Oxygen, gastrin-releasing peptide, and pediatric lung disease: life in the balance
 Mary E. Sunday
Mary E. Sunday- Department of Pathology, Duke University Medical Center, Durham, NC, USA
Excessive oxygen (O2) can cause tissue injury, scarring, aging, and even death. Our laboratory is studying O2-sensing pulmonary neuroendocrine cells (PNECs) and the PNEC-derived product gastrin-releasing peptide (GRP). Reactive oxygen species (ROS) generated from exposure to hyperoxia, ozone, or ionizing radiation (RT) can induce PNEC degranulation and GRP secretion. PNEC degranulation is also induced by hypoxia, and effects of hypoxia are mediated by free radicals. We have determined that excessive GRP leads to lung injury with acute and chronic inflammation, leading to pulmonary fibrosis (PF), triggered via ROS exposure or by directly treating mice with exogenous GRP. In animal models, GRP-blockade abrogates lung injury, inflammation, and fibrosis. The optimal time frame for GRP-blockade and the key target cell types remain to be determined. The concept of GRP as a mediator of ROS-induced tissue damage represents a paradigm shift about how O2 can cause injury, inflammation, and fibrosis. The host PNEC response in vivo may depend on individual ROS sensing mechanisms and subsequent GRP secretion. Ongoing scientific and clinical investigations promise to further clarify the molecular pathways and clinical relevance of GRP in the pathogenesis of diverse pediatric lung diseases.
Introduction
Oxygen (O2) is essential for life. In aerobic animals, the lung evolved as a critical organ for gas exchange permitting species to move from water to land. Lungs are exposed to all the elements: air, earth, water, and fire/radiation. Homeostasis and health represent a natural equilibrium between opposing forces. Disease results when there is an imbalance between environmental exposures and host defense. Individual responses to diverse challenges can vary due to genetic factors.
Life hinges on a delicate balance. Too much or too little heat, humidity, or O2 can be lethal. Although O2 is essential for life, too much O2 can lead to tissue injury, fibrosis, senescence, and death (1–4). For several decades my research has focused on O2-sensing pulmonary neuroendocrine cells (PNECs) and their product gastrin-releasing peptide (GRP), a mammalian homolog of amphibian bombesin (5). GRP secretion can be induced by reactive oxygen species (ROS) from exposure to hyperoxia (6), ozone (7), or ionizing radiation (RT) (8). Furthermore, PNEC degranulation is known to be induced by hypoxia (9), which is also associated with increased ROS levels (10).
In the current review, I will introduce background information about PNECs as O2-sensing cells. The discussion will then summarize the highlights of over 25 years of work from my laboratory regarding the role of GRP in lung development and postnatal lung diseases, especially bronchopulmonary dysplasia (BPD). Cumulatively, these studies provide the foundation for future exploration of how GRP could mediate lung injury including acute and chronic inflammation and pulmonary fibrosis (PF) (7, 8, 11).
Oxygen-Sensing Cells: Pulmonary Neuroendocrine Cells
O2-sensing cells are important regulators of vascular tone and cardiac function. Historically, most research about O2-sensing cell biology and physiology has been focused on cardiomyocytes (12), vascular smooth muscle cells (13), and carotid body cells (glomus cells) (14), although interest in PNEC biology is growing (15). Much has been written about all of these cells (Figure 1) and their collective tissues, with numbers of PubMed citations on July 5, 2014 as follows: 228,676 for cardiomyocyte(s), cardiac muscle cell(s), or cardiac muscle (cells vs. tissue = 61,228 vs. 213,615); 77,151 for vascular smooth muscle cell(s), vascular smooth myocyte(s), or vascular smooth muscle (cells vs. tissue = 53,567 vs. 72,628); 13,068 for carotid body cell(s), glomus cells(s) or carotid body (cells vs. tissue = 6,462 vs. 11,987). However, relatively little is known about PNECs or their clusters in pulmonary epithelium, called neuroepithelial bodies (NEBs): 3,547 total citations, representing 3048 for PNECs and/or 624 for NEB(s). If cancer is excluded from the search for the cardiac, vascular, or carotid cells or tissues, the numbers drop modestly (Figure 1, lower panel) with the percentage of non-cancer citations: cardiac muscle or myocytes and vascular smooth muscle or myocytes, and 79–80% for carotid body or glomus cells. In contrast, the numbers of non-cancer citations for PNECs is only 26%, providing objective evidence that PNEC research has been largely focused on lung cancer, especially small cell carcinoma of the lung, a highly malignant cancer apparently derived from PNECs (16). Although a PubMed search for NEBs yielded only 624 citations, 508 (81%) of these are not related to cancer, possibly because PNEC-derived cancers do not develop as normal, slow-growing, innervated, and organoid NEBs (17). Of note, postnatally and in adults, NEBs represent an important stem cell niche involved in lung injury/repair as well as lung carcinogenesis (17–21). The biology of non-neoplastic/homeostatic PNEC responses to environmental challenges has been relatively under-explored. The low number of cancer-related publications is also likely due in part to challenges in culturing normal PNECs or NEBs, which have a low rate of cell proliferation both in vitro (22, 23) and in vivo (24), although PNEC proliferation can occur in vivo following acute injury (25).
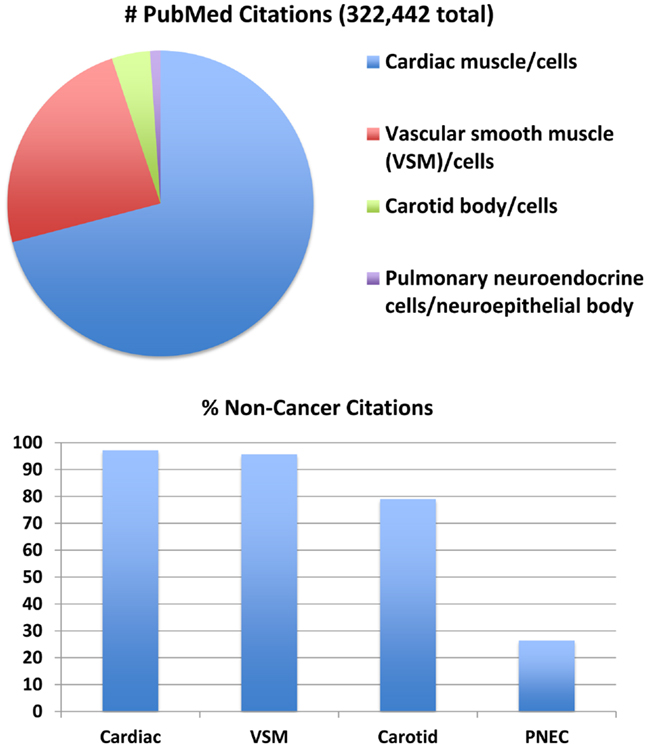
Figure 1. Major subsets of O2-sensing cells: proportions of the total number of 322,442 publications per subgroup. Top panel: relative numbers of citations per subgroup in July 2014. This comparison addresses four major subgroups of O2-sensing cells: cardiac muscle/cardiomyocytes, vascular smooth muscle (VSM)/myocytes, carotid body/glomus cells, and pulmonary neuroendocrine cells (PNECs)/neuroepithelial bodies. Actual numbers are given in the text. Lower panel: when citations including the keyword “cancer” are excluded, the number of citations is decreased per subset by 3–21% for cardiac muscle, vascular smooth muscle, and carotid body, but is decreased 74% for PNECs. (Mary E. Sunday, original unpublished data)
PNECs were first identified in the lung by Feyrter as part of a diffuse epithelial endocrine system (26, 27). Studying airway epithelium of human newborn lung, Lauweryns later identified clusters of similar amine-producing cells, which he called “NEBs,” containing dense-core neurosecretory vesicles (DCV) (28). He investigated physiological responses of PNECs to altered O2 and CO2 levels in a series of seminal experiments (9, 29–31). He first studied hypoxia- or hypercarbia-induced exocytosis of DCV from NEBs (9). Second, by using cross-circulation studies in rabbits, he observed that airway hypoxia but not hypoxemia induced exocytosis of DCV from NEBs (30). He postulated that NEB react to the composition of inhaled air and by releasing serotonin or peptides could produce a local vasoconstriction and/or bronchoconstriction in hypoxically aerated lung areas, thus enabling intrapulmonary regulation of the V/Q ratio (30). Innervation of single PNECs and NEBs is extensive in newborn rabbits (32), consisting predominantly of vagal afferent sensory nerves (15, 33). Although the function of NEB innervation remains unclear, evidence suggests a role in the generation of dyspnea (34).
