A breath of fresh air on the mesenchyme: impact of impaired mesenchymal development on the pathogenesis of bronchopulmonary dysplasia
 Cho-Ming Chao
Cho-Ming Chao Elie El Agha2,3
Elie El Agha2,3
- 1Department of General Pediatrics and Neonatology, University Children’s Hospital Giessen, Giessen, Germany
- 2Department of Internal Medicine II, Universities of Giessen and Marburg Lung Center, Giessen, Germany
- 3Member of the German Center for Lung Research (DZL), Giessen, Germany
- 4Division of Neonatology, Department of Pediatrics, Columbia University, New York, NY, USA
- 5Division of Newborn Medicine, Department of Pediatrics, Children’s Hospital Los Angeles, University of Southern California, Los Angeles, CA, USA
- 6Saban Research Institute, Childrens Hospital Los Angeles, University of Southern California, Los Angeles, CA, USA
- 7Kazan Federal University, Kazan, Russia
The early mouse embryonic lung, with its robust and apparently reproducible branching pattern, has always fascinated developmental biologists. They have extensively used this embryonic organ to decipher the role of mammalian orthologs of Drosophila genes in controlling the process of branching morphogenesis. During the early pseudoglandular stage, the embryonic lung is formed mostly of tubes that keep on branching. As the branching takes place, progenitor cells located in niches are also amplified and progressively differentiate along the proximo-distal and dorso-ventral axes of the lung. Such elaborate processes require coordinated interactions between signaling molecules arising from and acting on four functional domains: the epithelium, the endothelium, the mesenchyme, and the mesothelium. These interactions, quite well characterized in a relatively simple lung tubular structure remain elusive in the successive developmental and postnatal phases of lung development. In particular, a better understanding of the process underlying the formation of secondary septa, key structural units characteristic of the alveologenesis phase, is still missing. This structure is critical for the formation of a mature lung as it allows the subdivision of saccules in the early neonatal lung into alveoli, thereby considerably expanding the respiratory surface. Interruption of alveologenesis in preterm neonates underlies the pathogenesis of chronic neonatal lung disease known as bronchopulmonary dysplasia. De novo formation of secondary septae appears also to be the limiting factor for lung regeneration in human patients with emphysema. In this review, we will therefore focus on what is known in terms of interactions between the different lung compartments and discuss the current understanding of mesenchymal cell lineage formation in the lung, focusing on secondary septae formation.
Bronchopulmonary Dysplasia is Characterized by Impaired Alveologenesis
Bronchopulmonary dysplasia (BPD) is a chronic lung disease of prematurely born infants and remains a leading cause of morbidity and mortality. Currently, there is no curative therapy available. Based on the severity-based definition of BPD (inclusion of infants with mild BPD) 68% of premature infants born with a gestational age (GA) ≤28 weeks develop BPD (1–3). The risk of developing BPD correlates inversely with the GA and birth weight (BW) (4). Since premature infants (24–28 weeks of gestation) are born with a lung, which is in the canalicular or saccular stages of development, the lung structure (characterized by thickened airspace walls and surfactant deficiency) is therefore not adequate to provide sufficient ventilation and gas exchange. Thus, mechanical ventilation and high-oxygen concentration are often necessary at birth. Barotrauma induced by mechanical ventilation as well as oxygen toxicity and inflammation are major contributing factors responsible for the pulmonary damages in the morphological and functional immature lung. In addition, some studies have suggested a strong genetic component in BPD (5). For example, using genome-wide association study, it has been shown that polymorphisms (SNPs) in MMP16 and SPOCK2 might be associated with BPD (6). Due to remarkable advances in the management and therapy (e.g., gentle ventilation, restricted oxygen supplementation, antenatal steroids, and exogenous surfactant use) survival rate for premature infants has increased over the last decades. These advances in treatment have changed the histological characteristics of what is now called the old BPD since it was first described by Northway in 1967. The “old” BPD was mostly an airway disease characterized by interstitial fibrosis and squamous metaplasia of airways. The prominent histological findings in the lungs of “new” BPD are simplification of alveolar formation (fewer and larger alveoli) and dysmorphic pulmonary microvasculature (7, 8). Pulmonary hypertension is also a common complication in infants with BPD, resulting in high mortality (9). According to these findings, the “new” BPD is considered as a consequence of the premature lung interrupted in its development by postnatal lung injury leading to the growth arrest of the lung in the canalicular/saccular phase of normal lung development. BPD, as a chronic lung disease, leads to long-term morbidity (e.g., pulmonary infection, neurodevelopmental impairment) affecting quality of life during childhood and in some severely affected patients even into adulthood. Treatment for BPD represents a considerable health care burden (10–12).
The mechanisms responsible for alveolar simplification in BPD remain understudied and poorly understood. However, autopsy samples from premature infants from pre- and post-surfactant era, who died from BPD consistently showed abnormalities in the mesenchyme (interstitial fibrosis and dysmorphic microvasculature). In the new BPD, there is clear evidence for decreased number of secondary septae, a derivative of the lung mesoderm. Furthermore, animal models mimicking the premature lung and the risk factors for BPD provide more evidence that indeed the mesenchyme plays a pivotal role in late lung development/alveologenesis and therefore in BPD. This review will summarize the current understanding of the impaired mesenchymal compartment of the BPD lungs, with a focus on mesenchymal–endothelial and mesenchymal–epithelial crosstalk known to contribute to disease pathogenesis.
Normal Lung Development in Human and Mouse
In human and mouse, the lung arises from two germ layers: the gut endoderm gives rise to the lung epithelium and the splanchnic mesoderm is the origin of the lung mesenchyme. The human lung consists of three lobes on the right and two lobes on the left side; in mice four lobes form on the right (cranial, medial, caudal, and accessory lobe) and one on the left. Compared to the 12 airway generations observed in mice, human lungs comprise 23 airway generations.
In humans, lung development arises from the laryngo-tracheal groove and starts at week 4 of gestation as an outgrowth from the ventral wall of the caudal primitive foregut. During the further growth of the lung, the prospective trachea separates from the foregut by the formation of the so-called tracheo-esophageal septum. At the most distal part of the tracheal tube, two buds that will form the right and left primary bronchial buds appear. These primary buds are further ramified to form three secondary bronchial buds on the right and two secondary bronchial buds on the left side. These buds are the origin of the five lobes in the mature lung (13).
In mice, at embryonic day 8 (E8), signaling molecules and growth factors (e.g., Fgf1, Fgf2) emanate from the cardiac mesoderm and specify the prospective lung field in the primitive foregut endoderm, which is positive for the transcription factor Nkx2.1 (or Ttf1). These pre-lung epithelial progenitor cells represent the earliest and most likely the most pluripotent epithelial cells for the lung. At E9.5, the ventral foregut endoderm evaginates and elongates caudally dividing into two buds that form the prospective trachea and the first generation of bronchi (main bronchi). The process of lung development (human and mouse) has been divided into four distinct histological phases: pseudoglandular, canalicular, saccular, and alveolar (Figure 1).
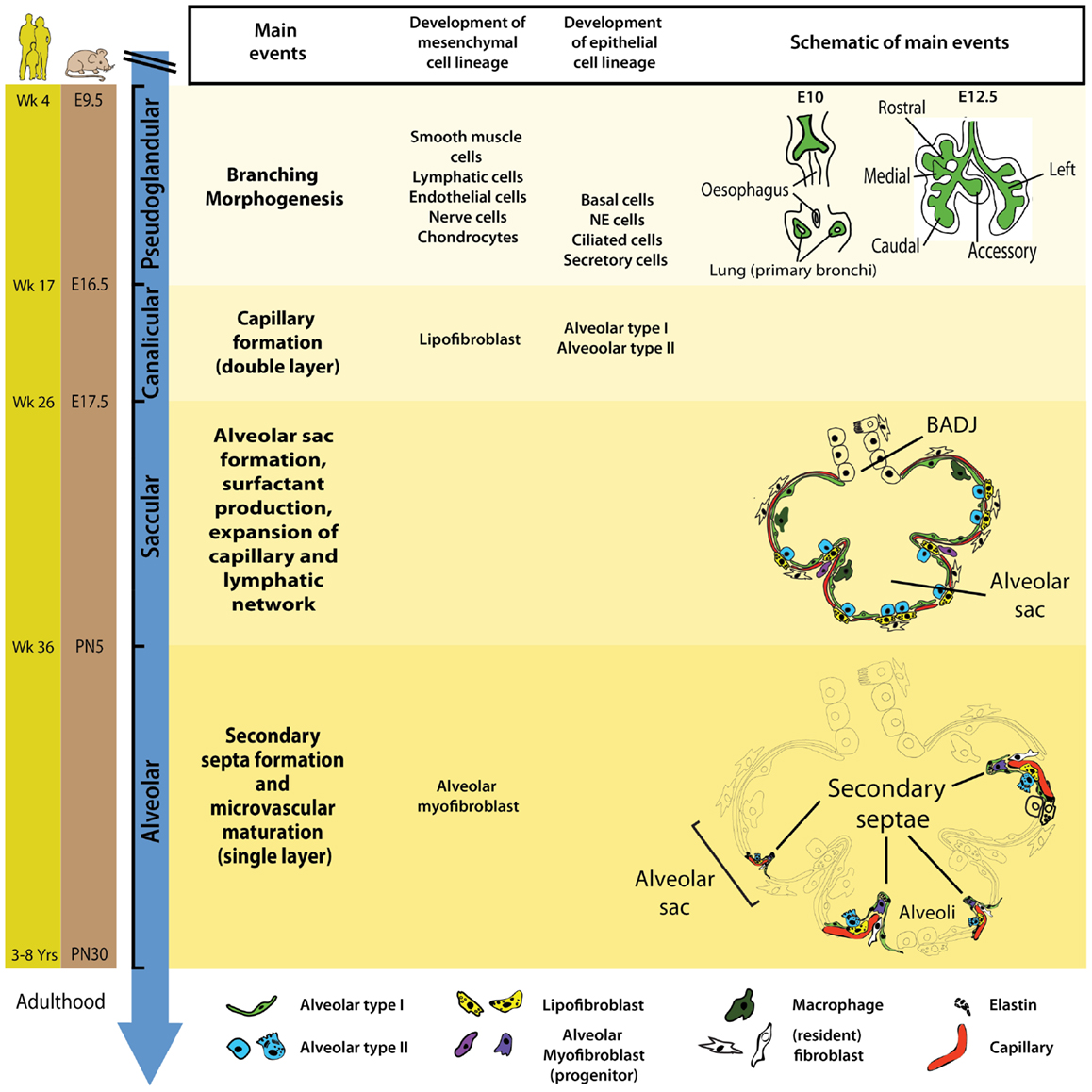
Figure 1. Timeline and stages of lung development in mice and humans. Lung development starts with the specification of the lung domain in the foregut endoderm followed by the formation of primary lung buds. These buds will later give rise to the respiratory tree via the process of branching morphogenesis. The latter is a characteristic of the pseudoglandular stage of lung development. Most epithelial and mesenchymal cell types start to form during the pseudoglandular stage. The canalicular stage is characterized by blood capillary formation and the appearance of AECI/II. During the saccular stage, primitive alveoli (sac-like structures) start to form and this is accompanied by surfactant production and the expansion of capillary and lymphatic networks. The alveolar stage of lung developments starts in utero in humans whereas in mice, it starts postnatally. Wk, week; E, embryonic; PN, postnatal; NE, neuroendocrine.
