 Ming Lei
Ming Lei Xin Wang
Xin Wang Yunbo Ke
Yunbo Ke R. John Solaro
R. John Solaro- 1Department of Pharmacology, University of Oxford, Oxford, UK
- 2Faculty of Life Science, University of Manchester, Manchester, UK
- 3Department of Physiology and Biophysics, Center for Cardiovascular Research, University of Illinois at Chicago, Chicago, IL, USA
Calcium transient in cardiomyocytes is regulated by multiple protein kinases and phosphatases. PP2A is a major protein phosphatase in the heart modulating Ca2+ handling through an array of ion channels, antiporters and pumps, etc. The assembly, localization/translocation, and substrate specificity of PP2A are controlled by different post-translational mechanisms, which in turn are linked to the activities of upstream signaling molecules. Abnormal PP2A expression and activities are associated with defective response to β-adrenergic stimulation and are indication and causal factors in arrhythmia and heart failure.
Introduction
Cyclic and effective cardiac contraction and relaxation depend on the appropriately timed generation and spread of cardiac electrical activity. At the cellular level, excitation-contraction (E-C) coupling is initiated by action potential depolarization resulting, via a cascade of events, in an increase in intracellular calcium concentration, which ultimately leads to activation of myofilament and muscle contraction; subsequent removal of intracellular calcium via a number of mechanisms results in detachment of myosin cross-bridges and relaxation. Excitation and contraction involve multiple trans-membrane (e.g., ion channels) and intracellular proteins (e.g., Ca2+ handling and sarcomeric proteins) and are highly regulated by multiple extra- and intra-cellular signaling pathways that frequently converge at protein phosphorylation.
Studies of reversible protein phosphorylation in the heart date back to early seventies of last century when it was reported that cardiac troponin I (cTnI) was phosphorylated and dephosphorylated in the same manner as the protein substrates involved in glycogen metabolism (England et al., 1972; Stull et al., 1972). cTnI is the inhibitory component of heterotrimeric troponin complex and a major phosphoprotein in ventricular myocytes. cAMP dependent protein kinase (PKA), a downstream effector of β-adrenergic stimulations, phosphorylates cTnI at serine 23 and 24 (Cole and Perry, 1975; Solaro et al., 1976). Phosphorylation of cTnI promotes Ca2+ release from the myofilament and promotes cardiac relaxation (Robertson et al., 1982; Kentish et al., 2001). PP2A came into spotlight of heart research following another line of observation in late 1980s and early 1990s. It was found that an extract from black sea sponge, okadaic acid, has positive inotropic effect on electro-mechanic properties of ventricular muscle and enhances pacemaker activities in rabbit SA node preparation (Kodama et al., 1986; Kondo et al., 1990). Okadaic acid inhibits protein phosphatase PP2A at very low concentration leading to increased phosphorylation in numerous proteins of mammalian cells, including a number of ion channels and myofilament regulatory proteins. Thus, PP2A coordinates cardiac excitation and contraction.
The catalytic subunit of PP2A is highly conserved from yeast to humans and is homologous to the counterpart of PP1 complex, another major protein phosphatase in mammalian cells, which consists of catalytic and regulatory/targeting subunit with more than more than 200 isoforms (Depaoli-Roach et al., 1994; Peti et al., 2013). PP1 and PP2A are responsible for greater than 90% of protein dephosphorylation in the heart and they often share the same protein substrates and serine/threonine sites of dephosphorylation (Luss et al., 2000). However, their relative contributions to specific protein substrates o are often different, which is reflected in dephosphoryation of L-type Ca2+ channels (PP2A preferred) and phospholamban (PP1 preferred). For a long time, mammalian protein phosphatases had been considered constitutively active with the regulatory function fulfilled solely by protein kinases. This notion has become obsolete with discovery of multiple regulatory mechanisms for protein phosphatases, especially those that link phosphatase activities to extracellular cues (Cohen, 1988). The importance of regulation of phosphatases in heart pathophysiology becomes more obvious when altered PP2A expression and activities are closely associated with heart diseases (Ai and Pogwizd, 2005; Ke et al., 2008; Wijnker et al., 2011).
PP2A and its Regulation by Upstream Signals in the Heart
PP2A actually refer to a large family of distinct heterotrimeric protein phosphatases that share a common core enzyme consisting of a scaffolding (A) and a catalytic (C) subunits that associate with a B subunit (Figure 1). A subunit contains multiple HEAT repeats and forms a horse shoe structure that bind to both B and C subunits (Groves et al., 1999). HEAT repeat exists in proteins with different functions that form helical structures and provide structural flexibility to PP2A-A subunit (Grinthal et al., 2010). Formation of the PP2A heterotrimer follows a sequential pattern in that the core enzyme AC arises first and then binds to the B subunit. The Tyrosine 307 and Leucine 309 show reversible phosphorylation and methylation that determine the phosphatase localization and substrate specificity (Chen et al., 1992; Chung et al., 1999). Methylation of Leucine 309 diverts the C-terminal carboxyl group from a repulsive negative charge interaction and facilitates assembly of ABC holoenzyme (Cho and Xu, 2007).
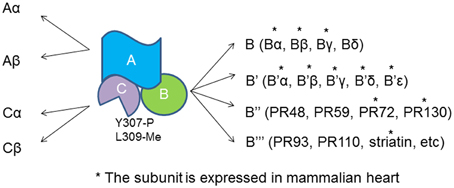
Figure 1. PP2A heterotrimer and the subunits. Both A and C subunit have two isoforms, α and β. The catalytic C subunit can be tyrosine phosphorylated at tyrosine 307 and methylated at Leucine 309. *The B subunit is expressed and identified in cardiomyocytes.
