Pole-to-Pole Connections: Similarities between Arctic and Antarctic Microbiomes and Their Vulnerability to Environmental Change
 Julia Kleinteich1‡
Julia Kleinteich1‡  Falk Hildebrand2‡
Falk Hildebrand2‡  Mohammad Bahram3,4
Mohammad Bahram3,4  Anita Y. Voigt2†
Anita Y. Voigt2†  Susanna A. Wood5,6
Susanna A. Wood5,6  Anne D. Jungblut7 Frithjof C. Küpper8,9
Anne D. Jungblut7 Frithjof C. Küpper8,9  Antonio Quesada10
Antonio Quesada10  Antonio Camacho11
Antonio Camacho11  David A. Pearce12,13
David A. Pearce12,13  Peter Convey13
Peter Convey13  Warwick F. Vincent14 Christiane Zarfl1 Peer Bork2,15,16
Warwick F. Vincent14 Christiane Zarfl1 Peer Bork2,15,16  Daniel R. Dietrich17*
Daniel R. Dietrich17*- 1Center for Applied Geosciences, University of Tübingen, Tübingen, Germany
- 2Structural and Computational Biology, European Molecular Biology Laboratory, Heidelberg, Germany
- 3Evolutionary Biology Centre, Uppsala University, Uppsala, Sweden
- 4Department of Botany, Institute of Ecology and Earth Sciences, University of Tartu, Tartu, Estonia
- 5Coastal and Freshwater, Cawthron Institute, Nelson, New Zealand
- 6Environmental Research Institute, University of Waikato, Hamilton, New Zealand
- 7London Natural History Museum, London, United Kingdom
- 8Scottish Association for Marine Science, Oban, United Kingdom
- 9Oceanlab, University of Aberdeen, Aberdeen, United Kingdom
- 10Department of Biology, Autonomous University of Madrid, Madrid, Spain
- 11Cavanilles Institute for Biodiversity and Evolutionary Biology, University of Valencia, Valencia, Spain
- 12Department of Applied Sciences, Faculty of Health and Life Sciences, University of Northumbria at Newcastle, Newcastle, United Kingdom
- 13British Antarctic Survey, Cambridge, United Kingdom
- 14Département de Biologie and Centre for Northern Studies, Université Laval, Quebec, QC, Canada
- 15Max Delbrück Centre for Molecular Medicine, Berlin, Germany
- 16Department of Bioinformatics, University of Wuerzburg, Wuerzburg, Germany
- 17Human and Environmental Toxicology, University of Konstanz, Konstanz, Germany
The global biogeography of microorganisms remains poorly resolved, which limits the current understanding of microbial resilience toward environmental changes. Using high-throughput 16S rRNA gene amplicon sequencing, we characterized the microbial diversity of terrestrial and lacustrine biofilms from the Arctic, Antarctic and temperate regions. Our analyses suggest that bacterial community compositions at the poles are more similar to each other than they are to geographically closer temperate habitats, with 32% of all operational taxonomic units (OTUs) co-occurring in both polar regions. While specific microbial taxa were confined to distinct regions, representing potentially endemic populations, the percentage of cosmopolitan taxa was higher in Arctic (43%) than in Antarctic samples (36%). The overlap in polar microbial OTUs may be explained by natural or anthropogenically-mediated dispersal in combination with environmental filtering. Current and future changing environmental conditions may enhance microbial invasion, establishment of cosmopolitan genotypes and loss of endemic taxa.
Introduction
In a time of rapid environmental change, the study of biogeographic patterns on global and local scales, and the identification of drivers behind the spatial distribution of species, has become increasingly important (Walther et al., 2002). Biogeographic zones are formed for most ecosystems and associated taxa, as increasing geographic distance leads to a decrease in the exchange of individuals between two localities (distance decay theory, Morlon et al., 2008). Consequently, distant and isolated habitats are expected to have a specific diversity with a high level of endemism, whereas geographically closer habitats have more exchange and a higher proportion of shared species with lower endemism. Such patterns have been observed in both macro- (Astorga et al., 2012) and micro-organisms (Tamames et al., 2010; Bahram et al., 2013; Hahn et al., 2015). The main drivers behind these processes are thought to be intrinsic dispersal limitations (Mittelbach and Schemske, 2015) in combination with environmental filtering by spatial gradients of physical, chemical and biological traits (Chong et al., 2015). However, microorganisms (including prokaryotes), in contrast to many macroorganisms, are considered to be less confined by geographical barriers due to more efficient dispersal capacities (Bell, 2010; Chong et al., 2015), so that even remote and isolated habitats should be under constant exchange with both near and distant habitats. In this case, local as well as more cosmopolitan species are expected to be present and species distributions are likely to reflect environmental filtering rather than dispersal limitations (global ubiquity hypothesis, Baas-Becking, 1934; O'Malley, 2008).
High Arctic islands and the Antarctic continent are the most geographically distant terrestrial bioregions on Earth. It has been speculated that, because of the isolated character of the Antarctic continent and the environmentally extreme conditions in both polar regions, “the limits of species' abilities to disperse and survive may become more apparent” (Cox et al., 2016). Yet, both regions are similar in their physical-chemical characteristics, showing continuously low temperatures, limited freshwater availability, extreme seasonality in light conditions and localized nutrient availability. The harsh conditions prevent the survival of most macroorganisms (Convey et al., 2014) but allow diverse microbial life to thrive and dominate (Vincent, 2000a; Vincent and Quesada, 2012). A common group of microbial communities found in the terrestrial and lacustrine environments of both polar regions are those forming “macro-biofilms,” which are highly organized structures which can have vertical differentiation of functional and physiological assemblages (Fernández-Valiente et al., 2007; Mohit et al., 2017).
Both the Arctic as well as large parts of the West Antarctic, including the Antarctic Peninsula, are facing rapid environmental changes such as increasing temperatures due to both regional and global processes (Turner et al., 2009, 2014; Stocker et al., 2013). It has been proposed that climate change has already led to a weakening of biogeographic boundaries and the merging of ecological niches in the polar regions (Walther et al., 2002; Post et al., 2009; Nielsen and Wall, 2013; Pointing et al., 2015). The polar regions therefore provide a unique opportunity to test biogeographic patterns of microbial diversity, providing predictors/indications of the future consequences of environmental change. The survival strategies and dispersal limitations, genetic identity and endemic character of polar microorganisms and, thus, their resilience and sensitivity are still under discussion (Vincent, 2000b; Taton et al., 2006; Jungblut et al., 2010; Vyverman et al., 2010). A comprehensive exploration of these microbial features using consistent methodology and sampling has been difficult to date due to the remoteness and inaccessibility of the polar regions.
We here evaluated the roles of two opposing mechanisms, geographic distance effects (distance decay theory) vs. environmental filtering (global ubiquity hypothesis), in structuring bacterial biogeography in the polar regions. To do so, we undertook a large-scale microbial community analysis using standardized methods. We applied this approach to 89 freshwater microbial mat samples from multiple Arctic and Antarctic locations, including glacier runoffs and small streams, meltwater ponds, and larger lakes, as well as cryoconite holes, which have all been recognized as hotspots for microbial diversity (Vincent, 2000a; Quesada et al., 2008). Similar reference sites were also examined in temperate regions from alpine or marine influenced habitats.
Materials and Methods
Study Sites and Sampling
Samples from microbial communities growing on wet soil, in small streams and ponds were collected during several field campaigns (three in each of the Arctic, the Antarctic and non-polar regions) between 2007 and 2014 (Supplementary Table 1). Samples were collected in sterile tubes or bags using a sterile spatula or similar equipment. All samples were stored frozen (−20°C) or freeze-dried and stored frozen until further use. Detailed information on the sampling sites and dates is given in Table 1 and the Supplementary Information (Supplementary Table 1).
