 Amanda J. Lee
Amanda J. Lee Ali A. Ashkar
Ali A. Ashkar- Department of Pathology and Molecular Medicine, McMaster Immunology Research Centre, McMaster University, Hamilton, ON, Canada
Type I and type II interferons (IFN) are central to both combating virus infection and modulating the antiviral immune response. Indeed, an absence of either the receptor for type I IFNs or IFN-y have resulted in increased susceptibility to virus infection, including increased virus replication and reduced survival. However, an emerging area of research has shown that there is a dual nature to these cytokines. Recent evidence has demonstrated that both type I and type II IFNs have immunoregulatory functions during infection and type II immune responses. In this review, we address the dual nature of type I and type II interferons and present evidence that both antiviral and immunomodulatory functions are critical during virus infection to not only limit virus replication and initiate an appropriate antiviral immune response, but to also negatively regulate this response to minimize tissue damage. Both the activating and negatively regulatory properties of type I and II IFNs work in concert with each other to create a balanced immune response that combats the infection while minimizing collateral damage.
Introduction
Type I and II interferons (IFN) are cytokines produced during virus infection that are integral for regulating the immune response. Type I IFNs are well known for their ability to directly induce an antiviral response within infected and surrounding cells through the upregulation of molecules that can antagonize virus replication (1). As they are produced rather early on during an infection, type I IFNs are also essential for activating the antiviral innate immune response, such as natural killer (NK) cell effector functions (2). Type II IFN, known as IFN-γ, while sharing a similar nomenclature to type I IFN, signals through a different receptor and has effects that are independent from type I IFN. As a part of the innate immune response, they are predominantly produced by natural killer cells during infection (2). IFN-γ, like type I IFN, promotes antiviral immunity through its regulatory effects on the innate immune response and acts as a key link between the innate immune response and activation of the adaptive immune response (3). Beyond their antiviral effects, a growing amount of evidence suggests that type I and type II IFNs have immunoregulatory functions that are critical for dampening immunopathogenic mechanisms and minimizing collateral damage from the infection. Altogether, this review will build a framework and provide evidence demonstrating that these two cytokines are both critical for limiting virus replication and promoting a beneficial virus limiting response, while simultaneously dampening immunopathology. If we consider the world outside of virus infections, however, this fundamental duality of type I and II IFNs can be applied to numerous pathological processes, ranging from allergy to autoimmune diseases.
Type I and II IFN Production and Signaling
Type I IFNs consist of a group of structurally similar cytokines and include 13–14 subtypes of IFN-α along with IFN-β, IFN-ε, IFN-κ, IFN-ω, IFN-δ, IFN-ζ, and IFN-τ (4, 5). As part of the innate immune antiviral response, these cytokines are rapidly produced after pattern-recognition receptor (PRR) stimulation (5). Current research suggests that an initial wave of IFN-β and IFN-α4 is produced and dependent upon IRF3 phosphorylation and NFκb activation (6–8). The initial type I IFN wave subsequently induces IRF7 phosphorylation and results in a positive feedback loop of increasing type I IFN release. Once produced, these cytokines all signal through the same receptor, the type I IFN receptor (IFNAR). IFNAR is composed of two subunits—IFNAR1 and IFNAR2—which when bound to type I IFN are endocytosed and activate their associated tyrosine kinases, Tyk2 and Jak1 (4, 9). The classical signaling cascade results in phosphorylation of STAT2 and STAT1, which forms a complex with IRF9, known as the IFN-stimulated gene factor 3 (ISGF3) (4). ISGF3 then leads to expression of IFN-stimulated genes (4). Beyond ISGF3, type I IFNs can also induce phosphorylation and dimerization of STAT3, STAT4, STAT5, and STAT6 and has been shown to induce activation of Rap1, CrkL, Map kinases, IRS-1 and -2, Vav, RAC1, and PI3-kinase signal transduction pathways (4, 10–14). Interestingly, IFN-β has been shown to additionally signal through the IFNAR1 subunit independent from IFNAR2 and carries through a non-canonical signaling pathway (15).
Type II IFN is predominantly produced by NK cells during the antiviral innate immune response (16). A multitude of evidence has shown that type I IFN, IL-12, IL-15, and IL-18 are all capable of inducing IFN-γ production from NK cells (17). NK cell IFN-γ is dependent upon STAT4 phosphorylation for its production. Once released, IFN-γ signals through the IFN-γ receptor (IFNGR), composed of IFNGR1 and IFNGR2 subunits. In the classical signaling pathway, ligation of IFN-γ to the IFNGR leads to activation of JAK1 and JAK2, resulting in homodimerization and phosphorylation of STAT1 (18). However, like type I IFN, IFN-γ has also been shown to signal through alternative pathways, including STAT4, Erk1/2, Pyk2, and CrkL, among others (18).
Type I IFN: Mastering the Antiviral Response
Type I IFN is one of the first cytokines produced during a virus infection. In the context of HSV-2 infection, for example, there is an initial wave of IFN-β production at 12 h post-infection, followed by both IFN-β and IFN-α production at 48 h post-infection (19, 20). This early production of type I IFN is critical for induction of both an antiviral response within infected and target cells, as well as activation of innate immune cells that will ultimately serve to control virus replication and activate the adaptive immune response to both clear the infection and generate memory to create a rapid response against future infections (21).
Type I IFN is a well-known stimulator of antiviral genes targeted against preventing virus replication from within target cells. When their production is stimulated by virus infection, type I IFN can act in an autocrine, paracrine, or systemic fashion. Their protective role during virus infection is highlighted by the increased mortality observed in mice deficient in the type I IFN receptor (Ifnar−/−) in comparison to their control counterparts when infected with a virus (22, 23). Upon ligation to its receptor, type I IFN has been shown to induce upwards of 300 ISGs. Of these 300 genes, 51 were found to contribute to host defense, while other genes contributed to inflammation, signaling, transcription, and immunomodulation, among other activities (24, 25). Further, De Veer et al. examined the ability of specific ISGs or combinations of ISGs to inhibit virus replication through overexpression of individual ISGs prior to virus infection (24). They found that many ISGs were capable of inhibiting virus replication, with some acting on a wide range of viruses, while others were only effective against particular viruses (24). Interestingly, they found that select ISGs enhanced virus replication in their experimental system (24). Antiviral ISGs can hinder virus replication through several mechanisms. Protein kinase R, for example, inhibits cellular translational functions (1). 2′5 OAS and RNaseL, on the other hand, degrade RNA (26, 27). Other ISG antiviral activities can prevent virion release, inhibit virus entry, and inhibit virus transcription (28).
