Targeted pharmacological treatment of autism spectrum disorders: fragile X and Rett syndromes
 Hansen Wang
Hansen Wang Sandipan Pati
Sandipan Pati Lucas Pozzo-Miller
Lucas Pozzo-Miller Laurie C. Doering
Laurie C. Doering- 1Faculty of Medicine, University of Toronto, 1 King’s College Circle, Toronto, ON, Canada
- 2Department of Neurology, Epilepsy Division, The University of Alabama at Birmingham, Birmingham, AL, USA
- 3Department of Neurobiology, Civitan International Research Center, The University of Alabama at Birmingham, Birmingham, AL, USA
- 4Faculty of Health Sciences, Department of Pathology and Molecular Medicine, McMaster University, Hamilton, ON, Canada
Autism spectrum disorders (ASDs) are genetically and clinically heterogeneous and lack effective medications to treat their core symptoms. Studies of syndromic ASDs caused by single gene mutations have provided insights into the pathophysiology of autism. Fragile X and Rett syndromes belong to the syndromic ASDs in which preclinical studies have identified rational targets for drug therapies focused on correcting underlying neural dysfunction. These preclinical discoveries are increasingly translating into exciting human clinical trials. Since there are significant molecular and neurobiological overlaps among ASDs, targeted treatments developed for fragile X and Rett syndromes may be helpful for autism of different etiologies. Here, we review the targeted pharmacological treatment of fragile X and Rett syndromes and discuss related issues in both preclinical studies and clinical trials of potential therapies for the diseases.
Introduction
Autism spectrum disorders (ASDs) encompass a group of neurodevelopmental disorders which are of different etiologies and characterized by impairments in socialization and communication, abnormalities in language development, restricted interests, and repetitive and stereotyped behaviors (Mefford et al., 2012; Zoghbi and Bear, 2012; Murdoch and State, 2013; Anagnostou et al., 2014b; Lai et al., 2014). These disorders are estimated to affect approximately 1% of the population. The genetic causes of ASDs show a high degree of heterogeneity, with hundreds of ASD-associated genes now identified (Mefford et al., 2012; Huguet et al., 2013; Murdoch and State, 2013; Lai et al., 2014; Ronemus et al., 2014). However, recent studies suggest that these ASD genes may functionally converge into a relatively smaller subset of cellular and biochemical pathways affecting distinct neuronal functions (Auerbach et al., 2011; Zoghbi and Bear, 2012; Ebert and Greenberg, 2013; Doll and Broadie, 2014; Krumm et al., 2014). In a subset of ASDs, such as the syndromic ASD fragile X syndrome, Rett syndrome (RTT) and tuberous sclerosis, mutations of genes have been found to be related to synaptic function, suggesting that abnormal neuronal homeostasis is a risk factor for ASD (Auerbach et al., 2011; Santoro et al., 2012; Zoghbi and Bear, 2012; Ebert and Greenberg, 2013; Banerjee et al., 2014). This subset of ASDs belong to a larger group of neurological conditions called “synaptopathies”, which refer to disorders with altered synaptic function and/or morphology as primary neuropathology (Auerbach et al., 2011; Zoghbi and Bear, 2012; Krumm et al., 2014). The functional convergence on particular signaling pathways and the shared synaptopathology of ASDs have raised the hope that similar therapeutic strategies may be effective for different forms of ASDs which are related, but genetically distinct (Zoghbi and Bear, 2012; Delorme et al., 2013; Wang and Doering, 2013).
Fragile X and Rett syndromes are two of the most widely and intensively studied monogenetic ASDs (Santoro et al., 2012; Zoghbi and Bear, 2012; Castro et al., 2013; Chapleau et al., 2013a; Banerjee et al., 2014). Numerous studies have investigated the possibility of treating fragile X and Rett syndromes in their relative animal models. Strategies to alleviate abnormal phenotypes include genetic manipulation, cellular therapy, pharmacological intervention and environmental stimulation (Wang and Doering, 2012; Zoghbi and Bear, 2012; Castro et al., 2013; Chapleau et al., 2013a; Delorme et al., 2013; Ebert and Greenberg, 2013). Most encouraging, some of these fundamental studies have led to the development of drugs that are in clinical trials.
In this review, we summarize the potential therapeutic targets and relative pharmacological interventions in fragile X and Rett syndromes. The challenges in preclinical studies and clinical trials, and the implications of these targeted pharmacological treatments for other ASDs and related neurodevelopmental disorders are also discussed.
Targeted Pharmacotherapy for Fragile X Syndrome
Fragile X syndrome is one of the most common genetic causes of intellectual disability and autism. It is mostly caused by the mutation of the trinucleotide CGG expansion in the 5′-untranslated region of the fragile X mental retardation (FMR1) gene, which eventually leads to the absence of its protein product, fragile X mental retardation protein (FMRP) (Garber et al., 2008; Bhakar et al., 2012; Santoro et al., 2012; Hagerman et al., 2014). FMRP is ubiquitous with the most abundance in the central nervous system (Santoro et al., 2012; Wang et al., 2012a; Sidorov et al., 2013) and exists in both neurons and glial cells (Wang et al., 2004; Pacey and Doering, 2007).
FMRP, a RNA binding protein, binds its mRNA targets and regulates the transport and translation of those mRNAs (Bear et al., 2004; Penagarikano et al., 2007; Bhakar et al., 2012; Santoro et al., 2012; Wang et al., 2012a; Sidorov et al., 2013; Abekhoukh and Bardoni, 2014). The mRNA targets of FMRP encode pre- and post-synaptic proteins and some of these proteins are implicated in other ASDs, suggesting a molecular overlap between fragile X syndrome and other neurodevelopmental disorders (Bhakar et al., 2012; Wang et al., 2012a; Sidorov et al., 2013). FMRP normally acts as a translational repressor, especially at synapses. Its absence has profound consequences on neural development and synaptic plasticity. Alterations in synaptic structure and function are believed to underlie the fragile X symptoms. Dysregulated protein synthesis is central to fragile X synaptopathy (Bear et al., 2004; Penagarikano et al., 2007; Bassell and Warren, 2008; Wang et al., 2010a; Bhakar et al., 2012; Santoro et al., 2012; Sidorov et al., 2013).
Over the past two decades, efforts have been made to elucidate the molecular and cellular events that give rise to synaptic dysfunction in fragile X syndrome. Studies in animal models have revealed defects in multiple neurotransmitter systems and relative signaling pathways/molecules that are responsible for fragile X synaptopathy (Wang et al., 2010b; Bhakar et al., 2012; Gross et al., 2012; Santoro et al., 2012; Zoghbi and Bear, 2012; Delorme et al., 2013; Ebert and Greenberg, 2013). Advances in neurobiology of fragile X syndrome have led to the development of therapeutic agents that target the underlying mechanisms of the disease (Summarized in Figure 1; Tables 1, 2).
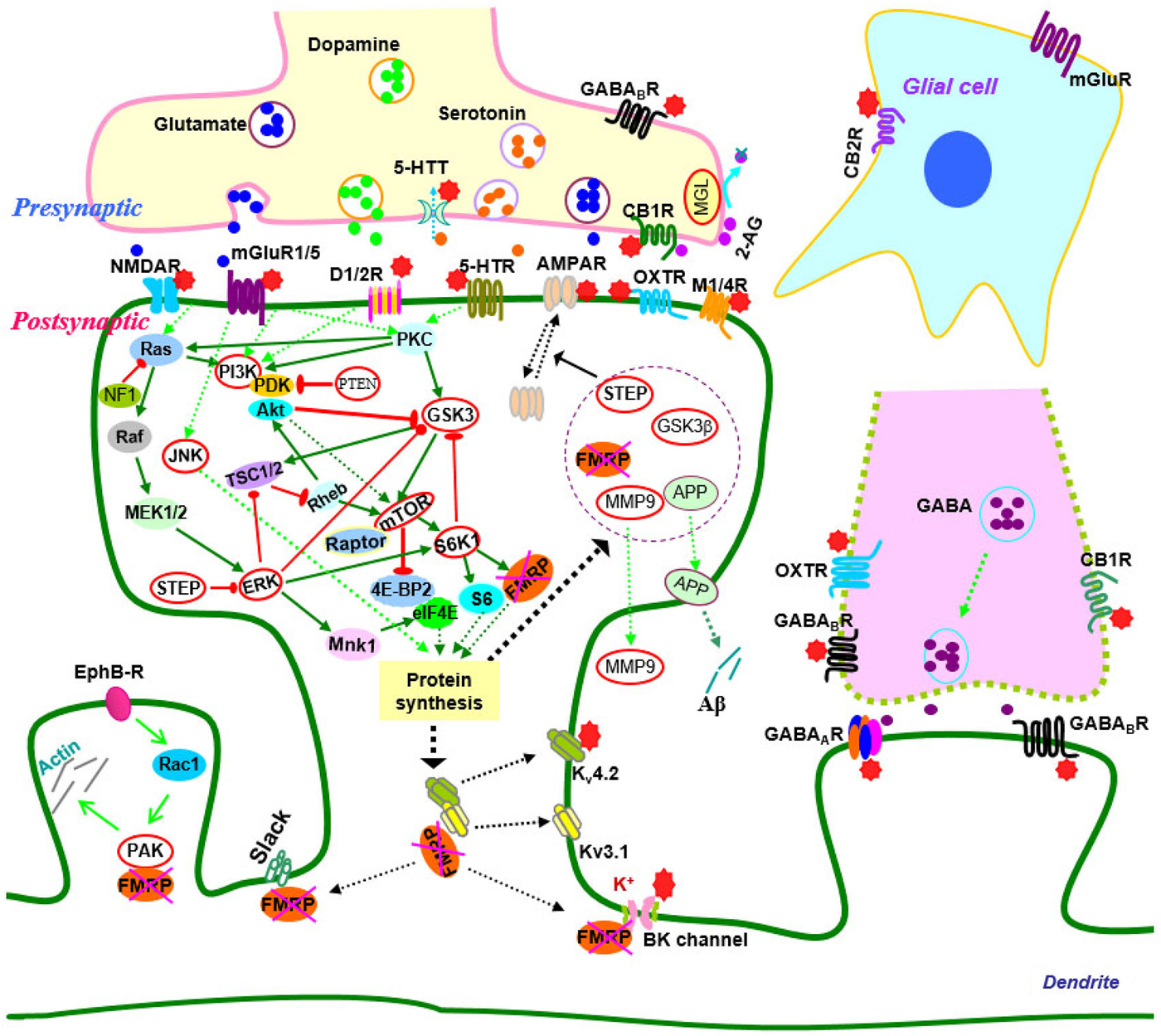
Figure 1. Signaling molecules and pathways in the neurobiology of fragile X syndrome. In signaling pathways, arrows indicate positive (green) or inhibitory (red) consequence on downstream components, but they do not necessarily represent direct interactions. Potential therapeutic targets which have been validated by genetic or pharmacological manipulation are indicated by red stars or highlighted by red circles. Abbreviations: 2-AG, 2-arachidonoyl-sn-glycerol; 4E-BP2, eIF4E-binding protein 2; 5-HT, serotonin; 5-HTR, serotonin receptors; 5-HTT, serotonin transporters; Akt (PKB), protein kinase B; AMPAR, α-amino-3-hydroxyl-4-isoxazole propionic acid receptors; APP, amyloid precursor protein; CB1(2)R, cannabinoid receptor 1(2); EphB-R, EphB receptors; ERK, extracellular signal related kinase; FMRP, fragile X mental retardation protein; GABA, gamma aminobutyric acid; GABAA(B)R, GABAA(B) receptors; GSK3, glycogen synthase kinase-3; JNK, c-Jun N-terminal kinase; M1/4R, muscarinic acetylcholine receptor 1/4; MEK, mitogen-activated protein kinase/ERK kinase; MGL, monoacylglycerol lipase; mGluR, metabotropic glutamate receptor; MMP9, matrix metalloproteinase 9; MSK, mitogen and stress-activated protein kinase; mTOR, mammalian target of rapamycin; NF1, neurofibromatosis 1; NMDAR, N-methyl-d-aspartate receptors; OXTR, oxytocin receptor; PAK, p21-activated kinase; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PP2A, protein phosphatase 2A; PTEN, Phosphatase and tensin homolog; Raptor, regulatory-associated protein of mTOR; RSK, p90 ribosomal S6 kinase; S6K1, p70 ribosomal kinase 1; STEP, striatal-enriched protein tyrosine phosphatase; TSC1/2, tuberous sclerosis complex 1/2.
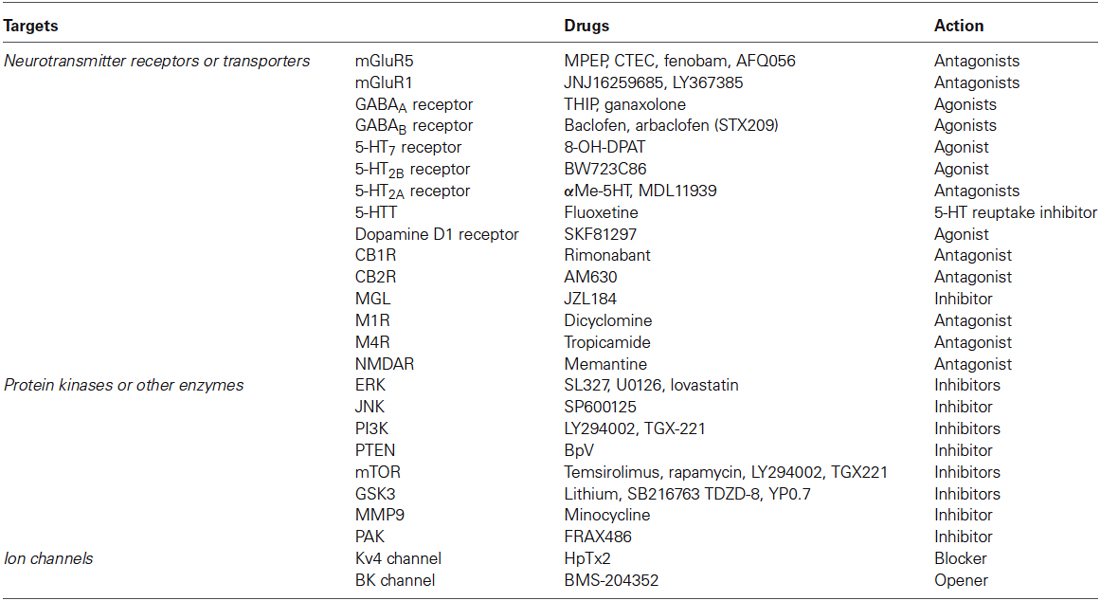
Table 1. Therapeutic targets and relative drugs in preclinical studies of fragile X syndrome.
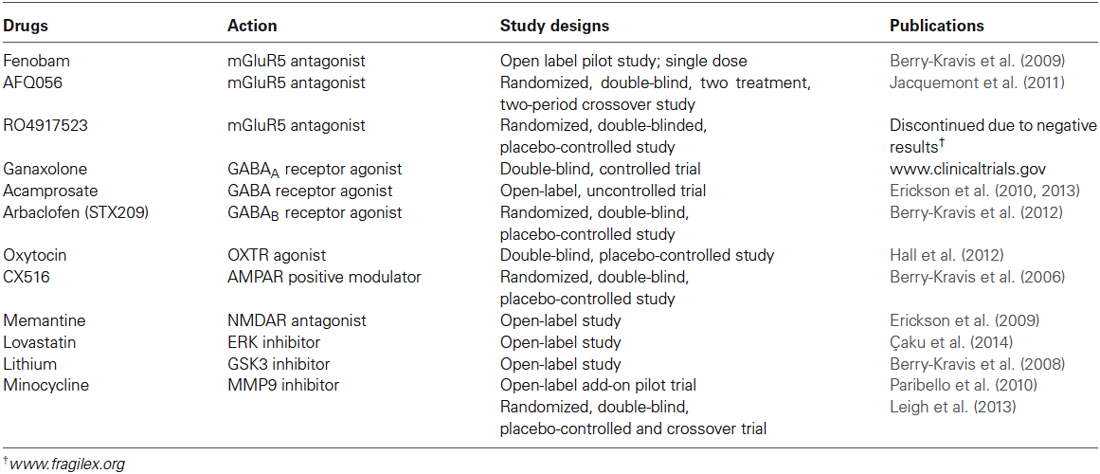
Table 2. Drugs in clinical trials of fragile X syndrome.
Targeting Neurotransmitter/Neuromodulator Systems
Metabotropic glutamate receptors
Metabotropic glutamate receptors (mGluRs) play important roles in synaptic plasticity, learning and memory (Bortolotto et al., 1999; Bear et al., 2004; Wang et al., 2008a; Wang and Zhuo, 2012; Mukherjee and Manahan-Vaughan, 2013). Activation of group 1 mGluRs (mGluR1s and mGluR5s) leads to local translation of pre-existing mRNAs and triggers long-term depression (LTD) (Weiler and Greenough, 1993; Bortolotto et al., 1999; Raymond et al., 2000; Penagarikano et al., 2007; Ronesi and Huber, 2008a; Upreti et al., 2013). FMRP is one of these newly synthesized proteins and serves as a repressor of the translation of other synaptic mRNAs that encode LTD proteins such as activity-regulated cytoskeleton-associated protein (Arc), microtubule associated protein (MAP) 1B and Striatal-enriched protein tyrosine phosphatase (STEP), which mediate a-amino-3-hydroxyl-4-isoxazole propionic acid receptor (AMPAR) internalization and LTD stabilization (Bhakar et al., 2012; Santoro et al., 2012; Wang et al., 2012a; Darnell and Klann, 2013; Sidorov et al., 2013). In fragile X conditions, protein synthesis is elevated and mGluR-LTD in the hippocampus is exaggerated in the absence of FMRP; the enhanced mGluR-LTD is no longer protein synthesis dependent as a result of increased basal protein synthesis. The mGluR theory of fragile X syndrome thus postulates that the core symptoms of the disease result from exaggerated group 1 mGluR mediated signaling including mGluR-LTD (Bear et al., 2004; Garber et al., 2008; Bhakar et al., 2012; Santoro et al., 2012; Sidorov et al., 2013).
The mGluR theory and its therapeutic significance have been validated in both genetic and pharmacological rescue studies (Dölen and Bear, 2008; Hays et al., 2011; Thomas et al., 2011; Gandhi et al., 2014; Michalon et al., 2014; Pop et al., 2014). In Fmr1 knockout mice, the mGluR5 antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP) stabilized hippocampal protein synthesis, increased the density or rescued the morphology of hippocampal dendritic spines, corrected altered brain network function, reduced audiogenic seizures and repetitive and/or perseverative behaviors (marble burying), rescued the deficits in prepulse inhibition of startle response, and improved the maze and motor learning (Yan et al., 2005; de Vrij et al., 2008; Hays et al., 2011; Thomas et al., 2012; Gandhi et al., 2014). Other mGluR5 antagonists tested in fragile X animal models include CTEC, fenobam and AFQ056. Acute treatment with CTEC corrected the elevated hippocampal LTD, protein synthesis, and audiogenic seizures; chronic treatment rescued cognitive deficits, auditory hypersensitivity, aberrant dendritic spine density, overactive extracellular signal regulated kinase (ERK) and mammalian target of rapamycin (mTOR) signaling, and partially corrected macroorchidism in adult fragile X mice (Michalon et al., 2012). Chronic CTEP treatment also corrected learning deficit in the inhibitory avoidance and extinction test, and partially normalized altered local brain activity in these animals (Michalon et al., 2014). Fenobam reversed some synaptic alterations in the cortex (Wang et al., 2014) and corrected deficits in associative motor learning and avoidance behaviors in fragile X mice (Vinueza Veloz et al., 2012). AFQ056 was found to be able to correct aberrant hippocampal dendritic spine morphology (Levenga et al., 2011; Pop et al., 2014), and rescue deficits in prepulse inhibition of acoustic startle response and abnormal social behaviors (Levenga et al., 2011; Gantois et al., 2013) in Fmr1 knockout mice. Treatment with mGluR1 antagonists (JNJ16259685 or LY367385) decreased repetitive and/or perseverative behaviors (Thomas et al., 2012), and rescued dysregulated synaptic protein synthesis in fragile X mice (Gross et al., 2010; Guo et al., 2012). These preclinical studies have been paving the way for treatments with mGluR antagonists in humans.
