 Ronald M. Clouse
Ronald M. Clouse Nicola Carraro
Nicola Carraro- 1Department of Bioinformatics and Genomics, University of North Carolina at Charlotte, Charlotte, NC, USA
- 2Department of Agronomy, Purdue University, West Lafayette, IN, USA
The PIN and ACO gene families present interesting questions about the evolution of plant physiology, including testing hypotheses about the ecological drivers of their diversification and whether unrelated genes have been recruited for similar functions. The PIN-formed proteins contribute to the polar transport of auxin, a hormone which regulates plant growth and development. PIN loci are categorized into groups according to their protein length and structure, as well as subcellular localization. An interesting question with PIN genes is the nature of the ancestral form and location. ACOs are members of a superfamily of oxygenases and oxidases that catalyze the last step of ethylene synthesis, which regulates many aspects of the plant life cycle. We used publicly available PIN and ACO sequences to conduct phylogenetic analyses. Third codon positions of these genes in monocots have a high GC content, which could be historical but is more likely due to a mutational bias. Thus, we developed methods to extract phylogenetic information from nucleotide sequences while avoiding this convergent feature. One method consisted in using only A-T transformations, and another used only the first and second codon positions for serine, which can only take A or T and G or C, respectively. We also conducted tree-searches for both gene families using unaligned amino acid sequences and dynamic homology. PIN genes appear to have diversified earlier than ACOs, with monocot and dicot copies more mixed in the phylogeny. However, gymnosperm PINs appear to be derived and not closely related to those from primitive plants. We find strong support for a long PIN gene ancestor with short forms subsequently evolving one or more times. ACO genes appear to have diversified mostly since the dicot-monocot split, as most genes cluster into a small number of monocot and dicot clades when the tree is rooted by genes from mosses. Gymnosperm ACOs were recovered as closely related and derived.
Introduction
The dramatic increase in the amount of publicly available genomic information has facilitated analyses of gene-family origins and evolution. Plant gene phylogenies have proliferated, but they are commonly made using distance matrices of amino acid sequences. This method is expected to amplify misleading information resulting from convergence (Farris, 1983; Simmons, 2000; Simmons et al., 2002), obscuring any signal of the true history.
Methodological approaches to studying gene family histories need focused attention, since gene phylogenies are inherently difficult to verify. Molecular phylogenies of species can be compared to their morphology and biogeography to ask whether resulting trees are plausible. Perfect congruence is not expected, but phylogenies at extreme variance cast doubt on the quality of the methods and underlying data. Because gene copies can undergo any combination of diversification, loss, and neofunctionalization in different taxonomic lineages, nearly any sort of phylogenetic hypothesis can be the reasonable outcome of an analysis. Gene copies in the same taxon or with the same morphology would be expected to have a higher probability of being closely related, but this is not necessarily so, and it is dependent on the timing of gene family diversification events and the phylogenetic placement of progenitor copies.
Here we employ phylogenetic methods designed to minimize the effects of convergence and amplify historical signal in the date. We apply these methods to the PIN-formed (PIN) auxin transporters and the ethylene-forming, ACC-oxidase enzymes (ACO). We chose to analyze these gene families because both are important plant development genes for which there are hypotheses about the timing and drivers of their diversification, but they differ in the amount of phylogenetic study thus far received. The PIN genes have been the subject of different phylogenetic analyses (Paponov et al., 2005; Křeček et al., 2009; De Smet et al., 2011; Carraro et al., 2012; Viaene et al., 2013; Bennett et al., 2014), but for ACOs no such analysis has been attempted. We thus are able to compare the results of our methods to prior analyses in PIN genes, and we lay the groundwork for a new discussion on the history of ACOs.
PIN-formed proteins polarly transport the plant hormone auxin, which regulates several aspects of plant growth and development (Robert and Friml, 2009; Zazímalová et al., 2010). Since the discovery and characterization of the first PIN mutant in Arabidopsis (Okada et al., 1991), several other PINs have been identified and characterized in different plant species. In the Arabidopsis genome, there are eight PIN loci, which are categorized into groups according to their protein length and structure, as well as subcellular localization (Paponov et al., 2005; Křeček et al., 2009). The first cladistic analysis of PIN genes (Carraro et al., 2012), which was rooted by moss and lycophyte copies, suggested that PIN genes diversified mostly since the rise of land plants, around the time of the monocot-dictot split. A subsequent analysis (Viaene et al., 2013) focused on the evolution of PIN gene morphology; the preferred topology—rooted by protist, animal, bacterial genes—suggested that the moss sequence “PpPIN1D” is sister to all other PIN genes, and the morphology evolved from short forms to long.
ACOs help in the synthesis of ethylene, which is a gaseous hormone under normal environmental conditions, and which regulates many aspects of the plant life cycle (Bleecker and Kende, 2000; Lin et al., 2009). In higher plants, ethylene is synthesized via two committed enzyme-catalyzed steps from S-adenosyl-L-methionine. The first step is catalyzed by 1-aminocyclopropane-1-carboxylic acid (ACC) synthase (ACS), and the second (and last) step is carried out by ACC oxidase (ACO). ACOs are members of a superfamily of oxygenases and oxidases, most of which utilize Fe (II) as a cofactor and 2-oxoglutarate (2OG) as a cosubstrate (Sato and Theologis, 1989; Bidonde et al., 1998; Wang et al., 2002). The subcellular localization of ACO proteins is prevalently cytosolic rather than membrane-bound (Chung et al., 2002; Hudgins et al., 2006; Lin et al., 2009). ACOs have high similarity throughout the protein coding sequences and expression analyses reveal that the ACO genes display a high degree of differential expression in tissues at various stages of development. A variety of plant species produce ethylene, including unicellular and multicellular algae, although angiosperms use a different biosynthetic pathway from primitive land plants and algae (Wang et al., 2002; De Paepe and Van der Straeten, 2005; Plettner et al., 2005; Yordanova et al., 2010; Wanke, 2011; Yasumura et al., 2012).
Materials and Methods
Assembling of the ACO and PIN Data Sets
For all phylogenetic analyses, unless otherwise specified, coding sequences (CDS) were used. All taxa with publicly available sequences were included, although a random subset of all available angiosperm sequences were taken, so as to generate a manageable data set size. For the PIN data set (Table 1), reported unique sequence identifiers were used to retrieve the corresponding sequences from the Phytozome v.9.1 (www.phytozome.org) (Goodstein et al., 2012), ConGenIE (congenie.org), and Genebank (www.ncbi.nlm.nih.gov/genbank/) (Benson et al., 2005) databases. The only major plant group that was not included in the PIN data set was ferns, as previously reported PIN genes from them are not publicly available (Viaene et al., 2013). ACO sequences (Table 2) were identified from previously published studies and via queries with the BLASTn algorithm at the National Centre for Biotechnology Information (NCBI) nucleotide collection and from the Phytozome v.9.1 (Goodstein et al., 2012). Only proteins that were annotated as aminocyclopropane-carboxylate oxidases were retained. Transmembrane profiles for PIN amino acid sequences were predicted querying the TMHMM Server v.2.0 (www.cbs.dtu.dk/services/TMHMM/) (Moller et al., 2001). PIN proteins were classified (1–5) according to their length, number of transmembrane domains, and length of the central hydrophilic loop (See Table 1). Generally, caution should be exerted when classifying PIN proteins according to their number of TMDs, as those are predicted protein domains that will need final confirmation by reconstruction of the tertiary structure by crystallography. In two cases where gene sequences showed no notable differences from long forms but were predicted to have only the N-terminal trans-membrane domain (OsPIN3a and Aco018694), we coded them as long.
