
- Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA
The generation of lipid products catalyzed by PI3K is critical for normal T cell homeostasis and a productive immune response. PI3K can be activated in response to antigen receptor, co-stimulatory, cytokine, and chemokine signals. Moreover, dysregulation of this pathway frequently occurs in T cell lymphomas and is implicated in lymphoproliferative autoimmune disease. Akt acts as a central mediator of PI3K signals, downstream of which is the mTOR pathway, controlling cell growth and metabolism. Members of the Foxo family of transcription factors are also regulated by Akt, thus linking control over homing and migration of T cells, as well cell cycle entry, apoptosis, and DNA damage and oxidative stress responses, to PI3K signaling. PTEN, first identified as a tumor suppressor gene, encodes a lipid phosphatase that, by catalyzing the reverse of the PI3K “reaction,” directly opposes PI3K signaling. However, PTEN may have other functions as well, and recent reports have suggested roles for PTEN as a tumor suppressor independent of its effects on PI3K signaling. Through the use of models in which Pten is deleted specifically in T cells, it is becoming increasingly clear that control over autoimmunity and lymphomagenesis by PTEN involves multi-faceted functions of this molecule at multiple stages within the T cell compartment.
Introduction
Class 1A PI3Ks are directly linked to lymphocyte activation mainly through receptor tyrosine kinases, such as the antigen and cytokine receptors (Engelman et al., 2006; Huang and Sauer, 2010; So and Fruman, 2012). Class 1A PI3Ks, hereafter referred to as PI3K, are comprised of a catalytic subunit of 110 kDa (of which there are three isoforms) that generates phosphatidylinositol 3,4,5 phosphate (PIP3) from its main substrate phosphatidylinositol 4,5 phosphate (PIP2), and a regulatory subunit (of which there are five isoforms). Pten encodes a protein with a lipid phosphatase function that directly opposes PI3K signaling by dephosphorylating PIP3 at the 3′ position to generate PIP2. Cells lacking PTEN have elevated levels of PIP3 and constitutive activation of PI3K signaling pathways (Stambolic et al., 1998; Cantley and Neel, 1999). With increasing age, mice heterozygous for Pten develop T cell lymphomas and cancers in multiple tissues, and develop a lethal polyclonal autoimmune disorder, similar to that seen in Fas-deficient mice (Di Cristofano et al., 1998, 1999; Podsypanina et al., 1999; Suzuki et al., 2001). Germline mutations in PTEN occur in a group of autosomal dominant syndromes known as the PTEN hamartoma tumor syndromes, which include Cowden syndrome, Proteus syndrome, Proteus-like syndrome, and Bannayan–Riley–Ruvalcaba syndrome, demonstrating the importance of PTEN as a tumor suppressor. Consistent with this, genomic amplification and mutation of either PI3K or Akt has been reported in a large number of cancers as well (Samuels et al., 2004; Lee et al., 2005; Carpten et al., 2007; Jaiswal et al., 2009), although the role of PTEN as a tumor suppressor is now believed to involve more than its ability to oppose PI3K signaling. While disruption of regulation by PTEN has the overt phenotype of cancer progression, PTEN also plays an important role in maintaining T cell tolerance at multiple stages within the T cell compartment.
Activation and Regulation of PI3K Class 1A in T Lymphocytes
Upon ligation of the T cell receptor (TCR) in the presence of co-stimulatory molecules, PI3K recruitment and activation leads to the production of lipid products which in turn recruit downstream PH-domain containing targets such as PDK1 and Akt (see Figure 1). Ultimately, these events lead to activation of the mTOR pathway and inactivation of members of the Foxo family, inducing growth and proliferation of T cells and acquisition of effector function (for review, see Engelman et al., 2006; Finlay and Cantrell, 2010; So and Fruman, 2012). In the absence of PTEN, TCR stimulation alone results in hyperactivation of the PI3K pathway, resulting in effective cytokine production and proliferation independent of co-stimulation (Buckler et al., 2006). Thus, put another way, negative regulation of PI3K signaling by PTEN enforces the requirement for co-stimulation in naïve T cells. However, the notion that PI3K signaling is required for co-stimulation to mediate its effects has been challenged by the generation of mice which lack all isoforms of the regulatory subunit of class 1A PI3K in T cells (Deane et al., 2007). T cells from these mice are able to proliferate under co-stimulatory conditions in the absence of detectable Akt signaling. These mice also maintained a normal anti-viral response upon MHV infection, although in vivo T helper function to B cell antibody responses was impaired. Whether the relative lack of defects is due to the fact that lack of PI3K was genetic, rather than acquired, is not known. However, these studies indicate that potential therapies targeting PI3K for inflammatory diseases and cancer may not compromise all aspects of cellular immunity, and further suggest that PI3K signaling has specialized functions in the context of T cell activation.
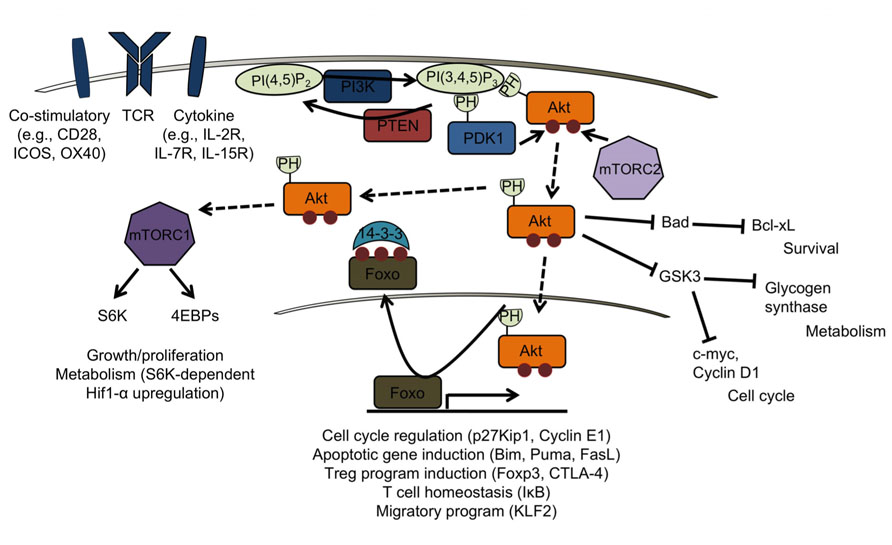
FIGURE 1. PTEN/PI3K signaling in mature T cells. Class 1A PI3Ks are activated by receptor tyrosine kinases that drive co-stimulatory, T cell receptor (TCR), and cytokine signaling pathways. PTEN directly opposes PI3K signaling by converting PI(3,4,5)P3 to PI(4,5)P2. Through their PH-domains, PDK1 and Akt bind PIP3, enabling Akt to be phosphorylated by PDK1 on Ser308. Full activation of Akt requires phosphorylation on Ser473 by mTORC2. Akt can then target downstream substrates such as Foxos in the nucleus, leading to their inactivation and export from the nucleus. This results in attenuation of a Foxo-dependent gene program normally acting to promote pathways involved in, but not limited to, T cell homeostasis (in resting T cells), apoptosis (upon, for example, cytokine withdrawal), Treg induction, and homing to secondary lymphoid organs (in circulating, naïve T cells). Akt also targets TSC2 and Pras40 (not shown), negative regulators of mTORC1, allowing for activation of mTORC1. Activation of S6K and 4EBPs by mTORC1 results in increased protein translation, allowing growth and proliferation, and upregulation of Hif-1α, important for glycolytic metabolism. Inhibitory targeting of Bad and GSK3 also contribute to Akt’s effect on cell cycle, survival and metabolism.