Fenobam, the first mGluR antagonist used in patients, showed beneficial effects, such as reduced anxiety and hyperarousal, improved prepulse inhibition of startle, and better accuracy on a continuous performance task, in adults with fragile X syndrome (Berry-Kravis et al., 2009). The pharmacokinetics and side effects of fenobam are currently tested in adult healthy volunteers.1 AFQ056 also showed improvements in inappropriate speech, stereotypic behavior, and hyperactivity and efficacy in adult patients with fully methylated FMR1 (Jacquemont et al., 2011). However, the mGluR5 negative allosteric modulator RG7090 (RO4917523) in fragile X was recently discontinued by Roche due to negative phase II clinical study results from fragile X patients.2 More drugs targeting mGluRs with higher efficacy and better safety need to be developed in the future (Berry-Kravis, 2014; Hagerman et al., 2014).
GABAergic receptors
The γ-aminobutyric acid (GABA) is the main inhibitory neurotransmitter in brain and signals through GABAA and GABAB receptors (Ben-Ari et al., 2012; Gassmann and Bettler, 2012; Sigel and Steinmann, 2012). The deficiencies in GABA receptor expression and GABA receptor-mediated inhibition have been demonstrated in fragile X animal models, linking GABA receptors to fragile X phenotypes and leading to the hypothesis that fragile X syndrome may result from an imbalance between excitation and inhibition, and increasing inhibition may ameliorate some fragile X pathophysiologies, including dysregulated protein synthesis (D’Hulst et al., 2006, 2009; Chang et al., 2008; Pacey et al., 2009; Olmos-Serrano et al., 2010; Paluszkiewicz et al., 2011; He et al., 2014). The potential of GABA receptors as a therapeutic target for fragile X syndrome has been validated in animal models. Treatment with a selective GABAA receptor agonist THIP (gaboxadol) corrected neuronal hyperexcitability in the amygdala (Olmos-Serrano et al., 2010), and attenuated hyperactivity and deficits in prepulse inhibition of the acoustic startle response of fragile X mice (Olmos-Serrano et al., 2011). Treatment with ganaxolone, a positive allosteric modulator of GABAA receptors, could prevent audiogenic seizures in Fmr1 knockout mice (Heulens et al., 2012). The GABAB receptor agonist baclofen reduced locomotor activity and hyperactivity (Zupan and Toth, 2008), and ameliorated audiogenic seizure susceptibility of Fmr1 knockout mice (Pacey et al., 2009, 2011). Notably, arbaclofen (STX209), the R-isomer of baclofen, was also able to attenuate audiogenic seizures in fragile X mice (Henderson et al., 2012). In addition, arbaclofen corrected dysregulated protein synthesis in the hippocampus, restored the elevated AMPA receptor internalization and the increased spine density, and thus corrected synaptic abnormalities which are central to fragile X pathophysiologies, suggesting that arbaclofen could be potentially used to treat the core symptoms of fragile X patients (Henderson et al., 2012).
A large clinical trial with ganaxolone is currently ongoing in children and adolescents with fragile X syndrome between 6 to 17 years of age (Wijetunge et al., 2013; Berry-Kravis, 2014; Hagerman et al., 2014).3 A controlled trial of arbaclofen in 63 fragile X patients between 6 to 40 years of age has revealed significant beneficial treatment effects, such as improvements in socialization and social avoidance scores, particularly in those with more severe social impairments, suggesting that GABAB receptor agonists have potential to improve social function and behaviors in fragile X patients (Berry-Kravis et al., 2012). However, this clinical trial for arbaclofen failed to fulfill its initial endpoints and had to be terminated by Seaside Therapeutics due to resource limitations.4 Acamprosate, with agonist properties on both GABAA and GABAB receptors, was well tolerated and was found to significantly improve social behavior and reduce inattention/hyperactivity in 12 fragile X children aged 6–17 years in a prospective open-label 10-week trial. Additionally, an increase in brain-derived neurotrophic factor (BDNF) in blood was observed with treatment and might become a useful biomarker for future clinical studies (Erickson et al., 2010, 2013). A double-blind, placebo-controlled study will be needed to further assess the effect of acamprosate. As a whole, it is hopeful that some of the GABA receptor agonists may eventually become clinically applicable.
Serotonin receptors/transporters
The serotonin (5-Hydroxytryptamine, 5-HT) system is involved in various brain functions, including synaptic plasticity, learning and memory (Matthys et al., 2011; Lesch and Waider, 2012; Gellynck et al., 2013). It has been shown that 5-HT7 receptor (agonist 8-OH-DPAT) activation could reverse mGluR-induced AMPA receptor internalization and correct excessive mGluR-LTD in hippocampal neurons of Fmr1 knockout mice, suggesting that 5-HT receptors may represent a novel therapeutic target for fragile X syndrome (Costa et al., 2012). Interestingly, a recent study has demonstrated that compounds activating 5-HT2B receptors or inhibiting 5-HT2A receptors could enhance phosphoinositide 3-kinase (PI3K) signaling transduction and AMPA GluR1 dependent synaptic plasticity, and restore learning in Fmr1 knockout mice, further linking 5-HT receptors to fragile X syndrome (Lim et al., 2014).
The alteration of 5-HT and its transporters have been known to associate with autism (Chugani, 2002; Devlin et al., 2005; Prasad et al., 2009; Veenstra-VanderWeele et al., 2012; Harrington et al., 2013). 5-HT transporters are the main target for widely used antidepressant agents including fluoxetine (Wong et al., 2005). Fluoxetine binds to 5-HT transporters and blocks 5-HT uptake from the synaptic cleft into presynaptic vesicles in the central nervous system (Wong et al., 2005; Tavoulari et al., 2009). Studies have suggested that fluoxetine may be beneficial to individuals with autism (Hollander et al., 2005, 2012; Kolevzon et al., 2006). Treatment with fluoxetine has shown some effect in fragile X mice and patients (Hagerman et al., 1999; Uutela et al., 2014). However, fluoxetine has some possible side effects in clinic, such as mood changes, agitation, restlessness, and aggression, and may not be suitable to all individuals with fragile X syndrome and other ASDs (Wernicke, 2004; Kolevzon et al., 2006; Uutela et al., 2014).
Dopamine receptors
Dopamine plays critical roles in synaptic plasticity, cognitive functioning and neuropsychiatic pathologies (Jay, 2003; Seamans and Yang, 2004; Surmeier et al., 2009; Cerovic et al., 2013). Deficits in the dopamine system have been demonstrated in both fragile X animal models and patients (Roberts et al., 2005; Wang et al., 2008b; Weinshenker and Warren, 2008; Fulks et al., 2010; Paul et al., 2013; Rogers et al., 2013). Electrophysiological studies have found that dopaminergic modulation of synaptic transmission and potentiation are impaired in fragile X mice (Wang et al., 2008b; Paul et al., 2013). Biochemical studies have further revealed that dopamine D1 receptor mediated synapse-associated protein synthesis, AMPA GluR1 receptor surface expression and subsequent internalization are defective in these mice (Wang et al., 2008b, 2010a). Application of the D1 receptor agonist and/or D2 receptor antagonist could promote PI3K signaling and AMPA GluR1 receptor delivery, and improve learning behaviors in Fmr1 knockout mice (Lim et al., 2014). The dopamine D1 receptor agonist also partially rescued the hyperactivity and enhanced motor function of fragile X mice (Wang et al., 2008b). These findings suggest that dopamine receptors could be a potential target for effectively treating cognitive impairment associated with fragile X syndrome.
Cannabinoid receptors
The endocannabinoids N-arachidonoyl ethanolamine and 2- arachidonoyl glycerol (2-AG) activate cannabinoid receptors (CB1R and CB2R), and modulate synaptic plasticity and cognitive function (Chevaleyre et al., 2006; Heifets and Castillo, 2009; Oudin et al., 2011). Alterations in endocannabinoid signaling contribute to cognitive dysfunction associated with fragile X syndrome. Various abnormalities in endocannabinoid signaling, such as CB1R-driven long-term regulation of synaptic strength due to mGluR5 activation, have been observed in several brain areas of Fmr1 knockout mice (Maccarrone et al., 2010; Zhang and Alger, 2010; Busquets-Garcia et al., 2013). FMRP exerts a regulatory control over the endocannabinoid system at central synapses (Maccarrone et al., 2010). Loss of FMRP affects endocannabinoid signaling, possibly through local 2-AG production (Straiker et al., 2013). In fragile X mice, the linkage between mGluR5 and the 2-AG producing enzyme, diacylglycerol lipase-α (DGL-α), is disrupted and mGluR5-dependent 2-AG formation is compromised, leading to impairment of endocannabinoid-mediated LTD in the ventral striatum and prefrontal cortex (Jung et al., 2012).
Pharmacological enhancement of 2-AG signaling by inhibiting 2-AG-deactivating enzyme monoacylglycerol lipase (MGL) with JZL184, normalized this synaptic defect and corrected behavioral abnormalities (hyperactivity and abnormal anxiety) in fragile X mice (Jung et al., 2012). Interestingly, dampening of endocannabinoid signaling through pharmacological or genetic approaches also benefit these mice. CB1R blockade with rimonabant or genetic reduction of CB1R, normalized cognitive impairment, nociceptive desensitization, susceptibility to audiogenic seizures, overactivated mTOR signaling and altered spine morphology in the male Fmr1 knockout (Fmr1(-/y)) mice, whereas CB2R antagonism with AM630 normalized anxiolytic-like behaviors in those mice (Busquets-Garcia et al., 2013). These studies thus demonstrate that targeting endocannabinoid signaling might provide a new therapeutic strategy for fragile X syndrome. Further investigation is needed to clarify the therapeutic value of this potential target.
Muscarinic acetylcholine receptors
The G-protein coupled muscarinic acetylcholine receptors (mAChRs, subtypes M1–M5) are widely expressed in the central nervous system and mediate the metabotropic actions of acetylcholine (Volpicelli and Levey, 2004; Picciotto et al., 2012). M1 mAChR activation facilitates synaptic plasticity, learning and memory. Activation of M1 mAChRs stimulates synthesis of synaptic proteins including FMRP, and induces protein synthesis dependent LTD similar to group 1 mGluR-LTD (McCoy and McMahon, 2007; Volk et al., 2007). In the absence of FMRP, hippocampal M1 mAChR-dependent LTD is enhanced, indicating overactive mAChR signaling in Fmr1 knockout mice (Volk et al., 2007). Genetic reduction of M4 mAChRs corrected the analgesic response and partly rescued the acoustic startle response in Fmr1 knockout mice (Veeraragavan et al., 2012). These studies suggest new therapeutic strategies for using mAChR antagonists in fragile X syndrome.
The subtype selective mAChR modulators, including the M1 receptor antagonist dicyclomine and M4 receptor antagonist tropicamide, have been tested in the fragile X mouse model. The M1 receptor antagonist dicyclomine decreased repetitive and/or perseverative behavior (marble burying) and reduced susceptibility to audiogenic seizures in Fmr1 knockout mice (Veeraragavan et al., 2011b). Similarly, the M4 receptor antagonist tropicamide also attenuated audiogenic seizure susceptibility in Fmr1 knockout mice (Veeraragavan et al., 2011a). Noteworthy, treatment with tropicamide reduced repetitive and/or perseverative behaviors, improved performance in the passive avoidance task in both wild-type and fragile X mice, and reduced audiogenic seizures in fragile X mice (Veeraragavan et al., 2011a). These studies indicate that pharmacological inhibition of mAChRs modulates specific behavioral responses and further support these receptors as therapeutic targets for fragile X syndrome.
Oxytocin receptors
Oxytocin acts as a neuromodulator through its receptors in various brain areas and regulates social cognition and behaviors (Neumann, 2008; Meyer-Lindenberg et al., 2011; Neumann and Landgraf, 2012; Knobloch and Grinevich, 2014). It is emerging as a target for treatment of anxiety and depression-related diseases or social dysfunction including autism (Neumann, 2008; Meyer-Lindenberg et al., 2011; Neumann and Landgraf, 2012; Anagnostou et al., 2014a; Preti et al., 2014). The perinatal excitatory-to-inhibitory shift of GABA is mediated by oxytocin receptors. Oxytocin-mediated GABA inhibition during delivery could attenuate autism pathogenesis in rodent offsprings. Application of the oxytocin receptor antagonist SSR126768A in naïve mothers could produce offspring which have the electrophysiological and behavioral autistic-like features (Tyzio et al., 2014). In fragile X mice, oxytocin is reduced in some brain regions and the oxytocin-mediated neuroprotective GABA excitatory-inhibitory shift during delivery is absent (Tyzio et al., 2014). This study thus indicates the importance of the oxytocin system in the pathogenesis of autism and fragile X sydrome.
The clinical actions of oxytocin have been validated in ASD patients, providing preliminary evidence that oxytocin is able to enhance brain function and improve social behaviors in autistic patients (Andari et al., 2010; Domes et al., 2013; Gordon et al., 2013; Anagnostou et al., 2014a; Preti et al., 2014; Scheele et al., 2014; Watanabe et al., 2014). Intranasal administration of oxytocin could ameliorate symptoms of social anxiety in children with fragile X syndrome (Hall et al., 2012). The double-blind placebo-controlled studies of oxytocin will be needed for further validation of its effect on fragile X patients.
AMPA receptors
The surface expression and synaptic delivery of AMPA receptors (GluR1) is impaired in Fmr1 knockout mice, altering signal transmission and synaptic plasticity (Nakamoto et al., 2007; Hu et al., 2008; Suvrathan et al., 2010; Wang et al., 2010a). AMPA receptor modulators have been tested in fragile X patients. CX516 is a positive allosteric modulator that potentiates glutamate activation with the final outcome of strengthening synapses (O’Neill et al., 2004; O’Neill and Witkin, 2007). In a double-blind placebo-controlled trial in adult patients with fragile X syndrome, 4-week treatment with CX516 produced no significant improvement in cognitive or behavioral measures (Berry-Kravis et al., 2006). The failure could be due to the potency of CX516 or its dosage which may be inadequate for a therapeutic effect. It is thus unclear whether modulation of AMPA receptors is a feasible therapeutic strategy for treatment of fragile X syndrome.
NMDA receptors
The defects in NMDA receptors observed in the hippocampus, prefrontal cortex and other brain areas of fragile X mice are believed to contribute to fragile X phenotypes (Pilpel et al., 2009; Suvrathan et al., 2010; Krueger et al., 2011; Yun and Trommer, 2011; Eadie et al., 2012; Gocel and Larson, 2012). Memantine is an uncompetitive NMDA receptor antagonist and has been shown to improve language function and social behavior in autistic patients (Erickson and Chambers, 2006; Chez et al., 2007; Niederhofer, 2007). A study in cultured cerebellar granule cells from Fmr1 knockout mice suggested that memantine may exert therapeutic capacity for fragile X syndrome through a stimulatory effect on dendritic spine maturation and excitatory synapse formation (Wei et al., 2012). Although a pilot study showed that memantine was modestly effective in several patients with fragile X syndrome, a systematic clinical study is needed to further evaluate its effectiveness (Erickson et al., 2009).
Targeting Signaling Pathways Downstream of Neurotransmitter Receptors
Studies in fragile X animal models have revealed defects in intracellular signaling pathways [Mitogen-activated protein kinases (MAPKs), PI3K, mTOR and glycogen synthase kinase-3 (GSK3)] which could be downstream of neurotransmitter receptors such as glutamate, GABA, endocannabinoid, 5-HT, dopamine or mACh receptors. These signaling pathways may serve as therapeutic targets for fragile X syndrome (Figure 1; Tables 1, 2).
Mitogen-activated protein kinases
MAPKs are a family of serine/threonine protein kinases, including ERKs, p38 MAPKs, and c-Jun N-terminal kinase (JNK; Wang and Zhuo, 2012). The ERK1/2 pathway, which is activated by the Ras-mitogen-activated protein kinase/ERK kinase (MEK) and eventually leads to gene transcription or mRNA translation, plays critical roles in synaptic plasticity (Kelleher et al., 2004; Thomas and Huganir, 2004; Wiegert and Bading, 2011; Wang and Zhuo, 2012). A number of studies have investigated ERK signaling under basal conditions or upon mGluR-induction using brain tissues from fragile X animal models or patients (Kim et al., 2008; Weng et al., 2008; Wang et al., 2012b; Curia et al., 2013). Although these studies sometimes have presented conflicting results, most of them have shown that the ERK1/2 pathway is altered in fragile X conditions, suggesting that the ERK pathway is likely to have translational implications for fragile X syndrome (Weng et al., 2008; Wang et al., 2012b; Curia et al., 2013). Indeed, treatment with MEK1/2 inhibitor SL327 could prevent audiogenic seizures in Fmr1 knockout mice (Wang et al., 2012b). Inhibition of ERK1/2 with the MEK1/2-ERK1/2 inhibitor U0126 also reduced elevated protein synthesis in hippocampus of fragile X mice (Osterweil et al., 2010). These findings further support that ERK1/2 pathway and the neurotransmitter systems that stimulate ERK1/2 may represent additional therapeutic targets for fragile X syndrome. Interestingly, statins, drug widely prescribed to treat hypercholesterolemia, have shown potentials for treating fragile X syndrome. Lovastatin corrected the excess protein synthesis and the exaggerated mGluR-LTD in the hippocampal slices of Fmr1 knockout mice, attenuated hyperexcitability in visual cortex, and reduced audiogenic seizures in those mice (Osterweil et al., 2013). In a recent Phase I clinical trial, lovastatin was found to improve aberrant behaviors (social avoidance/unresponsiveness, stereotypy, hyperactivity and irritability) in majority (12/15) of fragile X patients after 12-weeks of treatment; the effect was significant after 4-week treatment at the lower dose, with further improvement during the 4–12 week treatment (Çaku et al., 2014). Although it was suggested that statins may alter membrane cholesterol and lipid rafts, thus modulating group I mGluRs or/and other neurotransmitter systems to correct fragile X phenotypes, the drugs might also exert the effects through direct inhibition of Ras–ERK activity (Kumari et al., 2013; Osterweil et al., 2013; Wang, 2014).
The JNK pathway is known to regulate mGluR-dependent gene transcription (Wang and Zhuo, 2012). A recent study has shown that JNK is essential for mGluR-dependent expression of FMRP target proteins. In addition, JNK activity is upregulated in synapses of Fmr1 knockout mice, and inhibition of JNK with SP600125 decreased elevated postsynaptic protein synthesis in these mice, suggesting that JNK could be a key signaling downstream of mGluR in regulating FMRP-dependent protein synthesis and may provide a strategy to restore the deficits in fragile X syndrome (Schmit et al., 2013).