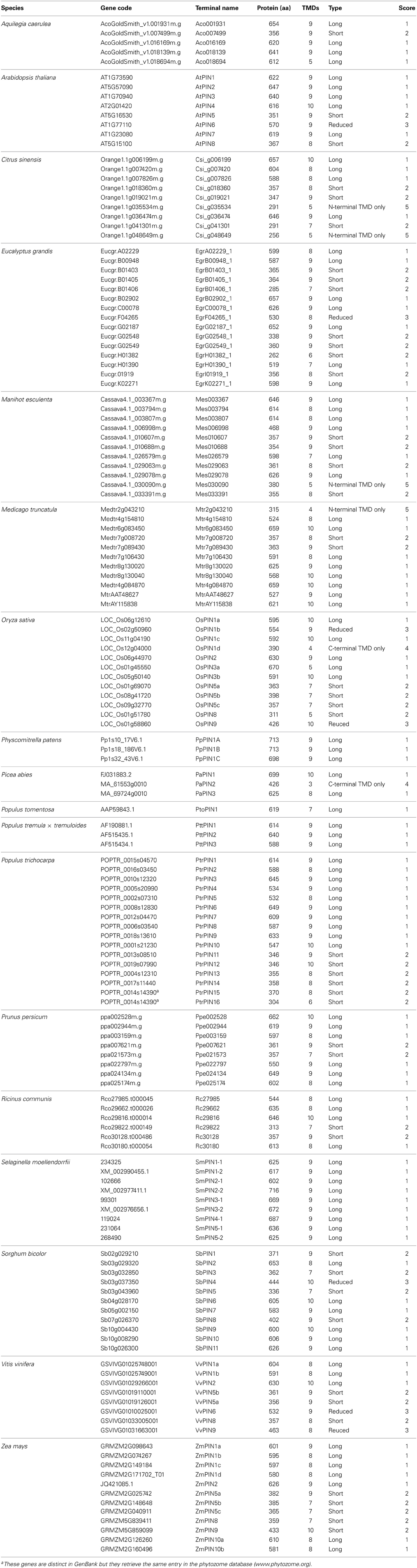
Table 1. PIN genes analyzed in this study.
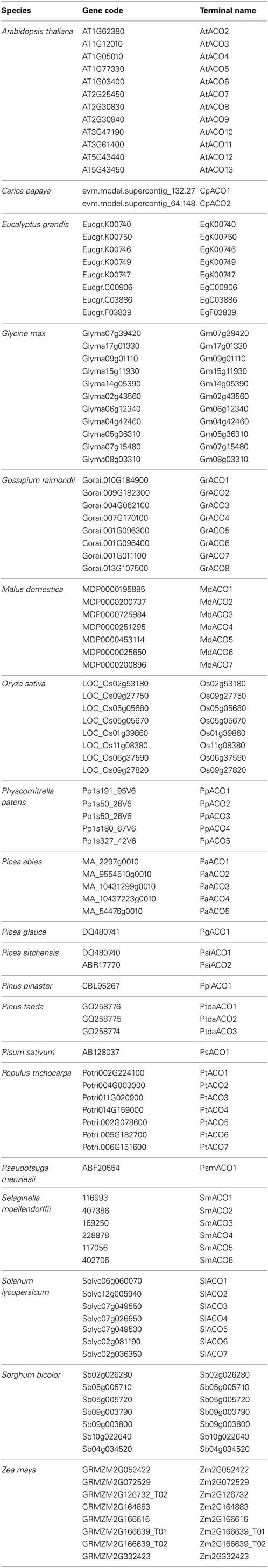
Table 2. ACO genes analyzed in this study.
Alignment and Phylogenetic Analysis
The moss PIN gene “PpPIN1D” (Gene Code “Pp1s79_126V6.1”) was excluded from analysis due to suspicion of it being a pseudogene; although not the most distant gene in the data set, it has only about half the number of nucleotides relative to other moss PIN genes (with the gaps appearing in the middle), and in preliminary phylogenies it was recovered on a very long branch (usually twice as long as its sister). This can also be seen in our previous phylogeny of PIN genes (Figure 2A in Carraro et al., 2012). Likewise, we did not include purported PIN homologs from non-plants (Viaene et al., 2013), as we had no evidence for their homology.
We aligned the amino acids with ClustalW2 (Larkin et al., 2007) in SeaView (Gouy et al., 2010) (-gapopen parameter set to 15) and then back-translated the amino acids to nucleotides (Figures 1A,B). The resulting nucleotide alignments for the PIN and ACO data sets are available as supplemental material. SeaView retains original nucleotide data through the process of amino acid alignment and thus allows an accurate back-translation after alignment. SeaView also has the alignment program MAFFT (Katoh et al., 2009) implemented, but, with the -gapopen parameter defined, it failed to run with our data set. Clustal misaligned one of the short genes, which was fixed by hand, although tree-searching on the uncorrected version of the alignment resulted in nearly identical topologies. We noticed that monocots tended to have a very high GC content, especially in the third codon position (Figure 2). Thus, using the program Mega v. 4.0.2 (Tamura et al., 2007), we removed the third codon positions of these nucleotide alignments, and in the program BioEdit (Hall, 2007) we replaced G and C with N (Figures 1C,D). Also in BioEdit, we translated nucleotide alignments and replaced all amino acids but Serine with N (Figure 1G); this required several steps, as BioEdit uses a default codon translation for amino acids in back-translation. We also used the program Mesquite (Maddison and Maddison, 2009) to convert fasta files into files that can be read by the phylogenetic program TNT (Goloboff et al., 2008). (Mesquite changes N to “?” automatically when generating TNT files and so these were returned to N using the search-and-replace function in BioEdit.) We used TNT because it clearly reads N as “any nucleotide,” preserving the gap information. We analyzed the same alignments under likelihood bootstrap in the program RAxML (Stamatakis et al., 2008) on the Cipres (Miller et al., 2010) computing cluster. RAxML does not recognize gaps and treats them and N as simply missing data.