PI3K activation is critical for optimal responses of T cells to cytokines which utilize the common gamma chain. For example, engagement of the IL-2R on activated T cells results in activation of the Jak/Stat pathway, as well as the PI3K and MAPK pathways, and the combination of these signals is required for the observed proliferation and cell survival in response to IL-2. Thus, appropriate responses to TCR ligation and cytokines require downregulation of PTEN, which is constitutively expressed in naïve T cells. This normally occurs as a consequence of TCR stimulation itself, which terminates detectable PTEN expression within 24–48 h. The importance of this is demonstrated by two findings. First, retroviral mediated “enforced” expression of PTEN renders activated IL-2R+ T cells unable to fully respond to IL-2 stimulation. Second, regulatory T cells, which normally do not divide in response to IL-2 alone, have complete responses and signaling to IL-2 restored solely by genetic ablation of Pten (Bensinger et al., 2004; Walsh et al., 2006; Locke et al., 2009). This demonstrates that control of PI3K through PTEN plays an important role not only in modulating the degree of activation signals within lymphocytes, but in maintaining traits of a specific lineage.
PI3K-related signals can modulate ongoing responses as well. Deletion of Pten, or expression of constitutively active myristoylated-Akt, within the CD8+ T cell compartment inhibits the development and survival of memory CD8+ T cells (Hand et al., 2010). On the contrary, Akt is required for a transcriptional program leading to upregulation of cytolytic effector, chemokine, and adhesion molecules (Macintyre et al., 2011). Conditional deletion of Pten using OX40-Cre has demonstrated that PTEN plays a critical role in limiting the expansion of Tfh cells and in maintaining control over GC reactions, indicating that T cell intrinsic roles of PTEN are crucial for maintaining global levels of tolerance (Rolf et al., 2010). Lastly, the promotion of iTreg generation and maintenance by PD-L1 is associated with downregulation of Akt signaling and concomitant upregulation of PTEN expression (Francisco et al., 2009). These examples all provide evidence that the balance of Akt signaling within multiple stages of development and differentiation states helps determine cellular fate.
PTEN also has been shown to play an important role in central tolerance and in regulating proliferation of developing cells in the thymus (Suzuki et al., 2001; Hagenbeek et al., 2004). In the absence of IL-7R and pre-TCR signaling, loss of Pten allows cells to bypass the β-selection checkpoint, indicating that PI3K signaling is crucial for the ongoing development of early T cell precursors. Interestingly, thymic cellularity and subset percentages are relatively unperturbed prior to tumor development, which likely occurs at the DP stage during thymic maturation, suggesting that secondary events emerge in this context of Pten loss to promote transformation (discussed below; Hagenbeek et al., 2004; Hagenbeek and Spits, 2008; Xue et al., 2008; Guo et al., 2011).
Signaling Downstream of Pten-PI3K-Akt: A Critical Role for Foxos
Members of the Foxo family of transcription factors play a critical role in DNA damage and oxidative stress responses (Tran et al., 2002; Miyamoto et al., 2007; Tothova et al., 2007; Choi et al., 2009) and in preventing entry into cell cycle (Medema et al., 2000), acting as bona fide tumor suppressors as demonstrated in hematopoietic and epithelial tissues (Coffer, 2003; Accili and Arden, 2004; Paik et al., 2007). It is becoming increasingly appreciated that Foxos play important roles within the T cell compartment as well, regulating homing (Fabre et al., 2008; Sinclair et al., 2008; Finlay et al., 2009; Kerdiles et al., 2009; Finlay and Cantrell, 2010), survival (Hedrick, 2009; Kerdiles et al., 2009; Dejean et al., 2011), and the development and function of effector and memory subsets (Kerdiles et al., 2010; Ouyang et al., 2010; Rao et al., 2012). In quiescent cells, Foxos are active within the nucleus, where they maintain the pattern of chemokine and adhesion molecule expression, and expression of the IL-7R, needed for the migration and survival of circulating T cells (Fabre et al., 2008; Finlay et al., 2009; Kerdiles et al., 2010). Akt phosphorylation on Ser473, mediated by mTORC2, is critical for Foxo inactivation, implicating mTOR in control over Foxos, and placing PTEN upstream in this pathway. Importantly, mTORC2 has been shown to be critical for the phenotype in models of PTEN loss. Prostate cancer caused by conditional Pten deletion in prostate epithelium requires mTORC2, and deletion of one copy of Rictor, required for mTORC2 complex assembly and activity, was sufficient to protect Pten heterozygous mice from prostate cancer (Guertin et al., 2009).