Phosphoinositide 3-kinase
PI3K transduces signals from cell surface receptors to the Akt/mTOR pathway and is essential for dendritic spine development and synaptic plasticity underlying learning and memory (Horwood et al., 2006; Hu et al., 2008; Cuesto et al., 2011). The PI3K catalytic subunit p110beta can be regulated by FMRP. Both p110beta level and PI3K activity are elevated and insensitive to mGluR stimulation in Fmr1 knockout neurons, suggesting that dysregulated PI3K signaling may underlie synaptic deficits in fragile X syndrome (Gross et al., 2010; Gross and Bassell, 2014). PI3K inhibitor LY294002 corrected dysregulated synaptic protein synthesis, excess AMPA receptor internalization and the increased spine density in Fmr1 knockout neurons, supporting PI3K as a potential therapeutic target for fragile X syndrome (Gross et al., 2010). Development of specific inhibitors for PI3K subunits may help to translate this strategy to patients since selective inhibition of the p110b-subunit with TGX-221 has been found to rescue excess protein synthesis in synaptoneurosomes from fragile X mice and in patient cells (Gross et al., 2010; Gross and Bassell, 2012, 2014). Conversely, one recent study showed that promoting PI3K signaling by dipotassium bisperoxo (5-hydroxypyridine-2-carboxyl)oxovanadate (BpV), a phosphatase and tensin homolog (PTEN) inhibitor, reversed deficits in both basal turnover and activity-mediated spine stabilization in hippocampal slices, restored defective long-term potentiation (LTP) mechanisms in slices and improved reversal learning in Fmr1 knockout mice (Boda et al., 2014). Thus, the specific role of PI3K signaling in fragile X syndrome needs to be further investigated due to the complexity of its upstream cell surface receptors and interactions with other signaling pathways.
Mammalian target of rapamycin
The mTOR signaling cascade controls initiation of cap-dependent translation (Hay and Sonenberg, 2004; Narayanan et al., 2008; Hoeffer and Klann, 2010). Its downstream effector ribosomal protein S6 kinase (S6K1) is a regulator of translation initiation and elongation in cap-dependent protein synthesis (Narayanan et al., 2008; Hoeffer and Klann, 2010). S6K1 is a major kinase which phosphorylates and regulates FMRP following group 1 mGluR or dopamine D1 receptor activation (Narayanan et al., 2008; Wang et al., 2010a). The basal levels of mTOR phosphorylation and activity were found to be elevated in the hippocampus of Fmr1 knockout mice (Sharma et al., 2010). In addition, group 1 mGluR activation of mTOR is absent is those mice (Ronesi and Huber, 2008b). The misregulation of mTOR signaling was also observed in fragile X patients (Hoeffer et al., 2012), suggesting that mTOR could be a target or biomarker for treatment of fragile X syndrome.
Treatment of Fmr1 knockout mice with temsirolimus, an mTOR inhibitor, prevented object recognition memory deficits and reduced audiogenic seizure susceptibility in those mice (Busquets-Garcia et al., 2013). Elevated phosphorylation of translational control molecules and exaggerated protein synthesis in fragile X mice were corrected through the targeting of S6K1. Genetic deletion of S6K1 also prevented a broad range of fragile X phenotypes, including exaggerated translation, enhanced mGluR-LTD, abnormal dendritic spine morphology, several behavioral characteristics and peripheral features (weight gain and macroorchidism) (Bhattacharya et al., 2012). Notably, administration of the mTORC1 (mTOR complex 1) inhibitor rapamycin improved sociability in the BTBR mouse model of ASDs (Burket et al., 2014); targeting downstream mTOR signaling such as eukaryotic translation initiation factor 4E (eIF4E) also reversed autism (Gkogkas et al., 2013; Wang and Doering, 2013). These studies thus support that mTOR signaling represents a potential therapeutic target for fragile X syndrome and other ASDs.
Glycogen synthase kinase-3
GSK3 is a serine/threonine kinase that exists in two isoforms (GSK3α and GSK3β) and regulates many cellular processes through phosphorylation of their substrates. The activity of GSK3 itself is controlled by inhibitory serine phosphorylation induced by various intracellular pathways including PI3K/Akt and MEK/ERK that converge on GSK3 (Cohen and Goedert, 2004; Jope and Roh, 2006; Sugden et al., 2008). In Fmr1 knockout mice, GSK3 phosphorylation was reduced in several brain regions resulting in elevated GSK3 signaling (Min et al., 2009; Mines et al., 2010; Yuskaitis et al., 2010). Lithium, a classical drug for psychiatric disorders, is a GSK3 inhibitor that both increases the inhibitory serine-phosphorylation of GSK3 and directly inhibits GSK3 activity (Chiu and Chuang, 2010; Mines and Jope, 2011). Since lithium has many other off-target effects including inhibition of inositol monophosphatase, more selective GSK3 inhibitors have been developed (Chiu and Chuang, 2010; Mines and Jope, 2011). Lithium, as well as the selective GSK3β inhibitor SB216763, was found to be able to correct mutant phenotypes (audiogenic seizure susceptibility and hyperactivity) of Fmr1 knockout mice. Particularly, lithium remained effective with chronic administration (Min et al., 2009). Acute or chronic lithium treatment could increase inhibitory serine phosphorylation of GSK3 in mouse brain; chronic treatment ameliorated alterations in open-field activity, elevated plus-maze and passive avoidance and impaired cognition in fragile X mice (Yuskaitis et al., 2010; King and Jope, 2013). Chronic treatment with SB216763 corrected hippocampus-dependent learning deficits as well as defects in adult neurogenesis in the hippocampus of fragile x mice (Guo et al., 2012). The specific GSK3 inhibitors TDZD-8 and VP0.7 corrected impairments in hippocampus-related cognitive tasks. Furthermore, the improvements in behaviors correlated to the rescue of deficits in NMDA receptor dependent LTP in the hippocampus of Fmr1 knockout mice as a result of GSK3 inhibition (Franklin et al., 2014). These studies thus support that targeting GSK3 may provide therapeutic benefits for fragile X syndrome.
In an open-label trial in 15 patients, the two month treatment with lithium was well-tolerated and had positive effects on behavior and adaptive skills in fragile X syndrome (Berry-Kravis et al., 2008). The placebo-controlled trials of lithium or other GSK3 inhibitors in fragile X syndrome will be warranted.
Targeting Proteins Regulated by FMRP
Matrix metalloproteinase 9
Matrix metalloproteinases (MMPs) are a family of extracellular proteases that are involved in synaptogenesis, neurotransmission and synaptic plasticity (Ethell and Ethell, 2007; Wright and Harding, 2009; Huntley, 2012). MPP9 mRNA is among the putative mRNA targets of FMRP with increased translation in the absence of FMRP (Janusz et al., 2013). MMP9 is necessary for the development of fragile X phenotypes. Genetic deletion of MMP9 rescued key features of fragile X syndrome, including dendritic spine abnormalities, exaggerated mGluR-LTD, aberrant cognitive and social behaviors as well as macroorchidism in the mouse model (Sidhu et al., 2014). Minocycline, a tetracycline analogue with anti-inflammatory and antiapoptotic activity, has been used to inhibit MMP9 (Bilousova et al., 2009; Siller and Broadie, 2012). Treatment of young mice with minocycline increased phosphorylation and subsequent membrane insertion of AMPA GluR1 receptors (Imbesi et al., 2008). Minocycline rescued maturation of dendritic spines in the hippocampus thereby correcting the spine phenotype in Fmr1 knockout mice (Bilousova et al., 2009). In addition, chronic minocycline reduced anxiety-related behavior, improved cognitive function in young Fmr1 knockout mice (Bilousova et al., 2009), and reversed impaired social communication during mating among fragile X mice (Rotschafer et al., 2012). In comparison of the effect of minocycline on young and adult fragile X mice, it was found that minocycline could reduce locomotor activity in both young and adult mice, some behavioral improvements could be long-lasting in young mice, but not in adults. In addition, minocycline reduced audiogenic seizure susceptibility in young mice (Dansie et al., 2013). This study provides further evidence that minocycline can produce long-lasting benefits in the fragile X animal model.
Clinical trials have been conducted to evaluate the safety and efficacy of minocycline. An open-label add-on pilot trial demonstrated that minocycline is well tolerated and can provide significant functional benefits to fragile X patients (Paribello et al., 2010). In a controlled clinical trial, minocycline treatment was observed to lower the elevated plasma activity of MMP9 in individuals with fragile X syndrome. In some cases, changes in MMP9 activity were found to be positively associated with improvement in clinical measures (Dziembowska et al., 2013). In another randomized, double-blind, placebo-controlled and crossover trial, minocycline treatment administered for 3 months was found to be safe and produce greater global improvement than a placebo in children with fragile X syndrome (Leigh et al., 2013). However, longer trials are warranted to further assess the benefits and side effects related to minocycline.
p21-activated kinase
The small GTPase Rac1 and its effector p-21 activated kinase (PAK) are critical for regulation of actin polymerization, dendritic spine morphogenesis and synaptic plasticity (Hayashi et al., 2004; Zhang et al., 2005; Kreis and Barnier, 2009; Murata and Constantine-Paton, 2013). In hippocampal synapses of Fmr1 knockout mice, physiological activation of the Rac-PAK signaling pathway is impaired (Chen et al., 2010). FMRP directly interacts with PAK1. Inhibition of PAK activity by expression of the dominant negative PAK (dnPAK) transgene results in a dendritic spine phenotype opposite to that of fragile X syndrome (Hayashi et al., 2007). A genetic expression of dnPAK in Fmr1 knockout mice at least partially corrected abnormalities in spine length and density in the cortex, and fully restored deficits in cortical LTP. Additionally, several behavioral abnormalities including hyperactivity, stereotypy, anxiety, and deficits in trace fear memory were ameliorated (Hayashi et al., 2007). This genetic rescue of fragile X phenotypes in the mouse model suggests that the PAK signaling pathway could be a novel intervention site for fragile X syndrome and autism. Pharmacological treatment with the PAK inhibitor FRAX486 reversed dendritic spine phenotypes, and rescued seizures and behavioral abnormalities such as hyperactivity and repetitive movements in Fmr1 knockout mice, further supporting PAK as a therapeutic target. Importantly, these effects could be achieved in adult Fmr1 knockout mice with a single administration of FRAX486, demonstrating the potential for therapy in adults with fragile X syndrome (Dolan et al., 2013).
Amyloid precursor protein
Amyloid precursor protein (APP) is a transmembrane protein that plays roles in synaptogenesis and synaptic plasticity (Gralle and Ferreira, 2007; Randall et al., 2010; Nalivaeva and Turner, 2013). APP is translated upon mGluR5 activation. FMRP binds to APP mRNA and controls its translation (Westmark and Malter, 2007). Absence of FMRP leads to APP overexpression and diminished mGluR-induced synthesis (Westmark and Malter, 2007). Cleavage of APP can produce β-amyloid (Aβ), which is overexpressed in the brain of Fmr1 knockout mice, suggesting a pathogenic role in fragile X syndrome (Westmark et al., 2010). Genetic reduction of APP/Aβ could partially or completely correct characteristic fragile X phenotypes, including audiogenic seizures, anxiety, dendritic spine morphology and exaggerated mGluR-LTD (Westmark et al., 2011), suggesting drugs directed at reducing Aβ in Alzheimer disease such as the secretase inhibitors or β-site APP cleaving enzyme (BACE-1) inhibitors may be applicable to fragile X syndrome (Malter et al., 2010; Westmark, 2013; Westmark et al., 2013).
Striatal-enriched protein tyrosine phosphatase
STEP, a brain-specific protein tyrosine phosphatase, dephosphorylates key signaling proteins including ERK1/2, p38 MAPK, the tyrosine kinase Fyn, and surface AMPA and NMDA receptors, thereby inactivating the kinases or promoting endocytosis of the receptors and opposing development of synaptic strengthening (Braithwaite et al., 2006; Goebel-Goody et al., 2012b). STEP is translated upon mGluR5 activation and mediates AMPA receptor internalization during mGluR-LTD (Zhang et al., 2008; Goebel-Goody and Lombroso, 2012). FMRP interacts with the transcript encoding STEP. The basal level of STEP is elevated and mGluR-dependent STEP synthesis is absent in Fmr1 knockout mice (Goebel-Goody et al., 2012a). It is possible that the synaptic deficits and behavioral abnormalities in fragile X syndrome may be linked to the dysregulation of STEP (Goebel-Goody and Lombroso, 2012). Genetic reduction of STEP could attenuate audiogenic seizures, improve characteristic social abnormalities, and reverse anxiety-related behaviors in Fmr1 knockout mice, suggesting that STEP might be a therapeutic target for treating fragile X patients (Goebel-Goody et al., 2012a).
Potassium channels
Potassium channels control the resting membrane potential and modulate the action potential waveform, mediating homeostasis of neuronal excitability (Misonou, 2010; Lee and Jan, 2012; Kim and Kaczmarek, 2014). In fragile X animal models, defects have been found in several types of potassium channels, such as the sodium-activated potassium Slack (Brown et al., 2010; Zhang et al., 2012), voltage-gated potassium channels Kv3.1b (Strumbos et al., 2010) and Kv4.2 (Gross et al., 2011; Lee et al., 2011), and large-conductance calcium-activated potassium (BK) channels (Deng et al., 2013; Zhang et al., 2014b).
FMRP interacts directly with Slack channels to enhance the channel activity, raising the possibility that Slack-FMRP interaction may link patterns of neuronal firing with changes in protein translation (Brown et al., 2010; Zhang et al., 2012). Disturbance of this link when FMRP is absent may underlie altered neuronal networks in fragile X syndrome. FMRP binds the mRNAs encoding Kv3.1b and is required for rapid experience-dependent regulation of Kv3.1b (Strumbos et al., 2010). FMRP also regulates mRNA translation and protein expression of Kv4.2; absence of FMRP-mediated control of Kv4.2 might contribute to excess neuronal excitability in Fmr1 KO mice (Gross et al., 2011). Recovery of Kv4.2 after NMDA receptor-mediated degradation also requires FMRP, probably due to NMDA receptor activation-induced FMRP dephosphorylation, which turns off FMRP suppression of Kv4.2 (Lee et al., 2011). Significantly, treating hippocampal slices of fmr1 KO mice with Kv4 channel blocker HpTx2 restored LTP induced by moderate stimuli, suggesting that potassium channels as an FMRP target could be of potential relevance to fragile X therapy (Lee et al., 2011). FMRP modulates action potential duration via its interaction with beta4 subunits of BK channels, thus regulating neurotransmitter release and synaptic transmission. Loss of these FMRP functions might be responsible for dysregulation of synaptic transmission in fragile X syndrome (Deng et al., 2013). Interestingly, a recent study showed that a selective BK channel opener (BMS-204352) could rescue multiple behavioral impairments (social, emotional and cognitive) in fragile X mice (Hebert et al., 2014), further demonstrating that potassium channels may open up new opportunities for treating fragile X syndrome.
In summary, multiple targeted pharmacological treatments have been found to rescue the phenotypes of fragile X animal models, but few have been beneficial to patients. Acamprosate, lovastatin, lithium and minocycline are the drugs that can be currently prescribed and have shown benefits to patients with fragile X syndrome. However, each single drug may not be effective for all patients. The combination of different drug therapies, together with behavioral interventions, will be necessary for better efficacy in treating fragile X syndrome (Braat and Kooy, 2014; Hagerman et al., 2014; Hagerman and Polussa, 2015).
Targeted Pharmacotherapy for Rett Syndrome
RTT is a postnatal neurodevelopmental disorder that occurs mainly in females and is the leading cause of intellectual disability in this gender (Hagberg et al., 1983; Laurvick et al., 2006; Neul et al., 2010). Clinical presentation occurs in stages with an initial developmental stagnation that is followed by a rapid regression during which RTT individuals have autistic features, stereotypic hand movements, loss of language, and develop aggressive behaviors (Chahrour and Zoghbi, 2007; Percy et al., 2010). RTT girls also have seizures during childhood, daytime breathing arrythmias, dysautonomia, and develop scoliosis at later stages, which also include Parkinsonian features and loss of mobility (Glaze et al., 2010; Neul et al., 2014).
Loss-of-function mutations in the X-linked gene MECP2 (methyl-CpG-binding protein-2) are responsible for RTT (Amir et al., 1999). In 95% of classic RTT cases, MECP2 mutations occur de novo in germ cells and are usually on the paternal side (Girard et al., 2001; Trappe et al., 2001; Bienvenu and Chelly, 2006). There is a spectrum of mutations ranging from missense, nonsense, and frameshift mutations (Moretti and Zoghbi, 2006). RTT individuals appear to have a typical development until 6–18 months of age, when they start missing developmental milestones. The MECP2 gene encodes a predominantly nuclear protein that is expressed ubiquitously but with predominance in the brain (Klose and Bird, 2006). The expression of MECP2 follows a temporal pattern: it peaks in the early postnatal period when maturation of neurons and activity dependent refinement of synapses occurs (LaSalle et al., 2001; Shahbazian et al., 2002; Cohen et al., 2011). Its expression remains elevated during adulthood, when it is involved in the maintenance of existing neurons and synapses. MeCP2 was initially identified as a transcriptional repressor through its binding to methylated CpG sites in gene promoters (Nan et al., 1998; Jung et al., 2003), although it regulates gene expression through multiple mechanisms (Guy et al., 2011).
From multiple studies using different knockout mouse models, converging data indicates that the pathological deficit in RTT is at the microcircuit level involving the structure and function of synapses that are critical for synaptic transmission and plasticity (Chen et al., 2001; Guy et al., 2001, 2007; Figure 2). Therefore, most therapeutic treatments revolve around the restoration of synaptic function and maturation (Gadalla et al., 2011). Current preclinical research on therapeutic strategies and human clinical trials can be divided into drugs that target the primary cause, i.e., loss-of-function mutations in MECP2, and drugs that target downstream consequences, including neurotransmitter receptor systems, neurotrophins, and their intracellular signaling pathways (Tables 3, 4). Since this review is focused on pharmacological treatments, genetic manipulations will not be discussed here and readers are directed to recent reviews on this topic (Cobb et al., 2010; Gadalla et al., 2011; Guy et al., 2011). Treatment paradigms that have targeted pathways downstream of the Mecp2 gene in mice, and the current human clinical trials are discussed in detail below.