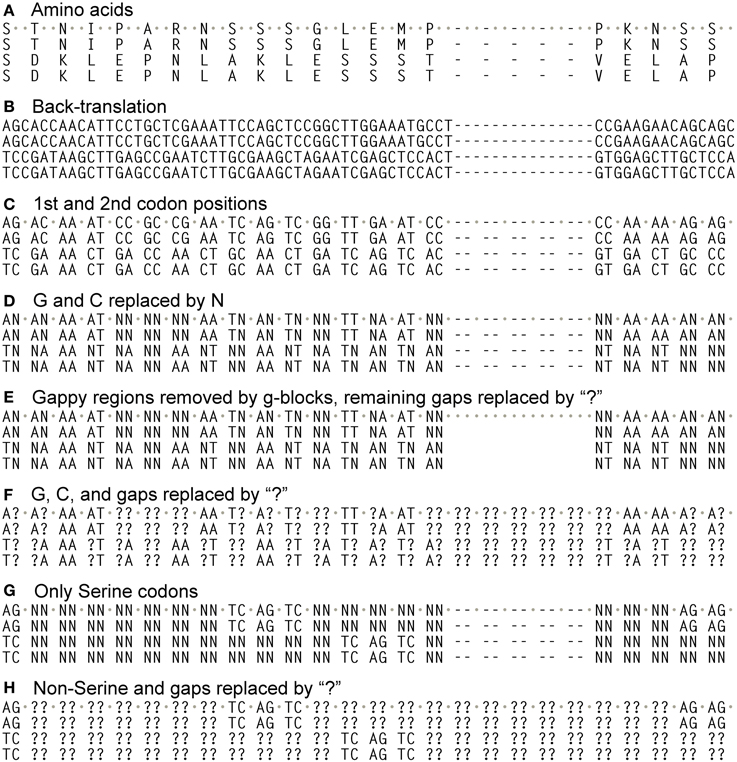
Figure 1. Different alignments used in this study. Alignments began by translating coding sequences and aligning them with ClustalW2 (“gapopen” option set to 15), and then back-translating them into nucleotides (A,B). From these only the 1st and 2nd codon positions were taken (C), and then all G and C nucleotides converted to “N” (D), which is read as “any nucleotide” in the phylogenetic program TNT. This alignment then had gappy regions removed by the program Gblocks (E) or had all of its GC bases and gaps converted to “?” (F), which is read as “any nucleotide or gap” in TNT. The alignment in B also had all codon positions except those coding for Serine converted to “N”, then the 3rd codon positons removed (G). This also had all Ns and gaps converted to “?” (H).
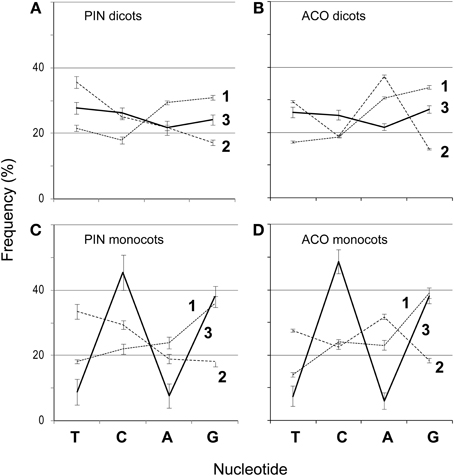
Figure 2. Frequencies of nucleotides (T, C, A, and G) at each codon position (1–3) for 151 PIN and 193 ACO genes in dicots (A,B), and monocots (C,D). Bars represent 95% confidence intervals.
For searches in TNT, we used the “new technology” function (which combines several strategies for exploring tree space) with 100 initial builds, and we followed with calculations of bootstrap support using 1000 pseudoreplicates. We took the strict consensus of the shortest trees, which is reported here. For searches of amino acids in POY, we started with 100 Wagner tree builds and conducted SPR and TBR swapping, selecting the shortest trees (with zero-length branches collapsed) and reporting the strict consensus.
Bootstrap support, which is a measure of the redundancy of signal for optimal clades, was not expected to be high, given the nature of these analyses. Single-gene phylogenies with large portions of their content removed to avoid convergence are unlikely to contain enough information to support every clade in resampling analyses, but we do report resampling support for the largest alignments (all 1st and 2nd codon positions, with GC replaced by N).
To mimic the way RAxML treats gaps and missing data, we also replaced all Ns and gaps with “?” (Figures 1F,H) and reran them in TNT, which reads “?” as “any nucleotide or gap.” We also removed gappy regions in the program Gblocks (Castresana, 2000) (Figure 1E), replacing the remaining gaps (which were allowed in half of the positions) with N. We also searched the unaligned amino acid sequences in the phylogenetic program POY v. 4 and 5 (Varón et al., 2009), which optimizes the multiple sequence alignment and tree searching simultaneously. When completed, PIN trees were uploaded to Mesquite with a character matrix of their protein lengths (Figure 3; coded as 1–5), and parsimony ancestral reconstructions traced over the trees. Characters were treated as unordered.
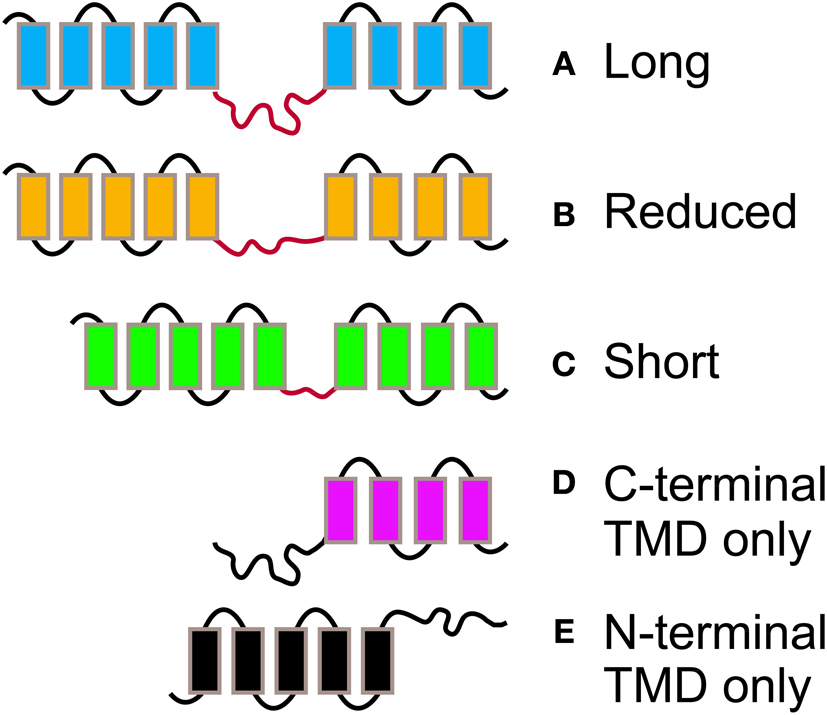
Figure 3. The different morphologies postulated for PIN proteins. PIN proteins are classified into 5 groups according to their length and structure. PINs with two complete TMDs and a long (A), reduced (B), or short (C) central hydrophylic loop. PINs with reduced protein length and presence of a TMD at the C-terminal (D), or N-Terminal end (E) only.
Results
PIN phylogenies recovered from a variety of sequence alignments and under parsimony or likelihood reconstructed the evolution of these genes from long to short (Figures 4, 5). The phylogeny recovered with full gap information under parsimony showed a clearer evolution from long, through intermediate, and to short forms, and the short versions were recovered as two clades under likelihood and parsimony when gaps were replaced by “?” or gappy regions removed by Gblocks (Figures 4B, 5A,B). Although several smaller clades remained stable throughout the analysis, and the moss and most of the lycophyte sequences tended to remain as sister to the remaining PIN genes, the relationships among the major lineages were generally unresolved. Genes having the trans-membrane domain only on the C-terminal end appeared to have evolved from long-form PINs, possibly twice, and those having this domain only on the N-terminal appear to have evolved from short-form genes, perhaps more than once. Monocot PINs were recovered mostly in small stable clades mixed among the dicot genes. Lycophyte genes were monophyletic under likelihood and under parsimony when the alignment was trimmed by Gblocks (Figure 5B); otherwise they tended to form a paraphyletic grade at the base of the tree, near the Pyscomitrella patens (moss) genes, or several small clades, only some of which were near the moss genes. Gymnosperm genes were found monophyletic only under likelihood (Figure 4B), and at the base of the tree, after the lycophytes, under parsimony (Figure 4A).