The role of Foxos in T cells was first described using mice in which retroviral gene-trap targeting of embryonic stem cells was used to generate a null Foxo3 allele, although the authors could not rule out the possibility that undetectable levels of a truncated form of Foxo3 was produced (Lin et al., 2004). These mice exhibited spontaneous lymphoproliferation and multi-organ lymphocyte infiltration, indicating that Foxo3 is important for the control of T cell tolerance and homeostasis. This hyper-activated phenotype was correlated with decreased expression of IκBε and IκBβ, leading to increased NF-κB activation. As IKKβ has been shown to phosphorylate and inactivate Foxos (Hu et al., 2004), this implicates Foxos in a critical negative feedback loop that may serve to limit inflammatory responses in certain settings, and is potentially important in the context of tumors which maintain low levels of Akt activation. Other studies using Foxo3-deficient strains did not find the same immunological defects (Hosaka et al., 2004; Dejean et al., 2009), attesting first to the redundancy of members of this family and, second, to the possibility of dominant-negative effects of undetected Foxo3 gene products in the aforementioned mice. T cell-specific loss of Foxo1 was shown to severely disrupt T cell homeostasis, resulting in multi-organ lymphocyte infiltration as well as exocrine pancreatitis and hind limb paralysis (Kerdiles et al., 2010). This was attributed to a defect in both the development and function of Foxp3+ regulatory T cells, and this phenotype was exacerbated by combined deletion of Foxo1 and Foxo3 (Kerdiles et al., 2010; Ouyang et al., 2010). More recently, Foxo3a has been mechanistically linked to anergy induction in T cells through upregulation of Sirt1 (Gao et al., 2012). IL-2 was shown to reverse T cell anergy through the PI3K pathway by inactivation of Foxos, which prevented transcriptional upregulation of Sirt1.
In mouse embryonic fibroblasts and lymphoid cells, growth factor and cytokine withdrawal upregulates BH3-only pro-apoptotic mediators Puma and Bim in a Foxo3a-dependent manner that requires downregulation of PI3K/Akt signaling (You et al., 2006). These findings strongly support the idea that impaired downregulation of PI3K activity, as seen in T cells lacking PTEN, results in survival effects and apoptotic resistance in response to cytokine deprivation through inactivation of Foxo family members. FasL has been shown to be another important target of Foxo (Brunet et al., 1999). Given that Pten heterozygous mice display decreased sensitivity to Fas-mediated AICD, similar to lpr and gld mice (Van Parijs and Abbas, 1996), constitutive inactivation of Foxos due to loss of PTEN is likely a partial mechanism underlying the phenotype observed in these mice. Additionally, the finding that a deficiency in Foxos is sufficient to drive the development of hemangiomas, similar to what is seen in patients with Cowden disease and Bannayan–Zonana syndrome, and thymic lymphomas, similar to mice with a T cell-specific deletion of Pten, as well suggests that inactivation of Foxos may be critical for the phenotype brought on by loss of Pten (Paik et al., 2007).
Lymphoma and Dysregulated Immune Function in the Absence of PTEN
Genetic studies in mice expressing conditional alleles of Pten have been crucial for studying the tissue specific role of PTEN in tumorigenesis. Deletion of Pten during hematopoiesis using Mx-1-Cre results in myeloproliferative disease and transplantable leukemia, and has shown that PTEN is required for maintaining the hematopoietic stem cell (HSC) compartment (Yilmaz et al., 2006; Lee et al., 2010). Similar to the Mx-1-Cre model of PTEN loss, Vec-Cre-mediated PTEN loss, in which nearly 40% of fetal liver HSCs were subject to deletion of Pten, led to impaired HSC self-renewal and the development of a myeloproliferative disorder followed by leukemia (Guo et al., 2008). It is notable that this phenotype shares striking similarity to mice in which all six alleles of Foxo1/3a/4 were deleted by Mx-1-Cre (Tothova et al., 2007), and to Foxo3a-/- mice (Miyamoto et al., 2007). Importantly, this study reported the same t(14;15) chromosomal translocation (seen within a subset of human T-ALL), in all blast-crisis samples analyzed from these mice, suggesting a critical genomic destabilizing event potentially independent of PTEN’s role in controlling PI3K/Akt signaling. Similarly, this translocation, involving the c-myc and TCRα/δ loci and resulting in constitutively high levels of c-myc, was found to recur with 100% incidence in T cell lymphomas from mice in which Pten was deleted specifically in T cells (CD4-Cre × Ptenfl/fl mice, hereafter referred to as PTEN-ΔT mice; Liu et al., 2010). Thus, well after T lineage commitment in mice subject to lymphoma and lymphoproliferative autoimmune disease, PTEN is critical to prevent the emergence of the same genetic abnormality present in HSCs that correlates with the selective pressure of these cells for the development of leukemia.
A deficiency in Rag1 or TCRα prevented the t(14;15) translocation event in PTEN-ΔT mice, but did not inhibit malignant transformation, although lymphomagenesis developed with delayed onset and was primarily restricted to the thymus (Liu et al., 2010). Interestingly, mature T cells from 3-week-old PTEN-ΔT mice that had not undergone transformation, when transferred into either immunocompetent or immunoincompetent recipients, did not develop a malignant phenotype throughout the duration of the host’s life, suggesting that malignancy arises within the thymus after a period of latency in these mice, consistent with an earlier report demonstrating that transplanted Pten-/- thymocytes gave rise to T cell lymphomas in immunodeficient recipients (Hagenbeek and Spits, 2008). Supporting this, PTEN-ΔT mice thymectomized at 3 weeks of age did not develop lymphoma, although later in life these mice exhibited signs of systemic autoimmunity. Thus, within distinct developmental stages, PTEN is required for the prevention of lymphoma and autoimmunity.
In the context of DNA damage, loss of PTEN enables cells to bypass the normal G2/M checkpoint enforced by CHK1 as a result of Akt-mediated CHK1 sequestration in the cytoplasm (Puc et al., 2005). Akt has also been shown to promote prosurvival responses following DNA double-strand breaks (DSBs; Bozulic et al., 2008). It is possible that secondary mutations or other genetic alterations occurred in Rag1-/- or TCRα-/- × PTEN-ΔT mice as a result of genetic instability due to PTEN loss, and that hyper-active Akt maintains the survival of these cells that would normally undergo apoptosis. Spectral karyotyping analyses, however, failed to detect any chromosomal translocations in these mice to support this idea. Induction of the Notch pathway in malignant cells from these mice, however, was shown to increase cellular levels of c-myc, suggesting that PI3K and c-myc cooperate in multiple models of PTEN loss to promote lymphomagenesis and lymphoproliferative autoimmune disorder.