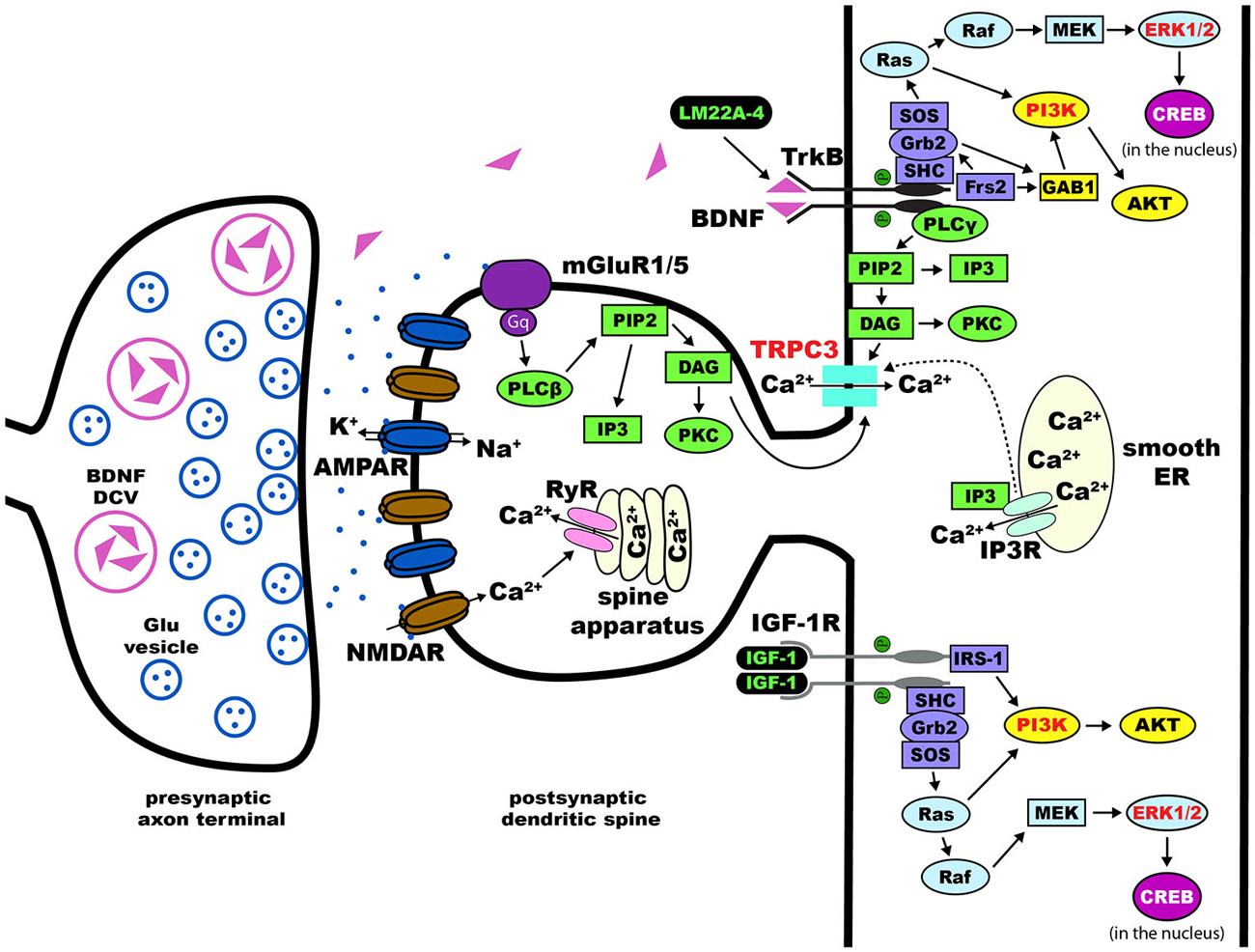
Figure 2. Dendritic spines in Rett syndrome (RTT). Proposed intracellular mechanisms that mediate the effects of BDNF/TrkB on dendritic spine density and morphology. Trk receptors are activated upon binding of neurotophic factors, leading to dimerization and auto-phosphorylation. This process allows for the intracellular binding of adaptor proteins to Trk and activation of major pathways including Ras/ERK, PI3K, and PLCγ. Components of each of these three pathways have been implicated in the effects of BDNF on dendritic spines (highlighted in red text). Potential therapies for the treatment of RTT act on these pathways (highlighted in green text)—LM22A-4 directly phosphorylates TrkB and [1,3]IGF-1 activates the PI3K and Ras/ERK pathways. Abbreviations: AKT(PKB), protein kinase B; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; BDNF, brain-derived neurotrophic factor; CaMK, Ca2+/calmodulin-dependent protein kinase; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; DAG, diacylglycerol; Frs2, fibroblast growth factor receptor substrate 2; GAB1, GRB2-associated-binding protein 1; Grb2, growth factor receptor-binding protein 2; IGF-1, insulin-like growth factor 1; IGF-1R, IGF-1 receptor; IP3, inositol triphosphate; MAPK, mitogen-activated protein kinase; MEK, MAPK//ERK kinase; NMDAR, N-methyl-D-aspartate receptor; PI3K, phosphoinositide 3-kinase; PIP2, phospohatidylinositol 4, 5 bisphosphate; PKC, protein kinase C; PLCγ, phospholipase C-γ; Raf, proto-oncogenic serine/threonine protein kinase; Ras, rat sarcoma proto-oncogenic G-protein; SHC, SH-2-containing protein; SH-2, src homology domain 2; SOS, nucleotide exchange factor son of sevenless; TrkB, tropomyosin related kinase B receptor; TRPC, transient receptor potential channel.
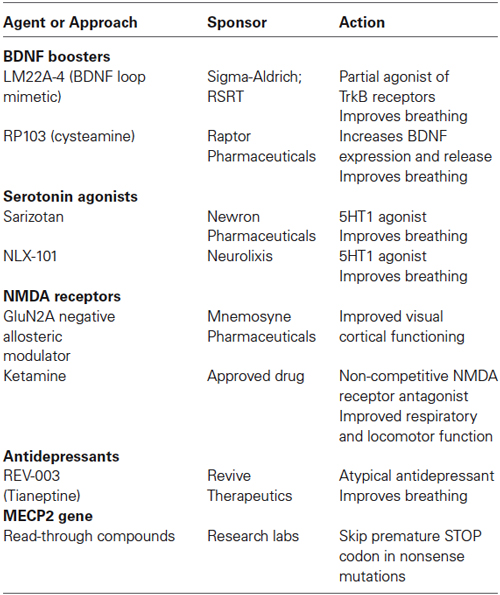
Table 3. Current preclinical trials in Mecp2 deficient mice.
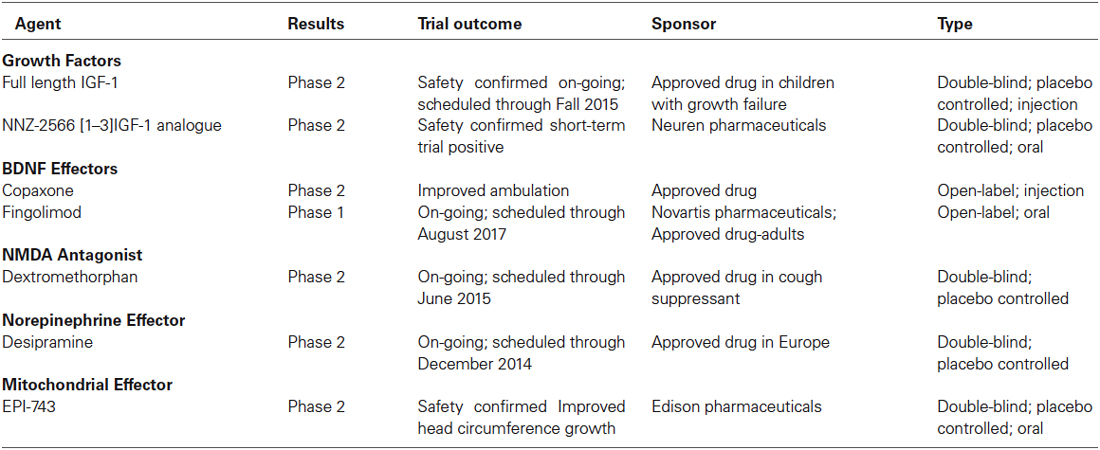
Table 4. Current clinical trials in RTT individuals.
Growth and Neurotrophic Factors
Brain-derived neurotrophic factor
Bdnf, the gene encoding the BDNF protein, is one of the first recognized direct targets of MeCP2 transcriptional regulation (Chen et al., 2003; Martinowich et al., 2003). BDNF binds to the high affinity receptor tropomyosin-related kinase B (TrkB), which activates intracellular signaling cascades critical for neuronal development, synaptic maturation, and learning and memory, like PLCγ-IP3R, PI3K-Akt, and MAPK-CREB (Segal and Greenberg, 1996). Bdnf expression is controlled by MeCP2 through complex interactions (Chen et al., 2003; Chang et al., 2006; Li and Pozzo-Miller, 2014), and reduced levels of BDNF mRNA and protein are considered to contribute to the pathophysiological mechanisms of RTT disease progression (Katz, 2014). Over-expression of BDNF in excitatory forebrain neurons of Mecp2 deficient mice improved their RTT-like neurological phenotypes (Chang et al., 2006). Over-expression of BDNF rescued dendritic atrophy caused by shRNA-mediated Mecp2 knockdown in cultured hippocampal neurons (Chapleau et al., 2009). The main limitation of recombinant BDNF is its low blood-brain barrier permeability, which prompted the search for BDNF “boosters” or mimetics with sufficient bioavailability in brain.
Currently, there are two clinical trials in RTT individuals testing compounds that boost BDNF levels: a Phase-1 open label trial of Fingolimod, and a Phase-2 open label trial of glatiramer acetate (Copaxone), both FDA-aproved drugs for the treatment of multiple sclerosis. Fingolimod is a modulator of the sphingosine-1 phosphate receptor, which leads to an increase in BDNF expression and activation of TrkB downstream signaling pathways (Deogracias et al., 2012). Glatiramer acetate is an immunomodulatory agent based on the amino acid structure of myelin basic protein (MBP) that is currently used for the treatment of relapsing-remitting multiple sclerosis. One of the proposed mechanisms of action of Copaxone is the increased expression and release of BDNF by autoreactive T-cells (Ziemssen et al., 2002).
Additonal potential leads with preclinical evidence include ampakines, which are known to increase BDNF expression by their action on AMPA-type glutamate receptors (Lauterborn et al., 2003). Peripheral treatment with ampakines significantly improved respiratory dysfunction in male Mecp2 knockout mice (Ogier et al., 2007) that model the recurrent apneas suffered by RTT girls. Cysteamine and its dimer cystamine increase BDNF levels (Borrell-Pagès et al., 2006), which supports the Phase-2/3 clinical trial of RP103 for Huntington’s disease. More recently, a small molecule BDNF loop mimetic (LM22A-4) designed in silico to interact with the BDNF binding pocket in the TrkB receptor (Massa et al., 2010), restored respiratory regularity in female Mecp2 heterozygous mice (Schmid et al., 2012; Kron et al., 2014).
Insulin Growth factor-1
Insulin Growth factor-1 (IGF-1) is a growth factor that, by binding to the IGF-1 receptor, activates similar intracellular signaling cascades to those triggered by BDNF activation of TrkB receptors. Indeed, IGF-1 modulates synaptic plasticity and neuronal maturation through a tyrosine kinase signaling pathway that includes PI3K-Akt and MAPK (Zheng and Quirion, 2004). Unlike BDNF, IGF-1 permeability through the blood brain barrier makes it an attractive compound for therapy. Intraperitoneal injection of the active IGF-1 tripeptide (also known as [1–3]IGF-1, or Glypromate) in male Mecp2 knockout mice improved survival, locomotor activity, as well as social and anxiety behaviors (Tropea et al., 2009). Full-length IGF-1 (Mecasermin) is already approved by the FDA for the treatment of growth failure in children, and it was shown to have similar effects in male Mecp2 knockout mice, as well as in female Mecp2 heterozygous mice (Castro et al., 2014); a cautionary note is that full-length IGF-1 can worsen the metabolic syndrome of Mecp2 deficient mice (Pitcher et al., 2013). The effects of full-length IGF-1 are due to the direct activation of IGF-1 receptors and its downstream signaling cascades, while the effects of the [1–3]IGF-1 tripeptide may reflect increased expression of IGF-1 (Corvin et al., 2012), although its molecular mechanism-of-action is currently unknown. Based on these promising leads, a Phase-2 double-blinded placebo-controlled clinical trail is underway to treat 3–10 years old RTT patients with full-length IGF-1 (Khwaja et al., 2014). More recently, a Phase-2 double-blinded placebo-controlled clinical trail in 16–45 years old RTT individuals has been initiated to test the efficacy of NNZ-2566, a protease-resistant analogue of the [1–3]IGF-1 tripeptide.
Neurotransmitter Systems
Monoamines
Multiple studies have documented low levels of monoamine markers (dopamine, 5-HT, noradrenaline) in autopsy RTT brains and in Mecp2 deficient mice (Brücke et al., 1987; Lekman et al., 1989; Roux et al., 2008). Desipramine is an antidepressant that blocks the uptake of noradrenaline, and has been shown to reverse the depletion of tyrosine hydroxylase in the brainstem, helping with the regulation of breathing and extending the life-span of male Mecp2 knockout mice (Roux et al., 2007; Zanella et al., 2008). Desipramine is currently in a Phase-2 double blinded, placebo-controlled clinical trial for RTT. The atypical tricyclic antidepressant tianeptine (REV-003) also improved respiratory activity in Mecp2 deficient mice, although this effect may reflect modulation of monoamine levels, glutamate receptor function, or BDNF levels (McEwen et al., 2010). Recently, preclinical studies in Mecp2 deficient mice have demonstrated that the 5-HT1a agonist Sarizotan inhibited expiratory neuron activity, which significantly improved breathing patterns and reduced the frequency of apneas (Abdala et al., 2014).
Glutamate
The NMDA-type of glutamate receptors is altered in Mecp2 knockout mice (Blue et al., 1999, 2011; Maliszewska-Cyna et al., 2010). Dextromethorphan is a NMDA receptor antagonist that has been tried in an open label clinical trial without any significant benefit. The FDA-approved NMDA receptor antagonist ketamine has been useful in Mecp2 knockout mice to improve RTT-like phenotypes (Kron et al., 2012); based on these encouraging results, a clinical trial of low dose ketamine is currently planned. A delay in the developmental switch in the expression of GluN2 subunits of the NMDA receptor in the visual cortex contributes to visual acuity deficits in Mecp2 deficient mice, which were improved by genetic deletion of the GluN2A subunit (Durand et al., 2012); a negative allosteric modulator selective for GluN2A-containing NMDA receptors is currently in preclinical trials in Mecp2 deficient mice.
GABA
Several studies have shown that GABAergic signaling is impaired in Mecp2 deficient mice, which alters the excitatory/inhibitory balance. Impaired GABAergic inhibition was described in the brainstem (Medrihan et al., 2008), thalamus (Zhang et al., 2010) and hippocampus (Calfa et al., 2015), and involved impaired evoked and spontaneous inhibitory synaptic transmission and numbers of GABAergic synapses on principal neurons. Furthermore, selective deletion of Mecp2 in GABAergic neurons led to impaired GABAergic transmission, cortical hyperexcitability and several neurological features of RTT and ASDs (Chao et al., 2010). Also, deletion of Mecp2 specifically in excitatory neurons caused impaired GABAergic transmission on cortical pyramidal neurons, which led to seizures and cortical hyperexcitation (Zhang et al., 2014a). A preclinical study in female Mecp2 heterozygous mice demonstrated that increasing ambient GABA levels by inhibiting the GABA reuptake transporter improved respiratory patterns (Abdala et al., 2010). Vigabatrin is an antiepileptic drug that irreversibly inhibits GABA transaminase, inhibits GABA catabolism and thereby increases GABA levels (Connelly, 1993). The drug is already FDA approved for use in epilepsy syndromes. Planning for a clinical trial for RTT is underway. However, retinal toxicity may limit the chronic use of this medication.
Mitochondrial Function
RTT is associated with high levels of systemic oxidative stress and alteration in mitochondrial morphology, while plasma levels of oxidative stress biomarkers correlate with disease severity and progression (De Felice et al., 2012). Based on these observations, the small molecule EPI-743 was tested in a phase-2 placebo controlled trial of RTT individuals. Results from this exploratory trial revealed improvement in head growth but not in other core features of RTT. The structure of EPI-743 is based on vitamin E and its proposed mechanisms-of-action includes augmenting glutathione synthesis and acting at the mitochondrial level to regulate electron transport.
“Read-Through” Compounds to Increase MECP2 Expression
In approximately one-third of RTT individuals, a nonsense mutation in MECP2 (e.g., R168X, R255X) leads to the premature termination of transcripton due to a premature STOP codon (Schanen et al., 2004; Percy et al., 2007). Clinically, these RTT individuals have a more severe presentation than RTT individuals with missense MECP2 mutations that result in single amino acid substitutions. Aminoglycoside antibiotics like gentamycin are so-called “read-through” compounds because they allow ribosomal read-through of the premature STOP codon during translation, yielding a full-length functional protein. Aminoglycoside and non-aminoglycoside “read-through” compounds have been tested for therapeutic efficacy in Duchenne muscular dystrophy and cystic fibrosis (Zingman et al., 2007). Preclinical studies relevant to RTT have demonstrated that either gentamycin or geneticin were effective in translating a full-length functional MeCP2 protein in a lymphocyte cell line derived from an individual with a R255X nonsense mutation (Popescu et al., 2010), and in transgenic mice expressing a R168X nonsense mutation (Brendel et al., 2011). Furthermore, gentamycin increased dendritic spine density in neurons derived from induced pluripotent stem cells (iPSC) that were obtained from a RTT individual with a Q244X nonsense mutation (Marchetto et al., 2010). However, the renal and auditory toxicity of gentamycin has limited its progress toward human clinical trials and prompted the development of new “read-through” compounds with better safety profiles.
Open Questions and Challenges
Design and Quality of Preclinical Studies
Although many targeted treatments have shown efficacy across multiple aspects in ASD animal models, none has thus far demonstrated the same effectiveness in patients (Katz et al., 2012; Hagerman et al., 2014). Hurdles to translational success may include lack of rigorous standards in assessing the effect of treatment, lack of transparency in reporting preclinical data, as well as publication bias caused by disregarding negative results in preclinical studies (Katz et al., 2012). The translational success stories in fragile X and Rett syndromes indicates that the effectiveness of targeted treatments in animal models can turn into clinical efficacy in humans, provided that studies are well designed, the models are reliable and robust, and that preclinical outcome measures are relevant to patients (Anagnostou, 2012; Lipton and Sahin, 2013; Castro et al., 2014; Khwaja et al., 2014). To achieve more translational success in ASDs, efforts are needed to improve the quality of preclinical studies in the future. Strict standards must be implemented for preclinical study designs and result reporting to ensure that clinical trials are grounded on reliable preclinical data (Katz et al., 2012; Landis et al., 2012).
One of the key issues in preclinical studies is that almost every study has applied its own animal behavioral experiment battery to evaluate the effects of genetic or pharmacological manipulations, making comparison of efficacy among different treatments a challenge. This situation will be greatly improved if animal behavioral test batteries for identifying potential therapy could be standardized. Despite the discrepancies in test batteries from different laboratories, many of the targeted treatments achieve predicted effect and rescue at least part of phenotypic features of the disorders in animal models (Braat and Kooy, 2014; Hagerman et al., 2014).
Challenges in Clinical Trials
Despite progress made in clinical trials, one of the major concerns for these studies is the lack of appropriate outcome measures for an objective assessment of patients’ daily performance (Berry-Kravis et al., 2011; Wijetunge et al., 2013; Berry-Kravis, 2014; Braat and Kooy, 2014; Jacquemont et al., 2014). As a result, some improvements following therapeutic intervention might not be observed without the right measurements. It is helpful that researchers have been trying to formulate suitable outcome measures, which could be adjusted accordingly for patients (Berry-Kravis et al., 2013), such as quantitative event related potential and expressive language outcome measure which have been developed for fragile X patients (Hessl et al., 2009). It is anticipated that these measures will better demonstrate improvements in language and cognition in individual patients. In addition, molecular markers, such as ERK, BDNF and MMP9, can be measured to provide a direct biochemical evidence of improvement with targeted treatments (Hagerman et al., 2014).
Differential responses to one specific treatment have been observed in individuals with ASDs. Some subgroups within the patient cohorts respond to the therapy where others do not, as evident from clinical trials in fragile X syndrome (Jacquemont et al., 2011; Berry-Kravis et al., 2012). It is therefore important and helpful to develop biomarkers for selection of patients that will benefit from a specific therapy depending on their genotypes and/or neurobiological phenotypes. For example, if follow-up studies would confirm that AFQ056 treatment significantly improves behavior in fully methylated patients, detecting the methylation status of Fmr1 promoter might help identify a subgroup of fragile X patients that will respond well to this treatment (Jacquemont et al., 2011; Braat and Kooy, 2014).