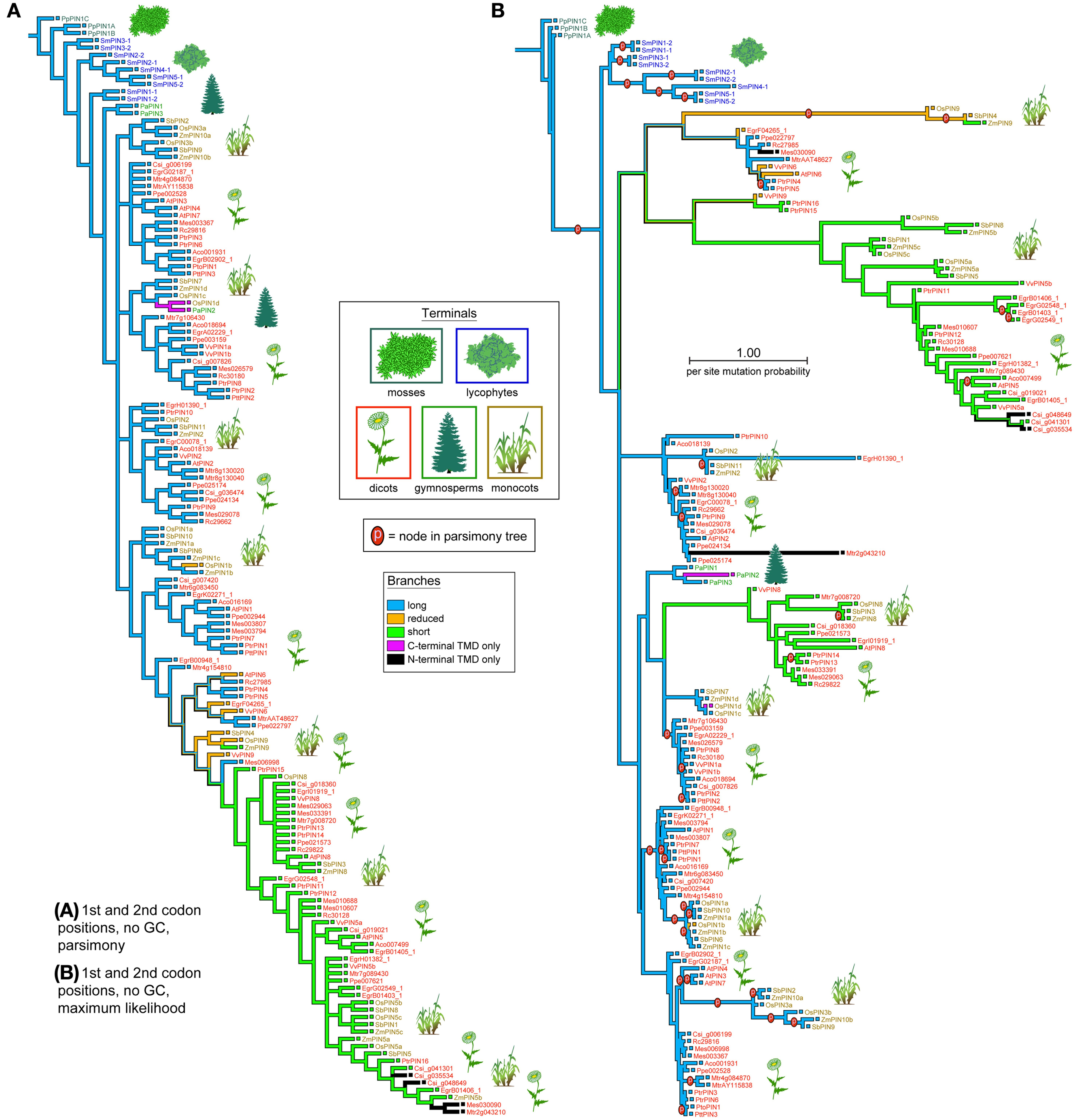
Figure 4. Phylogeny of the PIN genes, using only the 1st and 2nd codon positions and with al Gs and Cs converted to “N.” The strict consensus of 85 equally parsimonious trees (A), and the most likely tree under the GTR model (B). Branches are colored according to gene morphology, with parsimony-based historical reconstructions in both trees; equally parsimonious ancestral reconstructions are shown by multi-colored branches. Terminals are colored according to plant taxon, with icons used as guides. Clades recovered under the likelihood optimality criterion (B) which were also recovered under parsimony (A) are noted with the letter “p.”
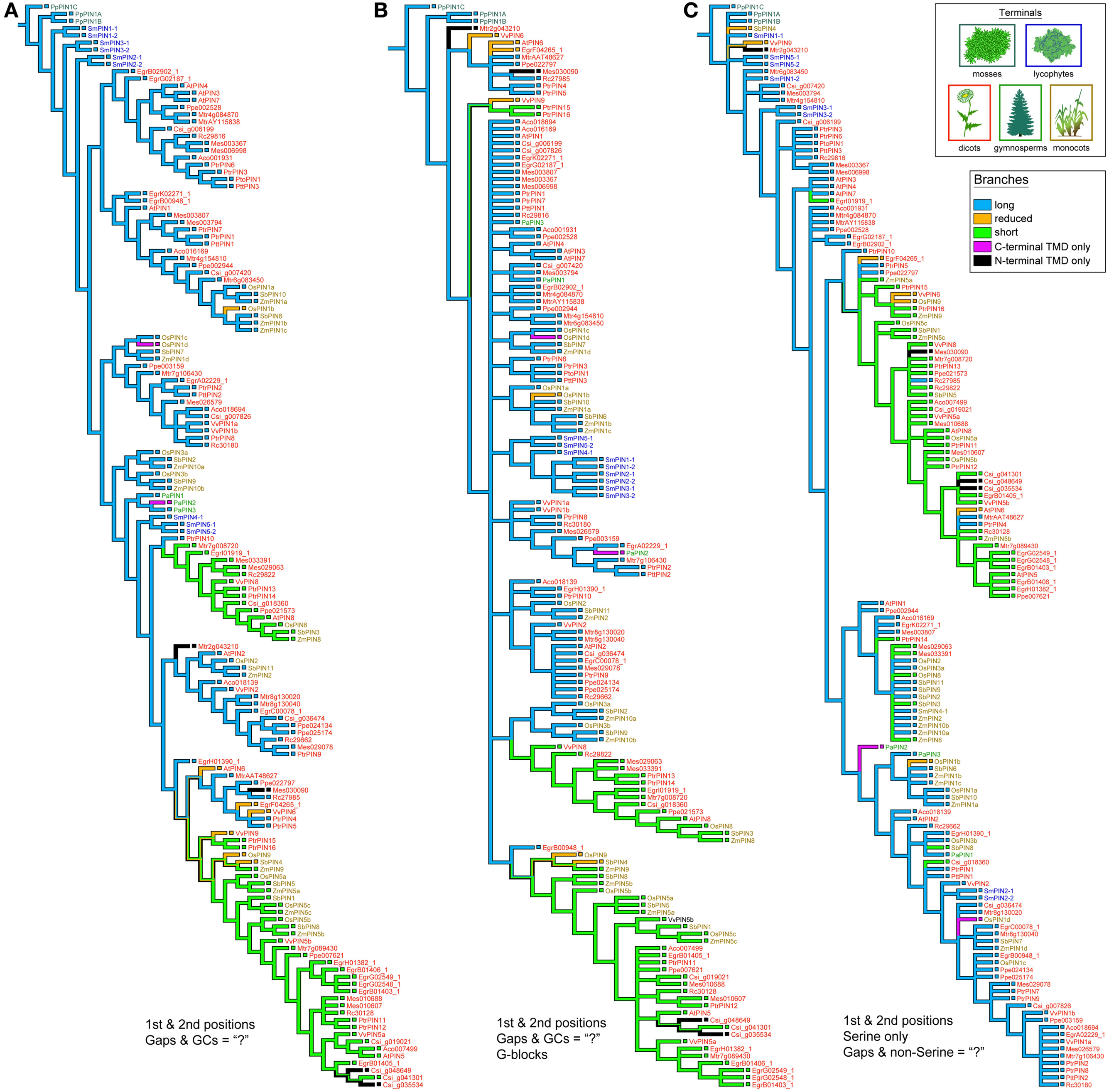
Figure 5. Strict consensuses of equally parsimonious trees using various alignments of the PIN gene data set with only 1st and 2nd codon positions. With Gs and Cs removed, the influence of gaps was minimized so as to mimic how gaps and Ns are treated in likelihood programs by replacing them with “?” (A). Alternatively, the alignment had gappy regions removed using the program Gblocks, and then all Gs, Cs, and remaining gaps replaced by “?” (B). A Serine-only alignment was also used, with only 1st and 2nd positions and all gaps and non-Serine postions replaced with “?” (C).
