
- 1 Saga Medical School, Health Administration Center, Saga, Japan
- 2 Division of Hepatology, Saga Medical School, Department of Internal Medicine, Saga University, Saga, Japan
Hepatocellular carcinoma (HCC) often arises from chronically diseased livers. Persistent liver inflammation causes the accumulation of excessive extracellular matrix (ECM) proteins and impairs the liver function, finally leading to the development of HCC. A pleiotropic cytokine, transforming growth factor (TGF)-β1, plays critical roles throughout the process of fibrogenesis and hepatocarcinogenesis. In the liver, TGF-β1 inhibits the proliferation of hepatocytes and stimulates the production of ECM from hepatic stellate cells (HSCs) to maintain tissue homeostasis. During disease progression, both growth factors/cytokines and the ECM alter the TGF-β1 signals by modifying the phosphorylation of Smad proteins at their C-terminal and linker regions. TGF-β1 stimulates the expression of integrins, cellular receptors for ECM, along with an increase in ECM accumulation. The activation of integrins by the ECM modulates the response to TGF-β1 in hepatic cells, resulting in their resistance to TGF-β1-induced growth suppression in hepatocytes and the sustained production of ECM proteins in activated HSCs/myofibroblasts. Both growth factor receptors and integrins modify the expression and/or functions of the downstream effectors of TGF-β1, resulting in the escape of hepatocytes from TGF-β1-induced apoptosis. Recent studies have revealed that the alterations of Smad phosphorylation that occur as the results of the crosstalk between TGF-β1, growth factors and integrins could change the nature of TGF-β1 signals from tumor suppression to promotion. Therefore, the modification of Smad phosphorylation could be an attractive target for the prevention and/or treatment of HCC.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignant neoplasms in the world and it is closely associated with many risk factors, including hepatitis B and C virus infection, alcoholic liver disease, non-alcoholic fatty liver diseases related to obesity and/or diabetes, aflatoxin B1 contamination of food, and less frequently, genetic predisposition. The incidence of HCC is increasing worldwide, mainly due to the spread of chronic infections with hepatitis B and C viruses (Okuda, 2000; LIovet et al., 2003; El-Serag and Rudolph, 2007). Most of these risk factors cause persistent inflammation in the liver, leading to the formation of liver cirrhosis characterized by an excessive accumulation of the collagen-rich extracellular matrix (ECM). The progression of liver fibrosis to cirrhosis is closely associated with the development of HCC based on both clinical observations and evidence in experimental animal models (Tsukuma et al., 1993; Sakaida et al., 1998; Yoshida et al., 1999; El-Serag and Rudolph, 2007; Giannelli et al., 2011).
Transforming growth factor (TGF)-β1 plays critical roles in hepatic fibrogenesis, hepatocarcinogenesis, and HCC progression (Inagaki and Okazaki, 2007; Friedman, 2008; Giannelli et al., 2011). TGF-β is a member of a large family of multifunctional cytokines, and consists of three isoforms called TGF-β1, β2, and β3, which share 80% homology in their amino acid sequences. TGF-β1 binds to its specific TGF-β receptor complex, consisting of the TGF-β type I receptor (TβRI) and type II receptor (TβRII), then TβRI phosphorylates the Smad2 and Smad3 transcription factors (TFs; Moses et al., 1990; Heldin et al., 1997; Massague et al., 2005). Phosphorylated Smad2 and Smad3 subsequently bind to Smad4 and translocate to the nucleus to initiate target gene expression. Several groups have reported the presence of a Smad-independent TGF-β1 activation pathway (Bhowmick et al., 2001; Derynck and Zhang, 2003; Moustakas and Heldin, 2005; Yi et al., 2005; Giannelli et al., 2011). TGF-β1 has been shown to rapidly activate ras and several members of the mitogen-activated protein kinase (MAPK) pathways, such as extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK) in several cell types. TGF-β1 can also stimulate the phosphoinositide-3 kinase (PI3K)-Akt pathway and Rho-like GTPases (Figure 1).
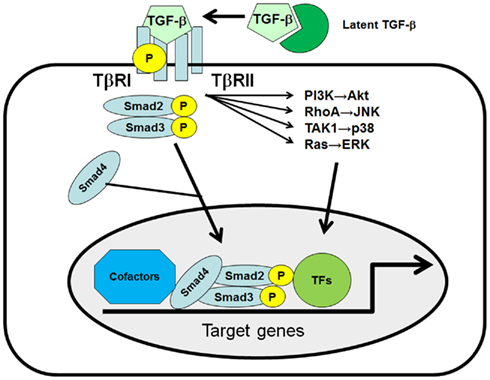
Figure 1. The TGF-β signaling pathway. Three distinct forms of TGF-β, TGF-β1, -β2, and -β3, can bind to the heterodimeric TGF-β receptor complex, consisting of the type I receptor (TβRI) and type II receptor (TβRII). Once TGF-β1 binds to the receptor complex, intracellular Smad2 and Smad3 are recruited and phosphorylated by TβRI at their C-terminal SSXS motif. Phosphorylated Smad2 and Smad3 subsequently bind to Smad4 and translocate to the nucleus to initiate target gene expression in collaboration with co-factors and other transcription factors (TFs). TGF-β can also activate the PI3K, RhoA, TAK1, and Ras/MAPK pathways in a Smad-independent manner.
In the liver TGF-β1 is a potent growth inhibitor of normal hepatocytes and HCC cells, and induces apoptosis and cellular senescence in these cells (Lin and Chou, 1992; Oberhammer et al., 1992; Siegel and Massague, 2003; Senturk et al., 2010). However, TGF-β1 is highly expressed in many malignant tumors, including HCC, and tumor cells are frequently thought to have lost their sensitivity to TGF-β1 (Ito et al., 1991; Bedossa et al., 1995; Tsai et al., 1997; Caja et al., 2007). During the disease progression, numerous growth factors/inflammatory cytokines such as TGF-β1, hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), tumor necrosis factor-α (TNF-α), interleukins (ILs), and interferons (IFNs) are released from various types of hepatic cells and participate in apoptosis, inflammation, cell proliferation, tissue fibrosis/remodeling, and HCC development/progression (Inagaki and Okazaki, 2007; Friedman, 2008). While TGF-β1 inhibits hepatocyte proliferation in the early stage of liver injury, it stimulates the proliferation of fibroblasts and induces the expression of ECM genes, including fibrillar collagens, to repair the injured tissue. When injurious stimulation is repeated, as occurs during persistent hepatitis virus infection, the production of ECM continues, and fibrous materials accumulate, thus leading to the formation of cirrhosis that predisposes the liver to HCC (El-Serag and Rudolph, 2007; Giannelli et al., 2011; Figure 2). Along with the progression of fibrogenesis, an increased expression of integrin, a cell surface receptor for the ECM, is observed (Volpes et al., 1991; Couvelard et al., 1993; Levine et al., 2000). Integrins interact with growth factor receptors, including TGF-β1 as well as with ECM proteins, and can modulate the signal transduction pathways (Hayashida, 2010; Margadant and Sonnenberg, 2010). Recent data indicate that during the progression of chronic hepatitis, from cirrhosis to HCC, changes in the TGF-β1-induced Smad phosphorylation pattern occur, and the alternatively phosphorylated Smad proteins (pSmadL) exert distinct transcriptional activities (Matsuzaki et al., 2007; Matsuzaki, 2011).