Interference with the molecular pathways disturbed in ASDs has led to the initiation of clinical trials. The fragile X and Rett syndromes are the prototypes of neurodevelopmental disorders for which targeted treatments are becoming realities (Samaco and Neul, 2011; Chapleau et al., 2013a,b; Braat and Kooy, 2014). However, it is unlikely that a single compound will be effective in all individuals with ASDs. Thus, it will be necessary to test combinations of multiple drugs that each rescue part of clinical presentations to find a combination of drugs that works more efficiently than each on its own. Importantly, pharmaceutical interventions need to be paired with appropriate behavioral and cognitive training to maximize the efficacy. This represents a significant step towards more personalized approaches to treating fragile X, RTT and other ASDs in future, with a better chance of success (Castro et al., 2013; Wijetunge et al., 2013; Braat and Kooy, 2014; Hagerman et al., 2014).
The Timing of Treatment Initiation
It is generally believed that earlier interventions in developmental disorders will have better outcomes. Abnormalities occurring during early development have usually been considered irreversible in adulthood. However, studies in mouse models of neurodevelopmental disorders, including fragile X and Rett syndromes, suggest that many pathophysiological aspects associated with the disorders can be reversed by genetic or pharmacological manipulations performed during adulthood (Castrén et al., 2012). Improvements have also been observed in adult patients with fragile X syndrome treated with arbaclofen and AFQ056 (Jacquemont et al., 2011; Henderson et al., 2012). These findings suggest that the pathophysiology associated with the loss of FMRP or MeCP2 may not arise from irreversible developmental brain abnormalities, but result from functional disturbances of neural circuits that could be corrected in adulthood, providing a potential rational basis for treatment of neurodevelopmental disorders in adulthood (Castrén et al., 2012; Wijetunge et al., 2013; Hagerman et al., 2014). Despite these exciting advances, translation from animal experimentation to clinical practice and finding out the right initiation timing for treating individual patients will be challenging issues in future investigation (Castrén et al., 2012; Wijetunge et al., 2013).
Conclusions
The increasing need for effective treatments of fragile X and Rett syndromes, coupled with the availability of animal models and iPSC-derived neurons from human individuals with these disorders (Marchetto et al., 2010; Wang and Doering, 2012), has promoted translational studies towards identifying potential therapeutics. New opportunities are now emerging that might lead to development of novel pharmacotherapies for patients with fragile X and Rett syndromes. The development of mechanism-based targeted treatments will require more extensive multidisciplinary researches (Chapleau et al., 2013a; Berry-Kravis, 2014; Braat and Kooy, 2014; Hagerman et al., 2014). It is the responsibility of the research community to rigorously validate disease models, outcome measures and study designs that will produce robust and reproducible preclinical findings with clear relevance to human diseases (Katz et al., 2012; Landis et al., 2012).
The rational therapeutics for ASDs requires the knowledge of an entire spectrum of symptoms that relate to each specific disorder. The complicated pathophysiology of fragile X and Rett syndromes should be taken fully into account in designing preclinical and clinical studies. It should be recognized that loss of FMRP or MeCP2 will affect a number of downstream targets which exist not only on neurons, but on glial cells and other tissues (McCauley et al., 2011; Cheng et al., 2012; Derecki et al., 2013; Ausió et al., 2014); that the loss can have direct, as well as indirect and cumulative effects on development and function of the central nervous system or other organs, leading to distinct phenotypic consequences that possibly require different treatment strategies. Understanding these complexities will be essential for selecting relative therapeutics for patients (Chapleau et al., 2013a,b; Delorme et al., 2013; Berry-Kravis, 2014; Hagerman et al., 2014). It is inspiring to see that fragile X and Rett syndromes are becoming role models for how research in animal models could be translated into patients and how management of symptoms of the diseases could extend and improve the quality of life, providing further insights into understanding and treating ASDs and other neurodevelopmental diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
HW was supported by the National Natural Science Foundation of China (NSFC, No.30200152) for Rett syndrome studies and the Fragile X Research Foundation of Canada. LP-M was supported by NIH grants NS-065027 and HD-074418, Rettsyndrome.org (former International Rett Syndrome Foundation), and the Rett Syndrome Research Trust (RSRT). LCD was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Brain Canada/Azrieli Neurodevelopmental Research Program.
Footnotes
References
Abdala, A. P., Dutschmann, M., Bissonnette, J. M., and Paton, J. F. (2010). Correction of respiratory disorders in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U S A 107, 18208–18213. doi: 10.1073/pnas.1012104107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Abdala, A. P., Lioy, D. T., Garg, S. K., Knopp, S. J., Paton, J. F., and Bissonnette, J. M. (2014). Effect of Sarizotan, a 5-HT1a and D2-like receptor agonist, on respiration in three mouse models of Rett syndrome. Am. J. Respir. Cell Mol. Biol. 50, 1031–1039. doi: 10.1165/rcmb.2013-0372OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Abekhoukh, S., and Bardoni, B. (2014). CYFIP family proteins between autism and intellectual disability: links with Fragile X syndrome. Front. Cell. Neurosci. 8:81. doi: 10.3389/fncel.2014.00081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anagnostou, E. (2012). Translational medicine: mice and men show the way. Nature 491, 196–197. doi: 10.1038/491196a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anagnostou, E., Soorya, L., Brian, J., Dupuis, A., Mankad, D., Smile, S., et al. (2014a). Intranasal oxytocin in the treatment of autism spectrum disorders: a review of literature and early safety and efficacy data in youth. Brain Res. 1580, 188–198. doi: 10.1016/j.brainres.2014.01.049.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anagnostou, E., Zwaigenbaum, L., Szatmari, P., Fombonne, E., Fernandez, B. A., Woodbury-Smith, M., et al. (2014b). Autism spectrum disorder: advances in evidence-based practice. CMAJ 186, 509–519. doi: 10.1503/cmaj.121756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andari, E., Duhamel, J. R., Zalla, T., Herbrecht, E., Leboyer, M., and Sirigu, A. (2010). Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc. Natl. Acad. Sci. U S A 107, 4389–4394. doi: 10.1073/pnas.0910249107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Auerbach, B. D., Osterweil, E. K., and Bear, M. F. (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68. doi: 10.1038/nature10658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ausió, J., de Paz, A. M., and Esteller, M. (2014). MeCP2: the long trip from a chromatin protein to neurological disorders. Trends Mol. Med. 20, 487–498. doi: 10.1016/j.molmed.2014.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Banerjee, S., Riordan, M., and Bhat, M. A. (2014). Genetic aspects of autism spectrum disorders: insights from animal models. Front. Cell. Neurosci. 8:58. doi: 10.3389/fncel.2014.00058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bassell, G. J., and Warren, S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. doi: 10.1016/j.neuron.2008.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. doi: 10.1016/j.tins.2004.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ben-Ari, Y., Khalilov, I., Kahle, K. T., and Cherubini, E. (2012). The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18, 467–486. doi: 10.1177/1073858412438697
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berry-Kravis, E. (2014). Mechanism-based treatments in neurodevelopmental disorders: fragile X syndrome. Pediatr. Neurol. 50, 297–302. doi: 10.1016/j.pediatrneurol.2013.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berry-Kravis, E., Hessl, D., Abbeduto, L., Reiss, A. L., Beckel-Mitchener, A., and Urv, T. K. (2013). Outcome measures for clinical trials in fragile X syndrome. J. Dev. Behav. Pediatr. 34, 508–522. doi: 10.1097/DBP.0b013e31829d1f20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berry-Kravis, E., Hessl, D., Coffey, S., Hervey, C., Schneider, A., Yuhas, J., et al. (2009). A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J. Med. Genet. 46, 266–271. doi: 10.1136/jmg.2008.063701
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berry-Kravis, E. M., Hessl, D., Rathmell, B., Zarevics, P., Cherubini, M., Walton-Bowen, K., et al. (2012). Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci. Transl. Med. 4:152ra127. doi: 10.1126/scitranslmed.3004214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berry-Kravis, E., Knox, A., and Hervey, C. (2011). Targeted treatments for fragile X syndrome. J. Neurodev. Disord. 3, 193–210. doi: 10.1007/s11689-011-9074-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berry-Kravis, E., Krause, S. E., Block, S. S., Guter, S., Wuu, J., Leurgans, S., et al. (2006). Effect of CX516, an AMPA-modulating compound, on cognition and behavior in fragile X syndrome: a controlled trial. J. Child Adolesc. Psychopharmacol. 16, 525–540. doi: 10.1089/cap.2006.16.525
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berry-Kravis, E., Sumis, A., Hervey, C., Nelson, M., Porges, S. W., Weng, N., et al. (2008). Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J. Dev. Behav. Pediatr. 29, 293–302. doi: 10.1097/DBP.0b013e31817dc447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhakar, A. L., Dolen, G., and Bear, M. F. (2012). The pathophysiology of fragile, X (and what it teaches us about synapses). Annu. Rev. Neurosci. 35, 417–443. doi: 10.1146/annurev-neuro-060909-153138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhattacharya, A., Kaphzan, H., Alvarez-Dieppa, A. C., Murphy, J. P., Pierre, P., and Klann, E. (2012). Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic and behavioral phenotypes in fragile X syndrome mice. Neuron 76, 325–337. doi: 10.1016/j.neuron.2012.07.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bienvenu, T., and Chelly, J. (2006). Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 7, 415–426. doi: 10.1038/nrg1878
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bilousova, T. V., Dansie, L., Ngo, M., Aye, J., Charles, J. R., Ethell, D. W., et al. (2009). Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J. Med. Genet. 46, 94–102. doi: 10.1136/jmg.2008.061796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blue, M. E., Kaufmann, W. E., Bressler, J., Eyring, C., O’driscoll, C., Naidu, S., et al. (2011). Temporal and regional alterations in NMDA receptor expression in Mecp2-null mice. Anat. Rec. (Hoboken) 294, 1624–1634. doi: 10.1002/ar.21380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blue, M. E., Naidu, S., and Johnston, M. V. (1999). Development of amino acid receptors in frontal cortex from girls with Rett syndrome. Ann. Neurol. 45, 541–545. doi: 10.1002/1531-8249(199904)45:4<541::aid-ana21>3.0.co;2-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boda, B., Mendez, P., Boury-Jamot, B., Magara, F., and Muller, D. (2014). Reversal of activity-mediated spine dynamics and learning impairment in a mouse model of Fragile X syndrome. Eur. J. Neurosci. 39, 1130–1137. doi: 10.1111/ejn.12488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Borrell-Pagès, M., Canals, J. M., Cordelières, F. P., Parker, J. A., Pineda, J. R., Grange, G., et al. (2006). Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J. Clin. Invest. 116, 1410–1424. doi: 10.1172/jci27607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bortolotto, Z. A., Fitzjohn, S. M., and Collingridge, G. L. (1999). Roles of metabotropic glutamate receptors in LTP and LTD in the hippocampus. Curr. Opin. Neurobiol. 9, 299–304. doi: 10.1016/s0959-4388(99)80044-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Braat, S., and Kooy, R. F. (2014). Fragile X syndrome neurobiology translates into rational therapy. Drug Discov. Today 19, 510–519. doi: 10.1016/j.drudis.2014.01.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Braithwaite, S. P., Paul, S., Nairn, A. C., and Lombroso, P. J. (2006). Synaptic plasticity: one STEP at a time. Trends Neurosci. 29, 452–458. doi: 10.1016/j.tins.2006.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brendel, C., Belakhov, V., Werner, H., Wegener, E., Gärtner, J., Nudelman, I., et al. (2011). Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model. J. Mol. Med. (Berl.) 89, 389–398. doi: 10.1007/s00109-010-0704-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, M. R., Kronengold, J., Gazula, V. R., Chen, Y., Strumbos, J. G., Sigworth, F. J., et al. (2010). Fragile X mental retardation protein controls gating of the sodium-activated potassium channel slack. Nat. Neurosci. 13, 819–821. doi: 10.1038/nn.2563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brücke, T., Sofic, E., Killian, W., Rett, A., and Riederer, P. (1987). Reduced concentrations and increased metabolism of biogenic amines in a single case of Rett-syndrome: a postmortem brain study. J. Neural Transm. 68, 315–324. doi: 10.1007/bf02098506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burket, J. A., Benson, A. D., Tang, A. H., and Deutsch, S. I. (2014). Rapamycin improves sociability in the BTBR T(+)Itpr3(tf)/J mouse model of autism spectrum disorders. Brain Res. Bull. 100, 70–75. doi: 10.1016/j.brainresbull.2013.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Busquets-Garcia, A., Gomis-González, M., Guegan, T., Agustín-Pavón, C., Pastor, A., Mato, S., et al. (2013). Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat. Med. 19, 603–607. doi: 10.1038/nm.3127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Çaku, A., Pellerin, D., Bouvier, P., Riou, E., and Corbin, F. (2014). Effect of lovastatin on behavior in children and adults with fragile X syndrome: an open-label study. Am. J. Med. Genet. A 164A, 2834–2842. doi: 10.1002/ajmg.a.36750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calfa, G., Li, W., Rutherford, J. M., and Pozzo-Miller, L. (2015). Excitation/inhibition imbalance and impaired synaptic inhibition in hippocampal area CA3 of Mecp2 knockout mice. Hippocampus 25, 159–168. doi: 10.1002/hipo.22360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Castrén, E., Elgersma, Y., Maffei, L., and Hagerman, R. (2012). Treatment of neurodevelopmental disorders in adulthood. J. Neurosci. 32, 14074–14079. doi: 10.1523/JNEUROSCI.3287-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Castro, J., Garcia, R. I., Kwok, S., Banerjee, A., Petravicz, J., Woodson, J., et al. (2014). Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U S A 111, 9941–9946. doi: 10.1073/pnas.1311685111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Castro, J., Mellios, N., and Sur, M. (2013). Mechanisms and therapeutic challenges in autism spectrum disorders: insights from Rett syndrome. Curr. Opin. Neurol. 26, 154–159. doi: 10.1097/WCO.0b013e32835f19a7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cerovic, M., d’Isa, R., Tonini, R., and Brambilla, R. (2013). Molecular and cellular mechanisms of dopamine-mediated behavioral plasticity in the striatum. Neurobiol. Learn. Mem. 105, 63–80. doi: 10.1016/j.nlm.2013.06.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, S., Bray, S. M., Li, Z., Zarnescu, D. C., He, C., Jin, P., et al. (2008). Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat. Chem. Biol. 4, 256–263. doi: 10.1038/nchembio.78
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, Q., Khare, G., Dani, V., Nelson, S., and Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348. doi: 10.1016/j.neuron.2005.12.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chapleau, C. A., Lane, J., Larimore, J., Li, W., Pozzo-Miller, L., and Percy, A. K. (2013b). Recent progress in Rett syndrome and MeCP2 Dysfunction: assessment of potential treatment options. Future Neurol. 8, 21–28. doi: 10.2217/fnl.12.79
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chapleau, C. A., Lane, J., Pozzo-Miller, L., and Percy, A. K. (2013a). Evaluation of current pharmacological treatment options in the management of Rett syndrome: from the present to future therapeutic alternatives. Curr. Clin. Pharmacol. 8, 358–369. doi: 10.2174/15748847113086660069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chapleau, C. A., Larimore, J. L., Theibert, A., and Pozzo-Miller, L. (2009). Modulation of dendritic spine development and plasticity by BDNF and vesicular trafficking: fundamental roles in neurodevelopmental disorders associated with mental retardation and autism. J. Neurodev. Disord. 1, 185–196. doi: 10.1007/s11689-009-9027-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C., et al. (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889. doi: 10.1126/science.1086446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, L. Y., Rex, C. S., Babayan, A. H., Kramár, E. A., Lynch, G., Gall, C. M., et al. (2010). Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J. Neurosci. 30, 10977–10984. doi: 10.1523/JNEUROSCI.1077-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheng, C., Sourial, M., and Doering, L. C. (2012). Astrocytes and developmental plasticity in fragile X. Neural Plast. 2012:197491. doi: 10.1155/2012/197491
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chevaleyre, V., Takahashi, K. A., and Castillo, P. E. (2006). Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 29, 37–76. doi: 10.1146/annurev.neuro.29.051605.112834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chez, M. G., Burton, Q., Dowling, T., Chang, M., Khanna, P., and Kramer, C. (2007). Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: an observation of initial clinical response and maintenance tolerability. J. Child Neurol. 22, 574–579. doi: 10.1177/0883073807302611