Apart from their induction of antiviral ISGs, type I IFNs are key regulators of the innate immune response. Within the type I IFN literature, a theme has emerged wherein acute type I IFN production promotes beneficial antiviral responses, while chronic type I IFN production can have a suppressive and deleterious effect on the immune response. Within this section, we will examine the ability of type I IFN to promote antiviral functions in dendritic cells (DC), monocytes, and NK cells.
Dendritic cells are critical for activation of antiviral T-cells (29). Type I IFN stimulation has been shown to enhance MHC II expression and presentation of antigens as well as upregulate co-stimulatory molecules and promote DC maturation (29–32), Further evidence suggests that type I IFN is able to increase differentiation of plasmacytoid DCs into myeloid-derived DCs to increase T-cell activation (33).
Inflammatory monocytes are rapidly recruited to sites of infection, where they can then stimulate local and migrating immune cell antiviral function, promote inflammation, and differentiate into macrophages and DCs (34). At sites of inflammation, type I IFNs induce production of CCL2 to recruit inflammatory monocytes (2, 34). Type I IFN produced during vaginal HSV-1 infection induces tissue resident macrophages and DCs to produce CCL2 to recruit and initial population of inflammatory monocytes, which then enact a positive feedback loops to produce more CCL2 to attract further inflammatory monocytes (35). A similar phenomenon has been observed during vaginal HSV-2 infection, influenza infection, and inflammatory monocyte recruitment to the brain during LPS-induced systemic inflammation (2, 36, 37), With influenza infection, absence of IFNAR resulted in differentiation of Ly6C intermediate expressing monocytes rather than Ly6Chi inflammatory monocytes, which additionally had a different phenotype (36). Further, Seo et al. demonstrated that Ifnar−/− bone marrow had a significantly decreased differentiation of hematopoietic cells into inflammatory monocytes in the presence of influenza infection (38). In regards to macrophages, Type I IFN has more of a suppressive function and will be discussed below.
Type I IFN and antiviral NK cell functionality are tightly interwoven, where type I IFN has emerged as a key NK cell regulator. Like their monocyte counterparts, type I IFN has been implicated in NK cell recruitment to sites of inflammation. During a vaginal HSV-1 infection, type I IFN was required to induce epithelium production of CCL3, CCL4, and CCL5 to recruit NK cells to the vaginal mucosa (35). Further, type I IFN has been implicated in the activation of NK cell antiviral functions. During an infection, NK cells have several weapons under their belt that they can utilize to combat infection. When activated, they can release IFN-γ, cytotoxic granules, and induce cell death of infected cells. Type I IFN has been implicated in both NK cell cytotoxicity and NK cell IFN-γ production. Mice deficient in STAT1, a key transcription factor downstream of type I IFN receptor, have been shown to have decreased NK cell cytotoxicity and increased virus-induced mortality in comparison to control mice (39). In the context of NK cell IFN-γ production, type I IFN is essential for this process in multiple virus infections, including MCMV, adenovirus, vaccinia virus, and HSV (2, 40–43). Type I IFN has been shown to act directly on NK cells to induce their release of IFN-γ in the context of adenovirus, vaccinia virus, and LCMV infections, whereas other evidence suggests that type I IFN stimulates DCs to trans-present IL-15 to activate NK cells in MCMV infection (2, 40–44). Recently, we have provided evidence demonstrating that NK cell IFN-γ production relies on type I IFN induction of IL-18 from inflammatory monocytes, rather than DCs in a mucosal HSV-2 infection (2). Our differing results may stem from the route of infection, where previous evidence used in vitro systems or non-mucosal routes of infection.
Type I IFN Negative Regulation: Beyond Interfering With Infection
As more evidence emerges, there is a greater understanding and appreciation for the suppressive and negative regulatory aspects of type I IFN. Early on, studies had shown that type I IFN exerted anti-proliferative effects on immune cells and cell lines (45, 46). Recently, Thomas et al. elegantly demonstrated that while all type I IFN subtypes were capable of inducing an intracellular antiviral response, the affinity of an individual type I IFN subtype to the type I IFN receptor largely determined the ability of type I IFN to inhibit cellular proliferation (47). The antiproliferation effects of type I IFN required higher binding affinities to IFNAR (47). Beyond proliferation, type I IFN can suppress innate immune cell functions as well.
While an acute infection and upregulation of type I IFN is beneficial for enhancing DC activation of T-cell adaptive functions, a chronic infection with sustained type I IFN production has been shown to dampen DC expansion and induce a suppressive phenotype. In chronic LCMV infection, a persistent type I IFN signature prevented BM differentiation and proliferation of conventional DCs (48, 49). Further, stimulation of splenic DCs with IFN-β, in vivo, resulted in a decrease in total CD11c+ cell number. In addition to reducing DC expansion, a chronic type I IFN signature was shown to upregulate PD-L1 expression and IL-10 in both DCs and macrophages (50, 51).
Type I IFN largely has a suppressive effect on macrophages. The literature largely suggests that it downregulates their expression of the IFN-γ receptor, making them less sensitive to IFN-γ stimulation (52). In certain bacterial infections, such as francisella tularensis and mycobacterium tuberculosis, type I IFN signaling is detrimental to the host (53–56). The ability of type I IFN to downregulate the IFN-γ receptor on macrophages likely contributes to this phenomenon.
As mentioned previously, type I IFN has been shown to be critical for inducing the antiviral functions of NK cells. Conversely, and almost paradoxically, type I IFN has also been shown to suppress the very functions that it enables. During LCMV infection, Teijaro et al. found that blocking the type I IFN receptor rescued IFN-γ production from NK cells (48). Further, persistent type I IFN production can induce expression of PD-L1 ligands, which is a mechanism that can suppress NK cell antiviral function (48). Though administration of pegylated IFN-α2 therapy resulted in an increased NK cell activation, TRAIL, and CD107a receptor expression in HCV-infected individuals, there was a concomitant reduction in IFN-γ+ NK cells within the PBMC compartment (57, 58). This contradictory effect of type I IFN may stem from the timing and magnitude of type I IFN produced or a shift in transcription factor association with the type I IFN receptor. In a listeria monocytogenes infection, exogenous IFN-β administered at an earlier time point during infection was able to activate NK cells and promote clearance of the infection, whereas the endogenous IFN-β produced at 24 h post-infection resulted in an impaired NK cell response (59). Further, Marshall et al. found that stimulation of NK cells with supernatants from CpG-stimulated pDCs in addition to IFN-α suppressed IFN-γ release from NK cells (60). In a seminal study from Miyagi et al. they demonstrated that stimulation of NK cells with type I IFN shifted the balance of transcription factors from a STAT4 association with the type I IFN receptor, which upon phosphorylation and nuclear translocation resulted in an initial burst of IFN-γ, to a STAT1 association that subsequently led to inhibition of NK cell IFN-γ production (61). Thus, as increasing amounts of type I IFNs are released during infection, this leads to an increasing shift in association between STAT1 and IFNAR and ultimately inhibition of IFN-γ production from NK cells.