During the pseudoglandular stage (human: week 4–17; mouse: E9.5–E16.5), the process of branching morphogenesis generates the basic tree-like structure of the lung including the conducting airways and the numerous terminal bronchioles surrounded by thick mesenchyme. Concurrently, epithelial cell progenitors undergo differentiation to give rise to basal, neuroendocrine, ciliated, and secretory cells. The mesodermal lung compartment serves as progenitors for the smooth muscle, lymphatic, endothelial, nerve, and chondrocytic cells.
In the subsequent canalicular stage (human: week 17–26; mouse: E16.5–E17.5), the lung undergoes further subdivision of the respiratory bronchioles accompanied by thinning of the surrounding mesenchyme and the massive formation of capillaries. For the first time during development, a primitive respiratory epithelium competent of gas exchange is formed by differentiation of distal lung epithelial progenitors. Recently, it has been shown that type I and type II alveolar epithelial cells (AEC I and II) emerge from a common alveolar bipotential progenitor (14). In mice, interstitial fibroblasts containing cytoplasmic lipid droplets (so called lipofibroblast, LIF) emerge in the mesenchyme. Additionally, this is the earliest time point of pregnancy (23–24 weeks of gestation) where a preterm infant can be born with a chance to survive. Those who died from BPD showed pathologic characteristics of the lung (interstitial fibrosis and dysmorphic microvasculature) similar to the morphology of the immature lung at this developmental stage thus reinforcing the concept that BPD results from interruption of normal lung development by deleterious environmental events. The introduction of antenatal steroids treatment and exogenous surfactant supplementation drastically increased survival of premature infants born at this stage (15).
The saccular stage of lung development occurs approximately between 26 and 36 weeks of gestation (mouse: E17.5–PN5). This stage is characterized by the formation of alveolar sacs, surfactant production, and thinning of the mesenchyme to facilitate gas exchange. Kresch demonstrated that the thinning of the mesenchyme results from apoptosis of mesenchymal cells (16). Furthermore, the capillary and lymphatic networks also expand in the saccular stage of lung development.
The last stage of lung development is termed alveolar stage (human: ~36 weeks to 8 years; mouse: PN5–PN30). During this stage, the alveolar surface area increases massively at the expense of the mesenchyme through subdividing the alveolar sacs (also called primitive alveoli) into mature alveoli by a process termed alveolarization (or alveologenesis) (Figure 2). This process starts with the deposition of elastin in primary septae (wall of alveolar sacs) and subsequently secondary septae emerge at the place of elastin and elongate toward the alveolar sac airspace to subdivide it into the smallest respiratory units of the lung – the mature alveoli. Importantly, concomitant with this process, primary septae, still containing a double layer of capillaries, become thinner and a single capillary network emerges allowing more efficient gas exchange (microvascular maturation). The bulk of alveolarization takes place during the first 6 months after birth in humans (mouse: PN5–PN15) (17). The alveolar myofibroblast (MYF), localized in the mesenchyme at the tip of the emerging secondary septae, is the cell responsible for secondary septae formation. A more detailed description of this mesenchymal cell lineage will be provided in the following sections.
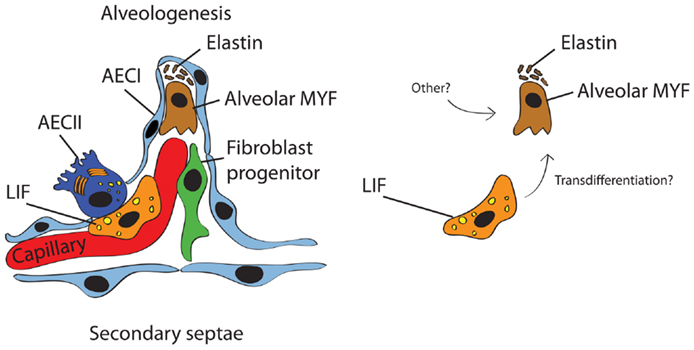
Figure 2. Schematic representation of the secondary septum during alveologenesis. Most of the alveolar surface is occupied by AECI (gas exchange) whereas a minor surface is occupied by AECII (surfactant production). The alveolar wall consists of the blood capillary, LIF, resident fibroblast progenitor, alveolar MYF, and ECM (mostly elastin). It has been proposed that alveolar MYF can originate from LIF (right panel) but this concept needs further validation.
In summary, the lung is a complex ramified organ that develops through continuous and elaborate interaction among the epithelium, mesenchyme, mesothelium, and endothelium. During this process, an intricate signaling network controls the amplification, proliferation, migration, and differentiation of diverse progenitor cells to populate these different compartments. Importantly, most of the epithelial and mesenchymal cell types in the lung are formed during the late pseudoglandular stage (E13.5–E16.6). This means that any deleterious factors present prenatally (such as inflammations due to chorioamnionitis) or postnatally (such as barotrauma injury and subsequent inflammation due to oxygen or mechanical ventilation), interfering with normal lung development at that time, could lead to impaired pulmonary function postnatally. Since preterm infants who die from BPD commonly display abnormal mesenchyme, a better understanding of aberrant signaling pathways in the lung mesenchyme of BPD lungs is important for improving the existing, and may facilitate the development of new preventive and curative therapies. In the next section, the current knowledge, mostly obtained from animal models of BPD, about abnormalities occurring in the lung mesenchyme will be reviewed.
The Embryonic Lung Mesenchyme
During the pseudoglandular stage of lung development (~E13.5), the distal lung bud is composed of three morphologically distinguishable layers: the mesothelium (outer layer), the mesenchyme (middle layer), and the epithelium (inner layer). The mesenchyme can be further divided into two domains, the submesothelial mesenchyme (SMM) and the subepithelial mesenchyme (SEM). Whereas mesenchymal cells constituting the SEM display high density and circumferential orientation, those of the SMM display low density and organization. Lineage-tracing experiments have identified markers for some mesenchymal progenitors such as Wnt2/Gli1/Isl1 (originating from the heart and invading the lung), Ret, Pdgfrα, Vegfr2, Prox1, and Fgf10 (18–20). Progenitors in these two compartments give rise to various cell types such as airway smooth muscle cells (ASMCs), vascular smooth muscle cells (VSMCs), resident mesenchymal stem cells (MSCs), LIFs, endothelial cells, chondrocytes, nerve cells, alveolar MYFs, lymphatic cells, and others. Mesenchymal progenitor cells are believed to play important roles not only in development but also in homeostasis and regeneration after injury.
Epithelial–Mesenchymal Crosstalk in Normal Lung Development and BPD
During development, the lung is formed through an elaborated epithelial–mesenchymal crosstalk that drives lung specification, budding, and branching. Signaling molecules like fibroblast growth factors (Fgf), Wnt (wingless and int), Sonic hedgehog (Shh), and bone morphogenetic proteins (Bmp) are key ligands initiating the pulmonary cell fate and specifying the early lung domain at the ventral foregut endoderm (21). So far, the most convincing evidence for epithelial–mesenchymal interactions during lung development came from recombination studies where distal lung mesenchyme, grafted on the tracheal epithelium led to ectopic budding accompanied by expression of surfactant protein C as a distal epithelial marker (22–24).
The mammalian Fgf family consists of 22 members subdivided in 7 subfamilies, based on phylogenetic as well as gene loci analyses (25). Fgfs acts in a paracrine, endocrine, or intracrine fashion and have diverse biological activities during embryonic organogenesis. These growth factors act via seven main receptors (Fgfrs 1b, 1c, 2b, 2c, 3b, 3c, and 4), exhibiting different ligand-binding specificity. The Fgf receptors are encoded by four Fgfr genes (Fgfr1–Fgfr4), which undergo alternative splicing to produce the different isoforms. Each receptor comprises an extracellular ligand-binding domain with three immunoglobulin-like loops (D I, D II, D III), a transmembrane domain and an intracellular tyrosine kinase domain. Human diseases involving gain or loss of function mutations have been described. For example, loss of function of FGF3 causes deafness, heterozygous loss of function of FGF10 results in lacrimo-auriculo-dento-digital syndrome (LADD syndrome), FGF10 haploinsufficiency is also associated with chronic obstructive pulmonary disease and FGF23 gain of function leads to autosomal dominant hypophosphataemic rickets (26–29). During early (E12.5) embryonic mouse lung development, Fgf9 and Fgf10 have been shown to play an important role in branching morphogenesis and the associated differentiation of the epithelium and mesenchyme. Fgf9 is expressed in the mesothelium and the epithelium and acts through Fgfr2c- and Fgfr1c-expressing cells in the mesenchyme to maintain Fgf10 expression as well as mesenchymal progenitors proliferative and undifferentiated (30). It also can signal directly to the epithelium to promote epithelial branching by induction of Dkk1 expression and inhibition of Wnt signaling (31). Fgf10 is a diffusible key molecule orchestrating branching morphogenesis during early lung development in mice (32, 33) but the exact mechanism of action remains unknown. During the early pseudoglandular stage, Fgf10 is secreted by cells located adjacent to the mesothelium in the distal mesenchyme and signals in a paracrine manner mainly through fibroblast growth factor receptor 2-IIIb (Fgfr2b) expressed on epithelial cells. Fgf10 has a high affinity for heparan sulfate and is therefore unlikely to diffuse over a long distance. Instead, Fgf10 promotes outgrowth of the distal epithelium via a chemotactic mechanism. Several studies using transgenic mouse lines that display abnormal Fgf10/Fgfr2b signaling confirmed the importance of this pathway (Table 1). Fgf10 and Fgfr2b knockout pups display similar phenotypes. The mutant pups die shortly after birth due to lung agenesis and multiple organ agenesis/defects (salivary gland, limb, inner ear, teeth, skin, pancreas, kidney, thyroid, pituitary gland, mammary gland) (34–38).