Disruption of Foxo function has been shown to accelerate c-myc-driven lymphomagenesis (Bouchard et al., 2007), and constitutively active mutants of Foxo3a resistant to inactivation by Akt directly repress multiple target genes of c-myc and block c-myc-dependent proliferation and transformation (Bouchard et al., 2004; Jensen et al., 2011). Together, this offers the possibility that c-myc and PI3K cooperate in tumorigenesis through inactivation of Foxos. Whether or not restoration of Foxo signaling in mice lacking PTEN in T cells has an effect on metabolism, survival, infiltrative capacity, lymphomagenesis, or the prevention of autoimmunity remains to be seen.
A recent study in which Ptenfl/fl × CD4-Cre mice were crossed to PDK1fl/fl mice has shown that while lymphoma did not develop in this model of T cell-specific Pten deletion, PDK1/Akt signaling was dispensable for the survival, proliferation and differentiation of T cell progenitors, and in vivo expansion of peripheral T cells (Finlay et al., 2009). The mechanism for control of cell metabolism independent of PDK1 is unclear in these mice, although thymocyte proliferation in the absence of PTEN was shown to require RhoA-dependent pathways, and a dependence on c-myc, which was recently shown to play a pivotal role in T cell metabolism following activation (Wang et al., 2011), was not ruled out. Mice which express a single hypomorphic PDK1 allele similarly prevented a wide range of tumors when crossed to Pten heterozygous mice, indicating that the requirement for PDK1 in the context of tumor formation brought on by loss of PTEN is not limited to its role in controlling migratory capacity of T cells (Bayascas et al., 2005). Additionally, deletion of S6K1 in the Mx-1-Cre model of Pten loss resulted in delayed development of leukemia, indicating that an mTORC1-mediated pathway, in this context shown to involve induction of a Hif-1α-dependent glycolytic program, contributes to leukemogenesis in Pten-deficient cells (Tandon et al., 2011).
Akt-Independent Roles of PTEN
PTEN nuclear function, independent of its lipid phosphatase activity, has been shown to contribute to its tumor-suppressive effects and, in particular, in maintaining genomic stability. Control of DNA stability and DSB repair has in part been attributed to interaction of PTEN with an integral kinetochore protein, CENP-C. Mutants originally identified in Cowden disease patients demonstrated that the C-terminus of PTEN, but not the phosphatase domain, was required for interaction with centromeres and with CENP-C, and that DNA instability and DSBs could be prevented by a phosphatase-dead PTEN mutant which retained its ability to bind to CENP-C (Shen et al., 2007). Additionally, nuclear exclusion of PTEN, but not a phosphatase-inactive mutant, was shown to impair the tumor-suppressive APC-CDH1 nuclear complex, again demonstrating tumor-suppressive capability of catalytically inactive PTEN (Song et al., 2011). Additional protein–protein interactions within the nucleus, independent of PTEN catalytic activity, have been shown to increase the activity and stability of p53 (Li et al., 2006; Salmena et al., 2008), and upregulate expression of Rad51, an essential component of the DSB repair machinery (Baker, 2007; Shen et al., 2007). A lysine to glutamate mutation in PTEN (K289E) identified in a Cowden syndrome family, which lead to a dramatic decrease in nuclear import without disrupting phosphatase activity or membrane localization of PTEN, as well strongly supports the idea that nuclear PTEN can be critical for tumor suppression independent of its role in opposing PI3K signaling in certain contexts (Trotman et al., 2007 See Figure 2).
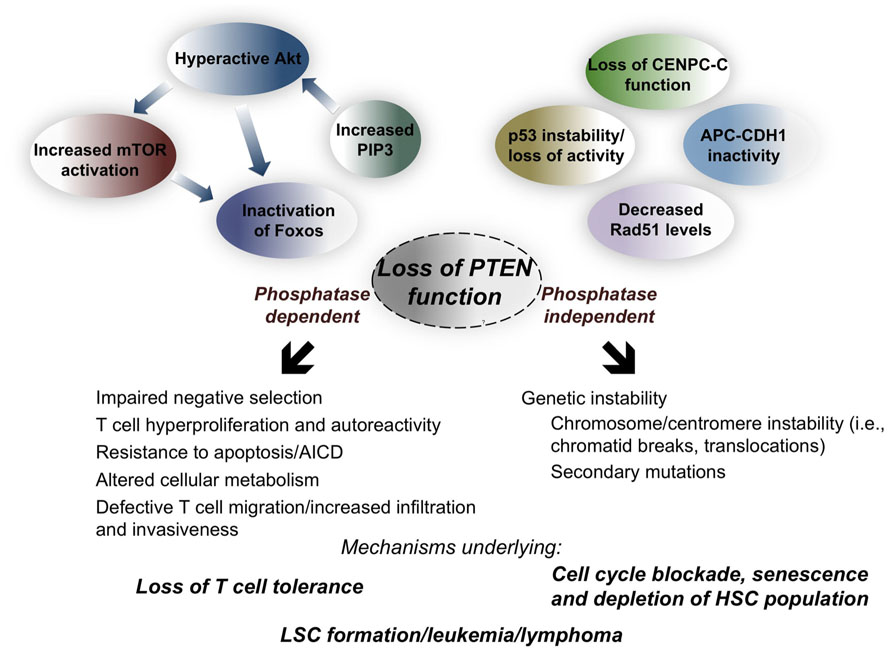
FIGURE 2. Molecular and phenotypic consequences of loss of PTEN phosphatase-dependent and -independent functions during hematopoiesis and within the T cell compartment. Loss of PTEN phosphatase activity results in increased levels of PIP3, causing hyperactivation of the Akt pathway and subsequent activation of the mTOR pathway, and inactivation of the Foxo family of transcription factors. Disruption of PTEN phosphatase-independent function accounts for loss of much of its nuclear activity, compromising genetic stability and DSB repair, leading to chromatid breaks and translocations. That mature T cells do not undergo transformation, and that lymphoma in T cell-specific PTEN knockout mice is of thymic origin, suggests that PTEN phosphatase-independent function contributes less to the phenotype at the mature T cell stage, and that loss of T cell tolerance can likely be attributed to loss of PTEN phosphatase activity. Whether depletion of hematopoietic stem cells (HSCs) and development of leukemia stem cells (LSCs), as well lymphomagenesis, depends on the absence of PTEN phosphatase activity (which likely maintains survival of genetically aberrant cells), PTEN phosphatase-independent activity (promoting genetic instability), or both, remains an open question.