The regulatory subunits of PP2A have many members with large sequence diversity and are coded by at least 17 distinct genes. At least 11 of them are expressed in cardiomyocytes with Bα and γ the most studied cardiac isoforms (Figure 1). Bα is abundant in cytoplasm in cardiomyocyte that associates with ankyrin-B, an adapter protein required for normal subcellular localization of the Na/Ca exchanger, Na/K ATPase (Bhasin et al., 2007). Overexpression of Bα leads to reduced phosphorylation cTnI, myosin-binding protein C and phospholamban, and repressed response of L-type Ca2+ channel current to stimulation of isoproterenol (Kirchhefer et al., 2014a). Bγ is expressed in the nuclear. In mouse model deficient in Bγ, an incomplete ventricular septum occurs during development. PR72 binds to Ca2+ resulting in conformational changes in the scaffolding subunit. Another Ca2+ responsive B subunit expressed in cariomyocytes is striatin that directly interacts with calmodulin (Chen et al., 2014; Hwang and Pallas, 2014). It remains unclear if PP2As containing these B subunits control cyclic dephophorylation on any protein substrates. A genome wide association studies has identified a deletion mutation that links abnormal striatin mRNA accumulation to arrhythmogenic right ventricular cardiomyopathy in canine model (Meurs et al., 2010).
Both PP1 and PP2A have native inhibitors in mammalian cells. Inhibitor I of PP1 is a phosphoprotein regulated by β-adrenergic stimulation and is important for modulation of Ca2+ re-uptake through phospholamban. I1 and I2 PP2A are specific PP2A inhibitors (Li et al., 1995). Their expression and functional role in cardiomyocytes is underexplored. PP2A is also up-regulated by small molecular weight chemicals, both native and artificial. C2 and C6 ceramides activates PP2A in different types of mammalian cells (Dobrowsky et al., 1993). FTY720 (fingolimod) is a synthetic analog of C2 and C6 ceramide and an immunosuppressor used for treatment of multiple schlerosis (Kappos et al., 2006). Like C2 and C6 ceramide, FTY720 activates PP2A without knowing exactly what the molecular mechanism of activation. P21 activated kinase-1 (Pak1), an upstream activator for PP2A, is activated by FTY720 and C2/C6 ceramides on vitro and in vivo (Ke and Solaro, 2008; Egom et al., 2010; Liu et al., 2011b).
Accumulating evidence has indicated that PP2A activities are up-regulated by stimulation of the inhibitory G proteins, Gi through different intermediate signaling processes (Ke et al., 2008). Treatment of ventricle cardiomyocytes with agonists that turn on receptors coupled to inhibitory G proteins (Gi/Go) leads to reduced phosphorylation on PKA substrates without any change in intracellular cAMP, suggesting phosphatases are responsible for reduction in protein phosphorylation (Gupta et al., 1993, 1994). In cardiomyocytes, methylation of PP2Ac is reduced when the cells are treated with pertussis toxin and the same result is generated by inhibition of p38 MAP kinase (Liu and Hofmann, 2002, 2003). Cdc42 and Rac1 have been shown to be the downstream effectors for Gi in cardiomycytes and other mammalian cells. The constitutively active Pak1, the downstream effectors for Cdc42 and Rac1 induces activation of PP2A and dephosphorylation of myofilament regulatory proteins (Ke et al., 2004). PI3K is another possible link between Gi and PP2A activities that enhances carboxylmethylation at leu309 (Longman et al., 2014) (Figure 2).
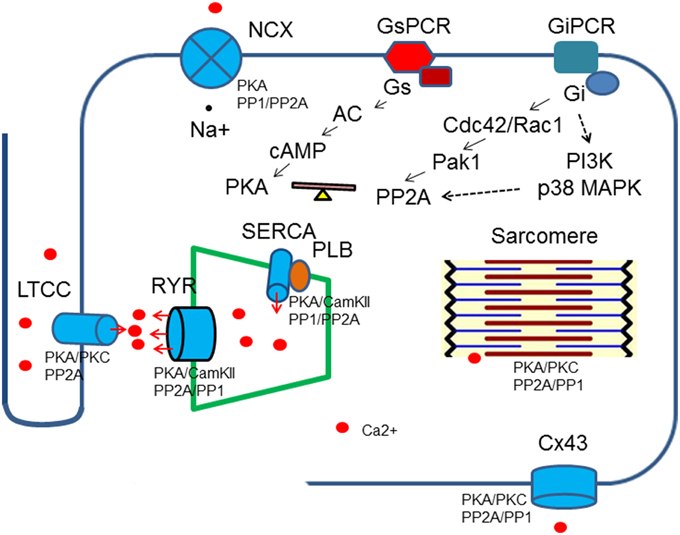
Figure 2. Regulation of Ca2+ transient by protein kinases and phosphatases. Protein kinases and phospahtases are associated with key Ca2+ transient regulatory proteins, which in turn are linked to upstream signaling cascades. A balance of protein kinase and phosphatase activities is required to maintain normal cardiac functions. Breakdown of the balance occurs at different levels: genetic mutations, gene expressions, post-translational modifications and excessive or deficient neuro-hormonal cues.
Regulation of Ca2+ Handling Proteins by PP2A
The calcium transient starts through depolarization-activated Ca2+ channels. The inward calcium current triggers Ca2+ release from the sarcoplasmic reticulum mediated primarily by ryanodine receptors. The Ca2+ binds to troponin C of troponin/tropomyosin complex and activates myofilaments. During relaxation, cytosolic Ca2+ is pumped back into sarcoplasmic reticulum by SR Ca ATPase (SERCA) and is removed from the cells by Na+/Ca2+ exchanger. Protein kinases and PP2A associate with all of these key regulatory machinery and shape the dynamics of Ca2+ flow (Table 1, Figure 2).