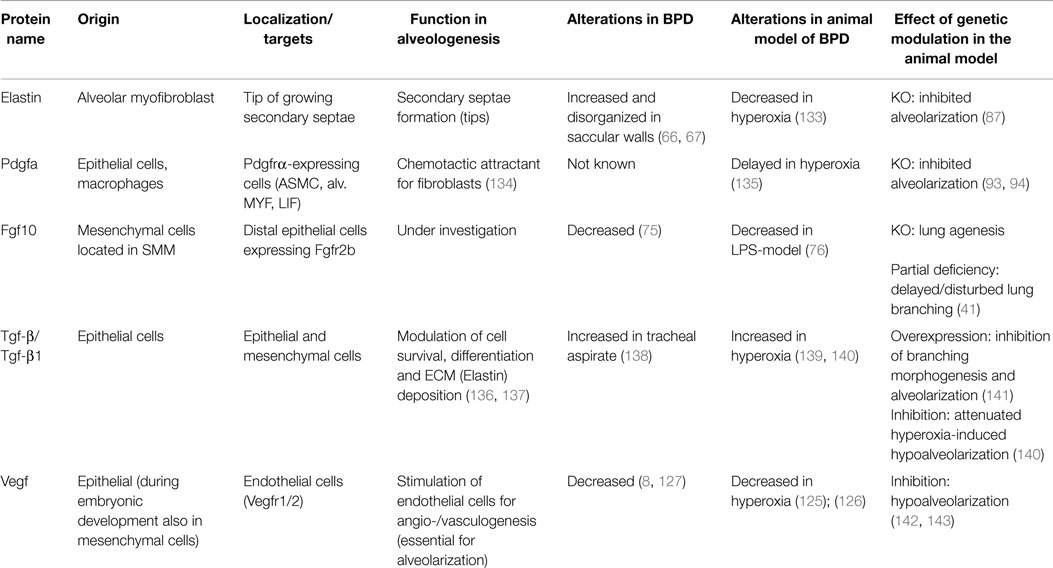
Table 1. Overview of proteins that are known to be involved in alveologenesis.
In order to identify epithelial-specific gene expressions mediated by recombinant human FGF10 during bud morphogenesis, Lu and colleagues (39) used mesenchyme-free epithelium in culture. By using microarray analysis, they identified a panel of transcriptional Fgf10 targets, which are associated with cell rearrangement, migration, inflammatory processes, lipid metabolism, cell cycle, and tumor invasion. Interestingly, the authors did not observe a remarkable induction of genes responsible for proliferation. Moreover, Fgf10 is proposed to control the angle of the mitotic spindle in distal epithelial cells during development. Thus, Tang et al. argued that Fgf10 signals via a Ras-regulated Erk1/2 signaling pathway to shape the lung tube (40). Fgf10 is also critical for the amplification of distal epithelial cell progenitors and for the formation of multiple mesenchymal lineages during lung development. Hypomorphic Fgf10lacZ/− pups expressing ~20% Fgf10 compared to wild type (WT) died within 24–48 h after birth due to lung defects, which included decreased branching, thickened primary septae, and vascular abnormalities with intrapulmonary hemorrhages. At the cellular level, Fgf10 deficiency led to decrease in Nkx2.1 and Sftpb-expressing cells, suggesting that adequate Fgf10 expression level is critical for the amplification of epithelial progenitors. Apart from the epithelium, constitutive decrease in Fgf10 expression also affects mesenchymal cell lineages as Pecam and αSma-positive cells are also diminished (41). Interestingly, recent experiments conducted in our lab to investigate the impact of Fgf10 levels on lung function demonstrate that even a 50% decrease in Fgf10 expression (Fgf10 heterozygous pups) leads to changes in the expression of genes relevant for lung development such as Epcam, Sftpc, Fgfr2b, Tgf-β, and Collagen. Additionally, Fgf10 heterozygous neonatal mice survive and do not display any obvious phenotypic differences compared to WT mice. However, when exposed to hyperoxia between PN0 and PN8 to trigger lung injury and mimic some of the clinical manifestations of BPD (impaired alveologenesis and inflammation), Fgf10-deficient pups display drastically increased mortality compared to WT controls. Further analysis indicates that under physiological conditions, Fgf10-deficient mice already show structural abnormalities during embryonic lung development supporting that Fgf10-deficient pups carry congenital defects. These findings suggest that Fgf10-deficient lung epithelium is more susceptible to oxygen toxicity and does not undergo normal repair after injury (Chao and Bellusci, in preparation). Additionally, recent studies suggest that Fgf10 may control basal cell density in the tracheal epithelium (42–45). This is not surprising as it has already been previously shown, by our group and others, that Fgf10 is part of the stem cell niche in the lung (20, 46, 47).
Wnt (Wnt2, Wnt2b) ligands are expressed in the mesenchyme and are important for lung domain specification of the foregut endoderm from E9.0 to E10.5. Wnt signaling is also essential for the proximo-distal patterning of the epithelium during embryonic lung development. Genetic deletion of Wnt2/2b or β-catenin leads to lung agenesis due to loss of Nkx2.1 (48, 49). Wnt2 null mice display lung hypoplasia and abnormal development of ASMCs (50). Furthermore, Mucenski and colleagues demonstrated, by using Spc-rtTA;tet(O)Cre double transgenic mice, that loss of function of β-catenin in the distal lung epithelium leads to the inhibition of distal airway formation (51). The authors showed an opposite phenotype by inducing gain of function of β-catenin signaling (52). The absence of Wnt7b results in a phenotype similar to Wnt2 null mice (53) but a combination of Wnt7b and Wnt2 loss of function leads to a more severe phenotype with decreased branching and abnormal distal endoderm patterning (54). The constitutive deletion of Wnt5a – a non-canonical Wnt ligand expressed in the mesenchyme and the epithelium – leads to increased proliferation of the mesenchyme and the distal epithelium as well as disrupted lung maturation (55). β-catenin inactivation in the mesenchyme (Dermo1-Cre line) leads to abnormal mesenchyme development with disrupted amplification of ASMC progenitors and defects in angioblast differentiation (56). Kumar and colleagues demonstrated by using a clonal cell labeling approach that ASMC progenitors are located exclusively at the tip mesenchyme and that mesenchymal Wnt signaling is able to prime the stalk mesenchyme to form an ASMC progenitor pool at the tip (57).
Bmp4 is dynamically expressed in the endoderm and in the mesenchyme during early embryonic lung development (E11.5). It is also expressed at the distal epithelial buds and has been shown to be an inhibitor of Fgf10-induced chemotaxis in the epithelium. Bmp4 controls intraepithelial crosstalk to form ASMCs. It has been shown that Fgf10 is able to upregulate Bmp4 mRNA expression. In vitro experiments demonstrated that exogenous recombinant human BMP4 inhibits Fgf10-induced bud outgrowth, providing evidence that Bmp4 is acting downstream of Fgf10 to inhibit its signaling cascade (58–61).
Other Fgf10 inhibitors are Sonic hedgehog (Shh) and Sprouty homolog 2 (Spry2), both expressed in the epithelium of the outgrowing buds. Shh is a secreted growth factor that acts through its mesenchymal receptor Patched (Ptc) to induce mesenchymal cell proliferation and differentiation. In E11.5 lung explants, exogenous recombinant SHH is able to induce expression of mesenchymal markers (Noggin, Acta2, Myosin) (19, 62). Spry2 is an intracellular inhibitor of receptor tyrosine kinase signaling (63, 64); (32). Using in vitro approaches, it has been shown that Spry2 reduction leads to increased epithelial branching and vice versa.
Apart from its important role in development, the mesenchyme is crucial in disease pathogenesis. Indeed, it has been reproducibly shown that the lung mesenchyme in preterm infants dying from BPD includes interstitial fibrosis and thickening with increased total collagen content (65–67). Similar findings were obtained in diverse animal models (rat, mice, baboon) recapitulating the conditions of preterm infants after birth (mechanical ventilation, oxygen supplementation, exogenous surfactant) leading to a human BPD-like phenotype (68–72). These pathological changes in the lung mesenchyme in BPD strengthen the concept that alveolarization depends on an intact and normally developed mesenchyme. Several studies using animal models of BPD to identify molecules located in the altered lung mesenchyme contributed to our understanding of disease pathogenesis. Some of them will be reviewed in the following section.
