ClC-1 chloride channels: state-of-the-art research and future challenges
 Paola Imbrici1*
Paola Imbrici1*  Concetta Altamura1
Concetta Altamura1  Mauro Pessia2
Mauro Pessia2  Renato Mantegazza3
Renato Mantegazza3  Jean-François Desaphy1
Jean-François Desaphy1  Diana Conte Camerino1
Diana Conte Camerino1- 1Department of Pharmacy - Drug Sciences, University of Bari “Aldo Moro”, Bari, Italy
- 2Department of Experimental Medicine, School of Medicine, University of Perugia, Perugia, Italy
- 3Neuroimmunology and Neuromuscular Diseases Unit, IRCCS Fondazione Istituto Neurologico “Carlo Besta”, Milano, Italy
The voltage-dependent ClC-1 chloride channel belongs to the CLC channel/transporter family. It is a homodimer comprising two individual pores which can operate independently or simultaneously according to two gating modes, the fast and the slow gate of the channel. ClC-1 is preferentially expressed in the skeletal muscle fibers where the presence of an efficient Cl- homeostasis is crucial for the correct membrane repolarization and propagation of action potential. As a consequence, mutations in the CLCN1 gene cause dominant and recessive forms of myotonia congenita (MC), a rare skeletal muscle channelopathy caused by abnormal membrane excitation, and clinically characterized by muscle stiffness and various degrees of transitory weakness. Elucidation of the mechanistic link between the genetic defects and the disease pathogenesis is still incomplete and, at this time, there is no specific treatment for MC. Still controversial is the subcellular localization pattern of ClC-1 channels in skeletal muscle as well as its modulation by some intracellular factors. The expression of ClC-1 in other tissues such as in brain and heart and the possible assembly of ClC-1/ClC-2 heterodimers further expand the physiological properties of ClC-1 and its involvement in diseases. A recent de novo CLCN1 truncation mutation in a patient with generalized epilepsy indeed postulates an unexpected role of this channel in the control of neuronal network excitability. This review summarizes the most relevant and state-of-the-art research on ClC-1 chloride channels physiology and associated diseases.
Introduction
CLC proteins are ubiquitous chloride channels/transporters playing important roles in many physiological and physiopathological processes (Hille, 1992). Their story began with the electric fish, which expresses a high number of voltage-gated chloride channels in the cell of an electric organ originated from the skeletal muscle tissue (White and Miller, 1979). Due to the lack of specific inhibitors and the difficulties of biochemical purification, it was only in the 1990s that a molecular identification of voltage-gated chloride channels was made. In these years, the chloride channel from Torpedo marmorata, called ClC-0, was cloned with an elegant but extremely sophisticated expression cloning strategy (Jentsch et al., 1990). This allowed the description of a large family of CLC proteins homologous to ClC-0 and expressed in organisms within the kingdoms of life from eubacteria to animals (Jentsch et al., 1999; Maduke et al., 2000). These proteins exhibit a high degree of evolutionary conservation and function as homodimers (Middleton et al., 1996). In mammalian tissues, nine different CLC proteins have been identified, divided into plasma membrane Cl- channels and vesicular Cl-/H+-exchangers (Jentsch, 2015).
The ClC-1 channel was the first member of the CLC family cloned in mammals (Steinmeyer et al., 1991a). In humans, the CLCN1 gene is located on chromosome 7q35 and codes for a protein of ∼990 amino acids in length. This channel is mainly expressed in the skeletal muscle, where it supports the large chloride conductance of sarcolemma. Its physiological role was discovered by analyzing a mouse model of myotonia congenita (MC), a genetic disease of the skeletal muscle characterized by a reduced sarcolemma chloride conductance. A plethora of mutations in the CLCN1 gene are known to produce dominant and recessive myotonia in humans and other animals. At lower levels it has been detected in kidney, heart, smooth muscle, and, more recently, in the central nervous system (Steinmeyer et al., 1991a; Chen et al., 2013). This review describes our current knowledge regarding ClC-1 channels, as a example for using human genetic diseases and mouse models to facilitate the elucidation of the cellular roles of ion channels, and the research challenges these proteins continue to offer twenty-five years after the cloning of the first CLC.
Molecular Structure and Function of ClC-1
The ClC-1 voltage-gated chloride channel belongs to the CLC family, which comprises nine members in humans, named ClC-1 through ClC-7, plus ClC-Ka and ClC-Kb. They were initially assumed to behave as chloride ion channels, but successive experiments revealed that five out of the nine hClCs (ClC-3 through ClC-7) rather function as anion-proton exchangers (Picollo and Pusch, 2005; Scheel et al., 2005; Neagoe et al., 2010; Leisle et al., 2011; Guzman et al., 2013). Experimental data also support a close structural similarity among the members of the CLC family. Actually, most of the current information about the 3D structure of ClC-1 channels have been derived from the analysis of CLC anion-proton exchangers crystallized from several prokaryotic (EcCLC) and eukaryotic species (CmCLC; Figure 1A; Dutzler et al., 2002; Accardi et al., 2006; Lobet and Dutzler, 2006; Feng et al., 2010; Robertson et al., 2010; Jayaram et al., 2011; Lim et al., 2012). The ClC-1 channel is a dimer of two homologous subunits, each forming a chloride ion conducting pore independent from the other, allowing ClC-1 to function as a “double-barrel” channel (Saviane et al., 1999). Each subunit consists of 18 domains (helices A-R) and two tandem cystathionine-β-synthase (CBS) domains located in the intracellular C-terminal (Meyer and Dutzler, 2006; Markovic and Dutzler, 2007). According to the 3D structures, membrane helices D, F, N, and R contribute to the ClC ion transport pathway, helices H, I, P, and Q form part of the interface between the two monomers, and helix R connects the transmembrane segments of the channel to the C-terminal domain (Dutzler et al., 2002, 2003; Feng et al., 2010). Each pore contains three separate chloride ion binding sites, named Sint, Scen, and Sext according to their position relative to the extra- or intracellular side of the plasma membrane. The Scen position forms a significant part of the selectivity filter where conserved and less well conserved aminoacids coordinate the chloride ion within the conduction pathway (Dutzler et al., 2002, 2003; Lobet and Dutzler, 2006).
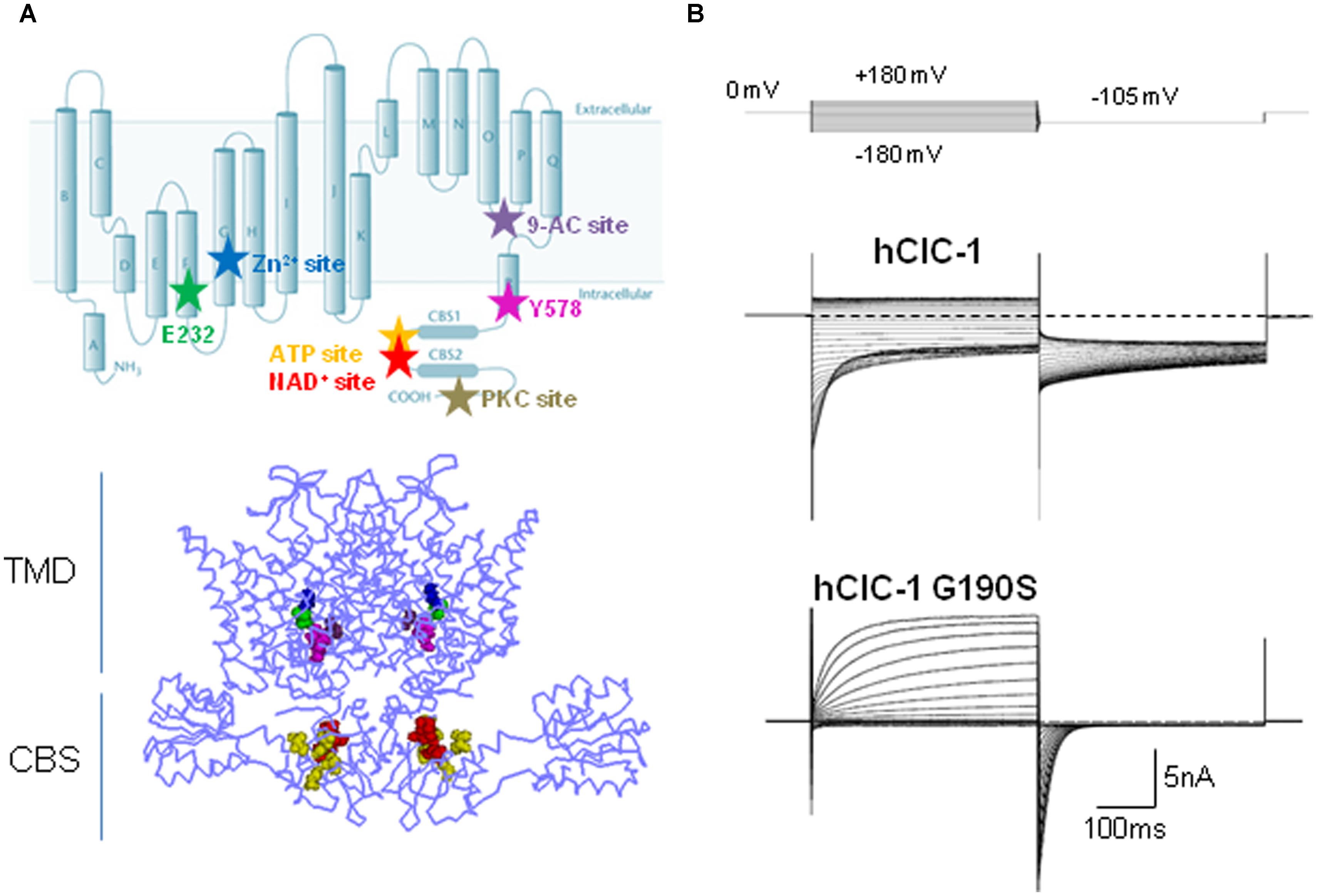
FIGURE 1. Structure, function, and modulation of hClC-1 channels. (A) Upper panel, diagram showing residues relevant for ClC-1 channel gating, E232 (green) and Y578 (magenta), and putative binding sites for Zn2+ (C277, blue), 9-AC (S537, violet), ATP (V613, V634, V860, E865, yellow), NAD+ (T636, H847, L848, red), PKC (Thr891, Ser892, Thr893, gray). Lower panel, three dimensional front view of hClC-1 channel modeled upon the structure of CmClC, including relevant residues for channel function and regulation. (B) Representative chloride currents recorded from tsA cells transfected with cDNAs coding for hClC-1 WT (up) and for a MC mutant hClC-1 G190S (down), in the whole-cell configuration of patch clamp. Note that G190S shows a strong outward rectification differently from WT. Cells were held at 0 mV and 400 ms voltage pulses were applied from -180 to +180 mV in 10-mV intervals every 3 s, in high chloride intracellular solution.
Early single channel recordings of ClC-1 channels expressed in heterologous systems, demonstrate two conductance states and confirmed the existence of two ion conduction pathways for homodimeric ClC channel, as shown for ClC-0 (Miller, 1982; Miller and White, 1984; Pusch et al., 1994; Saviane et al., 1999). Whole cell recordings of hClC-1 channels in high chloride intracellular solution show chloride currents that rapidly activate and deactivate at negative potentials and that saturate at positive potentials (Figure 1B).
Channel opening in ClC-1 is regulated by two distinct gating patterns: fast (or single-pore) and slow (or common-pore) gating (Accardi and Pusch, 2000; Dutzler, 2006), fast gating being about 10-fold faster than common gating (Miller, 1982; Saviane et al., 1999; Stölting et al., 2014b). The slow gate controls the opening and closing of the two subunits simultaneously, whereas the fast gate regulates the function of independent pores (Saviane et al., 1999). The carboxyl side chain of glutamate residue E232 of helix F is the main responsible for the fast gating: it projects into the extracellular-facing portion of the ion conduction pathway and functions as the voltage-, Cl-- and H+-activated fast gate, disclosing Sext when protonated (Zifarelli and Pusch, 2007). This residue is part of the Gly-Lys/Arg-Glu-Gly-Pro sequence motif that is conserved in all CLC channels apart from the kidney channels ClC-Ka and CIC-Kb. The molecular identity of the slow gate has still to be elucidated, and how the slow gate closes simultaneously both pores is still unknown. Far from simply involving helices and residues at the dimer interface, the emerging view on the mechanism of cooperative gating predicts the occurrence of concerted conformational changes involving the residue E232 in the channel pore, the intra-membrane interface between two monomers, and the C-terminal CBS domains via helix R (Duffield et al., 2003; Estévez et al., 2004; Fialho et al., 2007; Cederholm et al., 2010; Feng et al., 2010; Ma et al., 2011; Bennetts and Parker, 2013). Recently, it has been proposed that the slow channel closure depends on the H-bond formation between the E232 of pore helix F and the Y578 of the linker helix R (Bennetts and Parker, 2013). This H-bond interaction would enable the occlusion of the central anion binding site of the channel, thus hindering Cl- flow.