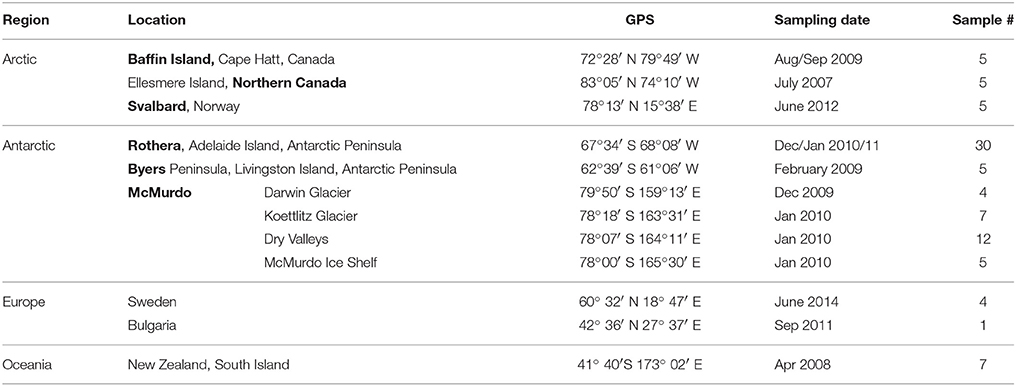
Table 1. Sampling sites, dates and number of samples.
DNA Extraction and 16S rRNA Gene Sequencing
Microbial samples for DNA extraction were gently thawed and DNA extracted from 0.05 to 0.1 g subsamples using the PowerSoil® DNA Isolation Kit (Qiagen, Germantown, USA) following the manufacturer's recommendations and DNA eluted in sterile DNAse-free water. The DNA quality and quantity was assessed using a NanoDrop (NanoDrop 3300 Fluorospectrometer, ThermoScientific). DNA extracts were stored frozen (−20°C) for no longer than three months before subsequent processing. Samples from Northern Canada were extracted as described in Jungblut et al. (2010), dried and stored frozen.
DNA obtained from 90 environmental samples was amplified using primers targeting the V3-V4 region of the 16S rRNA gene (F319 5′-ACTCCTACGGGAGGCAGCAG-3′, R806 5′-GGACTACHVGGGTWTCTAAT-3′, Fadrosh et al., 2014). We used a dual multiplexing approach and a “heterogeneity spacer” of 0–3 bp length between the Illumina adapter and the forward/reverse primer sequence to increase read quality. PCR was carried out according to the manual of the Q5 high-fidelity polymerase (New England BioLabs, Ipswich, USA) with a final primer concentration of 0.2 μM and an annealing temperature of 65°C for 15 cycles.
The PCR product (1 μl) was used in the second PCR. This PCR was performed using the forward and barcoded reverse primers from the NEXTflex™ 16S V1-V3 Amplicon-Seq Kit (Bioo Scientific, Austin, Texas, USA) at final concentrations of 0.15 μM and an annealing temperature of 65°C for 25 cycles. The remaining PCR conditions were as indicated in manufacturer's instructions for the Q5 high-fidelity polymerase. PCR products were cleaned up with the Agencourt AMPure XP—PCR Purification system (Beckman Coulter, Brea, USA) according to the manufacturer's instructions, and multiplexed at equal concentration. Sequencing was performed using a 300 bp paired-end sequencing protocol on the Illumina MiSeq platform (Illumina, San Diego, USA) at the Genomics Core Facility, European Molecular Biology Laboratory, Heidelberg. After quality filtering, 83,165 ± 68,277 sequences per sample were identified.
Bioinformatic Analysis
The LotuS 1.45 (Hildebrand et al., 2014) pipeline was used to process the sequencing data. Reads were demultiplexed with modified quality filtering to accommodate for the increased miSeq sequence length (“minSeqLength 210, TruncateSequenceLength 210, maxAccumulatedError 1, RejectSeqWithoutRevPrim T, RejectSeqWithoutFwdPrim T”), requiring unique reads to be present at least six times in one sample or three times in two separate samples. Operational taxonomic units (OTUs) were clustered and denoised with UPARSE (Edgar, 2013) while chimeric OTUs were removed against the RDP reference database (http://drive5.com/uchime/rdp_gold.fa) with the UCHIME algorithm (Edgar et al., 2011). Finally, reads were merged with FLASH (Magoč and Salzberg, 2011) and a taxonomy assigned against the SILVA 123 rRNA database (Yilmaz et al., 2014) with Lambda (Hauswedell et al., 2014), using the following optional LotuS command line options: “-p miSeq -id 0.97 -doBlast lambda -amplicon_type SSU -tax_group bacteria -derepMin 6:1,3:2 -CL uparse -thr 14.” OTUs were summed to genus, family, class and phylum level per sample, according to their taxonomic classification. Additionally we created a set of high-fidelity OTUs, using swarm clustering (Mahé et al., 2014) to enable fine resolution discrimination of cosmopolitan, Arctic and Antarctic phylotypes and with the following LotuS command line options: “-p miSeq -keepTmpFiles 0 -id 0.97 -doBlast lambda -amplicon_type SSU -tax_group bacteria -derepMin 6:1,3:2 -CL swarm -swarm_distance 1 -thr 14.” This increased the overall number of OTUs to 14,253, when rarefying to seven samples and 576,150 reads per region. Raw 16S data are available from http://www.ebi.ac.uk/ena/ under accession number PRJEB22851.
Statistical Analysis
All statistical calculations were conducted in R 3.3.2. For all univariate tests, taxa with less than five reads over all samples were excluded from this analysis to avoid including artifacts. The taxa abundance matrix on each respective taxonomic level was normalized, dividing each feature by the respective total sample sum, and transformed with log10(x+1), where x is the normalized feature abundance.
We calculated the among-habitat differences of streams, terrestrial ponds and ice-based ponds (beta-diversity) for the microbiota using Permutational Multivariate Analysis of Variance (PERMANOVA; Anderson, 2001) as implemented in the R-package vegan. This test compares the intragroup distances to the intergroup distances in a permutation scheme and from this assesses significance. PERMANOVA tests used 1,000 permutations of Bray–Curtis dissimilarities among samples. PERMANOVA post-hoc P-values were corrected for multiple testing using the Benjamini–Hochberg correction (Benjamini and Hochberg, 1995).
To determine the number of shared taxa between major regions, for each region the same number of samples with the largest number of reads were pooled. This pooled matrix was rarefied to the same depth for all regions and the overlap in OTUs shared between the regions calculated using the vennCounts function from the R-package limma.
Several environmental parameters (geographic location, yearly average for temperature, solar irradiation, and precipitation) for the specific sampling locations were collected from public databases such as http://www.yr.no/, http://www.weatherbase.com as well as selected publications (Bañón et al., 2013). Differences in environmental parameters between selected sampling sites were correlated to microbial beta-diversity, the significance being determined using a Mantel's test (Mantel, 1967) as implemented in the R-package vegan, using 4,999 repetitions and reporting the mantel statistic as correlation. Microbial beta-diversity was calculated from the log10 transformed metagenomic feature abundance matrix using a Bray–Curtis dissimilarity. Similarly, the difference in mean annual temperature, solar irradiation and precipitation of each sample were correlated to the microbial beta diversity for all bacterial OTUs between sites, excluding samples from the same locality to reduce the bias of local similarity.
Rarefaction
Sample count matrices were rarefied using the R implementation of rtk toolkit (Saary et al., 2017). Depending on the comparison, samples were rarefied to include all samples (e.g., richness comparisons including each sample) or to include groups of samples (e.g., calculating shared OTUs between Arctic and Antarctic samples) with the rarefaction depth being adapted to the number of samples as mentioned in the text. The number of different taxa was calculated for each rarefied sample. This was repeated five times per sample, and the average is the reported richness. Chao1 richness estimates (Chao, 1984) and Shannon diversity estimates (Shannon, 1948) were calculated from the rarefied OTU matrix. Correlations between richness and environmental factors were tested using a Spearman correlation test for continuous variables and a Kruskal–Wallis test for discrete variables; both were multiple testing corrected using Benjamini Hochberg false discovery rate (q-value).