Along with promoting antiviral functions (and later limiting these very same functions), type I IFN has been shown to limit damaging immune responses that can lead to tissue pathology and collateral damage. In a model of influenza infection, absence of the type I IFN receptor resulted in significant virus-induced immunopathology. Duerr et al. demonstrated that this pathology was mediated by an upregulation of type 2 cytokines from unregulated innate lymphoid type 2 cells (ILC2s) (62). Thus, type I IFN suppresses ILC2 function during virus infection. Type I IFN was also found to suppress pro-inflammatory NOS2+ Ly6Clo monocyte function (36). Moreover, type I IFN dampens recruitment of neutrophils by suppressing epithelial CXCL1 and CXCL2 production during virus infection (35, 38, 63). Not only can neutrophils produce a multitude of molecules and proteases that can promote inflammation and tissue damage, they have been shown to instigate rhinovirus-induced asthma exacerbations in mice (64, 65). A table comparing the effects of type I IFN on the innate immune response is summarized in Table 1.
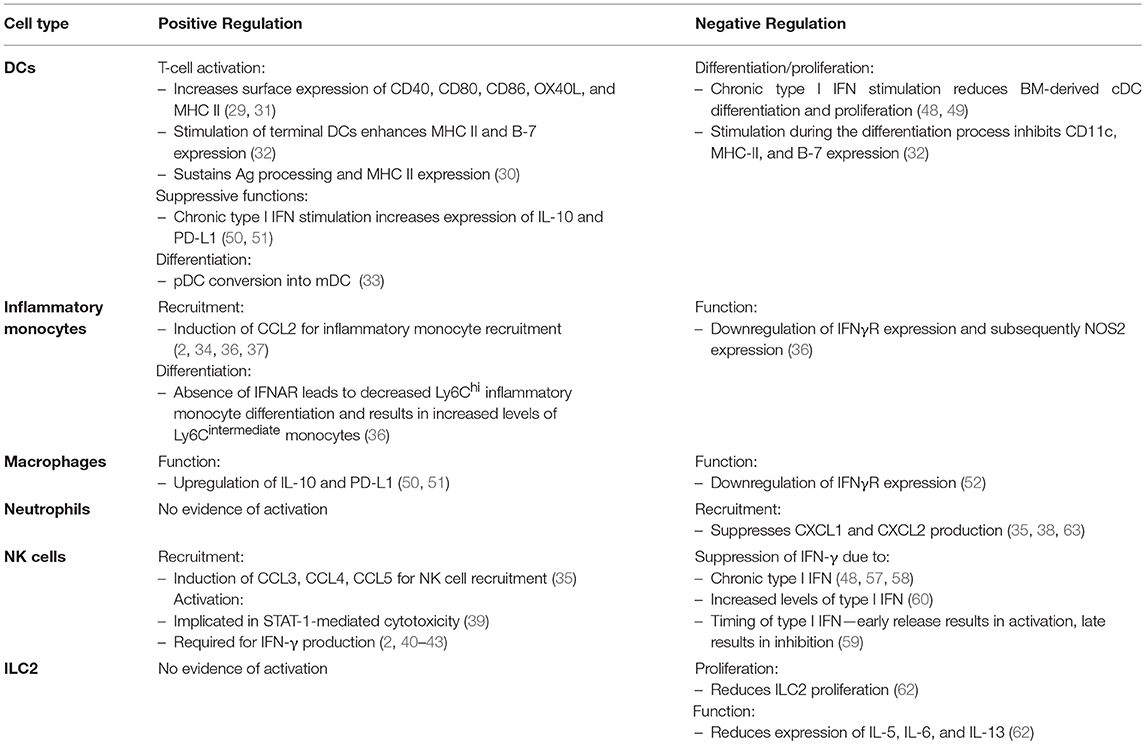
Table 1. The role of type I IFN in regulating the antiviral innate immune response.
Unweaving the Dual Nature of Type I IFNs
Within the literature, various themes are emerging that provide an explanation for this underlying dual functionality of type I IFN. First, acute virus infections and transient type I IFN production appears to promote antiviral responses from innate immune cells, while chronic infections with persistent type I IFN signatures result in a dampened antiviral response (66). This is particularly evident in the cases of chronic LCMV, which led to deterioration of the lymphoid architecture and T-cell suppression mediated by increased PD-L1 expression on DCs (48, 49). In simian immunodeficiency virus (SIV) infection, early administration of type I IFN resulted in a reduction in viral load, while chronic administration of type I IFN resulted in an increased level of virus and CD4+ T-cell depletion (67, 68). Second, the timing and magnitude of type I IFN produced can result in differing type I IFN responses, as previously discussed.
A growing body of evidence has revealed that individual subtypes of type I IFN can have differing effects, despite signaling through the same receptor. Indeed, stimulation of DCs with different subtypes of type I IFN resulted in varying profiles of receptor expression and cytokine production (69). Additionally, pre-treatment of influenza-infected mice with the same dose of different type I IFN subtypes resulted in varying levels of virus replication, with IFN-α5 and IFN-α6 having the greatest reduction in viral load (70). Their differing affinities for the type I IFN receptor, length of receptor binding, level of type I IFN receptor expression, and innate cellular differences may underlie the ability of these type I IFN subtypes to induce different responses (71). This is outlined in greater and more elegant detail in a review by Gideon Schreiber (71). In the context of virus infection, however, we hypothesize that type I IFN acts to optimize the antiviral response by both activating and enhancing beneficial innate immune cell function, while limiting detrimental and pathological immune responses that can cause unnecessary tissue damage.
Type II IFN: An Antiviral State of Mind
IFN-γ is an important component of the innate antiviral response and is predominantly produced by NK cells or innate lymphoid type 1 cells (2, 72, 73). In the context of HSV-2 infection, absence of IFN-γ production results in increased virus replication and decreased survival (74, 75). Indeed, IFN-γ has been shown to induce NO production, a potent inhibitor of virus replication, from surrounding cells (72, 76). As well, IFN-γ can induce intracellular antiviral programs, including PKR, as a resultant overlap in their gene expression with type I IFNs (77). Beyond that, however, IFN-γ itself has been demonstrated to impact the function of the surrounding innate immune cells, including macrophages and DCs.