The intracellular C-terminal of ClC-1 is a poorly characterized region. This portion, comprising two tandem CBS domains, is common to the eukaryotic CLC members, and appears to be relevant for ClC-1 expression, slow gating and intracellular modulation (Figure 1A; Meyer and Dutzler, 2006; Markovic and Dutzler, 2007; Tseng et al., 2007; Zhang et al., 2008; Hsiao et al., 2010; Bennetts et al., 2012; Bennetts and Parker, 2013). The crystal structure of the eukaryotic CLC protein demonstrates that CBS2 lies functionally close to the intramembrane domains while CBS1 faces into the cytoplasm (Feng et al., 2010). CBS2 interacts extensively with the R-helix linker and helix D, and with an intracellular loop linking helices H and I. By virtue of its direct connection to the C-terminus, the R helix could provide a pathway by which intracellular domain conformational changes regulate CLC activity during cell signaling events or modulate slow gating (Dutzler et al., 2002; Bykova et al., 2006; Garcia-Olivares et al., 2008; Feng et al., 2010; Ma et al., 2011; Stölting et al., 2014b). The presumed regulatory role of the CBS linker, with its highly variable and largely unstructured sequence, has not been assessed jet.
To date, no report exists showing that ClC-1 can form heteromers with members of the same family in vivo. However, as ClC-2 is ubiquitously expressed, the spontaneous formation of heterodimeric ClC-1-ClC-2 channels would be not surprising in native human tissues, where both channels co-exist under physiological conditions. ClC-1 and ClC-2 chloride channels differ profoundly in the voltage dependence of fast and slow gating. Whereas fast and slow opening of ClC-1 is stimulated by membrane depolarization (Stölting et al., 2014a), fast and cooperative gates of ClC-2 are closed at depolarized potentials and open upon membrane hyperpolarization (Stölting et al., 2014a). Therefore, an attempt to clarify the mechanism of cooperative interaction between subunits has been the functional characterization of “artificial” heterodimeric ClC-1-ClC-2 channels consisting of two functionally distinct ClC pores (Lorenz et al., 1996; Stölting et al., 2014a). The observation that ClC-1-ClC-2 heterodimeric channels lack cooperative gating (Stölting et al., 2014a) supports the idea that slow gating of CLC channels ultimately arises from conformational changes within an individual pore (Cederholm et al., 2010; Bennetts and Parker, 2013; Stölting et al., 2014b). Therefore, fast and slow gating are now supposed to be closely coupled and not independent processes, as previously thought.
Modulatory Pathways
The structural bases of ClC-1 gating have been extensively studied as well as the contribution of ClC-1 channels to skeletal muscle physiology. How ClC-1 proteins are modulated by cell signaling pathways is, however, less well understood. There are compelling evidence that ClC-1 channel is finely regulated by the metabolic state of skeletal muscle fibers under physiological conditions, and its physiological contribution to skeletal muscle excitability must be read in the context of intracellular adenosine triphosphate (ATP) content, changes of the cytosolic redox state, alteration of the β-nicotinamide adenine dinucleotide (NAD+/NADH) ratio, membrane depolarization, acidification in exercised muscle, and protein kinases modulation.
The direct effect of cytoplasmic ATP on ClC-1 channels has been studied by several laboratories with apparently controversial results (Bennetts et al., 2007; Tseng et al., 2007; Zifarelli and Pusch, 2008). Indeed, Tseng et al. (2007) and Bennetts et al. (2007) provided evidence that ATP directly inhibits recombinant ClC-1 channels by shifting the voltage dependence of slow gating to positive potentials, an effect that is enhanced by low intracellular pH (pHi). This experimental finding provided a reasonable explanation for previously published effects of pHi on chloride currents in different biological systems (Saviane et al., 1999; Pedersen et al., 2004, 2005). Indeed, reducing pHi was shown to slow deactivation and increase the open probability of heterologously expressed ClC-1 channels (Rychkov et al., 1996; Saviane et al., 1999; Accardi and Pusch, 2000). Conversely, the acidification of skeletal muscle cells was reported to decrease sarcolemmal chloride conductance, thus maintaining the essential excitability in the partially depolarized sarcolemma and limiting muscle fatigue (Pedersen et al., 2004, 2005). ATP binding to the intracellular side of ClC-1 C-terminus could be the prerequisite required to observe a reduction of the ClC-1 conductance at low pHi in intact muscle fibers, thus reconciling these apparently divergent results (Bennetts et al., 2007; Tseng et al., 2007). Despite these exhaustive experiments, Zifarelli and Push questioned the direct pHi-dependent ATP modulation of ClC-1, as they failed to detect any ATP block in inside-out patches from Xenopus oocytes expressing ClC-1 channels (Zifarelli and Pusch, 2008). The hidden culprit underlying this further controversy seems to rely on the oxidation of ClC-1. Indeed, it seems that the ATP-mediated inhibition of ClC-1 channels is dependent on the redox-state of the cell, disappearing in the oxidizing condition of excised inside-out patches whereas being evident when adding reducing agents to the patch (Zhang et al., 2008). Oxidation of ClC-1 might thus be one of the mechanisms that control the muscle response to fatigue, by regulating the ATP inhibition of ClC-1 (Zhang et al., 2008). As ATP, also NAD+ has been shown to affect the slow channel gating in a pHi-dependent mode by binding to the CBS domains of ClC-1 (Bennetts et al., 2012). Through mutagenesis experiments, relevant residues within both CBS domains forming a putative binding site for both ATP and NAD+ have been highlighted (Figure 1A; Bennetts et al., 2007; Tseng et al., 2007). Zn2+ ions are also known to block ClC-1 channels by facilitating closing of the slow gate of ClC-1 (Duffield et al., 2005). This effect is eliminated by the mutation C277S which greatly increases the minimal open probability of the slow gate in ClC-1 channels, and is facilitated by the mutant V321A, which reduces the open probability of slow gating (Accardi et al., 2001; Duffield et al., 2005).
Several pharmacological and molecular studies indicate that ClC-1 is regulated by protein phosphorylation events (Brinkmeier and Jockusch, 1987; Bryant and Conte-Camerino, 1991; Tricarico et al., 1991; Rosenbohm et al., 1999; Papponen et al., 2005; Camerino et al., 2014). The external application of 4β-phorbol esters on HEK 293 cells transfected with hClC-1 channels reduced the instantaneous whole-cell current amplitude (Rosenbohm et al., 1999). This effect was abolished when the cells were intracellularly perfused with a specific protein kinase C (PKC) inhibitor, chelerythrine, suggesting that the effect of 4β-phorbol esters was mediated by PKC. The C-terminal region seems at least in part responsible for the effect of PKC activation on ClC-1 channels: indeed, several serine residues have been identified in the C-terminal domain which mediate PKC-dependent inhibition of ClC-1 chloride currents (Figure 1A; Hsiao et al., 2010). Consistently, the chloride conductance of sarcolemma at rest (gCl), which is mainly supported by ClC-1, was shown to be reduced by activation of PKC or increased by inhibitors of PKC. Such an effect may have important implications for skeletal muscle function (Bryant and Conte-Camerino, 1991; De Luca et al., 1998; Pierno et al., 2007; Pedersen et al., 2009b; Camerino et al., 2014). Phosphorylation–dephosphorylation signaling was shown to be relevant also for the correct targeting of ClC-1 to sarcolemma in skeletal muscle fibers (Papponen et al., 2005). Furthermore, electrophysiological studies showed an increase of gCl in the PKCθ-null mice with respect to wild-type, and a consequent reduction of muscle excitability (Camerino et al., 2014). Regulation of ClC-1 channels through PKC-dependent phosphorylation appeared to be involved in the muscular effects of a number of growth factors, hormones, and drugs, including the growth hormone, insulin-like growth factor-1 (IGF-1), ghrelin, angiotensin II, growth hormone secretagogues, and statins (De Luca et al., 1994b, 1998; Pierno et al., 2003; Cozzoli et al., 2014).
The hypothesis that accessory subunits may exist and bind to the C-terminal region, as shown for other channels, cannot be excluded as well as the possible interaction of the carboxyl tail of ClC-1 with cytoskeletal elements in muscle cells. Such interactions may be necessary for the integrity of the cytoskeleton as well as for the homeostatic control of sarcolemmal ClC-1 content or function.
Physiological Roles of ClC-1
Muscle Excitability
The exact localization of ClC-1 channels in the skeletal muscle membrane is still debated (Lueck et al., 2010; Lamb et al., 2011). Studies performed on skinned muscle fibers provided evidence that the majority of the chloride conductance is located in the T-system (Dutka et al., 2008). This result is not surprising if one considers that a large Cl- conductance would be expected to dampen membrane depolarization due to K+ accumulation in the narrow T-tubular space during repetitive muscle stimulation. However another study, by recording chloride currents in the same dissociated mouse flexor digitorum brevis (FDB) muscle fibers before and after disconnection of the T-tubules by osmotic shock, argued for an exclusive expression in the surface membrane (Lueck et al., 2010). A particularly strong point of this latter study is that the authors did not see any significant reduction of ClC-1 currents after detubulation. In this case, the distantly localized sarcolemmal gCl would exert a “remote control” on the T-system guaranteeing an efficient control of the tubular membrane potential (Lueck et al., 2010; Zifarelli and Pusch, 2010). Several arguments supported this view. First, an exclusive sarcolemmal localization of ClC-1 would maintain a very negative chloride equilibrium potential allowing a higher buffering capacity. Second, in the absence of a large gCl in the T-tubules, the resting conductance of the T-tubular membrane would be small, allowing faster and less energy consuming action potential propagation. The more recent study performed using a potentiometric dye to measure the T-tubular potential in a region of a dissociated mouse FDB muscle fiber under two-electrode voltage-clamp, presents evidence for a homogeneous distribution across T-tubules and the outer sarcolemmal membrane (DiFranco et al., 2011). Regardless of their exact localization, ClC-1 channels constitute the main support of the shunting chloride conductance in the skeletal muscle, allowing the fibers to electrically tolerate T-tubular membrane invaginations and the extracellular K+ accumulation subsequent to membrane depolarization.
The study of myotonia pathomechanisms in muscle fibers from genetically myotonic goats has paved the way for the understanding of the physiological role of ClC-1 channels (Bryant, 1969; Bryant and Morales-Aguilera, 1971; Adrian and Bryant, 1974). Differently from other excitable tissues, the sarcolemma of adult skeletal muscle fibers at rest presents a characteristic high chloride conductance (gCl) that is more than fourfold the potassium conductance (Hodgkin and Horowicz, 1959; Bretag, 1987). Such a large inhibitory Cl- conductance provides the appropriate balance to the excitatory Na+ currents to guarantee a constant voltage in any particular region of the fiber. When an action potential is triggered, it is propagated along the surface sarcolemma and deep into the interior of fibers via the T-tubule system. Membrane repolarization initiates after sodium channel fast inactivation and is completed by potassium efflux through delayed rectifier potassium channels. In normal skeletal muscle, potassium accumulation in the T-system causes a small, transient after-depolarization at the end of muscle contraction, but the high resting chloride conductance prevents propagation of this depolarization along the sarcolemma (Dutka et al., 2008). Conversely, in myotonic muscle fibers, where the gCl is dramatically reduced by more than 50%, this potassium accumulation can trigger autonomous muscle fiber action potentials, that result in a prolonged electrical activity even after the end of neuronal input (the so-called myotonic after-discharges). This represents the electrophysiological basis for muscle stiffness (Figure 2; Adrian and Bryant, 1974; Steinmeyer et al., 1991b).