ACO genes clustered more distinctly by taxonomic group, under both parsimony and likelihood, except for the moss and lycophyte genes, which were mixed at the base of the tree (Figure 6). Gymnosperm ACOs were recovered as closely related but not near the basal plants. Under parsimony gymnosperms ACOs were recovered as monophyletic, with the exception of the Norway spruce gene PaABO4, and under likelihood they were recovered in two clades that formed a paraphyletic grade between two angiosperm diversifications. In both analysis, ACOs of monocotyledonous species (Oryza sativa, Sorghum bicolor, and Zea mays) cluster into three groups of closely related copies; under likelihood these groups are each monophyletic. Genes resulting from duplications are recovered as closest in both analysis (for example Sb05g005710 and Sb05g005720). ACOs from dicotyledonous species form several clades or paraphyletic grades, each containing copies from a mixture of species, except one which contains exclusively Arabidopsis sequences. As in monocots, dicot species that underwent relatively recent whole genome duplications (Malus domestica, Populus thricocarpa) present tightly related gene copies with high sequence similarity.
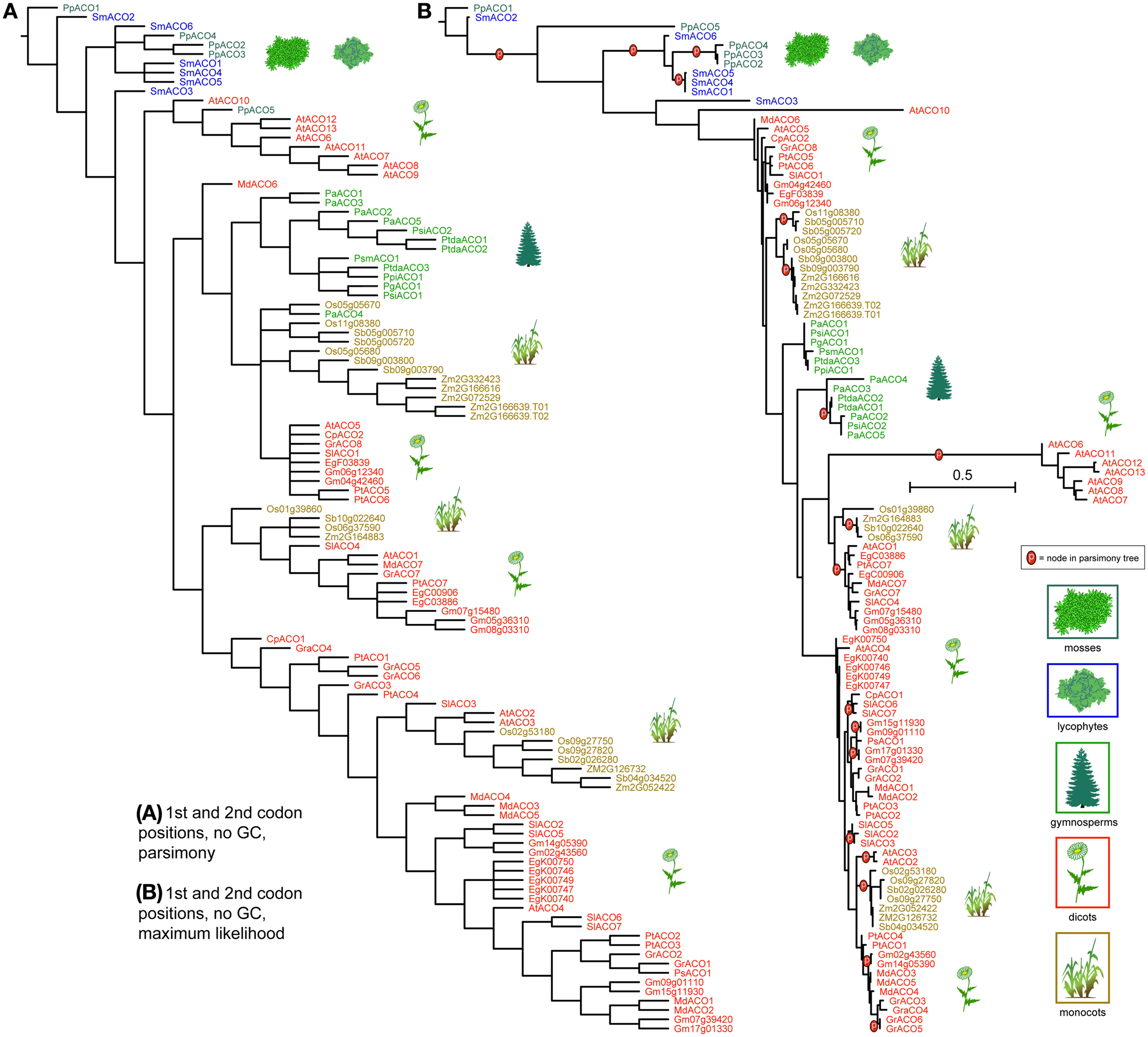
Figure 6. Phylogeny of the ACO genes, using only the 1st and 2nd codon positions and with al Gs and Cs converted to “N.” The strict consensus of 42 equally parsimonious trees is shown in (A), and the most likely tree under the GTR model is shown in (B). Terminals are colored according to plant taxon. Clades recovered under the likelihood optimality criterion (B) which were also recovered under parsimony (A) are noted with the letter “p.” Scale bar equals per-site mutation probability.
Trees recovered under dynamic homology using amino acids were less organized by taxonomic group than those recovered using the nucleotide alignments above (Figure 7). With PIN genes, copies from the moss P. patens remained sister to all other PINs, but the lycophyte copies were recovered throughout the rest of the tree. This would make any ancestral reconstruction of gene length ambiguous, as the lycophyte copies are classified as long. With the ACOs, the mixing of gene copies by taxonomic group occurred mostly at the base of the tree, although it was more complete.