To estimate the number of OTUs shared between the Antarctic, Arctic, and non-polar sampling locations, first we randomly selected seven samples from each region and subsequently rarefied these to 32,540 reads per sample. The “relative occurrence in region” was calculated pairwise between any two regions, by dividing the number of occurrences in one region by the sum of occurrences in both regions.
Results
Composition of Arctic and Antarctic Microbial Communities
In total, 3,556 OTUs and 1,008 genera from 549 families were identified (Supplementary Figure 1). Out of these, 77% of OTUs could not be unambiguously assigned to the 16S rRNA of known species (having either 100% identity or >97% sequence identity and unambiguous species assignments) and another 21% were assigned to uncultured species. This is in contrast to 98.6% of OTUs being assigned to phylum level and indicates that most of the OTUs described here can be considered novel. The median OTU richness was highest in Northern Canada with 161 ± 66 OTUs per sample when compared to all other polar and non-polar samples (between 58 ± 64 OTUs per sample in New Zealand and 99 ± 16 in Byers, Figure 1, Supplementary Table 2, rarefied to 620 reads per sample). The richness was neither significantly different between Arctic and Antarctic sites, nor in the polar regions relative to temperate sites (p > 0.05, Wilcox rank sum test).
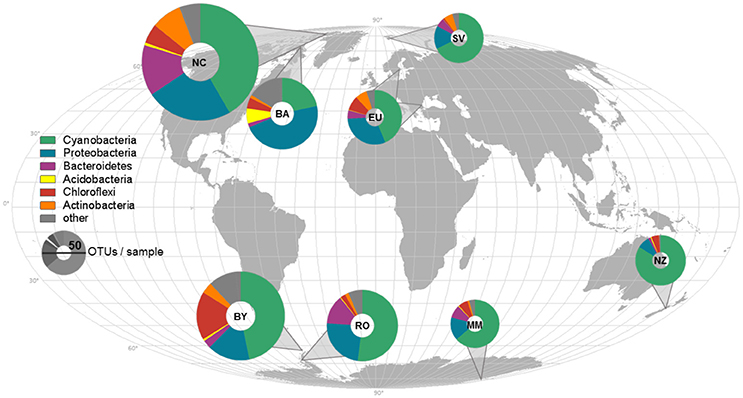
Figure 1. Sample locations, average composition of phyla, and species richness displayed on a world map. The average composition of bacterial phyla for each sampling location is represented and indicated by different colors. The richness was rarefied to the smallest sample size for each location and the same number of reads and is displayed by the size of the icons. NC, Northern Canada; BA, Baffin; EU, Europe; SV, Svalbard; BY, Byers; RO, Rothera; MM, McMurdo; NZ, New Zealand.
Cyanobacteria and Proteobacteria were the dominant phyla and Chloroflexi, Actinobacteria, and Firmicutes were present in low abundance in all sampled regions (Figure 1). The four most abundant genera among all samples were assigned to the Cyanobacteria phylum (Figure 2), with Leptolyngbya (Oscillatoriales) being followed by unclassified members of the family Oscillatoriaceae and genus Nostoc (Nostocaceae). Non-cyanobacterial OTUs were assigned to unclassified members of the family Comamonadaceae, and the genera Polymorphobacter and Leptothrix (Proteobacteria). Flavobacterium (Bacteriodetes), Blastocatella (Acidobacteria), as well as members of the family Anaerolinaceae (Chloroflexi) were also present. Comparative BLAST searches of the 10 most abundant OTUs (Supplementary Table 3) revealed a distinct strain population of Phormidium sp., previously identified in Antarctic habitats (Kleinteich et al., 2012; Zhang et al., 2015), as the most abundant OTU. This was followed by OTUs assigned to isolated strains of the genus Oscillatoria KNUA009 and to Leptolyngbya antarctica ANT.LAC.1.
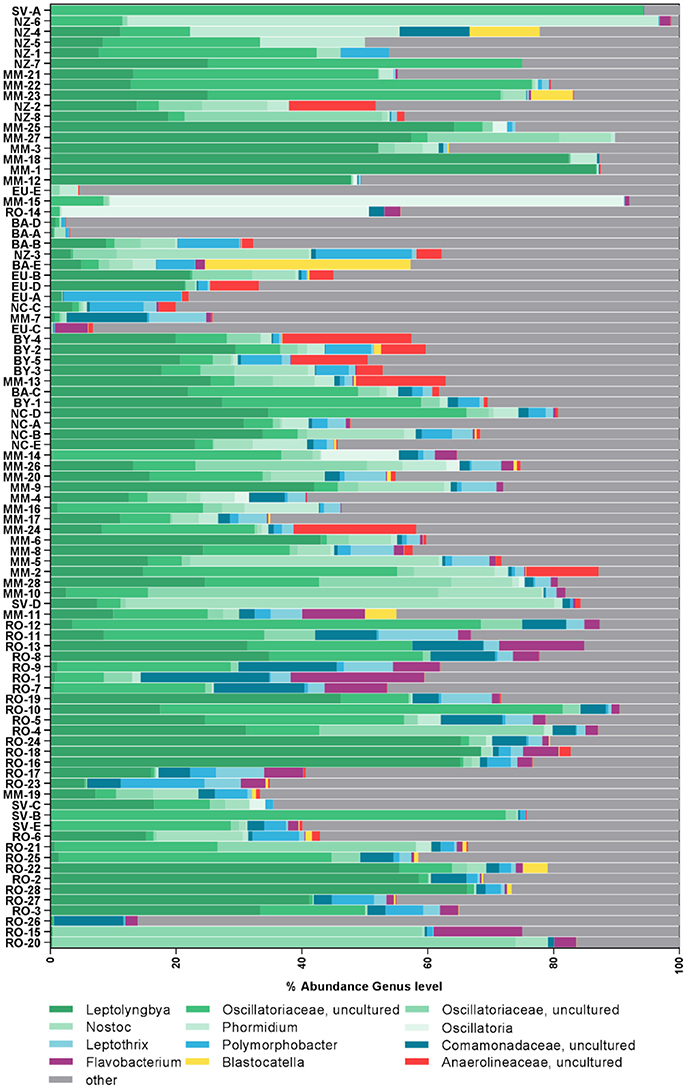
Figure 2. Composition of most abundant bacterial genera in the samples. Samples are sorted according to similarity. Genera were assigned according to the SILVA database.
Biogeographic Patterns and Environmental Selection in the Arctic and Antarctic
To compare between the major regions (Arctic, Antarctic, and non-polar regions), we pooled OTU abundances within these and rarefied them to the same number of samples and 545,665 reads per region. Of the resulting 2,769 OTUs in total, relatively more bacterial genera (590) and OTUs (1,678) were detected in the Antarctic than in the Arctic (525 genera and 1,392 OTUs) (Figure 3A, Supplementary Table 4). The number of cosmopolitan OTUs (present in all major regions) was high: 599 OTUs (22% of the total OTUs) were present in all three regions. The proportion of these cosmopolitan OTUs was somewhat higher within samples from the Arctic (43%) than the Antarctic (36%) and non-polar regions (37%). Interestingly, 11% (293) of all OTUs were detected at both poles but not at temperate sites. This cosmopolitan trend was stronger for cyanobacterial OTUs, 58 of which were detected in all three regions (34% of the total cyanobacterial OTUs, Supplementary Table 5). Also using the high-fidelity swarm clustering (Mahé et al., 2014) with a higher clustering stringency the fraction of OTUs overlapping between Arctic and Antarctic habitats remained the same, whereas the percentage of cosmopolitan OTUs decreased more than two-fold to 1,145 (8% of total swarmOTUs, Supplementary Table 4). Excluding the cosmopolitan fraction, 1,632 swarmOTUs (11%) were shared between the Arctic and the Antarctic, suggesting a biogeographic connection and a regular exchange between both polar regions, as shown in Figures 3, 4.