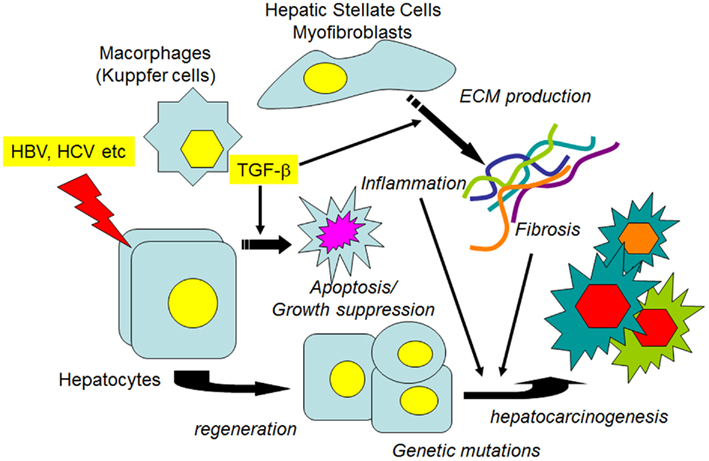
Figure 2. Transforming growth factor-β1, fibrosis, and HCC. When hepatocytes are injured by agents such as hepatitis viruses, various growth factors, and proinflammatory cytokines, as well as TGF-1β1 are released from non-parenchymal liver cells. In the earlier stages, growth factors stimulate the proliferation of hepatocytes to restore the liver function, while TGF-β1 induces the apoptosis of injured hepatocytes to maintain tissue homeostasis. If the injurious stimuli are persistent, ECM protein production from the activated HSCs/myofibroblasts is continuously stimulated. Both persistent inflammation and the accumulated ECM proteins contribute to the alteration of signal transduction pathways in regenerated hepatocytes, along with the accumulation of genetic mutations, which leads to the preneoplastic changes of hepatocytes and the eventual development of HCC.
In this manuscript we review the effects of growth factor-mediated signals and integrin-mediated signals on the tumor suppressive signals induced by TGF-β1 in liver epithelial cells and discuss the mechanisms regulating the phosphorylation of the Smad protein as potential therapeutic targets.
Liver Fibrosis, HCC, and Integrins
Among the various growth factors and proinflammatory cytokines involved in the long-lasting process of liver fibrogenesis and hepatocarcinogenesis, TGF-β1 plays critical roles throughout the progression of the disease (Inagaki and Okazaki, 2007; Friedman, 2008; Giannelli et al., 2011; Matsuzaki, 2011). Once the liver injury is initiated and continued, hepatic stellate cells (HSCs), the main source of ECM production in the liver, are activated by TGF-β1 and proinflammatory cytokines. Sustained expression of ECM genes by TGF-β1 in the activated HSCs and myofibroblasts leads to the accumulation of collagenous ECM proteins (Furukawa et al., 2003; Inagaki and Okazaki, 2007; Matsuzaki, 2011).
TGF-β1 is known to increase the expression of integrins, major cellular receptors for ECM proteins, as well as its ligands, the ECM proteins (Hayashida, 2010; Margadant and Sonnenberg, 2010). With the progression of liver fibrosis, the expression of integrins is also increased (Couvelard et al., 1993; Volpes et al., 1993; Levine et al., 2000). The integrins are large family of heterodimeric cell surface receptors consisting of 18 α- and 8 β-transmembrane subunits. Integrins can directly bind to ECM components and transmit the signals from the ECM to the cell interior and vice versa (Hynes, 2002). The short cytoplasmic tails of integrins are associated with adaptor proteins including Src, focal adhesion kinase (FAK), integrin-linked kinase (ILK), kindlin1, paxilin, talin, vinculin, and PINCH. These integrin-associated proteins not only transmit signals from the extracellular environment, but also increase integrins activation. Integrin also interacts with cytoskeletal proteins and can regulate the cellular shape and motility upon activation (Figure 3). Among the various integrin-associated proteins, kinases such as Src, FAK, or ILK activate the cellular signal transduction pathways including MAP kinase pathways (ERK, p38, JNK), PI3K/Akt pathways, and protein kinase C (PKC) cascades (Hynes, 2002; Hehlgans et al., 2007; Bottcher et al., 2009; Millard et al., 2011). Integrins have also been shown to interact with growth factor receptors including IGF-IR, PDGFR-β, c-Met, and EGFR (Alam et al., 2007; Desgrosellier and Cheresh, 2010; Ivaska and Heino, 2010). Similarly, integrins can modulate the TGF-β1/Samad pathways directly or indirectly via different mechanisms, while TGF-β1 stimulates the expression of integrins (Margadant and Sonnenberg, 2010).
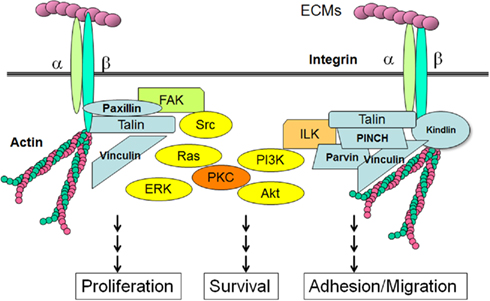
Figure 3. Integrins, adaptor proteins, and signaling pathways. When ECMs bind to integrin receptors and form focal adhesions, the cytoplasmic tails of β-integrins are associated with a number of adaptor proteins. Talin forms a contact with both the β-integrin tail and the actin cytoskeleton. ILK associates with actin through its partners such as parvin, PINCH and kindling. FAK is responsible for the binding of integrin-associated proteins like paxillin and talin. These integrin-associated proteins involve non-receptor type kinases, such as Src, FAK, or ILK, which activate cellular transduction pathways, including the MAP kinase pathways, PI3K/Akt pathways and PKCs. Integrins are also associated with cytoskeletal proteins through the adaptor proteins, and can regulate cellular shape and motility.