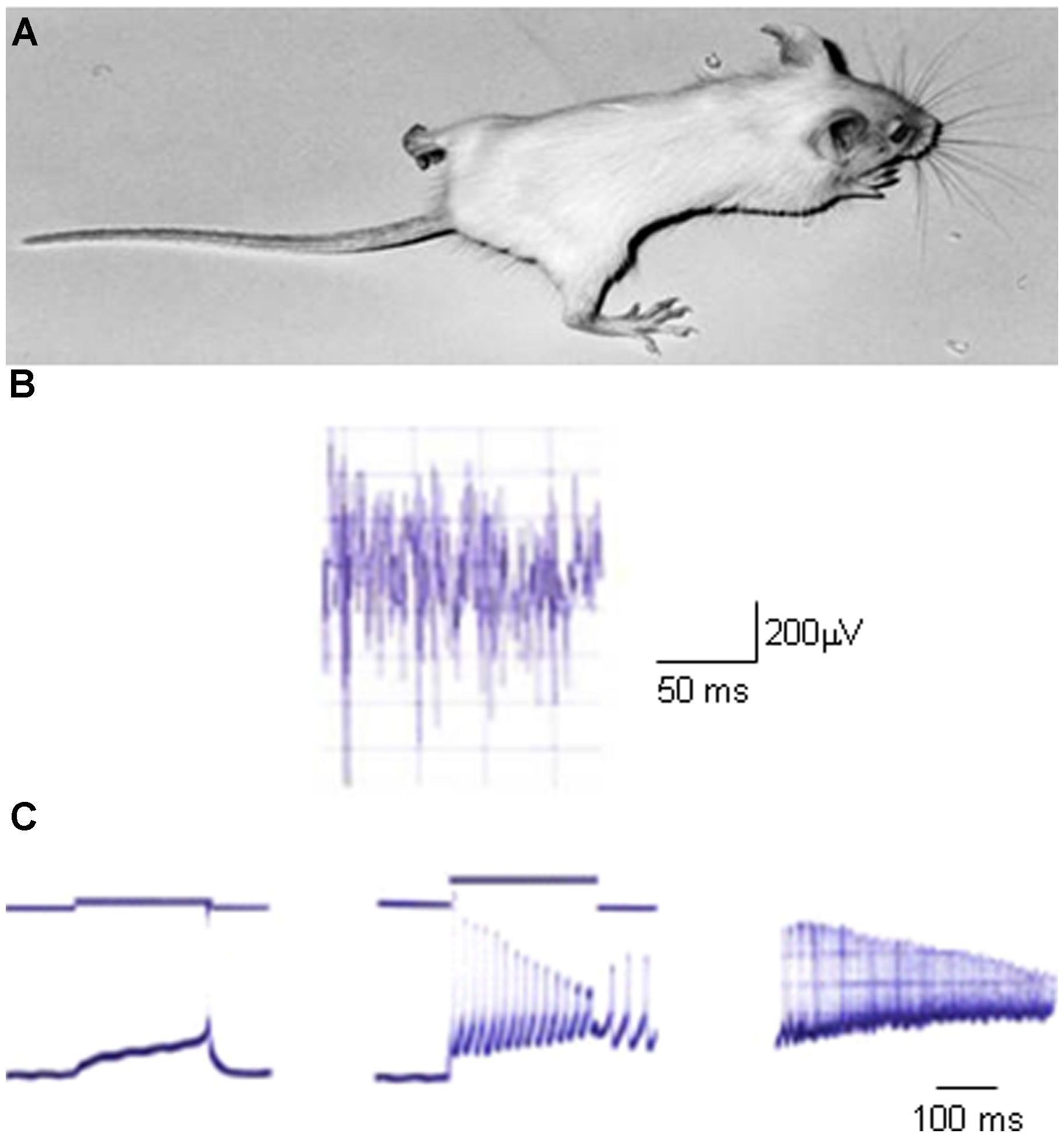
FIGURE 2. An animal model of myotonia congenita. (A) Picture of a homozygous adr/adr mouse carrying a loss-of-function mutation in the clcn1 gene, and showing stiffness in the hindlimb. (B) Typical electromyographic activity recorded in vivo with needle electrode in the hindlimb musculature of adr/adr mouse. (C) Typical electrophysiological recordings of excitability characteristics of adr/adr mouse intercostal muscle fibers with two-microelectrodes current-clamp technique. Left trace shows a single action potential elicited by a threshold current. Note the very long latency between the beginning of current stimulus and action potential onset, due to the reduced sarcolemmal chloride conductance. The middle trace shows the typical myotonic after-discharges, when the fiber fires action potentials after the end of current stimulus. The right trace shows typical myotonic spontaneous activity in absence of stimulus.
Recently, a series of elegant studies suggested that during a train of action potentials, the gCl is decreased at the onset of stimulation mainly due to PKC activation, allowing the working muscle to maintain neuromuscular transmission and T-system excitability (Pedersen et al., 2009a,b; Fraser et al., 2011). Nevertheless, after a determined number of action potentials during sustained activity, the gCl is largely increased together with the potassium conductance supported by ATP-sensitive K+ channels, most probably in order to terminate muscle activity and prevent metabolic stress that otherwise would damage the muscle fiber (Pedersen et al., 2009a). Interestingly, the rising phase of sarcolemma conductance during the action potentials train was not observed in the slow-twitch soleus muscle fibers, which is consistent with the elevated metabolic capacity of slow-twitch fibers that are rather involved in prolonged contractile activity (Pedersen et al., 2009b). The molecular mechanism responsible for the increased gCl is not well defined. It is likely that the consumption of ATP during prolonged activity may contribute to such an effect, as suggested also by the acceleration of this effect in glucose-free conditions (Pedersen et al., 2009b). The reduction of ATP might increase the gCl by completely removing the ATP- and/or phosphorylation-dependent inhibition of ClC-1 channels. Altogether these data suggest a pivotal role for ClC-1 channels for the modulation of muscular fatigue. Accordingly, the depression of muscle excitability provoked by intense exercise can be antagonized by inhibition of ClC-1 channels through intracellular acidification, addition of lactate, direct pharmacological block by 9-anthracen carboxylic acid (9-AC), or by removal of external Cl- (Nielsen et al., 2001; Pedersen et al., 2004, 2005; de Paoli et al., 2010). The more recent study confirms a biphasic relationship between the gCl and contractile endurance in isolated rat muscles (de Paoli et al., 2013). On one side, the reduction of gCl increases muscle excitability but, on the other side, reduces the capacity to maintain K+ homeostasis and membrane potential during contractions.
Muscle Plasticity
The ClC-1 channels contribute to determine skeletal muscle phenotype and to muscle plasticity. At birth, the sarcolemma gCl is very low, while it increases during the first weeks of postnatal development under the control of nerve input (Conte Camerino et al., 1989). The change in gCl is linearly correlated with changes in ClC-1 channel expression (Steinmeyer et al., 1991a). In adults, the resting gCl is about 10-fold greater than the gK in the fast-twitch muscles, while the slow-twitch muscle phenotype is characterized by a ∼50% lower gCl (Bretag, 1987). This difference is due to a reduced ClC-1 channel expression and an increased basal PKC-dependent phosphorylation of the channel in the slow-twitch muscle (Pierno et al., 2007). As anticipated above, such a phenotypic difference is of critical relevance for the specific modulation of fiber excitability and the consequent contractile function. Several evidences also indicate that the gCl contributes to the determination of muscle phenotype. Indeed, the pharmacological block of ClC-1 channels during the postnatal development forces the fast-twitch muscle to acquire a slow phenotype (De Luca et al., 1990a). Also in adult animals, the block of ClC-1 channels either through natural mutations or pharmacological block favors the transition of fast-twitch muscles toward a less fast phenotype (Salviati et al., 1986; Goblet and Whalen, 1995). Conversely, during the unloading condition, such as that encountered in microgravity condition or in the hindlimb-unloaded rodent model, the slow-to-fast phenotype shift of the postural slow-twitch soleus muscle is preceded by an increase of the gCl (Pierno et al., 2002). Such an increase stems from a reduced activity of the PKC likely due to changes in nerve input (Pierno et al., 2007). This indicates that the increase of gCl constitutes an early event during the adaptation of postural muscle to unloading, which may be necessary to complete the phenotype transition.
Aging
In the advanced age (>24 months for rats), the gCl can be dramatically reduced in the fast-twitch muscles (De Luca et al., 1990b, 1994a; De Luca and Conte Camerino, 1992). This reduction is attributable to a decrease of ClC-1 channel expression and an increased PKC-dependent phosphorylation of expressed ClC-1 channels (De Luca et al., 1997a; Pierno et al., 1999). The later effect may be related to the aging-related increase of calcium concentration in the muscle fiber (Fraysse et al., 2006). Chronic treatment of aged rats with the growth hormone was able to restore the gCl in fast-twitch muscles, likely through the induction of IGF-1 (De Luca et al., 1994b). In turn, IGF-1 was shown to increase the gCl in aged muscles, at least partially through the activation of an okadaic acid-dependent phosphatase able to de-phosphorylate the channel (De Luca et al., 1997a). All the effects of aging and GH were remarkably fiber type dependent, being less pronounced in the slow-twitch soleus muscle (Fraysse et al., 2006). Interestingly, chronic treatments of aged rats with the amino acid taurine or an olive oil-derived mixture, both with known antioxidant activity, were able to partially counteract the age-related decline of gCl, suggesting that the oxidative challenge during the aging process may be involved in the reduction of the gCl (Pierno et al., 1998, 2014). Based on the role of gCl in the adult fast-twitch muscle, a reduction of gCl in the aged muscle would be expected to increase excitability and force endurance. Nevertheless, the alteration of the entire electrical machinery of sarcolemma, including sodium, potassium, and calcium channels, actually impairs excitability and E-C coupling in the aged muscle (De Luca et al., 1990b, 1992; Tricarico and Camerino, 1994; Delbono et al., 1995; Renganathan et al., 1997; Desaphy et al., 1998).
Genetic Diseases Associated to ClC-1
Myotonia Congenita
Mutations in the CLCN1 gene, leading to partial or complete loss of ClC-1 channel function, are responsible for the pathogenesis of MC (Koch et al., 1992; George et al., 1993; Burge and Hanna, 2012). This is the most common skeletal muscle hereditary channelopathy in humans, characterized by muscle stiffness after a voluntary movement, that is worse after rest, and improves with repeated activity according to the so-called warm-up phenomenon. Inheritance can be dominant (Thomsen’s disease) or recessive (Becker’s disease), with a more severe phenotype in the latter form (Colding-Jørgensen, 2005; Fialho et al., 2007; Raja Rayan and Hanna, 2010). Patients with recessive MC may also have transient weakness at the onset of voluntary contraction, that may lead to falls. Different degrees of myotonic symptoms can be reported among family members with identical ClC-1 mutations and among different families (Colding-Jørgensen, 2005). Chloride channel myotonia also occurs in animals, including goat, mouse, dog, cat, poney, and water buffalo (Figure 2; Steinmeyer et al., 1991b; Rhodes et al., 1999; Wijnberg et al., 2012; Borges et al., 2013; Gandolfi et al., 2014). The study of myotonia in goats and mice proved to be decisive to the understanding of the pathomechanism, and eventually allowed the cloning of ClC-1 channel. The electrophysiological correlate of clinical myotonia is a burst of autonomous muscle action potentials that lasts a few seconds after the cessation of motor neuron activity and delays relaxation. Given the role played by ClC-1 channels in muscle fibers, the reduced chloride conductance resulting from ClC-1 mutations will predispose the sarcolemma to spontaneous action potential runs or abnormal after-discharges that hamper muscle relaxation after contraction, causing myotonia (Lossin and George, 2008).
So far more than 130 mutations in CLCN1 have been identified (Lossin and George, 2008; Matthews et al., 2010) over the entire length of the channel, and the analysis of a number of ClC-1 naturally occurring mutations in heterologous expression systems has contributed to the understanding of the molecular structure and function of the channel and of the disease pathogenesis. The CLCN1 mutations include small deletions, insertions, frame-shifts, stop codons, missense and splice-site mutations (Esteban et al., 1998; Sangiuolo et al., 1998; Brugnoni et al., 1999, 2013; de Diego et al., 1999; Pusch, 2002; Wu et al., 2002; Fialho et al., 2007; Lossin and George, 2008; Modoni et al., 2011; Mazón et al., 2012). Recently, it was demonstrated that also CLCN1 exon deletions or duplications in patients with a single identified recessive mutation, can cause recessive MC (Raja Rayan et al., 2012). The majority of MC mutations are predicted to reduce channel expression by defective trafficking or to cause various alterations of channel function including shifts of voltage dependence, reduced single channel conductance, altered ion selectivity (Figure 1B, Table 1; Tsujino et al., 2011; Ulzi et al., 2012; Weinberger et al., 2012; Desaphy et al., 2013). A novel mechanism has been proposed for the G200R and Y261C mutations, which abolished the potentiation of NAD+-induced ClC-1 channel inhibition by low pHi (Bennetts et al., 2012).
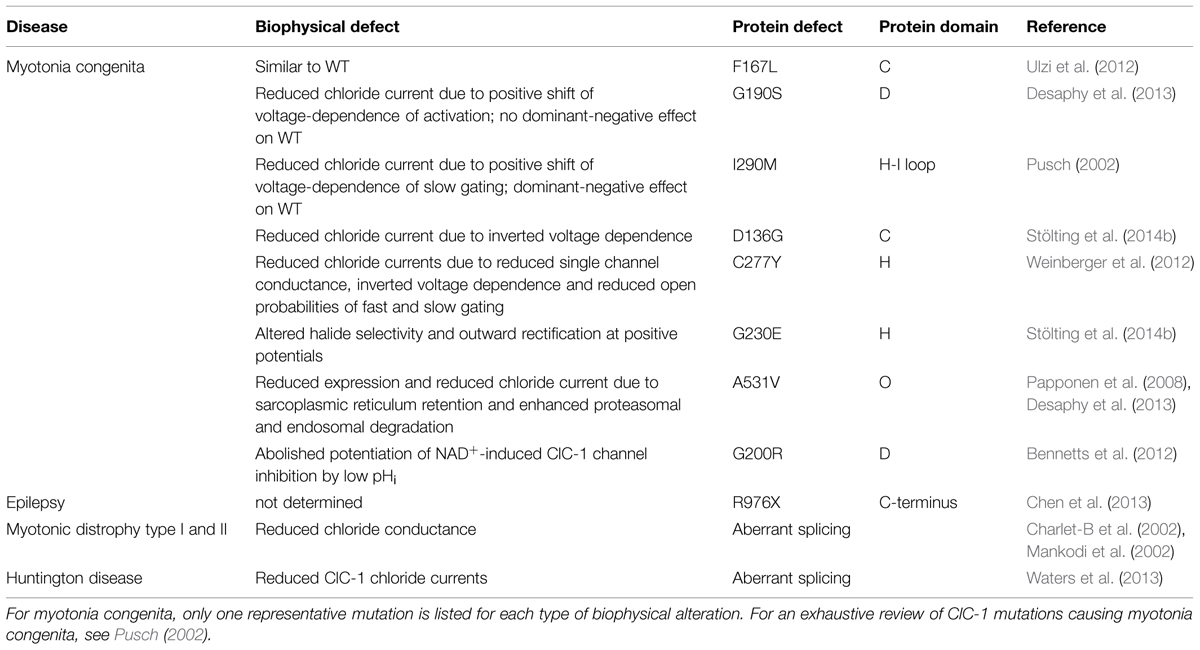
TABLE 1. Diseases associated to dysfunction of CLCN1 gene.