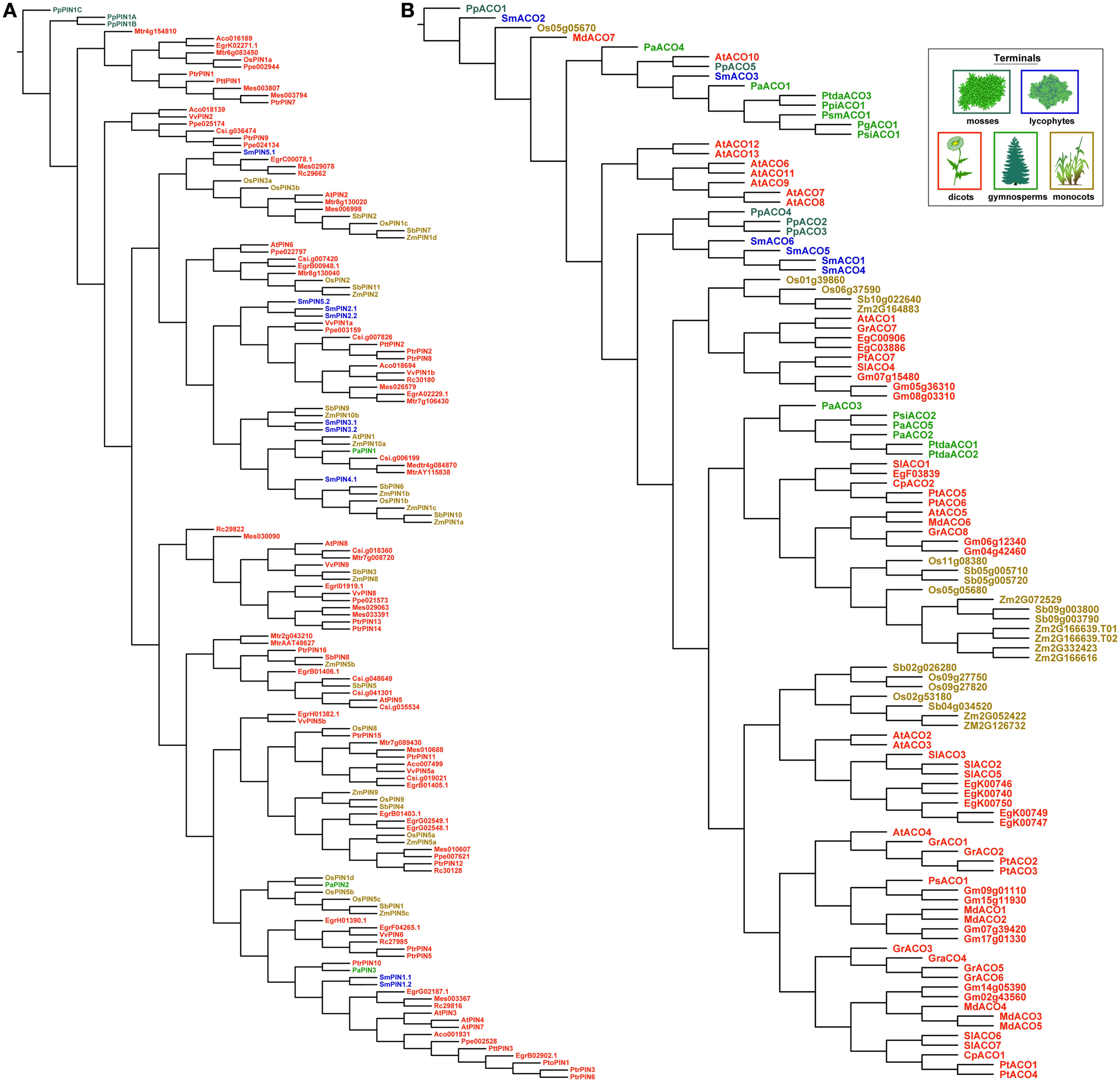
Figure 7. The most parsimonious reconstructions of the PIN (A) and ACO (B) gene family histories, using amino acid sequences analyzed using dynamic homology (alignment and tree-search optimized simultaneously). Terminals are colored according to plant taxon.
Tree lengths, numbers, and likelihoods are provided in Table 3. The full alignment of 1st and 2nd codon positions, with all GC content replaced by N resulted in a large number of equally parsimonious trees for PINs and ACOs, mostly due to equally parsimonious resolutions among the main lineages in PINs, and among members of small, derived clades in ACOs. These unstable regions of each gene family's phylogeny also received little bootstrap support (Figures S1, S2).

Table 3. The number of most parsimonious trees found and their lengths, or the log likelihoods, for each of the alignments searched.
Discussion
We find strong support for shorter PINs evolving during the diversification of angiosperms, and the basal plants (here represented by the moss Physcomitrella patens and lycophyte Selaginella moellendorfii) retaining only long PINs. Whether originating more than once or not, short PINs have evolved more recently and their number increased in monocots and dicots following genome duplications in species such as Oryza Sativa, Zea mays, Populus trichocarpa. This finding contradicts that of Viaene et al. (2013). The difference results from how our phylogenies are rooted, and rooting rests homology assessments, alignment methods, the way phylogenetic programs handle missing data, and assumptions about whether primitive plants are more likely to retain primitive gene copies. There are several tests for homology (Patterson, 1988), and the only one presently available for analyzing gene families is similarity; thus for highly dissimilar sequences some justification should be offered for their inclusion. To illustrate how divergent the outgroups of Viaene et al. (2013) are, their supposed PIN homolog from ants has a shorter uncorrected p-distance relative to the ingroups when reversed than in its original direction. Homology for the animal sequences cannot be justified based on auxin transport, since animals do not have auxin, so it seems likely such sequences are simply not homologous. For such divergent sequences, even if homologous, they will generate large gappy sections in the alignment, only some of which can be removed by hand, and they will likely root randomly. Another source of alignment difficulties is the partial nature of the algal PIN sequences which Viaene et al. (2013) took from ESTs. If the phylogenetic programs they employed treated gaps and missing data the same, algal sequences among the outgroups would be inclined to pull short PIN genes to the base of the tree. Indeed, it is not surprising that non-homologous and partial sequences in the outgroups attracted the strange and truncated PpPIN1D sequence to the outside of the ingroups—more through shared exclusion than similarity—thus strengthening the appearance of a short-to-long evolution of PIN genes.
In the absence of a suitable outgroup for plant PIN genes, we have simply made the assumption that plants recovered as sister to all the others (what one might call “primitive” or “basal” plants) carry gene copies that are also likely to be sister to all the others. This may not be correct, as evidenced by the placement of some gymnosperm copies as more derived than angiosperm copies. However, perhaps the most stable and supported relationship we recovered with PINs and ACOs was the clear distinction between moss (and usually lycophyte) copies and all the others. Thus, these gene families either diversified very early, and mosses and lycophytes retained only the most derived copies, or, more parsimoniously, the gene families simply diversified after the rise of spermatophytes. In any case, we note that the reconstruction of a long-to-short evolution of PIN genes is not merely the result of putting moss copies as the outgroup, for even in generally unresolved trees, like the one recovered from the alignment trimmed of gaps by Gblocks (Figure 5B), the main clades of short-form PINs are closely related to each other and are derived from angiosperm long forms. Only rerooting specifically by a short-form copy would change this, and in reconstructions with two origins of short-form copies, one clade would remain derived.