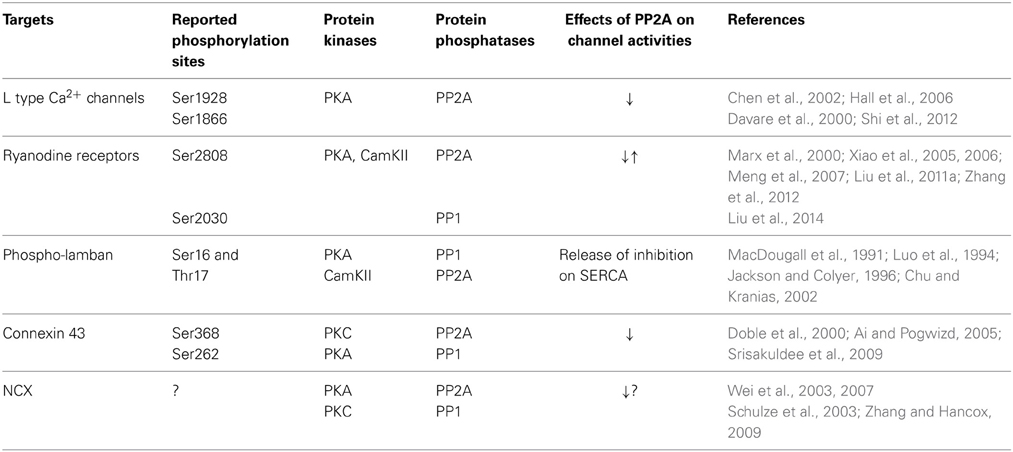
Table 1. Major targets regulating Ca2+ transient and regulated by PP2A.
PP2A is a Major Phosphatase for L-Type Ca2+ Channels (LTCC)
The voltage gated influx of Ca2+ through LTCC is highly responsive to β-adrenergic stimulation. PKA phosphorylates LTCC at the cytoplasmic, carboxyl end of alpha subunit of LTCC at Ser1928, Ser1866 (Chen et al., 2002; Hall et al., 2006), phosphorylation of S1512 and S1570 by Cam Kinase II may also play an auxiliary role modulating the channel activities (Blaich et al., 2010). The β-adrenergic effect on LTCC is reversed by PP2A, which associates with the channels at the PKA sites (Davare et al., 2000). In pacemaker cells, activation of PP2A by its upstream signal, Pak1, represses isoproterenol stimulated enhancement of the channel activities (Ke et al., 2007).
The Roles of PP2A on Ryanodine Receptor (RyR) Regulation
Ca2+ induced Ca2+ release through LTCC and ryanodine receptors is enhanced by β-adrenergic signaling cascades. Ser2808 and Ser2030 are considered as the PKA sites. Early studies suggest that hyperphosphorylation of RyR at Ser 2808 is responsible for increased leak for Ca2+ and associated with heart failure. Surprisinglya recent study has shown that in genetically modified mice with Ser2808 rendered unphosphorylatable, Ca2+ leak increases, instead of decrease with exacerbation of Ca2+-dependent cardiomyopathy (Liu et al., 2014). On the other hand, Yang et al. recently indicate that a reduced degradation of β2-AR due to Rnd3 deficiency results in enhanced PKA activities and increased Ca2+ leak from RyR (Yang et al., 2015). PP1 and PP2A form complexes on ryanodine receptors. In saponin permeabilized myocytes, exposure of PP1 and PP2A dramatically increased Ca sparks with a significant decrease of SR Ca store (Terentyev et al., 2003). On the other hand, targeting of PP2A regulatory subunit B56α by microRNA miR-1 leads to hyperphosphorylation of RyR at the CamKII sites and increases Ca2+ release and promote cardiac arrhythmogenesis (Terentyev et al., 2009; Belevych et al., 2011). PP2A is also responsible for dephosphorylation of RyR from the CamKII sites which have now been considered to play an even more important roles enhancing Ca2+ leak from the channel.
PP2A is not a Major Protein Phosphatase for Phospholamban
SERCA, a calcium transport ATPase for Ca2+ reuptake from cytosol to SR partners with phospholamban that is phosphorylated at Ser16 and threonine17 by PKA and CamKII, respectively. Phospholamban inhibits SERCA activities and the inhibition is released by PKA phosphorylation and Ser16. PP1 is the major phosphatase that removes phosphate from both locations. PP2A plays a minor role (30%) of dephosphorylation (MacDougall et al., 1991). In mice with overexpression of the regulatory subunit of PP2A, the isoproterenol stimulated phosphorylation of phosholamban and cTnI is partially reduced with increased basal contractility of the heart, likely due to elevated diastolic Ca2+ level and increased myofilament activities (Kirchhefer et al., 2014a).
The Activities of Connexin 43 are Inhibited by PP2A
The gap junction channel protein connexin 43 conducts ions and other small molecules between two adjacent myocytes. The conductivity of connexin 43 is enhanced by PKA and reduced by PP2A as demonstrated by intercellular dye coupling (Ai and Pogwizd, 2005; Ai et al., 2011).