Some mutations exhibit both a dominant and recessive inheritance pattern, that complicates our understanding of the genotype–phenotype correlation and mode of inheritance. Remarkably, defects in the fast and slow gating modes of the channel have been proposed to explain the molecular mechanisms for dominant and recessive MC forms (Accardi and Pusch, 2000; Simpson et al., 2004; Weinberger et al., 2012). Furthermore, structure-function relationships in the ClC-1 channel provided relevant information to the understanding of the molecular pathogenesis of MC (Skálová et al., 2013). As a general rule, dominant mutations located on one subunit of the dimer may disrupt the slow gate, thereby exerting a dominant-negative effect on the unaffected associated wild-type subunit. As the slow gate involves subunits interaction, and helices at the dimer interface primarily contribute to cooperative gating (Duffield et al., 2003), this mechanistic hypothesis easily fits to dominant mutations residing on the boundary region between two monomers (Fialho et al., 2007). The homology model of hClC-1 based on the crystallographic structure of CmCLC allowed to identify residues constituting the dimer interface and the ion conducting pore, and indeed to validate experimental data and mechanistic hypotheses (Feng et al., 2010; Skálová et al., 2013). Indeed, most dominant MC mutations proven to cause a dominant-negative effect when co-expressed with WT subunits effectively reside at the boundary region between two monomers or in its proximity. Conversely, mutations occurring in the channel pore show no dominant-negative effect and can cause the disease by directly affecting the channel activity or the local structure of one subunit, or induce misfolding of the mutant subunit (Skálová et al., 2013). On the other hand, recessive Becker mutations present in both alleles may limit expression or affect the fast gate of individual subunits, thereby causing different degrees of haploinsufficiency. Variation in symptoms amongst MC patients cannot be fully explained by the different effect of ClC-1 mutations, since relatives carrying identical ClC-1 mutations can exhibit markedly different phenotypes (Colding-Jørgensen, 2005). In addition, a number of ClC-1 mutations found in myotonic carriers show little or no evidence of defective channel function in heterologous expression systems (Pusch, 2002; Simpson et al., 2004; Desaphy et al., 2013; Lucchiari et al., 2013). One possibility is that these variants might actually modify the phenotype of a co-existing MC mutation without being frankly pathogenic. Alternatively, these mutations might impair channel modulation by signaling pathways or accessory proteins in the muscle context. In addition, difference in allelic expression of CLCN1 may influence the clinical phenotype (Duno et al., 2004). Last but not least, other genes may be involved in the phenotype variability, acting as disease modifiers, although such factors have not been identified so far (Colding-Jørgensen, 2005). Myotonic symptoms are known to worsen during pregnancy and with use of diuretics (Bretag et al., 1980; Colding-Jørgensen, 2005; Basu et al., 2009), both conditions being associated with increased risk of hypomagnesemia and hypocalcemia (Raman et al., 1991; Skov et al., 2013). Indeed, the elevation of [Mg2+]o or [Ca2+]o, within their physiological ranges, dampened 9-AC-induced myotonia in isolated rat and human muscle fibers (Skov et al., 2013, 2015).
Myotonic Dystrophy
Conversely to MC, myotonic dystrophy is an autosomal dominant multisystemic disease due to expansion of nucleotide repeats in the DMPK (DM1 or Steinert disease) or ZNF9 (DM2 or proximal myotonic myopathy or PROMM) genes (Meola, 2013; Meola and Cardani, 2014). It is the commonest form of muscular dystrophy in adults. In both forms, the nucleotide expansion is transcribed in RNAs that accumulate in the cell nucleus and constitute the so-called foci, which affects the transcription and splicing of various genes. In particular, DMPK- and ZNF9-induced alteration of CLCN1 transcription have been demonstrated in several mouse models of DM1 and DM2, and is likely responsible for the observed myotonia (Charlet-B et al., 2002; Mankodi et al., 2002; Chen et al., 2007; Lueck et al., 2007; Wheeler et al., 2007; Hao et al., 2008).
In addition, ClC-1 mutations have been found in diagnosed DM2 patients, which worsen the disease. For instance, the ClC-1 mutation R894X was shown to enhance myotonia and pain in DM2 patients (Ursu et al., 2012). In another DM2 patient, the F167L ClC-1 mutation might explain the unusual early onset of the disease (Cardani et al., 2012). Thus, it has been hypothesized that DM2 patients with co-segregating CLCN1 mutation have an increased likelihood to be referred for molecular diagnostic testing owing to the exacerbation of the clinical phenotype (Suominen et al., 2008).
The gCl is also an index of the dystrophic condition of dystrophin-deficient muscles in the mdx mouse, a model of Duchenne muscular dystrophy (De Luca et al., 1995). Indeed, the diaphragm and extensor digitorum longus (EDL) muscles from mdx mice show a decrease of gCl and an increase of gK. These changes were very dramatic in diaphragm fibers suggesting that the impairment of gCl in the degenerating diaphragm could account for some of the symptoms of the human disease (De Luca et al., 1997b).
Epilepsy and other Neurological Disorders
Epileptic phenotypes resulting from defects in a number of ion channel types have been recently classified as “channelepsies” (D’Adamo et al., 2013). Evidence for a role of ClC-1 channels in the pathogenesis of “Cl- -channelepilepsy” is derived from large-scale exome analysis of ion channel variants and expression studies (Chen et al., 2013). Indeed, the CLCN1 gene has been recently proposed as a candidate gene for epilepsy. The analysis of a cohort of patients suffering from idiopathic epilepsy showed that the occurrence of single nucleotide polymorphisms in CLCN1 was threefold higher in affected patients compared to controls. Moreover, a novel ClC-1 premature stop codon (R976X), resulting in truncation of the distal C-terminus of the protein, was identified by exomic sequencing in one patient with generalized pharmacoresistant epilepsy and mild myotonic features (Chen et al., 2013). The same study also showed unexpected ClC-1 mRNA transcripts and ClC-1 protein bands in several human brain areas, including hippocampus, cerebellar Purkinje cell layer, brainstem nuclei, frontal neocortex and thalamic nuclei.
This novel localization of ClC-1 might open new perspectives regarding the role of CLC channels in the central nervous system. First, if ClC-1 is expressed in human brain, then mutations that reduce chloride conductance could contribute to enhanced network excitability and increased susceptibility to seizures or other neurologic phenotypes. A second and intriguing observation is the presence of ClC-1 in subcortical structures, such as the basal ganglia, subthalamus, and cerebellar Purkinje cell layer, which raises the possibility that dysfunction of brain ClC-1 might contribute, in some individuals carrying ClC-1mutations, to the dystonia phenotype which has previously been regarded as being exclusively of muscular origin.
However, patients with MC and related animal models do not experience central neurological symptoms, and to date only one single case of ClC-1 mutation associated to seizures has been reported. An hypothesis to explain the low occurrence of ClC-1-associated epilepsy phenotype considers a compensatory mechanism possibly involving ClC-2, given the extensive overlapping in the regional brain expression of both channels, and the possible formation of heteromeric channels (Chen et al., 2013; Stölting et al., 2014a).
The association between hereditary myotonic disorders and brain diseases has been rarely reported in the literature. The first case of association between familial Thomsen myotonia and epilepsy has been presented very recently, with the description of one individual belonging to a two-generation family suffering from ClC-1-linked myotonia, who experienced epileptic seizures due to limbic encephalitis with antibodies against glutamate decarboxylase (Licchetta et al., 2014). Another recent paper have reported in a boy the co-existence of a ClC-1 mutation causing recessive MC and of a PMP22 mutation causing dominant Charcot-Marie-Tooth disease type 1A (Ardissone et al., 2013). These few “double trouble” cases do not allow to define whether the concomitant genetic condition may influence the severity and progression of the diseases.
A reduction of ClC-1 channel activity has recently been described in skeletal muscles of a mouse model of Huntington disease (HD; Waters et al., 2013). As in myotonic dystrophy, HD is due to the expansion of a trinucleotide repeat in the HD gene, and improper clcn1 mRNA splicing was shown to compromise ClC-1 channel expression and function. The muscle fibers of HD mouse were hyper-excitable, with a reduction of threshold to trigger action potentials, an increased number of fired action potentials, and the occurrence of spontaneous action potentials, eventually causing involuntary and prolonged contractions. It was thus proposed that ClC-1 impairment likely contribute to the chorea, stiffness, and dystonia that characterize HD (Waters et al., 2013).
Pharmacology of ClC-1
Much of the information regarding ClC-1 channel pharmacology has been obtained by studying skeletal muscle chloride conductance and heterologously expressed ClC-1 channels, leading to the identification of a series of poorly selective blockers and a few activators (Table 2).
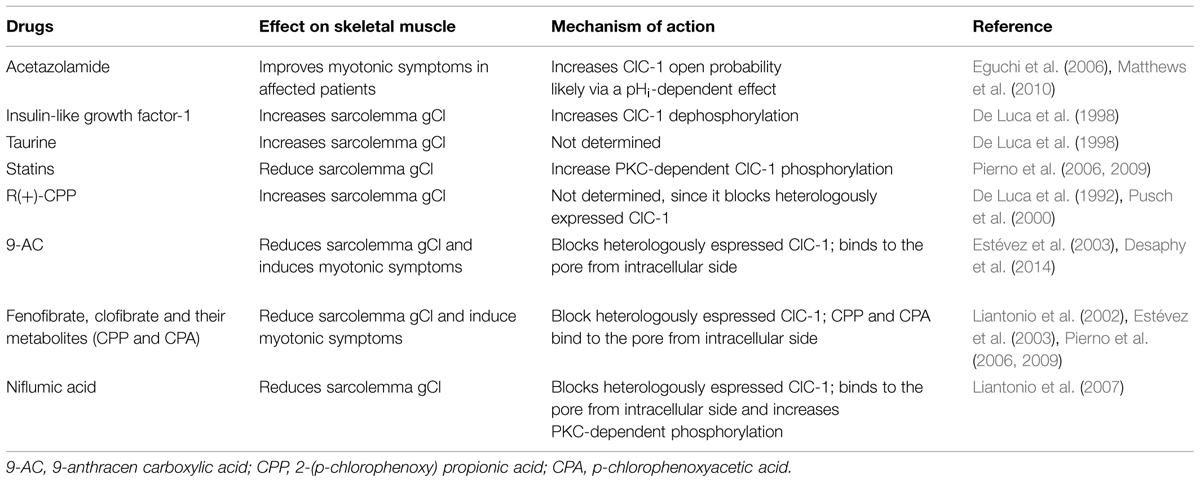
TABLE 2. Drugs acting on skeletal muscle gCl and ClC-1.
The interest in ClC-1 pharmacology stems primarily from the lack of a specific pharmacological treatment for MC patients. Today, mexiletine, a sodium channel blocker, represents the first line therapy for the myotonic syndromes, irrespective of the culprit gene. This drug can be effective in reducing stiffness and transient weakness in MC and myotonic dystrophy, but its use is limited by country availability, side effects and suboptimal or negative response in some patients (Logigian et al., 2010; Statland et al., 2012; Lo Monaco et al., 2015). The ideal drug to treat ClC-1 channelopathy should be one able to increase chloride currents. However, this goal is far from being achieved. Two carbonic anhydrase inhibitors, acetazolamide and diclorfenamide, have been used empirically for their beneficial action on a variety of neurological disorders including myotonia with variable results (Griggs et al., 1978; D’Adamo et al., 1999, 2012, 2014, 2015; Imbrici et al., 2008; Matthews et al., 2010; Markhorst et al., 2014). One possible mechanism accounting for membrane electrical stabilization by acetazolamide consists in the opening of calcium-activated potassium channels (Tricarico et al., 2000). More recently, it was proposed that this drug is also able to shift ClC-1 open probability toward more negative voltage, probably through change in pHi, resulting in an increased chloride current and consequently accounting for an antimyotonic effect (Eguchi et al., 2006; Desaphy et al., 2013). It is, however, possible that the significant percentage of non responders stems from a reduced sensitivity of some mutant channels to the drug (Desaphy et al., 2013).
