 Tobias Link
Tobias Link Christian Seibel
Christian Seibel Ralf T. Voegele
Ralf T. Voegele- Fachgebiet Phytopathologie, Institut für Phytomedizin, Fakultät Agrarwissenschaften, Universität Hohenheim, Stuttgart, Germany
Uromyces fabae is a major pathogen of broad bean, Vicia faba. U. fabae has served as a model among rust fungi to elucidate the development of infection structures, expression and secretion of cell wall degrading enzymes and gene expression. Using U. fabae, enormous progress was made regarding nutrient uptake and metabolism and in the search for secreted proteins and effectors. Here, we present results from a genome survey of U. fabae. Paired end Illumina sequencing provided 53 Gb of data. An assembly gave 59,735 scaffolds with a total length of 216 Mb. K-mer analysis estimated the genome size to be 329 Mb. Of a representative set of 23,153 predicted proteins we could annotate 10,209, and predict 599 secreted proteins. Clustering of the protein set indicates families of highly likely effectors. We also found new homologs of RTP1p, a prototype rust effector. The U. fabae genome will be an important resource for comparative analyses with U. appendiculatus and P. pachyrhizi and provide information regarding the phylogenetic relationship of the genus Uromyces with respect to other rust fungi already sequenced, namely Puccinia graminis f. sp. tritici, P. striiformis f. sp. tritici, Melampsora lini, and Melampsora larici-populina.
Introduction
The order Pucciniales is dominated by two genera: Puccinia with about 4,000 species and Uromyces with around 600 species (Maier et al., 2003). Though the phylogeny of the two genera has not been entirely disentangled, there is a clear tendency that Puccinia species mainly infect grass hosts, whereas Uromyces species seem to be concentrated on legumes. There is now sequence information available for two species within the genus Puccinia [Puccinia graminis f. sp. tritici (Pgt), and Puccinia striiformis f. sp. tritici (Pst)] (Duplessis et al., 2011; Cantu et al., 2013; Zheng et al., 2013). Recently, re-sequencing of several Pst strains was undertaken, enabling the search for accelerated evolution among secreted proteins, an interesting means for identifying highly likely effector candidates (Cantu et al., 2013). At the same time no genome information is available for the second largest genus Uromyces. Therefore, a reference genome for Uromyces enabling comparisons between these two closely related genera would be highly desirable.
Rust fungi cannot truly be called model species since their obligate biotrophic lifestyle makes basic research on these species very difficult. General information regarding fungi or basidiomycetes can much easier be obtained with other species. Among the 7,000 rust fungi, research is concentrated on a very limited selection of species. One reason that brought these species into focus is the high economic losses associated with them. This is true for the cereal rusts Pgt and Pst, or the Asian Soybean Rust, Phakopsora pachyrhizi. The other reason why some rust species are prominent in research is their historical significance. The most important example here is Melampsora lini on Linum usitatissimum, the system upon which Flor developed the gene-for-gene hypothesis (Flor, 1956). These early successes have been picked up in modern molecular research, for example making the connection between Avr genes and proteins secreted from haustoria (Catanzariti et al., 2006), or providing information on the interaction between Avr and R-proteins (Dodds et al., 2006), up to contributions as to how effectors may reach their targets (Rafiqi et al., 2010).
Among Uromyces species a similar role can be assigned to U. fabae. Over the years especially morphologic studies using light and electron microscopy contributed to the elucidation of infection structures of rust fungi (Kapooria and Mendgen, 1985). Later, biochemical studies contributed to the understanding of the physiology of early infection (Deising et al., 1991). The development of a method to isolate haustoria from infected leaves by Hahn and Mendgen (1992) made it possible to study these hallmark structures of obligate biotrophic pathogens. Building on the study of PIGs (in planta induced genes, genes found highly expressed in haustoria) (Hahn and Mendgen, 1997), more molecular and biochemical studies followed. These studies provided proof for the importance of haustoria in nutrient uptake (Voegele et al., 2001), as well as the generation of energy (Sohn et al., 2000). Among the PIGs also the first non-avirulence protein shown to be transferred from the fungus into the host cytoplasm (Uf-RTP1p) was discovered (Kemen et al., 2005). Current research on U. fabae is focused on searching novel candidate effectors among secreted proteins (Link and Voegele, 2008), augmenting the knowledge on carbohydrate uptake and metabolism, and the quest for a generally applicable method to stably transform rust fungi (Djulic et al., 2011). Keeping up this tradition of research on this species, U. fabae was the logical choice for generating a reference genome for the genus Uromyces. However, U. fabae could serve as a model not only for Uromyces species but also for other legume rusts, the most important of which at the moment is P. pachyrhizi.
