Striatal cholinergic interneurons in isolated generalized dystonia—rationale and perspectives for stem cell-derived cellular models
 Philipp Capetian
Philipp Capetian Martje Gesine Pauly1
Martje Gesine Pauly1 - 1Institute of Neurogenetics, University of Lübeck, Lübeck, Germany
- 2Laboratory of Stereotaxy and Interventional Neuroscience, Department of Stereotactic and Functional Neuroscience, University Medical Center, Freiburg, Germany
Interneurons comprise a minority of the striatal neuronal population of roughly 5%. However, this heterogeneous population is of particular interest as it fulfills an important relay function in modulating the output of the only type of striatal projection neurons, i.e., the medium spiny neuron (MSN).One subtype of this heterogenous group, the cholinergic interneuron, is of particular scientific interest as there is a relevant body of evidence from animal models supporting its special significance in the disease process. The development of protocols for directed differentiation of human pluripotent stem cells (PSC) into striatal interneurons provides a unique opportunity to derive in vitro those cell types that are most severely affected in dystonia.In this review we first aim to give a concise overview about the normal function of striatal interneurons and their dysfunction in dystonia in order to identify the most relevant interneuronal subtype for the pathogenesis of dystonia. Secondly we demonstrate how knowledge about the embryonic development of striatal interneurons is of particular help for the development of differentiation protocols from PSC and by this depict potential ways of deriving in vitro disease models of dystonia. We furthermore address the question as to whether cell replacement therapies might represent a beneficial approach for the treatment of dystonia.
Introduction
Dystonias are a heterogeneous group of movement disorders characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both. Dystonic movements are typically patterned and twisting and may be tremulous. Dystonia is often initiated or worsened by voluntary action and associated with overflow muscle activation (Albanese et al., 2013). Distribution of dystonia ranges from focal (one body region), segmental (two or more adjacent regions) to generalized forms (Geyer and Bressman, 2006). While complex dystonias arise from either brain damage or pharmacological/toxicological alterations, isolated dystonias have either no evident cause (previously referred to as idiopathic dystonias) or are due to inherited or spontaneous mutations in the genome (familial dystonias). In contrast, combined dystonias show additional symptoms (such as parkinsonism or myoclonus) in addition to the dystonia (Klein et al., 2014). Isolated dystonias can be further substratified with respect to body distribution into generalized, segmental, multifocal or focal forms. In the present review, we will focus on generalized, isolated dystonia which usually starts in childhood in a limb and progresses to generalize, however usually spares the neck and face (Warner and Jarman, 1998).
Although Oppenheim already presumed an organic cause of generalized dystonia in 1911 and even identified “hereditary burden to play a major role” (Oppenheim, 1911; Klein and Fahn, 2013), the bizarre nature of some manifestations and the absence of any structural abnormalities or degeneration in the isolated dystonias led to the hypothesis of dystonia being an entirely psychogenic group of disorders (Lanska, 2010). The first described mutation for generalized isolated dystonia was a GAG deletion in the Tor1a gene causing DYT-TOR1A (also known as DYT1) (Ozelius et al., 1997). Although additional genes have been implicated in generalized forms of dystonia in the meantime, DYT-TOR1A accounts for roughly 40% of the generalized dystonias (Valente et al., 1998) and reaches up to 90% in Ashkenazi Jews due to a founder effect (Kramer et al., 1994).
The emergence of animal models of DYT-TOR1A shed some light on the disease mechanism of dystonias and highlighted the role of striatal cholinergic interneurons in this process (Pisani et al., 2007). The advent of induced pluripotent stem cells (iPS) as a source of patient-derived and patient-specific cell lines (Takahashi et al., 2007; Park et al., 2008) also accelerated the development of neural induction and differentiation protocols offering the potential to generate regional specified human neurons in vitro (Chambers et al., 2009; Crompton et al., 2013; Liu et al., 2013b), paving the way for “disease in a dish” modeling.
Since no stem cell-derived in vitro model of dystonias has become available to date, our main goal in this review is to provide a potential “roadmap” for achieving this goal. The development of stem cell-derived in vitro models of neurological diseases requires a broad perspective including knowledge about the physiological function and pathological dysfunction, as well as insight into the embryogenesis of the affected cell type, since these serve the basis for the successful generation of directed differentiation protocols and their subsequent evaluation.
When it comes to modeling of diseases with iPS, the best suited model for this purpose are inherited conditions which are based on a known gene or mutations. Since all somatic cells of the organism carry this mutation, so will the cells being reprogrammed and the derived iPS. We will limit this review on the best-characterized form of inherited dystonia, i.e., isolated generalized DYT-TOR1A and will focus on findings from the corresponding animal model and the striatal cholinergic interneuron as the cell type with the best documented functional alterations in this context.
As another possibility of employing stem cell-derived striatal interneurons, we will discuss whether cell replacement strategies might represent a beneficial approach in treating certain subgroups of dystonia patients.
Striatal Interneurons
The human striatum is composed of two morphologically but not functionally distinct parts: the putamen and the caudate ncl. It is an integral part of the basal ganglia and serves as a relay station integrating glutamatergic input from the cortex and thalamus and dopaminergic input from the substantia nigra for selection, facilitation or suppression of action and movement (Groenewegen, 2003). The majority of striatal neurons (95%) are GABAergic projection neurons, the so called medium spiny neurons (MSNs).
The remaining 5% of striatal neurons are formed by a heterogeneous population of interneurons that modulate the striatal output of the MSN. At least five classes of interneurons can be subdivided in the mammalian striatum based on size, neurotransmitter, (immuno)cytochemical, and electrophysiological properties (Kawaguchi, 1993): neurons of the first group are medium-sized, GABAergic, express the calcium-binding protein parvalbumin and show a fast spiking activity (FS). The second group is also medium-sized and GABAergic; based on its electrophysiological characteristics cells of this group are termed persistent and low-threshold spike (PLTS) cells. They express the neuropeptides somatostatin (SOM), neuropeptide Y (NPY) and the nitric oxide synthase (NOS; Figueredo-Cardenas et al., 1996). Interneurons of the third group are small- to medium-sized, GABAergic, and express the calcium-binding protein calretinin (Tepper et al., 2010). The fourth group contains the largest cells of the striatum, which are, in contrast to all other striatal neurons, cholinergic and show a long-lasting afterhyperpolarization (LA) after excitation (Kawaguchi et al., 1995). The fifth subtype is comprised of medium-sized neurons, which are also GABAergic despite their positivity for tyrosine hydroxylase (TH), electrophysiologically, the latter can be subdivided into at least four subgroups (Ibáñez-Sandoval et al., 2010).