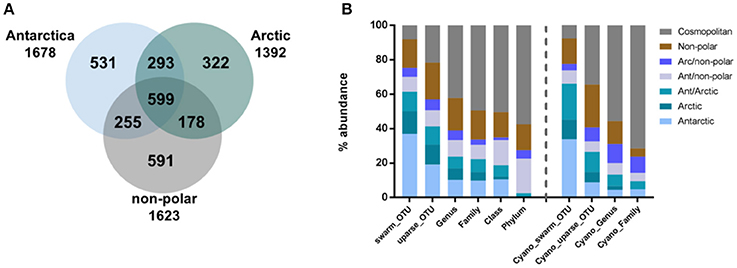
Figure 3. Geographic distribution of operational taxonomic units (OTUs). Numbers of OTUs are based on the UPARSE-algorithm and rarefied to the same number of samples per region and 546,002 reads per sample. The number of shared bacterial OTUs between the three regions is displayed in (A) and the geographic distribution of bacteria (left) and cyanobacterial (right) sequences relative to their phylogenetic level in (B).
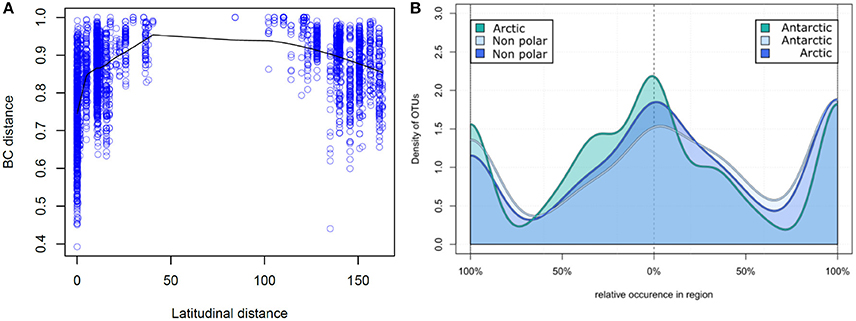
Figure 4. Relative occurrence of operational taxonomic units: (A) Correlation of latitudinal distance and sample dissimilarity. The Bray–Curtis dissimilarity between two samples is displayed correlating to their distance in latitude degrees. Samples closer to the poles share a higher number of OTUs and therefore have a higher community similarity. (B) Relative occurrence of OTUs between major biomes. Unique and shared relative occurrence of OTUs are compared between each pair of the three major biomes (Arctic, Antarctic, and non-polar regions). For better comparability the number of samples was rarefied to seven per region and for each OTU the proportion of occurrence between two regions calculated, so that local OTUs are on the left and right of the diagram whereas shared OTUs are in the center.
In contrast to these cosmopolitan OTUs, more than half of the total OTUs (52%) were assigned exclusively to one specific region (Arctic, Antarctic, or non-polar) and can be considered as potentially endemic. The relative percentage of these OTUs was highest in non-polar regions (36%, 591 OTUs) followed by the Antarctic (32%, 531 OTUs) and the Arctic (23%, 322 OTUs) (Supplementary Tables 4, 5). The relative fraction occurring exclusively in one specific region declined when moving from OTU to higher phylogenetic levels (Figure 3B, Supplementary Table 4). This decline can be expected since endemic species and genera may belong to cosmopolitan families. Nevertheless, the decline was stronger for the Arctic and Antarctic than for non-polar sequences or those shared between polar and non-polar sites. The decline concurred with an increase in cosmopolitans. Despite only 8% of swarmOTUs being cosmopolitan, 42% of genera and almost 50% of families were distributed globally. When considering only cyanobacteria, the cosmopolitan fraction of OTUs (34%), genera (55%) and families (51%) was higher than for all bacteria as described above (Figure 3B).
On a spatial scale the polar regions were more similar in their OTU composition (based on Bray–Curtis dissimilarity) to each other than to temperate regions (Figure 4A). Bray–Curtis dissimilarity was less within a biogeographic region than between biogeographic regions (p < 0.001, PERMANOVA, Supplementary Figure 2). This spatial correlation was also observed when considering only samples from the same region (Supplementary Figure 3) and is in contrast to a random dispersal, where all similarities would be equal an a spatial scale. While community dissimilarity and latitudinal distance were correlated as expected due to proximity (p = 0.001, Mantel r = 0.311), distance from the equator had a stronger correlation with community dissimilarity (p = 0.001, Mantel r = 0.433). Thus, samples located at a similar latitude, irrespective of the hemisphere, shared a high number of OTUs, whereas the number of shared OTUs decreased with increasing difference in latitudinal position.
We further investigated the relative pattern of OTU distribution by focusing on the fraction of OTUs shared to different degrees between two of each regions (Figure 4B). In this distribution we note three distinct peaks of OTU density, indicating that there is a clear separation into OTUs uniquely present in each region, and those equally distributed between two regions. This again is in contrast to a random dispersal, in which all OTUs would be distributed equal. Non-polar regions had a lower fraction of uniquely present OTUs in contrast to the two polar regions, where the fraction of unique OTUs was more similar. The fraction of shared OTUs was highest when comparing the two polar regions and lower for each polar region compared to non-polar sites. This further demonstrates that the non-polar regions are more dissimilar to both poles, and that the poles share relatively more OTUs with higher community similarity.
Average annual precipitation, monthly and annual temperature as well as solar radiation did not correlate, and local monthly average precipitation only weakly correlated, to community richness (p < 0.05, Spearman correlations test, Supplementary Table 6). Differences in temperature (monthly and annual average) as well as annual solar radiation between locations were significantly correlated (p < 0.05, Mantel test) to community beta diversity (Bray–Curtis dissimilarity), indicating that these factors influence community assembly (Supplementary Figure 4, Supplementary Table 6). In contrast, differences in average precipitation and monthly solar radiation were not significantly correlated. Some habitat types differed significantly in their composition (p < 0.05, PERMANOVA, Supplementary Table 7), with ice-based ponds being dissimilar in composition to streams and terrestrial ponds at lower taxonomic levels.
Discussion
This study provides the first large-scale comparative study of a unique set of microbial communities of the Arctic, Antarctic, and temperate areas using high-throughput sequencing. Due to the extreme remoteness and harsh environmental conditions, most polar microbial habitats and regions remain under-sampled and, comparative studies are limited due to the use of heterogeneous methodologies (Pearce et al., 2012). Thus, total polar bacterial biodiversity is currently underestimated, as is also suggested by the rarefaction analysis in the present study. The number of OTUs found in this study exceeded the number of OTUs and species detected using more traditional methods such as clone libraries or microscopic identification as summarized by Pearce et al. (2012). The high sample number combined with the high throughput sequencing used in this study enabled a more realistic and in-depth analysis of true (bi)polar microbial diversity.