Investigating how PNECs sense hypoxia, Cutz et al. carried out patch-clamp analysis of intact NEBs stained with a vital dye. They found the key players in rabbit and human lung are a membrane-bound O2-binding NADPH oxidase coupled to an H2O2-sensitive K+ channel protein (35, 36), later confirmed in knockout mice as Nox2 (37). Although NEBs express multiple NADPH oxidases and diverse voltage-gated potassium channels (Kv) and tandem pore acid-sensing K+ channels (TASK) (38), there is molecular complex formation between NOX2 (gp91 phox) and Kv but not TASK1. This observation implicates NOX2/Kv as the major O2 sensor complex in PNECs (39, 40).
Gastrin-Releasing Peptide during Physiological Hypoxia and Physiological Hyperoxia
Ernest Cutz is a pediatric pathologist who has carried out much of the seminal work on PNECs and GRP in pediatric lung diseases (17, 41). Writing a chapter together, we explored temporal and spatial expression of GRP expression during perinatal physiological processes versus postnatal disease states (42). This dichotomy can be viewed as functions of GRP in fetal lung development and perinatal transitioning (physiological hypoxia and physiological hyperoxia) versus GRP mediating pathological responses to sustained hyperoxic exposure, such as BPD.
In utero development can be considered a state of “physiological hypoxia.” Peak PNECs occur during the canalicular stage of development (at midgestation in primates and during late gestation in rodents), during which the foundation of the pulmonary capillary bed is established. At term, the umbilical artery pO2 is ~16 mm Hg (~24% O2 saturation), and umbilical vein pO2 is ~27 mm Hg (~55% O2 saturation), in contrast to postnatal arterial pO2 of ~100 mm Hg with O2 saturation >90% for term infants on room air (43).
Peak GRP mRNA levels are present in human fetal lung at midgestation (44), in the setting of physiological hypoxia (43). GRP (also known as bombesin, bombesin-like peptide or BLP) is initially synthesized as a 138–148 amino acid pro-hormone composed of three isoforms (45). These are all cleaved at methionine #27. This Met becomes the carboxy terminus of GRP that must be amidated to form the bioactive GRP peptide with GRP (14–27) amino acid sequence of – Met-Tyr-Pro-Arg-Gly-Asn-His-Trp-Ala-Val-Lys-His-Leu-Met-NH2.
Intrigued by this prenatal abundance of GRP gene expression, my laboratory began testing whether GRP alters fetal lung development. Our approach has focused on mouse, human, and baboon fetal lung organ cultures, and developing mice in utero. Cumulatively, we have determined that GRP or its amphibian homolog bombesin can promote widespread cell proliferation and accelerated differentiation of type 2 pneumocytes and PNECs (46–50). These observations were later confirmed by Fraslon and Bourbon in France (51) and Asokananthan and Cake in Australia (52), with the additional observation of GRP-induced surfactant secretion (52). We also determined that bombesin and a related frog peptide, Leu8-phyllolitorin, promote branching morphogenesis and cell proliferation in embryonic mouse lung buds (53).
In contrast to in utero development, postnatal adaptation is often referred to as “physiological hyperoxia” in recognition of the sudden change in O2 levels in the infant from ~27 mm Hg in utero to 100 mm Hg on room air (43). Room air is essentially hyperoxic to the newborn lungs. It has been recognized since the 1950s that postnatal lung development in premature infants is a unique medical situation, as first defined in pioneering work by Mel Avery that led to the discovery that respiratory distress syndrome (RDS) is due to a deficiency of surfactant. Consequently, preterm infants cannot readily expand their lungs with air to allow breathing (54). Before the arrival of surfactant therapy, premature infants often needed high levels of O2 therapy to survive. New challenges arose because prematurity is also associated with inadequate antioxidant defenses (55). Chronic lung disease of newborns, called BPD (56), was linked to O2 therapy, a mainstay of treatment for premature infants (57). The severity of BPD has decreased thanks to surfactant therapy and modern medical management such as low-barotrauma high-frequency ventilation and CPAP (58, 59). Despite improved medical care, the incidence of BPD has paradoxically increased or remains unchanged, which is puzzling regardless of how BPD is defined (57, 60, 61).
BPD: Neuroendocrine Cells and Gastrin-Releasing Peptide
Bronchopulmonary dysplasia remains a major cause of morbidity and mortality in very low birth weight infants with gestational age <28 weeks (60, 62). BPD is associated with persistent respiratory morbidity including increased hospital admissions for respiratory distress, bronchiolitis, status asthmaticus, and pneumonia (59). BPD is also associated with other complications including pulmonary hypertension, systemic hypertension, intraventricular hemorrhage, periventricular leukomalacia, neurocognitive delay, and cerebral palsy (62–65).
Early prediction of BPD has proven challenging. Relative numbers of GRP-positive PNECs normally decrease over the first postnatal months, and are markedly decreased in premature infants dying of RDS at postnatal day (PND) 1–7, thought to reflect PNEC degranulation (66). In contrast, PNECs are increased in bronchioles of infants dying with BPD at 2 weeks to 6 months of age (66). We hypothesized that elevated urine GRP levels precede BPD. One hundred thirty-two infants born at 28-weeks gestation or less, were studied. Urine GRP levels, determined by radioimmunoassay, were normalized for creatinine. BPD was defined as O2 dependence at 36 weeks post-menstrual age. Consistent with the increased number of PNECs, urine GRP was also elevated in a first urine sample at PND 1–5 in ≤28-week gestation infants who later developed BPD (67). GRP is excreted as a stable peptide in the urine; urine GRP levels are positively correlated with bronchoalveolar lavage (BAL) GRP levels (68). In the analysis by Anne Cullen (now Anne Cullen Twomey), a first urine specimen with GRP level greater than 20,000 pg/mg creatinine between PND 1–5 occurred among 54% of the infants who later developed BPD (p < 0.001), versus 10% among non-BPD infants (specificity 90%). Multivariable logistic regression analyses demonstrated that elevated urine GRP levels were associated with a 10-fold increased risk of BPD (p < 0.001) after adjusting for all confounding factors. Furthermore, urine GRP elevation occurs in parallel with markedly increased levels of GRP mRNA in newborn baboon lung (69). Utilizing urine GRP for screening might permit early therapeutic interventions to reduce disease progression and could provide a target for new preventive therapies.
We tested the hypothesis that GRP is linked to the pathogenesis of BPD through analysis of two baboon models of BPD: hyperoxia (140-day-old animals [∼32 weeks human gestational equivalent] given 100% O2 for 10 days, vs. non-BPD 140-day-old animals given PRN O2) and barotrauma (125-day-old animals [∼26 weeks human gestational equivalent] given PRN O2 for 14 days) in collaboration with Jackie Coalson and the NIH Program in BPD (70–72). In both BPD models, GRP was elevated at 24–72 h after birth. This GRP elevation was closely correlated with impaired respiratory function with increased oxygenation index, and also arrested alveolar number with alveolar wall thickening, decreased secondary alveolar septa, and blunted capillary tubulogenesis (69, 73). Remarkably, postnatal inhibition of GRP with a blocking anti-GRP antibody prevented the functional and histological changes of BPD in these animal models (69, 73). These observations suggest that GRP could be an important therapeutic target to decrease BPD prevalence and later pulmonary morbidity.