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiu, C. T., and Chuang, D. M. (2010). Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol. Ther. 128, 281–304. doi: 10.1016/j.pharmthera.2010.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chugani, D. C. (2002). Role of altered brain serotonin mechanisms in autism. Mol. Psychiatry 7(Suppl. 2), S16–S17. doi: 10.1038/sj.mp.4001167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cobb, S., Guy, J., and Bird, A. (2010). Reversibility of functional deficits in experimental models of Rett syndrome. Biochem. Soc. Trans. 38, 498–506. doi: 10.1042/BST0380498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cohen, S., Gabel, H. W., Hemberg, M., Hutchinson, A. N., Sadacca, L. A., Ebert, D. H., et al. (2011). Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 72, 72–85. doi: 10.1016/j.neuron.2011.08.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cohen, P., and Goedert, M. (2004). GSK3 inhibitors: development and therapeutic potential. Nat. Rev. Drug Discov. 3, 479–487. doi: 10.1038/nrd1415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Corvin, A. P., Molinos, I., Little, G., Donohoe, G., Gill, M., Morris, D. W., et al. (2012). Insulin-like growth factor 1 (IGF1) and its active peptide (1–3)IGF1 enhance the expression of synaptic markers in neuronal circuits through different cellular mechanisms. Neurosci. Lett. 520, 51–56. doi: 10.1016/j.neulet.2012.05.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Costa, L., Spatuzza, M., D’Antoni, S., Bonaccorso, C. M., Trovato, C., Musumeci, S. A., et al. (2012). Activation of 5-HT7 serotonin receptors reverses metabotropic glutamate receptor-mediated synaptic plasticity in wild-type and Fmr1 knockout mice, a model of Fragile X syndrome. Biol. Psychiatry 72, 924–933. doi: 10.1016/j.biopsych.2012.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cuesto, G., Enriquez-Barreto, L., Caramés, C., Cantarero, M., Gasull, X., Sandi, C., et al. (2011). Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J. Neurosci. 31, 2721–2733. doi: 10.1523/JNEUROSCI.4477-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Curia, G., Gualtieri, F., Bartolomeo, R., Vezzali, R., and Biagini, G. (2013). Resilience to audiogenic seizures is associated with p-ERK1/2 dephosphorylation in the subiculum of Fmr1 knockout mice. Front. Cell. Neurosci. 7:46. doi: 10.3389/fncel.2013.00046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dansie, L. E., Phommahaxay, K., Okusanya, A. G., Uwadia, J., Huang, M., Rotschafer, S. E., et al. (2013). Long-lasting effects of minocycline on behavior in young but not adult Fragile X mice. Neuroscience 246, 186–198. doi: 10.1016/j.neuroscience.2013.04.058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Darnell, J. C., and Klann, E. (2013). The translation of translational control by FMRP: therapeutic targets for FXS. Nat. Neurosci. 16, 1530–1536. doi: 10.1038/nn.3379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Felice, C., Signorini, C., Leoncini, S., Pecorelli, A., Durand, T., Valacchi, G., et al. (2012). The role of oxidative stress in Rett syndrome: an overview. Ann. N Y Acad. Sci. 1259, 121–135. doi: 10.1111/j.1749-6632.2012.06611.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Delorme, R., Ey, E., Toro, R., Leboyer, M., Gillberg, C., and Bourgeron, T. (2013). Progress toward treatments for synaptic defects in autism. Nat. Med. 19, 685–694. doi: 10.1038/nm.3193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deng, P. Y., Rotman, Z., Blundon, J. A., Cho, Y., Cui, J., Cavalli, V., et al. (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. doi: 10.1016/j.neuron.2012.12.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deogracias, R., Yazdani, M., Dekkers, M. P., Guy, J., Ionescu, M. C., Vogt, K. E., et al. (2012). Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U S A 109, 14230–14235. doi: 10.1073/pnas.1206093109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Derecki, N. C., Cronk, J. C., and Kipnis, J. (2013). The role of microglia in brain maintenance: implications for Rett syndrome. Trends Immunol. 34, 144–150. doi: 10.1016/j.it.2012.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Devlin, B., Cook, E. H. Jr., Coon, H., Dawson, G., Grigorenko, E. L., McMahon, W., et al. (2005). Autism and the serotonin transporter: the long and short of it. Mol. Psychiatry 10, 1110–1116. doi: 10.1038/sj.mp.4001724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Vrij, F. M., Levenga, J., van der Linde, H. C., Koekkoek, S. K., De Zeeuw, C. I., Nelson, D. L., et al. (2008). Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol. Dis. 31, 127–132. doi: 10.1016/j.nbd.2008.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D’Hulst, C., De Geest, N., Reeve, S. P., Van Dam, D., De Deyn, P. P., Hassan, B. A., et al. (2006). Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 1121, 238–245. doi: 10.1016/j.brainres.2006.08.115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D’Hulst, C., Heulens, I., Brouwer, J. R., Willemsen, R., De Geest, N., Reeve, S. P., et al. (2009). Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 1253, 176–183. doi: 10.1016/j.brainres.2008.11.075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dolan, B. M., Duron, S. G., Campbell, D. A., Vollrath, B., Shankaranarayana Rao, B. S., Ko, H. Y., et al. (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. U S A 110, 5671–5676. doi: 10.1073/pnas.1219383110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dölen, G., and Bear, M. F. (2008). Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J. Physiol. 586, 1503–1508. doi: 10.1113/jphysiol.2008.150722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doll, C. A., and Broadie, K. (2014). Impaired activity-dependent neural circuit assembly and refinement in autism spectrum disorder genetic models. Front. Cell. Neurosci. 8:30. doi: 10.3389/fncel.2014.00030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Domes, G., Heinrichs, M., Kumbier, E., Grossmann, A., Hauenstein, K., and Herpertz, S. C. (2013). Effects of intranasal oxytocin on the neural basis of face processing in autism spectrum disorder. Biol. Psychiatry 74, 164–171. doi: 10.1016/j.biopsych.2013.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Durand, S., Patrizi, A., Quast, K. B., Hachigian, L., Pavlyuk, R., Saxena, A., et al. (2012). NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron 76, 1078–1090. doi: 10.1016/j.neuron.2012.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dziembowska, M., Pretto, D. I., Janusz, A., Kaczmarek, L., Leigh, M. J., Gabriel, N., et al. (2013). High MMP-9 activity levels in fragile X syndrome are lowered by minocycline. Am. J. Med. Genet. A 161A, 1897–1903. doi: 10.1002/ajmg.a.36023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eadie, B. D., Cushman, J., Kannangara, T. S., Fanselow, M. S., and Christie, B. R. (2012). NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus 22, 241–254. doi: 10.1002/hipo.20890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ebert, D. H., and Greenberg, M. E. (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. doi: 10.1038/nature11860
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Erickson, C. A., and Chambers, J. E. (2006). Memantine for disruptive behavior in autistic disorder. J. Clin. Psychiatry 67:1000. doi: 10.4088/jcp.v67n0619h
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Erickson, C. A., Mullett, J. E., and McDougle, C. J. (2009). Open-label memantine in fragile X syndrome. J. Autism Dev. Disord. 39, 1629–1635. doi: 10.1007/s10803-009-0807-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Erickson, C. A., Mullett, J. E., and McDougle, C. J. (2010). Brief report: acamprosate in fragile X syndrome. J. Autism Dev. Disord. 40, 1412–1416. doi: 10.1007/s10803-010-0988-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Erickson, C. A., Wink, L. K., Ray, B., Early, M. C., Stiegelmeyer, E., Mathieu-Frasier, L., et al. (2013). Impact of acamprosate on behavior and brain-derived neurotrophic factor: an open-label study in youth with fragile X syndrome. Psychopharmacology (Berl) 228, 75–84. doi: 10.1007/s00213-013-3022-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ethell, I. M., and Ethell, D. W. (2007). Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J. Neurosci. Res. 85, 2813–2823. doi: 10.1002/jnr.21273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Franklin, A. V., King, M. K., Palomo, V., Martinez, A., McMahon, L. L., and Jope, R. S. (2014). Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol. Psychiatry 75, 198–206. doi: 10.1016/j.biopsych.2013.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fulks, J. L., O’Bryhim, B. E., Wenzel, S. K., Fowler, S. C., Vorontsova, E., Pinkston, J. W., et al. (2010). Dopamine release and uptake impairments and behavioral alterations observed in mice that model fragile X mental retardation syndrome. ACS Chem. Neurosci. 1, 679–690. doi: 10.1021/cn100032f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gadalla, K. K., Bailey, M. E., and Cobb, S. R. (2011). MeCP2 and Rett syndrome: reversibility and potential avenues for therapy. Biochem. J. 439, 1–14. doi: 10.1042/bj20110648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gandhi, R. M., Kogan, C. S., and Messier, C. (2014). 2-Methyl-6-(phenylethynyl) pyridine (MPEP) reverses maze learning and PSD-95 deficits in Fmr1 knock-out mice. Front. Cell. Neurosci. 8:70. doi: 10.3389/fncel.2014.00070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gantois, I., Pop, A. S., de Esch, C. E., Buijsen, R. A., Pooters, T., Gomez-Mancilla, B., et al. (2013). Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav. Brain Res. 239, 72–79. doi: 10.1016/j.bbr.2012.10.059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garber, K. B., Visootsak, J., and Warren, S. T. (2008). Fragile X syndrome. Eur. J. Hum. Genet. 16, 666–672. doi: 10.1038/ejhg.2008.61
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gassmann, M., and Bettler, B. (2012). Regulation of neuronal GABA(B) receptor functions by subunit composition. Nat. Rev. Neurosci. 13, 380–394. doi: 10.1038/nrn3249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gellynck, E., Heyninck, K., Andressen, K. W., Haegeman, G., Levy, F. O., Vanhoenacker, P., et al. (2013). The serotonin 5-HT7 receptors: two decades of research. Exp. Brain Res. 230, 555–568. doi: 10.1007/s00221-013-3694-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Girard, M., Couvert, P., Carrié, A., Tardieu, M., Chelly, J., Beldjord, C., et al. (2001). Parental origin of de novo MECP2 mutations in Rett syndrome. Eur. J. Hum. Genet. 9, 231–236. doi: 10.1038/sj.ejhg.5200618
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gkogkas, C. G., Khoutorsky, A., Ran, I., Rampakakis, E., Nevarko, T., Weatherill, D. B., et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. doi: 10.1038/nature11628
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glaze, D. G., Percy, A. K., Skinner, S., Motil, K. J., Neul, J. L., Barrish, J. O., et al. (2010). Epilepsy and the natural history of Rett syndrome. Neurology 74, 909–912. doi: 10.1212/wnl.0b013e3181d6b852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gocel, J., and Larson, J. (2012). Synaptic NMDA receptor-mediated currents in anterior piriform cortex are reduced in the adult fragile X mouse. Neuroscience 221, 170–181. doi: 10.1016/j.neuroscience.2012.06.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goebel-Goody, S. M., Baum, M., Paspalas, C. D., Fernandez, S. M., Carty, N. C., Kurup, P., et al. (2012b). Therapeutic implications for striatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders. Pharmacol. Rev. 64, 65–87. doi: 10.1124/pr.110.003053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goebel-Goody, S. M., and Lombroso, P. J. (2012). Taking STEPs forward to understand fragile X syndrome. Results Probl. Cell Differ. 54, 223–241. doi: 10.1007/978-3-642-21649-7_12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goebel-Goody, S. M., Wilson-Wallis, E. D., Royston, S., Tagliatela, S. M., Naegele, J. R., and Lombroso, P. J. (2012a). Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav. 11, 586–600. doi: 10.1111/j.1601-183x.2012.00781.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gordon, I., Vander Wyk, B. C., Bennett, R. H., Cordeaux, C., Lucas, M. V., Eilbott, J. A., et al. (2013). Oxytocin enhances brain function in children with autism. Proc. Natl. Acad. Sci. U S A 110, 20953–20958. doi: 10.1073/pnas.1312857110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gralle, M., and Ferreira, S. T. (2007). Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog. Neurobiol. 82, 11–32. doi: 10.1016/j.pneurobio.2007.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gross, C., and Bassell, G. J. (2012). Excess protein synthesis in FXS patient lymphoblastoid cells can be rescued with a p110beta-selective inhibitor. Mol. Med. 18, 336–345. doi: 10.2119/molmed.2011.00363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gross, C., and Bassell, G. J. (2014). Neuron-specific regulation of class I PI3K catalytic subunits and their dysfunction in brain disorders. Front. Mol. Neurosci. 7:12. doi: 10.3389/fnmol.2014.00012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gross, C., Berry-Kravis, E. M., and Bassell, G. J. (2012). Therapeutic strategies in fragile X syndrome: dysregulated mGluR signaling and beyond. Neuropsychopharmacology 37, 178–195. doi: 10.1038/npp.2011.137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gross, C., Nakamoto, M., Yao, X., Chan, C. B., Yim, S. Y., Ye, K., et al. (2010). Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci. 30, 10624–10638. doi: 10.1523/jneurosci.0402-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gross, C., Yao, X., Pong, D. L., Jeromin, A., and Bassell, G. J. (2011). Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J. Neurosci. 31, 5693–5698. doi: 10.1523/jneurosci.6661-10.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, W., Murthy, A. C., Zhang, L., Johnson, E. B., Schaller, E. G., Allan, A. M., et al. (2012). Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum. Mol. Genet. 21, 681–691. doi: 10.1093/hmg/ddr501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guy, J., Cheval, H., Selfridge, J., and Bird, A. (2011). The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 27, 631–652. doi: 10.1146/annurev-cellbio-092910-154121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hagberg, B., Aicardi, J., Dias, K., and Ramos, O. (1983). A progressive syndrome of autism, dementia, ataxia and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann. Neurol. 14, 471–479. doi: 10.1002/ana.410140412
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hagerman, R. J., Des-Portes, V., Gasparini, F., Jacquemont, S., and Gomez-Mancilla, B. (2014). Translating molecular advances in fragile X syndrome into therapy: a review. J. Clin. Psychiatry 75, e294–e307. doi: 10.4088/jcp.13r08714
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hagerman, R. J., Hills, J., Scharfenaker, S., and Lewis, H. (1999). Fragile X syndrome and selective mutism. Am. J. Med. Genet. 83, 313–317. doi: 10.1002/(sici)1096-8628(19990402)83:4<313::aid-ajmg15>3.0.co;2-f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hagerman, R. J., and Polussa, J. (2015). Treatment of the psychiatric problems associated with fragile X syndrome. Curr. Opin. Psychiatry 28, 107–112. doi: 10.1097/yco.0000000000000131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hall, S. S., Lightbody, A. A., McCarthy, B. E., Parker, K. J., and Reiss, A. L. (2012). Effects of intranasal oxytocin on social anxiety in males with fragile X syndrome. Psychoneuroendocrinology 37, 509–518. doi: 10.1016/j.psyneuen.2011.07.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harrington, R. A., Lee, L. C., Crum, R. M., Zimmerman, A. W., and Hertz-Picciotto, I. (2013). Serotonin hypothesis of autism: implications for selective serotonin reuptake inhibitor use during pregnancy. Autism Res. 6, 149–168. doi: 10.1002/aur.1288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hay, N., and Sonenberg, N. (2004). Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945. doi: 10.1101/gad.1212704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, M. L., Choi, S. Y., Rao, B. S., Jung, H. Y., Lee, H. K., Zhang, D., et al. (2004). Altered cortical synaptic morphology and impaired memory consolidation in forebrain-specific dominant-negative PAK transgenic mice. Neuron 42, 773–787. doi: 10.1016/j.neuron.2004.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, M. L., Rao, B. S., Seo, J. S., Choi, H. S., Dolan, B. M., Choi, S. Y., et al. (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. U S A 104, 11489–11494. doi: 10.1073/pnas.0705003104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hays, S. A., Huber, K. M., and Gibson, J. R. (2011). Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J. Neurosci. 31, 14223–14234. doi: 10.1523/jneurosci.3157-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, Q., Nomura, T., Xu, J., and Contractor, A. (2014). The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci. 34, 446–450. doi: 10.1523/jneurosci.4447-13.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hebert, B., Pietropaolo, S., Meme, S., Laudier, B., Laugeray, A., Doisne, N., et al. (2014). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by a BKCa channel opener molecule. Orphanet J. Rare Dis. 9:124. doi: 10.1186/s13023-014-0124-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heifets, B. D., and Castillo, P. E. (2009). Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 71, 283–306. doi: 10.1146/annurev.physiol.010908.163149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henderson, C., Wijetunge, L., Kinoshita, M. N., Shumway, M., Hammond, R. S., Postma, F. R., et al. (2012). Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4:152ra128. doi: 10.1126/scitranslmed.3004218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hessl, D., Nguyen, D. V., Green, C., Chavez, A., Tassone, F., Hagerman, R. J., et al. (2009). A solution to limitations of cognitive testing in children with intellectual disabilities: the case of fragile X syndrome. J. Neurodev. Disord. 1, 33–45. doi: 10.1007/s11689-008-9001-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heulens, I., D’Hulst, C., Van Dam, D., De Deyn, P. P., and Kooy, R. F. (2012). Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav. Brain Res. 229, 244–249. doi: 10.1016/j.bbr.2012.01.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoeffer, C. A., and Klann, E. (2010). mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75. doi: 10.1016/j.tins.2009.11.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoeffer, C. A., Sanchez, E., Hagerman, R. J., Mu, Y., Nguyen, D. V., Wong, H., et al. (2012). Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 11, 332–341. doi: 10.1111/j.1601-183x.2012.00768.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hollander, E., Phillips, A., Chaplin, W., Zagursky, K., Novotny, S., Wasserman, S., et al. (2005). A placebo controlled crossover trial of liquid fluoxetine on repetitive behaviors in childhood and adolescent autism. Neuropsychopharmacology 30, 582–589. doi: 10.1038/sj.npp.1300627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hollander, E., Soorya, L., Chaplin, W., Anagnostou, E., Taylor, B. P., Ferretti, C. J., et al. (2012). A double-blind placebo-controlled trial of fluoxetine for repetitive behaviors and global severity in adult autism spectrum disorders. Am. J. Psychiatry 169, 292–299. doi: 10.1176/appi.ajp.2011.10050764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horwood, J. M., Dufour, F., Laroche, S., and Davis, S. (2006). Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur. J. Neurosci. 23, 3375–3384. doi: 10.1111/j.1460-9568.2006.04859.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, H., Qin, Y., Bochorishvili, G., Zhu, Y., van Aelst, L., and Zhu, J. J. (2008). Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J. Neurosci. 28, 7847–7862. doi: 10.1523/jneurosci.1496-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huguet, G., Ey, E., and Bourgeron, T. (2013). The genetic landscapes of autism spectrum disorders. Annu. Rev. Genomics Hum. Genet. 14, 191–213. doi: 10.1146/annurev-genom-091212-153431
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huntley, G. W. (2012). Synaptic circuit remodelling by matrix metalloproteinases in health and disease. Nat. Rev. Neurosci. 13, 743–757. doi: 10.1038/nrn3320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imbesi, M., Uz, T., Manev, R., Sharma, R. P., and Manev, H. (2008). Minocycline increases phosphorylation and membrane insertion of neuronal GluR1 receptors. Neurosci. Lett. 447, 134–137. doi: 10.1016/j.neulet.2008.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacquemont, S., Berry-Kravis, E., Hagerman, R., von Raison, F., Gasparini, F., Apostol, G., et al. (2014). The challenges of clinical trials in fragile X syndrome. Psychopharmacology (Berl) 231, 1237–1250. doi: 10.1007/s00213-013-3289-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacquemont, S., Curie, A., des Portes, V., Torrioli, M. G., Berry-Kravis, E., Hagerman, R. J., et al. (2011). Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci. Transl. Med. 3:64ra61. doi: 10.1126/scitranslmed.3001708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Janusz, A., Milek, J., Perycz, M., Pacini, L., Bagni, C., Kaczmarek, L., et al. (2013). The fragile X mental retardation protein regulates matrix metalloproteinase 9 mRNA at synapses. J. Neurosci. 33, 18234–18241. doi: 10.1523/JNEUROSCI.2207-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jay, T. M. (2003). Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog. Neurobiol. 69, 375–390. doi: 10.1016/s0301-0082(03)00085-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jope, R. S., and Roh, M. S. (2006). Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr. Drug Targets 7, 1421–1434. doi: 10.2174/1389450110607011421
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jung, B. P., Jugloff, D. G., Zhang, G., Logan, R., Brown, S., and Eubanks, J. H. (2003). The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J. Neurobiol. 55, 86–96. doi: 10.1002/neu.10201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jung, K. M., Sepers, M., Henstridge, C. M., Lassalle, O., Neuhofer, D., Martin, H., et al. (2012). Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat. Commun. 3:1080. doi: 10.1038/ncomms2045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Katz, D. M. (2014). Brain-derived neurotrophic factor and Rett syndrome. Handb. Exp. Pharmacol. 220, 481–495. doi: 10.1007/978-3-642-45106-5_18
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Katz, D. M., Berger-Sweeney, J. E., Eubanks, J. H., Justice, M. J., Neul, J. L., Pozzo-Miller, L., et al. (2012). Preclinical research in Rett syndrome: setting the foundation for translational success. Dis. Model. Mech. 5, 733–745. doi: 10.1242/dmm.011007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelleher, R. J. 3rd, Govindarajan, A., Jung, H. Y., Kang, H., and Tonegawa, S. (2004). Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116, 467–479. doi: 10.1016/s0092-8674(04)00115-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khwaja, O. S., Ho, E., Barnes, K. V., O’Leary, H. M., Pereira, L. M., Finkelstein, Y., et al. (2014). Safety, pharmacokinetics and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc. Natl. Acad. Sci. U S A 111, 4596–4601. doi: 10.1073/pnas.1311141111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, G. E., and Kaczmarek, L. K. (2014). Emerging role of the KCNT1 Slack channel in intellectual disability. Front. Cell. Neurosci. 8:209. doi: 10.3389/fncel.2014.00209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, S. H., Markham, J. A., Weiler, I. J., and Greenough, W. T. (2008). Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc. Natl. Acad. Sci. U S A 105, 4429–4434. doi: 10.1073/pnas.0800257105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