Based on this information, it has been speculated that an increased expression of integrins on the hepatocytes in a fibrotic liver could also modulate the intracellular signaling pathways and contribute to the development of HCC. The expression of several integrin subunits, including the α1, α2, α3, α5, α6, and β1 chains, is also upregulated in HCC cells, and their expression patterns are distinct from those of non-cancerous liver tissue (Volpes et al., 1993; Begum et al., 1995; Ozaki et al., 1998; Lai et al., 2011). Furthermore integrin α6 chain expression is correlated with a poor prognosis for HCC patients (Ke et al., 2011). Integrinsα1β1, α2β1 α3β1, and α6β1 play key roles in the invasion of HCC cells (Carloni et al., 2001; Giannelli et al., 2001; Torimura et al., 2001; Yang et al., 2006) and integrinsα3β1 and α6β4 promote the growth of HCC cells upon activation by their ligand, laminin-5 (Bergamini et al., 2007). These data strongly suggest the important roles of the cell-ECM interactions through integrin receptors in the progression of HCC.
Mechanisms of Resistance to TGF-β1-Induced Apoptosis in Liver Epithelial Cells
Transforming growth factor-β1 has a dual role in the development and progression of HCC. TGF-β1 is a potent growth inhibitor of normal hepatocytes and HCC cells and induces apoptosis in these cells, and as a result, it is regarded as a tumor-suppressive cytokine. However, TGF-β1 is able to promote the growth, invasion, and metastasis of HCC cells through the epithelial-mesenchymal transition (EMT; Bierie and Moses, 2006; Massague, 2008; Matsuzaki, 2011). The ability of TGF-β1 to inhibit or stimulate cancer progression is partly dependent on the tumor microenvironment, including the presence of growth factors/inflammatory cytokines and/or ECM proteins. During the progression of liver fibrogenesis with chronic inflammation, the hepatocyte responsiveness to TGF-β1 changes, and cells become resistant to TGF-β1-induced apoptosis, thus suggesting the existence of preneoplastic changes of hepatocytes in cirrhotic livers (Ito et al., 1991).
Growth Factors and TGF-β1-Induced Apoptosis
The presence of growth factors such as HGF and EGF that stimulate hepatocyte proliferation has been shown to affect the TGF-β1-induced growth-suppressive signal in hepatocytes (Fabregat et al., 1996; Murillo et al., 2005). Similar effects of growth factors on TGF-β1 signals were observed in TGF-β1-sensitive HCC cells (Mori et al., 2004; Caja et al., 2007). When TGF-β1 induces apoptosis in hepatic cells, several downstream molecules are involved in the induction of apoptosis. For example, the activation of p38 by GADD45β (Yoo et al., 2003) and JNK activation by the Daxx protein (Perlman et al., 2001) were reported to be involved in TGF-β1-induced apoptosis in hepatocytes. In the human HCC cell line, Hep3B, DAP kinase was reported to be induced by TGF-β1 in a Smad-dependent manner, and to promote the apoptosis by releasing cytochrome c from the mitochondria (Jang et al., 2002). We recently reported that programmed cell death 4 (PDCD4) is induced by TGF-β1 in a Smad-dependent manner, and promotes apoptosis in Huh7 human HCC cells (Zhang et al., 2006). The addition of EGF or the tumor promoter TPA inhibits TGF-β1-induced apoptosis by suppressing the induction of PDCD4 by TGF-β1. When TGF-β1 induces apoptosis, the activities of ERK, PI3K/Akt/S6K are attenuated (Petritsch et al., 2000). Activation of these pathways by EGF or TPA, both of which involve the activation of PKCs, abolished TGF-β1-induced apoptosis in Huh7 cells, suggesting the involvement of PKCs, as well as the ERK and PI3K/Akt pathways, in the resistance to TGF-β1-induced growth inhibition in HCC cells (Nakashima et al., 2010).
The Interaction between Growth Factor Receptors and Integrins
Interestingly, integrins have been reported to interact with several growth factor receptors, including IGF-IR, PDGFR-β, c-Met, VEGFR, and EGFR, as well as TGF-β receptors, and to modulate their downstream signals by different mechanisms (Alam et al., 2007; Desgrosellier and Cheresh, 2010; Ivaska and Heino, 2010). PDGFR-β, a receptor tyrosine kinase receptor involved in the activation of profibrogenic HCS cells, is regulated by integrins. Detachment of cells from the ECM decreases PDGFR autophosphorylation and protein expression, and re-plating of cells on fibronectin prevents the proteasomal degradation of the PDGFR (Baron and Schwartz, 2000). The EGFR, a tyrosine kinase transmembrane glycoprotein that is regulated by cell adhesion through β1-integrin (Reginato et al., 2003), interacts with many integrins. Integrin β1 and the EGFR, as well as αvβ3 and the EGFR, co-precipitate with Src kinase and p130 Cas (Moro et al., 2002; Cabodi et al., 2004).
Integrins also modulates the activity of c-MET, a receptor for HGF, by regulating its ligand-independent tyrosine phosphorylation (Wang et al., 2001). Although HGF is a potent stimulator of hepatocyte proliferation, it is known to inhibit the proliferation of certain HCC cell lines, such as HepG2 (Tajima et al., 1991; Shiota et al., 1992). When β1-integrin is overexpressed in HepG2 cells, HGF promotes the growth of the cells overexpressing β1-integrin, while the growth of wild-type and mock-transfected cells are suppressed by HGF (Zhang et al., 2003). β1-integrin suppressed the induction of p27 by HGF, a CDK inhibitor that inhibits cell cycle progression, and also promoted the proteasomal degradation of p27 by increasing the expression of its E3-ubiquitin ligase, Skp2. These findings indicate that β1-integrin-mediated signals from the ECM and growth factor receptor-mediated signals are cooperatively controlled and regulate the cellular functions and behavior.