Despite the heterogeneity of the sampled habitats, Arctic, Antarctic, and non-polar microbial mat communities in this study had a similar OTU richness and composition in terms of the dominant phyla. They do not show a declining latitudinal diversity gradient as described for most macro-organisms (Chown et al., 2004). The detection of the same bacterial OTUs (32% of all) in both polar regions as well as in temperate sites suggests a potentially ubiquitous distribution for at least some bacterial taxa, which supports the global ubiquity theory. Cosmopolitan distribution of prokaryotes and eukaryotes has been discussed for organisms smaller than one millimeter and usually decreases with increasing organism size (Finlay and Fenchel, 2004; Smith and Wilkinson, 2007; Yang et al., 2010). Finlay and Fenchel (2004) state that the cosmopolitan fraction of organisms in the bacterial size range is usually about 2.5-fold higher than detected in the present study. Cox et al. (2016) also found a percentage of ~35–60% cosmopolitan OTUs in Antarctic fungi and Petz et al. (2007) showed that 13% of freshwater ciliate species were shared between the Arctic and the Antarctic. For larger organisms Griffiths et al. (2009) suggest a general level of endemism of 50% in Southern Ocean benthos, and even greater levels of endemism (up to possibly 100% at species level) are reported across a range of Antarctic terrestrial biota (Convey et al., 2008; Pugh and Convey, 2008). This indicates that other mechanisms, such as geographical and climatic isolation and environmental selection play a particular role in structuring biogeography in the polar regions (Chown et al., 2004).
Since even 98% 16S rRNA gene similarity can represent >10 million years of bacterial evolution (Kuo and Ochman, 2009), we tested this result using a more stringent single linkage clustering sequences with less than two nucleotide differences (Mahé et al., 2014), that results in more OTUs with less average dissimilarity each, which confirmed the cosmopolitan and bi-polar distribution of 19% of the swarmOTUs. The high percentage of shared OTUs at a high 16S rRNA gene similarity between both polar regions suggests at least a periodic dispersal and exchange of individuals between both regions. This shows that geographical distance and barriers are not rigid and can be overcome by many microbial species, which is in line with the ubiquity hypothesis (Baas-Becking, 1934; O'Malley, 2008; Biersma et al., 2017). The underlying mechanisms could include efficient dispersal strategies for microorganisms via aerial (Hughes et al., 2004; Pearce et al., 2010) or vector-based transport including animals. Beyond these natural dispersal mechanisms, anthropogenic mediated dispersal for example via ships and planes (Cowan et al., 2011; Chown et al., 2012) could facilitate the dispersal of microorganisms. The high cosmopolitan fraction of cyanobacteria, in contrast to other bacterial taxa, suggests this phylum employs the above mentioned dispersal strategies, likely profiting from broad tolerances to extreme physical and chemical conditions (Vincent, 2000a; Vincent and Quesada, 2012).
In contrast to the cosmopolitan fraction, in our dataset more than half of all OTUs were uniquely present in one of the three regions (Arctic, Antarctic, or non-polar) and there was a decay in community similarity with increasing distance on a local and regional scale as described previously in Antarctica for microorganisms (Yergeau et al., 2007) and macroorganisms (Griffiths et al., 2009) supporting the distance decay theory. The percentage of these uniquely present OTUs was highest in non-polar samples in our dataset and likely reflects the higher similarity of the polar compared to non-polar regions. The somewhat larger fraction of endemic OTUs in the Antarctic compared to the Arctic may partially be explained by more extreme environmental conditions in Antarctica (Vyverman et al., 2010), but may also reflect the geographical situation of the two regions. The terrestrial Arctic has broad north-south connections over continental land masses, ocean sectors and ecoclimatic zones, whereby wind, water currents and migrating animals may serve as vectors for species dispersal. In contrast, the Antarctic continent is oceanographically as well as meteorologically isolated from the rest of the globe (Clarke et al., 2005; Barnes et al., 2006; Pointing et al., 2015). This geographic isolation has been speculated to cause a higher degree of genetic separation and thus be the driver for microbial endemism on the Antarctic continent (Vincent, 2000b; Vyverman et al., 2010; Hahn et al., 2015) as well as for Southern Ocean benthos (Griffiths et al., 2009). Isolated microhabitats in combination with local and regional distance-decay on the wide-ranging Antarctic continent may additionally provide ecological niches for a high microbial diversity and a high level of endemism (Yergeau et al., 2007; Vyverman et al., 2010). The uniqueness of Antarctic habitats and the underrepresentation in public databases of OTUs found here in combination with the high percentage of potentially endemic microbial OTUs detected in the south polar region, highlights the importance of specially protected areas for the conservation of microbial diversity in the Antarctic (Hughes et al., 2015). The rapid disappearance of unique habitats in the High Arctic (Vincent, 2010) and the potential for loss of endemic northern taxa indicate the need for a concerted global effort in protecting polar microbial ecosystems.
Antarctic as well as Arctic microbial communities were more similar to each other than to the temperate reference sites, and more than half of the potential ubiquitous swarmOTUs occurred exclusively in the polar regions and were not detected in temperate sites. Even though temperate habitats from alpine areas in New Zealand and marine influenced sites in Europe are not fully comparable to polar habitats, these results are corroborated by studies on eukaryote biogeography finding bi-polar distributions of fungal OTUs (Cox et al., 2016), testate amoebae (Yang et al., 2010), and bryophyte species (Biersma et al., 2017). These findings suggest that the global dispersal of certain (microbial) species is combined with environmental selection shaping bacterial communities in the polar regions (Vyverman et al., 2010; Chong et al., 2015; Cox et al., 2016), though temporal (Rochera et al., 2010) or spatial (Villaescusa et al., 2013) variability in environmental conditions can cause differences among prokaryotic meta-communities in polar habitats. Highly seasonal conditions in the polar regions (Chong et al., 2015), such as the absence of light in winter (Alonso-Saez et al., 2012) freezing and limited freshwater availability (Convey et al., 2014; Mohit et al., 2017) and low temperatures (Yergeau et al., 2007, 2012; Kleinteich et al., 2012) may cause the observed differences between polar and non-polar communities, since non-polar regions exhibit more moderate environmental conditions. In fact, large differences in temperature and annual solar radiation but not regional precipitation explained community differences in the present dataset. A broad scale phylogenetic meta-analysis recently suggested the presence of cold-tolerant cyanobacteria in all cyanobacterial taxonomic clades (Chrismas et al., 2015). The latter is also reflected in our dataset, in which bacterial species and strains surviving in polar environments belong to cosmopolitan groups at higher taxonomic levels. Cold tolerance, among other features, therefore seems to be a characteristic that has evolved several times in diverse taxa, and thus appears to be a trait of polyphyletic adaptation, given the complex optimizations required across the bacterial genome (Amico et al., 2006). We propose that, in addition to endemic taxa, there is a pool of cosmopolitan bacteria with widespread dispersal capacities, from which certain species and strains are selected for their capability to sustain themselves in polar environments.
Polar communities may therefore be disproportionately influenced by future physical, chemical and biological environmental changes (Stocker et al., 2013). Both temperature and precipitation (frequency, intensity and duration of snow or rain deposition (Convey et al., 2014), as well as the length of the ice melting period) are predicted to increase with climate change in polar regions (Turner et al., 2009, 2014; Stocker et al., 2013) providing more liquid freshwater but also less predictable conditions in some polar habitats (Vincent et al., 2017). Based on our results and those of others, both, the availability of liquid freshwater (Convey et al., 2014) and temperature (Yergeau et al., 2007, 2012; Kleinteich et al., 2012) can induce a shift in the community composition of polar microbial communities. Communities with a higher diversity often show a higher resilience toward perturbations, as functional redundancy lowers the chance for key species to vanish during perturbations (Yachi and Loreau, 1999). Similarly, increased richness might also be beneficial to a community by enabling a community to react with several distinct responses to a perturbation, increasing the resilience (Elmqvist et al., 2003; Folke et al., 2004). Higher temperatures in the polar regions will probably directly influence microbial communities by an elongation of the growth season, the availability of freshwater and daily temperature cycles with less freeze-thaw events. Lower environmental filtering induced by climate change as described here may support the establishment of cosmopolitan taxa, capable of long-distance dispersal, over more specialized and endemic taxa and may support the invasion of species on other trophic levels which could impact benthic microbial biofilm biology and biodiversity (Nielsen and Wall, 2013). In the longer term it can be expected that multicellular organisms such as mosses and grasses will replace microbial primary producers in currently microbial dominated habitats (Convey et al., 2003; Biersma et al., 2017), with microbial-dominated communities increasingly restricted to the highest latitudes (Nielsen and Wall, 2013).