PP2A and Na/Ca Exchanger
The cardiac Na/Ca2+ exchanger (NCX) is involved in the extrusion of cytosolic Ca2+ with a major role in the decay phase of the intracellular Ca2+ transient. PP1 and PP2A form complex with Na/Ca exchanger (Schulze et al., 2003). Stimulation of PKA activities by dibutyryl cyclic AMP and inhibition of PP2A by okadaic acid inhibits NCX activities (Lin et al., 1994). However, studies from other groups reported mixed results regarding the role of β-adrenergic stimulation on NCX activities (Zhang and Hancox, 2009). Wei et al. indicated that hyperphosphorylation of NCX is associated with an increased NCX current. In failing heart, low phosphatase activity and hyperphosphorylation is responsible for impaired sensitivity to β-adrenergic stimulation (Wei et al., 2007).
Aberrant Expression, Localization, and Activities of PP2A in Arrhythmia and Heart Failure
The importance of PP2A in the heart resides in its capacity to antagonize the effects of β-adrenergic stimulation with reduction of the amplitude of Ca transient and meanwhile increasing the Ca2+ sensitivity of myofilament in force development. Therefore, abnormality in PP2A expression, localization and activities are frequently associated with heart failure. However, the role of PP2A as a causal or beneficial factor in heart failure remains unclear.
Expression and Activities of PP2A in Heart Failure
In a rat model with chronic isoproterenol infusion that lead to cardiac hypertrophy and heart failure, PP2A activities increased significantly at day 2 (Boknik et al., 2000). In HF induced by tachypacing in sheep, increased PP1 and PP2A activities are associated with diminished response to β-adrenergic stimulation in amplitude of Ca2+ transient compared to normal heart (Briston et al., 2011). Overexpression of the catalytic subunit of PP2A (PP2A-C) by transgenic approach in mouse heart leads to left ventricular hypertrophy and reduced contractility along with an increase of PP2A activities in myocardium (Gergs et al., 2004). A more detailed analysis of expression and localization of different PP2A B subunits in cardiomyocytes from normal and failing hearts indicate that proper targeting and localization of PP2A holoenzyme are important for normal cardiac functions (DeGrande et al., 2013). On the other hand, in human heart with ischemic cardiomyopathy (ICM) and dilated cardiomyopathy (DCM), expression of both PP2A-C and PP2A-B α are reduced by half or more compared to the non-failing heart. Studies in transgenic mice over-expressing the regulatory subunit Bα indicate that this subunit directs PP2A core enzyme to Ca2+ release channels and myofilament regulatory proteins (Kirchhefer et al., 2014a). Although there is no change in PP2A activities in the ICM and DCM samples, the total protein phosphatase activities and PP1 activities increases with reduced phosphorylation on cTnI (Wijnker et al., 2011). Hyperphosphorylation of ryanodine by enhanced β-adrenergic stimulation and reduced phosphatase activities results in “Ca2+ leak” from sarcoplasmic reticulum in failing heart (Marx et al., 2000; Reiken et al., 2001).
Reduced PP2A Activities are Associated with Arrhythmia and Atrial Fibrillation (AF)
As reduced density of L-type Ca2+ current is characteristic of AF, increased PP2A activities were considered as an cause for the cardiac condition (Christ et al., 2004). Further analysis indicates that reduction of L-type calcium current density is due to a transcriptional downregulation of the pore forming alpha (1c)-subunit of LTCC, while single channel peak average current is 1.7-fold higher in AF than the control due to a 3.1-fold higher open probability of LCC. Inhibition of PP2A by okadaic acid only increases Ica in control but not in AF, suggesting phosphorylation of LCC in AF is high (Klein et al., 2003). Down regulation of PP2A-Bα by microRNA miR-1 is associated with elevated phosphorylation of RyR at CamKII site, but not the PKA sites with enhanced frequency of spontaneous Ca2+ sparks and arrhythmogenic oscillations of intracellular Ca2+(Terentyev et al., 2009).
Post-Translational Modifications and Mutation of PP2A Associated with Heart Failure
Kirchhefer et al. reported that Bα of PP2A is phosphorylated at Ser41 by PKC α and phosphorylation at this site lead to reduction of the phosphatase activities. In failing human heart, phosphorylation of Bα is 7-fold higher (Kirchhefer et al., 2014b). The A subunit is also phosphorylated and phosphorylation attenuates assembly of PP2A heterotrimer and reduces PP2A activities characterized by increased phosphorylation occurred to a large number of proteins in cells expressing the peudophosphorylated constructs. Unlike phosphorylated Bα, in a rat model of heart failure phosphorylation at this subunit is reduced leading to higher PP2A activities. In transgenic mice expressing a truncated A subunit that is a dominant negative mutant disrupting normal PP2a assembly, dilated cardiomyopathy developed with increased end-diastolic and end-systolic dimensions and decreased fractional shortening (Brewis et al., 2000).
The Roles of PP2A in Sensitizing β-Adrenergic Stimulation
Loss of response to β-adrenergic stimulation is a hall mark of end stage heart failure. Previously, it is believed that increased phosphatase activity is a major cause for desensitizing β-adrenergic stimulation as the β-adrenergic stimulation are effectively and rapidly damped by enhanced phosphatase activities. Accumulating evidence suggest that this may not be true because in failing heart, phosphorylation on L-type Ca2+ channels, ryanodine receptors and NCX are usually high. Phosphatases, especially PP2A can make them more responsive to β-adrenergic signals by bringing down phosphorylation levels. Recent studies by Zheng et al suggest that pyruvate restores β-adrenergic sensitivity of L-type Ca2+ channels in failing rat heart through PP2A (Zheng et al., 2013).