The impact of antiviral IFN-γ on antigen presenting cells (APCs) is to enhance stimulation of the adaptive antiviral response to both clear the infection and generate memory as a safe-guard for future infections (78, 79). Thus, it is a critical propellant of the Th1 response. IFN-γ stimulation enhances the antigen presentation process during T-cell priming. It has been shown to increase various aspects of antigen presentation, including efficiency, quantity, quality, and diversity of peptides being loaded into the MHC I receptor (80). Along with MHC I, IFN-γ increases MHC II expression and maturation of DCs (81). Further, it induces the expression of IL-12 and co-stimulatory CD80 in antigen-presenting cells, which is a critical component of Th1 polarization (82–84).
With respect to macrophages, IFN-γ induced NO production from these cells not only inhibits virus replication, but also potently vasodilates blood vessels to decrease blood flow and allow for increased extravasation of recruited immune cells to the site of infection and inflammation (80). Further, IFN-γ has been shown to “prime” macrophages to release reactive oxygen species, through the upregulation of cellular components required for this function (85). IFN-γ also appears to increase macrophage receptor-mediated phagocytosis through the upregulation of complement receptors, though this has been observed more so in bacterial infections, rather than viral (86). Further, IFN-γ promotes polarization of macrophages to an M1 phenotype and primes these cells to produce pro-inflammatory cytokines IL-12, TNF-α, and IL-1β (87, 88).
Type II IFN Negative Regulation: An Emerging Role
IFN-γ has many overlapping features with type I IFNs, including suppression of type 2 immune responses and inhibition of proliferation. In the context of virus infection, however, we believe that IFN-γ released during the innate immune response has more of a supportive role in this respect as it is less potent in its effects in comparison to type I IFNs. Aside from type I IFN, IFN-γ has a number of immunoregulatory functions that serve to optimize the antiviral response and limit overzealous responses that could lead to collateral damage.
An optimal antiviral response involves both activating beneficial immune responses, while simultaneously inhibiting impractical and potentially damaging responses. In the context of virus infection, IFN-γ is a prototypical Th1 promoting cytokine. Further, evidence from Kang et al. demonstrates that IFN-γ plays a critical role in not only polarizing macrophages to an M1 phenotype, but actively suppresses the M2 polarization pathway (87). However, recent evidence has revealed that type I IFN is capable of suppressing type 2 immunity. Independently, both Duerr et al. and Moro et al. demonstrated that, similar to type I IFN, IFN-γ is able to suppress ILC2 proliferation and type 2 cytokine production (62, 89). Indeed, in vivo administration of IFN-γ potently suppressed IL-33-induced ILC2 proliferation, which was dependent upon STAT1 signaling (62). In the context of RSV infection, Stat1−/− mice, a transcription factor downstream of both type I and type II IFNs, led to increased lung pathology because of increased cytokine production from ILC2s and ILC3s (90). Further, in a mouse model of influenza infection, administration of IFN-γ suppressed ILC2 function while deficiency of IFN-γ led to increased IL-5 and amphiregulin release from ILC2s. These authors ultimately found that the suppressive effects of IFN-γ on ILC2 function led to increased lung pathology (91).
Along with dampening immune responses, there is evidence demonstrating that IFN-γ can indirectly induce immunoregulatory effects through the upregulation of PD-L1 and differentiation of myeloid derived suppressive cells. In conjunction with GM-CSF, IFN-γ was shown to differentiate monocytes into myeloid derived suppressor cells (MDSCs) in vitro (92). In a mouse model of endotoxemia, IFN-γ has also been shown to upregulate PD-L1 on neutrophils (93). A table comparing the effects of IFN-γ on the innate immune response is summarized in Table 2.
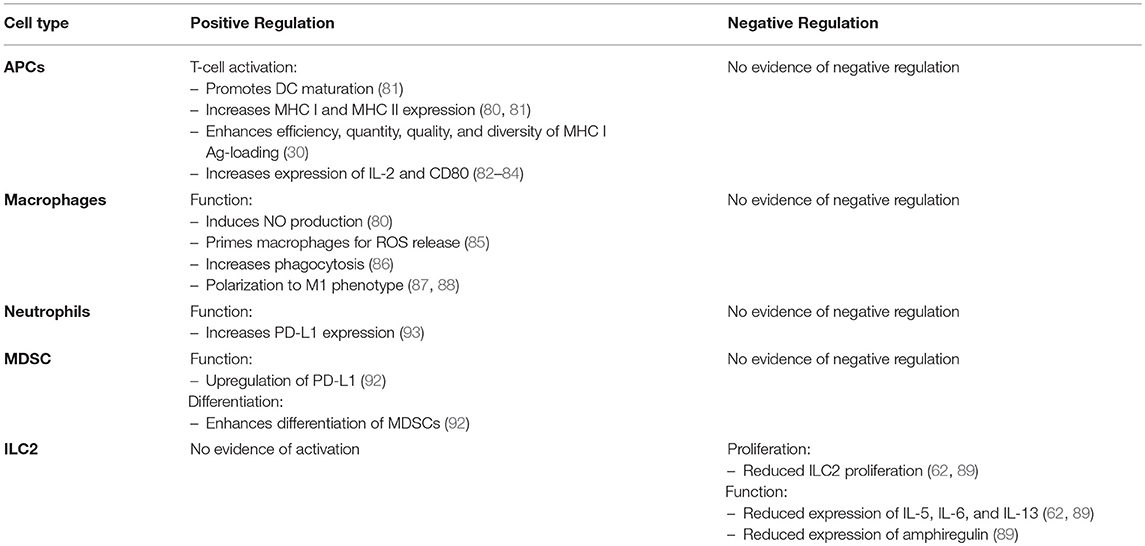
Table 2. The role of IFN-γ in regulating the antiviral innate immune response.
Understanding the Dual Nature of IFN-γ: Unraveling the Paradox
Similar to type I IFNs, IFN-γ has both seemingly paradoxical activating and suppressive functions on the innate antiviral response. These functions can be teased apart if we examine the cell type that IFN-γ is acting upon and bring other cytokines into the picture with IFN-γ. If we consider macrophages, IFN-γ has complementary effects on inducing an antiviral macrophage function. IFN-γ induces NO production, enhances macrophage antigen presenting function, and an overall M1 phenotype while actively suppressing the M2 phenotype (72, 80, 87). Similar to macrophages, IFN-γ predominantly increases antigen presentation function of DCs. Further, IFN-γ has a predominantly suppressive effect on ILC2 cells (62).