In contrast to other striatal cells, striatal cholinergic interneurons show a spontaneous spiking acitivity, leading to the term “tonically active neurons” (TAN; Bennett and Wilson, 1999). In vivo recordings in primates demonstrated that salient stimuli evoked a short bursting of these neurons followed by a pause in firing, a response also elicited by conditioned stimuli after classical conditioning (Aosaki et al., 1994). These findings were detailed by in vitro recordings of acute rodent brain slices consisting of the cortico-striatal and thalamo-striatal connections: glutamatergic thalamo-striatal projections on cholinergic interneurons evoke the burst-pause response which suppresses transiently cortico-striatal projections on MSN, followed however by a phase of enhanced responsiveness of MSNs to cortical inputs in terms of an enhanced connectivity (Ding et al., 2010). It is therefore assumed that those interneurons mediate a “stop and no-go” response to salient stimuli followed by a redirection of attention. This response is modulated by dopaminergic nigro-striatal projections innervating the cholinergic interneurons by D2 receptors (D2R; Reynolds et al., 2004; Smith and Villalba, 2008). As G-protein-coupled receptors, D2R inhibit voltage-gated sodium channels (NaV 1.1, 1.2 and 1.6) and reduce the spontaneous spiking activity of cholinergic interneurons (Carr et al., 2003). They, however, also act in an inhibitory manner on N-type CaV 2.2 calcium channels that promote the opening of hyperpolarizing inward-rectifying potassium channels (Kir3) (Goldberg and Wilson, 2005). Under physiological conditions the resulting net effect of an activation of D2R is a reduction of spiking activity (see Figure 1). For a summary of the different interneuronal subtypes, see Table 1.
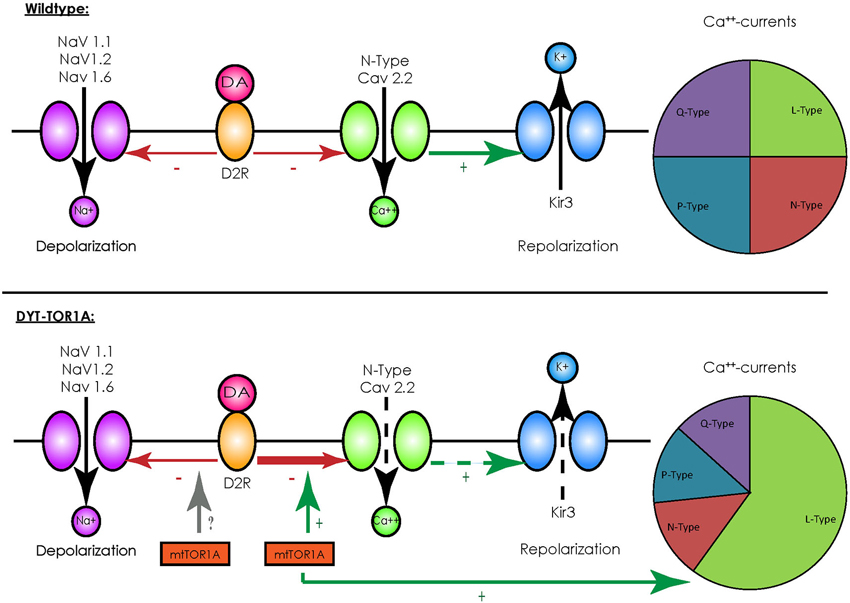
Figure 1. Overview of D2-receptor coupling in striatal cholinergic interneurons. Under physiological conditions (upper panel) binding of dopamine (DA) to D2 receptors (D2R) has an inhibitory effect on voltage-activated sodium channels (NaV 1.1, 1.2 and 1.6), which depolarize the cell membrane, and to N-type calcium channels (CaV 2.2), which activate inward-rectifying sodium channels (Kir3) and hyperpolarize the cell membrane. Although this represents two opposing effects, the net effect of D2R-activity results in a reduction of spiking and the spontaneous firing rate. In the presence of mutated TorsinA (mtTor1A) (lower panel), the inhibitory coupling of D2R to CaV 2.2 is enhanced and the amount of currents through the N-type channels in proportion to the total cellular calcium currents is increased (represented by the changes in the pie charts). This shifts the net effect of D2R towards enhanced activity of cholinergic interneurons. Whether mutant TorsinA also affects the coupling of D2R to NaVs is currently unknown.
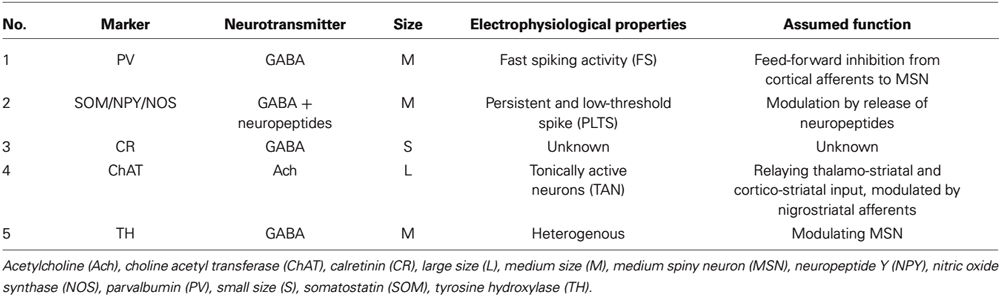
Table 1. Overview of striatal interneurons.
Striatal Cholinergic Interneurons in DYT-TOR1A
In order to study isolated dystonias on an experimental basis, appropriate animal models are needed. Animal models of dystonia are manifold and can be divided into a toxic and a genetic category: the genetic category better reproduces molecular and neurophysiologic features, however, the motor phenotype is typically more subtle (for a complete review see Oleas et al., 2013; Wilson and Hess, 2013). Although there are many known genes, mutations in which lead to dystonia (Lohmann and Klein, 2013) animal models are largely unavailable.
As mentioned above, DYT-TOR1A dystonia is caused by a heterozygous 3-bp GAG deletion in the Tor1a gene (Ozelius et al., 1997). The product of the gene (TorsinA) is as a member of the AAA+ (ATPases associated with a variety of cellular activities) protein family and fulfills a widespread role in vesicle trafficking and serves as a chaperone in protein processing (Hanson and Whiteheart, 2005). The deletion of GAG results in the loss of a c-terminal glutamic acid residue and is thought to destabilize the tertiary structure of the protein acting as to a loss-of-function (Kock et al., 2006). Moreover mutant torsinA impairs the function of wildtype protein and is therefore exerting a dominant negative effect (Torres et al., 2004). The neuron-specific effect is believed to be the result of a lack of other forms of torsins, notably torsinB in neurons, that could compensate for the relative shortage of functional torsin (Jungwirth et al., 2010). Certain levels of normal torsinA are obviously crucial for the embryonic development as complete knock-outs, but also combinations of knock-out of wildtype and knock-in of mutant torsinA, resulting in neonatal lethality (Goodchild et al., 2005; Yokoi et al., 2012).
Animal models for DYT-TOR1A have been established for invertebrates (namely C. elegans and D. melanogaster) (Caldwell et al., 2003; Koh et al., 2004; Cao et al., 2005) which are suitable models for studying basic principles of the disease mechanisms on a multi-cellular level. However, the vast differences to the mammalian central nervous system limit the explanatory value for complex neuronal networks.