Inactivation of PTEN phosphatase activity is sufficient to abrogate its tumor-suppressive effects, emphasizing the importance of PTEN’s role in downregulation of PI3K signaling. This has been demonstrated by a subset of Cowden disease patients that harbor a missense mutation at a cysteine residue (C124) critical for phosphatase activity. A C124S mutant has been shown to form a stable complex with PIP3, potentially protecting it from dephosphorylation by other lipid phosphatases to account for higher cellular levels of PIP3 compared to conditions in which PTEN is completely absent (Myers et al., 1998). As well, the importance of Akt activity in tumor development induced by loss of PTEN has been reported in a number of studies (Stiles et al., 2002; Bayascas et al., 2005; Chen et al., 2006). Constitutive Akt activation in T cells and thymocytes has been shown to be sufficient to drive autoimmunity and lymphoma (Rathmell et al., 2003) and thymomas (Malstrom et al., 2001), respectively, and bone marrow chimera experiments have demonstrated that enforced expression of constitutively active Akt in HSCs was sufficient for the development of myeloproliferative disease, T cell lymphoma, or AML (Kharas et al., 2010).
PTEN-ΔT mice crossed onto the Rag1-/- background, which do not harbor the t(14;15) translocation, potentially serve as an example in which Akt signaling is not sufficient to drive cancer progression (Liu et al., 2010). In vitro studies using malignant cells from these mice demonstrated a dependence on Notch, suggesting that Akt at the very least must cooperate with other oncogenic pathways to allow tumor growth in this model, similar to what has been suggested in an MMTV-myrAkt breast cancer model with respect to tumorigenesis induced by loss of PTEN (Blanco-Aparicio et al., 2007). In contrast to this, in a model of Notch-induced tumorigenesis, genetic loss of Pten induced an oncogene addiction switch that rendered T-ALL cells resistant to Notch inhibition through GSIs and dependent on PI3K/Akt signaling to maintain tumor growth (Palomero et al., 2007). Thus, the reliance on Akt signaling differs among models of PTEN loss. This idea is perhaps emphasized by the observation that PTEN-ΔT mice, regardless of whether the t(14;15) translocation event occurs, maintain constitutively high levels of c-myc.
The reliance on c-myc in the context of PTEN loss remains an important question. In tumors driven by inducible c-myc activation in a zebrafish model of T-ALL, constitutive activation of the Akt pathway through genetic disruption of Pten or transgenichexpression of Akt2 rendered cells in this model independent of c-myc for tumor progression (Gutierrez et al., 2011). Conversely, conditional genetic disruption of both Pten and c-myc in mice, while still subject to myeloproliferative and lymphoproliferative disorders, prevented the development of hematopoietic malignancies, highlighting an important dependence on c-myc in this context of PTEN loss (Zhang et al., 2011). Whether loss of PTEN allows the survival of cells that, through other mechanisms, have acquired genetic aberrations, such as translocations resulting in constitutive c-myc, or whether PTEN, independent of its role in PI3K downregulation, prevents genetic instability that collaborates with dysregulation of PI3K to allow cancer progression, remains an open question that needs to be addressed in a context-dependent manner, with consideration of what function of PTEN is lost, changes in levels of expression, and in which tissues abnormal growth originates.
Future Directions
Pten is the one of the most frequently mutated or lost genes in human cancer. Given the emerging roles of PI3K in immune system regulation, it is possible that PTEN is also playing a prominent role in the prevention of autoimmune disease and inflammatory/lymphoproliferative syndromes. The finding that PTEN did not play a role in lymphomagenesis in mature T cells from mice which lack PTEN specifically in T cells suggested that PTEN has a specific function in preventing lymphoma within a distinct time frame in this compartment. That these non-malignant T cells were able to provoke autoimmunity demonstrates that well beyond the stage at which Pten is required for prevention of lymphomagenesis in this model, PTEN plays a critical role in the maintenance of T cell tolerance. It will be important to determine which biochemical requirements PTEN fulfills in each stage of protection, and whether these requirements are distinct within different stages from cells of the same lineage, or whether lack of particular features leads to different outcomes depending on developmental stage. Given the importance of PI3K signaling in both normal immunity and the development of cancer, it will be interesting to see how closely PTEN’s role as a lipid phosphatase is tied to prevention of disease, and how integral PI3K signaling remains throughout disease progression. Uncovering the specific biochemical functions of PTEN within these contexts will be key to the development of targeted therapies for the prevention and treatment of T cell malignancies and autoimmune disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Accili, D., and Arden, K. C. (2004). FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 117, 421–426.
Bayascas, J. R., Leslie, N. R., Parsons, R., Fleming, S., and Alessi, D. R. (2005). Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN(+/-) mice. Curr. Biol. 15, 1839–1846.
Bensinger, S. J., Walsh, P. T., Zhang, J., Carroll, M., Parsons, R., Rathmell, J. C., Thompson, C. B., Burchill, M. A., Farrar, M. A., and Turka, L. A. (2004). Distinct IL-2 receptor signaling pattern in CD4+CD25+ regulatory T cells. J. Immunol. 172, 5287–5296.
Blanco-Aparicio, C., Renner, O., Leal, J. F., and Carnero, A. (2007). PTEN, more than the AKT pathway. Carcinogenesis 28, 1379–1386.
Bouchard, C., Lee, S., Paulus-Hock, V., Loddenkemper, C., Eilers, M., and Schmitt, C. A. (2007). FoxO transcription factors suppress Myc-driven lymphomagenesis via direct activation of Arf. Genes Dev. 21, 2775–2787.
Bouchard, C., Marquardt, J., Bras, A., Medema, R. H., and Eilers, M. (2004). Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. 23, 2830–2840.
Bozulic, L., Surucu, B., Hynx, D., and Hemmings, B. A. (2008). PKBalpha/Akt1 acts downstream of DNA-PKh in the DNA double-strand break response and promotes survival. Mol. Cell 30, 203–213.
Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., Anderson, M. J., Arden, K. C., Blenis, J., and Greenberg, M. E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868.