We conclude that the microbiota of both polar regions may be more connected than previously assumed and our data suggest a biogeographical connection between both poles, consistent with some previous studies (Pearce et al., 2007; Jungblut et al., 2010; Chrismas et al., 2015; Cox et al., 2016; Biersma et al., 2017; Mohit et al., 2017). The polar microbial communities in our study are an assemblage of ubiquitously-distributed and potentially endemic taxa. This suggests a combined model of global dispersal of bacterial species in combination with habitat-specific selection pressure in cold environments (global ubiquity hypothesis) and distance-related biogeography in isolated Arctic and Antarctic communities (distance-decay theory). In both cases, the severe climate of the polar regions as well as their spatial and climatic isolation from the rest of the globe selects for a specialized community and leads to a high level of potentially endemic species. These organisms are potentially vulnerable to ongoing climate change in both polar regions, which is likely to favor cosmopolitan generalist species at the expense of distinct taxa that currently occur in specific polar habitats.
Author Contributions
JK, FH, and DD designed the study. Laboratory research and experiments were performed by JK, FH, and AV with the methodological and material support of DD, PB, SW, and CZ. FH and MB performed the bioinformatics and statistical analyzes. Field work and sample collection were carried out by JK, SW, AJ, FK, AQ, and AC. Field trips were organized by WV, AC, AQ, DP, and PC. JK, FH, and DD wrote the first draft of the manuscript with substantial help of CZ, MB, and PB, and all other authors contributed substantially to revisions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
JK acknowledges the Carl Zeiss foundation for PhD funding, the Marie-Curie COFUND-BEIPD PostDoc fellowship for PostDoc funding, FNRS travel funding and the logistical and financial support by UNIS. JK and FK acknowledge the Natural Environment Research Council (NERC) Antarctic Funding Initiative AFI-CGS-70 (collaborative gearing scheme) and logistic support from the British Antarctic Survey (BAS) for field work in Antarctica. JK and CZ acknowledge the Excellence Initiative at the University of Tübingen funded by the German Federal Ministry of Education and Research and the German Research Foundation (DFG). FH, AV, and PB received funding from MetaHIT (HEALTH-F4-2007-201052), Microbios (ERC-AdG-502 669830) and the European Molecular Biology Laboratory (EMBL). We thank members of the Bork group at EMBL for helpful discussions. We acknowledge the EMBL Genomics Core Facility for sequencing support and Y. P. Yuan and the EMBL Information Technology Core Facility for support with high-performance computing and EMBL for financial support. PC is supported by NERC core funding to the BAS “Biodiversity, Evolution and Adaptation” Team. MB was funded by Helge Ax:son Johnsons Stiftelse and PUT1317. DRD acknowledges the DFG funded project DI698/18-1 Dietrich and the Marie Curie International Research Staff Exchange Scheme Fellowship (PIRSES-GA-2011-295223). Operations in the Canadian High Arctic were supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), ArcticNet and the Polar Continental Shelf Program (PCSP). We are also grateful to the TOTAL Foundation (Paris) and the UK NERC (WP 4.3 of Oceans 2025 core funding to FCK at the Scottish Association for Marine Science) for funding the expedition to Baffin Island and within this context Olivier Dargent and Dr. Pieter van West for sample collection, and the Spanish Ministry of Science and Technology through project LIMNOPOLAR (POL200606635 and CGL2005-06549-C02-01/ANT to AQ as well as CGL2005-06549-C02-02/ANT to AC, the last of these co-financed by European FEDER funds). We are grateful for funding from the MASTS pooling initiative (The Marine Alliance for Science and Technology for Scotland), funded by the Scottish Funding Council (HR09011) and contributing institutions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2017.00137/full#supplementary-material
References
Alonso-Saez, L., Waller, A. S., Mende, D. R., Bakker, K., Farnelid, H., Yager, P. L., et al. (2012). Role for urea in nitrification by polar marine Archaea. Proc. Natl. Acad. Sci. U.S.A. 109, 17989–17994. doi: 10.1073/pnas.1201914109
Amico, S. D., Collins, T., Marx, J., Feller, G., and Gerday, C. (2006). Psychrophilic microorganisms: challenges for life. EMBO Rep. 7, 5–9. doi: 10.1038/sj.embor.7400662
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austr. Ecol. 26, 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x
Astorga, A., Oksanen, J., Luoto, M., Soininen, J., Virtanen, R., and Muotka, T. (2012). Distance decay of similarity in freshwater communities: do macro- and microorganisms follow the same rules? Glob. Ecol. Biogeogr. 21, 365–375. doi: 10.1111/j.1466-8238.2011.00681.x
Baas-Becking, L. (1934). Geobiologie of Inleiding Tot de Milieukunde. Den Haag: Stockum and Zoon, WP.
Bahram, M., Kõljalg, U., Courty, P.-E., Diédhiou, A. G., Kjøller, R., Põlme, S., et al. (2013). The distance decay of similarity in communities of ectomycorrhizal fungi in different ecosystems and scales. J. Ecol. 101, 1335–1344. doi: 10.1111/1365-2745.12120
Bañón, M., Justel, A., Velázquez, D., and Quesada, A. (2013). Regional weather survey on Byers Peninsula, Livingston Island, South Shetland Islands, Antarctica. Antarct. Sci. 25, 146–156. doi: 10.1017/S0954102012001046
Barnes, D. K. A., Hodgson, D. A., Convey, P., Allen, C. S., and Clarke, A. (2006). Incursion and excursion of Antarctic biota: past, present and future. Glob. Ecol. Biogeogr. 15, 121–142. doi: 10.1111/j.1466-822X.2006.00216.x
Bell, T. (2010). Experimental tests of the bacterial distance-decay relationship. ISME J. 4, 1357–1365. doi: 10.1038/ismej.2010.77
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300.
Biersma, E. M., Jackson, A., Hyvönen, J., Koskinen, S., Linse, K., Griffiths, H., et al. (2017). Global biogeographic patterns in bipolar moss species. R. Soc. Open Sci. 4:170147. doi: 10.1098/rsos.170147
Chao, A. (1984). Nonparametric-estimation of the number of classes in a population. Scand. J. Stat. 11, 265–270.
Chong, C. W., Pearce, D. A., and Convey, P. (2015). Emerging spatial patterns in Antarctic prokaryotes. Front. Microbiol. 6:1058. doi: 10.3389/fmicb.2015.01058
Chown, S. L., Huiskes, A. H. L., Gremmen, N. J. M., Lee, J. E., Terauds, A., Crosbie, K., et al. (2012). Continent-wide risk assessment for the establishment of nonindigenous species in Antarctica. Proc. Natl. Acad. Sci. U.S.A. 109, 6–11. doi: 10.1073/pnas.1119787109
Chown, S. L., Sinclair, B. J., Leinaas, H. P., and Gaston, K. J. (2004). Hemispheric asymmetries in biodiversity - a serious matter for ecology. PLoS Biol. 2:e406. doi: 10.1371/journal.pbio.0020406
Chrismas, N. A. M., Anesio, A. M., and Sánchez-Baracaldo, P. (2015). Multiple adaptations to polar and alpine environments within cyanobacteria: a phylogenomic and Bayesian approach. Front. Microbiol. 6:1070. doi: 10.3389/fmicb.2015.01070
Clarke, A., Barnes, D. K. A., and Hodgson, D. A. (2005). How isolated is Antarctica? Trends Ecol. Evol. 20, 1–3. doi: 10.1016/j.tree.2004.10.004
Convey, P., Chown, S. L., Clarke, A., Barnes, D. K. A., Bokhorst, S., Cummings, V., et al. (2014). The spatial structure of Antarctic biodiversity. Ecol. Monogr. 84, 203–244. doi: 10.1890/12-2216.1
Convey, P., Gibson, J. A. E., Hillenbrand, C.-D., Hodgson, D. A., Pugh, P. J. A., Smellie, J. L., et al. (2008). Antarctic terrestrial life-challenging the history of the frozen continent? Biol. Rev. Camb. Philos. Soc. 83, 103–117. doi: 10.1111/j.1469-185X.2008.00034.x
Convey, P., Scott, D., and Fraser, W. R. (2003). “Biophysical and habitat changes in response to climate alteration in the Arctic and Antarctic,” in Monographic Series Advances in Applied Biodiversity Science. No. 4: Climate Change and Biodiversity: Synergistic Impacts (Washington D.C.: Conservation International), 79–84.