Here, we report first results from a genome survey on U. fabae. Based on the results of this survey we expect to get a better understanding of the physiology of U. fabae. We found more candidate effectors, and we did and will do more comparisons of gene content against Pgt and Pst and also Melampsora larici-populina (Mlp). We plan to expand this survey into a full genome sequencing project.
Sequencing
DNA was prepared from urediospores of U. fabae isolate I2. This isolate has been in use in the Mendgen lab (Universität Konstanz, Germany) and the Voegele lab for many years. Virtually all experiments published on U. fabae were made using this strain. DNA was isolated from germinated urediospores using a protocol modified from Kolmer et al. (1995). Urediospores were washed for 30 min and germinated for 3.5 h. Germinated spores were homogenized by grinding in liquid N2 and acid washed sand, and incubated in CTAB solution. Phenol-chloroform extraction, chloroform extraction, precipitation with 2-propanol, and RNaseA digest were performed to purify the DNA. Quality assessments showed only minor degradation and a slight bacterial contamination.
Paired end sequencing using Illumina HiSeq2000 with a 500 nt library was performed by BGI TECH SOLUTIONS (HONGKONG) CO. LIMITED (16 Dai Fu Street, Tai Po Industrial Estate, Tai Po, N.T., Hong Kong) who also supplied the assembly that is presented. 593,062,170 reads with a length of 90 bp were produced giving 53,375 Mb in raw data. 8.8% of the data were removed during filtering, leaving 48,661 Mb of clean data. The sequence reads were deposited in NCBI SRA in experiment SRX547322 corresponding to BioProject PRJNA248166.
Using SOAPdenovo reads were assembled into 59,735 scaffolds with a total length of 215,710,123 bp. N50 for the scaffolds is 5873 bp, the longest scaffold spans 72,118 bp, the shortest one 1,000 bp. These scaffolds were built from 95,847 contigs with a total length of 209,504,160 bp. N50 for the contigs is 4,171 bp, the longest contig is 45,252 bp, the shortest 200 bp. This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession JNCO00000000. The version described in this paper is version JNCO01000000.
The original assembly with 1,191,649 scaffolds that was used for prediction of proteins, is very fragmented, so it is not possible to draw definite conclusions. We hope to improve our data by further sequencing (i.e., pacbio sequencing) and performing new assemblies for example with realizing mate pair sequence. One objective of this publication is to spark interest in this genome so that other groups or organizations could add their expertise into the project. However, as mentioned above, we were most interested in the gene complement, especially in distinct metabolic pathways and secreted proteins as effector candidates, so nevertheless, we set out to do further analyses that are presented below. Here we want to supply the reader with this caveat: All analyses are based on a provisional assembly.
Genome Size
Our data so far gave us three estimates on the size of the U. fabae genome. On the one hand we have two different k-mer analyses, a 15mer analysis that estimates the genome size to 329 Mb and a 17mer analysis that calculates to 330 Mb. On the other hand, there is the original assembly size of 422 Mb and the filtered assembly with 216 Mb. Apart from this, the genome size of U. fabae isolate I2 was recently measured using flow cytometry on isolated nuclei from germinated urediospores, giving an estimate of 379 Mb (Tavares et al., 2014). Given the preliminary nature of the assemblies we consider the k-mer analysis and the flow cytometric data as most reliable and thus estimate the actual genome size in the range between 330 and 379 Mb.