Oxidative Stress, Neuroendocrine Cells, and Gastrin-Releasing Peptide
PNEC hyperplasia occurs in weanling rat lungs in response to cigarette smoke (74) or hyperoxia (75). Elevated GRP has been associated with oxidative stress in humans including cystic fibrosis (CF) patients (76), asymptomatic smokers (68, 77), and patients with chronic obstructive pulmonary disease (78).
ROS, also known as oxygen free radicals, have been implicated in the pathogenesis of BPD. In the hyperoxic baboon model of BPD, inhibition of oxidative stress using a catalytically active metalloporphyrin (AEOL10113) decreased the number of PNEC cells, decreased GRP levels, and diminished BPD severity pathologically (6). The antioxidant not only decreased PNECs, but abrogated parenchymal mast cells and eosinophils (6). Subsequent work determined a direct link between GRP and mast cell accumulation (79). Despite the epidemiologic evidence that oxidative stress is linked to risk for BPD, this knowledge has not yet been translated into validated biomarkers for disease, or into mechanism-specific therapies to mitigate BPD morbidity.
Notably, several urine biomarkers of oxidative stress have been shown to be elevated in BPD in published clinical studies: F2-isoprostane (80, 81), 8-hydroxydeoxyguanosine (82, 83), and allantoin (84). F2-isoprostanes are increased in term infants ventilated with FiO2 of 1.0 for severe pulmonary disease due to meconium aspiration, neonatal pneumonia, or primary pulmonary hypertension (85) or in preterm infants with BPD. 8-hydroxy-2′-deoxyguanosine is an established marker of in vitro and in vivo oxidative stress and is increased in preterm infants (82), is greater in sick vs. stable preterm infants (83), and is increased in patients with chronic obstructive pulmonary disease (86), smokers (87), and workers exposed to traffic exhaust (88).
The question arose whether administration of GRP alone during perinatal transition could lead to histopathological and functional perturbations similar to BPD, even in a clinical setting free of abnormal oxidative stress. To test this hypothesis, we turned to a mouse model, considering that basic molecular mechanisms of lung development have often been explored in mice (89–94).
Model of Newborn Mice Treated with Exogenous GRP
Extending Koch’s postulates (95) to a non-infectious disease process, we tested whether exogenous GRP would alter lung development in newborn mice. To recapitulate elevated GRP levels shortly after birth, as observed in infants with BPD, we treated newborn mice with bombesin or GRP twice daily from PND 1–3 (11). On Day 14, when alveolarization is normally about half complete, we observed pathological effects similar to BPD induced by bombesin or GRP: alveolar myofibroblast proliferation, increased alveolar wall thickness and diminished alveolarization. Compared with wild-type littermates, bombesin or GRP-treated GRP receptor (GRPR)-null mice (96) had reduced defects in alveolarization, although bombesin-induced interstitial fibrosis was the same as in wild-type littermates. Neuromedin B (NMB) receptor-null (97), and bombesin receptor subtype 3-null (98) mice had the same responses as their wild-type littermates (11). Neither NMB nor a synthetic bombesin receptor type 3 ligand had any effect, consistent with effects of GRP being abrogated in GRPR-null mice. Bombesin/GRP can induce features of BPD, including interstitial fibrosis and diminished alveolarization. GRPR appears to mediate all effects of GRP, but only part of the bombesin effect on alveolarization, suggesting that novel receptors may transduce some effects of amphibian bombesin in newborn lung.
These observations in newborn mice indicate that excessive GRP alone can alter normal lung development, potentially mediating a cascade leading to abnormal pulmonary structure and function weeks to months later. GRP levels are elevated in urine and BAL of asymptomatic smokers (68), who also have elevated oxidative stress markers in urine (99). Maternal smoking is associated with many pediatric lung diseases, including asthma (100). It was hypothesized by Sam Aguayo that GRP could mediate tobacco-related lung diseases (77). We began to explore whether GRP can mediate lung injury due to oxidative stress in older patients, such as that occurring secondary to radiation (RT) exposure.
GRP and Radiation-Induced Pulmonary Fibrosis
RT-induced lung injury is a clinically relevant model for studying PF in humans, including idiopathic pulmonary fibrosis (IPF). RT produces ROS in target tissues, inducing acute and chronic radiation pneumonitis, and ultimately leading to interstitial fibrosis. In mice and other experimental animals, PF is similar to the human disease caused by environmental exposures or autoimmune diseases, and idiopathic PF. In humans, PF is progressive and irreversible, usually developing over 6–12 months post-RT. The mean survival of patients following the diagnosis of idiopathic PF is 3–5 years. There is no cure for PF except for lung transplantation, which has limited accessibility and has its own set of morbidities. We seek to reverse fibrotic responses in lung by identifying new pathways and bridges preserving organ integrity and homeostasis.
Long-term survivors of childhood malignancies, especially those treated with RT for thoracic tumors, are at a ninefold increased risk of developing PF (101). Post-treatment pulmonary disease is becoming less common with newer modalities of RT therapy such as high-resolution RT and proton beam therapy. In contrast, children undergoing total body irradiation (TBI) prior to bone marrow transplantation frequently develop serious pulmonary sequelae including interstitial fibrosis (102). Like IPF, there is no effective treatment for this post-TBI PF. Similarly, accidental nuclear exposure of children can lead to significant interstitial (restrictive) lung disease that is greater in those individuals exposed to the highest doses of radioactivity (103). Analysis of GRP+ PNECs or urine GRP levels in patients post-RT could clarify the disease pathogenesis and potentially set the stage for GRP-blockade treatment to prevent the chronic lung disease in similar clinical settings.
Considering that GRP-blockade abrogates pulmonary inflammation and fibrosis in the hyperoxic baboon model of BPD, we sought to determine whether GRP contributes to inflammatory and fibrotic phases of RT induced lung injury. Using a well-characterized mouse model of PF developing ~20 weeks after high-dose thoracic RT (15 Gy) (104), we injected GRP blocking small molecule 77427 1 h after RT then twice weekly for up to 20 weeks (8). Mice given RT plus PBS had increased interstitial CD68+ macrophages 4 weeks later and increased GRP+/PGP9.5+ PNECs 6 weeks later. Ten weeks post-RT, PBS controls had increased pSmad2/3+ nuclei indicating active TGFβ signaling. GRP-blockade with 77427 abrogated or significantly diminished CD68+, GRP+, and pSmad2/3+ cells. Twenty weeks post-RT interstitial fibrosis was demonstrated by α-smooth muscle actin (SMA) immunostaining for myofibroblasts (105, 106), which execute organ fibrosis, and also by Masson’s trichrome histochemical staining for interstitial collagen deposition (107, 108). Treatment with 77427 abrogated both interstitial SMA and collagen. Sham mice given 77427 did not differ significantly from PBS controls (8). These observations indicate that GRP-blockade decreases inflammatory and fibrotic responses to RT in mice. Similar to our experiments with hyperoxia and ozone, we propose a general working hypothesis, summarized in Figure 2. Environmental exposures generating ROS trigger PNECs to secrete GRP, which can act directly on target cells bearing cognate receptors, including airway smooth muscle cells (109), macrophages (7), CD4+ T cells (7), neutrophils (7, 110), endothelial cells (69), and pulmonary fibroblasts (69). Secondary effects could be due to GRP-induced cell differentiation (46, 50) and/or secretion of cytokines by macrophages and T cells (7, 111). Novel approaches to interrupting GRP signaling could prevent or reverse lung injury and fibrosis caused by RT, hyperoxia, or ozone.