More relevant in terms of understanding alterations in neuronal networks in DYT-TOR1A are of course rodent models. Of these, the most relevant one is the Dyt1 ΔGAG, which consists of a heterozygous GAG deletion in the murine TOR1A gene, the same recurrent mutation observed in DYT-TOR1A patients. Another group of models are brain region-specific knock-outs of TOR1A based on the CRE-lox-system (Yokoi et al., 2008; Sciamanna et al., 2012a). These models are particularly helpful for studying the contribution of different cerebral regions to the disease process. The third group is comprised of transgenic models engineered to overexpress human mutant or wildtype torsinA driven by different promoters, such as the neuron-specific enolase (NSE; Shashidharan et al., 2005) or the constitutively active human cytomegalovirus (hCMV) promoter (Sharma et al., 2005). However, the results of this latter group of models need to be interpreted with care as observed effects could also be attributed to ectopic expression or unphysiologically high levels of torsinA.
A particular motor phenotype was found in the mouse model expressing the human mutated TOR1A gene under control of the NSE promoter (Shashidharan et al., 2005): transgenic animals showed enhanced motor activity in the open field test which increased significantly over time, and some animals displayed an abnormal shaking of the head from one side to the other or even tonic lateralized deviations of it. Another feature was “self clasping” of the limbs when hung from the tail. Admittedly, this phenotype differs from the pronounced generalized dystonia known from patients suffering from DYT-TOR1A, however, it must be kept in mind that—as we learned from other transgenic models of neurodegenerative diseases (like the transgenic models of Huntington’s disease (Figiel et al., 2012))—motor abnormalities in rodents often differ considerably from those seen in humans.
Despite these drawbacks, animal models have helped to identify alterations in neuronal networks possibly leading to dystonia. Their value was further emphasized by the fact that a reduction of the dopamine-binding capacity of striatal type 2 dopamine-receptors (D2R; Dang et al., 2012), as well as alterations in the cerebellothalamocortical pathways could be observed (Uluğ et al., 2011), findings already known from human mutation carriers (Asanuma et al., 2005; Carbon et al., 2010a,b). The transgenic knock-in models have been extensively characterized on an electrophysiological level and broadened our understanding of the neuronal network alterations potentially leading to dystonia: for example, recordings of MSNs in mice overexpressing mutant human torsinA showed a pronounced alteration of glutamatergic synaptic transmission. While a long-term depression (LTD) could not be elicited, long-term potentiation (LTP) was more pronounced and synaptic depotentiation (SD) to low-frequency stimuli (LFS) could not be evoked (Martella et al., 2009). These alterations could be reverted by either lowering acetylcholine levels or antagonizing muscarinic receptors of the M1-type linking these changes to altered cholinergic transmission. Recordings of striatal cholinergic interneurons did indeed reveal an altered response to thalamic stimulation, as the evoked burst-pause response was significantly shorter and followed by an abnormal spiking activity (Sciamanna et al., 2012b). As the pause response is a D2R-dependent process (Goldberg and Reynolds, 2011), a link between the already mentioned altered dopaminergic response in DYT-TOR1A and this phenomenon seems likely. Accordingly, a profound alteration of the D2R-mediated response could be demonstrated in striatal cholinergic interneurons, as their activation caused a paradoxical excitation instead the expected inhibition (Pisani et al., 2006). The cause of this alteration is an enhanced inhibitory coupling of D2R to the previously mentioned N-type Cav 2.2 channels and an increased proportion of their currents to the overall cellular calcium currents, leading to a rise in spiking activity (Figure 1). The cause of this enhanced inhibitory coupling is at the moment unclear, however, it is tempting to speculate that the regulator of G-protein signaling 9–2 (RGS 9–2), which was shown to modulate the coupling between D2R and Cav 2.2, might play a role in this regard. However, this does not answer the question whether the inhibitory coupling to the voltage-gated sodium channels (which should counterbalance this effect) is not enhanced (Cabrera-Vera et al., 2004). In addition, these results need to be interpreted with caution as the ubiquitous over expression of mutant torsinA in this model does not closely correspond to the actual situation in DYT-TOR1A. However, these findings were reproduced in a mouse model with a targeted deletion of TOR1A in cholinergic interneurons, supporting the findings that mutant torsinA exerts a dominant-negative effect on wildtype torsinA (Sciamanna et al., 2012a).
The key point is, however, how these findings explain the occurrence of the dystonic movement disorder? Current models of basal ganglia function are based on the presence of parallel distinct loops connecting the cortex with different parts of the basal ganglia and thus facilitating wanted and suppressing unwanted movement. In contrast, dystonia results from a co-contraction of muscle groups usually not activated during a particular task, a phenomenon termed “dystonic overflow” (Mink, 2003). As the striatum plays an important role in the basal ganglia circuitry, it is tempting to speculate that enhanced LTP and reduced LTD based on an increased cholinergic tone in dystonia favors the promotion of aberrant and contradicting motor commands (Sciamanna et al., 2012b), however, more experimental data will be needed to further clarify this matter.
While the role of GABAergic interneurons in DYT1 has not yet been studied in depth, one study found altered intracellular calcium dynamics in heterozygous ΔE-torsinA knock-in mice leading to a higher excitability of those cells (Iwabuchi et al., 2013).
Embryonal Development of Striatal Interneurons
The striatum (caudate nucleus and pallidum) develops from the lateral (LGE) and medial ganglionic eminence (MGE) located in the basal part of the telecephalon. While striatal projection neurons (namely MSN) arise from the lateral ganglionic eminence (Deacon et al., 1994), almost all striatal interneurons as well as cortical interneurons, cholinergic basal forebrain projection neurons, projection neurons of the globus pallidus, and telencephalic oligodendrocytes are generated in the MGE and reach their final destination either by migrating tangentially or radially (Anderson et al., 1999; Marin et al., 2000; Kessaris et al., 2006). The ventricular zone (VZ) of the MGE comprises neural progenitors expressing the NKX2.1 homeodomain protein, required for their specification (Butt et al., 2008), their patterning being in turn dependent on the morphogen sonic hedgehog (SHH; Xu et al., 2010). After exiting the cell cycle, postmitotic NKX2.1+ progenitors start migrating out of the MGE and express the LIM-homeodomain-transcription factor LHX6 (Du et al., 2008). This process is dependent on the transcription factors DLX1/2 and MASH1 (Marin et al., 2000). These immature neurons also contain GABA and share certain functional characteristics with mature GABAergic neurons, however, lack other molecular markers of specified interneuronal populations (like PV, SOM, NOS), so that they are termed proto-GABAergic (Flames and Marín, 2005). These cells are bipotential precursors being able to terminally differentiate either into the GABAergic or cholinergic lineage. Terminal differentiation into GABAergic interneurons is dependent on sustained expression of LHX6 and thus representing a sort of “default” maturation pathway (Liodis et al., 2007; Zhao et al., 2008). Differentiation into the cholinergic lineage requires expression of LHX7 (also known as L3/LHX8) which induces expression of another LIM-homeodomain-transcription factor islet-1 (ISL1), which directly downregulates LHX6 expression (Fragkouli et al., 2009). Interestingly, the cholinergic phenotype remains dependent on ongoing LHX7 expression even in postmitotically committed cholinergic neurons, as deletion of the LHX7 gene leads to a switch of phenotype to GABAergic interneurons (Lopes et al., 2012). The processes described above are summarized in Figure 2.