Analysis on other rust fungi has shown that compared to other fungi the genomes of rusts are fairly inflated. U. fabae seems to be no exception. The pioneering genome sequences of Mlp and Pgt also revealed a reason or the mechanism for these big genome sizes—a high amount of transposable elements (TE) (Duplessis et al., 2011). We assume that U. fabae likewise has a large amount of TE, probably more than Mlp and Pgt. The high amount of predicted proteins that were annotated as transposon related (see below) also seems to point in this direction. So far, an analysis of repeats and TE could not be performed, but given the large genome size, this will be an important part in the analysis of later assemblies.
A Preliminary View on the Gene Complement
All data regarding predicted proteins (annotation, prediction as secreted, clustering) as described below are integrated in Supplementary Table 1.
Protein Prediction and Annotation
To have an idea, how useful the original assembly could be despite its fragmented state, we analyzed it with CEGMA (core eukaryotic genes mapping approach, Parra et al. (2009). This analysis showed that of the 248 highly conserved CEGs 95% were at least partially, and 89% completely present. Thus, this indicates that a large portion of the gene complement should be represented in the current assembly. Without preceding prediction and masking of repeats we used the Augustus Web server (Hoff and Stanke, 2013) with the gene structure file from the CEGMA output as a training set and 590 available ESTs (Jakupovic et al., 2006; Link and Voegele, 2008) as “hints” to predict 70,913 proteins. Compared to the 17,773 protein coding genes predicted for Pgt, and the 16,399 for Mlp (Duplessis et al., 2011), this is a gross over-prediction—most likely due in large part to TEs. For a more accurate gene prediction a better assembly, prediction of repeats, and more information on gene structure and especially more cDNA sequence information will be necessary.
To get a workable dataset steps were taken to reduce the set of predicted proteins closer to realistic numbers. First, all predicted sequences were truncated to the first methionine and all sequences shorter than 80 aa were removed. To remove redundancy among the remaining 56,594 predicted proteins they were clustered using the cd-hit-suite web server. Clustering with 0.7% ID as cutoff yielded 23,153 clusters, which seemed adequate. The representative proteins from this clustering were used for subsequent analyses.
Proteins were annotated using the Blast2GO suite. BLAST results could be obtained for 20,153 proteins, Gene Ontology (GO) terms were mapped for 14,085 proteins, and after integrating the InterProScan results and running Annex, 10,209 proteins could be annotated according to the Blast2GO rule.
The most important species among the BLAST hits in the NCBI nr database is Puccinia graminis, both for all hits and for best hits. The rest of the list is dominated by other fungal species, though surprisingly also plant and animal species are represented. Despite the result of the PCR on rDNA that predicted a bacterial contamination (and despite the omission of steps to remove this contamination), no bacterial species was prominent among the best hits. Almost half of the annotated proteins are transposon related indicating again that a new assembly will be necessary that should be masked against repeats with RepeatMasker.
Families of Secreted Proteins/Candidate Effectors
Using SignalP4 760 signal peptides were predicted. Of the proteins carrying a signal peptide 135 had additional transmembrane domains (predicted by TMHMM), two were predicted as mitochondrial by TargetP, and 33 carry a predicted glycophosphatidylinositol (GPI) anchor (predGPI); six proteins have both transmembrane domains and a GPI anchor. 599 predicted secreted proteins remained.
To identify candidate effectors, which we assume to be specific to rust fungi or even to a single species, all proteins were blasted (blast+) against the protein complement of 10 basidiomycete species, among them four rust fungi, a hemibiotrophic smut fungus, a biotrophic mutualistic symbiont, a close relative of rust and smut fungi and three saprotrophic fungi (see Supplementary Tables 1, 3). Using spectral clustering (SCPS), 1,315 clusters containing at least two proteins were formed. 18,908 proteins fell into these clusters. This way, several families of secreted proteins, and also specific to rust fungi or lineage specific could be identified. For a better overview a smaller clustering just for proteins with predicted signal peptide was performed, including secretome results for U. fabae and predicted secreted proteins of U. appendiculatus and P. pachyrhizi (Link and Voegele, 2008; Link et al., 2014). Of the clusters that were formed 191 contained predicted secreted proteins from U. fabae. 44 of these families contained proteins that were found secreted with the signal sequence trap (Link and Voegele, 2008), 121 also contained U. appendiculatus proteins, 66 P. pachyrhizi proteins. Table 1 shows an overview of the 10 biggest families. Remarkably, only one of these families could be assigned a function, cluster 9, which is a family of expansins.