Integrins and TGF-β1-Induced Apoptosis
The ECM has been used as a substrate for hepatocyte culture, and the presence of cell-matrix interactions in vitro was found to contribute to the survival and the maintenance of the differentiated functions of primary hepatocytes (Gómez-Lechón et al., 1998; Hansen and Albrecht, 1999; Cukierman et al., 2001; Godoy et al., 2009). Godoy et al. (2009) reported that the presence of ECM collagen inhibited the apoptosis of primary cultured hepatocytes induced by TGF-β1 in a Src/FAK-mediated Akt and p38-dependent manner. During the progression of liver fibrosis, TGF-β1 stimulates the expression of both ECMs and integrins (Inagaki and Okazaki, 2007; Hayashida, 2010). The activation of integrins by the ECM in the liver reciprocally alters the response of cells to TGF-β1, inducing resistance to TGF-β1-induced growth suppression in hepatocytes, and the sustained production of ECM proteins in activated HSCs and myofibroblasts. This interaction might further enhance the survival of hepatocytes and ECM production from HSCs by establishing a feed-forward loop between apoptosis resistance and fibrosis, and may therefore contribute to the progression from liver fibrosis to cirrhosis, and the development of preneoplastic changes in hepatocytes (Figure 4).
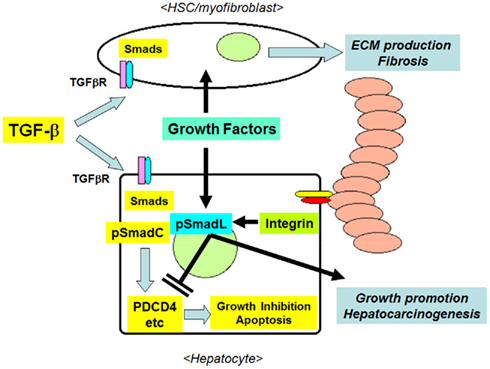
Figure 4. The interactions between TGF-β1 and ECM/integrin-mediated signaling in hepatocytes in the fibrotic liver. Initially, TGF-β1 inhibits the growth of hepatocytes by inducing the expression of pro-apoptotic molecules, such as PDCD4, via the formation of pSmadC. TGF-β1 stimulates the production of fibrous ECM from activated HSCs/myofibroblasts. The accumulated ECM proteins in turn activate integrins, then these activated integrins and co-existing growth factors inhibit the TGF-β1-induced growth-suppressive signals via the formation of pSmadL, leading to the resistance of hepatocytes to apoptosis, and the promotion of hepatocarcinogenesis.
The resistance to apoptosis induced by ECM/integrins is also observed in cancer cells. The presence of the ECM and activation of integrin-mediated signals has been shown to inhibit cytotoxic agent-induced apoptosis in a MAP kinase-dependent manner (Sethi et al., 1999; Zhang et al., 2002). We have previously shown that human HCC cells overexpressing β1-integrin become resistant to TGF-β1-induced apoptosis compared to mock-transfected cells in a MAP kinase-dependent manner (Zhang et al., 2004). TGF-β1 is known to transiently activate the MAP kinase pathway in a Smad-independent manner (Derynck and Zhang, 2003; Moustakas and Heldin, 2005; Giannelli et al., 2011). An overexpression of β1-integrin in HCC cells was shown to induce the activation of the MAP kinase pathway, and TGF-β1-induced the further sustained activation of ERK, p38, and JNK. The inhibition of these pathways reversed the TGF-β1-induced apoptosis, suggesting that the MAP kinase pathway has critical roles in the escape of cells from TGF-β1-induced apoptosis (Zhang et al., 2004).
Furthermore, recent reports have suggested that the physical properties of the ECM, including the matrix stiffness, modulate the intracellular signaling and cellular behavior (Wells, 2008; Tilghman et al., 2010; Zhao et al., 2010; Schrader et al., 2011). Clinical observations indicated that the liver stiffness, as measured by transient elastography, was closely associated with the degree of liver fibrosis and cirrhosis, and could predict the development of HCC in patients with viral hepatitis (Masuzaki et al., 2009; Jung et al., 2011). Primary cultured hepatocytes with a stiff collagen matrix become resistant to TGF-β1-induced apoptosis by decreasing caspase activation through FAK-mediated Akt signaling, while hepatocytes on soft collagen gels are sensitive to apoptosis (Godoy et al., 2009). Matrix stiffness modulates the growth of Huh7 and HepG2 HCC cells, as well as cisplatin-induced apoptosis. A stiff matrix enhances the mitogenic activity through β1-integrin and FAK, activating the ERK, Akt, and STAT3 pathways, and promoting the survival of HCC cells after cisplatin treatment. Furthermore, an increased stiffness promotes the EMT of HCC cells, suggesting the contribution of the mechanical properties of the EMC to tumor invasion and metastasis (Schrader et al., 2011).
Alterations of the Smad Phosphorylation Status during the Progression of Chronic Liver Disease
When TGF-β1 exerts growth-inhibitory effects on epithelial cells, the TβRI-mediated phosphorylation of Smad TFs (Smad2/3) occurs at their C-terminal region (SSXS motif). Recent studies have shown that the linker region of Smad proteins could be phosphorylated by various kinases, including MAP kinases (ERK, p38, JNK), CDK, and ROCK (Kretzschmar et al., 1999; Matsuura et al., 2004; Mori et al., 2004; Kamaraju and Roberts, 2005; Wrighton et al., 2009; Burch et al., 2011) to modulate the transcriptional activity of the Smad complex, resulting in the escape of cells from apoptosis and the induction of EMT that contributed to tumor development and progression.
During the chronic inflammation of the liver, altered phosphorylation patterns of the Smad2 and Smad3 proteins are observed. Along with the progression of chronic liver disease, the site of phosphorylation in the Smad proteins induced by TGF-β1 is shifted from the C-terminal region (pSmadC) to the linker region (pSmadL) in activated HSCs and hepatocytes (Yoshida et al., 2005; Matsuzaki et al., 2007; Murata et al., 2009; Matsuzaki, 2011). Phosphorylation of the linker region of Smad2/3 in HCSs is caused by the activation of JNK through PDGF, leading to the sustained activation of HSCs, differentiation to myofibroblast-like cells and continued production of collagen-rich ECM. In hepatocytes in the earlier stages of chronic liver disease, TGF-β1 induces the C-terminal phosphorylation of Smad (pSmadC). However, in the later stage of chronic liver disease when fibrosis in the liver has advanced, the linker-pSmadL in hepatocytes become prominent, and TGF-β1-induced growth inhibition is attenuated. The loss of TGF-β1-induced growth inhibition might be a marker for the preneoplastic changes of hepatocytes, and the dominance of pSmadL could be a useful indicator of the development of HCC (Matsuzaki et al., 2007; Murata et al., 2009; Matsuzaki, 2011).