IFN-γ as a cytokine rarely acts alone and its effects should be considered in conjunction with other cytokines present in the local microenvironment. The combinatorial effect between IFN-γ and other cytokines likely plays a role in the ultimate outcome of IFN-γ stimulation. Indeed, both IFN-γ and TNF-α have been shown to synergize in the upregulation of iNOS in macrophages. Salim et al. used mathematical modeling to dissect out the roles of each cytokine and found that TNF-α was largely responsible for the timing of iNOS induction by inducing a rapid response, whereas IFN-γ impacted the levels and concentrations of NO production (94). Further, the role of IFN-γ in the in vitro differentiation process of MDSCs required an initial priming with GM-CSF. Ribechini et al. found that GM-CSF altered the signaling pathway of IFN-γ allowing it to differentiate monocytes into MDSCs (92). In a recent article by Zha et al. they found that IFN-γ was able to suppress the functions of gp130 cytokines, particularly the ability of OSM, to differentiate mesenchymal stem cells through the upregulation of STAT1, concomitant decrease in STAT3 activation, and internalization of the gp130 receptor (95). Thus, IFN-γ can both be altered by additional cytokine signaling as well as regulate the signaling pathways of other cytokines.
Putting the Pieces of the Puzzle Together
As we start to put the pieces of this type I and type II IFN puzzle together, we can see that these two cytokines act in concert with one another to limit virus replication and encourage an antiviral adaptive immune response while suppressing detrimental functions of other immune cells to limit tissue pathology. Using vaginal HSV-2 infection as an example, we find that there are multiple waves of type I IFN production, starting with IFN-β at 12 h post-infection (20). This early wave of IFN-β is likely responsible for the induction of MCP-1-mediated inflammatory monocyte recruitment, ultimately leading to IL-18-induced NK cell IFN-γ production (2). From there, we've observed a second wave of type I IFN production, both IFN-α and IFN-β, at 48 h post-infection (19). Along with type I IFN, there's a sharp increase in IFN-γ from NK cells at 48 h post-infection (16). The IFN-γ released from NK cells is also negatively regulated by type I IFN, as NK cells lacking IFNAR have increased IFN-γ production in the context of HSV-2 infection (2). This second wave of type I and II IFNs likely work in concert with each other to promote APC maturation, upregulation of co-stimulatory molecules, and antigen processing and presentation to promote Th1 polarization, while simultaneously suppressing ILC2-mediated immunopathology (Figure 1).
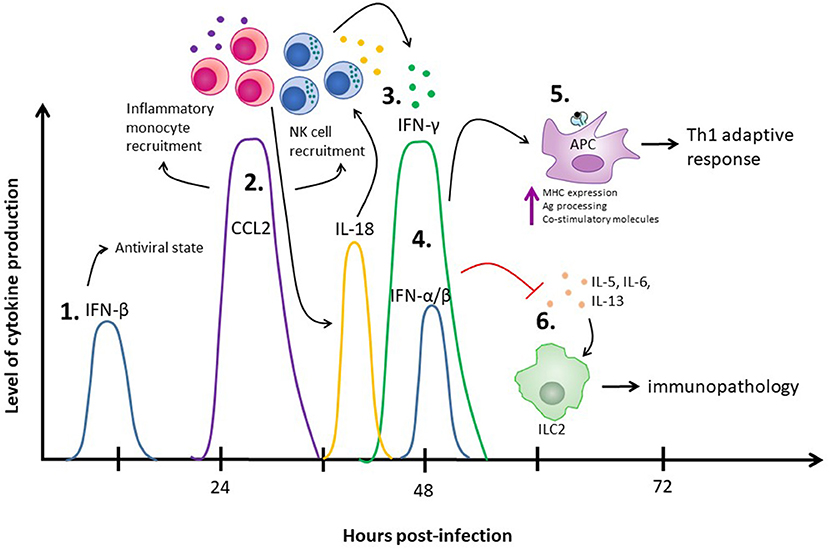
Figure 1. The role of IFNs in the innate immune response to HSV-2 infection. (1) IFN-β is produced at 12 h post-infection and through autocrine and paracrine signaling places surrounding cells into an antiviral state. (2) The IFN-β produced at 12 h post-infection also increases production of CCL2 between days 1 and 2 post-infection, which results in inflammatory monocyte recruitment and has been implicated in NK cell recruitment. (3) The recruited inflammatory monocytes result in release of IL-18, which stimulates NK cells to produce IFN-γ at 48 h post-infection. (4) A second wave of type I IFNs, including both IFN-α and IFN-β, are detected at 48 h post-infection. (5) Both IFN-γ and the type I IFNs produced at 48 h post-infection enhance APC antigen presentation capacities to stimulate a Th1 adaptive immune response. (6) Simultaneously, the type I IFNs at 48 h inhibit ILC2-mediated virus-induced immunopathology. IFN-γ, supporting the negative regulatory effects of type I IFN, also suppresses ILC2-mediated immunopathology.
Without type I IFN, and potentially type II IFN, there is uncontrolled virus replication coupled with uncontrolled inflammation that work together to cause tissue demise. On the other hand, a chronic type I IFN signature is detrimental as it can result in immunosuppression and increased virus replication. Thus, we believe a balanced and appropriate type I IFN response is required to regulate an optimal and advantageous antiviral innate immune response.
Clinical Implications: Going Beyond Infection
While the focus of this review has been on type I and II IFNs and their ability to control the innate immune response, IFNs have been implicated in several non-infectious pathological conditions. Select autoimmune diseases, the most prominent being systemic erythematous lupus (SLE), have high type I IFN signatures associated with their pathology (96). An antibody targeting human IFNAR has recently been developed to block this signature with therapeutic benefit (97). On the other hand, IFN-β therapy has had success in treating multiple sclerosis (98). Indeed, the concepts discussed in this review are relevant in the context of pharmacotherapies targeting the type I and type II IFN pathways. This begs the question: what is the role of type I IFN outside of virus infection? A growing amount of evidence has shown that type I IFN production is not isolated to infectious disease stimuli, it can be produced during any inflammatory insult. Thus, our fundamental understanding of the innate immune response during virus infection has an underlying application to many disease processes, beyond virus infection.
Author Contributions
AL generated themes and ideas and wrote the manuscript. AA guided and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR) awarded to Ali Ashkar. Ali Ashkar is also a recipient of a CIHR Tier 1 Canada Research Chair.