Perspective
Structural diversity and complex regulation of PP2A constitute a significant challenge in understanding its function in the heart. Emerging evidence begins to point out connections between specific PP2A heterotrimers and their protein substrates in cardiomyocytes, but definitive results are still scarce. Application of general PP2A inhibitors for heart diseases may not be applicable as these inhibitors usually are tumorigenic. However, cardiac conditions including heart failure may become ameliorated by elevating PP2A activities. FTY720 (fingolimod), a FDA recently approved drug activates PP2A and target novel anti-adrenergic signaling pathways mediated by Pak1 (Egom et al., 2010). FTY720 protect heart against ischemia-reperfusion induced arrhythmia and has demonstrated anti-hypertrophic effect in mouse TAC model (Liu et al., 2011b, 2013). Its roles in modulation of Ca2+ transient in failing heart in animal models and in humans deserve further investigation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work is supported by NIH Grant HL 064035 (R. John Solaro), PO1 HL 062426 (R. John Solaro) and the Medical Research Council (G10002647: Ming Lei, Xin Wang, Elizabeth J. Cartwright, R. John Solaro, Yunbo Ke).
References
Ai, X., Jiang, A., Ke, Y., Solaro, R. J., and Pogwizd, S. M. (2011). Enhanced activation of p21-activated kinase 1 in heart failure contributes to dephosphorylation of connexin 43. Cardiovasc. Res. 92, 106–114. doi: 10.1093/cvr/cvr163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ai, X., and Pogwizd, S. M. (2005). Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ. Res. 96, 54–63. doi: 10.1161/01.RES.0000152325.07495.5a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belevych, A. E., Sansom, S. E., Terentyeva, R., Ho, H. T., Nishijima, Y., Martin, M. M., et al. (2011). MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS ONE 6:e28324. doi: 10.1371/journal.pone.0028324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhasin, N., Cunha, S. R., Mudannayake, M., Gigena, M. S., Rogers, T. B., and Mohler, P. J. (2007). Molecular basis for PP2A regulatory subunit B56α targeting in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 293, H109–H119. doi: 10.1152/ajpheart.00059.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blaich, A., Welling, A., Fischer, S., Wegener, J. W., Kostner, K., Hofmann, F., et al. (2010). Facilitation of murine cardiac L-type Ca(v)1.2 channel is modulated by calmodulin kinase II-dependent phosphorylation of S1512 and S1570. Proc. Natl. Acad. Sci. U.S.A. 107, 10285–10289. doi: 10.1073/pnas.0914287107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boknik, P., Fockenbrock, M., Herzig, S., Knapp, J., Linck, B., Luss, H., et al. (2000). Protein phosphatase activity is increased in a rat model of long-term beta-adrenergic stimulation. Naunyn Schmiedebergs. Arch. Pharmacol. 362, 222–231. doi: 10.1007/s002100000283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brewis, N., Ohst, K., Fields, K., Rapacciuolo, A., Chou, D., Bloor, C., et al. (2000). Dilated cardiomyopathy in transgenic mice expressing a mutant A subunit of protein phosphatase 2A. Am. J. Physiol. Heart Circ. Physiol. 279, H1307–H1318.
Briston, S. J., Caldwell, J. L., Horn, M. A., Clarke, J. D., Richards, M. A., Greensmith, D. J., et al. (2011). Impaired beta-adrenergic responsiveness accentuates dysfunctional excitation-contraction coupling in an ovine model of tachypacing-induced heart failure. J. Physiol. (Lond). 589, 1367–1382. doi: 10.1113/jphysiol.2010.203984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, C., Shi, Z., Zhang, W., Chen, M., He, F., Zhang, Z., et al. (2014). Striatins contain a noncanonical coiled coil that binds protein phosphatase 2A A subunit to form a 2:2 heterotetrameric core of striatin-interacting phosphatase and kinase (STRIPAK) complex. J. Biol. Chem. 289, 9651–9661. doi: 10.1074/jbc.M113.529297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, J., Martin, B. L., and Brautigan, D. L. (1992). Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 257, 1261–1264. doi: 10.1126/science.1325671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, X., Piacentino, V. III, Furukawa, S., Goldman, B., Margulies, K. B., and Houser, S. R. (2002). L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 91, 517–524. doi: 10.1161/01.RES.0000033988.13062.7C
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cho, U. S., and Xu, W. (2007). Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 445, 53–57. doi: 10.1038/nature05351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christ, T., Boknik, P., Wohrl, S., Wettwer, E., Graf, E. M., Bosch, R. F., et al. (2004). L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 110, 2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chu, G., and Kranias, E. G. (2002). Functional interplay between dual site phospholambam phosphorylation: insights from genetically altered mouse models. Basic Res. Cardiol. 97(Suppl. 1), I43–I48. doi: 10.1007/s003950200028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chung, H., Nairn, A. C., Murata, K., and Brautigan, D. L. (1999). Mutation of Tyr307 and Leu309 in the protein phosphatase 2A catalytic subunit favors association with the alpha 4 subunit which promotes dephosphorylation of elongation factor-2. Biochemistry 38, 10371–10376. doi: 10.1021/bi990902g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cohen, P. (1988). Protein phosphorylation and hormone action. Proc. R. Soc. Lond. B Biol. Sci. 234, 115–144. doi: 10.1098/rspb.1988.0040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cole, H. A., and Perry, S. V. (1975). The phosphorylation of troponin I from cardiac muscle. Biochem. J. 149, 525–533.
