Optimization of a 96-well electroporation assay for postnatal rat CNS neurons suitable for cost–effective medium-throughput screening of genes that promote neurite outgrowth
- 1 Neurorestoration Group, Wolfson Centre for Age-Related Diseases, King’s College London, London, UK
- 2 The Miami Project to Cure Paralysis, University of Miami Miller School of Medicine, Miami, FL, USA
Following an injury, central nervous system (CNS) neurons show a very limited regenerative response which results in their failure to successfully form functional connections with their original target. This is due in part to the reduced intrinsic growth state of CNS neurons, which is characterized by their failure to express key regeneration-associated genes (RAGs) and by the presence of growth inhibitory molecules in CNS environment that form a molecular and physical barrier to regeneration. Here we have optimized a 96-well electroporation and neurite outgrowth assay for postnatal rat cerebellar granule neurons (CGNs) cultured upon an inhibitory cellular substrate expressing myelin-associated glycoprotein or a mixture of growth inhibitory chondroitin sulfate proteoglycans. Optimal electroporation parameters resulted in 28% transfection efficiency and 51% viability for postnatal rat CGNs. The neurite outgrowth of transduced neurons was quantitatively measured using a semi-automated image capture and analysis system. The neurite outgrowth was significantly reduced by the inhibitory substrates which we demonstrated could be partially reversed using a Rho Kinase inhibitor. We are now using this assay to screen large sets of RAGs for their ability to increase neurite outgrowth on a variety of growth inhibitory and permissive substrates.
Introduction
The transfection of primary cells with cDNA plasmids encoding transgenes or short hairpin RNAs (shRNAs) is a widely used method to investigate the effects of gene over-expression or knockdown on cultured cells or cellular networks. However, post-mitotic cells such as neurons remain notoriously difficult to transfect efficiently. In mitotic cells, transfection delivers cDNA to the cytoplasm which then enters into the nucleus during cell division when the nuclear envelope breaks down. The latter does not occur in post-mitotic cells resulting in lower transfection efficiencies.
Most non-viral methods of transfection such as calcium phosphate precipitation, cationic lipids, biolistics, and microinjection are inefficient at transfecting primary neurons, resulting in low transfection efficiencies (1–10%) and high cytotoxicity (Washbourne and McAllister, 2002). In contrast, when optimized, electroporation can provide an efficient method of transfecting primary neurons with reported transfection efficiencies of up to 50% (Washbourne and McAllister, 2002; Zeitelhofer et al., 2007).
Electroporation is achieved by applying an external electric field that surpasses the capacitance of the cell membrane, leading to cell permeabilization via the formation of reversible hydrophilic pores in the lipid bilayer (Neumann et al., 1999; Teissie et al., 2005). Further characterization has shown that the area and degree of membrane permeabilization and hence transfection efficiency and viability can be controlled by the electric pulse amplitude (voltage), length, or number (Wolf et al., 1994; Gabriel and Teissie, 1997). Consequently it is essential to optimize the voltage, pulse length, and pulse number for every cell type used (Wolf et al., 1994; Gabriel and Teissie, 1995; Rols and Teissie, 1998).
Technical advancements including the availability of square-wave pulse generators that allow precise control over the parameters of the electric pulse have greatly increased the transfection efficiency of post-mitotic cells. The use of high voltages facilitates DNA entry in to the nucleus independent of cell division by either generating transient holes in the nuclear envelope or by increasing the permeability of the endogenous nuclear pores (Zeitelhofer et al., 2007). Other recent advances include the introduction of optimized electroporation buffers and protocols that have lead to a variety of primary neurons including retinal ganglion cells (RGCs), dorsal root ganglion neurons (DRGs), cortical neurons, hippocampal neurons, and murine cerebellar granule neurons (CGNs) being transfected with relatively high transfection efficiencies and viabilities (Mertz et al., 2002; Dityateva et al., 2003; Leclere et al., 2005; Buchser et al., 2006; Gartner et al., 2006; Blackmore et al., 2010).