In HCC cells, alterations in the Smad phosphorylation patterns modify the cellular responses to TGF-β1. Blockade of the C-terminal phosphorylation of Smad2 in HCC cells by transfecting mutant plasmids in which the C-terminal serine residues are substituted with alanine inhibited TGF-β1-induced pro-apoptotic Smad-mediated transcriptional activities (Sugano et al., 2003). Both growth factors and integrins regulate the phosphorylation pattern of Smads and modulate the cellular behavior. The presence of ECM proteins or overexpression of β1-integrin both induced the phosphorylation of the linker region of Smad2 and Smad3 and blocked the TGF-β1-induced C-terminal phosphorylation (Hamajima et al., 2009). Accordingly, increased phosphorylation at the linker region of Smad3 in the hepatocytes adjacent to the fibrous area in livers infected with chronic hepatitis C has been observed, thus suggesting a role for the ECM on the linker phosphorylation of Smad3 (Matsuzaki et al., 2007).
When integrins attenuate the activation of TGF-β1-induced apoptosis, the expression of PDCD4, a pro-apoptotic molecule downstream of TGF-β1, is repressed (unpublished data). ERK and JNK inhibitors, but not a p38 inhibitor, restored the formation of pSmad2C. All three inhibitors restored pSmad3C formation, and the transfection of a linker region mutated Smad3, but not Smad2, restored the TGF-β1-induced transcriptional activity, suggesting the differential roles of Smad2 and Smad3 (Hamajima et al., 2009; Matsuzaki, 2011).
Future Perspectives for Prevention and Therapy
Recent studies have shown that the phosphorylation state of the Smad TFs plays a pivotal role in converting the tumor suppressor signal of TGF-β1 to a tumor promoting signal. Thus, the inhibition of the linker phosphorylation of Smads could be an attractive approach to inhibit the development and progression of HCC. Among the kinases that phosphorylate the linker region of Smads directly or indirectly, JNK has been shown to phosphorylate the linker regions of both Smad2 and Smad3. Indeed, administration of a JNK inhibitor to rats successfully inhibited the development of HCC in a DEN-induced HCC model (Nagata et al., 2009). Integrins can propagate the multiple signals that promote the survival and growth of HCC cells by interacting with growth factor receptors, including the TGF-β receptor. Therefore inhibitors of integrin activation, which might modulate the phosphorylation of Smad indirectly, could represent an alternative approach for the prevention and/or treatment of HCC and tumor progression. In fact, antisense-mediated inhibition of integrin αv or integrin β3 suppressed the growth of HCC cells injected subcutaneously in a mouse model (Li et al., 2007). Various types of integrin inhibitors are currently being examined in preclinical and clinical studies (Desgrosellier and Cheresh, 2010; Millard et al., 2011). Further studies are necessary to clarify the interaction between integrins and TGF-β1, and will contribute to the development of novel therapeutics for HCC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alam, N., Goel, H. L., Zarif, M. J., Butterfield, J. E., Perkins, H. M., Sansoucy, B. G., Sawyer, T. K., and Languino, L. R. (2007). The integrin-growth factor receptor duet. J. Cell. Physiol. 123, 649–653.
Baron, V., and Schwartz, M. (2000). Cell adhesion regulates ubiquitin-mediated degradation of the platelet-derived growth factor receptor beta. J. Biol. Chem. 275, 39318–39323.
Bedossa, P., Peltier, E., Terris, B., Franco, D., and Polynard, T. (1995). Transforming growth factor-beta 1(TGF-beta 1) and TGF-beta 1 receptors in normal, cirrhotic and neoplastic human livers. Hepatology 21, 760–766.
Begum, N. A., Mori, M., Matsumata, T., Takenaka, K., Sugimachi, K., and Barnard, G. F. (1995). Differential display and integrin alpha 6 messenger RNA overexpression in hepatocellular carcinoma. Hepatology 22, 1447–1455.
Bergamini, C., Sgarra, C., Trerotoli, P., Lupo, L., Azzariti, A., Antonaci, S., and Giannelli, G.. (2007). Laminin-5 stimulates hepatocellular carcinoma growth through a different function of alpha6beta4 and alpha3beta1 integrins. Hepatology 46, 1801–1809.
Bhowmick, N. A., Ghiassi, M., Bakin, A., Aakre, M., Lundquist, C. A., Engel, M. E., Arteaga, C. L., and Moses, H. L. (2001). Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 12, 27–36.
Bierie, B., and Moses, H. L. (2006). TGFβ: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 6, 506–520.
Bottcher, R. T., Lange, A., and Fassler, R. (2009). How ILK and kindlins cooperate to orchestrate integrin signaling. Curr. Opin. Cell Biol. 21, 670–675.
Burch, M. L., Zheng, W., and Little, P. J. (2011). Smad linker phosphorylation in the regulation of extracellular matrix synthesis. Cell Mol. Life Sci. 68, 97–107.
Cabodi, S., Moro, L., Bergatto, E., Boeri Erba, E., Di Stefano, P., Turco, E., Tarone, G., and Defilippi, P. (2004). Integrin regulation of epidermal growth factor (EGF) receptor and of EGF-dependent responses. Biochem. Soc. Trans. 32, 438–442.
Caja, L., Ortiz, C., Bertran, E., Murillo, M. M., Miró-Obradors, M. J., Palacios, E., and Fabregat, I. (2007). Differential intracellular signaling induced by TGF-beta in rat adult hepatocytes and hepatoma cells: implications in liver carcinogenesis. Cell. Signal. 19, 683–694.
Carloni, V., Mazzocca, A., Pantaleo, P., Cordella, C., Laffi, G., and Gentilini, P. (2001). The integrin, alpha6beta1, is necessary for the matrix-dependent activation of FAK and MAP kinase and the migration of human hepatocarcinoma cells. Hepatology 34, 42–49.
Couvelard, A., Scoazec, J.-Y., and Feldmann, G. (1993). Expression of cell-cell and cell-matrix adhesion proteins by sinusoidal endothelial cells in the normal and cirrhotic human liver. Am. J. Pathol. 143, 738–752.
Cukierman, E., Pankov, R., Stevens, D. R., and Yamada, K. M. (2001). Taking cell-matrix adhesions to the third dimension. Science 294, 1708–1712.
Derynck, R., and Zhang, Y. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signaling. Nature 425, 577–584.