King, M. K., and Jope, R. S. (2013). Lithium treatment alleviates impaired cognition in a mouse model of fragile X syndrome. Genes Brain Behav. 12, 723–731. doi: 10.1111/gbb.12071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klose, R. J., and Bird, A. P. (2006). Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97. doi: 10.1016/j.tibs.2005.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Knobloch, H. S., and Grinevich, V. (2014). Evolution of oxytocin pathways in the brain of vertebrates. Front. Behav. Neurosci. 8:31. doi: 10.3389/fnbeh.2014.00031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kolevzon, A., Mathewson, K. A., and Hollander, E. (2006). Selective serotonin reuptake inhibitors in autism: a review of efficacy and tolerability. J. Clin. Psychiatry 67, 407–414. doi: 10.4088/jcp.v67n0311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kreis, P., and Barnier, J. V. (2009). PAK signalling in neuronal physiology. Cell. Signal. 21, 384–393. doi: 10.1016/j.cellsig.2008.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kron, M., Howell, C. J., Adams, I. T., Ransbottom, M., Christian, D., Ogier, M., et al. (2012). Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J. Neurosci. 32, 13860–13872. doi: 10.1523/JNEUROSCI.2159-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kron, M., Lang, M., Adams, I. T., Sceniak, M., Longo, F., and Katz, D. M. (2014). A BDNF loop-domain mimetic acutely reverses spontaneous apneas and respiratory abnormalities during behavioral arousal in a mouse model of Rett syndrome. Dis. Model. Mech. 7, 1047–1055. doi: 10.1242/dmm.016030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krueger, D. D., Osterweil, E. K., Chen, S. P., Tye, L. D., and Bear, M. F. (2011). Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. U S A 108, 2587–2592. doi: 10.1073/pnas.1013855108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krumm, N., O’Roak, B. J., Shendure, J., and Eichler, E. E. (2014). A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 37, 95–105. doi: 10.1016/j.tins.2013.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumari, R., Castillo, C., and Francesconi, A. (2013). Agonist-dependent signaling by group I metabotropic glutamate receptors is regulated by association with lipid domains. J. Biol. Chem. 288, 32004–32019. doi: 10.1074/jbc.M113.475863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lai, M. C., Lombardo, M. V., and Baron-Cohen, S. (2014). Autism. Lancet 383, 896–910. doi: 10.1016/S0140-6736(13)61539-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Landis, S. C., Amara, S. G., Asadullah, K., Austin, C. P., Blumenstein, R., Bradley, E. W., et al. (2012). A call for transparent reporting to optimize the predictive value of preclinical research. Nature 490, 187–191. doi: 10.1038/nature11556
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
LaSalle, J. M., Goldstine, J., Balmer, D., and Greco, C. M. (2001). Quantitative localization of heterogeneous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum. Mol. Genet. 10, 1729–1740. doi: 10.1093/hmg/10.17.1729
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laurvick, C. L., de Klerk, N., Bower, C., Christodoulou, J., Ravine, D., Ellaway, C., et al. (2006). Rett syndrome in Australia: a review of the epidemiology. J. Pediatr. 148, 347–352. doi: 10.1016/j.jpeds.2005.10.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lauterborn, J. C., Truong, G. S., Baudry, M., Bi, X., Lynch, G., and Gall, C. M. (2003). Chronic elevation of brain-derived neurotrophic factor by ampakines. J. Pharmacol. Exp. Ther. 307, 297–305. doi: 10.1124/jpet.103.053694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H. Y., Ge, W. P., Huang, W., He, Y., Wang, G. X., Rowson-Baldwin, A., et al. (2011). Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 72, 630–642. doi: 10.1016/j.neuron.2011.09.033.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H. Y., and Jan, L. Y. (2012). Fragile X syndrome: mechanistic insights and therapeutic avenues regarding the role of potassium channels. Curr. Opin. Neurobiol. 22, 887–894. doi: 10.1016/j.conb.2012.03.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leigh, M. J., Nguyen, D. V., Mu, Y., Winarni, T. I., Schneider, A., Chechi, T., et al. (2013). A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J. Dev. Behav. Pediatr. 34, 147–155. doi: 10.1097/DBP.0b013e318287cd17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lekman, A., Witt-Engerstrom, I., Gottfries, J., Hagberg, B. A., Percy, A. K., and Svennerholm, L. (1989). Rett syndrome: biogenic amines and metabolites in postmortem brain. Pediatr. Neurol. 5, 357–362. doi: 10.1016/0887-8994(89)90049-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lesch, K. P., and Waider, J. (2012). Serotonin in the modulation of neural plasticity and networks: implications for neurodevelopmental disorders. Neuron 76, 175–191. doi: 10.1016/j.neuron.2012.09.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levenga, J., Hayashi, S., de Vrij, F. M., Koekkoek, S. K., van der Linde, H. C., Nieuwenhuizen, I., et al. (2011). AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol. Dis. 42, 311–317. doi: 10.1016/j.nbd.2011.01.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, W., and Pozzo-Miller, L. (2014). BDNF deregulation in Rett syndrome. Neuropharmacology 76(Pt. C), 737–746. doi: 10.1016/j.neuropharm.2013.03.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lim, C. S., Hoang, E. T., Viar, K. E., Stornetta, R. L., Scott, M. M., and Zhu, J. J. (2014). Pharmacological rescue of Ras signaling, GluA1-dependent synaptic plasticity and learning deficits in a fragile X model. Genes Dev. 28, 273–289. doi: 10.1101/gad.232470.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lipton, J., and Sahin, M. (2013). Fragile X syndrome therapeutics: translation, meet translational medicine. Neuron 77, 212–213. doi: 10.1016/j.neuron.2013.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maccarrone, M., Rossi, S., Bari, M., De Chiara, V., Rapino, C., Musella, A., et al. (2010). Abnormal mGlu 5 receptor/endocannabinoid coupling in mice lacking FMRP and BC1 RNA. Neuropsychopharmacology 35, 1500–1509. doi: 10.1038/npp.2010.19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maliszewska-Cyna, E., Bawa, D., and Eubanks, J. H. (2010). Diminished prevalence but preserved synaptic distribution of N-methyl-D-aspartate receptor subunits in the methyl CpG binding protein 2(MeCP2)-null mouse brain. Neuroscience 168, 624–632. doi: 10.1016/j.neuroscience.2010.03.065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malter, J. S., Ray, B. C., Westmark, P. R., and Westmark, C. J. (2010). Fragile X syndrome and Alzheimer’s disease: another story about APP and beta-amyloid. Curr. Alzheimer Res. 7, 200–206. doi: 10.2174/156720510791050957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marchetto, M. C., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y., et al. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. doi: 10.1016/j.cell.2010.10.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., et al. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893. doi: 10.1126/science.1090842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Massa, S. M., Yang, T., Xie, Y., Shi, J., Bilgen, M., Joyce, J. N., et al. (2010). Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Invest. 120, 1774–1785. doi: 10.1172/JCI41356
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matthys, A., Haegeman, G., Van Craenenbroeck, K., and Vanhoenacker, P. (2011). Role of the 5-HT7 receptor in the central nervous system: from current status to future perspectives. Mol. Neurobiol. 43, 228–253. doi: 10.1007/s12035-011-8175-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCauley, M. D., Wang, T., Mike, E., Herrera, J., Beavers, D. L., Huang, T. W., et al. (2011). Pathogenesis of lethal cardiac arrhythmias in Mecp2 mutant mice: implication for therapy in Rett syndrome. Sci. Transl. Med. 3:113ra125. doi: 10.1126/scitranslmed.3002982
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCoy, P. A., and McMahon, L. L. (2007). Muscarinic receptor dependent long-term depression in rat visual cortex is PKC independent but requires ERK1/2 activation and protein synthesis. J. Neurophysiol. 98, 1862–1870. doi: 10.1152/jn.00510.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McEwen, B. S., Chattarji, S., Diamond, D. M., Jay, T. M., Reagan, L. P., Svenningsson, P., et al. (2010). The neurobiological properties of tianeptine (Stablon): from monoamine hypothesis to glutamatergic modulation. Mol. Psychiatry 15, 237–249. doi: 10.1038/mp.2009.80
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Medrihan, L., Tantalaki, E., Aramuni, G., Sargsyan, V., Dudanova, I., Missler, M., et al. (2008). Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 99, 112–121. doi: 10.1152/jn.00826.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mefford, H. C., Batshaw, M. L., and Hoffman, E. P. (2012). Genomics, intellectual disability and autism. N. Engl. J. Med. 366, 733–743. doi: 10.1056/NEJMra1114194
Meyer-Lindenberg, A., Domes, G., Kirsch, P., and Heinrichs, M. (2011). Oxytocin and vasopressin in the human brain: social neuropeptides for translational medicine. Nat. Rev. Neurosci. 12, 524–538. doi: 10.1038/nrn3044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michalon, A., Bruns, A., Risterucci, C., Honer, M., Ballard, T. M., Ozmen, L., et al. (2014). Chronic metabotropic glutamate receptor 5 inhibition corrects local alterations of brain activity and improves cognitive performance in fragile X mice. Biol. Psychiatry 75, 189–197. doi: 10.1016/j.biopsych.2013.05.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michalon, A., Sidorov, M., Ballard, T. M., Ozmen, L., Spooren, W., Wettstein, J. G., et al. (2012). Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56. doi: 10.1016/j.neuron.2012.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Min, W. W., Yuskaitis, C. J., Yan, Q., Sikorski, C., Chen, S., Jope, R. S., et al. (2009). Elevated glycogen synthase kinase-3 activity in fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 56, 463–472. doi: 10.1016/j.neuropharm.2008.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mines, M. A., and Jope, R. S. (2011). Glycogen synthase kinase-3: a promising therapeutic target for fragile x syndrome. Front. Mol. Neurosci. 4:35. doi: 10.3389/fnmol.2011.00035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mines, M. A., Yuskaitis, C. J., King, M. K., Beurel, E., and Jope, R. S. (2010). GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One 5:e9706. doi: 10.1371/journal.pone.0009706
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Misonou, H. (2010). Homeostatic regulation of neuronal excitability by K(+) channels in normal and diseased brains. Neuroscientist 16, 51–64. doi: 10.1177/1073858409341085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moretti, P., and Zoghbi, H. Y. (2006). MeCP2 dysfunction in Rett syndrome and related disorders. Curr. Opin. Genet. Dev. 16, 276–281. doi: 10.1016/j.gde.2006.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mukherjee, S., and Manahan-Vaughan, D. (2013). Role of metabotropic glutamate receptors in persistent forms of hippocampal plasticity and learning. Neuropharmacology 66, 65–81. doi: 10.1016/j.neuropharm.2012.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murata, Y., and Constantine-Paton, M. (2013). Postsynaptic density scaffold SAP102 regulates cortical synapse development through EphB and PAK signaling pathway. J. Neurosci. 33, 5040–5052. doi: 10.1523/jneurosci.2896-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murdoch, J. D., and State, M. W. (2013). Recent developments in the genetics of autism spectrum disorders. Curr. Opin. Genet. Dev. 23, 310–315. doi: 10.1016/j.gde.2013.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakamoto, M., Nalavadi, V., Epstein, M. P., Narayanan, U., Bassell, G. J., and Warren, S. T. (2007). Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl. Acad. Sci. U S A 104, 15537–15542. doi: 10.1073/pnas.0707484104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nalivaeva, N. N., and Turner, A. J. (2013). The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett. 587, 2046–2054. doi: 10.1016/j.febslet.2013.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389. doi: 10.3410/f.718020287.793478774
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Narayanan, U., Nalavadi, V., Nakamoto, M., Thomas, G., Ceman, S., Bassell, G. J., et al. (2008). S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J. Biol. Chem. 283, 18478–18482. doi: 10.1074/jbc.c800055200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neul, J. L., Kaufmann, W. E., Glaze, D. G., Christodoulou, J., Clarke, A. J., Bahi-Buisson, N., et al. (2010). Rett syndrome: revised diagnostic criteria and nomenclature. Ann. Neurol. 68, 944–950. doi: 10.1002/ana.22124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neul, J. L., Lane, J. B., Lee, H. S., Geerts, S., Barrish, J. O., Annese, F., et al. (2014). Developmental delay in Rett syndrome: data from the natural history study. J. Neurodev. Disord. 6:20. doi: 10.1186/1866-1955-6-20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neumann, I. D. (2008). Brain oxytocin: a key regulator of emotional and social behaviours in both females and males. J. Neuroendocrinol. 20, 858–865. doi: 10.1111/j.1365-2826.2008.01726.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neumann, I. D., and Landgraf, R. (2012). Balance of brain oxytocin and vasopressin: implications for anxiety, depression and social behaviors. Trends Neurosci. 35, 649–659. doi: 10.1016/j.tins.2012.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niederhofer, H. (2007). Glutamate antagonists seem to be slightly effective in psychopharmacologic treatment of autism. J. Clin. Psychopharmacol. 27, 317–318. doi: 10.1097/01.jcp.0000270082.30500.69
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogier, M., Wang, H., Hong, E., Wang, Q., Greenberg, M. E., and Katz, D. M. (2007). Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J. Neurosci. 27, 10912–10917. doi: 10.1523/jneurosci.1869-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Olmos-Serrano, J. L., Corbin, J. G., and Burns, M. P. (2011). The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev. Neurosci. 33, 395–403. doi: 10.1159/000332884
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Olmos-Serrano, J. L., Paluszkiewicz, S. M., Martin, B. S., Kaufmann, W. E., Corbin, J. G., and Huntsman, M. M. (2010). Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J. Neurosci. 30, 9929–9938. doi: 10.1523/jneurosci.1714-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Neill, M. J., Bleakman, D., Zimmerman, D. M., and Nisenbaum, E. S. (2004). AMPA receptor potentiators for the treatment of CNS disorders. Curr. Drug Targets CNS Neurol. Disord. 3, 181–194. doi: 10.2174/1568007043337508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
O’Neill, M. J., and Witkin, J. M. (2007). AMPA receptor potentiators: application for depression and Parkinson’s disease. Curr. Drug Targets 8, 603–620. doi: 10.2174/138945007780618517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Osterweil, E. K., Chuang, S. C., Chubykin, A. A., Sidorov, M., Bianchi, R., Wong, R. K., et al. (2013). Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77, 243–250. doi: 10.1016/j.neuron.2012.01.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Osterweil, E. K., Krueger, D. D., Reinhold, K., and Bear, M. F. (2010). Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci. 30, 15616–15627. doi: 10.1523/jneurosci.3888-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oudin, M. J., Hobbs, C., and Doherty, P. (2011). DAGL-dependent endocannabinoid signalling: roles in axonal pathfinding, synaptic plasticity and adult neurogenesis. Eur. J. Neurosci. 34, 1634–1646. doi: 10.1111/j.1460-9568.2011.07831.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pacey, L. K., and Doering, L. C. (2007). Developmental expression of FMRP in the astrocyte lineage: implications for fragile X syndrome. Glia 55, 1601–1609. doi: 10.1002/glia.20573
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pacey, L. K., Heximer, S. P., and Hampson, D. R. (2009). Increased GABA(B) receptor-mediated signaling reduces the susceptibility of fragile X knockout mice to audiogenic seizures. Mol. Pharmacol. 76, 18–24. doi: 10.1124/mol.109.056127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pacey, L. K., Tharmalingam, S., and Hampson, D. R. (2011). Subchronic administration and combination metabotropic glutamate and GABAB receptor drug therapy in fragile X syndrome. J. Pharmacol. Exp. Ther. 338, 897–905. doi: 10.1124/jpet.111.183327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paluszkiewicz, S. M., Martin, B. S., and Huntsman, M. M. (2011). Fragile X syndrome: the GABAergic system and circuit dysfunction. Dev. Neurosci. 33, 349–364. doi: 10.1159/000329420
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paribello, C., Tao, L., Folino, A., Berry-Kravis, E., Tranfaglia, M., Ethell, I. M., et al. (2010). Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 10:91. doi: 10.1186/1471-2377-10-91
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paul, K., Venkitaramani, D. V., and Cox, C. L. (2013). Dampened dopamine-mediated neuromodulation in prefrontal cortex of fragile X mice. J. Physiol. 591, 1133–1143. doi: 10.1113/jphysiol.2012.241067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Penagarikano, O., Mulle, J. G., and Warren, S. T. (2007). The pathophysiology of fragile x syndrome. Annu. Rev. Genomics Hum. Genet. 8, 109–129. doi: 10.1146/annurev.genom.8.080706.092249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Percy, A. K., Lane, J. B., Childers, J., Skinner, S., Annese, F., Barrish, J., et al. (2007). Rett syndrome: North American database. J. Child Neurol. 22, 1338–1341. doi: 10.1177/0883073807308715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Percy, A. K., Neul, J. L., Glaze, D. G., Motil, K. J., Skinner, S. A., Khwaja, O., et al. (2010). Rett syndrome diagnostic criteria: lessons from the natural history study. Ann. Neurol. 68, 951–955. doi: 10.1002/ana.22154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Picciotto, M. R., Higley, M. J., and Mineur, Y. S. (2012). Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76, 116–129. doi: 10.1016/j.neuron.2012.08.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pilpel, Y., Kolleker, A., Berberich, S., Ginger, M., Frick, A., Mientjes, E., et al. (2009). Synaptic ionotropic glutamate receptors and plasticity are developmentally altered in the CA1 field of Fmr1 knockout mice. J. Physiol. 587, 787–804. doi: 10.1113/jphysiol.2008.160929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pitcher, M. R., Ward, C. S., Arvide, E. M., Chapleau, C. A., Pozzo-Miller, L., Hoeflich, A., et al. (2013). Insulinotropic treatments exacerbate metabolic syndrome in mice lacking MeCP2 function. Hum. Mol. Genet. 22, 2626–2633. doi: 10.1093/hmg/ddt111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pop, A. S., Levenga, J., de Esch, C. E., Buijsen, R. A., Nieuwenhuizen, I. M., Li, T., et al. (2014). Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/Mavoglurant. Psychopharmacology (Berl) 231, 1227–1235. doi: 10.1007/s00213-012-2947-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Popescu, A. C., Sidorova, E., Zhang, G., and Eubanks, J. H. (2010). Aminoglycoside-mediated partial suppression of MECP2 nonsense mutations responsible for Rett syndrome in vitro. J. Neurosci. Res. 88, 2316–2324. doi: 10.1002/jnr.22409
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prasad, H. C., Steiner, J. A., Sutcliffe, J. S., and Blakely, R. D. (2009). Enhanced activity of human serotonin transporter variants associated with autism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 163–173. doi: 10.1098/rstb.2008.0143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Preti, A., Melis, M., Siddi, S., Vellante, M., Doneddu, G., and Fadda, R. (2014). Oxytocin and autism: a systematic review of randomized controlled trials. J. Child Adolesc. Psychopharmacol. 24, 54–68. doi: 10.1089/cap.2013.0040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Randall, A. D., Witton, J., Booth, C., Hynes-Allen, A., and Brown, J. T. (2010). The functional neurophysiology of the amyloid precursor protein (APP) processing pathway. Neuropharmacology 59, 243–267. doi: 10.1016/j.neuropharm.2010.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raymond, C. R., Thompson, V. L., Tate, W. P., and Abraham, W. C. (2000). Metabotropic glutamate receptors trigger homosynaptic protein synthesis to prolong long-term potentiation. J. Neurosci. 20, 969–976.