Davare, M. A., Horne, M. C., and Hell, J. W. (2000). Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J. Biol. Chem. 275, 39710–39717. doi: 10.1074/jbc.M005462200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DeGrande, S. T., Little, S. C., Nixon, D. J., Wright, P., Snyder, J., Dun, W., et al. (2013). Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J. Biol. Chem. 288, 1032–1046. doi: 10.1074/jbc.M112.426957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Depaoli-Roach, A. A., Park, I. K., Cerovsky, V., Csortos, C., Durbin, S. D., Kuntz, M. J., et al. (1994). Serine/threonine protein phosphatases in the control of cell function. Adv. Enzyme Regul. 34, 199–224. doi: 10.1016/0065-2571(94)90017-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doble, B. W., Ping, P., and Kardami, E. (2000). The epsilon subtype of protein kinase C is required for cardiomyocyte connexin-43 phosphorylation. Circ. Res. 86, 293–301. doi: 10.1161/01.RES.86.3.293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dobrowsky, R. T., Kamibayashi, C., Mumby, M. C., and Hannun, Y. A. (1993). Ceramide activates heterotrimeric protein phosphatase 2A. J. Biol. Chem. 268, 15523–15530.
Egom, E. E., Ke, Y., Musa, H., Mohamed, T. M., Wang, T., Cartwright, E., et al. (2010). FTY720 prevents ischemia/reperfusion injury-associated arrhythmias in an ex vivo rat heart model via activation of Pak1/Akt signaling. J. Mol. Cell. Cardiol. 48, 406–414. doi: 10.1016/j.yjmcc.2009.10.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
England, P. J., Stull, J. T., and Krebs, E. G. (1972). Dephosphorylation of the inhibitor component of troponin by phosphorylase phosphatase. J. Biol. Chem. 247, 5275–5277.
Gergs, U., Boknik, P., Buchwalow, I., Fabritz, L., Matus, M., Justus, I., et al. (2004). Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J. Biol. Chem. 279, 40827–40834. doi: 10.1074/jbc.M405770200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grinthal, A., Adamovic, I., Weiner, B., Karplus, M., and Kleckner, N. (2010). PR65, the HEAT-repeat scaffold of phosphatase PP2A, is an elastic connector that links force and catalysis. Proc. Natl. Acad. Sci. U.S.A. 107, 2467–2472. doi: 10.1073/pnas.0914073107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Groves, M. R., Hanlon, N., Turowski, P., Hemmings, B. A., and Barford, D. (1999). The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell 96, 99–110. doi: 10.1016/S0092-8674(00)80963-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gupta, R. C., Neumann, J., Boknik, P., and Watanabe, A. M. (1994). M2-specific muscarinic cholinergic receptor-mediated inhibition of cardiac regulatory protein phosphorylation. Am. J. Physiol. 266, H1138–H1144.
Gupta, R. C., Neumann, J., Durant, P., and Watanabe, A. M. (1993). A1-adenosine receptor-mediated inhibition of isoproterenol-stimulated protein phosphorylation in ventricular myocytes. Evidence against a cAMP-dependent effect. Circ. Res. 72, 65–74. doi: 10.1161/01.RES.72.1.65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hall, D. D., Feekes, J. A., Arachchige Don, A. S., Shi, M., Hamid, J., Chen, L., et al. (2006). Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry 45, 3448–3459. doi: 10.1021/bi051593z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hwang, J., and Pallas, D. C. (2014). STRIPAK complexes: structure, biological function, and involvement in human diseases. Int. J. Biochem. Cell Biol. 47, 118–148. doi: 10.1016/j.biocel.2013.11.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jackson, W. A., and Colyer, J. (1996). Translation of Ser16 and Thr17 phosphorylation of phospholamban into Ca2+-pump stimulation. Biochem. J. 316(Pt 1), 201–207.
Kappos, L., Antel, J., Comi, G., Montalban, X., O'Connor, P., Polman, C. H., et al. (2006). Oral fingolimod (FTY720) for relapsing multiple sclerosis. N. Engl. J. Med. 355, 1124–1140. doi: 10.1056/NEJMoa052643
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ke, Y., Lei, M., Collins, T. P., Rakovic, S., Mattick, P. A., Yamasaki, M., et al. (2007). Regulation of L-type calcium channel and delayed rectifier potassium channel activity by p21-activated kinase-1 in guinea pig sinoatrial node pacemaker cells. Circ. Res. 100, 1317–1327. doi: 10.1161/01.RES.0000266742.51389.a4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ke, Y., Lei, M., and Solaro, R. J. (2008). Regulation of cardiac excitation and contraction by p21 activated kinase-1. Prog. Biophys. Mol. Biol. 98, 238–250. doi: 10.1016/j.pbiomolbio.2009.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ke, Y., and Solaro, R. J. (2008). Use of a decoy peptide to purify p21 activated kinase-1 in cardiac muscle and identification of ceramide-related activation. Biologics 2, 903–909.