The recent production of 96-well plate electroporators allows the rapid, efficient, and relatively in-expensive screening of large numbers of genes in primary neurons (Buchser et al., 2010). The 96-well plates reduce the time, volume, and number of primary neurons required for each electroporation. In theory 96 genes can be tested simultaneously under identical conditions, although screening smaller numbers of genes with replicates improves assay precision (Malo et al., 2006). Following electroporation, the neurons can be cultured on a number of different inhibitory or permissive substrates to challenge and evaluate their ability to overcome a range of conditions. Semi-automated image acquisition of transfected and immunostained cells can then be obtained using computer-controlled microscope imaging systems (e.g., GE’s InCell Analyzer or Cellomics KineticScan). These images can then undergo semi-automated quantitative analysis for multiple morphological parameters such as neurite length (Buchser et al., 2006, 2010; Mitchell et al., 2007; Moore et al., 2009; Blackmore et al., 2010).
In an ongoing project, we have identified genes whose levels are increased in rat central nervous system (CNS) neurons when they regenerate an axon in vivo (into a permissive cellular transplant; unpublished data). Our next goal is to perform medium-throughput screening using a 96-well electroporation system to identify which of these genes, when over-expressed, increases neurite outgrowth on various growth inhibitory and growth-permissive substrates in vitro.
In the present study, we have optimized a method to efficiently transfect and assess the neurite outgrowth of postnatal rat CGNs on two growth inhibitory substrates using a 96-well electroporation assay. This is the first study to optimize an electroporation screening assay for postnatal rat CGNs and to use a growth inhibitory substrate of Chinese hamster ovary cells expressing myelin associated glycoprotein (CHO-MAG) suitable for identifying molecules that enable a neuron to overcome an inhibitory cellular substrate.
Materials and Methods
Assay Plate Preparation
Poly-L-Lysine (PLL)
A 96-well assay plate (Nunc, Rochester, NY, USA) was coated with 30 μl 0.01% PLL (Sigma, Gillingham, UK) in PBS (Invitrogen, Paisley, UK) for 2 h and washed twice with PBS (Invitrogen).
Chondroitin sulfate proteoglycans
A 96-well assay plate (Nunc) pre-coated with PLL was incubated with 45 μl 5 μg/ml Laminin (Sigma) and 1 μg/ml chondroitin sulfate proteoglycans (CSPGs) extracted from embryonic chicken brain and composted of a mixture of CSPGs including neurocan, phosphacan, versican, and aggrecan (Millipore, Watford, UK) overnight at 4°C and then washed four times with PBS (Invitrogen).
Chinese hamster ovary cells
The CHO-MAG and CHO-R2 cell lines were a kind gift from Prof. Marie Filbin (Hunter College, The City University of New York). CHO cell lines were established previously (Mukhopadhyay et al., 1994; DeBellard et al., 1996) by stably transfecting with the pSHL plasmid containing the sequence for the large (L)-MAG isoform followed by gene amplification using the dihydrofolate reductase/methotrexate (MTX) strategy. The CHO-MAG cells therefore express the L-MAG isoform on their cell surface, while the control CHO cells (CHO-R2) do not express L-MAG on their cell surface. Twenty four hours prior to electroporation a PLL coated 96-well assay plate (Nunc) was incubated with 30 μl 10 μg/ml fibronectin from bovine plasma (Sigma) in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) for 2 h at 37°C and washed once with DMEM (Invitrogen). CHO-MAG and CHO-R2 cells were trypsinized using 5 ml 0.05% trypsin-ethylenediaminetetraacetic acid (EDTA; Sigma) in DMEM (Invitrogen) for 2 min and deactivated with an equal volume of CHO cell media containing DMEM supplemented with 10% heat inactivated fetal bovine serum (FBS; Invitrogen), 2 mM L-glutamine (Sigma), 0.04 mg/ml L-Proline (Sigma), 7.5 μg/ml Glycine (Sigma), 0.73 μg/ml Thymidine (Sigma), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). The trypsinized CHO-MAG and CHO-R2 cells were pelleted by centrifugation at 100 × g for 5 min. The trypsin EDTA was removed and the CHO cells resuspended in 5 ml CHO cell media. After counting using a hemocytometer the CHO-MAG and CHO-R2 cells were plated at a density of 5 × 104 cells per well in 100 μl CHO cell media and incubated overnight at 37°C, 5% CO2.