In the past, two electrode voltage clamp recordings from muscle fibers revealed that the R(+)-isomer of 2-(p-chlorophenoxy) propionic acid (CPP) was able to increase the skeletal muscle chloride conductance at concentrations as low as 1–5 μM, making it a promising compound in therapy (Conte-Camerino et al., 1988; De Luca et al., 1992). However, the opener activity of R-CPP was not observed when applied to heterologously expressed ClC-1 channels (Aromataris et al., 1999; Pusch et al., 2000), suggesting an indirect action through a muscle-specific component in the native system. Another attempt to enhance chloride currents has been to indirectly increase ClC-1 channel expression or function by stimulating intracellular biochemical pathways. For instance, muscle chloride conductance increases in vivo and in vitro after application of IGF-1 or taurine (De Luca et al., 1996, 1998). PKC inhibitors, such as staurosporine or chelerythrine, has been shown to modulate ClC-1 channel (Rosenbohm et al., 1999) and increase gCl (De Luca et al., 1994a; Pierno et al., 2003), as above described. Some of them already used in therapy to treat cancer (i.e., enzastaurin) might be considered for ameliorating ClC-1 channel loss of function. Only recently, lubiprostone, a bicyclic fatty acid derivative, approved for the treatment of idiopathic chronic constipation, has been shown to selectively activate ClC-2 channels without activation of prostaglandin receptors (Lacy and Levy, 2007). Further studies would be necessary to verify whether such compound may serve as lead compound for developing future ClC-1 openers.
Although they lack a direct therapeutic interest, compounds able to block gCl have proved useful pharmacological tools to develop animal models of myotonia or to investigate hClC-1 structure-function relationship. For instance, through the reduction of gCl, 9-AC has been largely used to reproduce myotonia in isolated muscles or in vivo (Bryant and Morales-Aguilera, 1971; Skov et al., 2013; Desaphy et al., 2014). Also, clofibrate induces a myotonic-like state in rodents blocking gCl in a dose-dependent manner (De Luca et al., 1992). A considerable number of clofibric acid derivatives have been developed and tested in native skeletal muscle fibers and in heterologously expressed hClC-1 channels in order to find the structural determinants for a selective channel block. The most active compound resulted to be the S(-) isomer of CPP, capable of inhibiting ClC-0 and ClC-1 with higher potency than ClC-2 (Pusch et al., 2000; Liantonio et al., 2002). This compound induces a voltage-dependent block, reducing currents at negative voltages, thus indicating a higher affinity toward the closed state of the channel (Aromataris et al., 1999; Pusch et al., 2001). The p-chlorophenoxyacetic acid (CPA), as 9-AC, exerts its blocking activity by binding to an hydrophobic pocket within the channel pore (Accardi and Pusch, 2003; Estévez et al., 2003). Like clofibrate, fenofibrate directly blocks heterologous-expressed hClC-1 channels, and determined a reduction of gCl in rat skeletal muscles after in vivo treatment (Pierno et al., 2006, 2009).
Also the widely-used cholesterol lowering medications, statins, have been found to reduce the gCl in rat muscles, which may contribute to the common side effects of the drugs on muscles. The effects of statins on the gCl is, however, indirect, involving the PKC-dependent phosphorylation of ClC-1 channels or a reduction of ClC-1 expression (Pierno et al., 2006, 2009).
Interestingly, niflumic acid, a non steroidal anti-inflammatory drug, has been found to decrease muscle gCl by blocking ClC-1 channels both through direct binding and through Ca2+-dependent phosphorylation by PKC (Liantonio et al., 2007).
Challenges
A wealth of information about ClC-1 channels have been acquired during the 25 years since the cloning of the first member of the CLC family. However, several questions still await an exhaustive answer and represent major challenges to next research. The molecular identity and mechanism of the slow gating is still debated and the functions of the two CBS domains is still under investigation. Little is known about ClC-1 modulation by intracellular and extracellular factors and, despite the fact that the role of gCl in skeletal muscle plasticity has been assessed, the underlying molecular mechanisms are not completely understood. Future investigation will be committed to understand the possible role of ClC-1 in the brain and its involvement in the etiopathogenesis of epilepsy, as well as to face the still unclear aspects of MC, such as the lack of a precise genetic diagnosis for several patients, the exact molecular mechanisms for dominant and recessive inheritance, the mechanisms underlying myotonic symptoms in patients carrying apparently silent mutations. One major challenge regards the possibility to guarantee an appropriate and personalized therapy to MC patients through the identification of specific pharmacological strategies able to counteract the reduction in chloride conductance that causes MC (Trivedi et al., 2014). It is expected that the experimental data already available on ClC-1 blockers, through molecular dynamics simulations and functional studies, may offer a solid pharmacophore model for developing selective ClC-1 openers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Italian Department of Health (GR-2009-1580433 to PI), the Italian Telethon Foundation (grants GGP10101 and GGP14096 to DC), the French “Association Française contre les Myopathies” (grant #15020 to DC).
References
Accardi, A., Ferrera, L., and Pusch, M. (2001). Drastic reduction of the slow gate of human muscle chloride channel (ClC-1) by mutation C277S. J. Physiol. 534, 745–752. doi: 10.1111/j.1469-7793.2001.00745.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Accardi, A., Lobet, S., Williams, C., Miller, C., and Dutzler, R. (2006). Synergism between halide binding and proton transport in a CLC-type exchanger. J. Mol. Biol. 362, 691–699. doi: 10.1016/j.jmb.2006.07.081
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Accardi, A., and Pusch, M. (2000). Fast and slow gating relaxations in the muscle chloride channel ClC-1. J. Gen. Physiol. 116, 433–444. doi: 10.1085/jgp.116.3.433
Accardi, A., and Pusch, M. (2003). Conformational changes in the pore of CLC-0. J. Gen. Physiol. 122, 277–293. doi: 10.1085/jgp.200308834
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Adrian, R. H., and Bryant, S. H. (1974). On the repetitive discharge in myotonic muscle fibres. J. Physiol. 240, 505–515. doi: 10.1113/jphysiol.1974.sp010620
Ardissone, A., Bragato, C., Caffi, L., Blasevich, F., Maestrini, S., Bianchi, M. L., et al. (2013). Novel PTRF mutation in a child with mild myopathy and very mild congenital lipodystrophy. BMC Med. Genet. 14:89. doi: 10.1186/1471-2350-14-89
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aromataris, E. C., Astill, D. S., Rychkov, G. Y., Bryant, S. H., Bretag, A. H., and Roberts, M. L. (1999). Modulation of the gating of CIC-1 by S-(-) 2- (4-chlorophenoxy) propionic acid. Br. J. Pharmacol. 126, 1375–1382. doi: 10.1038/sj.bjp.0702459
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Basu, A., Nishanth, P., and Ifaturoti, O. (2009). Pregnancy in women with myotonia congenita. Int. J. Gynaecol. Obstet. 106, 62–63. doi: 10.1016/j.ijgo.2009.01.031
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bennetts, B., and Parker, M. W. (2013). Molecular determinants of common gating of a ClC chloride channel. Nat. Commun. 4, 2507. doi: 10.1038/ncomms3507
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bennetts, B., Parker, M. W., and Cromer, B. A. (2007). Inhibition of skeletal muscle ClC-1 chloride channels by low intracellular pH and ATP. J. Biol. Chem. 282, 32780–32791. doi: 10.1074/jbc.M703259200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bennetts, B., Yu, Y., Chen, T. Y., and Parker, M. W. (2012). Intracellular β-nicotinamide adenine dinucleotide inhibits the skeletal muscle ClC-1 chloride channel. J. Biol. Chem. 287, 25808–25820. doi: 10.1074/jbc.M111.327551
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Borges, A. S., Barbosa, J. D., Resende, L. A. L., Mota, L. S. L. S., Amorim, R. M., Carvalho, T. L., et al. (2013). Clinical and molecular study of a new form of hereditary myotonia in Murrah water buffalo. Neuromuscul. Disord. 23, 206–213. doi: 10.1016/j.nmd.2012.11.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bretag, A. H., Dawe, S. R., Kerr, D. I., and Moskwa, A. G. (1980). Myotonia as a side effect of diuretic action. Br. J. Pharmacol. 71, 467–471. doi: 10.1111/j.1476-5381.1980.tb10959.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brinkmeier, H., and Jockusch, H. (1987). Activators of protein kinase C induce myotonia by lowering chloride conductance in muscle. Biochem. Biophys. Res. Comm. 148, 1383–1389. doi: 10.1016/S0006-291X(87)80285-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brugnoni, R., Galantini, S., Confalonieri, P., Balestrini, M. R., Cornelio, F., and Mantegazza, R. (1999). Identification of three novel mutations in the major human skeletal muscle chloride channel gene (CLCN1), causing myotonia congenita. Hum. Mutat. 14, 447. doi: 10.1002/(SICI)1098-1004(199911)14:5<447::AID-HUMU13>3.0.CO;2-Z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brugnoni, R., Kapetis, D., Imbrici, P., Pessia, M., Canioni, E., Colleoni, L., et al. (2013). A large cohort of myotonia congenita probands: novel mutations and a high-frequency mutation region in exons 4 and 5 of the CLCN1 gene. J. Hum. Genet. 58, 581–587. doi: 10.1038/jhg.2013.58
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bryant, S. H. (1969). Cable properties of external intercostal muscle fibres from myotonic and nonmyotonic goats. Muscle membrane of normal and myotonic goats in normal and low external chloride. J. Physiol. 204, 539–550. doi: 10.1113/jphysiol.1969.sp008930
Bryant, S. H., and Conte-Camerino, D. (1991). Chloride channel regulation in the skeletal muscle of normal and myotonic goats. Pflugers. Arch. 417, 605–610. doi: 10.1007/BF00372958
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bryant, S. H., and Morales-Aguilera, A. (1971). Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. J. Physiol. 219, 367–383. doi: 10.1113/jphysiol.1971.sp009667
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Burge, J. A., and Hanna, M. G. (2012). Novel insights into the pathomechanisms of skeletal muscle channelopathies. Curr. Neurol. Neurosci. Rep. 12, 62–69. doi: 10.1007/s11910-011-0238-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bykova, E. A., Zhang, X. D., Chen, T. Y., and Zheng, J. (2006). Large movement in the C terminus of CLC-0 chloride channel during slow gating. Nat. Struct. Mol. Biol. 13, 1115–1119. doi: 10.1038/nsmb1176
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Camerino, G. M., Bouchè, M., De Bellis, M., Cannone, M., Liantonio, A., Musaraj, K., et al. (2014). Protein kinase C theta (PKCθ) modulates the ClC-1 chloride channel activity and skeletal muscle phenotype: a biophysical and gene expression study in mouse models lacking the PKCθ. Pflugers. Arch. 466, 2215–28. doi: 10.1007/s00424-0141495-1.
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cardani, R., Giagnacovo, M., Botta, A., Rinaldi, F., Morgante, A., Udd, B., et al. (2012). Co-segregation of DM2 with a recessive CLCN1 mutation in juvenile onset of myotonic dystrophy type 2. J. Neurol. 259, 2090–2099. doi: 10.1007/s00415-012-6462-1.
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cederholm, J. M., Rychkov, G. Y., Bagley, C. J., and Bretag, A. H. (2010). Inter-subunit communication and fast gate integrity are important for common gating in hClC-1. Int. J. Biochem. Cell Biol. 42, 1182–1188. doi: 10.1016/j.biocel.2010.04.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Charlet-B, N., Savkur, R. S., Singh, G., Philips, A. V., Grice, E. A., and Cooper, T. A. (2002). Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell 10, 45–53. doi: 10.1016/S1097-2765(02)00572-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, T. T., Klassen, T. L., Goldman, A. M., Marini, C., Guerrini, R., and Noebels, J. L. (2013). Novel brain expression of ClC-1 chloride channels and enrichment of CLCN1 variants in epilepsy. Neurology 80, 1078–1085. doi: 10.1212/WNL.0b013e31828868e7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, W., Wang, Y., Abe, Y., Cheney, L., Udd, B., and Li, Y. P. (2007). Haploinsufficiency for Znf9 in Znf9+/- mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J. Mol. Biol. 368, 8–17. doi: 10.1016/j.jmb.2007.01.088
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colding-Jørgensen, E. (2005). Phenotypic variability in myotonia congenita. Muscle Nerve 32, 19–34. doi: 10.1002/mus.20295
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Conte Camerino, D., De Luca, A., Mambrini, M., and Vrbovà, G. (1989). Membrane ionic conductances in normal and denervated skeletal muscle of the rat during development. Pflugers. Arch. 413, 568–570. doi: 10.1007/BF00594192
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Conte-Camerino, D., Mambrini, M., De Luca, A., Tricarico, D., Bryant, S. H., Tortorella, V., et al. (1988). Enantiomers of clofibric acid analogs have opposite actions on rat skeletal muscle chloride channels. Pflugers. Arch. 413, 105–107. doi: 10.1007/BF00581238
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cozzoli, A., Liantonio, A., Conte, E., Cannone, M., Massari, A. M., Giustino, A., et al. (2014). Angiotensin II modulates mouse skeletal muscle resting conductance to chloride and potassium ions and calcium homeostasis via the AT1 receptor and NADPH oxidase. Am. J. Physiol. Cell Physiol. 307, C634–C647. doi: 10.1152/ajpcell.00372.2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
D’Adamo, M. C., Catacuzzeno, L., Di Giovanni, G., Franciolini, F., and Pessia, M. (2013). K+ Channelepsy: progress in the neurobiology of potassium channels and epilepsy. Front. Cell. Neurosci. 7:134. doi: 10.3389/fncel.2013.00134
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
D’Adamo, M. C., Di Giovanni, G., and Pessia, M. (2014). “Animal models of episodic ataxia type 1 (EA1),” in Movement Disorders: Genetics and Models, 2nd Edn, eds Mark and S. LeDoux, (San Diego, CA: Academic Press Inc./Elsevier Science Publishing Co. Inc.), 797–807.