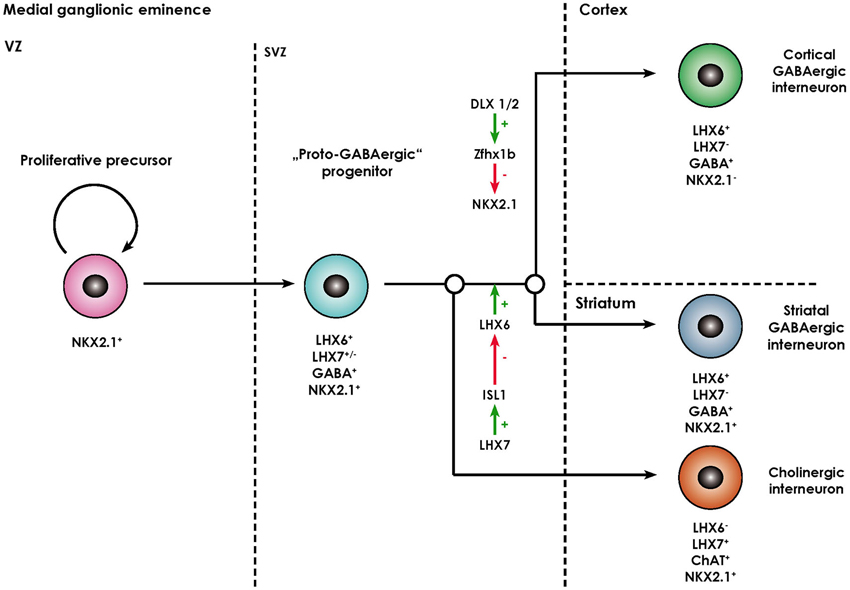
Figure 2. Overview of the molecular processes involved in the specification of MGE-derived forebrain interneurons (adapted from Fragkouli et al., 2009). Forebrain interneurons are derived from NKX2.1+ proliferative progenitors located in the ventricular zone (VZ) of the MGE. Once they exit the cell cycle they move to the subventricular zone (SVZ) and start expressing LHX6. As they contain GABA, they are termed “proto-GABAergic”. When LHX6 expression is maintained, they either continue to express NKX2.1 and differentiate towards striatal GABAergic interneurons. Alternatively, the zinc finger homeobox gene Zfhx1b, which acts downstream of DLX1/2, represses NKX2.1 expression, in which case cortical GABAergic interneurons arise. Through ISL1, LHX7 expression leads to a repression of LHX6 and thus differentiation towards cholinergic neurons positive for choline acetyl transferase (ChAT).
Deriving Striatal Interneurons from Pluripotent Stem Cells
Although animal models have been of great help in elucidating altered cellular mechanisms in dystonia and will most likely remain unsurpassed when it comes to the study of complex neural networks, obtaining human neurons is highly desirable in order to confirm findings from animal models or gaining new insights into mechanisms of the human basal ganglia. Pluripotent stem cells (PSC) can be differentiated into cells of the entire body, including neurons. iPS even allow generating patient-specific cells, sharing all possible genetic deviations and variants of the donor patient (Yamanaka, 2009).
In order to obtain a neuronal population enriched for a desired regional phenotype, it is necessary to recapitulate the in vivo developmental processes under in vitro conditions.
Patterning primitive neuroectoderm or neuroepithelial precursors in order to obtain a desired regional phenotype requires the presence of morphogens mediating the expression of downstream effectors, the most important in this respect being SHH, fibroblast growth factors (FGFs), bone morphogenetic proteins (BMPs), and agonists of the wingless/Int-1 pathway (WNT; Hébert and Fishell, 2008; Sousa and Fishell, 2010; Cho et al., 2013). Patterning neural cells in vitro requires the addition of morphogens in a timed manner and at appropriate concentrations. Since almost all forebrain interneurons arise from the MGE in the ventral forebrain, the addition of morphogens required for ventral forebrain specification to the cell culture should promote a corresponding phenotype. Accordingly, all published protocols utilize the strong ventralizing activity of SHH or its small molecule agonists (Bissonnette et al., 2011; Crompton et al., 2013; Germain et al., 2013; Liu et al., 2013a,b; Nicholas et al., 2013; Duan et al., 2014). In this respect, a dose dependency was demonstrated, as increasing the dose of SHH increased the number of NKX 2.1+ MGE-progenitors with doses ranging up to 1000 ng/ml (Li et al., 2009; Liu et al., 2013b). Another common component for MGE patterning is FGF8 due to its important role in ventral forebrain patterning (Storm et al., 2006). Although many published protocols include FGF8 (Bissonnette et al., 2011; Danjo et al., 2011), there are also reports omitting FGF8. As SHH concentrations used are higher in these protocols, it can be speculated that FGF8 can be replaced by high SHH concentrations (Liu et al., 2013a,b). The canonical WNT/β-catenin-pathway plays an important role in forebrain patterning both on the rostro-caudal- and ventro-dorsal axis: patterning of the dorsal telencephalon depends on activation of canonical WNT signaling (Backman et al., 2005). Conversely, cells of the ventrally localized VZ express the dickkopf proteins (a family of WNT-antagonists) (DKK; Diep et al., 2004) and combined SHH and DKK1 exposure enhances ventral telencephalic identity (Li et al., 2009; Nicoleau et al., 2013). Many protocols achieving successful MGE patterning however do not employ WNT modulation. A potential reason for this could be the fact that DKK alone has no pronounced ventralizing effect but rather potentiates SHH activity (Li et al., 2009), so that high levels of SHH alone could also be sufficient. The pronounced rostralizing effect of DKK1 (Caneparo et al., 2007) is obviously not necessary for successful patterning of in vitro ventral-forebrain patterning, since neural induction of PSCs leads to an anterior-dorsal identity by default (Pankratz et al., 2007).
Under optimal conditions, over 90% of the cells express NKX 2.1, thereby demonstrates MGE identity. These neurons are 45% cholinergic and 55% GABAergic (Liu et al., 2013b). This underlines the general fact that in vitro patterning can indeed specify the regional identity of cells in a highly effective manner, however, full specification to a single cell type (e.g., solely cholinergic) cannot be achieved in this way. One more recent protocol reported achievement of 94% forebrain cholinergic interneurons by transient overexpression of GBX1 and LHX7 (L3/LHX8) (Bissonnette et al., 2011). This example demonstrates how knowledge of molecular pathways in neural differentiation can help increase the specificity of directed differentiation beyond the scope of regional patterning.