One of the major causes of BPD is believed to be inflammation. Inflammation is caused prenatally by chorioamnionitis and postnatally by mechanical stretch (ventilation), oxygen toxicity, as well as infection. Emerging evidence gained from in vitro and in vivo studies support this hypothesis (73–77). For example, it has been shown that lipopolysaccharides (LPS from Escherichia coli) inhibit branching morphogenesis in vitro (73). Blackwell et al. published similar results using activated resident macrophages to inhibit epithelial branching. The proposed mechanism is that LPS activates nuclear factor kappa beta (NF-kappa B), which is then accompanied by increased expression of interleukin-1beta (IL-1β) and tumor necrosis factor-a (TNF-a) in resident macrophages (74). This branching inhibitory effect caused by macrophage-mediated inflammation has been confirmed by a macrophage-depletion study in the lung. Benjamin et al. explained this inhibitory effect by linking Fgf10 signaling with inflammatory signals. Using in vitro experiments, they demonstrated that NF-kappa B, IL-1β, and TNF-a are capable of reducing Fgf10 expression in LPS-treated primary mesenchymal cells. The mechanism involved activation of toll-like receptors 2 and 4 (TLR2/4). The authors showed that FGF10-positive cells were decreased in lung samples of premature infants who died from BPD (75).
Tgf-β1 has been demonstrated to induce epithelial–mesenchymal transition (EMT) of AEC to MYF-like cells leading to extracellular matrix (ECM) deposition and thereby contributing to fibrosis and destruction of alveolar structure (78–80) (see also Table 1). Endogenous nitric oxide is proposed to attenuate EMT in AECs in an in vitro approach using primary culture of AEC II (81).
As previously mentioned, the alveologenesis phase leads to a dramatic increase in alveolar surface, which is essential for gas exchange. The current consensus is that this process is interrupted by exogenous deleterious factors leading to simplification of alveoli in BPD. Many studies confirmed that the alveolar MYF, located in the mesenchyme, is the unique cell type responsible for secondary septae formation. During alveologenesis, the alveolar myofibroblast is characterized by expression of alpha-smooth-muscle-actin (αSMA or Acta2) compared to other mesenchymal fibroblast population. By deposition of elastin and collagen, the alveolar myofibroblast initiates the process of secondary septation (82, 83). Both elastin and alveolar myofibroblast have been shown to be critical for secondary septae formation (83, 84). Expression of tropoelastin starts in the pseudoglandular stage of lung development and reaches the highest level during the alveolar stage (85, 86). The strongest evidence so far showing the importance of elastin for secondary septae formation came from the Elastin-knock-out mice that reveal a complete failure of alveologenesis leading to an emphysematous-like phenotype (87, 88) (Table 1). Interestingly, both hyperoxia and mechanical ventilation lead to increased expression of Elastin (89–91). Fgfr3 and Fgfr4 have been shown to direct alveologenesis in the murine lung by controlling elastogenesis (92). By using mice homozygous for Pdgfa-null allele, Boström and colleagues demonstrated failed alveolar formation due to loss of alveolar myofibroblasts and consequent loss of Elastin fibers (93, 94). Likewise, blocking antibody against Pdgfrα in newborn mice (PN1–PN7) led to aberrant Elastin fiber deposition and impaired alveolar septation, resulting in long-term failure in alveologenesis that lasted into adulthood. Pdgfa is expressed in the epithelium and targets its receptor (Pdgfrα) on mesenchymal cells such as alveolar myofibroblast and LIF (Table 1). Given the many mesenchymal targets of Pdgfa, it is not clear whether the impact of Pdgfa or Pdgfrα deletion on myofibroblast formation is via a direct effect of Pdgfa on alveolar myofibroblasts (and or alveolar myofibroblast progenitors) or indirectly via Pdgfa action on other targets (ASMCs and LIF). Gain and loss of function for Pdgfa/Pdgfrα signaling using cell autonomous-based approaches in specific lineages should be carried out in the future to sort out these issues.
Interestingly, increased levels of Fgf signaling in the mesenchyme also leads to arrested development of terminal airways accompanied by reduced Elastin deposition (95). The authors achieved this condition by taking advantage of Fgfr2c+/Δ mice that develop an autocrine Fgf10–Fgfr2b signaling loop in the mesenchyme due to a splicing switch, resulting in the ectopic expression of Fgfr2b instead of Fgfr2c. The proposed mechanism of action is that mesenchymal Fgf signaling suppresses the differentiation of alveolar myofibroblast progenitors. Furthermore, the blockade of Fgfr2b ligands in the lung from E14.5 to E18.5 by overexpression of a soluble dominant negative receptor of Fgfr2b (Sftpc-rtTA/+;tetOsolFgfr2b/+) blocking all Fgfr2b ligands also leads to arrest in secondary septae formation and alveolar simplification (96) suggesting that Fgfr2b ligands, during this time period, are also important for the formation of alveolar myofibroblasts. Subsequent treatment with retinoic acid (RA, biologically active derivative of vitamin A) induced re-alveolarization and was accompanied by increased Pdgfra-positive cells and decreased αSma/Acta2-positive cells. Concurrent induction of the dominant negative Fgfr2b in these experimental conditions is able to prevent the RA-mediated alveolar regeneration. These data suggest that re-alveolarization is dependent on Fgfr2b ligands. Furthermore, the authors proposed a conceptual model that alveolar myofibroblasts (αSma/Acta2-positive) arise from Pdgfra-positive LIF. Specific lineage-tracing studies targeting subsets of lung fibroblasts (e.g., Adrp for LIF) are needed to validate this model. Chen and colleagues also demonstrated that Fgfr2b ligands are necessary for alveolar myofibroblast formation during compensatory lung growth after pneumonectomy (97). However, the blockade of Fgfr2b ligands by soluble Fgfr2b (decoy receptor) postnatally during alveolarization does not impair alveologenesis in mice. Recently, it has been shown that reduced Pdgfra expression is a primary feature of human BPD. The authors showed decreased mRNA and protein expression of PDGFR-α and PDGFR-β in MSCs isolated from tracheal aspirates of premature neonates with BPD. Similarly, lungs of infants dying from BPD display less PDGFRα-positive cells in the alveolar septae. These findings were confirmed using a BPD mouse model exposed to hyperoxia (75% oxygen) for 14 days (98).
The LIF remains a poorly characterized lipid-containing interstitial cell located in the mesenchyme in close proximity to AEC II. LIFs, which accumulate lipid vacuoles (83, 99), are abundant in the early postnatal lung and regress significantly in number after alveolar septation. The presence of LIF in rodent lungs has been demonstrated extensively. However, whether LIF reside in adult human lung remains controversial (100, 101). Because of their close localization to AEC II, LIF have been proposed to interact with AEC II. Indeed, it has been shown convincingly that LIF are involved in the trafficking of lipids to the AEC II for surfactant production (102, 103). Apart from triglycerides, LIF also secrete leptin and retinoic acid, both important for surfactant production and alveolar septation (104, 105). On the other hand, AECII secrete parathyroid hormone-related protein (Pthrp) to signal through Pthrp receptor expressed on LIF to induce expression of adipose differentiation-related protein (Adrp) via peroxisome proliferator-activated receptor gamma (Pparg) pathway (Figure 3). The current consensus is that this signaling pathway is essential for the maintenance of the LIF phenotype as well as for regulation of surfactant production (104, 106–108). By performing co-culture experiments it has been proposed recently that LIF constitute a niche for AECII cells postnatally. Co-culture of LIF with AECII cells allows the formation of alveolospheres (109).
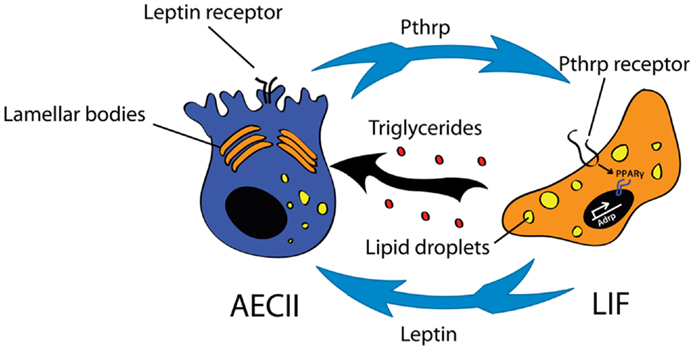
Figure 3. Interaction between type II alveolar epithelial cells (AEC II) and lipofibroblasts (LIF) for surfactant production. The Pthrp (parathyroid hormone-related protein)/Pparg (peroxisome proliferator-activated receptor gamma) axis is important for LIF formation and maintenance. LIF secrete triglycerides and leptin that are essential for surfactant production.
The contribution of LIF to lung regeneration and structural maintenance in later phases of life is currently unknown. Several lung injury models including cigarette-smoke exposure induce the transdifferentiation of LIF to αSma/Acta2+ MYF in vitro. These αSma/Acta2+ MYF are highly proliferative and express high levels of collagen (110). For this reason, it has been proposed that LIF are progenitors for alveolar MYF (Figure 2). However, an alternative and more plausible possibility is that LIF give rise to the activated MYF. Activated MYF, unlike the alveolar MYF, is involved in pathological situations and is responsible for fibrosis. Supporting this possibility, we have recently shown that during alveologenesis, Fgf10-positive cells give rise to LIF rather than alveolar MYF and during adult life, a subpopulation of Fgf10-expressing cells represents a pool of resident MSCs (Cd45− Cd31− Sca-1+) (20). In addition, the LIF-to-“activated MYF” transdifferentiation would translate indeed into loss of pulmonary integrity by smoke in vivo. Such transdifferentiation can be prevented and reversed in vitro using Pparg agonists such as rosiglitazone (111). However, it remains unclear whether such transdifferentiation occurs in vivo. Of note, exposure of premature neonates to hyperoxia induces arrest of alveolar septation and thickened primary septae due to MYF hyperplasia and excessive ECM production. Therefore, LIFs are unlikely progenitors for alveolar MYF. In the future, these results will have to be confirmed by lineage-tracing experiments in the context of injury using more specific knock-in lines to target the LIF and determine their fate.