Desgrosellier, J. S., and Cheresh, D. A. (2010). Integrins in cancer:biological implications and therapeutic opportunities. Nat. Rev. Cancer 10, 9–22.
El-Serag, H. B., and Rudolph, K. L. (2007). Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–2576.
Fabregat, I., Sánchez, A., Alvarez, A. M., Nakamura, T., and Benito, M. (1996). Epidermal growth factor, but not hepatocyte growth factor, suppresses the apoptosis induced by transforming growth factor-beta in fetal hepatocytes in primary culture. FEBS Lett. 384, 14–16.
Furukawa, F., Matsuzaki, K., Mori, S., Tahashi, Y., Yoshida, K., Sungano, Y., Yamagata, H., Matsushita, M., Seki, T., Inagaki, Y., Nishizawa, M., Fujisawa, J., and Inoue, K. (2003). p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 38, 879–889.
Giannelli, G., Bergamini, C., Fransvea, E., Marinosci, F., Quaranta, V., and Antonaci, S. (2001). Human hepatocellular carcinoma (HCC) cells require both alpha3beta1 integrin and matrix metalloproteinases activity for migration and invasion. Lab. Invest. 81, 613–627.
Giannelli, G., Mazzocca, A., Fransvea, E., Lahn, M., and Antonaci, S. (2011). Inhibiting TGF-β signaling in hepatocellular carcinoma. Biochem. Biophys. Acta 1815, 214–223.
Godoy, Pl., Hengstler, J. G., Ilkavets, I., Bachmann, A., Muller, A., Tuschl, G., Mueller, S. O., and Dooley, S. (2009). Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid differentiation and transforming growth factor beta-induced apoptosis. Hepatology 49, 2031–2043.
Gómez-Lechón, M. J., Jover, R., Donato, T., Ponsoda, X., Rodriguez, C., Stenzel, K. G., Klocke, R., Paul, D., Guillén, I., Bort, R., and Castell, J. V. (1998). Long-term expression of differentiated functions in hepatocytes cultured in three-dimensional collagen matrix. J. Cell. Physiol. 177, 553–562.
Hamajima, H., Ozaki, I., Zhang, H., Iwane, S., Kawaguchi, Y., Eguchi, Y., Matsuhashi, S., Mizuta, T., Matsuzaki, K., and Fujimoto, K. (2009). Modulation of the transforming growth factor-beta1-induced Smad phosphorylation by the extracellular matrix receptor beta1-integrin. Int. J. Oncol. 35, 1441–1447.
Hansen, L. K., and Albrecht, J. H. (1999). Regulation of the hepatocyte cell cycle by type I collagen matrix: role of cyclin D1. J. Cell Sci. 112, 2971–2981.
Hayashida, T. (2010). Integrins modulate cellular fibrogenesis at multiple levels; regulation of TGF-β signaling. Endocr. Metab. Immune Disord. Drug Targets 10, 302–319.
Hehlgans, S., Haase, M., and Cordes, N. (2007). Signaling via integrins: Implications for cell survival and anticancer strategies. Biochem. Biophys. Acta 1775, 163–180.
Heldin, C. H., Miyazono, K., and ten Dijke, P. (1997). TGF-β signaling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471.
Inagaki, Y., and Okazaki, I. (2007). Emerging insights into transforming growth factor β Smad signaling in hepatic fibrogenesis. Gut 56, 284–292.
Ito, N., Kawata, S., Tamura, S., Takaishi, K., Shirai, Y., Kiso, S., Yabuuchi, I., Matsuda, Y., Nishioka, M., and Tarui, S. (1991). Elevated levels of transforming growth factor beta messenger RNA and its polypeptide in human hepatocellular carcinoma. Cancer Res. 51, 4080–4083.
Ivaska, J., and Heino, J. (2010). Interplay between cell adhesion and growth factor receptors: from the plasma membrane to the endosomes. Cell. Tissue Res. 339, 111–120.
Jang, C. W., Chen, C. H., Chen, C. C., Chen, J. Y., Su, Y. H., and Chen, R. H. (2002). TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 4, 51–58.
Jung, K. S., Kim, S. U., Ahn, S. H., Park, Y. N., Kim do, Y., Park, J. Y., Chon, C. Y., Choi, E. H., and Han, K. H. (2011). Risk assessment of hepatitis B virus-related hepatocellular carcinoma development using liver stiffness measurement (FibroScan). Hepatology 53, 885–894.
Kamaraju, A. K., and Roberts, A. B. (2005). Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-β-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J. Biol. Chem. 280, 1024–1036.
Ke, A. W., Shi, G. M., Zhou, J., Huang, X. Y., Shi, Y. H., Ding, Z. B., Wang, X. Y., Devbhandari, R. P., and Fan, J. (2011). CD151 amplifies signaling by integrin α6β1 to PI3K and induces the epithelial–mesenchymal transition in HCC Cells. Gastroenterology 140, 1629–1641.
Kretzschmar, M., Doody, J., Timokhina, I., and Massagué, J. (1999). A mechanism of repression of TGF-β/Smad signaling by oncogenic Ras. Genes Dev. 13, 804–816.
Lai, K. K., Shang, S., Lohia, N., Booth, G. C., Masse, D. J., Fausto, N., Campbell, J. S., and Beretta, L. (2011). Extracellular matrix dynamics in hepatocarcinogenesis: a comparative proteomics study of PDGFC transgenic and Pten null mouse models. PLoS Genet. 7, e1002147. doi:10.1371/journal.pgen.1002147
Levine, D., Rockey, D. C., Milner, T. A., Breuss, T., Fallon, J. T., and Schnapp, L. M. (2000). Expression of α8β1 integrin during pulmonary and hepatic fibrosis. Am. J. Pathol. 156, 1927–1935.
Li, J., Tan, H., Dong, X., Xu, Z., Shi, C., Han, X., Jiang, H., Krissansen, G. W., and Sun, X. (2007). Antisense integrin alphaV and beta3 gene therapy suppresses subcutaneously implanted hepatocellular carcinomas. Dig. Liver Dis. 39, 557–565.
Lin, J. K., and Chou, C. K. (1992). In vitro apoptosis in the human hepatoma cell line induced by transforming growth factor beta 1. Cancer Res. 52, 385–388.
LIovet, J. M., Burroughs, A., and Bruix, J. (2003). Hepatocellular carcinoma. Lancet 362, 1907–1917.
Margadant, C., and Sonnenberg, A. (2010). Integrin-TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 11, 97–105.
Massague, J., Seonae, J., and Wotton, D. (2005). Smad transcription factors. Genes Dev. 19, 2783–2810.