Rationale for Cell Replacement Strategies in Dystonia
Cell replacement strategies as potential treatment for neurodegenerative disorders have been carried out for a couple of decades with promising results in animal models but mixed outcome in clinical trials (Bjorklund and Kordower, 2013; Kim et al., 2013; Lindvall, 2013). While replacing a subpopulation of neurons seems at least in theory a feasible approach for treating neurodegenerative disorders, isolated dystonias as the DYT-TOR1A lack an overt cell loss. Accordingly, there are no reports of neural cell transplantation approaches in the current literature. Assuming the area of application for neural cell transplantation is neurodegenerative disorders with predominant affection of a confined cell type, isolated dystonias do obviously not qualify for such an approach due to the lack of neurodegeneration. On the other hand, complex dystonias frequently show too widespread a damage (Klein et al., 2014). Possibly the only potentially promising predominantly dystonic movement disorder is the combined dystonia DYT-TAF1 (X-linked dystonia parkinsonism = XDP). This disease is characterized by an adult-onset initial phase of generalized dystonia with oromandibular predominance, followed by a second predominantly hypokinetic-rigid phase (Klein et al., 2014). This disease is caused by a pronounced degeneration of MSNs inside the striosome compartment in the first stage of the disease, followed by a degeneration of MSNs inside the matrix compartment during the second phase, while relatively sparing the striatal interneurons (Goto et al., 2005; Kaji et al., 2005). Therefore, a possible neurorestaurative approach to DYT-TAF1 would indeed be the transplantation of precursors for MSNs out of the LGE, similarly as in Huntington’s disease.
Conclusions
The absence of neurodegeneration identifies isolated dystonias as neuronal network diseases. Interneurons representing roughly 5% of the striatal neural population play an important role in modifying the output of the MSN as projection neurons and relaying thalamo-striatal and cortico-striatal inputs, especially cholinergic interneurons play an important role in integrating sensory input and redirecting attention. Using the example of DYT-TOR1A as the best studied form of isolated dystonia, we present the current knowledge as to how striatal cholinergic interneurons may play a central role in the pathophysiology of dystonia through an altered response to dopaminergic input leading to overactivity and dysfunctional gating of thalamostriatal input.
We summarized the origins and developmental processes of forebrain interneurons in combination with the molecular mechanisms that switch between GABAergic and cholinergic phenotypes.
We demonstrated on the basis of recent protocols, how knowledge of the developmental mechanisms can be employed for directed neural differentiation of PSC in order to obtain human forebrain interneurons in vitro. Although to date no published study employed PSC-derived striatal interneurons in order to study dystonia, this technique holds great potential for deciphering the underlying disease mechanisms of dystonia, as the necessary protocols have already been published and just await their application for in vitro modeling of DYT-TOR1A. We finally discuss potential neurorestaurative approaches for different subtypes of dystonias: while isolated dystonias most likely are no suitable candidates for this procedure due to the lack of neurodegeneration and complex dystonias are commonly characterized by too widespread a neural damage, transplantation of MSN precursors could be a potential field of application in XDP or DYT-TAF1 with its relatively confined degeneration of striosomes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Albanese, A., Bhatia, K., Bressman, S. B., Delong, M. R., Fahn, S., Fung, V. S. C., et al. (2013). Phenomenology and classification of dystonia: a consensus update. Mov. Disord. 28, 863–873. doi: 10.1002/mds.25475
Anderson, S., Mione, M., Yun, K., and Rubenstein, J. L. (1999). Differential origins of neocortical projection and local circuit neurons: role of Dlx genes in neocortical interneuronogenesis. Cereb. Cortex 9, 646–654. doi: 10.1093/cercor/9.6.646
Aosaki, T., Tsubokawa, H., Ishida, A., Watanabe, K., Graybiel, A. M., and Kimura, M. (1994). Responses of tonically active neurons in the primate’s striatum undergo systematic changes during behavioral sensorimotor conditioning. J. Neurosci. 14, 3969–3984.
Asanuma, K., Ma, Y., Okulski, J., Dhawan, V., Chaly, T., Carbon, M., et al. (2005). Decreased striatal D2 receptor binding in non-manifesting carriers of the DYT1 dystonia mutation. Neurology 64, 347–349. doi: 10.1212/01.wnl.0000149764.34953.bf
Backman, M., Machon, O., Mygland, L., van den Bout, C. J., Zhong, W., Taketo, M. M., et al. (2005). Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev. Biol. 279, 155–168. doi: 10.1016/j.ydbio.2004.12.010
Bennett, B. D., and Wilson, C. J. (1999). Spontaneous activity of neostriatal cholinergic interneurons in vitro. J. Neurosci. 19, 5586–5596.
Bissonnette, C. J., Lyass, L., Bhattacharyya, B. J., Belmadani, A., Miller, R. J., and Kessler, J. A. (2011). The controlled generation of functional basal forebrain cholinergic neurons from human embryonic stem cells. Stem Cells 29, 802–811. doi: 10.1002/stem.626
Bjorklund, A., and Kordower, J. H. (2013). Cell therapy for Parkinson’s disease: what next? Mov. Disord. 28, 110–115. doi: 10.1002/mds.25343
Butt, S. J. B., Sousa, V. H., Fuccillo, M. V., Hjerling-Leffler, J., Miyoshi, G., Kimura, S., et al. (2008). The requirement of Nkx2–1 in the temporal specification of cortical interneuron subtypes. Neuron 59, 722–732. doi: 10.1016/j.neuron.2008.07.031
Cabrera-Vera, T. M., Hernandez, S., Earls, L. R., Medkova, M., Sundgren-Andersson, A. K., Surmeier, D. J., et al. (2004). RGS9–2 modulates D2 dopamine receptor-mediated Ca2+ channel inhibition in rat striatal cholinergic interneurons. Proc. Natl. Acad. Sci. U S A 101, 16339–16344. doi: 10.1073/pnas.0407416101
Caldwell, G. A., Cao, S., Sexton, E. G., Gelwix, C. C., Bevel, J. P., and Caldwell, K. A. (2003). Suppression of polyglutamine-induced protein aggregation in Caenorhabditis elegans by torsin proteins. Hum. Mol. Genet. 12, 307–319. doi: 10.1093/hmg/12.3.307
Caneparo, L., Huang, Y.-L., Staudt, N., Tada, M., Ahrendt, R., Kazanskaya, O., et al. (2007). Dickkopf-1 regulates gastrulation movements by coordinated modulation of Wnt/beta catenin and Wnt/PCP activities, through interaction with the Dally-like homolog Knypek. Genes Dev. 21, 465–480. doi: 10.1101/gad.406007