References
1. Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, et al. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell (1990) 62:379–90. doi: 10.1016/0092-8674(90)90374-N
2. Lee AJ, Chen B, Chew MV, Barra NG, Shenouda MM, Nham T, et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J Exp Med. (2017) 214:1153–67. doi: 10.1084/jem.20160880
3. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. (2008) 9:503–10. doi: 10.1038/ni1582
4. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
5. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi: 10.1126/science.1232458
6. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity (2006) 25:349–60. doi: 10.1016/j.immuni.2006.08.009
7. Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. (2008) 26:535–84. doi: 10.1146/annurev.immunol.26.021607.090400
8. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A, and Type I interferons in infectious disease Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
9. Marchetti M, Monier MN, Fradagrada A, Mitchell K, Baychelier F, Eid P, et al. Stat-mediated signaling induced by type I and type II interferons (IFNs) is differentially controlled through lipid microdomain association and clathrin-dependent endocytosis of IFN receptors. Mol Biol Cell (2006) 17:2896–909. doi: 10.1091/mbc.e06-01-0076
10. Uddin S, Lekmine F, Sharma N, Majchrzak B, Mayer I, Young PR, et al. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J Biol Chem. (2000) 275:27634–40. doi: 10.1074/jbc.M003170200
11. Uddin S, Yenush L, Sun XJ, Sweet ME, White MF, Platanias LC. Interferon-alpha engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3'-kinase. J Biol Chem. (1995) 270:15938–41. doi: 10.1074/jbc.270.27.15938
12. Uddin S, Majchrzak B, Woodson J, Arunkumar P, Alsayed Y, Pine R, et al. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. (1999) 274:30127–31. doi: 10.1074/jbc.274.42.30127
13. Platanias LC, Sweet ME. Interferon alpha induces rapid tyrosine phosphorylation of the vav proto-oncogene product in hematopoietic cells. J Biol Chem. (1994) 269:3143–6.
14. Ahmad S, Alsayed YM, Druker BJ, Platanias LC. The type I interferon receptor mediates tyrosine phosphorylation of the CrkL adaptor protein. J Biol Chem. (1997) 272:29991–4. doi: 10.1074/jbc.272.48.29991
15. de Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, Braniff SJ, et al. Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR1. Nat Immunol. (2013) 14:901–7. doi: 10.1038/ni.2667
16. Gill N, Chenoweth MJ, Verdu EF, Ashkar AA. NK cells require type I IFN receptor for antiviral responses during genital HSV-2 infection. Cell Immunol. (2011) 269:29–37. doi: 10.1016/j.cellimm.2011.03.007
17. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol. (2011) 89:216–24. doi: 10.1038/icb.2010.78
18. Gotthardt D, Sexl V. STATs in NK-Cells: The good, the bad, and the ugly. Front Immunol. (2016) 7:694. doi: 10.3389/fimmu.2016.00694
19. Oh JE, Kim BC, Chang DH, Kwon M, Lee SY, Kang D, et al. Dysbiosis-induced IL-33 contributes to impaired antiviral immunity in the genital mucosa. Proc Natl Acad Sci USA. (2016) 113:E762–71. doi: 10.1073/pnas.1518589113
20. Gill N, Deacon PM, Lichty B, Mossman KL, Ashkar AA. Induction of innate immunity against herpes simplex virus type 2 infection via local delivery of Toll-like receptor ligands correlates with beta interferon production. J Virol. (2006) 80:9943–50. doi: 10.1128/JVI.01036-06
21. Lee AJ, Ashkar AA. Herpes simplex virus-2 in the genital mucosa: insights into the mucosal host response and vaccine development. Curr Opin Infect Dis. (2012) 25:92–9. doi: 10.1097/QCO.0b013e32834e9a56
22. Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science (1994) 264:1918–21. doi: 10.1126/science.8009221
23. van den Broek MF, Muller U, Huang S, Zinkernagel RM, Aguet M. Immune defence in mice lacking type I and/or type II interferon receptors. Immunol Rev. (1995) 148:5–18. doi: 10.1111/j.1600-065X.1995.tb00090.x
24. de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. (2001) 69:912–20. doi: 10.1189/jlb.69.6.912
25. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature (2011) 472:481–5. doi: 10.1038/nature09907
26. Mangan NE, Fung KY. Type I interferons in regulation of mucosal immunity. Immunol Cell Biol. (2012) 90:510–9. doi: 10.1038/icb.2012.13
27. Carroll SS, Chen E, Viscount T, Geib J, Sardana MK, Gehman J, et al. Cleavage of oligoribonucleotides by the 2',5'-oligoadenylate- dependent ribonuclease L. J Biol Chem. (1996) 271:4988–92. doi: 10.1074/jbc.271.9.4988
28. Liu SY, Sanchez DJ, Cheng G. New developments in the induction and antiviral effectors of type I interferon. Curr Opin Immunol. (2011) 23:57–64. doi: 10.1016/j.coi.2010.11.003
29. Kurche JS, Haluszczak C, McWilliams JA, Sanchez PJ, Kedl RM. Type I IFN-dependent T cell activation is mediated by IFN-dependent dendritic cell OX40 ligand expression and is independent of T cell IFNR expression. J Immunol. (2012) 188:585–93. doi: 10.4049/jimmunol.1102550
30. Simmons DP, Wearsch PA, Canaday DH, Meyerson HJ, Liu YC, Wang Y, et al. Type I IFN drives a distinctive dendritic cell maturation phenotype that allows continued class II MHC synthesis and antigen processing. J Immunol. (2012) 188:3116–26. doi: 10.4049/jimmunol.1101313
31. Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, et al. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood (2002) 99:3263–71. doi: 10.1182/blood.V99.9.3263
32. Hahm B, Trifilo MJ, Zuniga EI, Oldstone MB. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity (2005) 22:247–57. doi: 10.1016/j.immuni.2005.01.005
33. Zuniga EI, McGavern DB, Pruneda-Paz JL, Teng C, Oldstone MB. Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat Immunol. (2004) 5:1227–34. doi: 10.1038/ni1136
34. Iijima N, Mattei LM, Iwasaki A. Recruited inflammatory monocytes stimulate antiviral Th1 immunity in infected tissue. Proc Natl Acad Sci USA. (2011) 108:284–9. doi: 10.1073/pnas.1005201108
35. Uyangaa E, Kim JH, Patil AM, Choi JY, Kim SB, Eo SK. Distinct upstream role of type I IFN signaling in hematopoietic stem cell-derived and epithelial resident cells for concerted recruitment of Ly-6Chi monocytes and NK cells via CCL2-CCL3 cascade. PLoS Pathog (2015) 11:e1005256. doi: 10.1371/journal.ppat.1005256
36. Stifter SA, Bhattacharyya N, Pillay R, Florido M, Triccas JA, Britton WJ, et al. Functional interplay between type I and II interferons is essential to limit influenza A virus-induced tissue inflammation. PLoS Pathog (2016) 12:e1005378. doi: 10.1371/journal.ppat.1005378
37. Peralta Ramos JM, Bussi C, Gaviglio EA, Arroyo DS, Baez NS, Rodriguez-Galan MC, et al. Type I IFNs are required to promote central nervous system immune surveillance through the recruitment of inflammatory monocytes upon systemic inflammation. Front Immunol. (2017) 8:1666. doi: 10.3389/fimmu.2017.01666
38. Seo SU, Kwon HJ, Ko HJ, Byun YH, Seong BL, Uematsu S, etal Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog. (2011) 7:e1001304. doi: 10.1371/journal.ppat.1001304
39. Liang S, Wei H, Sun R, Tian Z. IFNalpha regulates NK cell cytotoxicity through STAT1 pathway. Cytokine (2003) 23:190–9. doi: 10.1016/S1043-4666(03)00226-6