Buckler, J. L., Walsh, P. T., Porrett, P. M., Choi, Y., and Turka, L. A. (2006). Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J. Immunol. 177, 4262–4266.
Cantley, L. C., and Neel, B. G. (1999). New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 4240–4245.
Carpten, J. D., Faber, A. L., Horn, C., Donoho, G. P., Briggs, S. L., Robbins, C. M., Hostetter, G., Boguslawski, S., Moses, T. Y., Savage, S., Uhlik, M., Lin, A., Du, J., Qian, Y. W., Zeckner, D. J., Tucker-Kellogg, G., Touchman, J., Patel, K., Mousses, S., Bittner, M., Schevitz, R., Lai, M. H., Blanchard, K. L., and Thomas, J. E. (2007). A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444.
Chen, M. L., Xu, P. Z., Peng, X. D., Chen, W. S., Guzman, G., Yang, X., Di Cristofano, A., Pandolfi, P. P., and Hay, N. (2006). The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/- mice. Genes Dev. 20, 1569–1574.
Choi, J., Oh, S., Lee, D., Oh, H. J., Park, J. Y., Lee, S. B., and Lim, D. S. (2009). Mst1-FoxO signaling protects Naive T lymphocytes from cellular oxidative stress in mice. PLoS ONE 4, e8011. doi: 10.1371/journal.pone.0008011
Coffer, P. J. (2003). Transcriptional regulation of lymphocyte quiescence: as cunning as a FOX. Trends Immunol. 24, 470–471; author reply 471.
Deane, J. A., Kharas, M. G., Oak, J. S., Stiles, L. N., Luo, J., Moore, T. I., Ji, H., Rommel, C., Cantley, L. C., Lane, T. E., and Fruman, D. A. (2007). T-cell function is partially maintained in the absence of class IA phosphoinositide 3-kinase signaling. Blood 109, 2894–2902.
Dejean, A. S., Beisner, D. R., Ch’en, I. L., Kerdiles, Y. M., Babour, A., Arden, K. C., Castrillon, D. H., DePinho, R. A., and Hedrick, S. M. (2009). Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nat. Immunol. 10, 504–513.
Dejean, A. S., Hedrick, S. M., and Kerdiles, Y. M. (2011). Highly specialized role of Forkhead box O transcription factors in the immune system. Antioxid. Redox Signal. 14, 663–674.
Di Cristofano, A., Kotsi, P., Peng, Y. F., Cordon-Cardo, C., Elkon, K. B., and Pandolfi, P. P. (1999). Impaired Fas response and autoimmunity in Pten+/- mice. Science 285, 2122–2125.
Di Cristofano, A., Pesce, B., Cordon-Cardo, C., and Pandolfi, P. P. (1998). Pten is essential for embryonic development and tumour suppression. Nat. Genet. 19, 348–355.
Engelman, J. A., Luo, J., and Cantley, L. C. (2006). The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7, 606–619.
Fabre, S., Carrette, F., Chen, J., Lang, V., Semichon, M., Denoyelle, C., Lazar, V., Cagnard, N., Dubart-Kupperschmitt, A., Mangeney, M., Fruman, D. A., and Bismuth, G. (2008). FOXO1 regulates L-Selectin and a network of human T cell homing molecules downstream of phosphatidylinositol 3-kinase. J. Immunol. 181, 2980–2989.
Finlay, D., and Cantrell, D. (2010). Phosphoinositide 3-kinase and the mammalian target of rapamycin pathways control T cell migration. Ann. N. Y. Acad. Sci. 1183, 149–157.
Finlay, D. K., Sinclair, L. V., Feijoo, C., Waugh, C. M., Hagenbeek, T. J., Spits, H., and Cantrell, D. A. (2009). Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. J. Exp. Med. 206, 2441–2454.
Francisco, L. M., Salinas, V. H., Brown, K. E., Vanguri, V. K., Freeman, G. J., Kuchroo, V. K., and Sharpe, A. H. (2009). PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029.
Gao, B., Kong, Q., Kemp, K., Zhao, Y. S., and Fang, D. (2012). Analysis of sirtuin 1 expression reveals a molecular explanation of IL-2-mediated reversal of T-cell tolerance. Proc. Natl. Acad. Sci. U.S.A. 109, 899–904.
Guertin, D. A., Stevens, D. M., Saitoh, M., Kinkel, S., Crosby, K., Sheen, J. H., Mullholland, D. J., Magnuson, M. A., Wu, H., and Sabatini, D. M. (2009). mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15, 148–159.
Guo, W., Lasky, J. L., Chang, C. J., Mosessian, S., Lewis, X., Xiao, Y., Yeh, J. E., Chen, J. Y., Iruela-Arispe, M. L., Varella-Garcia, M., and Wu, H. (2008). Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature 453, 529–533.
Guo, W., Schubbert, S., Chen, J. Y., Valamehr, B., Mosessian, S., Shi, H., Dang, N. H., Garcia, C., Theodoro, M. F., Varella-Garcia, M., and Wu, H. (2011). Suppression of leukemia development caused by PTEN loss. Proc. Natl. Acad. Sci. U.S.A. 108, 1409–1414.
Gutierrez, A., Grebliunaite, R., Feng, H., Kozakewich, E., Zhu, S., Guo, F., Payne, E., Mansour, M., Dahlberg, S. E., Neuberg, D. S., den Hertog, J., Prochownik, E. V., Testa, J. R., Harris, M., Kanki, J. P., and Look, A. T. (2011). Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J. Exp. Med. 208, 1595–1603.
Hagenbeek, T. J., Naspetti, M., Malergue, F., Garcon, F., Nunes, J. A., Cleutjens, K. B., Trapman, J., Krimpenfort, P., and Spits, H. (2004). The loss of PTEN allows TCR alphabeta lineage thymocytes to bypass IL-7 and Pre-TCR-mediated signaling. J. Exp. Med. 200, 883–894.
Hagenbeek, T. J., and Spits, H. (2008). T-cell lymphomas in T-cell-specific Pten-deficient mice originate in the thymus. Leukemia 22, 608–619.
Hand, T. W., Cui, W., Jung, Y. W., Sefik, E., Joshi, N. S., Chandele, A., Liu, Y., and Kaech, S. M. (2010). Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc. Natl. Acad. Sci. U.S.A. 107, 16601–16606.