Ke, Y., Wang, L., Pyle, W. G., de Tombe, P. P., and Solaro, R. J. (2004). Intracellular localization and functional effects of P21-activated kinase-1 (Pak1) in cardiac myocytes. Circ. Res. 94, 194–200. doi: 10.1161/01.RES.0000111522.02730.56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kentish, J. C., McCloskey, D. T., Layland, J., Palmer, S., Leiden, J. M., Martin, A. F., et al. (2001). Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ. Res. 88, 1059–1065. doi: 10.1161/hh1001.091640
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kirchhefer, U., Brekle, C., Eskandar, J., Isensee, G., Kucerova, D., Mueller, F. U., et al. (2014a). Cardiac function is regulated by B56α-mediated targeting of PP2A to contractile relevant substrates. J. Biol. Chem. 289, 33862–33873. doi: 10.1074/jbc.M114.598938
Kirchhefer, U., Heinick, A., Konig, S., Kristensen, T., Muller, F. U., Seidl, M. D., et al. (2014b). Protein phosphatase 2A is regulated by protein kinase Calpha (PKCalpha)-dependent phosphorylation of its targeting subunit B56α at Ser41. J. Biol. Chem. 289, 163–176. doi: 10.1074/jbc.M113.507996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klein, G., Schroder, F., Vogler, D., Schaefer, A., Haverich, A., Schieffer, B., et al. (2003). Increased open probability of single cardiac L-type calcium channels in patients with chronic atrial fibrillation. role of phosphatase 2A. Cardiovasc. Res. 59, 37–45. doi: 10.1016/S0008-6363(03)00357-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kodama, I., Kondo, N., and Shibata, S. (1986). Electromechanical effects of okadaic acid isolated from black sponge in guinea-pig ventricular muscles. J. Physiol. (Lond). 378, 359–373. doi: 10.1113/jphysiol.1986.sp016224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kondo, N., Kodama, I., Kotake, H., and Shibata, S. (1990). Electrical effects of okadaic acid extracted from black sponge on rabbit sinus node. Br. J. Pharmacol. 101, 241–246. doi: 10.1111/j.1476-5381.1990.tb12694.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, M., Guo, H., and Damuni, Z. (1995). Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 34, 1988–1996. doi: 10.1021/bi00006a020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, L. F., Kao, L. S., and Westhead, E. W. (1994). Agents that promote protein phosphorylation inhibit the activity of the Na+/Ca2+ exchanger and prolong Ca2+ transients in bovine chromaffin cells. J. Neurochem. 63, 1941–1947. doi: 10.1046/j.1471-4159.1994.63051941.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, B., Ho, H. T., Velez-Cortes, F., Lou, Q., Valdivia, C. R., Knollmann, B. C., et al. (2014). Genetic ablation of ryanodine receptor 2 phosphorylation at Ser-2808 aggravates Ca(2+)-dependent cardiomyopathy by exacerbating diastolic Ca2+ release. J. Physiol. (Lond). 592, 1957–1973. doi: 10.1113/jphysiol.2013.264689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, J., Sirenko, S., Juhaszova, M., Ziman, B., Shetty, V., Rain, S., et al. (2011a). A full range of mouse sinoatrial node AP firing rates requires protein kinase A-dependent calcium signaling. J. Mol. Cell. Cardiol. 51, 730–739. doi: 10.1016/j.yjmcc.2011.07.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Q., and Hofmann, P. A. (2002). Antiadrenergic effects of adenosine A(1) receptor-mediated protein phosphatase 2a activation in the heart. Am. J. Physiol. Heart Circ. Physiol. 283, H1314–H1321.
Liu, Q., and Hofmann, P. A. (2003). Modulation of protein phosphatase 2a by adenosine A1 receptors in cardiomyocytes: role for p38 MAPK. Am. J. Physiol. Heart Circ. Physiol. 285, H97–H103.
Liu, W., Zi, M., Naumann, R., Ulm, S., Jin, J., Taglieri, D. M., et al. (2011b). Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart. Circulation 124, 2702–2715. doi: 10.1161/CIRCULATIONAHA.111.048785
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, W., Zi, M., Tsui, H., Chowdhury, S. K., Zeef, L., Meng, Q. J., et al. (2013). A novel immunomodulator, FTY-720 reverses existing cardiac hypertrophy and fibrosis from pressure overload by targeting NFAT (Nuclear Factor of Activated T-cells) signaling and periostin. Circ. Heart Fail. 6, 833–844. doi: 10.1161/CIRCHEARTFAILURE.112.000123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Longman, M. R., Ranieri, A., Avkiran, M., and Snabaitis, A. K. (2014). Regulation of PP2AC carboxylmethylation and cellular localisation by inhibitory class G-protein coupled receptors in cardiomyocytes. PLoS ONE 9:e86234. doi: 10.1371/journal.pone.0086234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, W., Grupp, I. L., Harrer, J., Ponniah, S., Grupp, G., Duffy, J. J., et al. (1994). Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ. Res. 75, 401–409. doi: 10.1161/01.RES.75.3.401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luss, H., Klein-Wiele, O., Boknik, P., Herzig, S., Knapp, J., Linck, B., et al. (2000). Regional expression of protein phosphatase type 1 and 2A catalytic subunit isoforms in the human heart. J. Mol. Cell. Cardiol. 32, 2349–2359. doi: 10.1006/jmcc.2000.1265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
MacDougall, L. K., Jones, L. R., and Cohen, P. (1991). Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur. J. Biochem. 196, 725–734. doi: 10.1111/j.1432-1033.1991.tb15871.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/S0092-8674(00)80847-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meng, X., Xiao, B., Cai, S., Huang, X., Li, F., Bolstad, J., et al. (2007). Three-dimensional localization of serine 2808, a phosphorylation site in cardiac ryanodine receptor. J. Biol. Chem. 282, 25929–25939. doi: 10.1074/jbc.M704474200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meurs, K. M., Mauceli, E., Lahmers, S., Acland, G. M., White, S. N., and Lindblad-Toh, K. (2010). Genome-wide association identifies a deletion in the 3′ untranslated region of striatin in a canine model of arrhythmogenic right ventricular cardiomyopathy. Hum. Genet. 128, 315–324. doi: 10.1007/s00439-010-0855-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peti, W., Nairn, A. C., and Page, R. (2013). Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 280, 596–611. doi: 10.1111/j.1742-4658.2012.08509.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reiken, S., Gaburjakova, M., Gaburjakova, J., He Kl, K. L., Prieto, A., Becker, E., et al. (2001). beta-adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation 104, 2843–2848. doi: 10.1161/hc4701.099578
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robertson, S. P., Johnson, J. D., Holroyde, M. J., Kranias, E. G., Potter, J. D., and Solaro, R. J. (1982). The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J. Biol. Chem. 257, 260–263.