Masuzaki, R., Tateishi, R., Yoshida, H., Goto, E., Sato, T., Ohki, T., Imamura, J., Goto, T., Kanai, F., Kato, N., Ikeda, H., Shiina, S., Kawabe, T., and Omata, M. (2009). Prospective risk assessment for hepatocellular carcinoma development in patients with chronic hepatitis C by transient elastography. Hepatology 49, 1954–1961.
Matsuura, I., Denissova, N. G., Wang, G., He, D., Long, J., and Liu, F. (2004). Cyclin-dependent kinases regulated the antiproliferative function of Smads. Nature 430, 226–231.
Matsuzaki, K. (2011). Smad phosphoisoform signals in acute and chronic liver injury: similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res. doi: 10.1007/s00441-011-1178-6. [Epub ahead of print]..
Matsuzaki, K., Murata, M., Yoshida, K., Sekimoto, G., Uemura, Y., Sakaida, N., Kaibori, M., Kamiyama, Y., Nishizawa, M., Fujisawa, J., Okazaki, K., and Seki, T. (2007). Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 46, 48–57.
Millard, M., Odde, S., and Neamati, N. (2011). Integrin targeted therapeutics. Theranostics 1, 154–188.
Mori, S., Matsuzaki, K., Yoshida, K., Furukawa, F., Tahashi, Y., Yamagata, H., Sekimoto, G., Seki, T., Matsui, H., Nishizawa, M., Fujisawa, J., and Okazaki, K. (2004). TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at linker regions. Oncogene 23, 7416–7429.
Moro, L., Dolce, L., Cabodi, S., Bergatto, E., Boeri-Erba, E., Smeriglio, M., Turco, E., Retta, S. F., Giuffrida, M. G., Venturino, M., Godovac-Zimmermann, J., Conti, A., Schaefer, E., Beguinot, L., Tacchetti, C., Gaggini, P., Silengo, L., Tarone, G., and Defilippi, P. (2002). Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J. Biol. Chem. 277, 9405–9414.
Moses, H. L., Yang, E. Y., and Pietenpol, J. A. (1990). TGF-β stimulation and inhibition of cell proliferation: new mechanistic insights. Cell 63, 245–247.
Murata, M., Matsuzaki, K., Yoshida, K., Sekimoto, G., Tahashi, Y., Mori, S., Uemura, Y., Sakaida, N., Fujisawa, J., Seki, T., Kobayashi, K., Yokote, K., Koike, K., and Okazaki, K. (2009). Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology 49, 1203–1217.
Murillo, M. M., del Castillo, G., Sánchez, A., Fernández, M., and Fabregat, I. (2005). Involvement of EGF receptor and c-Src in the survival signals induced by TGF-beta1 in hepatocytes. Oncogene 24, 4580–4587.
Nagata, H., Hatano, E., Tada, M., Murata, M., Kitamura, K., Asechi, H., Narita, M., Tamaki, N., Yagi, S., Ikai, I., Matsuzaki, K., and Uemoto, S. (2009). Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesisi to tumor-suppression in rat hepatocellular carcinoma. Hepatology 49, 1944–1953.
Nakashima, M., Hamajima, H., Xia, J., Iwane, S., Kwaguchi, Y., Eguchi, Y., Mizuta, T., Fujimoto, K., Ozaki, I., and Matsuhashi, S. (2010). Regulation of tumor suppressor PDCD4 by novel protein kinase C isoforms. Biochim. Biophys. Acta 1803, 1020–1027.
Oberhammer, F. A., Pavelka, M., Sharma, S., Tiefenbacher, R., Purchio, A. F., Bursch, W., and Shulte-Hermann, R. (1992). Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc. Natl. Acad. Sci. U.S.A. 89, 5408–5412.
Ozaki, I., Yamamoto, K., Mizuta, T., Kajihara, S., Fukushima, N., Setoguchi, Y., Morito, F., and Sakai, T. (1998). Differential expression of laminin receptors in human hepatocellular carcinoma. Gut 43, 837–842.
Perlman, R., Schiemann, W. P., Brooks, M. W., Lodish, H. F., and Weinberg, R. A. (2001). TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat. Cell Biol. 3, 708–714.
Petritsch, C., Beug, H., Balmain, A., and Oft, M. (2000). TGF-beta inhibits p70 S6 kinase via protein phosphatase 2A to induce G(1) arrest. Genes Dev. 14, 3093–3101.
Reginato, M. J., Mills, K. R., Paulus, J. K., Lynch, D. K., Sgroi, D. C., Debnath, J., Muthuswamy, S. K., and Brugge, J. S. (2003). Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 5, 733–740.
Sakaida, I., Hironaka, K., Uchida, K., Suzuki, C., Kayano, K., and Okita, K. (1998). Fibrosis accelerates the development of enzyme-altered lesions in rat liver. Hepatology 28, 1247–1252.
Schrader, J., Gordon-Walker, T. T., Aucott, R. L., van Deemter, M., Quaas, A., Walsh, S., Benten, D., Forbes, S. J., Wells, R. G., and Iredale, J. P. (2011). Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 53, 1192–1205.
Senturk, S., Mumcuoglu, M., Gursoy-Yuzugullu, O., Cingoz, B., Akcali, K. C., and Ozturk, M. (2010). Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology 52, 966–974.
Sethi, T., Rintoul, R. C., Moore, S. M., MacKinnon, A. C., Salter, D., Choo, C., Chilvers, E. R., Dransfield, I., Donnelly, S. C., Strieter, R., and Haslett, C. (1999). Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 5, 662–668.
Shiota, G., Rhoads, D. B., Wang, T. C., Nakamura, T., and Schmidt, E. V. (1992). Hepatocyte growth factor inhibits growth of hepatocellular carcinoma cells. Proc. Natl. Acad. Sci. U.S.A. 89, 373–377.
Siegel, P. M., and Massague, J. (2003). Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 3, 807–821.
Sugano, Y., Matsuzaki, K., Tahashi, Y., Furukawa, F., Mori, S., Yamagata, H., Yoshida, K., Matsushita, M., Nishizawa, M., Fujisawa, J., and Inoue, K. (2003). Distortion of autocrine transforming growth factor b signal acceleratesmalignant potential by enhancing cell growth as well as PAI-1 and VEGF production in human hepatocellular carcinoma cells. Oncogene 22, 2309–2321.
Tajima, H., Matsumoto, K., and Nakamura, T. (1991). Hepatocyte growth factor has potent anti-proliferative activity in various tumor cell lines. FEBS Lett. 291, 229–232.