Hedrick, S. M. (2009). The cunning little vixen: Foxo and the cycle of life and death. Nat. Immunol. 10, 1057–1063.
Hosaka, T., Biggs, W. H. III, Tieu, D., Boyer, A. D., Varki, N. M., Cavenee, W. K., and Arden, K. C. (2004). Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc. Natl. Acad. Sci. U.S.A. 101, 2975–2980.
Hu, M. C., Lee, D. F., Xia, W., Golfman, L. S., Ou-Yang, F., Yang, J. Y., Zou, Y., Bao, S., Hanada, N., Saso, H., Kobayashi, R., and Hung, M. C. (2004). IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 117, 225–237.
Huang, Y. H., and Sauer, K. (2010). Lipid signaling in T-cell development and function. Cold Spring Harb. Perspect. Biol. 2, a002428.
Jaiswal, B. S., Janakiraman, V., Kljavin, N. M., Chaudhuri, S., Stern, H. M., Wang, W., Kan, Z., Dbouk, H. A., Peters, B. A., Waring, P., Dela Vega, T., Kenski, D. M., Bowman, K. K., Lorenzo, M., Li, H., Wu, J., Modrusan, Z., Stinson, J., Eby, M., Yue, P., Kaminker, J. S., de Sauvage, F. J., Backer, J. M., and Seshagiri, S. (2009). Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell 16, 463–474.
Jensen, K. S., Binderup, T., Jensen, K. T., Therkelsen, I., Borup, R., Nilsson, E., Multhaupt, H., Bouchard, C., Quistorff, B., Kjaer, A., Landberg, G., and Staller, P. (2011). FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 30, 4554–4570.
Kerdiles, Y. M., Beisner, D. R., Tinoco, R., Dejean, A. S., Castrillon, D. H., DePinho, R. A., and Hedrick, S. M. (2009). Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat. Immunol. 10, 176–184.
Kerdiles, Y. M., Stone, E. L., Beisner, D. R., McGargill, M. A., Ch’en, I. L., Stockmann, C., Katayama, C. D., and Hedrick, S. M. (2010). Foxo transcription factors control regulatory T cell development and function. Immunity 33, 890–904.
Kharas, M. G., Okabe, R., Ganis, J. J., Gozo, M., Khandan, T., Paktinat, M., Gilliland, D. G., and Gritsman, K. (2010). Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 115, 1406–1415.
Lee, J. W., Soung, Y. H., Kim, S. Y., Lee, H. W., Park, W. S., Nam, S. W., Kim, S. H., Lee, J. Y., Yoo, N. J., and Lee, S. H. (2005). PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene 24, 1477–1480.
Lee, J. Y., Nakada, D., Yilmaz, O. H., Tothova, Z., Joseph, N. M., Lim, M. S., Gilliland, D. G., and Morrison, S. J. (2010). mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 7, 593–605.
Li, A. G., Piluso, L. G., Cai, X., Wei, G., Sellers, W. R., and Liu, X. (2006). Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol. Cell 23, 575–587.
Lin, L., Hron, J. D., and Peng, S. L. (2004). Regulation of NF-kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity 21, 203–213.
Liu, X., Karnell, J. L., Yin, B., Zhang, R., Zhang, J., Li, P., Choi, Y., Maltzman, J. S., Pear, W. S., Bassing, C. H., and Turka, L. A. (2010). Distinct roles for PTEN in prevention of T cell lymphoma and autoimmunity in mice. J. Clin. Invest. 120, 2497–2507.
Locke, N. R., Patterson, S. J., Hamilton, M. J., Sly, L. M., Krystal, G., and Levings, M. K. (2009). SHIP regulates the reciprocal development of T regulatory and Th17 cells. J. Immunol. 183, 975–983.
Macintyre, A. N., Finlay, D., Preston, G., Sinclair, L. V., Waugh, C. M., Tamas, P., Feijoo, C., Okkenhaug, K., and Cantrell, D. A. (2011). Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity 34, 224–236.
Malstrom, S., Tili, E., Kappes, D., Ceci, J. D., and Tsichlis, P. N. (2001). Tumor induction by an Lck-MyrAkt transgene is delayed by mechanisms controlling the size of the thymus. Proc. Natl. Acad. Sci. U.S.A. 98, 14967–14972.
Medema, R. H., Kops, G. J., Bos, J. L., and Burgering, B. M. (2000). AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404, 782–787.
Miyamoto, K., Araki, K. Y., Naka, K., Arai, F., Takubo, K., Yamazaki, S., Matsuoka, S., Miyamoto, T., Ito, K., Ohmura, M., Chen, C., Hosokawa, K., Nakauchi, H., Nakayama, K., Nakayama, K. I., Harada, M., Motoyama, N., Suda, T., and Hirao, A. (2007). Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 1, 101–112.
Myers, M. P., Pass, I., Batty, I. H., Van der Kaay, J., Stolarov, J. P., Hemmings, B. A., Wigler, M. H., Downes, C. P., and Tonks, N. K. (1998). The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proc. Natl. Acad. Sci. U.S.A. 95, 13513–13518.
Ouyang, W., Beckett, O., Ma, Q., Paik, J. H., DePinho, R. A., and Li, M. O. (2010). Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat. Immunol. 11, 618–627.
Paik, J. H., Kollipara, R., Chu, G., Ji, H., Xiao, Y., Ding, Z., Miao, L., Tothova, Z., Horner, J. W., Carrasco, D. R., Jiang, S., Gilliland, D. G., Chin, L., Wong, W. H., Castrillon, D. H., and DePinho, R. A. (2007). FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128, 309–323.
Palomero, T., Sulis, M. L., Cortina, M., Real, P. J., Barnes, K., Ciofani, M., Caparros, E., Buteau, J., Brown, K., Perkins, S. L., Bhagat, G., Agarwal, A. M., Basso, G., Castillo, M., Nagase, S., Cordon-Cardo, C., Parsons, R., ZÚñiga-Pflücker, J. C., Dominguez, M., and Ferrando, A. A. (2007). Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 13, 1203–1210.
Podsypanina, K., Ellenson, L. H., Nemes, A., Gu, J., Tamura, M., Yamada, K. M., Cordon-Cardo, C., Catoretti, G., Fisher, P. E., and Parsons, R. (1999). Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc. Natl. Acad. Sci. U.S.A. 96, 1563–1568.