Cerebellar Granule Neuron Culture
Postnatal day 7–9 (P7–9) Long Evans rat pups were killed via decapitation. The cerebellum was dissected and the meninges removed in 3 ml calcium and magnesium free medium (CMF) containing 0.4 mg/ml KCl, 0.06 mg/ml KH2PO4, 7.65 mg/ml NaCl, 0.35 mg/ml NaHCO3, 0.048 mg/ml Na2HPO4, 2.38 mg/ml Hepes in sterile water, pH 7.2. The dissected cerebellum was then placed in 1 ml CMF and finely diced with a razor blade before being incubated with 5 ml 0.05% trypsin EDTA in CMF for 15 min at 37°C, inverting every few minutes. After the incubation the trypsin EDTA was deactivated using an equal volume of 10% FBS in CMF. The cell pellet was transferred to a new tube containing 0.5 ml 5 mg/ml DNase I (Sigma) in 2 ml CMF and mechanically triturated eight times using a 5-ml pipette and four times using a 2-ml pipette. The cells were left to settle for 5 min before 1.5 ml of supernatant was harvested and the cells collected by centrifugation at 100 × g for 5 min. The cell pellet was resuspended in 5 ml serum free media (SFM) containing neurobasal media (Invitrogen) supplemented with, 2% B27 (Invitrogen), 25 mM KCl (Sigma), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen), 3 mg/ml D-glucose (Sigma), 2 mM L-glutamine (Sigma), and then counted using a hemocytometer.
DNA Preparation
For the electroporation optimization experiments, 1 μg of the pmaxGFP plasmid (Lonza, Walkersville, MD, USA) was added per well. For dual transfection optimization experiments a range of 1–10 μg pCMVSPORT6 plasmid expressing the red fluorescent protein mCherry and 1 μg of the pmaxGFP plasmid (Lonza) was added per well. For the assessment of regeneration-associated genes (RAG) over-expression and neurite outgrowth experiments, 4 μg of the pCMVSPORT6 plasmid expressing ATF-3 (NM_007498.3; Source Bioscience, Nottingham, UK) or KLF-7 (NM_033563; Source Bioscience) and 1 μg of the pmaxGFP plasmid (Lonza) was added per well.
Electroporation
The desired amount of DNA was added to 30 μl internal neuronal buffer (INB) containing 135 mM KCl, 0.2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 5 mM ethylene glycol tetraacetic acid (EGTA), in sterile water, pH 7.3 (Buchser et al., 2006), and pipetted into the wells of the 96-well electroporation plate. The 250,000 CGNs/well were resuspended in 35 μl INB/well and then added to the 96-well electroporation plate wells, which already contained the DNA/INB solution and had a gap size of 2 mm (BTX Harvard Apparatus, Holliston, MA, USA). The 96-well electroporation plate was then placed in the HT-200 plate handler (BTX Harvard Apparatus) which was connected to a ECM 830 square-wave pulse generator (BTX Harvard Apparatus) that generates and delivers the specified electric pulse. The ECM 830 square-wave pulse generator was connected to a TDS 1002 oscilloscope (Tektronix, Beaverton, OR, USA) to monitor the delivered pulse parameters. For CGN electroporation optimization the ECM 830 square-wave pulse generator was set to deliver a range of parameters. For voltage optimization CGNs were electroporated with a single pulse with a duration of 1 ms and 1 of 11 different voltages (0, 200, 220, 240, 260, 280, 300, 320, 340, 360, or 380 V). For pulse length optimization CGNs were electroporated with a single 300 V pulse at a pulse length of either 0 (non-electroporated), 0.1, 0.2, 0.3, 0.5, 0.7, 0.9, 1, or 2 ms. Pulse number optimization was assessed using 300 V for 1 ms with either 0 (non-electroporated), 1, 2, or 3 pulses. In all further electroporation experiments the optimized electroporation pulse parameters (1 pulse at 300 V, 1 ms pulse length) were used. It has previously been shown that lower temperatures, e.g., 4°C or 21°C at the time of electroporation followed by immediate warming to 37°C increases transfection efficiency and cell viability (Rols et al., 1994). Therefore in all experiments, solutions were kept on ice or at room temperature and immediately following electroporation, 80 μl of Hibernate E (Brain Bits, Springfield, IL, USA), pre-warmed to 37°C was added per well. Electroporated CGNs were then plated at a density of 3.5 × 104 CGNs per well in a 96-well assay plate (Nunc). If the 96-well assay plate contained a monolayer of CHO-MAG and CHO-R2 cells then the electroporated CGNs were added to 180 μl pre-warmed SATO media/well containing DMEM supplemented with 2% FBS (Invitrogen), 1% N2 supplement (Invitrogen) and 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). If the assay plate was prepared with a PLL or CSPG substrate then the electroporated CGNs were added to 180 μl pre-warmed SFN/well. The 96-well assay plates were then incubated at 37°C for 48 h.