D’Adamo, M. C., Gallenmüller, C., Servettini, I., Hartl, E., Tucker, S. J., Arning, L., et al. (2015). Hyperthermia, short-sleep phenotype and severe migraine associated with episodic ataxia type 1. Front. Physiol. 5:525. doi: 10.3389/fphys.2014.00525
D’Adamo, M. C., Hanna, M. G., Di Giovanni, G., and Pessia, M. (2012). “Episodic Ataxia Type 1,” in GeneReviews [Internet], eds R. A. Pagon, T. C. Bird, C. R. Dolan, and K. Stephens (Seattle, WA: University of Washington).
D’Adamo, M. C., Imbrici, P., Sponcichetti, F., and Pessia, M. (1999). Mutations in the KCNA1 gene associated with episodic ataxia type-1 syndrome impair heteromeric voltage-gated K+ channel function. FASEB J. 13, 1335–1345.
de Diego, C., Gámez, J., Plassart-Schiess, E., Lasa, A., Del Río, E., Cervera, C., et al. (1999). Novel mutations in the muscle chloride channel CLCN1 gene causing myotonia congenita in Spanish families. J. Neurol. 246, 825–829. doi: 10.1007/s004150050462
De Luca, A., Conte Camerino, D., Connold, A., and Vrbovà, G. (1990a). Pharmacological block of chloride channels of developing rat skeletal muscle affects the differentiation of specific contractile properties. Pflugers. Arch. 416, 17–21. doi: 10.1007/BF00370216
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Luca, A., Mambrini, M., and Conte Camerino, D. (1990b). Changes in membrane ionic conductances and excitability characteristics of rat skeletal muscle during aging. Pflugers Arch. 415, 642–644. doi: 10.1007/BF02583519
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Luca, A., and Conte Camerino, D. (1992). Effects of aging on the mechanical threshold of rat skeletal muscle fibers. Pflugers Arch. 420, 407–409. doi: 10.1007/BF00374477
De Luca, A., Pierno, S., and Camerino, D. C. (1995). Changes of membrane electrical properties in extensor digitorum longus muscle from dystrophic (mdx) mice. Muscle Nerve 18, 1196–1198. doi: 10.1002/mus.880181016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Luca, A., Pierno, S., and Camerino, D. C. (1996). Effect of taurine depletion on excitation-contraction coupling and Cl- conductance of rat skeletal muscle. Eur. J. Pharmacol. 296, 215–222. doi: 10.1016/0014-2999(95)00702-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Luca, A., Pierno, S., Cocchi, D., and Conte Camerino, D. (1997a). Effects of chronic growth hormone treatment in aged rats on the biophysical and pharmacological properties of skeletal muscle chloride channels. Br. J. Pharmacol. 121, 369–374. doi: 10.1038/sj.bjp.0701129
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Luca, A., Pierno, S., and Camerino, D. C. (1997b). Electrical properties of diaphragm and EDL muscles during the life of dystrophic mice. Am. J. Physiol. 272, C333–C340.
De Luca, A., Pierno, S., Liantonio, A., Camerino, C., and Conte Camerino, D. (1998). Phosphorylation and IGF-1-mediated dephosphorylation pathways control the activity and the pharmacological properties of skeletal muscle chloride channels. Br. J. Pharmacol. 125, 477–482. doi: 10.1038/sj.bjp.0702107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Luca, A., Tricarico, D., Pierno, S., and Conte Camerino, D. (1994a). Aging and chloride channel regulation in rat fast-twitch muscle fibres. Pflugers. Arch. 427, 80–85. doi: 10.1007/BF00585945
De Luca, A., Pierno, S., Cocchi, D., and Conte Camerino, D. (1994b). Growth hormone administration to aged rats improves membrane electrical properties of skeletal muscle fibers. J. Pharmacol. Exp. Ther. 269, 948–953.
De Luca, A., Tricarico, D., Wagner, R., Bryant, S. H., Tortorella, V., and Conte Camerino, D. (1992). Opposite effects of enantiomers of clofibric acid derivative on rat skeletal muscle chlorideconductance: antagonism studies and theoretical modeling of two different receptor site interactions. J. Pharmacol. Exp. Ther. 260, 364–368.
de Paoli, F. V., Broch-Lips, M., Pedersen, T. H., and Nielsen, O. B. (2013). Relationship between membrane Cl- conductance and contractile endurance in isolated rat muscles. J. Physiol. 591, 531–545. doi: 10.1113/jphysiol.2012.243246
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
de Paoli, F. V., Ørtenblad, N., Pedersen, T. H., Jørgensen, R., and Nielsen, O. B. (2010). Lactate per se improves the excitability of depolarized rat skeletal muscle by reducing the Cl- conductance. J. Physiol. 588, 4785–4794. doi: 10.1113/jphysiol.2010.196568.
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Delbono, O., O’Rourke, K. S., and Ettinger, W. H. (1995). Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J. Membr. Biol. 148, 211–222. doi: 10.1007/BF00235039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Desaphy, J.-F., Carbonara, R., Costanza, T., and Conte Camerino, D. (2014). Preclinical evaluation of marketed sodium channel blockers in a rat model of myotonia discloses new promising antimyotonic drugs. Exp. Neurol. 255, 96–102. doi: 10.1016/j.expneurol.2014.02.023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Desaphy, J.-F., De Luca, A., Pierno, S., Imbrici, P., and Conte Camerino, D. (1998). Partial recovery of skeletal muscle sodium channel properties in aged rats chronically treated with growth hormone or the GH-secretagogue hexarelin. J. Pharmacol. Exp. Ther. 286, 903–912.
Desaphy, J.-F., Gramegna, G., Altamura, C., Dinardo, MM., Imbrici, P., George, A. L. Jr., et al. (2013). Functional characterization of ClC-1 mutations from patients affected by recessive myotonia congenita presenting with different clinical phenotypes. Exp. Neurol. 248, 530–540. doi: 10.1016/j.expneurol.2013.07.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DiFranco, M., Herrera, A., and Vergara, J. L. (2011). Chloride currents from the transverse tubular system in adult mammalian skeletal muscle fibers. J. Gen. Physiol. 137, 21–41. doi: 10.1085/jgp.201010496
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duffield, M., Rychkov, G., Bretag, A., and Roberts, M. (2003). Involvement of helices at the dimmer interface in ClC-1 common gating. J. Gen. Physiol. 121, 149–161. doi: 10.1085/jgp.20028741
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duffield, M. D., Rychkov, G. Y., Bretag, A. H., and Roberts, M. L. (2005). Zinc inhibits human ClC-1 muscle chloride channel by interacting with its common gating mechanism. J. Physiol. 568, 5–12. doi: 10.1113/jphysiol.2005.091777
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duno, M., Colding-Jorgensen, E., Grunnet, M., Jespersen, T., Vissing, J., and Schwartz, M. (2004). Difference in allelic expression of the CLCN1 gene and the possible influence on the myotonia congenita phenotype. Eur. J. Hum. Genet. 12, 738–743. doi: 10.1038/sj.ejhg.5201218
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dutka, T. L., Murphy, R. M., Stephenson, D. G., and Lamb, G. D. (2008). Chloride conductance in the transverse tubular system of rat skeletal muscle fibres: importance in excitation–contraction coupling and fatigue. J. Physiol. 586.3, 875–887. doi: 10.1113/jphysiol.2007.144667
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dutzler, R. (2006). The ClC family of chloride channels and transporters. Curr. Opin. Struct. Biol. 16, 439–446. doi: 10.1016/j.sbi.2006.06.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dutzler, R., Campbell, E. B., Cadene, M., Chait, B. T., and MacKinnon, R. (2002). X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 415, 287–294. doi: 10.1038/415287a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dutzler, R., Campbell, E. B., and MacKinnon, R. (2003). Gating the selectivity filter in ClC chloride channels. Science 300, 108–112. doi: 10.1126/science.10 82708
Eguchi, H., Tsujino, A., Kaibara, M., Hayashi, H., Shirabe, S., Taniyama, K., et al. (2006). Acetazolamide acts directly on the human skeletal muscle chloride channel. Muscle Nerve 34, 292–297. doi: 10.1002/mus.20585
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Esteban, J., Neumeyer, A. M., McKenna-Yasek, D., and Brown, R. H. (1998). Identification of two mutations and a polymorphism in the chloride channel CLCN-1 in patients with Becker’s generalized myotonia. Neurogenetics 1, 185–188. doi: 10.1007/s100480050027
Estévez, R., Pusch, M., Ferrer-Costa, C., Orozco, M., and Jentsch, T. J. (2004). Functional and structural conservation of CBS domains from CLC chloride channels. J. Physiol. 557, 363–378. doi: 10.1113/jphysiol.2003. 058453
Estévez, R., Schroeder, B. C., Accardi, A., Jentsch, T. J., and Pusch, M. (2003). Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron 38, 47–59. doi: 10.1016/S0896-6273(03)00168-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Feng, L., Campbell, E. B., Hsiung, Y., and MacKinnon, R. (2010). Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science 330, 635–641. doi: 10.1126/science.1195230
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fialho, D., Schorge, S., Pucovska, U., Davies, N. P., Labrum, R., Haworth, A., et al. (2007). Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions. Brain 130, 3265–3274. doi: 10.1093/brain/awm248
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fraser, J. A., Huang, C. L., and Pedersen, T. H. (2011). Relationships between resting conductances, excitability, and t-system ionic homeostasis in skeletal muscle. J. Gen. Physiol. 138, 95–116. doi: 10.1085/jgp.201110617
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fraysse, B., Desaphy, J.-F., Rolland. J.-F., Pierno, S., Liantonio, A., Giannuzzi, V., et al. (2006). Fiber type-related changes in rat skeletal muscle calcium homeostasis during aging and restoration by growth hormone. Neurobiol. Dis. 21, 372–380. doi: 10.1016/j.nbd.2005.07.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gandolfi, B., Daniel, R. J., O’Brien, D. P., Guo, L. T., Youngs, M. D., Leach, S. B., et al. (2014). A novel mutation in CLCN1 associated with feline myotonia congenita. PLoS ONE 9:e109926. doi: 10.1371/journal.pone.0109926
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garcia-Olivares, J., Alekov, A., Boroumand, M. R., Begemann, B., Hidalgo, P., and Fahlke, C. (2008). Gating of human ClC-2 chloride channels and regulation by carboxy-terminal domains. J. Physiol. 586, 5325–5336. doi: 10.1113/jphysiol.2008.158097
George, A. L. Jr., Crackower, M. A., Abdalla, J. A., Hudson, A. J., and Ebers, G. C. (1993). Molecular basis of Thomsen’s disease (autosomal dominant myotonia congenita) Nat. Genet. 3, 305–310. doi: 10.1038/ng0493-305
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goblet, C., and Whalen, R. G. (1995). Modifications of gene expression in myotonic murine skeletal muscle are associated with abnormal expression of myogenic regulatory factors. Dev. Biol. 170, 262–273. doi: 10.1006/dbio.1995.1213
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Griggs, R. C., Moxley, R. T. III, Riggs, J. E., and Engel, W. K. (1978). Effects of acetazolamide on myotonia. Ann. Neurol. 3, 531–537. doi: 10.1002/ana.410030614
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guzman, R. E., Grieschat, M., Fahlke, C., and Alekov, A. K. (2013). ClC-3 is an intracellular chloride/proton exchanger with large voltage-dependent nonlinear capacitance. ACS Chem. Neurosci. 4, 994–1003. doi: 10.1021/cn400032z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hao, M., Akrami, K., Wei, K., De Diego, C., Che, N., Ku, J. H., et al. (2008). Muscleblind-like 2 (Mbnl2) -deficient mice as a model for myotonic dystrophy. Dev. Dyn. 237, 403–410. doi: 10.1002/dvdy.21428
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hille, B. (1992). Axons, ions, and dons. Science 258, 144–145. doi: 10.1126/science.258.5079.144
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hodgkin, A. L., and Horowicz, P. (1959). The influence of potassium and chloride ions on the membrane potential of single muscle fibres. J. Physiol. 148, 127–160. doi: 10.1113/jphysiol.1959.sp006278
Hsiao, K. M., Huang, R. Y., Tang, P. H., and Lin, M. J. (2010). Functional study of CLC-1 mutants expressed in Xenopus ocytes reveals that a C-terminal region Thr891-Ser892-Thr893 is responsible for the effects of protein kinase C activator. Cell. Physiol. Biochem. 25, 687–694. doi: 10.1159/000315088