Cowan, D. A., Chown, S. L., Convey, P., Tuffin, M., Hughes, K., Pointing, S., et al. (2011). Non-indigenous microorganisms in the Antarctic: assessing the risks. Trends Microbiol. 19, 540–548. doi: 10.1016/j.tim.2011.07.008
Cox, F., Newsham, K. K., Bol, R., Dungait, J. A. J., and Robinson, C. H. (2016). Not poles apart: Antarctic soil fungal communities show similarities to those of the distant Arctic. Ecol. Lett. 19, 528–536. doi: 10.1111/ele.12587
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Elmqvist, T., Folke, C., Nyström, M., Peterson, G., Bengtsson, J., Walker, B., et al. (2003). Response diversity, ecosystem change, and resilience. Front. Ecol. Environ. 1, 488–494. doi: 10.1890/1540-9295(2003)001[0488:RDECAR]2.0.CO;2
Fadrosh, D. W., Ma, B., Gajer, P., Sengamalay, N., Ott, S., Brotman, R. M., et al. (2014). An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2:6. doi: 10.1186/2049-2618-2-6
Fernández-Valiente, E., Camacho, A., Rochera, C., Rico, E., Vincent, W. F., and Quesada, A. (2007). Community structure and physiological characterization of microbial mats in Byers Peninsula, Livingston Island (South Shetland Islands, Antarctica). FEMS Microbiol. Ecol. 59, 377–385. doi: 10.1111/j.1574-6941.2006.00221.x
Finlay, B. J., and Fenchel, T. (2004). Cosmopolitan metapopulations of free-living microbial eukaryotes. Protist 155, 237–244. doi: 10.1078/143446104774199619
Folke, C., Carpenter, S., Walker, B., Scheffer, M., Elmqvist, T., Gunderson, L., et al. (2004). Regime shifts, resilience, and biodiversity in ecosystem management. Annu. Rev. Ecol. Evol. Syst. 35, 557–581. doi: 10.1146/annurev.ecolsys.35.021103.105711
Griffiths, H. J., Barnes, D. K. A., and Linse, K. (2009). Towards a generalized biogeography of the Southern Ocean benthos. J. Biogeogr. 36, 162–177. doi: 10.1111/j.1365-2699.2008.01979.x
Hahn, M. W., Koll, U., Jezberová, J., and Camacho, A. (2015). Global phylogeography of pelagic P olynucleobacter bacteria: restricted geographic distribution of subgroups, isolation by distance and influence of climate. Environ. Microbiol. 17, 829–840. doi: 10.1111/1462-2920.12532
Hauswedell, H., Singer, J., and Reinert, K. (2014). Lambda: the local aligner for massive biological data. Bioinformatics 30, i349–i355. doi: 10.1093/bioinformatics/btu439
Hildebrand, F., Tadeo, R., Voigt, A. Y., Bork, P., and Raes, J. (2014). LotuS: an efficient and user-friendly OTU processing pipeline. Microbiome 2:30. doi: 10.1186/2049-2618-2-30
Hughes, K. A., Cowan, D. A., and Wilmotte, A. (2015). Protection of Antarctic microbial communities – “out of sight, out of mind.” Front. Microbiol. 6:151. doi: 10.3389/fmicb.2015.00151
Hughes, K. A., McCartney, H. A., Lachlan-Cope, T. A., and Pearce, D. A. (2004). A preliminary study of airborne microbial biodiversity over Peninsular Antarctica. Cell. Mol. Biol. 50, 537–542. doi: 10.1170/T543
Jungblut, A. D., Lovejoy, C., and Vincent, W. F. (2010). Global distribution of cyanobacterial ecotypes in the cold biosphere. ISME J. 4, 1–12. doi: 10.1038/ismej.2009.113
Kleinteich, J., Wood, S. A., Küpper, F. C., Camacho, A., Quesada, A., Frickey, T., et al. (2012). Temperature-related changes in polar cyanobacterial mat diversity and toxin production. Nat. Clim. Change 2, 356–360. doi: 10.1038/nclimate1418
Kuo, C.-H., and Ochman, H. (2009). Inferring clocks when lacking rocks: the variable rates of molecular evolution in bacteria. Biol. Direct 4:35. doi: 10.1186/1745-6150-4-35
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mahé, F., Rognes, T., Quince, C., de Vargas, C., and Dunthorn, M. (2014). Swarm: robust and fast clustering method for amplicon-based studies. Peer J. 2:e593. doi: 10.7717/peerj.593
Mantel, N. (1967). The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220.
Mittelbach, G. G., and Schemske, D. W. (2015). Ecological and evolutionary perspectives on community assembly. Trends Ecol. Evol. 30, 241–247. doi: 10.1016/j.tree.2015.02.008
Mohit, V., Culley, A., Lovejoy, C., Bouchard, F., and Vincent, W. F. (2017). Hidden biofilms in a far northern lake and implications for the changing Arctic. NPJ Biofilms Microbiomes 3, 17. doi: 10.1038/s41522-017-0024-3
Morlon, H., Chuyong, G., Condit, R., Hubbell, S., Kenfack, D., Thomas, D., et al. (2008). A general framework for the distance-decay of similarity in ecological communities. Ecol. Lett. 11, 904–917. doi: 10.1111/j.1461-0248.2008.01202.x
Nielsen, U. N., and Wall, D. H. (2013). The future of soil invertebrate communities in polar regions: different climate change responses in the Arctic and Antarctic? Ecol. Lett. 16, 409–419. doi: 10.1111/ele.12058
O'Malley, M. A. (2008). “Everything is everywhere: but the environment selects”: ubiquitous distribution and ecological determinism in microbial biogeography. Stud. Hist. Philos. Biol. Biomed. Sci. 39, 314–325. doi: 10.1016/j.shpsc.2008.06.005
Pearce, D. A., Cockell, C. S., Lindstrom, E. S., and Tranvik, L. J. (2007). First evidence for a bipolar distribution of dominant freshwater lake bacterioplankton. Antarct. Sci. 19, 245–252. doi: 10.1017/S0954102007000326
Pearce, D. A., Hughes, K. A., Lachlan-Cope, T., Harangozo, S. A., and Jones, A. E. (2010). Biodiversity of air-borne microorganisms at Halley station, Antarctica. Extremophiles 14, 145–159. doi: 10.1007/s00792-009-0293-8
Pearce, D. A., Newsham, K. K., Thorne, M. A. S., Calvo-Bado, L., Krsek, M., Laskaris, P., et al. (2012). Metagenomic analysis of a southern maritime Antarctic soil. Front. Microbiol. 3:403. doi: 10.3389/fmicb.2012.00403
Petz, W., Valbonesi, A., Schiftner, U., Quesada, A., and Ellis-Evans, J. C. (2007). Ciliate biogeography in Antarctic and Arctic freshwater ecosystems: endemism or global distribution of species? FEMS Microbiol. Ecol. 59, 396–408. doi: 10.1111/j.1574-6941.2006.00259.x
Pointing, S. B., Büdel, B., Convey, P., Gillman, L. N., Körner, C., Leuzinger, S., et al. (2015). Biogeography of photoautotrophs in the high polar biome. Front. Plant Sci. 6:692. doi: 10.3389/fpls.2015.00692
Post, E., Forchhammer, M. C., Bret-Harte, M. S., Callaghan, T. V., Christensen, T. R., Elberling, B., et al. (2009). Ecological dynamics across the Arctic associated with recent climate change. Science 325, 1355–1358. doi: 10.1126/science.1173113
Pugh, P. J. A., and Convey, P. (2008). Surviving out in the cold: Antarctic endemic invertebrates and their refugia. J. Biogeogr. 35, 2176–2186. doi: 10.1111/j.1365-2699.2008.01953.x
Quesada, A., Fernández-Valiente, E., Hawes, I., and Howard-Williams, C. (2008). “Benthic primary production in polar lakes and rivers,” in Polar Lakes And Rivers Limnology Of Arctic And Antarctic Ecosystems, eds W. F. Vincent and J. Laybourn-Parry (Oxford: Oxford University Press), 179–196.