Immunocytochemistry
The media was removed and cultures were fixed using 100 μl cold 4% paraformaldehyde in PBS, pH 7.4 for 20 min and then washed once with PBS (Invitrogen). The neurons were stained for beta-III-tubulin by incubating with 30 μl of a mouse monoclonal antibody (1:2000 Promega, Madison, WI, USA) in PBS (Invitrogen) with 10% normal goat serum (NGS; Invitrogen) and 0.3% Triton X (Sigma) on a shaker for 1 h at room temperature. Following the incubation the cultures were washed three times with PBS (Invitrogen) and then incubated with 30 μl goat anti-mouse Alexa Fluor 546 conjugated secondary antibody (1:1000, Molecular Probes) and 4′, 6-diamidino-2-phenylindole (DAPI) to visualize the nucleus in PBS (Invitrogen) with 0.3% Triton X-100 (Sigma) on a shaker for 45 min at room temperature. The cultures were then washed a further three times with PBS (Invitrogen) and left in 100 μl PBS containing 0.02% sodium azide (Sigma).
RNA Extraction
Forty eight hours after transfection, the medium was removed and total cellular RNA was isolated using 0.1 ml TRIZOL reagent (Invitrogen) per well, incubating for 5 min at room temperature. Twenty microliters chloroform (Sigma) was then added per sample and mixed for 15 s and then incubated for 2 min at room temperature. The samples were centrifuged at 12,000 × g for 15 min at 4°C, the aqueous phase was transferred to a fresh eppendorf and the RNA precipitated by addition of 50 μl isopropyl alcohol (Sigma). The samples were incubated at room temperature for 10 min and then centrifuged at 12,000 × g for 10 min at 4°C. The supernatant was removed and the RNA pellet washed with 100 μl 75% ethanol (Sigma) followed by centrifugation at 7500 × g for 5 min at 4°C. The RNA pellet was dried for 5 min and then resuspended in 20 μl RNase free water. The RNA concentration was measured using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, USA). Following isolation the extracted total RNA was treated with DNase I. Two-hundred fifty nanogram RNA was added to an eppendorf with 10× DNase I reaction buffer (Invitrogen), 0.25 U amplification grade DNase I (Invitrogen) and incubated for 15 min at room temperature. To inactivate the DNase I, 0.25 μl of 25 nM EDTA was added to the eppendorf and incubated for 10 min at 65°C.
Reverse Transcription
For first strand cDNA synthesis, 250 ng random primers (Invitrogen), 10 mM dNTP mix (Invitrogen) and 100 ng total RNA were added to an eppendorf and heated to 65°C for 5 min and then chilled on ice. 5× first strand buffer (Invitrogen), 0.1M DTT (Invitrogen) were then added and incubated for 2 min at room temperature. Hundred units of Superscript II Reverse Transcriptase (Invitrogen) was added and incubated for 10 min at room temperature, followed by heating to 42°C for 50 min. The enzyme was then inactivated by heating to 70°C for 15 min.