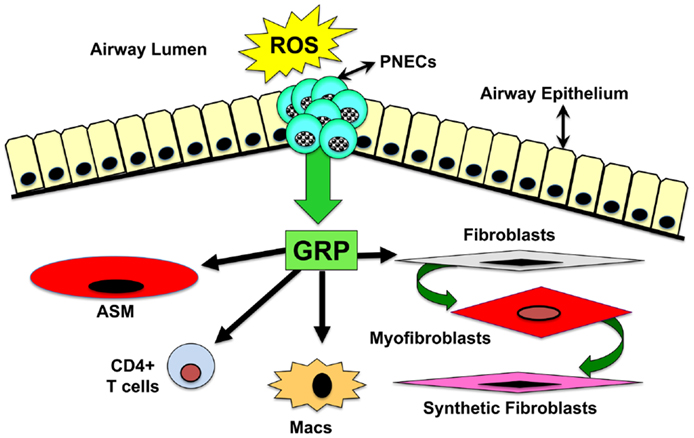
Figure 2. Schematic drawing of overall hypothesis: mechanisms by which GRP mediates lung injury and fibrosis. Environmental exposures generating ROS trigger PNECs to secrete GRP, which acts directly on target cells bearing GRPR or NMBR. These target cells include airway smooth muscle cells, macrophages, CD4+ T cells, neutrophils, endothelial cells, and pulmonary fibroblasts. Secondary effects could be due to GRP-induced cell differentiation or secretion of cytokines by macrophages and T cells.
Potential Relevance of GRP to Other Pediatric Lung Diseases
Additional pediatric lung diseases have been associated with altered numbers of GRP-positive PNECs (17). A large body of work has identified PNECs as airway O2 sensors that may function in perinatal adaptation, as detailed above (9, 112). In addition, PNEC and NEB are associated with a stem cell niche that is implicated in airway epithelial regeneration and possibly lung carcinogenesis (17–21). PNEC abnormalities have been described in seemingly unrelated lung diseases, especially PNEC hyperplasia or elevated GRP levels in association with inflammatory lung diseases (113, 114), a few of which will be briefly discussed here. It should be emphasized that increased numbers of PNEC may be due to cell differentiation rather than proliferation (24, 115), and this could represent a general adaptive response to injury or hypoxia. The clinical relevance and precise mechanisms leading to PNEC hyperplasia remain to be explored. Notch family genes (116, 117), human achaete–scute homolog (15, 118), and NeuroD (119) are likely to be involved, but specific signaling defects in patients are unknown.
Idiopathic neuroendocrine cell hyperplasia of infancy (NEHI) has been identified by Robin Deterding as a cause of chronic interstitial lung disease in young children (120–122). The cause of this disorder is unknown. Typically, patients present before 2 years of age with persistent tachypnea, hypoxia, retractions, or respiratory crackles. Lung biopsy findings are non-specific and non-diagnostic, with increased GRP-positive PNECs compared to age-matched controls. Radiographs demonstrate hyperinflation, interstitial markings, and ground-glass densities. Most patients have been treated with O2 for long periods of time, but symptoms are generally not eliminated by any medical treatment. Although there has been no mortality in over 5 years of follow-up, a few NEHI patients have improved (120). Thus, NEHI represents a distinct group of pediatric patients with clinical signs and symptoms of interstitial lung disease (120). NEHI may occur in families in some cases (122). However, the abundance of NECs may not fully explain the disease pathogenesis (115).
PNEC hyperplasia has been demonstrated in lungs of infants dying of sudden infant death syndrome (SIDS), possibly secondary to chronic hypoxia in infants at risk (123, 124). Considering that PNECs function as airway O2 sensors, Cutz suggested that GRP or another PNEC marker could herald airway chemoreceptor dysfunction as a risk factor for SIDS (125). However, GRP levels are low in SIDS victims, suggesting that another PNEC-derived product could play a role, such as calcitonin gene-related peptide (CGRP) (124). Moreover, parents of SIDS infants have a diminished ventilator response to acute hypercapnia (126), whereas hypercapnia has no effect on PNEC secretion (9).
Cystic fibrosis has also been associated with increased numbers of PNECs immunostaining for GRP, calcitonin, and serotonin (113). CF is a complex lung disease with altered mucus, chronic infection with lung inflammation, and destruction leading to bronchiectasis (127). Urine GRP levels are high postnatally in children with CF, in contrast to the decline in normal infants (76). PNECs express CFTR at the apical membrane, suggesting that NEBs could contribute to CF lung disease, including the early stages before establishment of chronic infection and progressive lung disease (128, 129). Although PNECs, airway innervation, and smooth muscle are altered in Cftr-null mice (130), it remains possible that PNEC abnormalities are secondary to infection and/or inflammation. For instance, NE cell differentiation can be induced by TNFα (131) or other cytokines. At this time, there is no clear-cut evidence for a pivotal role for GRP or PNEC in CF lung disease.
Time for a Paradigm Shift
Early and excessive GRP secretion is associated with chronic lung disease in infants. With regards to the variable interstitial fibrosis and arrested alveolarization that are characteristic of modern-day BPD, the body of evidence indicates a cause-and-effect relationship: elevated GRP can cause the clinical and pathological hallmarks of BPD in animal models. An NIH observational multicenter clinical investigation of premature infants is currently underway with Judy Voynow and Mike Cotten as PIs, with outcomes including urine levels of GRP and oxidative stress markers. The focus of this collaborative work has now intensified: to determine how transient, early GRP elevation triggers chronic lung disease with fibrosis weeks to months later. Last, but not least, we are actively seeking an optimal approach for GRP-blockade to most effectively prevent BPD in infants and PF in older children and adults.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I wish to acknowledge the major contributions of Drs. Jackie Coalson at UT San Antonio, Stella Kourembanas, Linda van Marter, and Mel Avery at Boston Children’s Hospital, and Shutang Zhou, Mike Cotten, Judy Voynow, and Mark Dewhirst at Duke University Medical Center for all they have done to make this work possible (Judy Voynow is now at Virginia Commonwealth University). This work has been supported by the following grants: NIH UO1-HL52638 (Mary E. Sunday), RO1-HL50045 (Mary E. Sunday), P50-HL67669 (Stella Kourembanas), UO1-AI-067798 (Mark Dewhirst), and R01-HL105702 (Judy Voynow and Mike Cotten), and an Established Investigator Award from the American Asthma Foundation (Mary E. Sunday).
References
1. Yee M, Buczynski BW, Lawrence BP, O’Reilly MA. Neonatal hyperoxia increases sensitivity of adult mice to bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol (2013) 48:258–66. doi: 10.1165/rcmb.2012-0238OC
2. Saretzki G, Feng J, von Zglinicki T, Villeponteau B. Similar gene expression pattern in senescent and hyperoxic-treated fibroblasts. J Gerontol A Biol Sci Med Sci (1998) 53:B438–42. doi:10.1093/gerona/53A.6.B438
3. Cataldi A, Di Giulio C. “Oxygen supply” as modulator of aging processes: hypoxia and hyperoxia models for aging studies. Curr Aging Sci (2009) 2:95–102. doi:10.2174/1874609810902020095
4. Gore A, Muralidhar M, Espey MG, Degenhardt K, Mantell LL. Hyperoxia sensing: from molecular mechanisms to significance in disease. J Immunotoxicol (2010) 7:239–54. doi:10.3109/1547691X.2010.492254
5. Wharton J, Polak JM, Bloom SR, Ghatei MA, Solcia E, Brown MR, et al. Bombesin-like immunoreactivity in the lung. Nature (1978) 273:769–70. doi:10.1038/273769a0
6. Chang L, Subramaniam M, Yoder BA, Day BJ, Coalson JJ, Sunday M, et al. A catalytic antioxidant attenuates alveolar structural remodeling in bronchopulmonary dysplasia. Am J Respir Cell Mol Biol (2003) 167:57–64.