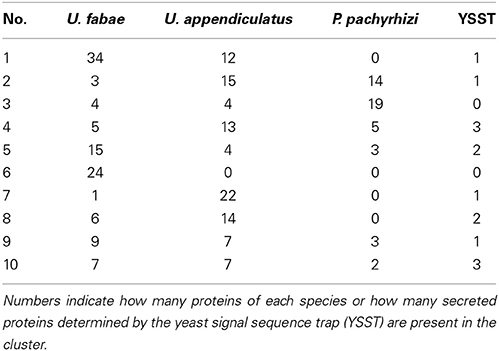
Table 1. The 10 largest clusters of predicted secreted proteins.
One effector family that has held our interest for some time now is the RTP (rust transferred protein) family. Our latest findings on RTP1p indicate that the protein has proteinase inhibitor function (Pretsch et al., 2013). Other findings show, that the protein forms fibrils in the extrahaustorial matrix as well as in the host cytoplasm (Kemen et al., 2013). This may be associated with slowing down cyclosis in the host cell and/or keeping the plant cell nucleus in the immediate vicinity of the haustorium, thus ensuring close contact and a better influence of the pathogen on its host. While these two functions are not mutually exclusive, as the fibrils around the haustorium could have a protective function, it is highly unusual that a protein should have both a structural and an enzymatic function. So far, structural and functional analyses have been limited to Uf-RTP1p.
That Uf-RTP1p is a member of an extended gene family was shown in recent sequencing projects. Search for RTP1 homologs using degenerate primers yielded additional information. The most recent summary of these homologs can be found in Pretsch et al. (2013). Using tblastn to search RTP1 homologs against our assembly we could find—in addition to Uf-RTP1—two more homologs. One, located on scaffold6572, shows highest similarity to Ua-RTP2 (Ua: Uromyces appendiculatus). According to the nomenclature proposed by Puthoff et al. (2008), it will be designated Uf-RTP2. The second homolog showed low similarity to several RTP1 homologs and could not be clearly assigned a best match. Using as query RTP homologs from other rust fungi, we also found seven homologs to Ua-RTP9, the highest scoring of these is located on scaffold2295 and was designated Uf-RTP9. The similarity between the Uf-RTP9 homologs and Uf-RTP1 was so low however, that they did not cluster. Therefore, we find it reasonable to designate a new family, the RTP9 family.
It will now be interesting to do corresponding experiments with the newly identified RTP homologs to check whether these have a similar duality of functions as Uf-RTP1p. This phenomenon seems all the more fascinating now that RTP was shown to be a gene family in U. fabae as well as in other rust species. It seems reasonable to presume that the different proteins could be expressed at different stages and secreted from different structures, as was shown for the Mlp-RTPs by Hacquard et al. (2012).
In additional analyses we want to systemize the families of predicted secreted proteins into those that have functional annotation, and those that are also lineage specific, which makes them likely effector candidates. We will search the secreted proteins and the protein families for common motifs, both novel motifs and motifs already linked to effector function and/or transfer into the host cytoplasm. We will also build phylogenies for both all secreted proteins and selected families of high interest. We will also build groups of orthologs and identify those genes that have formed a high number of paralogs. Alignments of protein families will also help us to sort out proteins that were predicted as secreted because of truncations. These predictions will lead to wet-lab experiments, i.e., phenotypical screens like cell death suppression assays, search for interaction partners, silencing experiments and localizations. As indicated earlier, we will try to improve the assemblies and gene prediction and, given the opportunity, expand this survey into a full genome sequencing project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sibylle Berger for technical assistance. We are also grateful to the reviewers and the editor Sébastien Duplessis for useful suggestions that will help us to improve our work in later analyzes.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2014.00587/abstract
References