QRT-PCR
ATF-3 expression was assessed using qRT-PCR, measuring the relative level of ATF-3 mRNA and normalizing to GAPDH, using standard curves. qRT-PCR was done using the RotorGene-3000 (Corbett Life Science, Australia). Each PCR reaction contained 20 ng of cDNA, 25 ng of the relevant forward and reverse primers and 4 μl SYBR® Green PCR Master Mix (Roche, USA). The PCR cycling conditions were as follows: hold; 95°C for 10 min. Cycle; 60°C for 10 s, 72°C for 20 s, 95°C 10 s repeated 40 times. Melt; 72°C increasing to 95°C 1°/step waiting 1 s at the first step and 5 s each step thereafter. Forward and reverse primers for ATF-3 and GAPDH were designed using NCBI Primer-BLAST tool. Sequences were as follows: ATF-3 forward 5′-CAG-ACC-CCT-GGA-GAT-GTC-AGT-3′ and reverse 5′-TTC-TTG-TTT-CGA-CAC-TTG-GCA-3′. GAPDH forward 5′-ATG-GGA-AGC-TGG-TCA-TCA-AC-3′ and reverse 5′-CCA-CAG-TCT-TCT-GAG-TGG-CA-3′. Standard curves were obtained for each of the target genes (ATF-3 and GAPDH) using threefold serial dilutions of embryonic day 15 (E15) rat head cDNA and melt curve analysis confirmed the specificity of the PCR primers; a single peak represents a specific product.
Imaging and Analysis
Immunostained 96-well assay plates were imaged; eight fields of view were taken per well using the IN Cell Analyzer 1000 semi-automated cell imager with a 10× Nikon ApoPlan objective (GE Healthcare Life Sciences, Buckinghamshire, UK). An example of a typical field of view showing electroporated CGNs cultured on a monolayer of CHO-R2 cells can be seen in Figure 1. The measurement of cell parameters was completed using the semi-automated IN Cell Developer Toolbox software (GE Healthcare Life Sciences) using a custom-written algorithm (available on request). The number of transfected neurons was determined by counting the number of GFP and beta-III-tubulin positive cells. The total number of neurons was determined by counting the total number of beta-III-tubulin cells. The mean neurite length per transfected neuron was determined by measuring the neurite length of GFP and beta-III-tubulin positive cells divided by the total number of transfected neurons. For each parameter, the total per well was obtained by multiplying the average per field by the total number of fields per well. Each experiment was carried out once using 4 or 8 wells per condition (n = 4/8). Percentage transfection efficiency was calculated by dividing the number of transfected neurons by the total number of surviving neurons multiplied by 100. Percentage viability was calculated by dividing the total number of electroporated neurons by the number of neurons in the non-electroporated control, multiplied by 100, n.b., the variable of interest is differences in survival due to electroporation: accordingly, survival expressed relative to the number of cells which survived dissociation and culturing following an electroporation pulse of zero (i.e., non-electroporated) and not relative to the number of neurons initially plated.
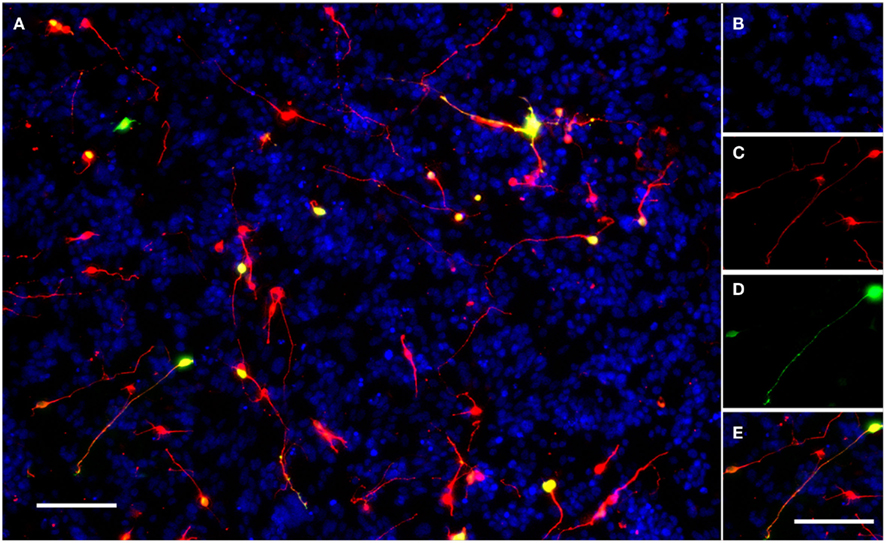
Figure 1. A typical field of view showing electroporated CGNs cultured on a monolayer of growth-permissive CHO-R2 cells. (A) A typical field of view taken using the IN Cell Analyzer 1000 semi-automated cell imager with a 10× Nikon ApoPlan objective. (B) DAPI stained nuclei of CHO-R2 cells and CGNs. (C) CGNs stained for beta-III-tubulin. (D) eGFP positive, transfected CGNs expressing GFP. (E) The merged image; transfected neurons appear yellow. Scale bars: 100 μm.