7. Zhou S, Potts EN, Cuttitta F, Foster WM, Sunday ME. Gastrin-releasing peptide blockade as a broad-spectrum anti-inflammatory therapy for asthma. Proc Natl Acad Sci U S A (2011) 108:2100–5. doi:10.1073/pnas.1014792108
8. Zhou S, Nissao E, Jackson IL, Leong W, Dancy L, Cuttitta F, et al. Radiation-induced lung injury is mitigated by blockade of gastrin-releasing peptide. Am J Pathol (2013) 182:1248–54. doi:10.1016/j.ajpath.2012.12.024
9. Lauweryns JM, Cokelaere M, Deleersynder M, Liebens M. Intrapulmonary neuro-epithelial bodies in newborn rabbits. Influence of hypoxia, hyperoxia, hypercapnia, nicotine, reserpine, L-DOPA and 5-HTP. Cell Tiss Res (1977) 182:425–40. doi:10.1007/BF00219827
10. Rascon B, Harrison JF. Lifespan and oxidative stress show a non-linear response to atmospheric oxygen in Drosophila. J Exp Biol (2010) 213:3441–8. doi:10.1242/jeb.044867
11. Ashour K, Shan L, Lee JH, Wada SJK, Wada E, Sunday ME. Bombesin inhibits alveolarization and promotes pulmonary fibrosis in newborn mice. Am J Respir Crit Care Med (2006) 173:1377–85. doi:10.1164/rccm.200507-1014OC
12. Cadenas S, Aragones J, Landazuri MO. Mitochondrial reprogramming through cardiac oxygen sensors in ischaemic heart disease. Cardiovasc Res (2010) 88:219–28. doi:10.1093/cvr/cvq256
13. Waypa GB, Schumacker PT. Hypoxia-induced changes in pulmonary and systemic vascular resistance: where is the O2 sensor? Respir Physiol Neurobiol (2010) 174:201–11. doi:10.1016/j.resp.2010.08.007
14. Gonzalez C, Conde SV, Gallego-Martin T, Olea E, Gonzalez-Obeso E, Ramirez M, et al. Fernando de Castro and the discovery of the arterial chemoreceptors. Front Neuroanat (2014) 8:25. doi:10.3389/fnana.2014.00025
15. Linnoila RI. Functional facets of the pulmonary neuroendocrine system. Lab Invest (2006) 86:425–44. doi:10.1038/labinvest.3700412
16. Koo CW, Baliff JP, Torigian DA, Litzky LA, Gefter WB, Akers SR. Spectrum of pulmonary neuroendocrine cell proliferation: diffuse idiopathic pulmonary neuroendocrine cell hyperplasia, tumorlet, and carcinoids. AJR Am J Roentgenol (2010) 195:661–8. doi:10.2214/AJR.09.3811
17. Cutz E, Yeger H, Pan J. Pulmonary neuroendocrine cell system in pediatric lung disease-recent advances. Pediatr Dev Pathol (2007) 10:419–35. doi:10.2350/07-04-0267.1
18. Reynolds SD, Hong KU, Giangreco A, Mango GW, Guron C, Morimoto Y, et al. Conditional Clara cell ablation reveals a self-renewing progenitor function of pulmonary neuroendocrine cells. Am J Physiol Lung Cell Mol Physiol (2000) 278:L1256–63.
19. Volckaert T, Dill E, Campbell A, Tiozzo C, Majka S, Bellusci S, et al. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J Clin Invest (2011) 121:4409–19. doi:10.1172/JCI58097
20. Giangreco A, Arwert EN, Rosewell IR, Snyder J, Watt FM, Stripp BR. Stem cells are dispensable for lung homeostasis but restore airways after injury. Proc Natl Acad Sci U S A (2009) 106:9286–91. doi:10.1073/pnas.0900668106
21. Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell (2005) 121:823–35. doi:10.1016/j.cell.2005.03.032
22. Linnoila RI, Nettesheim P, DiAugustine RP. Lung endocrine-like cells in hamsters treated with diethylnitrosamine: alterations in vivo and in cell culture. Proc Natl Acad Sci U S A (1981) 78:5170–4. doi:10.1073/pnas.78.8.5170
23. Speirs V, Wang YV, Yeger H, Cutz E. Isolation and culture of neuroendocrine cells from fetal rabbit lung using immunomagnetic techniques. Am J Respir Cell Mol Biol (1992) 6:63–7. doi:10.1165/ajrcmb/6.1.63
24. Sunday ME, Willett CG, Patidar K, Graham SA. Modulation of oncogene and tumor suppressor gene expression in a hamster model of chronic lung injury with varying degrees of pulmonary neuroendocrine cell hyperplasia. Lab Invest (1994) 70:875–88.
25. Stevens TP, McBride JT, Peake JL, Pinkerton KE, Stripp BR. Cell proliferation contributes to PNEC hyperplasia after acute airway injury. Am J Physiol (1997) 272:L486–93.
27. Feyrter F. Zur pathologie des argyrophilen helle-zellen-organes im bronchialbaum des menschen. Virchows Archives (1954) 325:723–32. doi:10.1007/BF00955103
28. Lauweryns JM, Peuskens JC. Argyrophil (kinin and amine producing?) cells in human infant airway epithelium. Life Sci (1969) 8:577–85. doi:10.1016/0024-3205(69)90019-8
29. Lauweryns JM, Cokelaere M. Hypoxia-sensitive neuro-epithelial bodies. Intrapulmonary secretory neuroreceptors, modulated by the CNS. Cell Tiss Res (1973) 145:521–40.
30. Lauweryns JM, Cokelaere M, Lerut T. Cross-circulation studies on the influence of hypoxia and hypoxaemia on neuro-epithelial bodies in young rabbits. Cell Tiss Res (1978) 193:373–86. doi:10.1007/BF00225336
31. Lauweryns JM, Van Lommel A. The intrapulmonary neuroepithelial bodies after vagotomy: demonstration of their sensory neuroreceptor-like innervation. Experientia (1983) 39:1123–4. doi:10.1007/BF01943141
32. Pan J, Yeger H, Cutz E. Innervation of pulmonary neuroendocrine cells and neuroepithelial bodies in developing rabbit lung. J Histochem Cytochem (2004) 52:379–89. doi:10.1177/002215540405200309
33. Brouns I, De Proost I, Pintelon I, Timmermans JP, Adriaensen D. Sensory receptors in the airways: neurochemical coding of smooth muscle-associated airway receptors and pulmonary neuroepithelial body innervation. Auton Neurosci 126- (2006) 127:307–19. doi:10.1016/j.autneu.2006.02.006
34. Domnik NJ, Cutz E. Pulmonary neuroepithelial bodies as airway sensors: putative role in the generation of dyspnea. Curr Opin Pharmacol (2011) 11:211–7. doi:10.1016/j.coph.2011.04.003
35. Youngson C, Nurse C, Yeger H, Cutz E. Oxygen sensing in airway chemoreceptors. Nature (1993) 365:153–5. doi:10.1038/365153a0
36. Wang D, Youngson C, Wong V, Yeger H, Dinauer MC, Vega-Saenz Miera E, et al. NADPH-oxidase and a hydrogen peroxide-sensitive K+ channel may function as an oxygen sensor complex in airway chemoreceptors and small cell lung carcinoma cell lines. Proc Natl Acad Sci U S A (1996) 93:13182–7. doi:10.1073/pnas.93.23.13182
37. Fu XW, Wang D, Nurse CA, Dinauer MC, Cutz E. NADPH oxidase is an O2 sensor in airway chemoreceptors: evidence from K+ current modulation in wild-type and oxidase-deficient mice. Proc Natl Acad Sci U S A (2000) 97:4374–9. doi:10.1073/pnas.97.8.4374
38. Buttigieg J, Pan J, Yeger H, Cutz E. NOX2 (gp91phox) is a predominant O2 sensor in a human airway chemoreceptor cell line: biochemical, molecular, and electrophysiological evidence. Am J Physiol Lung Cell Mol Physiol (2012) 303:L598–607. doi:10.1152/ajplung.00170.2012
39. Fu XW, Nurse C, Cutz E. Characterization of slowly inactivating KV{alpha} current in rabbit pulmonary neuroepithelial bodies: effects of hypoxia and nicotine. Am J Physiol Lung Cell Mol Physiol (2007) 293:L892–902. doi:10.1152/ajplung.00098.2007
40. Cutz E, Pan J, Yeger H. The role of NOX2 and “novel oxidases” in airway chemoreceptor O(2) sensing. Adv Exp Med Biol (2009) 648:427–38. doi:10.1007/978-90-481-2259-2_49
41. Cutz E. Neuro-endocrine cells of the lung: an overview of morphologic characteristics and development. Exp Lung Res (1982) 3:185–208. doi:10.3109/01902148209069653
42. Sunday ME, Cutz E. Role of neuroendocrine cells in fetal and postnatal lung. In: Mendelson CR editor. Endocrinology of the Lung. Totowa, NJ: Humana Press (2000). p. 299–336.