Endothelial–Mesenchymal Crosstalk in Normal Lung Development and BPD
In parallel to branching morphogenesis during early embryonic lung development the lung vasculature begins to form in the mesenchyme at around E10.0 (112). This process involves angiogenesis and vasculogenesis. Angiogenesis occurs when pre-existing endothelial cells sprout to form capillaries. In comparison, vasculogenesis is characterized by migration and differentiation of endothelial progenitor cells (or hemangioblasts) in the distal mesenchyme to form new blood vessels. DeMello and colleagues investigated the early fetal development of lung vasculature by employing light and transmission electron microscopy as well as vascular casts and scanning electron microscopy. They demonstrated three features of the lung vasculature occurring between E9.0 and E20.0 in mice: (1) angiogenesis occurs in the proximal (central) lung vasculature, (2) peripheral lung vessels are established by vasculogenesis, and (3) at E13.0/E14.0 the central and peripheral parts of lung vasculature begin to connect to each other via a lytic process (113, 114). Finally, the main event of microvascular maturation takes place during the alveolar stage of lung development where the transition from a double capillary network to a single capillary system within alveolar walls occurs.
Although BPD has long been regarded as an epithelial disease due to its emphysematous aspect, much emphasis has now been placed also on the role of the lung vasculature in this disease. The temporal–spatial proximity of lung vasculature development and branching morphogenesis suggests a close interaction between these two important structures via endothelial–epithelial tissue crosstalk. The better understanding of this crosstalk in development and disease condition might be highly relevant for future therapies. Vascular endothelial growth factor receptor 2 (Vegfr2 or Flk-1) is an early marker for endothelial progenitors located in the SEM (115, 116). However, it is not yet clear whether these progenitors arise from the mesothelium or the mesenchyme (117, 118). Progenitors for VSMCs are believed to arise from Fgf10+ cells (20), Wnt2+, Gli1+ and Isl1+ cells (coming from the second heart field) (18), Pdgfrb+ cells (119), and mesothelial cells (117, 118).
During murine embryonic development, Vegfr2-positive cells receive the Vegfa signal from epithelial and mesenchymal cells until E14.5, after which Vegfa expression becomes restricted to the epithelium (120) (Table 1). Furthermore, it has been shown that Shh and Fgf9, secreted by the epithelium, are able to induce expression of Vegfa in the mesenchyme (121). Reciprocally, mesenchymally secreted Fgf10 leads to the upregulation of Vegf in the distal epithelium (122). In our previous work, we showed that treatment of embryonic lung explants with recombinant Vegfa not only upregulates Vegfr2 in the mesenchyme but also induces branching of the epithelium (123). However, it is unclear whether the effect of Vegfa on epithelial branching is direct or indirect. This endothelial–epithelial tissue crosstalk has been extensively examined by using in vitro recombination studies (co-culture of epithelium and mesenchyme respectively and mesenchyme alone) as well as in in vivo lung agenesis model (β-catenin knockout) (112). Using an in vivo inducible decoy receptor of Vegfr1 (solubleVegfr1), Lazarus and colleagues demonstrated that Spry2 is upregulated in the epithelium upon inhibition of Vegfr1-mediated signaling, suggesting an inhibition of Fgf signaling (as mentioned before Spry2 is an inhibitor of Fgf10), which is essential for branching morphogenesis (124). Another link in the endothelial–epithelial crosstalk came from the Pecam1-deficient mice that display a failure in endothelial cell formation accompanied by simplified alveolarization (125).
During a pathological process, Vegfa has been found down-regulated in preterm infants with BPD (8, 126, 127). Furthermore, Thebaud and colleagues demonstrated that Vegf and Vegfr2 are decreased in the hyperoxia model of BPD in newborn rats and that adenoviral administration of VEGF improved alveolar architecture and promoted capillary formation (128, 129). Although the trophic and angiogenic potential of VEGF on the lung vasculature is known, the aforementioned study, and the studies from other groups, suggest that vascular growth serves as a driving force for alveolar growth and maturation, leading to improvement of lung structure, and promoting secondary septae formation. A recent report revealed the association of a VEGF polymorphism with BPD in Japanese preterm newborns (130).
Newborn mice that are hypomorphic for Fgf10 also display reduced expression of Vegfa and Pecam. These mice suffer from an oversimplified lung with an abnormally developed lung vasculature (41). Interestingly, Fgf10 expression is reduced in lungs from BPD patients (75). Whether the effect of mesenchyme-derived growth factors (such as Fgf10) on the lung endothelium is direct needs to be demonstrated. Another animal injury model demonstrating the importance of endothelial–epithelial interactions is the pneumonectomy model in mice. An inducible endothelium-specific deletion of Vegfr2 and Fgfr1 leads to reduction of Mmp14 secretion. Mmp14 is critical for expansion of epithelial progenitor cells during compensatory lung growth by unmasking Egfr ecto-domains. This was confirmed convincingly by rescue-experiments where EGF/MMP14 administration resulted in restored alveologenesis (131).
Conclusion
Although advances in pharmacotherapy and medical technology (e.g., gentle ventilation) have improved the management of premature infants, BPD remains the most common incurable chronic lung disease of infancy with considerable mortality and long-term morbidity. An unintended consequence of these advances has been the survival rate of premature infants born before the 24th week of gestation who represent the highest risk group for pathogenesis of BPD. This means that the number of infants born with an immature lung (in the canalicular stage of lung development) is increased, leading to an increase in the incidence of BPD. Thus, it is urgent to find a treatment for BPD. The treatment of BPD has been confounded by its multifactorial causes. Therefore, only a comprehensive and individualized therapy adjusted to the profile of risk factors of each prematurely born infant will likely be able to provide a meaningful and effective strategy. The early identification of infants predisposed to BPD is essential. Several studies have been conducted to detect biomarkers in premature infants indicating their level of risk for BPD (132). Yet, the results remain inconclusive due to the low numbers of infants considered for the study. Importantly, more effort should be made in establishing preventive therapy. For example, more research should be conducted to understand the pathogenesis of preterm labor, a common cause of preterm delivery, which is one of the main risk factor associated with BPD. In addition, the current knowledge about the mechanisms of alveolarization must be expanded. Due to lack of human lung samples, the establishment of animal models precisely resembling the disease condition of BPD in humans will be helpful. Single cell transcriptomic studies should be carried out on alveolar myofibroblasts in physiological and pathological conditions to unravel the aberrant gene expression patterns and/or gene mutations responsible for impaired secondary septae formation. Furthermore, the use of the pneumonectomy mouse model and cell specific lineage-tracing approaches to understand the process of de novo alveolarization should contribute significantly to our understanding of lung regeneration. Last, but not least, the knowledge about progenitor/stem cells located in niches of the postnatal lung will be a valuable source of information that would be useful in triggering lung regeneration subsequent to injury.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the members of the Bellusci lab for their input. We apologize to those colleagues whose references have been omitted from this discussion; due to space restrictions and our focus, we were unable to include all articles on this interesting and diverse subject matter. PM acknowledges the support from the Hastings foundation. PM and SB acknowledge the NHLBI support (HL107307). SB was also supported by grants from the DFG (BE4443/4-1 and BE4443/6-1), LOEWE and UKGM as well as the program of competitive growth of Kazan Federal University.
References
1. Northway WH Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med (1967) 276:357–68. doi: 10.1056/NEJM196702162760701
2. Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, et al. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics (2010) 126:443–56. doi:10.1542/peds.2009-2959
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med (2007) 357:1946–55. doi:10.1056/NEJMra067279
4. Bhandari A, Bhandari V. Pitfalls, problems, and progress in bronchopulmonary dysplasia. Pediatrics (2009) 123:1562–73. doi:10.1542/peds.2008-1962
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Shaw GM, O’Brodovich HM. Progress in understanding the genetics of bronchopulmonary dysplasia. Semin Perinatol (2013) 37:85–93. doi:10.1053/j.semperi.2013.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Hadchouel A, Durrmeyer X, Bouzigon E, Incitti R, Huusko J, Jarreau PH, et al. Identification of SPOCK2 as a susceptibility gene for bronchopulmonary dysplasia. Am J Respir Crit Care Med (2011) 184:1164–70. doi:10.1164/rccm.201103-0548OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Jobe AJ. The new BPD: an arrest of lung development. Pediatr Res (1999) 46:641–3. doi:10.1203/00006450-199912000-00007
8. Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med (2001) 164:1971–80. doi:10.1164/ajrccm.164.10.2101140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Rossor T, Greenough A. Advances in paediatric pulmonary vascular disease associated with bronchopulmonary dysplasia. Expert Rev Respir Med (2015) 9:35–43. doi:10.1586/17476348.2015.986470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Furman L, Baley J, Borawski-Clark E, Aucott S, Hack M. Hospitalization as a measure of morbidity among very low birth weight infants with chronic lung disease. J Pediatr (1996) 128:447–52. doi:10.1016/S0022-3476(96)70353-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Schmidt B, Asztalos EV, Roberts RS, Robertson CM, Sauve RS, Whitfield MF. Impact of bronchopulmonary dysplasia, brain injury, and severe retinopathy on the outcome of extremely low-birth-weight infants at 18 months: results from the trial of indomethacin prophylaxis in preterms. JAMA (2003) 289:1124–9. doi:10.1001/jama.289.9.1124
12. El Mazloum D, Moschino L, Bozzetto S, Baraldi E. Chronic lung disease of prematurity: long-term respiratory outcome. Neonatology (2014) 105:352–6. doi:10.1159/000360651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Moore KL, Persuad TVN. The Developing Human: Clinically Oriented Embryology. Philadelphia, PA: W.B. Saunders Co (2002).
14. Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature (2014) 509:371–5. doi:10.1038/nature13173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Roberts D, Dalziel S. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev (2006) 3:CD004454. doi:10.1002/14651858.CD004454.pub2
16. Kresch MJ, Christian C, Wu F, Hussain N. Ontogeny of apoptosis during lung development. Pediatr Res (1998) 43:426–31. doi:10.1203/00006450-199803000-00020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Schittny JC, Mund SI, Stampanoni M. Evidence and structural mechanism for late lung alveolarization. Am J Physiol Lung Cell Mol Physiol (2008) 294:L246–54. doi:10.1152/ajplung.00296.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Peng T, Tian Y, Boogerd CJ, Lu MM, Kadzik RS, Stewart KM, et al. Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature (2013) 500:589–92. doi:10.1038/nature12358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. El Agha E, Bellusci S. Walking along the fibroblast growth factor 10 route: a Key pathway to understand the control and regulation of epithelial and mesenchymal cell-lineage formation during lung development and repair after injury. Scientifica (Cairo) (2014) 2014:538379. doi:10.1155/2014/538379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. El Agha E, Herold S, Al Alam D, Quantius J, Mackenzie B, Carraro G, et al. Fgf10-positive cells represent a progenitor cell population during lung development and postnatally. Development (2014) 141:296–306. doi:10.1242/dev.099747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Hines EA, Sun X. Tissue crosstalk in lung development. J Cell Biochem (2014) 115:1469–77. doi:10.1002/jcb.24811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Shannon JM. Induction of alveolar type II cell differentiation in fetal tracheal epithelium by grafted distal lung mesenchyme. Dev Biol (1994) 166:600–14. doi:10.1006/dbio.1994.1340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Shannon JM, Nielsen LD, Gebb SA, Randell SH. Mesenchyme specifies epithelial differentiation in reciprocal recombinants of embryonic lung and trachea. Dev Dyn (1998) 212:482–94. doi:10.1002/(SICI)1097-0177(199808)212:4<482::AID-AJA2>3.0.CO;2-D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Hyatt BA, Shangguan X, Shannon JM. FGF-10 induces SP-C and Bmp4 and regulates proximal-distal patterning in embryonic tracheal epithelium. Am J Physiol Lung Cell Mol Physiol (2004) 287:L1116–26. doi:10.1152/ajplung.00033.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem (2011) 149:121–30. doi:10.1093/jb/mvq121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Tekin M, Hismi BO, Fitoz S, Ozdag H, Cengiz FB, Sirmaci A, et al. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am J Hum Genet (2007) 80:338–44. doi:10.1086/510920
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Milunsky JM, Zhao G, Maher TA, Colby R, Everman DB. LADD syndrome is caused by FGF10 mutations. Clin Genet (2006) 69:349–54. doi:10.1111/j.1399-0004.2006.00597.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Klar J, Blomstrand P, Brunmark C, Badhai J, Hakansson HF, Brange CS, et al. Fibroblast growth factor 10 haploinsufficiency causes chronic obstructive pulmonary disease. J Med Genet (2011) 48:705–9. doi:10.1136/jmedgenet-2011-100166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int (2001) 60:2079–86. doi:10.1046/j.1523-1755.2001.00064.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. del Moral PM, De Langhe SP, Sala FG, Veltmaat JM, Tefft D, Wang K, et al. Differential role of FGF9 on epithelium and mesenchyme in mouse embryonic lung. Dev Biol (2006) 293:77–89. doi:10.1016/j.ydbio.2006.01.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Ornitz DM, Yin Y. Signaling networks regulating development of the lower respiratory tract. Cold Spring Harb Perspect Biol (2012) 4:a008318. doi:10.1101/cshperspect.a008318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Bellusci S, Furuta Y, Rush MG, Henderson R, Winnier G, Hogan BL. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development (1997) 124:53–63.
33. Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development (1997) 124:4867–78.
34. Peters K, Werner S, Liao X, Wert S, Whitsett J, Williams L. Targeted expression of a dominant negative FGF receptor blocks branching morphogenesis and epithelial differentiation of the mouse lung. EMBO J (1994) 13:3296–301.
35. Arman E, Haffner-Krausz R, Gorivodsky M, Lonai P. Fgfr2 is required for limb outgrowth and lung-branching morphogenesis. Proc Natl Acad Sci U S A (1999) 96:11895–9. doi:10.1073/pnas.96.21.11895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, et al. Fgf10 is essential for limb and lung formation. Nat Genet (1999) 21:138–41. doi:10.1038/5096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development (2000) 127:483–92.
38. Mailleux AA, Spencer-Dene B, Dillon C, Ndiaye D, Savona-Baron C, Itoh N, et al. Role of FGF10/FGFR2b signaling during mammary gland development in the mouse embryo. Development (2002) 129:53–60.
39. Lu J, Izvolsky KI, Qian J, Cardoso WV. Identification of FGF10 targets in the embryonic lung epithelium during bud morphogenesis. J Biol Chem (2005) 280:4834–41. doi:10.1074/jbc.M410714200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Tang W, Wei Y, Le K, Li Z, Bao Y, Gao J, et al. Mitogen-activated protein kinases ERK 1/2- and p38-GATA4 pathways mediate the Ang II-induced activation of FGF2 gene in neonatal rat cardiomyocytes. Biochem Pharmacol (2011) 81:518–25. doi:10.1016/j.bcp.2010.11.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Ramasamy SK, Mailleux AA, Gupte VV, Mata F, Sala FG, Veltmaat JM, et al. Fgf10 dosage is critical for the amplification of epithelial cell progenitors and for the formation of multiple mesenchymal lineages during lung development. Dev Biol (2007) 307:237–47. doi:10.1016/j.ydbio.2007.04.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Hines EA, Jones MK, Verheyden JM, Harvey JF, Sun X. Establishment of smooth muscle and cartilage juxtaposition in the developing mouse upper airways. Proc Natl Acad Sci U S A (2013) 110:19444–9. doi:10.1073/pnas.1313223110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Turcatel G, Rubin N, Menke DB, Martin G, Shi W, Warburton D. Lung mesenchymal expression of Sox9 plays a critical role in tracheal development. BMC Biol (2013) 11:117. doi:10.1186/1741-7007-11-117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Sala FG, Del Moral PM, Tiozzo C, Alam DA, Warburton D, Grikscheit T, et al. FGF10 controls the patterning of the tracheal cartilage rings via Shh. Development (2011) 138:273–82. doi:10.1242/dev.051680
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Volckaert T, Campbell A, Dill E, Li C, Minoo P, De Langhe S. Localized Fgf10 expression is not required for lung branching morphogenesis but prevents differentiation of epithelial progenitors. Development (2013) 140:3731–42. doi:10.1242/dev.096560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Volckaert T, Dill E, Campbell A, Tiozzo C, Majka S, Bellusci S, et al. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J Clin Invest (2011) 121:4409–19. doi:10.1172/JCI58097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. McQualter JL, Yuen K, Williams B, Bertoncello I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc Natl Acad Sci U S A (2010) 107:1414–9. doi:10.1073/pnas.0909207107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Goss AM, Tian Y, Tsukiyama T, Cohen ED, Zhou D, Lu MM, et al. Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev Cell (2009) 17:290–8. doi:10.1016/j.devcel.2009.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Harris-Johnson KS, Domyan ET, Vezina CM, Sun X. Beta-catenin promotes respiratory progenitor identity in mouse foregut. Proc Natl Acad Sci U S A (2009) 106:16287–92. doi:10.1073/pnas.0902274106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Goss AM, Tian Y, Cheng L, Yang J, Zhou D, Cohen ED, et al. Wnt2 signaling is necessary and sufficient to activate the airway smooth muscle program in the lung by regulating myocardin/Mrtf-B and Fgf10 expression. Dev Biol (2011) 356:541–52. doi:10.1016/j.ydbio.2011.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Mucenski ML, Wert SE, Nation JM, Loudy DE, Huelsken J, Birchmeier W, et al. Beta-catenin is required for specification of proximal/distal cell fate during lung morphogenesis. J Biol Chem (2003) 278:40231–8. doi:10.1074/jbc.M305892200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Mucenski ML, Nation JM, Thitoff AR, Besnard V, Xu Y, Wert SE, et al. Beta-catenin regulates differentiation of respiratory epithelial cells in vivo. Am J Physiol Lung Cell Mol Physiol (2005) 289:L971–9. doi:10.1152/ajplung.00172.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Shu W, Jiang YQ, Lu MM, Morrisey EE. Wnt7b regulates mesenchymal proliferation and vascular development in the lung. Development (2002) 129:4831–42.
54. Miller MF, Cohen ED, Baggs JE, Lu MM, Hogenesch JB, Morrisey EE. Wnt ligands signal in a cooperative manner to promote foregut organogenesis. Proc Natl Acad Sci U S A (2012) 109:15348–53. doi:10.1073/pnas.1201583109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Li C, Xiao J, Hormi K, Borok Z, Minoo P. Wnt5a participates in distal lung morphogenesis. Dev Biol (2002) 248:68–81. doi:10.1006/dbio.2002.0729
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. De Langhe SP, Carraro G, Tefft D, Li C, Xu X, Chai Y, et al. Formation and differentiation of multiple mesenchymal lineages during lung development is regulated by beta-catenin signaling. PLoS One (2008) 3:e1516. doi:10.1371/journal.pone.0001516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Kumar ME, Bogard PE, Espinoza FH, Menke DB, Kingsley DM, Krasnow MA. Mesenchymal cells. Defining a mesenchymal progenitor niche at single-cell resolution. Science (2014) 346:1258810. doi:10.1126/science.1258810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Weaver M, Dunn NR, Hogan BL. Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development (2000) 127:2695–704.
59. Mailleux AA, Kelly R, Veltmaat JM, De Langhe SP, Zaffran S, Thiery JP, et al. Fgf10 expression identifies parabronchial smooth muscle cell progenitors and is required for their entry into the smooth muscle cell lineage. Development (2005) 132:2157–66. doi:10.1242/dev.01795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Weaver M, Yingling JM, Dunn NR, Bellusci S, Hogan BL. Bmp signaling regulates proximal-distal differentiation of endoderm in mouse lung development. Development (1999) 126:4005–15.
61. Weaver M, Batts L, Hogan BL. Tissue interactions pattern the mesenchyme of the embryonic mouse lung. Dev Biol (2003) 258:169–84. doi:10.1016/S0012-1606(03)00117-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Minowada G, Jarvis LA, Chi CL, Neubuser A, Sun X, Hacohen N, et al. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development (1999) 126:4465–75.