A recent phylogenetic analysis of PIN genes done independently of the analysis here (Bennett et al., 2014) uses a similar methodological approach and obtains results that are broadly congruent with our previous analysis (Carraro et al., 2012) and the analysis here. Bennett et al. (2014) use nucleotide sequences and root by bryophytes, and they recover the odd PpPIND in a derived position on a long branch. They also find multiple, later origins for short (or more specifically, what they term “non-canonical”) forms of PIN genes. It thus appears that whether improvements are made to PIN phylogenies by adding more sequences (Bennett et al., 2014) or excluding sources of convergence in the data, as we do here, PIN genes increasingly seem to have undergone shortening events multiple times.
In our phylogenies it appears that most modern ACOs arose subsequent to the monocot-dicot split [140–150 Ma ago, during the late Jurassic-early Cretaceous (Chaw et al., 2004)], but PIN genes diversified much earlier, as evidenced by their more thorough historical mixture of monocot and dicot copies. A broad diversification of ACOs during the Mesozoic is later than was previously hypothesized by John (1997), who did not consider them present in primitive land plants and believed their appearance was necessitated by droughts at the end of certain Permian periods (the Devonian at 360 Ma ago and the Carboniferous at 300 Ma ago). It appears that multiple ACO copies were present during the Permian and before the split between gymnosperms and angiosperms, and even before the split between mosses and lycophytes, but the later proliferation of copies in angiosperms requires a new ecological driver besides droughts in the Permian.
Gymnosperm PINs and ACOs appear to have derived from angiosperm gene copies. This is not corrected simply by using a gymnosperm root, since that renders moss and lycophyte copies derived. Rather it indicates that for both PINs and ACOs the ancestral copy in gymnosperms was a more derived copy than some of the copies inherited and retained in the ancestral angiosperm. A very limited number of gymnosperm sequences are available for both gene families, so the possibility remains that the history of gymnosperm sequences will become clearer as more of them are included in future analyses.
Amino acid sequences should present problems for historical reconstruction (Simmons, 2000), despite their popularity in plant gene family phylogenies. Genetic code degeneracy and selection pressure on protein function are sources of convergence, and although amino acids may correct for back-mutations in the third codon position, which can be another source of convergence, ignoring the third codon position removes information on recent divergences. Using amino acids with phenetic tree-building methods like Neighbor-Joining (algorithms that cluster sequences by overall similarity) (Saitou and Nei, 1987) will likely amplify convergence in amino acids, and using them with probabilistic optimality criteria requires a model of evolution both for the alignment step and tree-searching. We find here that trees made with amino acids and the most agnostic cladistic method for optimizing alignment and tree-searching (dynamic homology) produced trees with little in their favor relative to the trees made from GC-free nucleotide alignments, and we would not recommend using amino acids for future historical studies of gene families.
Although a few monocot PIN and ACO seqeunces do not have a high GC content, it seems likely that this quality is the result of an ongoing substitution bias and thus a source of misleading, convergent signal. Monocot copies are not monophyletic, and almost all of them present very high GC content, which argues against this being the result of a historical event no longer maintained in monocots. For example, in PINs almost half of the monocot copies have GC content at the third codon position above 50%, and all but four are over 40%. We notice in Meister and Barow's survey (2007) that monocots in general have statistically significantly higher GC content than dicots (using a t-test of the GC percentages, p < 0.001), and that they attain a maximum content around 50%, about 10% higher than the maximum GC content of dicots. However on average monocot genomes have only about 1% more GC content than dicots. We notice that the most GC-rich species in Meister and Barow (2007) are the grasses, which constitute all the monocots in our data set. Given how strong this bias appears in grass ACOs and PINs, it was perhaps trivial for us to retrieve monocot clades in our previous PIN phylogeny (Carraro et al., 2012), which was based on all three codon positions and did not have GC content removed. However, we still recover most monocots together (either still in clades or paraphyletic grades) and in the same combinations here as before. For example, the monocot group of nine short-form genes that includes OsPIN5b and SbPIN5 was recovered both previously and here (Figure 4B), but previously these were monopheletic, and here they form a grade out of which diversifies a clade of short-form and N-terminal-TMD-only genes. Even using the alignment that had only the first and second positions of serine, which should be immune from a GC mutational bias, we recovered clusters of monocots, a close relationship among primitive land plants, and a clade of mostly short PIN genes (Figure 5C).
Here we present new phylogenies for PIN and ACO genes, after working to improve the methods used to reconstruct the histories of gene families. First, we avoid the use of amino acids and distance (phenetic) algorithms, which have the potential to convey and amplify homoplasy. Next, in a further attempt to avoid homoplasy among genes with similar lengths and GC content, we avoid the use of indels and treat Gs and Cs or their transformations as missing data. We root trees by gene copies found in bryophytes, and we exclude sequences which are not clearly homologous. The results suggest an evolution from long to short PINs, perhaps multiple times, and a diversification of ACOs mostly after the dicot-monocot split. More sequences from a wider taxonomic range for these gene families are welcome for the continued development of their phylogenetic hypotheses and a deeper understanding of their histories.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Jon Halter and the University Research Computing staff at UNC Charlotte for assistance with cluster computing. This work was partially conducted under a postdoctoral position funded by a grant from Defense Advanced Research Projects Agency (DARPA, W911NF-05-1-0271).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2014.00296/abstract
References
Benson, D., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Wheeler, D. L. (2005). GenBank. Nucleic Acids Res. 33, D34–D38. doi: 10.1093/nar/gki063
Bennett, T., Brockington, S. F., Rothfels, C., Graham, S., Stevenson, D. and, Kutchan, T., et al. (2014). Paralagous radiations of PIN proteins with multiple origins of non-canonica PIN structure. Mol. Biol. Evol. doi: 10.1093/molbev/msu147. [Epub ahead of print].
Bleecker, B., and Kende, H. (2000). Ethylene: a gaseous signal molecule in plants. Annu. Rev. Cell Dev. Biol. 16, 1–18. doi: 10.1146/annurev.cellbio.16.1.1
Bidonde, S., Ferrer, M. A., Zegzouti, H., Ramassamy, S., Latché, A., Pech, J. C., et al. (1998). Expression and characterization of three tomato 1-aminocyclopropane-1-carboxylate oxidase cDNAs in yeast. Eur. J. Biochem. 253, 20–26. doi: 10.1046/j.1432-1327.1998.2530020.x
Carraro, N., Tisdale-Orr, T. E., Clouse, R. M., Knöller, A. S., and Spicer, R. (2012). Diversification and expression of the PIN, AUX/LAX, and ABCB families of putative auxin transporters in Populus. Front. Plant Sci. 3:17. doi: 10.3389/fpls.2012.00017
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chaw, S. M., Chang, C. C., Chen, H. L., and Li, W. H. (2004). Dating the monocot-dicot divergence and the origin of core eudicots using whole chloroplast genomes. J. Mol. Evol. 58, 424–441. doi: 10.1007/s00239-003-2564-9
Chung, M.-C., Chou, S.-J., Kuang, L.-Y., Charng, Y.-Y., and Yang, S. F. (2002). Subcellular localization of 1-aminocyclopropane-1-carboxylic acid oxidase in apple fruit. Plant Cell Physiol. 43, 549–554. doi: 10.1093/pcp/pcf067
De Paepe, A., and Van der Straeten, D. (2005). Ethylene biosynthesis and signaling: an overview. Vitam. Horm. 72, 399–430. doi: 10.1016/S0083-6729(05)72011-2
De Smet, I., Voss, U., Lau, S., Wilson, M., Shao, N., Timme, R. E., et al. (2011). Unraveling the evolution of auxin signaling. Plant Physiol. 155, 209–221. doi: 10.1104/pp.110.168161
Farris, J. S. (1983). “The logical basis of phylogenetic analysis,” in Advances in Cladistics; Proceedings of the Second Meeting of the Willi Hennig Society, eds N. I. Platnick and V. A. Funk (New York, NY: Columbia University Press), 7–36.