Cao, S., Gelwix, C. C., Caldwell, K. A., and Caldwell, G. A. (2005). Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J. Neurosci. 25, 3801–3812. doi: 10.1523/jneurosci.5157-04.2005
Carbon, M., Argyelan, M., and Eidelberg, D. (2010a). Functional imaging in hereditary dystonia. Eur. J. Neurol. 17(Suppl. 1), 58–64. doi: 10.1111/j.1468-1331.2010.03054.x
Carbon, M., Argyelan, M., Habeck, C., Ghilardi, M. F., Fitzpatrick, T., Dhawan, V., et al. (2010b). Increased sensorimotor network activity in DYT1 dystonia: a functional imaging study. Brain 133, 690–700. doi: 10.1093/brain/awq017
Carr, D. B., Day, M., Cantrell, A. R., Held, J., Scheuer, T., Catterall, W. A., et al. (2003). Transmitter modulation of slow, activity-dependent alterations in sodium channel availability endows neurons with a novel form of cellular plasticity. Neuron 39, 793–806. doi: 10.1016/s0896-6273(03)00531-2
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. doi: 10.1038/nbt.1529
Cho, G.-S., Choi, S.-C., and Han, J.-K. (2013). BMP signal attenuates FGF pathway in anteroposterior neural patterning. Biochem. Biophys. Res. Commun. 434, 509–515. doi: 10.1016/j.bbrc.2013.03.105
Crompton, L. A., Byrne, M. L., Taylor, H., Kerrigan, T. L., Bru-Mercier, G., Badger, J. L., et al. (2013). Stepwise, non-adherent differentiation of human pluripotent stem cells to generate basal forebrain cholinergic neurons via hedgehog signaling. Stem Cell Res. 11, 1206–1221. doi: 10.1016/j.scr.2013.08.002
Dang, M. T., Yokoi, F., Cheetham, C. C., Lu, J., Vo, V., Lovinger, D. M., et al. (2012). An anticholinergic reverses motor control and corticostriatal LTD deficits in Dyt1 ΔGAG knock-in mice. Behav. Brain Res. 226, 465–472. doi: 10.1016/j.bbr.2011.10.002
Danjo, T., Eiraku, M., Muguruma, K., Watanabe, K., Kawada, M., Yanagawa, Y., et al. (2011). Subregional specification of embryonic stem cell-derived ventral telencephalic tissues by timed and combinatory treatment with extrinsic signals. J. Neurosci. 31, 1919–1933. doi: 10.1523/JNEUROSCI.5128-10.2011
Deacon, T. W., Pakzaban, P., and Isacson, O. (1994). The lateral ganglionic eminence is the origin of cells committed to striatal phenotypes: neural transplantation and developmental evidence. Brain Res. 668, 211–219. doi: 10.1016/0006-8993(94)90526-6
Diep, D. B., Hoen, N., Backman, M., Machon, O., and Krauss, S. (2004). Characterisation of the Wnt antagonists and their response to conditionally activated Wnt signalling in the developing mouse forebrain. Brain Res. Dev. Brain Res. 153, 261–270. doi: 10.1016/j.devbrainres.2004.09.008
Ding, J. B., Guzman, J. N., Peterson, J. D., Goldberg, J. A., and Surmeier, D. J. (2010). Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron 67, 294–307. doi: 10.1016/j.neuron.2010.06.017
Du, T., Xu, Q., Ocbina, P. J., and Anderson, S. A. (2008). NKX2.1 specifies cortical interneuron fate by activating Lhx6. Development 135, 1559–1567. doi: 10.1242/dev.015123
Duan, L., Bhattacharyya, B. J., Belmadani, A., Pan, L., Miller, R. J., and Kessler, J. A. (2014). Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death. Mol. Neurodegener. 9:3. doi: 10.1186/1750-1326-9-3
Figueredo-Cardenas, G., Morello, M., Sancesario, G., Bernardi, G., and Reiner, A. (1996). Colocalization of somatostatin, neuropeptide Y, neuronal nitric oxide synthase and NADPH-diaphorase in striatal interneurons in rats. Brain Res. 735, 317–324. doi: 10.1016/0006-8993(96)00801-3
Flames, N., and Marín, O. (2005). Developmental mechanisms underlying the generation of cortical interneuron diversity. Neuron 46, 377–381. doi: 10.1016/j.neuron.2005.04.020
Fragkouli, A., van Wijk, N. V., Lopes, R., Kessaris, N., and Pachnis, V. (2009). LIM homeodomain transcription factor-dependent specification of bipotential MGE progenitors into cholinergic and GABAergic striatal interneurons. Development 136, 3841–3851. doi: 10.1242/dev.038083
Germain, N. D., Banda, E. C., Becker, S., Naegele, J. R., and Grabel, L. B. (2013). Derivation and isolation of NKX2.1-positive basal forebrain progenitors from human embryonic stem cells. Stem Cells Dev. 22, 1477–1489. doi: 10.1089/scd.2012.0264
Geyer, H. L., and Bressman, S. B. (2006). The diagnosis of dystonia. Lancet Neurol. 5, 780–790. doi: 10.1016/S1474-4422(06)70547-6
Goldberg, J. A., and Reynolds, J. N. J. (2011). Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience 198, 27–43. doi: 10.1016/j.neuroscience.2011.08.067
Goldberg, J. A., and Wilson, C. J. (2005). Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J. Neurosci. 25, 10230–10238. doi: 10.1523/jneurosci.2734-05.2005
Goodchild, R. E., Kim, C. E., and Dauer, W. T. (2005). Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron 48, 923–932. doi: 10.1016/j.neuron.2005.11.010
Goto, S., Lee, L. V., Munoz, E. L., Tooyama, I., Tamiya, G., Makino, S., et al. (2005). Functional anatomy of the basal ganglia in X-linked recessive dystonia-parkinsonism. Ann. Neurol. 58, 7–17. doi: 10.1002/ana.20513
Groenewegen, H. J. (2003). The basal ganglia and motor control. Neural Plast. 10, 107–120. doi: 10.1155/NP.2003.107
Hanson, P. I., and Whiteheart, S. W. (2005). AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 6, 519–529. doi: 10.1038/nrm1684
Hébert, J. M., and Fishell, G. (2008). The genetics of early telencephalon patterning: some assembly required. Nat. Rev. Neurosci. 9, 678–685. doi: 10.1038/nrn2463
Ibáñez-Sandoval, O., Tecuapetla, F., Unal, B., Shah, F., Koós, T., and Tepper, J. M. (2010). Electrophysiological and morphological characteristics and synaptic connectivity of tyrosine hydroxylase-expressing neurons in adult mouse striatum. J. Neurosci. 30, 6999–7016. doi: 10.1523/JNEUROSCI.5996-09.2010
Iwabuchi, S., Koh, J.-Y., Wang, K., Ho, K. W. D., and Harata, N. C. (2013). Minimal change in the cytoplasmic calcium dynamics in striatal GABAergic neurons of a DYT1 dystonia knock-in mouse model. PLoS One 8:e80793. doi: 10.1371/journal.pone.0080793. Available online at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3834333/
Jungwirth, M., Dear, M. L., Brown, P., Holbrook, K., and Goodchild, R. (2010). Relative tissue expression of homologous torsinB correlates with the neuronal specific importance of DYT1 dystonia-associated torsinA. Hum. Mol. Genet. 19, 888–900. doi: 10.1093/hmg/ddp557
Kaji, R., Goto, S., Tamiya, G., Ando, S., Makino, S., and Lee, L. V. (2005). Molecular dissection and anatomical basis of dystonia: X-linked recessive dystonia-parkinsonism (DYT3). J. Med. Invest. 52, 280–283. doi: 10.2152/jmi.52.280
Kawaguchi, Y. (1993). Physiological, morphological and histochemical characterization of three classes of interneurons in rat neostriatum. J. Neurosci. 13, 4908–4923.
