer Treat Rev (2013) 39(3):290–6. doi:10.1016/j.ctrv.2012.10.004
158. Borovski T, De Sousa EMF, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res (2011) 71(3):634–9. doi:10.1158/0008-5472.CAN-10-3220
159. Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature (2012) 481(7379):85–9. doi:10.1038/nature10694
160. Zhang B, Zhang Y, Bowerman NA, Schietinger A, Fu YX, Kranz DM, et al. Equilibrium between host and cancer caused by effector T cells killing tumor stroma. Cancer Res (2008) 68(5):1563–71. doi:10.1158/0008-5472.CAN-07-5324
161. Fridman WH, Teillaud JL, Sautes-Fridman C, Pages F, Galon J, Zucman-Rossi J, et al. The ultimate goal of curative anti-cancer therapies: inducing an adaptive anti-tumor immune response. Front Immunol (2011) 2:66. doi:10.3389/fimmu.2011.00066
162. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13(9):1050–9. doi:10.1038/nm1622
163. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol (2008) 8(1):59–73. doi:10.1038/nri2216
164. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
165. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
166. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
167. Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med (2012) 18(8):1254–61. doi:10.1038/nm.2883
168. Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer (2008) 8(5):351–60. doi:10.1038/nrc2373
169. Chen CS, Doloff JC, Waxman DJ. Intermittent metronomic drug schedule is essential for activating antitumor innate immunity and tumor xenograft regression. Neoplasia (2014) 16(1):84–96. doi:10.1593/neo.131910
Keywords: tumor-propagating cells, tumor immunology, tumor immunosurveillance, tumor dormancy, latency, tumor immune escape, cancer stem cells, cancer stem cell immunology
Citation: Bruttel VS and Wischhusen J (2014) Cancer stem cell immunology: key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 5:360. doi: 10.3389/fimmu.2014.00360
Received: 03 April 2014; Accepted: 13 July 2014;
Published online: 29 July 2014.
Edited by:
Salem Chouaib, Institut Gustave Roussy, FranceReviewed by:
Bozena Kaminska, Nencki Institute of Experimental Biology, PolandBenjamin Bonavida, University of California Los Angeles, USA
Copyright: © 2014 Bruttel and Wischhusen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jörg Wischhusen, Section for Experimental Tumor Immunology, Department of Obstetrics and Gynecology, School of Medicine, University of Würzburg, Josef-Schneider-Strasse 4, Würzburg 97080, Germany e-mail: wischhusen_J@ukw.de