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imbrici, P., Gualandi, F., D’Adamo, M. C., Taddei Masieri, M., Cudia, P., De Grandis, D., et al. (2008). A Novel KCNA1 mutation identified in an Italian family affected by episodic ataxia type 1. Neuroscience 157, 577–587. doi: 10.1016/j.neuroscience.2008.09.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jayaram, H., Robertson, J. L., Wu, F., Williams, C., and Miller, C. (2011). Structure of a slow CLC Cl-/H+ antiporter from a cyanobacterium. Biochemistry 50, 788–794. doi: 10.1021/bi1019258
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jentsch, T. J. (2015). Discovery of CLC transport proteins: cloning, structure, function and pathophysiology. J. Physiol. doi: 10.1113/JP270043 [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jentsch, T. J., Friedrich, T., Schriever, A., and Yamada, H. (1999). The CLC chloride channel family. Pflugers. Arch. 437, 783–795. doi: 10.1007/s004240050847
Jentsch, T. J., Steinmeyer, K., and Schwarz, G. (1990). Primary structure of Torpedo marmorata chloride channel isolated by expression cloning in Xenopus ocytes. Nature 348, 510–514. doi: 10.1038/348510a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koch, M. C., Steinmeyer, K., Lorenz, C., Ricker, K., Wolf, F., Otto, M., et al. (1992). The skeletal muscle chloride channel in dominant and recessive human myotonia. Science 257, 797–800. doi: 10.1126/science.1379744
Lacy, B. E., and Levy, L. C. (2007). Lubiprostone: a chloride channel activator. J. Clin. Gastroenterol. 41, 345–351. doi: 10.1097/01.mcg.0000225665.68920.df
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lamb, G. D., Murphy, R. M., and Stephenson, D. G. (2011). On the localization of ClC-1 in skeletal muscle fibers. J. Gen. Physiol. 137, 327–329. doi: 10.1085/jgp.201010580
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leisle, L., Ludwig, C. F., Wagner, F. A., Jentsch, T. J., and Stauber, T. (2011). ClC-7 is a slowly voltage-gated 2Cl(-)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J. 30, 2140–2152. doi: 10.1038/emboj.2011.137
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liantonio, A., Accardi, A., Carbonara, G., Fracchiolla, G., Loiodice, F., Tortorella, P., et al. (2002). Molecular requisites for drug binding to muscle CLC-1 and renal CLC-K channel revealed by the use of phenoxy-alkyl derivatives of 2-(p-chlorophenoxy)propionic acid. Mol. Pharmacol. 62, 265–271. doi: 10.1124/mol.62.2.265
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liantonio, A., Giannuzzi, V., Picollo, A., Babini, E., Pusch, M., and Conte Camerino, D. (2007). Niflumic acid inhibits chloride conductance of rat skeletal muscle by directly inhibiting the CLC-1 channel and by increasing intracellular calcium. Br. J. Pharmacol. 150, 235–247. doi: 10.1038/sj.bjp.0706954
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Licchetta, L., Bisulli, F., Naldi, I., Mainieri, G., and Tinuper, P. (2014). Limbic encephalitis with anti-GAD antibodies and Thomsen myotonia: a casual or causal association? Epileptic Disord. 16, 362–365. doi: 10.1684/epd.2014.0668
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lim, H.-H., Shane, T., and Miller, C. (2012). Intracellular proton access in a Cl-/H+ antiporter. PLoS Biol. 10:e1001441. doi: 10.1371/journal.pbio.1001441
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lo Monaco, M., D’Amico, A., Luigetti, M., Desaphy, J.-F., and Modoni, A. (2015). Effect of mexiletine on transitory depression of compound motor action potential in recessive myotonia congenita. Clin. Neurophysiol. 126, 399–403. doi: 10.1016/j.clinph.2014.06.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lobet, S., and Dutzler, R. (2006). Ion-binding properties of the ClC chloride selectivity filter. EMBO J. 25, 24–33. doi: 10.1038/sj.emboj.7600909
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Logigian, E. L., Martens, W. B., Moxley R. T. IV, McDermott, M. P., Dilek, N., Wiegner, A. W., et al. (2010). Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology 74, 1441–1448. doi: 10.1212/WNL.0b013e3181dc1a3a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lorenz, C., Pusch, M., and Jentsch, T. J. (1996). Heteromultimeric CLC chloride channels with novel properties. Proc. Natl. Acad. Sci. U.S.A. 93, 13362–13366. doi: 10.1073/pnas.93.23.13362
Lossin, C., and George, A. L. Jr. (2008). Myotonia congenita. Adv. Genet. 63, 25–55. doi: 10.1016/S0065-2660(08)01002-X
Lucchiari, S., Ulzi, G., Magri, F., Bucchia, M., Corbetta, F., Servida, M., et al. (2013). Clinical evaluation and cellular electrophysiology of a recessive CLCN1 patient. J. Physiol. Pharmacol. 64, 669–678.
Lueck, J. D., Lungu, C., Mankodi, A., Osborne, R. J., Welle, S. L., Dirksen, R. T., et al. (2007). Chloride channelopathy in myotonic dystrophy resulting from loss of posttranscriptional regulation for CLCN1. Am. J. Physiol. Cell Physiol. 292, C1291–C2917. doi: 10.1152/ajpcell.00336.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lueck, J. D., Rossi, A. E., Thornton, C. A., Campbell, K. P., and Dirksen, R. T. (2010). Sarcolemmal-restricted localization of functional ClC-1 channels in mouse skeletal muscle. J. Gen. Physiol. 136, 597–613. doi: 10.1085/jgp.201010526
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ma, L., Rychkov, G. Y., Bykova, E. A., Zheng, J., and Bretag, A. H. (2011). Movement of hClC-1 C-termini during common gating and limits on their cytoplasmic location. Biochem. J. 436, 415–428. doi: 10.1042/BJ20102153
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maduke, M., Miller, C., and Mindell, J. A. (2000). A decade of CLC chloride channels: structure, mechanism, and many unsettled questions. Annu. Rev. Biophys. Biomol. Struct. 29, 411–438. doi: 10.1146/annurev.biophys.29.1.411
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mankodi, A., Takahashi, M. P., Jiang, H., Beck, C. L., Bowers, W. J., Moxley, R. T., et al. (2002). Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 10, 35–44. doi: 10.1016/S1097-2765(02)00563-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Markhorst, J. M., Stunnenberg, B. C., Ginjaar, I. B., Drost, G., Erasmus, C. E., and Sie, L. T. (2014). Clinical Experience With Long-Term Acetazolamide Treatment in Children With Nondystrophic Myotonias: a three-case report. Pediatr. Neurol. 51, 537–541. doi: 10.1016/j.pediatrneurol.2014.05.027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Markovic, S., and Dutzler, R. (2007). The structure of the cytoplasmic domain of the chloride channel ClC-Ka reveals a conserved interaction interface. Structure 15, 715–725. doi: 10.1016/j.str.2007.04.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matthews, E., Fialho, D., Tan, S. V., Venance, S. L., Cannon, S. C., Sternberg, D., et al. (2010). The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain 133, 9–22. doi: 10.1093/brain/awp294
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mazón, M. J., Barros, F., De la Peña, P., Quesada, J. F., Escudero, A., Cobo, A. M., et al. (2012). Screening for mutations in Spanish families with myotonia. Functional analysis of novel mutations in CLCN1 gene. Neuromuscul. Disord. 22, 231–243. doi: 10.1016/j.nmd.2011.10.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meola, G. (2013). Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies. Acta Myol. 32, 154–165.
Meola, G., and Cardani, R. (2014). Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta 1852, 594–606. doi: 10.1016/j.bbadis.2014.05.019.
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meyer, S., and Dutzler, R. (2006). Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure 14, 299–307. doi: 10.1016/j.str.2005.10.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Middleton, R. E., Pheasant, D. J., and Miller, C. (1996). Homodimeric architecture of a ClC-type chloride ion channel. Nature 383, 337–340. doi: 10.1038/383337a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miller, C. (1982). Open-state substructure of single chloride channels from Torpedo electroplax. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 299, 401–411. doi: 10.1098/rstb.1982.0140
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miller, C., and White, M. M. (1984). Dimeric structure of single chloride channels from Torpedo electroplax. Proc. Natl. Acad. Sci. U.S.A. 81, 2772–2775. doi: 10.1073/pnas.81.9.2772
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Modoni, A., D’Amico, A., Dallapiccola, B., Mereu, M. L., Merlini, L., Pagliarani, S., et al. (2011). Low-rate repetitive nerve stimulation protocol in an Italian cohort of patients affected by recessive myotonia congenita. J. Clin. Neurophysiol. 28, 39–44. doi: 10.1097/WNP.0b013e31820510d7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neagoe, I., Stauber, T., Fidzinski, P., Bergsdorf, E. Y., and Jentsch, T. J. (2010). The late endosomal ClC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J. Biol. Chem. 285, 21689–21697. doi: 10.1074/jbc.M110.125971
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nielsen, O. B., de Paoli, F., and Overgaard, K. (2001). Protective effects of lactic acid on force production in rat skeletal muscle. J. Physiol. 536, 161–166. doi: 10.1111/j.1469-7793.2001.t01-1-00161.x
Papponen, H., Kaisto, T., Myllylä, V. V., Myllylä, R., and Metsikkö, K. (2005). Regulated sarcolemmal localization of the muscle-specific ClC-1 chloride channel. Exp. Neurol. 191, 163–173. doi: 10.1016/j.expneurol.2004.07.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Papponen, H., Nissinen, M., Kaisto, T., Myllylä, V. V., Myllylä, R., and Metsikkö, K. (2008). F413C and A531V but not R894X myotonia congenita mutations cause defective endoplasmic reticulum export of the muscle-specific chloride channel CLC-1. Muscle Nerve 37, 317–325. doi: 10.1002/mus.20922
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pedersen, T. H., de Paoli, F. V., Flatman, J. A., and Nielsen, O. B. (2009a). Regulation of ClC-1 and KATP channels in action potential-firing fast-twitch muscle fibers. J. Gen. Physiol. 134, 309–322. doi: 10.1085/jgp.200910290
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pedersen, T. H., Macdonald, W. A., de Paoli, F. V., Gurung, I. S., and Nielsen, O. B. (2009b). Comparison of regulated passive membrane conductance in action potential-firing fast- and slow-twitch muscle. J. Gen. Physiol. 134, 323–337. doi: 10.1085/jgp.200910291
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pedersen, T. H., de Paoli, F., and Nielsen, O. B. (2005). Increased excitability of acidified skeletal muscle: role of chloride conductance. J. Gen. Physiol. 125, 237–246. doi: 10.1085/jgp.200409173
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pedersen, T. H., Nielsen, O. B., Lamb, G. D., and Stephenson, D. G. (2004). Intracellular acidosis enhances the excitability of working muscle. Science 305, 1144–1147. doi: 10.1126/science.1101141
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Picollo, A., and Pusch, M. (2005). Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 436, 420–423. doi: 10.1038/nature03720
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pierno, S., Camerino, G. M., Cippone, V., Rolland, J.-F., Desaphy, J.-F., De Luca, A., et al. (2009). Statins and fenofibrate affect skeletal muscle chloride conductance in rats by differently impairing ClC-1 channel regulation and expression. Br. J. Pharmacol. 156, 1206–1215. doi: 10.1111/j.1476-5381.2008.00079.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pierno, S., De Luca, A., Beck, C. L., George, A.L. Jr., and Conte Camerino, D. (1999). Aging-associated down-regulation of ClC-1 expression in skeletal muscle: phenotypic-independent relation to the decrease of chloride conductance. FEBS Lett. 449, 12–16. doi: 10.1016/S0014-5793(99)00202-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pierno, S., De Luca, A., Camerino, C., Huxtable, R. J., and Camerino, D. C. (1998). Chronic administration of taurine to aged rats improves the electrical and contractile properties of skeletal muscle fibers. J. Pharmacol. Exp. Ther. 286, 1183–1190.