Schulze, D. H., Muqhal, M., Lederer, W. J., and Ruknudin, A. M. (2003). Sodium/calcium exchanger (NCX1) macromolecular complex. J. Biol. Chem. 278, 28849–28855. doi: 10.1074/jbc.M300754200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shi, J., Gu, P., Zhu, Z., Liu, J., Chen, Z., Sun, X., et al. (2012). Protein phosphatase 2A effectively modulates basal L-type Ca(2+) current by dephosphorylating Ca(v)1.2 at serine 1866 in mouse cardiac myocytes. Biochem. Biophys. Res. Commun. 418, 792–798. doi: 10.1016/j.bbrc.2012.01.105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Solaro, R. J., Moir, A. J., and Perry, S. V. (1976). Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature 262, 615–617. doi: 10.1038/262615a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Srisakuldee, W., Jeyaraman, M. M., Nickel, B. E., Tanguy, S., Jiang, Z. S., and Kardami, E. (2009). Phosphorylation of connexin-43 at serine 262 promotes a cardiac injury-resistant state. Cardiovasc. Res. 83, 672–681. doi: 10.1093/cvr/cvp142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stull, J. T., Brostrom, C. O., and Krebs, E. G. (1972). Phosphorylation of the inhibitor component of troponin by phosphorylase kinase. J. Biol. Chem. 247, 5272–5274.
Terentyev, D., Belevych, A. E., Terentyeva, R., Martin, M. M., Malana, G. E., Kuhn, D. E., et al. (2009). miR-1 overexpression enhances Ca(2+) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56α and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ. Res. 104, 514–521. doi: 10.1161/CIRCRESAHA.108.181651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Terentyev, D., Viatchenko-Karpinski, S., Gyorke, I., Terentyeva, R., and Gyorke, S. (2003). Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J. Physiol. (Lond). 552, 109–118. doi: 10.1113/jphysiol.2003.046367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wei, S. K., Ruknudin, A., Hanlon, S. U., McCurley, J. M., Schulze, D. H., and Haigney, M. C. (2003). Protein kinase A hyperphosphorylation increases basal current but decreases beta-adrenergic responsiveness of the sarcolemmal Na+-Ca2+ exchanger in failing pig myocytes. Circ. Res. 92, 897–903. doi: 10.1161/01.RES.0000069701.19660.14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wei, S. K., Ruknudin, A. M., Shou, M., McCurley, J. M., Hanlon, S. U., Elgin, E., et al. (2007). Muscarinic modulation of the sodium-calcium exchanger in heart failure. Circulation 115, 1225–1233.
Wijnker, P. J., Boknik, P., Gergs, U., Muller, F. U., Neumann, J., dos Remedios, C., et al. (2011). Protein phosphatase 2A affects myofilament contractility in non-failing but not in failing human myocardium. J. Muscle Res. Cell Motil. 32, 221–233. doi: 10.1007/s10974-011-9261-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xiao, B., Jiang, M. T., Zhao, M., Yang, D., Sutherland, C., Lai, F. A., et al. (2005). Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ. Res. 96, 847–855. doi: 10.1161/01.RES.0000163276.26083.e8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xiao, B., Zhong, G., Obayashi, M., Yang, D., Chen, K., Walsh, M. P., et al. (2006). Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem. J. 396, 7–16. doi: 10.1042/BJ20060116
Yang, X., Wang, T., Lin, X., Yue, X., Wang, Q., Wang, G., et al. (2015). Genetic deletion of Rnd3/RhoE results in mouse heart calcium leakage through upregulation of protein kinase a signaling. Circ. Res. 116, e1–e10. doi: 10.1161/CIRCRESAHA.116.304940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, H., Makarewich, C. A., Kubo, H., Wang, W., Duran, J. M., Li, Y., et al. (2012). Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ. Res. 110, 831–840. doi: 10.1161/CIRCRESAHA.111.255158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y. H., and Hancox, J. C. (2009). Regulation of cardiac Na+-Ca2+ exchanger activity by protein kinase phosphorylation—still a paradox? Cell Calcium 45, 1–10. doi: 10.1016/j.ceca.2008.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, M. Q., Li, X., Tang, K., Sharma, N. M., Wyatt, T. A., Patel, K. P., et al. (2013). Pyruvate restores beta-adrenergic sensitivity of L-type Ca(2+) channels in failing rat heart: role of protein phosphatase. Am. J. Physiol. Heart Circ. Physiol. 304, H1352–H1360. doi: 10.1152/ajpheart.00873.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: calcium handling, ion channels, phosphatase, FTY720, arrhythmia, heart failure
Citation: Lei M, Wang X, Ke Y and Solaro RJ (2015) Regulation of Ca2+ transient by PP2A in normal and failing heart. Front. Physiol. 6:13. doi: 10.3389/fphys.2015.00013
Received: 24 November 2014; Accepted: 09 January 2015;
Published online: 29 January 2015.
Edited by:
Antonius Baartscheer, Academic Medical Center, NetherlandsReviewed by:
Ravi C. Balijepalli, University of Wisconsin, USAThomas Hund, Ohio State University, USA
Copyright © 2015 Lei, Wang, Ke and Solaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunbo Ke, Department of Physiology and Biophysics, Center for Cardiovascular Research, University of Illinois at Chicago, 835 S. Wolcott Ave., Chicago, IL 60612, USA e-mail: yke@uic.edu