63. Mailleux AA, Tefft D, Ndiaye D, Itoh N, Thiery JP, Warburton D, et al. Evidence that SPROUTY2 functions as an inhibitor of mouse embryonic lung growth and morphogenesis. Mech Dev (2001) 102:81–94. doi:10.1016/S0925-4773(01)00286-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Tefft D, Lee M, Smith S, Crowe DL, Bellusci S, Warburton D. mSprouty2 inhibits FGF10-activated MAP kinase by differentially binding to upstream target proteins. Am J Physiol Lung Cell Mol Physiol (2002) 283:L700–6. doi:10.1152/ajplung.00372.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Husain AN, Siddiqui NH, Stocker JT. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol (1998) 29:710–7. doi:10.1016/S0046-8177(98)90280-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Thibeault DW, Mabry SM, Ekekezie II, Truog WE. Lung elastic tissue maturation and perturbations during the evolution of chronic lung disease. Pediatrics (2000) 106:1452–9. doi:10.1542/peds.106.6.1452
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Thibeault DW, Mabry SM, Ekekezie II, Zhang X, Truog WE. Collagen scaffolding during development and its deformation with chronic lung disease. Pediatrics (2003) 111:766–76. doi:10.1542/peds.111.4.766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Warner BB, Stuart LA, Papes RA, Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol (1998) 275:L110–7.
69. Dauger S, Ferkdadji L, Saumon G, Vardon G, Peuchmaur M, Gaultier C, et al. Neonatal exposure to 65% oxygen durably impairs lung architecture and breathing pattern in adult mice. Chest (2003) 123:530–8. doi:10.1378/chest.123.2.530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Auten RL, Mason SN, Auten KM, Brahmajothi M. Hyperoxia impairs postnatal alveolar epithelial development via NADPH oxidase in newborn mice. Am J Physiol Lung Cell Mol Physiol (2009) 297:L134–42. doi:10.1152/ajplung.00112.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Velten M, Heyob KM, Rogers LK, Welty SE. Deficits in lung alveolarization and function after systemic maternal inflammation and neonatal hyperoxia exposure. J Appl Physiol (1985) (2010) 108:1347–56. doi:10.1152/japplphysiol.01392.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med (1999) 160:1333–46. doi:10.1164/ajrccm.160.4.9810071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Prince LS, Dieperink HI, Okoh VO, Fierro-Perez GA, Lallone RL. Toll-like receptor signaling inhibits structural development of the distal fetal mouse lung. Dev Dyn (2005) 233:553–61. doi:10.1002/dvdy.20362
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Blackwell TS, Hipps AN, Yamamoto Y, Han W, Barham WJ, Ostrowski MC, et al. NF-kappaB signaling in fetal lung macrophages disrupts airway morphogenesis. J Immunol (2011) 187:2740–7. doi:10.4049/jimmunol.1101495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Benjamin JT, Smith RJ, Halloran BA, Day TJ, Kelly DR, Prince LS. FGF-10 is decreased in bronchopulmonary dysplasia and suppressed by toll-like receptor activation. Am J Physiol Lung Cell Mol Physiol (2007) 292:L550–8. doi:10.1152/ajplung.00329.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Benjamin JT, Carver BJ, Plosa EJ, Yamamoto Y, Miller JD, Liu JH, et al. NF-kappaB activation limits airway branching through inhibition of Sp1-mediated fibroblast growth factor-10 expression. J Immunol (2010) 185:4896–903. doi:10.4049/jimmunol.1001857
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Carver BJ, Plosa EJ, Stinnett AM, Blackwell TS, Prince LS. Interactions between NF-kappaB and SP3 connect inflammatory signaling with reduced FGF-10 expression. J Biol Chem (2013) 288:15318–25. doi:10.1074/jbc.M112.447318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A (2006) 103:13180–5. doi:10.1073/pnas.0605669103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, Du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol (2005) 166:1321–32. doi:10.1016/S0002-9440(10)62351-6
80. Phan SH. The myofibroblast in pulmonary fibrosis. Chest (2002) 122:286S–9S. doi:10.1378/chest.122.6_suppl.286S
81. Vyas-Read S, Shaul PW, Yuhanna IS, Willis BC. Nitric oxide attenuates epithelial-mesenchymal transition in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol (2007) 293:L212–21. doi:10.1152/ajplung.00475.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Noguchi A, Reddy R, Kursar JD, Parks WC, Mecham RP. Smooth muscle isoactin and elastin in fetal bovine lung. Exp Lung Res (1989) 15:537–52. doi:10.3109/01902148909069617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Vaccaro C, Brody JS. Ultrastructure of developing alveoli. I. The role of the interstitial fibroblast. Anat Rec (1978) 192:467–79. doi:10.1002/ar.1091920402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Dickie R, Wang YT, Butler JP, Schulz H, Tsuda A. Distribution and quantity of contractile tissue in postnatal development of rat alveolar interstitium. Anat Rec (Hoboken) (2008) 291:83–93. doi:10.1002/ar.20622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Mariani TJ, Sandefur S, Pierce RA. Elastin in lung development. Exp Lung Res (1997) 23:131–45. doi:10.3109/01902149709074026
86. Willet KE, McMenamin P, Pinkerton KE, Ikegami M, Jobe AH, Gurrin L, et al. Lung morphometry and collagen and elastin content: changes during normal development and after prenatal hormone exposure in sheep. Pediatr Res (1999) 45:615–25. doi:10.1203/00006450-199905010-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY. Impaired distal airway development in mice lacking elastin. Am J Respir Cell Mol Biol (2000) 23:320–6. doi:10.1165/ajrcmb.23.3.3906
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Shifren A, Durmowicz AG, Knutsen RH, Hirano E, Mecham RP. Elastin protein levels are a vital modifier affecting normal lung development and susceptibility to emphysema. Am J Physiol Lung Cell Mol Physiol (2007) 292:L778–87. doi:10.1152/ajplung.00352.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Bruce MC, Bruce EN, Janiga K, Chetty A. Hyperoxic exposure of developing rat lung decreases tropoelastin mRNA levels that rebound postexposure. Am J Physiol (1993) 265:L293–300.
90. Albertine KH, Jones GP, Starcher BC, Bohnsack JF, Davis PL, Cho SC, et al. Chronic lung injury in preterm lambs. Disordered respiratory tract development. Am J Respir Crit Care Med (1999) 159:945–58. doi:10.1164/ajrccm.159.3.9804027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Nakamura T, Liu M, Mourgeon E, Slutsky A, Post M. Mechanical strain and dexamethasone selectively increase surfactant protein C and tropoelastin gene expression. Am J Physiol Lung Cell Mol Physiol (2000) 278:L974–80.
92. Weinstein M, Xu X, Ohyama K, Deng CX. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development (1998) 125:3615–23.
93. Bostrom H, Willetts K, Pekny M, Leveen P, Lindahl P, Hedstrand H, et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell (1996) 85:863–73. doi:10.1016/S0092-8674(00)81270-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Lindahl P, Karlsson L, Hellstrom M, Gebre-Medhin S, Willetts K, Heath JK, et al. Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development (1997) 124:3943–53.
95. De Langhe SP, Carraro G, Warburton D, Hajihosseini MK, Bellusci S. Levels of mesenchymal FGFR2 signaling modulate smooth muscle progenitor cell commitment in the lung. Dev Biol (2006) 299:52–62. doi:10.1016/j.ydbio.2006.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Perl AK, Gale E. FGF signaling is required for myofibroblast differentiation during alveolar regeneration. Am J Physiol Lung Cell Mol Physiol (2009) 297:L299–308. doi:10.1152/ajplung.00008.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Chen L, Acciani T, Le Cras T, Lutzko C, Perl AK. Dynamic regulation of platelet-derived growth factor receptor alpha expression in alveolar fibroblasts during realveolarization. Am J Respir Cell Mol Biol (2012) 47:517–27. doi:10.1165/rcmb.2012-0030OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Popova AP, Bentley JK, Cui TX, Richardson MN, Linn MJ, Lei J, et al. Reduced platelet-derived growth factor receptor expression is a primary feature of human bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol (2014) 307:L231–9. doi:10.1152/ajplung.00342.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. O’Hare KH, Sheridan MN. Electron microscopic observations on the morphogenesis of the albino rat lung, with special reference to pulmonary epithelial cells. Am J Anat (1970) 127:181–205. doi:10.1002/aja.1001270205
100. Rehan VK, Sugano S, Wang Y, Santos J, Romero S, Dasgupta C, et al. Evidence for the presence of lipofibroblasts in human lung. Exp Lung Res (2006) 32:379–93. doi:10.1080/01902140600880257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Tahedl D, Wirkes A, Tschanz SA, Ochs M, Muhlfeld C. How common is the lipid body-containing interstitial cell in the mammalian lung? Am J Physiol Lung Cell Mol Physiol (2014) 307:L386–94. doi:10.1152/ajplung.00131.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Torday J, Hua J, Slavin R. Metabolism and fate of neutral lipids of fetal lung fibroblast origin. Biochim Biophys Acta (1995) 1254:198–206. doi:10.1016/0005-2760(94)00184-Z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Tordet C, Marin L, Dameron F. Pulmonary di-and-triacylglycerols during the perinatal development of the rat. Experientia (1981) 37:333–4. doi:10.1007/BF01959845
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Torday JS, Rehan VK. Stretch-stimulated surfactant synthesis is coordinated by the paracrine actions of PTHrP and leptin. Am J Physiol Lung Cell Mol Physiol (2002) 283:L130–5. doi:10.1152/ajplung.00380.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Simon DM, Mariani TJ. Role of PPARs and retinoid X receptors in the regulation of lung maturation and development. PPAR Res (2007) 2007:91240. doi:10.1155/2007/91240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Schultz CJ, Torres E, Londos C, Torday JS. Role of adipocyte differentiation-related protein in surfactant phospholipid synthesis by type II cells. Am J Physiol Lung Cell Mol Physiol (2002) 283:L288–96. doi:10.1152/ajplung.00204.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Rubin LP, Kovacs CS, De Paepe ME, Tsai SW, Torday JS, Kronenberg HM. Arrested pulmonary alveolar cytodifferentiation and defective surfactant synthesis in mice missing the gene for parathyroid hormone-related protein. Dev Dyn (2004) 230:278–89. doi:10.1002/dvdy.20058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Rehan VK, Torday JS. PPARgamma signaling mediates the evolution, development, homeostasis, and repair of the lung. PPAR Res (2012) 2012:289867. doi:10.1155/2012/289867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest (2013) 123:3025–36. doi:10.1172/JCI68782
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Rehan VK, Wang Y, Sugano S, Romero S, Chen X, Santos J, et al. Mechanism of nicotine-induced pulmonary fibroblast transdifferentiation. Am J Physiol Lung Cell Mol Physiol (2005) 289:L667–76. doi:10.1152/ajplung.00358.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR, Hogaboam CM, et al. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2008) 294:L891–901. doi:10.1152/ajplung.00333.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Gebb SA, Shannon JM. Tissue interactions mediate early events in pulmonary vasculogenesis. Dev Dyn (2000) 217:159–69. doi:10.1002/(SICI)1097-0177(200002)217:2<159::AID-DVDY3>3.3.CO;2-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. deMello DE, Sawyer D, Galvin N, Reid LM. Early fetal development of lung vasculature. Am J Respir Cell Mol Biol (1997) 16:568–81. doi:10.1165/ajrcmb.16.5.9160839
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Schachtner SK, Wang Y, Scott Baldwin H. Qualitative and quantitative analysis of embryonic pulmonary vessel formation. Am J Respir Cell Mol Biol (2000) 22:157–65. doi:10.1165/ajrcmb.22.2.3766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Kappel A, Ronicke V, Damert A, Flamme I, Risau W, Breier G. Identification of vascular endothelial growth factor (VEGF) receptor-2 (Flk-1) promoter/enhancer sequences sufficient for angioblast and endothelial cell-specific transcription in transgenic mice. Blood (1999) 93:4284–92.