Kawaguchi, Y., Wilson, C. J., Augood, S. J., and Emson, P. C. (1995). Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 18, 527–535. doi: 10.1016/0166-2236(95)98374-8
Kessaris, N., Fogarty, M., Iannarelli, P., Grist, M., Wegner, M., and Richardson, W. D. (2006). Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 9, 173–179. doi: 10.1038/nn1620
Kim, S. U., Lee, H. J., and Kim, Y. B. (2013). Neural stem cell-based treatment for neurodegenerative diseases. Neuropathology 33, 491–504. doi: 10.1111/neup.12020
Klein, C., and Fahn, S. (2013). Translation of Oppenheim’s 1911 paper on dystonia. Mov. Disord. 28, 851–862. doi: 10.1002/mds.25546
Klein, C., Marras, C., and Münchau, A. (2014). “Dystonia overview,” in GeneReviews®ledR [Internet], eds R. A. Pagon, M. P. Adam, H. H. Ardinger, T. D. Bird, C. R. Dolan, C.-T. Fong, et al. (Seattle, WA: University of Washington, Seattle). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1155/
Kock, N., Naismith, T. V., Boston, H. E., Ozelius, L. J., Corey, D. P., Breakefield, X. O., et al. (2006). Effects of genetic variations in the dystonia protein torsinA: identification of polymorphism at residue 216 as protein modifier. Hum. Mol. Genet. 15, 1355–1364. doi: 10.1093/hmg/ddl055
Koh, Y.-H., Rehfeld, K., and Ganetzky, B. (2004). A Drosophila model of early onset torsion dystonia suggests impairment in TGF-beta signaling. Hum. Mol. Genet. 13, 2019–2030. doi: 10.1093/hmg/ddh208
Kramer, P. L., Heiman, G. A., Gasser, T., Ozelius, L. J., de Leon, D., Brin, M. F., et al. (1994). The DYT1 gene on 9q34 is responsible for most cases of early limb-onset idiopathic torsion dystonia in non-Jews. Am. J. Hum. Genet. 55, 468–475.
Lanska, D. J. (2010). Chapter 33: the history of movement disorders. Handb. Clin. Neurol. 95, 501–546. doi: 10.1016/s0072-9752(08)02133-7
Li, X.-J., Zhang, X., Johnson, M. A., Wang, Z.-B., Lavaute, T., and Zhang, S.-C. (2009). Coordination of sonic hedgehog and Wnt signaling determines ventral and dorsal telencephalic neuron types from human embryonic stem cells. Development 136, 4055–4063. doi: 10.1242/dev.036624
Lindvall, O. (2013). Developing dopaminergic cell therapy for Parkinson’s disease—give up or move forward? Mov. Disord. 28, 268–273. doi: 10.1002/mds.25378
Liodis, P., Denaxa, M., Grigoriou, M., Akufo-Addo, C., Yanagawa, Y., and Pachnis, V. (2007). Lhx6 activity is required for the normal migration and specification of cortical interneuron subtypes. J. Neurosci. 27, 3078–3089. doi: 10.1523/jneurosci.3055-06.2007
Liu, Y., Liu, H., Sauvey, C., Yao, L., Zarnowska, E. D., and Zhang, S.-C. (2013a). Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nat. Protoc. 8, 1670–1679. doi: 10.1038/nprot.2013.106
Liu, Y., Weick, J. P., Liu, H., Krencik, R., Zhang, X., Ma, L., et al. (2013b). Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat. Biotechnol. 31, 440–447. doi: 10.1038/nbt.2565
Lohmann, K., and Klein, C. (2013). Genetics of dystonia: what’s known? What’s new? What’s next?. Mov. Disord. 28, 899–905. doi: 10.1002/mds.25536
Lopes, R., Verhey van Wijk, N., Neves, G., and Pachnis, V. (2012). Transcription factor LIM homeobox 7 (Lhx7) maintains subtype identity of cholinergic interneurons in the mammalian striatum. Proc. Natl. Acad. Sci. U S A 109, 3119–3124. doi: 10.1073/pnas.1109251109
Marin, O., Anderson, S. A., and Rubenstein, J. L. (2000). Origin and molecular specification of striatal interneurons. J. Neurosci. 20, 6063–6076.
Martella, G., Tassone, A., Sciamanna, G., Platania, P., Cuomo, D., Viscomi, M. T., et al. (2009). Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain 132, 2336–2349. doi: 10.1093/brain/awp194
Mink, J. W. (2003). The basal Ganglia and involuntary movements: impaired inhibition of competing motor patterns. Arch. Neurol. 60, 1365–1368. doi: 10.1001/archneur.60.10.1365
Figiel, M., Szlachcic, W. J., Switonski, P. M., Gabka, A., and Krzyzosiak, W. J. (2012). Mouse models of polyglutamine diseases: review and data table. Part I. Mol. Neurobiol. 46, 393–429. doi: 10.1007/s12035-012-8315-4
Nicholas, C. R., Chen, J., Tang, Y., Southwell, D. G., Chalmers, N., Vogt, D., et al. (2013). Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12, 573–586. doi: 10.1016/j.stem.2013.04.005
Nicoleau, C., Varela, C., Bonnefond, C., Maury, Y., Bugi, A., Aubry, L., et al. (2013). ES cells neural differentiation qualifies the role of Wnt/β-catenin signals in human telencephalic specification and regionalization. Stem Cells 31, 1763–1774. doi: 10.1002/stem.1462
Oleas, J., Yokoi, F., DeAndrade, M. P., Pisani, A., and Li, Y. (2013). Engineering animal models of dystonia. Mov. Disord. 28, 990–1000. doi: 10.1002/mds.25583
Oppenheim, H. (1911). Über eine eigenartige Krampfkrankheit des kindlichen und jugendlichen Alters (Dysbasia lordotica progressiva, Dystonia musculorum deformans). Neurol. Zentralbl. 30, 1090–1107.