Cantu, D., Segovia, V., Maclean, D., Bayles, R., Chen, X., Kamoun, S., et al. (2013). Genome analyses of the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal polymorphic and haustorial expressed secreted proteins as candidate effectors. BMC Genomics 14:270. doi: 10.1186/1471-2164-14-270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Catanzariti, A. M., Dodds, P. N., Lawrence, G. J., Ayliffe, M. A., and Ellis, J. G. (2006). Haustorially expressed secreted proteins from flax rust are highly enriched for avirulence elicitors. Plant Cell 18, 243–256. doi: 10.1105/tpc.105.035980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deising, H., Jungblut, P. R., and Mendgen, K. (1991). Differentiation-related proteins of the broad bean rust fungus Uromyces viciae-fabae, as revealed by high resolution two-dimensional polyacrylamide gel electrophoresis. Arch. Microbiol. 155, 191–198. doi: 10.1007/BF00248616
Djulic, A., Schmid, A., Lenz, H., Sharma, P., Koch, C., Wirsel, S. G., et al. (2011). Transient transformation of the obligate biotrophic rust fungus Uromyces fabae using biolistics. Fungal Biol. 115, 633–642. doi: 10.1016/j.funbio.2011.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dodds, P. N., Lawrence, G. J., Catanzariti, A. M., Teh, T., Wang, C. I., Ayliffe, M. A., et al. (2006). Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc. Natl. Acad. Sci. U.S.A. 103, 8888–8893. doi: 10.1073/pnas.0602577103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Duplessis, S., Cuomo, C. A., Lin, Y. C., Aerts, A., Tisserant, E., Veneault-Fourrey, C., et al. (2011). Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. U.S.A. 108, 9166–9171. doi: 10.1073/pnas.1019315108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Flor, H. H. (1956). The complementary genic systems in flax and flax rust. Adv. Genet. 8, 29–54. doi: 10.1016/S0065-2660(08)60498-8
Hacquard, S., Joly, D. L., Lin, Y. C., Tisserant, E., Feau, N., Delaruelle, C., et al. (2012). A comprehensive analysis of genes encoding small secreted proteins identifies candidate effectors in Melampsora larici-populina (poplar leaf rust). Mol. Plant Microbe Interact. 25, 279–293. doi: 10.1094/MPMI-09-11-0238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hahn, M., and Mendgen, K. (1992). Isolation by ConA binding of haustoria from different rust fungi and comparison of their surface qualities. Protoplasma 170, 95–103. doi: 10.1007/BF01378785
Hahn, M., and Mendgen, K. (1997). Characterization of in planta-induced rust genes isolated from a haustorium-specific cDNA library. Mol. Plant Microbe Interact. 10, 427–437. doi: 10.1094/MPMI.1997.10.4.427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoff, K. J., and Stanke, M. (2013). WebAUGUSTUS—a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucl. Acids Res. 41, W123–W128. doi: 10.1093/nar/gkt418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jakupovic, M., Heintz, M., Reichmann, P., Mendgen, K., and Hahn, M. (2006). Microarray analysis of expressed sequence tags from haustoria of the rust fungus Uromyces fabae. Fungal Genet. Biol. 43, 8–19. doi: 10.1016/j.fgb.2005.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kapooria, R. G., and Mendgen, K. (1985). Infection structures and their surface changes during differentiation in Uromyces fabae. J. Phytopathol. 113, 317–323. doi: 10.1111/j.1439-0434.1985.tb04832.x
Kemen, E., Kemen, A., Ehlers, A., Voegele, R., and Mendgen, K. (2013). A novel structural effector from rust fungi is capable of fibril formation. Plant J. 75, 767–780. doi: 10.1111/tpj.12237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kemen, E., Kemen, A. C., Rafiqi, M., Hempel, U., Mendgen, K., Hahn, M., et al. (2005). Identification of a protein from rust fungi transferred from haustoria into infected plant cells. Mol. Plant Microbe Interact. 18, 1130–1139. doi: 10.1094/MPMI-18-1130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kolmer, J. A., Liu, J. Q., and Sies, M. (1995). Virulence and molecular polymorphism in Puccinia recondita f. sp. tritici in Canada. Phytopathology 85, 276–285. doi: 10.1094/Phyto-85-276