116. Yamaguchi TP, Dumont DJ, Conlon RA, Breitman ML, Rossant J. flk-1, an flt-related receptor tyrosine kinase is an early marker for endothelial cell precursors. Development (1993) 118:489–98.
117. Que J, Wilm B, Hasegawa H, Wang F, Bader D, Hogan BL. Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development. Proc Natl Acad Sci U S A (2008) 105:16626–30. doi:10.1073/pnas.0808649105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Dixit R, Ai X, Fine A. Derivation of lung mesenchymal lineages from the fetal mesothelium requires hedgehog signaling for mesothelial cell entry. Development (2013) 140:4398–406. doi:10.1242/dev.098079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Greif DM, Kumar M, Lighthouse JK, Hum J, An A, Ding L, et al. Radial construction of an arterial wall. Dev Cell (2012) 23:482–93. doi:10.1016/j.devcel.2012.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Bhatt AJ, Amin SB, Chess PR, Watkins RH, Maniscalco WM. Expression of vascular endothelial growth factor and Flk-1 in developing and glucocorticoid-treated mouse lung. Pediatr Res (2000) 47:606–13. doi:10.1203/00006450-200005000-00009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. White AC, Lavine KJ, Ornitz DM. FGF9 and SHH regulate mesenchymal Vegfa expression and development of the pulmonary capillary network. Development (2007) 134:3743–52. doi:10.1242/dev.004879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Scott CL, Walker DJ, Cwiklinski E, Tait C, Tee AR, Land SC. Control of HIF-1{alpha} and vascular signaling in fetal lung involves cross talk between mTORC1 and the FGF-10/FGFR2b/Spry2 airway branching periodicity clock. Am J Physiol Lung Cell Mol Physiol (2010) 299:L455–71. doi:10.1152/ajplung.00348.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Del Moral PM, Sala FG, Tefft D, Shi W, Keshet E, Bellusci S, et al. VEGF-A signaling through Flk-1 is a critical facilitator of early embryonic lung epithelial to endothelial crosstalk and branching morphogenesis. Dev Biol (2006) 290:177–88. doi:10.1016/j.ydbio.2005.11.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Lazarus A, Del-Moral PM, Ilovich O, Mishani E, Warburton D, Keshet E. A perfusion-independent role of blood vessels in determining branching stereotypy of lung airways. Development (2011) 138:2359–68. doi:10.1242/dev.060723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. DeLisser HM, Helmke BP, Cao G, Egan PM, Taichman D, Fehrenbach M, et al. Loss of PECAM-1 function impairs alveolarization. J Biol Chem (2006) 281:8724–31. doi:10.1074/jbc.M511798200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Lassus P, Ristimaki A, Ylikorkala O, Viinikka L, Andersson S. Vascular endothelial growth factor in human preterm lung. Am J Respir Crit Care Med (1999) 159:1429–33. doi:10.1164/ajrccm.159.5.9806073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Lassus P, Turanlahti M, Heikkila P, Andersson LC, Nupponen I, Sarnesto A, et al. Pulmonary vascular endothelial growth factor and Flt-1 in fetuses, in acute and chronic lung disease, and in persistent pulmonary hypertension of the newborn. Am J Respir Crit Care Med (2001) 164:1981–7. doi:10.1164/ajrccm.164.10.2012036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Thebaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation (2005) 112:2477–86. doi:10.1161/CIRCULATIONAHA.105.541524
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Kunig AM, Balasubramaniam V, Markham NE, Morgan D, Montgomery G, Grover TR, et al. Recombinant human VEGF treatment enhances alveolarization after hyperoxic lung injury in neonatal rats. Am J Physiol Lung Cell Mol Physiol (2005) 289:L529–35. doi:10.1152/ajplung.00336.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Fujioka K, Shibata A, Yokota T, Koda T, Nagasaka M, Yagi M, et al. Association of a vascular endothelial growth factor polymorphism with the development of bronchopulmonary dysplasia in Japanese premature newborns. Sci Rep (2014) 4:4459. doi:10.1038/srep04459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell (2011) 147:539–53. doi:10.1016/j.cell.2011.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Bhandari A, Bhandari V. Biomarkers in bronchopulmonary dysplasia. Paediatr Respir Rev (2013) 14:173–9. doi:10.1016/j.prrv.2013.02.008
133. Bruce MC, Pawlowski R, Tomashefski JF Jr. Changes in lung elastic fiber structure and concentration associated with hyperoxic exposure in the developing rat lung. Am Rev Respir Dis (1989) 140:1067–74. doi:10.1164/ajrccm/140.4.1067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Prodhan P, Kinane TB. Developmental paradigms in terminal lung development. Bioessays (2002) 24:1052–9. doi:10.1002/bies.10177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Buch S, Han RN, Cabacungan J, Wang J, Yuan S, Belcastro R, et al. Changes in expression of platelet-derived growth factor and its receptors in the lungs of newborn rats exposed to air or 60% O(2). Pediatr Res (2000) 48:423–33. doi:10.1203/00006450-200010000-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. McGowan SE, McNamer R. Transforming growth factor-beta increases elastin production by neonatal rat lung fibroblasts. Am J Respir Cell Mol Biol (1990) 3:369–76. doi:10.1165/ajrcmb/3.4.369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. McGowan SE, Jackson SK, Olson PJ, Parekh T, Gold LI. Exogenous and endogenous transforming growth factors-beta influence elastin gene expression in cultured lung fibroblasts. Am J Respir Cell Mol Biol (1997) 17:25–35. doi:10.1165/ajrcmb.17.1.2686
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Kotecha S, Wangoo A, Silverman M, Shaw RJ. Increase in the concentration of transforming growth factor beta-1 in bronchoalveolar lavage fluid before development of chronic lung disease of prematurity. J Pediatr (1996) 128:464–9. doi:10.1016/S0022-3476(96)70355-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Alejandre-Alcazar MA, Kwapiszewska G, Reiss I, Amarie OV, Marsh LM, Sevilla-Perez J, et al. Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol (2007) 292:L537–49. doi:10.1152/ajplung.00050.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Nakanishi H, Sugiura T, Streisand JB, Lonning SM, Roberts JD Jr. TGF-beta-neutralizing antibodies improve pulmonary alveologenesis and vasculogenesis in the injured newborn lung. Am J Physiol Lung Cell Mol Physiol (2007) 293:L151–61. doi:10.1152/ajplung.00389.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Gauldie J, Galt T, Bonniaud P, Robbins C, Kelly M, Warburton D. Transfer of the active form of transforming growth factor-beta 1 gene to newborn rat lung induces changes consistent with bronchopulmonary dysplasia. Am J Pathol (2003) 163:2575–84. doi:10.1016/S0002-9440(10)63612-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol (2002) 283:L555–62. doi:10.1152/ajplung.00408.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. McGrath-Morrow SA, Cho C, Zhen L, Hicklin DJ, Tuder RM. Vascular endothelial growth factor receptor 2 blockade disrupts postnatal lung development. Am J Respir Cell Mol Biol (2005) 32:420–7. doi:10.1165/rcmb.2004-0287OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: lung development, alveologenesis, bronchopulmonary dysplasia, epithelial–mesenchymal interaction, endothelial–mesenchymal interaction, secondary septae formation
Citation: Chao C-M, El Agha E, Tiozzo C, Minoo P and Bellusci S (2015) A breath of fresh air on the mesenchyme: impact of impaired mesenchymal development on the pathogenesis of bronchopulmonary dysplasia. Front. Med. 2:27. doi: 10.3389/fmed.2015.00027
Received: 06 February 2015; Accepted: 11 April 2015;
Published: 28 April 2015
Edited by:
Anne Hilgendorff, Helmholtz Zentrum München, GermanyAndrew Bush, Royal Brompton Hospital and Imperial College London, UK
Reviewed by:
Koshika Yadava, Stanford University, USAPaschalis Sideras, Biomedical Research Foundation Academy of Athens, Greece
Copyright: © 2015 Chao, El Agha, Tiozzo, Minoo and Bellusci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saverio Bellusci, Department of Internal Medicine II, Universities of Giessen and Marburg Lung Center (UGMLC), Klinikstraβe 36, Giessen, Hessen 35392, Germany, saverio.bellusci@innere.med.uni-giessen.de