Statistical Analysis
Statistical analysis was carried out using SPSS 17.0 (SPSS Inc., Chicago, IL, USA). A threshold level of significance (α) of 0.05 was selected. Graphs show means and standard deviations (SD) and the text reports these to the nearest micrometer. The Kolmogorov–Smirnov and Levene’s tests were used to test for normality and the equality of variances. For the electroporation and inhibitory substrate optimization experiments a one way ANOVA was used to test for differences among the groups followed by Dunnett’s post hoc tests comparing with the relevant control. In the assessment of RAG over-expression experiment an independent samples Student’s t-test was used to compare the means of the two groups.
G*Power 3.1 (Faul et al., 2007) was used to estimate the minimum sample size that would be required during screening to identify a RAG that promoted at least a 40% increase in neurite length on CHO-MAG cells (comparing two groups: RAG vs. mCherry). Calculations were performed using means and SD drawn from the experiment that showed Y-27632 could overcome inhibition associated with CHO-MAG cells (Figure 3). A 40% increase in neurite length corresponds to a standardized effect size of 2.6. Calculations were performed using the algorithm for a priori sample size calculations [“difference between two independent means (two groups)”] and using the following parameters; type I error rate (α) ≤ 0.05 and power (1-β) ≥ 0.80.
Results
Voltage Optimization
Transfection efficiency and CGN viability was assessed with increasing voltage. P7–9 CGNs were electroporated using a single pulse with a pulse length of 1 ms at 1 of 11 different voltages (0, 200, 220, 240, 260, 280, 300, 320, 340, 360, or 380 V). The number of transfected neurons was significantly affected by voltage [df = 10(77), F = 21.50, P < 0.001, one way ANOVA, n = 8]. Dunnett’s post hoc test revealed that compared with the non-electroporated control the mean number of transfected CGNs significantly increased at voltages higher than 200 V (*P < 0.05, ***P < 0.001, n = 8; Figure 2A). The number of transfected neurons peaked at 300 V (190 ± 45 transfected CGNs; values represent mean ± SD, n = 8; Figure 2A). Percentage transfection efficiency at 300 V relative to the total number of surviving neurons was 28%. Cell viability, calculated by counting the total number of beta III tubulin positive CGNs per well, was significantly affected by voltage [df = 10(77), F = 38.67, P < 0.001, one way ANOVA, n = 8]. Dunnett’s post hoc test revealed that compared with the non-electroporated control the mean number of CGNs was significantly decreased at all voltages (*P < 0.05, ***P < 0.001, n = 8; Figure 2A). The percentage viability at 300 V was 49%. Using this data 300V was chosen as the optimal voltage as it gave both reasonable transfection efficiency and cell viability. All further optimization experiments were conducted at 300 V.