40. Martinez J, Huang X, Yang Y. Direct action of type I IFN on NK cells is required for their activation in response to vaccinia viral infection in vivo. J Immunol. (2008) 180:1592–7. doi: 10.4049/jimmunol.180.3.1592
41. Zhu J, Huang X, Yang Y. A critical role for type I IFN-dependent NK cell activation in innate immune elimination of adenoviral vectors in vivo. Mol Ther. (2008) 16:1300–7. doi: 10.1038/mt.2008.88
42. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity (2007) 26:503–17. doi: 10.1016/j.immuni.2007.03.006
43. Baranek T, Manh TP, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, et al. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe. (2012) 12:571–84. doi: 10.1016/j.chom.2012.09.002
44. Mack EA, Kallal LE, Demers DA, Biron CA. Type 1 interferon induction of natural killer cell gamma interferon production for defense during lymphocytic choriomeningitis virus infection. MBio (2011) 2:e00169–11. doi: 10.1128/mBio.00169-11
45. Paucker K, Cantell K, Henle W. Quantitative studies on viral interference in suspended L cells. III. Effect of interfering viruses and interferon on the growth rate of cells. Virology (1962) 17:324–34. doi: 10.1016/0042-6822(62)90123-X
46. Welsh RM, Bahl K, Marshall HD, Urban SL. Type 1 interferons and antiviral CD8 T-cell responses. PLoS Pathog. (2012) 8:e1002352. doi: 10.1371/journal.ppat.1002352
47. Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell (2011) 146:621–32. doi: 10.1016/j.cell.2011.06.048
48. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science (2013) 340:207–11. doi: 10.1126/science.1235214
49. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340:202–7. doi: 10.1126/science.1235208
50. Wilson EB, Kidani Y, Elsaesser H, Barnard J, Raff L, Karp CL, et al. Emergence of distinct multiarmed immunoregulatory antigen-presenting cells during persistent viral infection. Cell Host Microbe. (2012) 11:481–91. doi: 10.1016/j.chom.2012.03.009
51. Ng CT, Oldstone MB. Infected CD8alpha- dendritic cells are the predominant source of IL-10 during establishment of persistent viral infection. Proc Natl Acad Sci USA. (2012) 109:14116–21. doi: 10.1073/pnas.1211910109
52. Eshleman EM, Delgado C, Kearney SJ, Friedman RS, Lenz LL. Down regulation of macrophage IFNGR1 exacerbates systemic L. monocytogenes infection. PLoS Pathog. (2017) 13:e1006388. doi: 10.1371/journal.ppat.1006388
53. McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N, et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-gamma for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol. (2014) 193:3600–12. doi: 10.4049/jimmunol.1401088
54. Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol. (2011) 187:2540–7. doi: 10.4049/jimmunol.1100926
55. Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, et al. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol. (2010) 184:3755–67. doi: 10.4049/jimmunol.0902065
56. Metzger DW, Bakshi CS, Kirimanjeswara G. Mucosal immunopathogenesis of Francisella tularensis. Ann N Y Acad Sci. (2007) 1105:266–83. doi: 10.1196/annals.1409.007
57. Ahlenstiel G, Edlich B, Hogdal LJ, Rotman Y, Noureddin M, Feld JJ, et al. Early changes in natural killer cell function indicate virologic response to interferon therapy for hepatitis C. Gastroenterology (2011) 141:1231–9, 1239 e1-2. doi: 10.1053/j.gastro.2011.06.069
58. Werner JM, Serti E, Chepa-Lotrea X, Stoltzfus J, Ahlenstiel G, Noureddin M, et al. Ribavirin improves the IFN-gamma response of natural killer cells to IFN-based therapy of hepatitis C virus infection. Hepatology (2014) 60:1160–9. doi: 10.1002/hep.27092
59. Pontiroli F, Dussurget O, Zanoni I, Urbano M, Beretta O, Granucci F, et al. The timing of IFNbeta production affects early innate responses to Listeria monocytogenes and determines the overall outcome of lethal infection. PLoS ONE (2012) 7:e43455. doi: 10.1371/journal.pone.0043455
60. Marshall JD, Heeke DS, Abbate C, Yee P, Van Nest G. Induction of interferon-gamma from natural killer cells by immunostimulatory CpG DNA is mediated through plasmacytoid-dendritic-cell-produced interferon-alpha and tumour necrosis factor-alpha. Immunology (2006) 117:38–46. doi: 10.1111/j.1365-2567.2005.02261.x
61. Miyagi T, Gil MP, Wang X, Louten J, Chu WM, Biron CA. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J Exp Med. (2007) 204:2383–96. doi: 10.1084/jem.20070401
62. Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol. (2016) 17:65–75. doi: 10.1038/ni.3308
63. Stock AT, Smith JM, Carbone FR. Type I IFN suppresses Cxcr2 driven neutrophil recruitment into the sensory ganglia during viral infection. J Exp Med. (2014) 211:751–9. doi: 10.1084/jem.20132183
64. Galani IE, Andreakos E. Neutrophils in viral infections: Current concepts and caveats. J Leukoc Biol. (2015) 98:557–64. doi: 10.1189/jlb.4VMR1114-555R
65. Toussaint M, Jackson DJ, Swieboda D, Guedan A, Tsourouktsoglou TD, Ching YM, et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat Med. (2017) 23:681–91. doi: 10.1038/nm.4332
66. Teijaro JR. Type I interferons in viral control and immune regulation. Curr Opin Virol. (2016) 16:31–40. doi: 10.1016/j.coviro.2016.01.001
67. Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest. (2009) 119:3544–55. doi: 10.1172/JCI40093
68. Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature (2014) 511:601–5. doi: 10.1038/nature13554
69. Garcin G, Bordat Y, Chuchana P, Monneron D, Law HK, Piehler J, et al. Differential activity of type I interferon subtypes for dendritic cell differentiation. PLoS ONE (2013) 8:e58465. doi: 10.1371/journal.pone.0058465
70. James CM, Abdad MY, Mansfield JP, Jacobsen HK, Vind AR, Stumbles PA, et al. Differential activities of alpha/beta IFN subtypes against influenza virus in vivo and enhancement of specific immune responses in DNA vaccinated mice expressing haemagglutinin and nucleoprotein. Vaccine (2007) 25:1856–67. doi: 10.1016/j.vaccine.2006.10.038
71. Schreiber G. The molecular basis for differential type I interferon signaling. J Biol Chem. (2017) 292:7285–7294. doi: 10.1074/jbc.R116.774562
72. Karupiah G, Xie QW, Buller RM, Nathan C, Duarte C, MacMicking JD. Inhibition of viral replication by interferon-gamma-induced nitric oxide synthase. Science (1993) 261:1445–8. doi: 10.1126/science.7690156
73. Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, et al. ILC1 confer early host protection at initial sites of viral infection. Cell (2017) 171:795–808 e12. doi: 10.1016/j.cell.2017.09.052
74. Thapa M, Kuziel WA, Carr DJ. Susceptibility of CCR5-deficient mice to genital herpes simplex virus type 2 is linked to NK cell mobilization. J Virol. (2007) 81:3704–13. doi: 10.1128/JVI.02626-06
75. Ashkar AA, Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol. (2003) 77:10168–71. doi: 10.1128/JVI.77.18.10168-10171.2003
76. Croen KD. Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J Clin Invest. (1993) 91:2446–52. doi: 10.1172/JCI116479
77. Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schafer R, Kumar A, et al. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. (1995) 14:6095–106.