Link, T. I., Lang, P., Scheffler, B. E., Duke, M. V., Graham, M. A., Cooper, B., et al. (2014). The haustorial transcriptomes of Uromyces appendiculatus and Phakopsora pachyrhizi and their candidate effector families. Mol. Plant Pathol. 15, 379–393. doi: 10.1111/mpp.12099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Link, T. I., and Voegele, R. T. (2008). Secreted proteins of Uromyces fabae: similarities and stage specificity. Mol. Plant Pathol. 9, 59–66. doi: 10.1111/j.1364-3703.2007.00448.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maier, W., Begerow, D., Weiß, M., and Oberwinkler, F. (2003). Phylogeny of the rust fungi: an approach using nuclear large subunit ribosomal DNA sequences. Can. J. Bot. 81, 12–23. doi: 10.1139/b02-113
Parra, G., Bradnam, K., Ning, Z., Keane, T., and Korf, I. (2009). Assessing the gene space in draft genomes. Nucl. Acids Res. 37, 289–297. doi: 10.1093/nar/gkn916
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pretsch, K., Kemen, A. C., Kemen, E., Geiger, M., Mendgen, K., and Voegele, R. T. (2013). The rust transferred proteins—a new family of effector proteins exhibiting protease inhibitor function. Mol. Plant Pathol. 14, 96–107. doi: 10.1111/j.1364-3703.2012.00832.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Puthoff, D. P., Neelam, A., Ehrenfried, M. L., Scheffler, B. E., Ballard, L., Song, Q., et al. (2008). Analysis of expressed sequence tags from Uromyces appendiculatus hyphae and haustoria and their comparison to sequences from other rust fungi. Phytopathology 98, 1126–1135. doi: 10.1094/PHYTO-98-10-1126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rafiqi, M., Gan, P. H., Ravensdale, M., Lawrence, G. J., Ellis, J. G., Jones, D. A., et al. (2010). Internalization of flax rust avirulence proteins into flax and tobacco cells can occur in the absence of the pathogen. Plant Cell 22, 2017–2032. doi: 10.1105/tpc.109.072983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sohn, J., Voegele, R. T., Mendgen, K., and Hahn, M. (2000). High level activation of vitamin B1 biosynthesis genes in haustoria of the rust fungus Uromyces fabae. Mol. Plant Microbe Interact. 13, 629–636. doi: 10.1094/MPMI.2000.13.6.629
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tavares, S., Ramos, A. P., Pires, A. S., Azinheira, H. G., Caldeirinha, P., Link, T., et al. (2014). Genome size analysis of Pucciniales reveal the largest fungal genomes. Front. Plant Sci. 5:422. doi: 10.3389/fpls.2014.00422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Voegele, R. T., Struck, C., Hahn, M., and Mendgen, K. (2001). The role of haustoria in sugar supply during infection of broad bean by the rust fungus Uromyces fabae. Proc. Natl. Acad. Sci. U.S.A. 98, 8133–8138. doi: 10.1073/pnas.131186798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, W., Huang, L., Huang, J., Wang, X., Chen, X., Zhao, J., et al. (2013). High genome heterozygosity and endemic genetic recombination in the wheat stripe rust fungus. Nat. Commun. 4, 2673. doi: 10.1038/ncomms3673
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Uromyces fabae, rust fungus, genome survey, genome size, candidate effectors, RTP1 homologs
Citation: Link T, Seibel C and Voegele RT (2014) Early insights into the genome sequence of Uromyces fabae. Front. Plant Sci. 5:587. doi: 10.3389/fpls.2014.00587
Received: 31 May 2014; Accepted: 09 October 2014;
Published online: 29 October 2014.
Edited by:
Sébastien Duplessis, Institut National de la Recherche Agronomique, FranceReviewed by:
Claude Murat, Institut National de la Recherche Agronomique, FranceYao-Cheng Lin, Vlaams Instituut voor Biotechnologie/Ghent University, Belgium
Copyright © 2014 Link, Seibel and Voegele. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tobias Link and Ralf T. Voegele, Fachgebiet Phytopathologie, Institut für Phytomedizin, Fakultät Agrarwissenschaften, Universität Hohenheim, Otto-Sander-Str. 5, 70599 Stuttgart, Germany e-mail: tobias.link@uni-hohenheim.de; ralf.voegele@uni-hohenheim.de