Tilghman, R. W., Cowan, C. R., Mih, J. D., Koryakina, Y., Gioeli, D., Slack-Davis, J. K., Blackman, B. R., Tschumperlin, D. J., and Parsons, J. T. (2010). Matrix rigidity regulates cancer cell growth and cellular phenotype. PLoS ONE 5, e12905. doi:10.1371/journal.pone.0012905
Torimura, T., Ueno, T., Kin, M., Harada, R., Nakamura, T., Kawaguchi, T., Harada, M., Kumashiro, R., Watanabe, H., Avraham, R., and Sata, M. (2001). Autocrine motility factor enhances hepatoma cell invasion across the basement membrane through activation of beta1 integrins. Hepatology 34, 62–71.
Tsai, J. F., Jeng, J. E., Chuang, L. Y., Yang, M. L., Ho, M. S., Chang, W. Y., Hsieh, M. Y., Lin, Z. Y., and Tsai, J. H. (1997). Elevated urinary transforming growth factor-beta 1 level as a tumor marker and predictor of poor survival in cirrhotic hepatocellular carcinoma. Br. J. Cancer 76, 244–50.
Tsukuma, H., Hiyama, T., Tanaka, S., Nakao, M., Yabuuchi, T., Kitamura, T., Nakanishi, K., Fujimoto, I., Inoue, A., Yamazaki, H., and Kawashima, T. (1993). Risk factor for hepatocellular carcinoma among patients with chronic liver disease. N. Engl. J. Med. 328, 1797–1801.
Volpes, R., van den Oord, J. J., and Desmet, V. J. (1991). Distribution of the VLA family of integrins in normal and pathological human liver tissue. Gastroenterology 101, 200–206.
Volpes, R., van den Oord, J. J., and Desmet, V. J. (1993). Integrins as differential cell lineage markers of primary liver tumors. Am. J. Pathol. 142, 1483–1492.
Wang, R., Ferrell, L. D., Faouzi, S., Maher, J. J., and Bishop, J. M. (2001). Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J. Cell Biol. 153, 1023–1034.
Wells, R. G. (2008). The role of matrix stiffness in regulating cell behavior. Hepatology 47, 1394–1400.
Wrighton, K. H., Lin, X., and Fen, X. (2009). Phospho-control of TGF-β superfamily signaling. Cell Res. 19, 8–20.
Yang, Y. A., Zhang, G. M., Feigenbaum, L., and Zhang, Y. E. (2006). Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of Bcl-2. Cancer Cell 9, 445–457.
Yi, J. Y., Shin, I., and Arteaga, C. L. (2005). Type I transforming growth factor-beta receptor binds to and activates phosphatidylinositol 3-kinase. J. Biol. Chem. 280, 10870–10876.
Yoo, J., Ghiassi, M., Jirmanova, L., Balliet, A. G., Hoffman, B., Fornace, A. J. Jr., Liebermann, D. A., Bottinger, E. P., and Roberts, A. B. (2003). Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45β through p38 activation. J. Biol. Chem. 278, 43001–43007.
Yoshida, H., Shiratori, Y., Moriyama, M., Arakawa, Y., Ide, T., Sata, M., Inoue, O., Yano, M., Tanaka, M., Fujiyama, S., Nishiguchi, S., Kuroki, T., Imazeki, F., Yokosuka, O., Kinoyama, S., Yamada, G., and Omata, M. (1999). Interferon therapy reduces the risk for hepatocellular carcinoma: national surveillance program of cirrhotic and non-cirrhotic patients with chronic hepatitis C in Japan. IHIT Study Group. Inhibition of Hepatocarcinogenesis by interferon therapy. Ann. Intern. Med. 131, 174–181.
Yoshida, K., Matsuzaki, K., Mori, S., Tahashi, Y., Yamagata, H., Furukawa, F., Seki, T., Nishizawa, M., Fujisawa, J., and Okazaki, K. (2005). Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am. J. Pathol. 166, 1029–1039.
Zhang, H., Ozaki, I., Mizuta, T., Hamajima, H., Yasutake, T., Eguchi, Y., Ideguchi, H., Yamamoto, K., and Matsuhashi, S. (2006). Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene 25, 6101–6012.
Zhang, H., Ozaki, I., Mizuta, T., Matsuhashi, S., Yoshimura, T., Hisatomi, A., Tadano, J., Sakai, T., and Yamamoto, K.. (2002). Beta 1-integrin protects hepatoma cells from chemotherapy induced apoptosis via a mitogen-activated protein kinase dependent pathway. Cancer 95, 896–906.
Zhang, H., Ozaki, I., Mizuta, T., Yoshimura, T., Matsuhashi, S., Eguchi, Y., Yasutake, T., Hisatomi, A., Sakai, T., and Yamamoto, K. (2004). Transforming growth factor-beta 1-induced apoptosis is blocked by beta 1-integrin-mediated mitogen-activated protein kinase activation in human hepatoma cells. Cancer Sci. 95, 878–886.
Zhang, H., Ozaki, I., Mizuta, T., Yoshimura, T., Matsuhashi, S., Hisatomi, A., Tadano, J., Sakai, T., and Yamamoto, K. (2003). Mechanism of beta 1-integrin-mediated hepatoma cell growth involves p27 and S-phase kinase-associated protein 2. Hepatology 38, 305–313.
Keywords: TGF-β1, Smad, liver fibrosis, integrins, hepatocellular carcinoma
Citation: Ozaki I, Hamajima H, Matsuhashi S and Mizuta T (2011) Regulation of TGF-β1-induced pro-apoptotic signaling by growth factor receptors and extracellular matrix receptor integrins in the liver. Front. Physio. 2:78. doi: 10.3389/fphys.2011.00078
Received: 12 August 2011; Paper pending published: 06 September 2011;
Accepted: 11 October 2011; Published online: 24 October 2011.
Edited by:
Atsushi Masamune, Tohoku University Graduate School of Medicine, JapanReviewed by:
Arkaitz Carracedo, CIC bioGUNE, SpainDiego Francesco Calvisi, Ernst-Moritz-Arndt-Universität Greifswald, Germany
Copyright: © 2011 Ozaki, Hamajima, Matsuhashi and Mizuta. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Iwata Ozaki, Saga Medical School, Health Administration Center, Saga University, 5-1-1 Nabeshima, Saga 849-8501, Japan. e-mail: ozaki@cc.saga-u.ac.jp