Pierno, S., De Luca, A., Desaphy, J.-F., Fraysse, B., Liantonio, A., Didonna, M. P., et al. (2003). Growth hormone secretagogues modulate the electrical and contractile properties of rat skeletal muscle through a ghrelin-specific receptor. Br. J. Pharmacol. 139, 575–584. doi: 10.1038/sj.bjp.0705284
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pierno, S., Desaphy, J.-F., Liantonio, A., De Bellis, M., Bianco, G., De Luca, A., et al. (2002). Change of chloride ion channel conductance is an early event of slow-to-fast fibre type transition during unloading-induced muscle disuse. Brain 125, 1510–1521. doi: 10.1093/brain/awf162
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pierno, S., Desaphy, J.-F., Liantonio, A., De Luca, A., Zarrilli, A., Mastrofrancesco, L., et al. (2007). Disuse of rat muscle in vivo reduces protein kinase C activity controlling the sarcolemma chloride conductance. J. Physiol. 584, 983–995. doi: 10.1113/jphysiol.2007.141358
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pierno, S., Didonna, M. P., Cippone, V., De Luca, A., Pisoni, M., Frigeri, A., et al. (2006). Effects of chronic treatment with statins and fenofibrate on rat skeletal muscle: a biochemical, histological and electrophysiological study. Br. J. Pharmacol. 149, 909–919. doi: 10.1038/sj.bjp.0706917
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pierno, S., Tricarico, D., Liantonio, A., Mele, A., Digennaro, C., Rolland, J.-F., et al. (2014). An olive oil-derived antioxidant mixture ameliorates the age-related decline of skeletal muscle function. Age (Dordr.) 36, 73–88. doi: 10.1007/s11357-013-9544-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pusch, M. (2002). Myotonia caused by mutations in the muscle chloride channel gene ClCN1. Hum. Mut. 19, 423–434. doi: 10.1002/humu.10063
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pusch, M., Accardi, A., Liantonio, A., Ferrera, L., De Luca, A., Camerino, D. C., et al. (2001). Mechanism of block of single protopores of the Torpedo chloride channel ClC-0 by 2-(p chlorophenoxy)butyric acid (CPB). J. Gen. Physiol. 118, 45–62. doi: 10.1085/jgp.118.1.45
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pusch, M., Liantonio, A., Bertorello, L., Accardi, A., De Luca, A., Pierno, S., et al. (2000). Pharmacological characterization of chloride channels belonging to the ClC family by the use of chiral clofibric acid derivatives. Mol. Pharmacol. 58, 498–507.
Pusch, M., Steinmeyer, K., and Jentsch, T. J. (1994). Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophys. J. 66, 149–152. doi: 10.1016/S0006-3495(94)80753-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Raja Rayan, D. L., and Hanna, M. G. (2010). Skeletal muscle channelopathies: nondystrophic myotonias and periodic paralysis. Curr. Opin. Neurol. 23, 466–476. doi: 10.1097/WCO.0b013e32833cc97e
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Raja Rayan, D. L., Haworth, A., Sud, R., Matthews, E., Fialho, D., Burge, J., et al. (2012). A new explanation for recessive myotonia congenita: exon deletions and duplications in CLCN1. Neurology 78, 1953–1958. doi: 10.1212/WNL.0b013e318259e19c
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Raman, L., Yasodhara, P., and Ramaraju, L. A. (1991). Calcium and magnesium in pregnancy. Nutr. Res. 11, 1231–1236. doi: 10.1016/S0271-5317(05)80542-1
Renganathan, M., Messi, M. L., and Delbono, O. (1997). Dihydropyridine receptor-ryanodine receptor uncoupling in aged skeletal muscle. J. Membr. Biol. 157, 247–253. doi: 10.1007/s002329900233
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rhodes, T. H., Vite, C. H., Giger, U., Patterson, D. F., Fahlke, C., and George, A. L. Jr. (1999). A missense mutation in canine ClC-1 causes recessive myotonia congenita in the dog. FEBS Lett. 456, 54–58. doi: 10.1016/S0014-5793(99)00926-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Robertson, J. L., Kolmakova-Partensky, L., and Miller, C. (2010). Design, function and structure of a monomeric ClC transporter. Nature 468, 844–847. doi: 10.1038/nature09556
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosenbohm, A., Rüdel, R., and Fahlke, C. (1999). Regulation of the human skeletal muscle chloride channel hClC-1 by protein kinase C. J. Physiol. 514, 677–685. doi: 10.1111/j.1469-7793.1999.677ad.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rychkov, G. Y., Pusch, M., Astill, D. S., Roberts, M. L., Jentsch, T. J., and Bretag, A. H. (1996). Concentration and pH dependence of skeletal muscle chloride channel ClC-1. J. Physiol. 497, 423–435. doi: 10.1113/jphysiol.1996.sp021778
Salviati, G., Biasia, E., Betto, R., and Danieli Betto, D. (1986). Fast to slow transition induced by experimental myotonia in rat EDL muscle. Pflugers. Arch. 406, 266–272. doi: 10.1007/BF00640912
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sangiuolo, F., Botta, A., Mesoraca, A., Servidei, S., Merlini, L., Fratta, G., et al. (1998). Identification of five new mutations and three novel polymorphisms in the muscle chloride channel gene (CLCN1) in 20 Italian patients with dominant and recessive myotonia congenita. Hum. Mutat. 11, 331. doi: 10.1002/(SICI)1098-1004(1998)11:4<331::AID-HUMU13>3.0.CO;2-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Saviane, C., Conti, F., and Pusch, M. (1999). The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 113, 457–468. doi: 10.1085/jgp.113.3.457
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Scheel, O., Zdebik, A. A., Lourdel, S., and Jentsch, T. J. (2005). Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 436, 424–427. doi: 10.1038/nature03860
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Simpson, B. J., Height, T. A., Rychkov, G. Y., Nowak, K. J., Laing, N. G., Hughes, B. P., et al. (2004). Characterization of three myotonia-associated mutations of the CLCN1 chloride channel gene via heterologous expression. Hum. Mutat. 24, 185–191. doi: 10.1002/humu.9260
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Skálová, D., Zídková, J., Voháňka, S., Mazanec, R., Mušová, Z., Vondráček, P., et al. (2013). CLCN1 mutations in Czech patients with myotonia congenita, in silico analysis of novel and known mutations in the human dimeric skeletal muscle chloride channel. PLoS ONE 8:e82549. doi: 10.1371/journal.pone.0082549. eCollection 2013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Skov, M., De Paoli, F. V., Lausten, J., Nielsen, O. B., and Pedersen, T. H. (2015). Extracellular magnesium and calcium reduce myotonia in isolated ClC-1 chloride channel-inhibited human muscle. Muscle Nerve 51, 65–71. doi: 10.1002/mus.24260
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Skov, M., Riisager, A., Fraser, J. A., Nielsen, O. B., and Pedersen, T. H. (2013). Extracellular magnesium and calcium reduce myotonia in ClC-1 inhibited rat muscle. Neuromuscul. Disord. 23, 489–502. doi: 10.1016/j.nmd.2013.03.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Statland, J. M., Bundy, B. N., Wang, Y., Rayan, D. R., Trivedi, J. R., Sansone, V. A., et al. (2012). Mexiletine for symptoms and signs of myotonia in nondystrophic myotonia: a randomized controlled trial. JAMA 308, 1357–1365. doi: 10.1001/jama.2012.12607
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steinmeyer, K., Ortland, C., and Jentsch, T. J. (1991a). Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature 354, 301–304. doi: 10.1038/354301a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steinmeyer, K., Klocke, R., Ortland, C., Gronemeier, M., Jockusch, H., Grunder, S., et al. (1991b). Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature 354, 304–308. doi: 10.1038/354304a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stölting, G., Fischer, M., and Fahlke, C. (2014a). ClC-1 and ClC-2 form hetero-dimeric channels with novel protopore functions. Pflugers. Arch. 466, 2191–2204. doi: 10.1007/s00424-014-1490-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stölting, G., Fischer, M., and Fahlke, C. (2014b). CLC channel function and dysfunction in health and disease. Front. Physiol. 5:378. doi: 10.3389/fphys.2014.00378
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suominen, T., Schoser, B., Raheem, O., Auvinen, S., Walter, M., Krahe, R., et al. (2008). High frequency of co-segregating CLCN1 mutations among myotonic dystrophy type 2 patients from Finland and Germany. J. Neurol. 255, 1731–1736. doi: 10.1007/s00415-008-0010-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tricarico, D., Barbieri, M., and Camerino, D. C. (2000). Acetazolamide opens the muscular KCa2+ channel: a novel mechanism of action that may explain the therapeutic effect of the drug in hypokalemic periodic paralysis. Ann. Neurol. 48, 304–312. doi: 10.1002/1531-8249(200009)48:3<304::AID-ANA4>3.0.CO;2-A
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tricarico, D., and Camerino, D. C. (1994). ATP-sensitive K+ channels of skeletal muscle fibers from young adult and aged rats: possible involvement of thiol-dependent redox mechanisms in the age-related modifications of their biophysical and pharmacological properties. Mol. Pharmacol. 46, 754–761.
Tricarico, D., Conte Camerino. D., Govoni, S., and Bryant, S. H. (1991). Modulation of rat skeletal muscle chloride channels by activators and inhibitors of protein kinase C. Pflugers. Arch. 418, 500–503. doi: 10.1007/BF00497778
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Trivedi, J. R., Cannon, S. C., and Griggs, R. C. (2014). Nondystrophic myotonia: challenges and future directions. Exp. Neurol. 253, 28–30. doi: 10.1016/j.expneurol.2013.12.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tseng, P. Y., Bennetts, B., and Chen, T. Y. (2007). Cytoplasmic ATP inhibition of CLC-1 is enhanced by low pH. J. Gen. Physiol. 130, 217–221. doi: 10.1085/jgp.200709817
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsujino, A., Kaibara, M., Hayashi, H., Eguchi, H., Nakayama, S., Sato, K., et al. (2011). A CLCN1 mutation in dominant myotonia congenita impairs the increment of chloride conductance during repetitive depolarization. Neurosci. Lett. 494, 155–160. doi: 10.1016/j.neulet.2011.03.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ulzi, G., Lecchi, M., Sansone, V., Redaelli, E., Corti, E., Saccomanno, D., et al. (2012). Myotonia congenita: novel mutations in CLCN1 gene and functional characterizations in Italian patients. J. Neurol. Sci. 318, 65–71. doi: 10.1016/j.jns.2012.03.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ursu, S. F., Alekov, A., Mao, N. H., and Jurkat-Rott, K. (2012). ClC1 chloride channel in myotonic dystrophy type 2 and ClC1 splicing in vitro. Acta Myol. 31, 144–153.
Waters, C. W., Varuzhanyan, G., Talmadge, R. J., and Voss, A. A. (2013). Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc. Natl. Acad. Sci. U.S.A. 110, 9160–9165. doi: 10.1073/pnas.1220068110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weinberger, S., Wojciechowski, D., Sternberg, D., Lehmann-Horn, F., Jurkat-Rott, K., Becher, T., et al. (2012). Disease-causing mutations C277R and C277Y modify gating of human ClC-1 chloride channels in myotonia congenita. J. Physiol. 590, 3449–3464. doi: 10.1113/jphysiol.2012.232785
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wheeler, T. M., Lueck, J. D., Swanson, M. S., Dirksen, R. T., and Thornton, C. A. (2007). Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J. Clin. Invest. 117, 3952–3957. doi: 10.1172/JCI33355
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
White, M. M., and Miller, C. (1979). A voltage-gated anion channel from the electric organ of Torpedo californica. J. Biol. Chem. 254, 10161–10166.
Wijnberg, I. D., Owczarek-Lipska, M., Sacchetto, R., Mascarello, F., Pascoli, F., Grünberg, W., et al. (2012). A missense mutation in the skeletal muscle chloride channel 1 (CLCN1) as candidate causal mutation for congenital myotonia in a New Forest pony. Neuromuscul. Disord. 22, 361–367. doi: 10.1016/j.nmd.2011.10.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, F. F., Ryan, A., Devaney, J., Warnstedt, M., Korade-Mirnics, Z., Poser, B., et al. (2002). Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain 125, 2392–2407. doi: 10.1093/brain/awf246
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, X. D., Tseng, P. Y., and Chen, T. Y. (2008). ATP inhibition of CLC-1 is controlled by oxidation and reduction. J. Gen. Physiol. 132, 421–428. doi: 10.1085/jgp.200810023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zifarelli, G., and Pusch, M. (2007). CLC chloride channels and transporters: a biophysical and physiological perspective. Rev. Physiol. Biochem. Pharmacol. 158, 23–76. doi: 10.1007/112_2006_0605
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zifarelli, G., and Pusch, M. (2008). The muscle chloride channel ClC-1 is not directly regulated by intracellular ATP. J. Gen. Physiol. 131, 109–116. doi: 10.1085/jgp.200709899
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zifarelli, G., and Pusch, M. (2010). Relaxing messages from the sarcolemma. J. Gen. Physiol. 136, 593–596. doi: 10.1085/jgp.201010567
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: ClC-1 chloride channel, skeletal muscle physiology, myotonia congenita, ion channel pharmacology, skeletal muscle plasticity
Citation: Imbrici P, Altamura C, Pessia M, Mantegazza R, Desaphy J-F and Camerino DC (2015) ClC-1 chloride channels: state-of-the-art research and future challenges. Front. Cell. Neurosci. 9:156. doi: 10.3389/fncel.2015.00156
Received: 20 January 2015; Accepted: 08 April 2015;
Published online: 27 April 2015
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyReviewed by:
Maria Bykhovskaia, Universidad Central del Caribe, USAAlessio Accardi, Weill Cornell Medical College, USA
Copyright © 2015 Imbrici, Altamura, Pessia, Mantegazza, Desaphy and Camerino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Imbrici, Department of Pharmacy - Drug Sciences, University of Bari “Aldo Moro”, Via Orabona 4, 70124 Bari, Italy paola.imbrici@uniba.it