Goloboff, P. A., Farris, J. S., and Nixon, K. C. (2008). TNT, a free program for phylogenetic analysis. Cladistics 24, 774–786. doi: 10.1111/j.1096-0031.2008.00217.x
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView Version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Hall, T. (2007). BioEdit (Version 7.0.9) [Computer Program]. Available online at: www.ibisbiosciences.com/
Hudgins, J. W., Ralph, S. G., Franceschi, V. R., and Bohlmann, J. (2006). Ethylene in induced conifer defense: cDNA cloning, protein expression, and cellular and subcellular localization of 1-aminocyclopropane-1-carboxylate oxidase in resin duct and phenolic parenchyma cells. Planta 224, 865–877. doi: 10.1007/s00425-006-0274-4
John, P. (1997). Ethylene biosynthesis: the role of 1-aminocyclopropane-lcarboxylate (ACC) oxidase, and its possible evolutionary origin. Physiol. Plant. 100, 583–592. doi: 10.1111/j.1399-3054.1997.tb03064.x
Katoh, K., Asimenos, G., and Toh, H. (2009). Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 537, 39–64. doi: 10.1007/978-1-59745-251-9_3
Křeček, P., Skùpa, P., Libus, J., Naramoto, S., Tejos, R., Friml, J., et al. (2009). The PIN-FORMED (PIN) protein family of auxin transporters. Genome Biol. 10, 1–11. doi: 10.1186/gb-2009-10-12-249
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., Mcwilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lin, Z., Zhong, S., and Grierson, D. (2009). Recent advances in ethylene research. J. Exp. Bot. 60, 3311–3336. doi: 10.1093/jxb/erp204
Maddison, W. P., and Maddison, D. R. (2009). Mesquite: a Modular System for Evolutionary Analysis (Version 2.6) [Computer Program]. Available online at: http://mesquiteproject.org.
Meister, A., and Barow, M. (2007). “DNA base composition of plant genomes,” in Flow Cytometry with Plant Cells: Analysis of Genes, Chromosomes and Genomes, eds J. Doležel, J. Greilhuber, and J. Suda (Weinheim: Wiley–VCH), 177–215.
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE) (New Orleans, LA), 1–8.
Moller, S., Croning, M. D. R., and Apweiler, R. (2001). Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17, 646–653. doi: 10.1093/bioinformatics/17.7.646
Okada, K., Ueda, J., Komaki, M. K., Bell, C. J., and Shimura, Y. (1991). Requirement of the auxin polar transport system in early stages of Arabidopsis floral bud formation. Plant Cell 3, 677–684. doi: 10.1105/tpc.3.7.677
Paponov, I. A., Teale, W. D., Trebar, M., Blilou, I., and Palme, K. (2005). The PIN auxin efflux facilitators: evolutionary and functional perspectives. Trends Plant Sci. 10, 170–177. doi: 10.1016/j.tplants.2005.02.009
Plettner, I. N. A., Steinke, M., and Malin, G. (2005). Ethene (ethylene) production in the marine macroalga Ulva (Enteromorpha) intestinalis L. (Chlorophyta, Ulvophyceae): effect of light-stress and co-production with dimethyl sulphide. Plant Cell Environ. 28, 1136–1145. doi: 10.1111/j.1365-3040.2005.01351.x
Robert, H. S., and Friml, J. (2009). Auxin and other signals on the move in plants. Nat. Chem. Biol. 5, 325–332. doi: 10.1038/nchembio.170
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Sato, T., and Theologis, A. (1989). Cloning the mRNA encoding l-aminocyclopropane-l-carboxylate synthase, the key enzyme for ethylene biosynthesis in plants. Proc. Natl. Acad. Sci. U.S.A. 86, 6621–6625. doi: 10.1073/pnas.86.17.6621
Simmons, M. (2000). A fundamental problem with amino-acid-sequence characters for phylogenetic analyses. Cladistics 16, 274–282. doi: 10.1111/j.1096-0031.2000.tb00283.x
Simmons, M., Ochoterena, H., and Freudenstein, J. V. (2002). Conflict between amino acid and nucleotide characters. Cladistics 18, 200–206. doi: 10.1111/j.1096-0031.2002.tb00148.x
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Varón, A., Sy Vinh, L., and Wheeler, W. C. (2009). POY version 4: phylogenetic analysis using dynamic homologies. Cladistics 26, 72–85. doi: 10.1111/j.1096-0031.2009.00282.x
Viaene, T., Delwiche, C. F., Rensing, S. A., and Friml, J. (2013). Origin and evolution of PIN auxin transporters in the green lineage. Trends Plant Sci. 18, 5–10. doi: 10.1016/j.tplants.2012.08.009
Wang, K. L., Li, H., and Ecker, J. R. (2002). Ethylene biosynthesis and signaling networks. Plant Cell 14(Suppl.), S131–S152. doi: 10.1105/tpc.001768
Wanke, D. (2011). The ABA-mediated switch between submersed and emersed life-styles in aquatic macrophytes. J. Plant Res. 124, 467–475. doi: 10.1007/s10265-011-0434-x
Yasumura, Y., Pierik, R., Fricker, M. D., Voesenek, L. A., and Harberd, N. P. (2012). Studies of Physcomitrella patens reveal that ethylene-mediated submergence responses arose relatively early in land-plant evolution. Plant J. 72, 947–959. doi: 10.1111/tpj.12005
Yordanova, Z. P., Iakimova, E. T., Cristescu, S. M., Harren, F. J. M., Kapchina-Toteva, V. M., and Woltering, E. J. (2010). Involvement of ethylene and nitric oxide in cell death in mastoparan-treated unicellular alga Chlamydomonas reinhardtii. Cell Biol. Int. 34, 301–308. doi: 10.1042/CBI20090138
Keywords: ethylene synthesis, auxin, ancestral reconstruction, GC mutational bias, PIN-formed, 1-aminocyclopropane-1-carboxylate oxidase
Citation: Clouse RM and Carraro N (2014) A novel phylogeny and morphological reconstruction of the PIN genes and first phylogeny of the ACC-oxidases (ACOs). Front. Plant Sci. 5:296. doi: 10.3389/fpls.2014.00296
Received: 25 March 2014; Accepted: 06 June 2014;
Published online: 24 June 2014.
Edited by:
Serena Varotto, University of Padova, ItalyReviewed by:
Joseph Colasanti, University of Guelph, CanadaDavid G. Oppenheimer, University of Florida, USA
Copyright © 2014 Clouse and Carraro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronald M. Clouse, Department of Bioinformatics and Genomics, University of North Carolina at Charlotte, 9201 University City Blvd. Bioinformatics, Room 224, Charlotte, NC 28223, USA e-mail: rclouse@uncc.edu