Puc, J., Keniry, M., Li, H. S., Pandita, T. K., Choudhury, A. D., Memeo, L., Mansukhani, M., Murty, V. V., Gaciong, Z., Meek, S. E., Piwnica-Worms, H., Hibshoosh, H., and Parsons, R. (2005). Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 7, 193–204.
Rao, R. R., Li, Q., Gubbels Bupp, M. R., and Shrikant, P. A. (2012). Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity 36, 374–387.
Rathmell, J. C., Elstrom, R. L., Cinalli, R. M., and Thompson, C. B. (2003). Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur. J. Immunol. 33, 2223–2232.
Rolf, J., Bell, S. E., Kovesdi, D., Janas, M. L., Soond, D. R., Webb, L. M., Santinelli, S., Saunders, T., Hebeis, B., Killeen, N., Okkenhaug, K., and Turner, M. (2010). Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J. Immunol. 185, 4042–4052.
Salmena, L., Carracedo, A., and Pandolfi, P. P. (2008). Tenets of PTEN tumor suppression. Cell 133, 403–414.
Samuels, Y., Wang, Z., Bardelli, A., Silliman, N., Ptak, J., Szabo, S., Yan, H., Gazdar, A., Powell, S. M., Riggins, G. J., Willson, J. K., Markowitz, S., Kinzler, K. W., Vogelstein, B., and Velculescu, V. E. (2004). High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554.
Shen, W. H., Balajee, A. S., Wang, J., Wu, H., Eng, C., Pandolfi, P. P., and Yin, Y. (2007). Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128, 157–170.
Sinclair, L. V., Finlay, D., Feijoo, C., Cornish, G. H., Gray, A., Ager, A., Okkenhaug, K., Hagenbeek, T. J., Spits, H., and Cantrell, D. A. (2008). Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat. Immunol. 9, 513–521.
So, L., and Fruman, D. A. (2012). PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem. J. 442, 465–481.
Song, M. S., Carracedo, A., Salmena, L., Song, S. J., Egia, A., Malumbres, M., and Pandolfi, P. P. (2011). Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144, 187–199.
Stambolic, V., Suzuki, A., de la Pompa, J. L., Brothers, G. M., Mirtsos, C., Sasaki, T., Ruland, J., Penninger, J. M., Siderovski, D. P., and Mak, T. W. (1998). Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39.
Stiles, B., Gilman, V., Khanzenzon, N., Lesche, R., Li, A., Qiao, R., Liu, X., and Wu, H. (2002). Essential role of AKT-1/protein kinase B alpha in PTEN-controlled tumorigenesis. Mol. Cell. Biol. 22, 3842–3851.
Suzuki, A., Yamaguchi, M. T., Ohteki, T., Sasaki, T., Kaisho, T., Kimura, Y., Yoshida, R., Wakeham, A., Higuchi, T., Fukumoto, M., Tsubata, T., Ohashi, P. S., Koyasu, S., Penninger, J. M., Nakano, T., and Mak, T. W. (2001). T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity 14, 523–534.
Tandon, P., Gallo, C. A., Khatri, S., Barger, J. F., Yepiskoposyan, H., and Plas, D. R. (2011). Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency. Proc. Natl. Acad. Sci. U.S.A. 108, 2361–2365.
Tothova, Z., Kollipara, R., Huntly, B. J., Lee, B. H., Castrillon, D. H., Cullen, D. E., McDowell, E. P., Lazo-Kallanian, S., Williams, I. R., Sears, C., Armstrong, S. A., Passegué, E., DePinho, R. A., and Gilliland, D. G. (2007). FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128, 325–339.
Tran, H., Brunet, A., Grenier, J. M., Datta, S. R., Fornace, A. J. Jr., DiStefano, P. S., Chiang, L. W., and Greenberg, M. E. (2002). DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 296, 530–534.
Trotman, L. C., Wang, X., Alimonti, A., Chen, Z., Teruya-Feldstein, J., Yang, H., Pavletich, N. P., Carver, B. S., Cordon-Cardo, C., Erdjument-Bromage, H., Tempst, P., Chi, S. G., Kim, H. J., Misteli, T., Jiang, X., and Pandolfi, P. P. (2007). Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156.
Van Parijs, L., and Abbas, A. K. (1996). Role of Fas-mediated cell death in the regulation of immune responses. Curr. Opin. Immunol. 8, 355–361.
Walsh, P. T., Buckler, J. L., Zhang, J., Gelman, A. E., Dalton, N. M., Taylor, D. K., Bensinger, S. J., Hancock, W. W., and Turka, L. A. (2006). PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J. Clin. Invest. 116, 2521–2531.
Wang, R., Dillon, C. P., Shi, L. Z., Milasta, S., Carter, R., Finkelstein, D., McCormick, L. L., Fitzgerald, P., Chi, H., Munger, J., and Green, D. R. (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882.
Xue, L., Nolla, H., Suzuki, A., Mak, T. W., and Winoto, A. (2008). Normal development is an integral part of tumorigenesis in T cell-specific PTEN-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 105, 2022–2027.
Yilmaz, O. H., Valdez, R., Theisen, B. K., Guo, W., Ferguson, D. O., Wu, H., and Morrison, S. J. (2006). Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–482.
You, H., Pellegrini, M., Tsuchihara, K., Yamamoto, K., Hacker, G., Erlacher, M., Villunger, A., and Mak, T. W. (2006). FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J. Exp. Med. 203, 1657–1663.
Keywords: Pten, T cells, autoimmunity, lymphoma
Citation: Newton RH and Turka LA (2012) Regulation of T cell homeostasis and responses by Pten. Front. Immun. 3:151. doi: 10.3389/fimmu.2012.00151
Received: 12 April 2012; Accepted: 22 May 2012;
Published online: 15 June 2012.
Edited by:
Klaus Okkenhaug, Babraham Institute, UKReviewed by:
Hergen Spits, University of Amsterdam, NetherlandsKlaus Okkenhaug, Babraham Institute, UK
Copyright: © 2012 Newton and Turka. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Laurence A. Turka, Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, 330 Brookline Ave., Boston, MA 02215, USA. e-mail: lturka@bidmc.harvard.edu