Rochera, C., Justel, A., Fernández-Valiente, E., Ba-ón, M., Rico, E., Toro, M., et al. (2010). Interannual meteorological variability and its effects on a lake from maritime Antarctica. Polar Biol. 33, 1615–1628. doi: 10.1007/s00300-010-0879-8
Saary, P., Forslund, K., Bork, P., and Hildebrand, F. (2017). RTK: efficient rarefaction analysis of large datasets. Bioinformatics 33, 2594–2595. doi: 10.1093/bioinformatics/btx206
Shannon, C. E. (1948). A mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423. doi: 10.1002/j.1538-7305.1948.tb01338.x
Smith, H. G., and Wilkinson, D. M. (2007). Not all free-living microorganisms have cosmopolitan distributions? The case of Nebela (Apodera) vas Certes (Protozoa: Amoebozoa: Arcellinida). J. Biogeogr. 34, 1822–1831. doi: 10.1111/j.1365-2699.2007.01733.x
Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., et al. (2013). IPCC, 2013: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge; New York, NY.
Tamames, J., Abellán, J. J., Pignatelli, M., Camacho, A., and Moya, A. (2010). Environmental distribution of prokaryotic taxa. BMC Microbiol. 10:85. doi: 10.1186/1471-2180-10-85
Taton, A., Grubisic, S., Balthasart, P., Hodgson, D. A., Laybourn-Parry, J., and Wilmotte, A. (2006). Biogeographical distribution and ecological ranges of benthic cyanobacteria in East Antarctic lakes. FEMS Microbiol. Ecol. 57, 272–289. doi: 10.1111/j.1574-6941.2006.00110.x
Turner, J., Barrand, N. E., Bracegirdle, T. J., Convey, P., Hodgson, D. A., Jarvis, M., et al. (2014). Antarctic climate change and the environment: an update. Polar Rec. 50, 237–259. doi: 10.1017/S0032247413000296
Turner, J., Bindschadler, R., Convey, P., Di Prisco, G., Fahrbach, E., Gutt, J., et al. (eds.). (2009). Antarctic Climate Change and the Environment. Cambridge, UK: The Scientific Committee on Antarctic Research.
Villaescusa, J. A., Casamayor, E. O., Rochera, C., Quesada, A., Michaud, L., and Camacho, A. (2013). Heterogeneous vertical structure of the bacterioplankton community in a non-stratified Antarctic lake. Antarct. Sci. 25, 229–238. doi: 10.1017/S0954102012000910
Vincent, W. F. (2000a). “Cyanobacterial dominance in the polar regions,” in The Ecology of Cyanobacteria, eds B. A. Whitton and M. Potts (Dordrecht: Kluwer Academic Publishers; Springer), 321–340.
Vincent, W. F. (2000b). Evolutionary origins of Antarctic microbiota: invasion, selection and endemism. Antarct. Sci. 12, 374–385. doi: 10.1017/S0954102000000420
Vincent, W. F. (2010). Microbial ecosystem responses to rapid climate change in the Arctic. ISME J. 4, 1089–1091. doi: 10.1038/ismej.2010.108
Vincent, W. F., Lemay, M., and Allard, M. (2017). Arctic permafrost landscapes in transition: towards an integrated Earth system approach. Arct. Sci. 3, 39–64. doi: 10.1139/as-2016-0027
Vincent, W. F., and Quesada, A. (2012). |Cyanobacteria in High Latitude Lakes, Rivers and Seas,” in Ecology of Cyanobacteria II Their Diversity in Space and Time, ed B. A. Whitton (Dordrecht: Springer), 371–385.
Vyverman, W., Verleyen, E., Wilmotte, A., Hodgson, D. A., Willems, A., Peeters, K., et al. (2010). Evidence for widespread endemism among Antarctic micro-organisms. Polar Sci. 4, 103–113. doi: 10.1016/j.polar.2010.03.006
Walther, G.-R., Post, E., Convey, P., Menzel, A., Parmesan, C., Beebee, T. J. C., et al. (2002). Ecological responses to recent climate change. Nature 416, 389–395. doi: 10.1038/416389a
Yachi, S., and Loreau, M. (1999). Biodiversity and ecosystem productivity in a fluctuating environment: the insurance hypothesis. Proc. Natl. Acad. Sci. U.S.A. 96, 1463–1468. doi: 10.1073/pnas.96.4.1463
Yang, J., Smith, H. G., Sherratt, T. N., and Wilkinson, D. M. (2010). Is there a size limit for cosmopolitan distribution in free-living microorganisms? A biogeographical analysis of testate amoebae from polar areas. Microb. Ecol. 59, 635–645. doi: 10.1007/s00248-009-9615-8
Yergeau, E., Bokhorst, S., Kang, S., Zhou, J., Greer, C. W., Aerts, R., et al. (2012). Shifts in soil microorganisms in response to warming are consistent across a range of Antarctic environments. ISME J. 6, 692–702. doi: 10.1038/ismej.2011.124
Yergeau, E., Newsham, K. K., Pearce, D. A., and Kowalchuk, G. A. (2007). Patterns of bacterial diversity across a range of Antarctic terrestrial habitats. Environ. Microbiol. 9, 2670–2682. doi: 10.1111/j.1462-2920.2007.01379.x
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Keywords: biogeography, diversity, microbiology, polar regions, ecology, high-throughput sequencing, 16S rRNA gene
Citation: Kleinteich J, Hildebrand F, Bahram M, Voigt AY, Wood SA, Jungblut AD, Küpper FC, Quesada A, Camacho A, Pearce DA, Convey P, Vincent WF, Zarfl C, Bork P and Dietrich DR (2017) Pole-to-Pole Connections: Similarities between Arctic and Antarctic Microbiomes and Their Vulnerability to Environmental Change. Front. Ecol. Evol. 5:137. doi: 10.3389/fevo.2017.00137
Received: 03 August 2017; Accepted: 27 October 2017;
Published: 10 November 2017.
Edited by:
Kevin C. Burns, Victoria University of Wellington, New ZealandReviewed by:
Corey Garza, California State University, Monterey Bay, United StatesKaty Morgan, University of New Orleans, United States
Copyright © 2017 Kleinteich, Hildebrand, Bahram, Voigt, Wood, Jungblut, Küpper, Quesada, Camacho, Pearce, Convey, Vincent, Zarfl, Bork and Dietrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel R. Dietrich, daniel.dietrich@uni-konstanz.de
†Present Address: Anita Y. Voigt, The Jackson Laboratory for Genomic Medicine, Farmington, CT, United States
‡Shared first authors