43. Arikan GM, Scholz HS, Petru E, Haeusler MC, Haas J, Weiss PA. Cord blood oxygen saturation in vigorous infants at birth: what is normal? BJOG (2000) 107:987–94. doi:10.1111/j.1471-0528.2000.tb10401.x
44. Spindel ER, Sunday ME, Hofler H, Wolfe HJ, Habener JF, Chin WW. Transient elevation of mRNAs encoding gastrin-releasing peptide (GRP), a putative pulmonary growth factor, in human fetal lung. J Clin Invest (1987) 80:1172–9. doi:10.1172/JCI113176
45. Spindel ER, Chin WW, Price J, Rees LH, Besser GM, Habener JF. Cloning and characterization of cDNAs encoding human gastrin-releasing peptide. Proc Natl Acad Sci U S A (1984) 81:5699–703. doi:10.1073/pnas.81.18.5699
46. Sunday ME, Hua J, Dai HB, Nusrat A, Torday JS. Bombesin increases fetal lung growth and maturation in utero and in organ culture. Am J Respir Cell Mol Biol (1990) 3:199–205. doi:10.1165/ajrcmb/3.3.199
47. Sunday ME, Hua J, Torday JS, Reyes B, Shipp MA. CD10/neutral endopeptidase 24.11 in developing human fetal lung. Patterns of expression and modulation of peptide-mediated proliferation. J Clin Invest (1992) 90:2517–25. doi:10.1172/JCI116145
48. King KA, Hua J, Torday JS, Drazen JM, Graham SA, Shipp MA, et al. CD10/neutral endopeptidase 24.11 regulates fetal lung growth and maturation in utero by potentiating endogenous bombesin-like peptides. J Clin Invest (1993) 91:1969–73. doi:10.1172/JCI116417
49. Sunday ME, Hua J, Reyes B, Masui H, Torday JS. Anti-bombesin antibodies modulate fetal mouse lung growth and maturation in utero and in organ cultures. Anat Rec (1993) 236:25–32. doi:10.1002/ar.1092360107
50. Emanuel RL, Torday JS, Mu Q, Asokananthan N, Sikorski KA, Sunday ME. Bombesin-like peptides and receptors in normal fetal baboon lung: roles in lung growth and maturation. Am J Physiol (1999) 277:L1003–17.
51. Fraslon C, Bourbon JR. Comparison of effects of epidermal and insulin-like growth factors, gastrin releasing peptide and retinoic acid on fetal lung cell growth and maturation in vitro. Biochim Biophysica Acta (1992) 1123:65–75. doi:10.1016/0005-2760(92)90172-R
52. Asokananthan N, Cake MH. Stimulation of surfactant lipid secretion from fetal type II pneumocytes by gastrin-releasing peptide. Am J Physiol (1996) 270:L331–7.
53. King KA, Torday JS, Sunday ME. Bombesin and [leu8]phyllolitorin promote fetal mouse lung branching morphogenesis via a specific receptor-mediated mechanism. Proc Natl Acad Sci U S A (1995) 92:4357–61. doi:10.1073/pnas.92.10.4357
54. Avery ME. Surfactant deficiency in hyaline membrane disease: the story of discovery. Am J Respir Crit Care Med (2000) 161:1074–5. doi:10.1164/ajrccm.161.4.16142
55. Morton RL, Ikle D, White CW. Loss of lung mitochondrial aconitase activity due to hyperoxia in bronchopulmonary dysplasia in primates. Am J Physiol (1998) 274:L127–33.
56. Northway WH, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline membrane disease. N Engl J Med (1967) 276:357–68. doi:10.1056/NEJM196702162760701
57. Bancalari E, Claure N, Sosenko IR. Bronchopulmonary dysplasia: changes in pathogenesis, epidemiology and definition. Semin Neonatol (2003) 8:63–71. doi:10.1016/S1084-2756(02)00192-6
58. Avery ME, Tooley WH, Keller JB, Hurd SS, Bryan MH, Cotton RB, et al. Is chronic lung disease in low birth weight infants preventable? A survey of eight centers. Pediatrics (1987) 79:26–30.
59. Flor-de-Lima F, Rocha G, Guimaraes H. Impact of changes in perinatal care on neonatal respiratory outcome and survival of preterm newborns: an overview of 15 years. Crit Care Res Pract (2012) 2012:643246. doi:10.1155/2012/643246
60. Strueby L, Thebaud B. Advances in bronchopulmonary dysplasia. Expert Rev Respir Med (2014). doi:10.1586/17476348.2014.899907
61. Jensen EA, Schmidt B. Epidemiology of bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol (2014) 100:145–57. doi:10.1002/bdra.23235
62. Ambalavanan N, Mourani P. Pulmonary hypertension in bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol (2014) 100:240–6. doi:10.1002/bdra.23241
64. Coalson JJ. Pathology of bronchopulmonary dysplasia. Semin Perinatol (2006) 30:179–84. doi:10.1053/j.semperi.2006.05.004
65. Gien J, Kinsella JP. Pathogenesis and treatment of bronchopulmonary dysplasia. Curr Opin Pediatr (2011) 23:305–13. doi:10.1097/MOP.0b013e328346577f
66. Johnson DE, Lock JE, Elde RP, Thompson TR. Pulmonary neuroendocrine cells in hyaline membrane disease and bronchopulmonary dysplasia. Pediatr Res (1982) 16:446–54. doi:10.1203/00006450-198206000-00009
67. Cullen A, Van Marter LJ, Moore M, Parad R, Sunday ME. Urine bombesin-like peptide elevation precedes clinical evidence of bronchopulmonary dysplasia. Am J Respir Crit Care Med (2002) 165:1093–7. doi:10.1164/ajrccm.165.8.2108044
68. Aguayo SM, King TE Jr, Kane MA, Sherritt KM, Silvers W, Nett LM, et al. Urinary levels of bombesin-like peptides in asymptomatic cigarette smokers: a potential risk marker for smoking-related diseases. Cancer Res (1992) 52:2727s–31s.