78. Goldszmid RS, Caspar P, Rivollier A, White S, Dzutsev A, Hieny S, et al. NK cell-derived interferon-gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity (2012) 36:1047–59. doi: 10.1016/j.immuni.2012.03.026
79. Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. (2004) 5:1260–5. doi: 10.1038/ni1138
80. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. (2004) 75:163–89. doi: 10.1189/jlb.0603252
81. Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science (1994) 265:106–9. doi: 10.1126/science.8016643
82. Yokozeki H, Katayama I, Ohki O, Arimura M, Takayama K, Matsunaga T, et al. Interferon-gamma differentially regulates CD80 (B7-1) and CD86 (B7-2/B70) expression on human Langerhans cells. Br J Dermatol. (1997) 136:831–7. doi: 10.1111/j.1365-2133.1997.tb03921.x
83. Bauvois B, Nguyen J, Tang R, Billard C, Kolb JP. Types I and II interferons upregulate the costimulatory CD80 molecule in monocytes via interferon regulatory factor-1. Biochem Pharmacol. (2009) 78:514–22. doi: 10.1016/j.bcp.2009.05.005
84. Ma X, Chow JM, Gri G, Carra G, Gerosa F, Wolf SF, et al. The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J Exp Med. (1996) 183:147–57. doi: 10.1084/jem.183.1.147
85. MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. (1997) 15:323–50. doi: 10.1146/annurev.immunol.15.1.323
86. Drevets DA, Leenen PJ, Campbell PA. Complement receptor type 3 mediates phagocytosis and killing of Listeria monocytogenes by a TNF-alpha- and IFN-gamma-stimulated macrophage precursor hybrid. Cell Immunol. (1996) 169:1–6. doi: 10.1006/cimm.1996.0083
87. Kang K, Park SH, Chen J, Qiao Y, Giannopoulou E, Berg K, et al. Interferon-gamma represses M2 gene expression in human macrophages by disassembling enhancers bound by the transcription factor MAF. Immunity (2017) 47:235–250 e4. doi: 10.1016/j.immuni.2017.07.017
88. Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine (2018) 30:303–16. doi: 10.1016/j.ebiom.2018.02.009
89. Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. (2016) 17:76–86. doi: 10.1038/ni.3309
90. Stier MT, Goleniewska K, Cephus JY, Newcomb DC, Sherrill TP, Boyd KL, et al. STAT1 represses cytokine-producing group 2 and group 3 innate lymphoid cells during viral infection. J Immunol. (2017) 199:510–19. doi: 10.4049/jimmunol.1601984
91. Califano D, Furuya Y, Roberts S, Avram D, McKenzie ANJ, Metzger DW. IFN-gamma increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol. (2018) 11:209–19. doi: 10.1038/mi.2017.41
92. Ribechini E, Hutchinson JA, Hergovits S, Heuer M, Lucas J, Schleicher U, et al. Novel GM-CSF signals via IFN-gammaR/IRF-1 and AKT/mTOR license monocytes for suppressor function. Blood Adv. (2017) 1:947–60. doi: 10.1182/bloodadvances.2017006858
93. Langereis JD, Pickkers P, de Kleijn S, Gerretsen J, de Jonge MI, Kox M. Spleen-derived IFN-gamma induces generation of PD-L1(+)-suppressive neutrophils during endotoxemia. J Leukoc Biol. (2017) 102:1401–9. doi: 10.1189/jlb.3A0217-051RR
94. Salim T, Sershen CL, May EE. Investigating the role of TNF-alpha and IFN-gamma activation on the dynamics of iNOS gene expression in LPS stimulated macrophages. PLoS ONE (2016) 11:e0153289. doi: 10.1371/journal.pone.0153289
95. Zha Z, Bucher F, Nejatfard A, Zheng T, Zhang H, Yea K, et al. Interferon-gamma is a master checkpoint regulator of cytokine-induced differentiation. Proc Natl Acad Sci USA. (2017) 114:E6867–74. doi: 10.1073/pnas.1706915114
96. Lee-Kirsch MA. The type I interferonopathies. Annu Rev Med. (2017) 68:297–315. doi: 10.1146/annurev-med-050715-104506
97. Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:376–86. doi: 10.1002/art.39962
Keywords: type I interferon, interferon-γ, innate immunity, virus infection, immunoregulation
Citation: Lee AJ and Ashkar AA (2018) The Dual Nature of Type I and Type II Interferons. Front. Immunol. 9:2061. doi: 10.3389/fimmu.2018.02061
Received: 01 June 2018; Accepted: 21 August 2018;
Published: 11 September 2018.
Edited by:
Maziar Divangahi, McGill University, CanadaReviewed by:
Jianzhong Zhu, Yangzhou University, ChinaAlain Lamarre, Institut National de la Recherche Scientifique (INRS), Canada
Copyright © 2018 Lee and Ashkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ali A. Ashkar, ashkara@mcmaster.ca