Ozelius, L. J., Hewett, J. W., Page, C. E., Bressman, S. B., Kramer, P. L., Shalish, C., et al. (1997). The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 17, 40–48. doi: 10.1038/ng0997-40
Pankratz, M. T., Li, X.-J., Lavaute, T. M., Lyons, E. A., Chen, X., and Zhang, S.-C. (2007). Directed neural differentiation of human embryonic stem cells via an obligated primitive anterior stage. Stem Cells 25, 1511–1520. doi: 10.1634/stemcells.2006-0707
Park, I.-H., Arora, N., Huo, H., Maherali, N., Ahfeldt, T., Shimamura, A., et al. (2008). Disease-specific induced pluripotent stem cells. Cell 134, 877–886. doi: 10.1016/j.cell.2008.07.041
Pisani, A., Bernardi, G., Ding, J., and Surmeier, D. J. (2007). Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 30, 545–553. doi: 10.1016/j.tins.2007.07.008
Pisani, A., Martella, G., Tscherter, A., Bonsi, P., Sharma, N., Bernardi, G., et al. (2006). Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol. Dis. 24, 318–325. doi: 10.1016/j.nbd.2006.07.006
Reynolds, J. N. J., Hyland, B. I., and Wickens, J. R. (2004). Modulation of an afterhyperpolarization by the substantia nigra induces pauses in the tonic firing of striatal cholinergic interneurons. J. Neurosci. 24, 9870–9877. doi: 10.1523/jneurosci.3225-04.2004
Sciamanna, G., Hollis, R., Ball, C., Martella, G., Tassone, A., Marshall, A., et al. (2012a). Cholinergic dysregulation produced by selective inactivation of the dystonia-associated protein torsinA. Neurobiol. Dis. 47, 416–427. doi: 10.1016/j.nbd.2012.04.015
Sciamanna, G., Tassone, A., Mandolesi, G., Puglisi, F., Ponterio, G., Martella, G., et al. (2012b). Cholinergic dysfunction alters synaptic integration between thalamostriatal and corticostriatal inputs in DYT1 dystonia. J. Neurosci. 32, 11991–12004. doi: 10.1523/JNEUROSCI.0041-12.2012
Sharma, N., Baxter, M. G., Petravicz, J., Bragg, D. C., Schienda, A., Standaert, D. G., et al. (2005). Impaired motor learning in mice expressing torsinA with the DYT1 dystonia mutation. J. Neurosci. 25, 5351–5355. doi: 10.1523/jneurosci.0855-05.2005
Shashidharan, P., Sandu, D., Potla, U., Armata, I. A., Walker, R. H., McNaught, K. S., et al. (2005). Transgenic mouse model of early-onset DYT1 dystonia. Hum. Mol. Genet. 14, 125–133. doi: 10.1093/hmg/ddi012
Smith, Y., and Villalba, R. (2008). Striatal and extrastriatal dopamine in the basal ganglia: an overview of its anatomical organization in normal and Parkinsonian brains. Mov. Disord. 23(Suppl. 3), S534–S547. doi: 10.1002/mds.22027
Sousa, V. H., and Fishell, G. (2010). Sonic Hedgehog functions through dynamic changes in temporal competence in the developing ventral telencephalon. Curr. Opin. Genet. Dev. 20, 391–399. doi: 10.1016/j.gde.2010.04.008
Storm, E. E., Garel, S., Borello, U., Hebert, J. M., Martinez, S., McConnell, S. K., et al. (2006). Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 133, 1831–1844. doi: 10.1242/dev.02324
Takahashi, K., Okita, K., Nakagawa, M., and Yamanaka, S. (2007). Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2, 3081–3089. doi: 10.1038/nprot.2007.418
Tepper, J. M., Tecuapetla, F., Koos, T., and Ibanez-Sandoval, O. (2010). Heterogeneity and diversity of striatal GABAergic interneurons. Front. Neuroanat. 4:150. doi: 10.3389/fnana.2010.00150. . Available online at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3016690/
Torres, G. E., Sweeney, A. L., Beaulieu, J.-M., Shashidharan, P., and Caron, M. G. (2004). Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated ΔE-torsinA mutant. Proc. Natl. Acad. Sci. U S A 101, 15650–15655. doi: 10.1073/pnas.0308088101
Uluğ, A. M., Vo, A., Argyelan, M., Tanabe, L., Schiffer, W. K., Dewey, S., et al. (2011). Cerebellothalamocortical pathway abnormalities in torsinA DYT1 knock-in mice. Proc. Natl. Acad. Sci. U S A 108, 6638–6643. doi: 10.1073/pnas.1016445108
Valente, E. M., Warner, T. T., Jarman, P. R., Mathen, D., Fletcher, N. A., Marsden, C. D., et al. (1998). The role of DYT1 in primary torsion dystonia in Europe. Brain 121, 2335–2339. doi: 10.1093/brain/121.12.2335
Warner, T., and Jarman, P. (1998). The molecular genetics of the dystonias. J. Neurol. Neurosurg. Psychiatry 64, 427–429. doi: 10.1136/jnnp.64.4.427
Wilson, B. K., and Hess, E. J. (2013). Animal models for dystonia. Mov. Disord. 28, 982–989. doi: 10.1002/mds.25526
Xu, Q., Guo, L., Moore, H., Waclaw, R. R., Campbell, K., and Anderson, S. A. (2010). Sonic hedgehog signaling confers ventral telencephalic progenitors with distinct cortical interneuron fates. Neuron 65, 328–340. doi: 10.1016/j.neuron.2010.01.004
Yokoi, F., Dang, M. T., and Li, Y. (2012). Improved motor performance in Dyt1 ΔGAG heterozygous knock-in mice by cerebellar Purkinje-cell specific Dyt1 conditional knocking-out. Behav. Brain Res. 230, 389–398. doi: 10.1016/j.bbr.2012.02.029
Yokoi, F., Dang, M. T., Mitsui, S., Li, J., and Li, Y. (2008). Motor deficits and hyperactivity in cerebral cortex-specific Dyt1 conditional knockout mice. J. Biochem. 143, 39–47. doi: 10.1093/jb/mvm191
Keywords: striatal interneurons, development, dystonia, disease modeling, induced pluripotent neurons, differentiation
Citation: Capetian P, Pauly MG, Azmitia LM and Klein C (2014) Striatal cholinergic interneurons in isolated generalized dystonia—rationale and perspectives for stem cell-derived cellular models. Front. Cell. Neurosci. 8:205. doi: 10.3389/fncel.2014.00205
Received: 17 March 2014; Accepted: 07 July 2014;
Published online: 28 July 2014.
Edited by:
Màté Döbrössy, University Freiburg-Medical Center, GermanyReviewed by:
Yehezkel Ben-Ari, Institut National de la Santé et de la Recherche Médicale, FranceLaurens Bosman, Erasmus MC, Netherlands
Copyright © 2014 Capetian, Pauly, Azmitia and Klein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philipp Capetian, Institute of Neurogenetics, University of Lübeck, Ratzeburger Allee 160, 23538 Lübeck, Germany e-mail: Philipp.Capetian@neuro.uni-luebeck.de