69. Subramaniam M, Bausch C, Twomey A, Andreeva S, Yoder BA, Chang LY, et al. Bombesin-like peptides modulate alveolarization and angiogenesis in bronchopulmonary dysplasia. Am J Respir Crit Care Med (2007) 176:902–12. doi:10.1164/rccm.200611-1734OC
70. Coalson JJ, Kuehl TJ, Escobedo MB, Hilliard JL, Smith F, Meredith K, et al. A baboon model of bronchopulmonary dysplasia: II. Pathologic features. Exp Mol Pathol (1982) 37:335–50. doi:10.1016/0014-4800(82)90046-6
71. Escobedo MB, Hilliard JL, Smith F, Meredith K, Walsh W, Johnson D, et al. A baboon model of bronchopulmonary dysplasia: I. Clinical features. Exp Mol Pathol (1982) 37:323–34. doi:10.1016/0014-4800(82)90045-4
72. Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med (1999) 160:1333–46. doi:10.1164/ajrccm.160.4.9810071
73. Sunday ME, Yoder BA, Cuttitta F, Haley KJ, Emanuel RL. Bombesin-like peptide mediates lung injury in a baboon model of bronchopulmonary dysplasia. J Clin Invest (1998) 102:584–94. doi:10.1172/JCI2329
74. Joad JP, Ji C, Kott KS, Bric JM, Pinkerton KE. In utero and postnatal effects of sidestream cigarette smoke exposure on lung function, hyperresponsiveness, and neuroendocrine cells in rats. Toxicol Appl Pharmacol (1995) 132:63–71. doi:10.1006/taap.1995.1087
75. Shenberger JS, Shew RL, Johnson DE. Hyperoxia-induced airway remodeling and pulmonary neuroendocrine cell hyperplasia in the weanling rat. Pediatr Res (1997) 42:539–44. doi:10.1203/00006450-199710000-00020
76. Scher H, Miller YE, Aguayo SM, Johnson KJ, Miller JE, McCray PB Jr. Urinary bombesin-like peptide levels in infants and children with bronchopulmonary dysplasia and cystic fibrosis. Pediatr Pulmonol (1998) 26:326–31. doi:10.1002/(SICI)1099-0496(199811)26:5<326::AID-PPUL4>3.3.CO;2-J
77. Aguayo SM. Determinants of susceptibility to cigarette smoke. Am J Respir Crit Care Med (1994) 149:1692–8. doi:10.1164/ajrccm.149.6.7911710
78. Meloni F, Ballabio P, Pistorio A, Todarello C, Montoli C, Berrayah L, et al. Urinary levels of bombesin-related peptides in a population sample from northern Italy: potential role in the pathogenesis of chronic obstructive pulmonary disease. Am J Med Sci (1998) 315:258–65. doi:10.1097/00000441-199804000-00008
79. Subramaniam M, Sugiyama K, Coy DH, Kong Y, Miller YE, Weller PF, et al. Bombesin-like peptides and mast cell responses: relevance to bronchopulmonary dysplasia? Am J Respir Crit Care Med (2003) 168:601–11. doi:10.1164/rccm.200212-1434OC
80. Ahola T, Fellman V, Kjellmer I, Raivio KO, Lapatto R. Plasma 8-isoprostane is increased in preterm infants who develop bronchopulmonary dysplasia or periventricular leukomalacia. Pediatr Res (2004) 56:88–93. doi:10.1203/01.PDR.0000130478.05324.9D
81. Ahren B. Regulatory peptides in the thyroid gland-a review on their localization and function. Acta Endocrinol (1991) 124:225–32.
82. Nassi N, Ponziani V, Becatti M, Galvan P, Donzelli G. Anti-oxidant enzymes and related elements in term and preterm newborns. Pediatr Int (2009) 51:183–7. doi:10.1111/j.1442-200X.2008.02662.x
83. Tsukahara H, Toyo-Oka M, Kanaya Y, Ogura K, Kawatani M, Hata A, et al. Quantitation of glutathione S transferase-pi in the urine of preterm neonates. Pediatr Int (2005) 47:528–31. doi:10.1111/j.1442-200x.2005.02123.x
84. Ogihara T, Kim HS, Hirano K, Imanishi M, Ogihara H, Tamai H, et al. Oxidation products of uric acid and ascorbic acid in preterm infants with chronic lung disease. Biol Neonate (1998) 73:24–33. doi:10.1159/000013956
85. Goil S, Truog WE, Barnes C, Norberg M, Rezaiekhaligh M, Thibeault D. Eight-epi-PGF2alpha: a possible marker of lipid peroxidation in term infants with severe pulmonary disease. J Pediatr (1998) 132:349–51. doi:10.1016/S0022-3476(98)70459-7
86. Igishi T, Hitsuda Y, Kato K, Sako T, Burioka N, Yasuda K, et al. Elevated urinary 8-hydroxydeoxyguanosine, a biomarker of oxidative stress, and lack of association with antioxidant vitamins in chronic obstructive pulmonary disease. Respirology (2003) 8:455–60. doi:10.1046/j.1440-1843.2003.00490.x
87. Tamae K, Kawai K, Yamasaki S, Kawanami K, Ikeda M, Takahashi K, et al. Effect of age, smoking and other lifestyle factors on urinary 7-methylguanine and 8-hydroxydeoxyguanosine. Cancer Sci (2009) 100:715–21. doi:10.1111/j.1349-7006.2009.01088.x
88. Lai CH, Liou SH, Lin HC, Shih TS, Tsai PJ, Chen JS, et al. Exposure to traffic exhausts and oxidative DNA damage. Occup Environ Med (2005) 62:216–22. doi:10.1136/oem.2004.015107
89. Buckingham S, Avery ME. Time of appearance of lung surfactant in the foetal mouse. Nature (1962) 193:688–9. doi:10.1038/193688a0
90. Schuger L, Varani J, Mitra R Jr, Gilbride K. Retinoic acid stimulates mouse lung development by a mechanism involving epithelial-mesenchymal interaction and regulation of epidermal growth factor receptors. Dev Biol (1993) 159:462–73. doi:10.1006/dbio.1993.1256
91. Kim C, Nielsen HC. Hoxa-5 in mouse developing lung: cell-specific expression and retinoic acid regulation. Am J Physiol Lung Cell Mol Physiol (2000) 279:L863–71.
92. Costa RH, Kalinichenko VV, Lim L. Transcription factors in mouse lung development and function. Am J Physiol Lung Cell Mol Physiol (2001) 280:L823–38.
93. Auten RL, O’Reilly MA, Oury TD, Nozik-Grayck E, Whorton MH. Transgenic extracellular superoxide dismutase protects postnatal alveolar epithelial proliferation and development during hyperoxia. Am J Physiol Lung Cell Mol Physiol (2006) 290:L32–40. doi:10.1152/ajplung.00133.2005
94. Gonzalez N, Moody TW, Igarashi H, Ito T, Jensen RT. Bombesin-related peptides and their receptors: recent advances in their role in physiology and disease states. Curr Opin Endocrinol Diabetes Obes (2008) 15:58–64. doi:10.1097/MED.0b013e3282f3709b
96. Wada E, Watase K, Yamada K, Ogura H, Yamano M, Inomata Y, et al. Generation and characterization of mice lacking gastrin-releasing peptide receptor. Biochem Biophys Res Commun (1997) 239:28–33. doi:10.1006/bbrc.1997.7418
97. Ohki-Hamazaki H, Sakai Y, Kamata K, Ogura H, Okuyama S, Watase K, et al. Functional properties of two bombesin-like peptide receptors revealed by the analysis of mice lacking neuromedin B receptor. J Neurosci (1999) 19:948–54.
98. Ohki-Hamazaki H, Watase K, Yamamoto K, Ogura H, Yamano M, Yamada K, et al. Mice lacking bombesin receptor subtype-3 develop metabolic defects and obesity. Nature (1997) 390:165–9. doi:10.1038/36568
99. Campos C, Guzman R, Lopez-Fernandez E, Casado A. Urinary biomarkers of oxidative/nitrosative stress in healthy smokers. Inhal Toxicol (2011) 23:148–56. doi:10.3109/08958378.2011.554460
100. Manoli SE, Smith LA, Vyhlidal CA, An CH, Porrata Y, Cardoso WV, et al. Maternal smoking and the retinoid pathway in the developing lung. Respir Res (2012) 13:42. doi:10.1186/1465-9921-13-42
101. Landau DA, Azaria B, Fineman R, Barenboim E, Goldstein L. Long-term survivors of childhood malignancies – aeromedical dilemmas and implications. Aviat Space Environ Med (2006) 77:1266–70.