Roberts, J. E., Symons, F. J., Johnson, A. M., Hatton, D. D., and Boccia, M. L. (2005). Blink rate in boys with fragile X syndrome: preliminary evidence for altered dopamine function. J. Intellect. Disabil. Res. 49, 647–656. doi: 10.1111/j.1365-2788.2005.00713.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rogers, T. D., Dickson, P. E., McKimm, E., Heck, D. H., Goldowitz, D., Blaha, C. D., et al. (2013). Reorganization of circuits underlying cerebellar modulation of prefrontal cortical dopamine in mouse models of autism spectrum disorder. Cerebellum 12, 547–556. doi: 10.1007/s12311-013-0462-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ronemus, M., Iossifov, I., Levy, D., and Wigler, M. (2014). The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet. 15, 133–141. doi: 10.1038/nrg3585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ronesi, J. A., and Huber, K. M. (2008a). Metabotropic glutamate receptors and fragile x mental retardation protein: partners in translational regulation at the synapse. Sci. Signal. 1:pe6. doi: 10.1126/stke.15pe6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ronesi, J. A., and Huber, K. M. (2008b). Homer interactions are necessary for metabotropic glutamate receptor-induced long-term depression and translational activation. J. Neurosci. 28, 543–547. doi: 10.1523/jneurosci.5019-07.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rotschafer, S. E., Trujillo, M. S., Dansie, L. E., Ethell, I. M., and Razak, K. A. (2012). Minocycline treatment reverses ultrasonic vocalization production deficit in a mouse model of Fragile X syndrome. Brain Res. 1439, 7–14. doi: 10.1016/j.brainres.2011.12.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roux, J. C., Dura, E., Moncla, A., Mancini, J., and Villard, L. (2007). Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur. J. Neurosci. 25, 1915–1922. doi: 10.1111/j.1460-9568.2007.05466.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roux, J. C., Dura, E., and Villard, L. (2008). Tyrosine hydroxylase deficit in the chemoafferent and the sympathoadrenergic pathways of the Mecp2 deficient mouse. Neurosci. Lett. 447, 82–86. doi: 10.1016/j.neulet.2008.09.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Samaco, R. C., and Neul, J. L. (2011). Complexities of Rett syndrome and MeCP2. J. Neurosci. 31, 7951–7959. doi: 10.1523/jneurosci.0169-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Santoro, M. R., Bray, S. M., and Warren, S. T. (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 7, 219–245. doi: 10.1146/annurev-pathol-011811-132457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schanen, C., Houwink, E. J., Dorrani, N., Lane, J., Everett, R., Feng, A., et al. (2004). Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. Am. J. Med. Genet. A 126A, 129–140. doi: 10.1002/ajmg.a.20571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scheele, D., Kendrick, K. M., Khouri, C., Kretzer, E., Schläpfer, T. E., Stoffel-Wagner, B., et al. (2014). An oxytocin-induced facilitation of neural and emotional responses to social touch correlates inversely with autism traits. Neuropsychopharmacology 39, 2078–2085. doi: 10.1038/npp.2014.78
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmid, D. A., Yang, T., Ogier, M., Adams, I., Mirakhur, Y., Wang, Q., et al. (2012). A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J. Neurosci. 32, 1803–1810. doi: 10.1523/JNEUROSCI.0865-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmit, T. L., Dowell, J. A., Maes, M. E., and Wilhelm, M. (2013). c-Jun N-terminal kinase regulates mGluR-dependent expression of post-synaptic FMRP target proteins. J. Neurochem. 127, 772–781. doi: 10.1111/jnc.12453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seamans, J. K., and Yang, C. R. (2004). The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog. Neurobiol. 74, 1–58. doi: 10.1016/j.pneurobio.2004.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Segal, R. A., and Greenberg, M. E. (1996). Intracellular signaling pathways activated by neurotrophic factors. Annu. Rev. Neurosci. 19, 463–489. doi: 10.1146/annurev.neuro.19.1.463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shahbazian, M. D., Antalffy, B., Armstrong, D. L., and Zoghbi, H. Y. (2002). Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 11, 115–124. doi: 10.1093/hmg/11.2.115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sharma, A., Hoeffer, C. A., Takayasu, Y., Miyawaki, T., McBride, S. M., Klann, E., et al. (2010). Dysregulation of mTOR signaling in fragile X syndrome. J. Neurosci. 30, 694–702. doi: 10.1523/jneurosci.3696-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sidhu, H., Dansie, L. E., Hickmott, P. W., Ethell, D. W., and Ethell, I. M. (2014). Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile x syndrome in a mouse model. J. Neurosci. 34, 9867–9879. doi: 10.1523/jneurosci.1162-14.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sidorov, M. S., Auerbach, B. D., and Bear, M. F. (2013). Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 6:15. doi: 10.1186/1756-6606-6-15
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sigel, E., and Steinmann, M. E. (2012). Structure, function and modulation of GABA(A) receptors. J. Biol. Chem. 287, 40224–40231. doi: 10.1074/jbc.R112.386664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Siller, S. S., and Broadie, K. (2012). Matrix metalloproteinases and minocycline: therapeutic avenues for fragile X syndrome. Neural Plast. 2012:124548. doi: 10.1155/2012/124548
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Straiker, A., Min, K. T., and Mackie, K. (2013). Fmr1 deletion enhances and ultimately desensitizes CB(1) signaling in autaptic hippocampal neurons. Neurobiol. Dis. 56, 1–5. doi: 10.1016/j.nbd.2013.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strumbos, J. G., Brown, M. R., Kronengold, J., Polley, D. B., and Kaczmarek, L. K. (2010). Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J. Neurosci. 30, 10263–10271. doi: 10.1523/jneurosci.1125-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sugden, P. H., Fuller, S. J., Weiss, S. C., and Clerk, A. (2008). Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br. J. Pharmacol. 153(Suppl 1), S137–S153. doi: 10.1038/sj.bjp.0707659
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Surmeier, D. J., Plotkin, J., and Shen, W. (2009). Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr. Opin. Neurobiol. 19, 621–628. doi: 10.1016/j.conb.2009.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suvrathan, A., Hoeffer, C. A., Wong, H., Klann, E., and Chattarji, S. (2010). Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. U S A 107, 11591–11596. doi: 10.1073/pnas.1002262107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tavoulari, S., Forrest, L. R., and Rudnick, G. (2009). Fluoxetine (Prozac) binding to serotonin transporter is modulated by chloride and conformational changes. J. Neurosci. 29, 9635–9643. doi: 10.1523/jneurosci.0440-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomas, A. M., Bui, N., Graham, D., Perkins, J. R., Yuva-Paylor, L. A., and Paylor, R. (2011). Genetic reduction of group 1 metabotropic glutamate receptors alters select behaviors in a mouse model for fragile X syndrome. Behav. Brain Res. 223, 310–321. doi: 10.1016/j.bbr.2011.04.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomas, A. M., Bui, N., Perkins, J. R., Yuva-Paylor, L. A., and Paylor, R. (2012). Group I metabotropic glutamate receptor antagonists alter select behaviors in a mouse model for fragile X syndrome. Psychopharmacology (Berl) 219, 47–58. doi: 10.1007/s00213-011-2375-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183. doi: 10.1038/nrn1346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trappe, R., Laccone, F., Cobilanschi, J., Meins, M., Huppke, P., Hanefeld, F., et al. (2001). MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am. J. Hum. Genet. 68, 1093–1101. doi: 10.1086/320109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., et al. (2009). Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U S A 106, 2029–2034. doi: 10.1073/pnas.0812394106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679. doi: 10.1126/science.1247190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Upreti, C., Zhang, X. L., Alford, S., and Stanton, P. K. (2013). Role of presynaptic metabotropic glutamate receptors in the induction of long-term synaptic plasticity of vesicular release. Neuropharmacology 66, 31–39. doi: 10.1016/j.neuropharm.2012.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uutela, M., Lindholm, J., Rantamäki, T., Umemori, J., Hunter, K., Võikar, V., et al. (2014). Distinctive behavioral and cellular responses to fluoxetine in the mouse model for fragile X syndrome. Front. Cell. Neurosci. 8:150. doi: 10.3389/fncel.2014.00150
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veenstra-VanderWeele, J., Muller, C. L., Iwamoto, H., Sauer, J. E., Owens, W. A., Shah, C. R., et al. (2012). Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc. Natl. Acad. Sci. U S A 109, 5469–5474. doi: 10.1073/pnas.1112345109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veeraragavan, S., Bui, N., Perkins, J. R., Yuva-Paylor, L. A., Carpenter, R. L., and Paylor, R. (2011b). Modulation of behavioral phenotypes by a muscarinic M1 antagonist in a mouse model of fragile X syndrome. Psychopharmacology (Berl) 217, 143–151. doi: 10.1007/s00213-011-2276-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veeraragavan, S., Bui, N., Perkins, J. R., Yuva-Paylor, L. A., and Paylor, R. (2011a). The modulation of fragile X behaviors by the muscarinic M4 antagonist, tropicamide. Behav. Neurosci. 125, 783–790. doi: 10.1037/a0025202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veeraragavan, S., Graham, D., Bui, N., Yuva-Paylor, L. A., Wess, J., and Paylor, R. (2012). Genetic reduction of muscarinic M4 receptor modulates analgesic response and acoustic startle response in a mouse model of fragile X syndrome (FXS). Behav. Brain Res. 228, 1–8. doi: 10.1016/j.bbr.2011.11.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vinueza Veloz, M. F., Buijsen, R. A., Willemsen, R., Cupido, A., Bosman, L. W., Koekkoek, S. K., et al. (2012). The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav. 11, 325–331. doi: 10.1111/j.1601-183x.2011.00763.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Volk, L. J., Pfeiffer, B. E., Gibson, J. R., and Huber, K. M. (2007). Multiple Gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile X syndrome mental retardation. J. Neurosci. 27, 11624–11634. doi: 10.1523/jneurosci.2266-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Volpicelli, L. A., and Levey, A. I. (2004). Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog. Brain Res. 145, 59–66. doi: 10.1016/s0079-6123(03)45003-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H. (2014). Lipid rafts: a signaling platform linking cholesterol metabolism to synaptic deficits in autism spectrum disorders. Front. Behav. Neurosci. 8:104. doi: 10.3389/fnbeh.2014.00104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, L. W., Berry-Kravis, E., and Hagerman, R. J. (2010b). Fragile X: leading the way for targeted treatments in autism. Neurotherapeutics 7, 264–274. doi: 10.1016/j.nurt.2010.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, T., Bray, S. M., and Warren, S. T. (2012a). New perspectives on the biology of fragile X syndrome. Curr. Opin. Genet. Dev. 22, 256–263. doi: 10.1016/j.gde.2012.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., and Doering, L. C. (2012). Induced pluripotent stem cells to model and treat neurogenetic disorders. Neural Plast. 2012:346053. doi: 10.1155/2012/346053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., and Doering, L. C. (2013). Reversing autism by targeting downstream mTOR signaling. Front. Cell. Neurosci. 7:28. doi: 10.3389/fncel.2013.00028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., Kim, S. S., and Zhuo, M. (2010a). Roles of fragile X mental retardation protein in dopaminergic stimulation-induced synapse-associated protein synthesis and subsequent alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-4-propionate (AMPA) receptor internalization. J. Biol. Chem. 285, 21888–21901. doi: 10.1074/jbc.M110.116293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., Ku, L., Osterhout, D. J., Li, W., Ahmadian, A., Liang, Z., et al. (2004). Developmentally-programmed FMRP expression in oligodendrocytes: a potential role of FMRP in regulating translation in oligodendroglia progenitors. Hum. Mol. Genet. 13, 79–89. doi: 10.1093/hmg/ddh009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, G. X., Smith, S. J., and Mourrain, P. (2014). Fmr1 KO and fenobam treatment differentially impact distinct synapse populations of mouse neocortex. Neuron 84, 1273–1286. doi: 10.1016/j.neuron.2014.11.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X., Snape, M., Klann, E., Stone, J. G., Singh, A., Petersen, R. B., et al. (2012b). Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile x-syndrome. J. Neurochem. 121, 672–679. doi: 10.1111/j.1471-4159.2012.07722.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., Wu, L. J., Kim, S. S., Lee, F. J., Gong, B., Toyoda, H., et al. (2008b). FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron 59, 634–647. doi: 10.1016/j.neuron.2008.06.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., Wu, L. J., Zhang, F., and Zhuo, M. (2008a). Roles of calcium-stimulated adenylyl cyclase and calmodulin-dependent protein kinase IV in the regulation of FMRP by group I metabotropic glutamate receptors. J. Neurosci. 28, 4385–4397. doi: 10.1523/JNEUROSCI.0646-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., and Zhuo, M. (2012). Group I metabotropic glutamate receptor-mediated gene transcription and implications for synaptic plasticity and diseases. Front. Pharmacol. 3:189. doi: 10.3389/fphar.2012.00189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watanabe, T., Abe, O., Kuwabara, H., Yahata, N., Takano, Y., Iwashiro, N., et al. (2014). Mitigation of sociocommunicational deficits of autism through oxytocin-induced recovery of medial prefrontal activity: a randomized trial. JAMA Psychiatry 71, 166–175. doi: 10.1001/jamapsychiatry.2013.3181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wei, H., Dobkin, C., Sheikh, A. M., Malik, M., Brown, W. T., and Li, X. (2012). The therapeutic effect of memantine through the stimulation of synapse formation and dendritic spine maturation in autism and fragile X syndrome. PLoS One 7:e36981. doi: 10.1371/journal.pone.0036981
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weiler, I. J., and Greenough, W. T. (1993). Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc. Natl. Acad. Sci. U S A 90, 7168–7171. doi: 10.1073/pnas.90.15.7168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weinshenker, D., and Warren, S. T. (2008). Neuroscience: fragile dopamine. Nature 455, 607–608. doi: 10.1038/455607a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weng, N., Weiler, I. J., Sumis, A., Berry-Kravis, E., and Greenough, W. T. (2008). Early-phase ERK activation as a biomarker for metabolic status in fragile X syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 1253–1257. doi: 10.1002/ajmg.b.30765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wernicke, J. F. (2004). Safety and side effect profile of fluoxetine. Expert Opin. Drug Saf. 3, 495–504. doi: 10.1517/14740338.3.5.495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westmark, C. J. (2013). What’s hAPPening at synapses? The role of amyloid beta-protein precursor and β-amyloid in neurological disorders. Mol. Psychiatry 18, 425–434. doi: 10.1038/mp.2012.122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westmark, C. J., Berry-Kravis, E. M., Ikonomidou, C., Yin, J. C., and Puglielli, L. (2013). Developing BACE-1 inhibitors for FXS. Front. Cell. Neurosci. 7:77. doi: 10.3389/fncel.2013.00077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westmark, C. J., and Malter, J. S. (2007). FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 5:e52. doi: 10.1371/journal.pbio.0050052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westmark, C. J., Westmark, P. R., and Malter, J. S. (2010). MPEP reduces seizure severity in Fmr-1 KO mice over expressing human Abeta. Int. J. Clin. Exp. Pathol. 3, 56–68.
Westmark, C. J., Westmark, P. R., O’Riordan, K. J., Ray, B. C., Hervey, C. M., Salamat, M. S., et al. (2011). Reversal of fragile X phenotypes by manipulation of AβPP/Aβ levels in Fmr1KO mice. PLoS One 6:e26549. doi: 10.1371/journal.pone.0026549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wiegert, J. S., and Bading, H. (2011). Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 49, 296–305. doi: 10.1016/j.ceca.2010.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wijetunge, L. S., Chattarji, S., Wyllie, D. J., and Kind, P. C. (2013). Fragile X syndrome: from targets to treatments. Neuropharmacology 68, 83–96. doi: 10.1016/j.neuropharm.2012.11.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong, D. T., Perry, K. W., and Bymaster, F. P. (2005). Case history: the discovery of fluoxetine hydrochloride (Prozac). Nat. Rev. Drug Discov. 4, 764–774. doi: 10.1038/nrd1821
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wright, J. W., and Harding, J. W. (2009). Contributions of matrix metalloproteinases to neural plasticity, habituation, associative learning and drug addiction. Neural Plast. 2009:579382. doi: 10.1155/2009/579382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, Q. J., Rammal, M., Tranfaglia, M., and Bauchwitz, R. P. (2005). Suppression of two major fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49, 1053–1066. doi: 10.1016/j.neuropharm.2005.06.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yun, S. H., and Trommer, B. L. (2011). Fragile X mice: reduced long-term potentiation and N-Methyl-D-Aspartate receptor-mediated neurotransmission in dentate gyrus. J. Neurosci. Res. 89, 176–182. doi: 10.1002/jnr.22546
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yuskaitis, C. J., Mines, M. A., King, M. K., Sweatt, J. D., Miller, C. A., and Jope, R. S. (2010). Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem. Pharmacol. 79, 632–646. doi: 10.1016/j.bcp.2009.09.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zanella, S., Mebarek, S., Lajard, A. M., Picard, N., Dutschmann, M., and Hilaire, G. (2008). Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir. Physiol. Neurobiol. 160, 116–121. doi: 10.1016/j.resp.2007.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, L., and Alger, B. E. (2010). Enhanced endocannabinoid signaling elevates neuronal excitability in fragile X syndrome. J. Neurosci. 30, 5724–5729. doi: 10.1523/JNEUROSCI.0795-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y., Bonnan, A., Bony, G., Ferezou, I., Pietropaolo, S., Ginger, M., et al. (2014b). Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1(-/y) mice. Nat. Neurosci. 17, 1701–1709. doi: 10.1038/nn.3864
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y., Brown, M. R., Hyland, C., Chen, Y., Kronengold, J., Fleming, M. R., et al. (2012). Regulation of neuronal excitability by interaction of fragile X mental retardation protein with slack potassium channels. J. Neurosci. 32, 15318–15327. doi: 10.1523/JNEUROSCI.2162-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, W., Peterson, M., Beyer, B., Frankel, W. N., and Zhang, Z. W. (2014a). Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. J. Neurosci. 34, 2754–2763. doi: 10.1523/JNEUROSCI.4900-12.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y., Venkitaramani, D. V., Gladding, C. M., Kurup, P., Molnar, E., Collingridge, G. L., et al. (2008). The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J. Neurosci. 28, 10561–10566. doi: 10.1523/JNEUROSCI.2666-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, H., Webb, D. J., Asmussen, H., Niu, S., and Horwitz, A. F. (2005). A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J. Neurosci. 25, 3379–3388. doi: 10.1523/jneurosci.3553-04.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Z. W., Zak, J. D., and Liu, H. (2010). MeCP2 is required for normal development of GABAergic circuits in the thalamus. J. Neurophysiol. 103, 2470–2481. doi: 10.1152/jn.00601.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, W. H., and Quirion, R. (2004). Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J. Neurochem. 89, 844–852. doi: 10.1111/j.1471-4159.2004.02350.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ziemssen, T., Kümpfel, T., Klinkert, W. E., Neuhaus, O., and Hohlfeld, R. (2002). Glatiramer acetate-specific T-helper 1- and 2-type cell lines produce BDNF: implications for multiple sclerosis therapy. Brain-derived neurotrophic factor. Brain 125, 2381–2391. doi: 10.1093/brain/awf252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zingman, L. V., Park, S., Olson, T. M., Alekseev, A. E., and Terzic, A. (2007). Aminoglycoside-induced translational read-through in disease: overcoming nonsense mutations by pharmacogenetic therapy. Clin. Pharmacol. Ther. 81, 99–103. doi: 10.1038/sj.clpt.6100012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zoghbi, H. Y., and Bear, M. F. (2012). Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 4, pii:a009886. doi: 10.1101/cshperspect.a009886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zupan, B., and Toth, M. (2008). Inactivation of the maternal fragile X gene results in sensitization of GABAB receptor function in the offspring. J. Pharmacol. Exp. Ther. 327, 820–826. doi: 10.1124/jpet.108.143990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: fragile X syndrome, Rett syndrome, autism spectrum disorders, pharmacotherapy, treatment, synaptic deficits, FMRP, MeCP2
Citation: Wang H, Pati S, Pozzo-Miller L and Doering LC (2015) Targeted pharmacological treatment of autism spectrum disorders: fragile X and Rett syndromes. Front. Cell. Neurosci. 9:55. doi: 10.3389/fncel.2015.00055
Received: 18 January 2015; Accepted: 05 February 2015;
Published online: 26 February 2015.
Edited by:
Thomas Knöpfel, Imperial College London, UKCopyright © 2015 Wang, Pati, Pozzo-Miller and Doering. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hansen Wang, Faculty of Medicine, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada e-mail: hansen.wang@utoronto.ca;
Lucas Pozzo-Miller, Department of Neurobiology, Civitan International Research Center, The University of Alabama at Birmingham, Birmingham, AL, 35294 USA e-mail: lucaspm@uab.edu;
Laurie C. Doering, Faculty of Health Sciences, Department of Pathology and Molecular Medicine, McMaster University, HSC 1R1, 1280 Main Street West, Hamilton, ON L8S 4K1, Canada e-mail: doering@mcmaster.ca