102. Meyer S, Reinhard H, Gottschling S, Nunold H, Graf N. Pulmonary dysfunction in pediatric oncology patients. Pediatr Hematol Oncol (2004) 21:175–95. doi:10.1080/08880010490272894
103. Svendsen ER, Kolpakov IE, Stepanova YI, Vdovenko VY, Naboka MV, Mousseau TA, et al. 137Cesium exposure and spirometry measures in Ukrainian children affected by the Chernobyl nuclear incident. Environ Health Perspect (2010) 118:720–5. doi:10.1289/ehp.0901412
104. Haston CK, Travis EL. Murine susceptibility to radiation-induced pulmonary fibrosis is influenced by a genetic factor implicated in susceptibility to bleomycin-induced pulmonary fibrosis. Cancer Res (1997) 57:5286–91.
105. Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol (2012) 180:1340–55. doi:10.1016/j.ajpath.2012.02.004
106. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med (2012) 18:1028–40. doi:10.1038/nm.2807
107. Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol (1988) 41:467–70. doi:10.1136/jcp.41.4.467
108. Higashiyama H, Yoshimoto D, Okamoto Y, Kikkawa H, Asano S, Kinoshita M. Receptor-activated Smad localisation in bleomycin-induced pulmonary fibrosis. J Clin Pathol (2007) 60:283–9. doi:10.1136/jcp.2006.037606
109. Lach E, Haddad EB, Gies JP. Contractile effect of bombesin on guinea pig lung in vitro: involvement of gastrin-releasing peptide-preferring receptors. Am J Physiol (1993) 264:L80–6.
110. Czepielewski RS, Porto BN, Rizzo LB, Roesler R, Abujamra AL, Pinto LG, et al. Gastrin-releasing peptide receptor (GRPR) mediates chemotaxis in neutrophils. Proc Natl Acad Sci U S A (2012) 109:547–52. doi:10.1073/pnas.1110996109
111. Lemaire I. Bombesin-related peptides modulate interleukin-1 production by alveolar macrophages. Neuropeptides (1991) 20:217–23. doi:10.1016/0143-4179(91)90011-7
112. Kazemian P, Stephenson R, Yeger H, Cutz E. Respiratory control in neonatal mice with NADPH oxidase deficiency. Respir Physiol (2001) 126:89–101. doi:10.1016/S0034-5687(01)00205-5
113. Johnson DE, Wobken JD, Landrum BG. Changes in bombesin, calcitonin, and serotonin immunoreactive pulmonary neuroendocrine cells in cystic fibrosis and after prolonged mechanical ventilation. Am Rev Respir Dis (1988) 137:123–31. doi:10.1164/ajrccm/137.1.123
114. Lemaire I, Jones S, Khan MF. Bombesin-like peptides in alveolar macrophage: increased release in pulmonary inflammation and fibrosis. Neuropeptides (1991) 20:63–72. doi:10.1016/0143-4179(91)90041-G
115. Young LR, Brody AS, Inge TH, Acton JD, Bokulic RE, Langston C, et al. Neuroendocrine cell distribution and frequency distinguish neuroendocrine cell hyperplasia of infancy from other pulmonary disorders. Chest (2011) 139:1060–71. doi:10.1378/chest.10-1304
116. Kong Y, Glickman J, Subramaniam M, Shahsafaei A, Allamneni KP, Aster JC, et al. Functional diversity of notch family genes in fetal lung development. Am J Physiol Lung Cell Mol Physiol (2004) 286:L1075–83. doi:10.1152/ajplung.00438.2002
117. Shan L, Aster JC, Sklar J, Sunday ME. Notch-1 regulates pulmonary neuroendocrine cell differentiation in cell lines and in transgenic mice. Am J Physiol Lung Cell Mol Physiol (2007) 292:L500–9. doi:10.1152/ajplung.00052.2006
118. Jensen-Taubman S, Wang XY, Linnoila RI. Achaete-scute homologue-1 tapers neuroendocrine cell differentiation in lungs after exposure to naphthalene. Toxicol Sci (2010) 117:238–48. doi:10.1093/toxsci/kfq177
119. Neptune ER, Podowski M, Calvi C, Cho JH, Garcia JG, Tuder R, et al. Targeted disruption of NeuroD, a proneural basic helix-loop-helix factor, impairs distal lung formation and neuroendocrine morphology in the neonatal lung. J Biol Chem (2008) 283:21160–9. doi:10.1074/jbc.M708692200
120. Deterding RR, Pye C, Fan LL, Langston C. Persistent tachypnea of infancy is associated with neuroendocrine cell hyperplasia. Pediatr Pulmonol (2005) 40:157–65. doi:10.1002/ppul.20243
121. Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, Bean JA, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med (2007) 176:1120–8. doi:10.1164/rccm.200703-393OC
122. Popler J, Gower WA, Mogayzel PJ Jr, Nogee LM, Langston C, Wilson AC, et al. Familial neuroendocrine cell hyperplasia of infancy. Pediatr Pulmonol (2010) 45:749–55. doi:10.1002/ppul.21219
123. Perrin DG, McDonald TJ, Cutz E. Hyperplasia of bombesin-immunoreactive pulmonary neuroendocrine cells and neuroepithelial bodies in sudden infant death syndrome. Pediatr Pathol (1991) 11:431–47. doi:10.3109/15513819109064779
124. Porzionato A, Macchi V, Parenti A, Matturri L, De Caro R. Peripheral chemoreceptors: postnatal development and cytochemical findings in Sudden Infant Death Syndrome. Histol Histopathol (2008) 23:351–65.
125. Cutz E, Perrin DG, Pan J, Haas EA, Krous HF. Pulmonary neuroendocrine cells and neuroepithelial bodies in sudden infant death syndrome: potential markers of airway chemoreceptor dysfunction. Pediatr Dev Pathol (2007) 10:106–16. doi:10.2350/06-06-0113.1
126. Schiffman PL, Westlake RE, Santiago TV, Edelman NH. Ventilatory control in parents of victims of sudden-infant-death syndrome. N Engl J Med (1980) 302:486–91. doi:10.1056/NEJM198002283020903
127. Salvatore D, Buzzetti R, Baldo E, Forneris MP, Lucidi V, Manunza D, et al. An overview of international literature from cystic fibrosis registries. Part 3. Disease incidence, genotype/phenotype correlation, microbiology, pregnancy, clinical complications, lung transplantation, and miscellanea. J Cyst Fibros (2011) 10:71–85. doi:10.1016/j.jcf.2010.12.005
128. Yeger H, Pan J, Fu XW, Bear C, Cutz E. Expression of CFTR and Cl(-) conductances in cells of pulmonary neuroepithelial bodies. Am J Physiol Lung Cell Mol Physiol (2001) 281:L713–21.
129. Grasemann H, Ratjen F. Early lung disease in cystic fibrosis. Lancet Respir Med (2013) 1:148–57. doi:10.1016/S2213-2600(13)70026-2
130. Pan J, Luk C, Kent G, Cutz E, Yeger H. Pulmonary neuroendocrine cells, airway innervation, and smooth muscle are altered in Cftr null mice. Am J Respir Cell Mol Biol (2006) 35:320–6. doi:10.1165/rcmb.2005-0468OC
Keywords: oxygen-sensing cells, pulmonary neuroendocrine cells, pulmonary fibrosis, radiation injury, bronchopulmonary dysplasia, macrophages, fibroblasts
Citation: Sunday ME (2014) Oxygen, gastrin-releasing peptide, and pediatric lung disease: life in the balance. Front. Pediatr. 2:72. doi: 10.3389/fped.2014.00072
Received: 21 April 2014; Accepted: 25 June 2014;
Published online: 18 July 2014.
Edited by:
Heber C. Nielsen, Tufts University School of Medicine, USAReviewed by:
MaryAnn Volpe, Tufts University School of Medicine, USAJonathan Michael Klein, University of Iowa, USA
Copyright: © 2014 Sunday. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mary E. Sunday, Department of Pathology, Duke University Medical Center, 201 Trent Drive, Davison Room 210, Durham, NC 27710, USA e-mail: mary.sunday@duke.edu