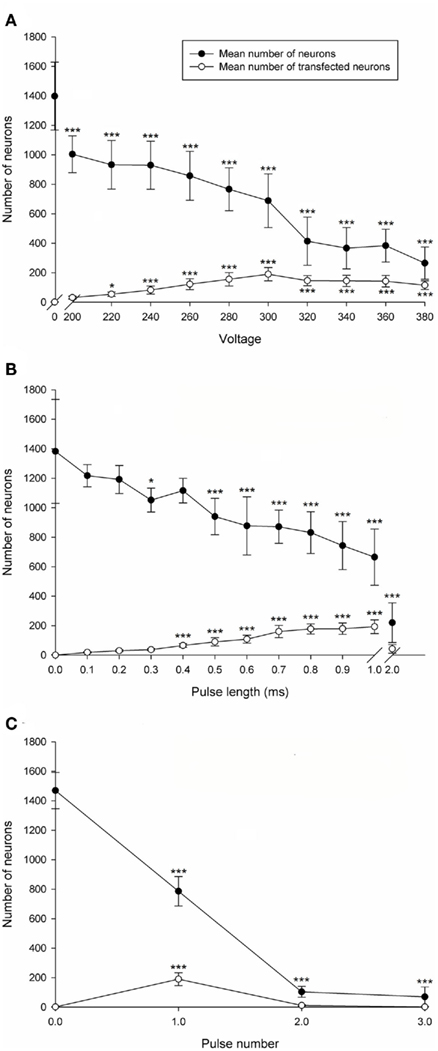
Figure 2. Electroporation optimization of postnatal rat CGNs. (A) Voltage optimization. The mean number of CGNs was significantly decreased at all voltages compared to the non-electroporated control. The mean number of transfected CGNs significantly increased at voltages higher than 200 V compared to the non-electroporated control. (B) Pulse length optimization. The mean number of CGNs was significantly decreased at pulse lengths higher than 0.4 ms. The mean number of transfected CGNs significantly increased using pulse lengths between 0.4 and 1 ms. (C) Pulse number optimization. The mean number of CGNs was significantly decreased at all pulse numbers. The mean number of transfected CGNs was significantly increased using 1 pulse. Values represent mean and SD; analysis was performed using one way ANOVA with Dunnett’s post hoc tests comparing with the non-electroporated control (*P < 0.05, ***P < 0.001), n = 8.
Pulse Length Optimization
Pulse length was then optimized for transfection efficiency and CGN survival. CGNs were electroporated with a single 300 V pulse at a pulse length of either 0 (non-electroporated), 0.1, 0.2, 0.3, 0.5, 0.7, 0.9, 1, or 2 ms (n.b., the square-wave generator does not deliver pulses intermediate in length between 1 and 2 ms). The number of transfected neurons was significantly affected by pulse duration [df = 11(84), F = 49.88, P < 0.001, one way ANOVA, n = 8]. Dunnett’s post hoc test revealed that compared with the non-electroporated control the mean number of transfected CGNs was significantly increased when using a pulse length between 0.4 and 1 ms (***P < 0.001, n = 8; Figure 2B). The highest number of transfected CGNs per well was obtained using a pulse length of 1 ms (193 ± 47 transfected CGNs; values represent mean ± SD, n = 8; Figure 2B), which generated a transfection efficiency of 29% relative to the total number of surviving neurons. Using 2 ms resulted in no significant difference compared to the non-electroporated control, which can be attributed to a considerable increase in cell death. The mean number of CGNs was significantly affected by pulse length [df = 11(84), F = 37.13, P < 0.001, one way ANOVA, n = 8]. Dunnett’s post hoc test revealed that compared with the non-electroporated control the mean number of CGNs was significantly decreased consistently at pulse lengths higher than 0.4 ms (*P < 0.05, ***P < 0.001, n = 8; Figure 2B). A substantial fall in cell survival was observed using a pulse length of 2 ms (Figure 2B). The percentage viability using 1 ms was 50% and therefore 1 ms was chosen as the optimized pulse length as it produced the highest number of transfected CGNs and a reasonable CGN viability.
Pulse Number Optimization
Transfection efficiency and viability of CGNs was assessed following electroporation using 300 V, 1 ms pulse duration with either 0 (non-electroporated), 1, 2, or 3 pulses and no interval between sequential pulses. The mean number of transfected neurons was significantly affected by the number of pulses [df = 3(28), F = 59.14, P < 0.001, one way ANOVA, n = 8]. Dunnett’s post hoc test revealed that the mean number of transfected CGNs was significantly increased using 1 pulse compared with the non-electroporated control (***P < 0.001, n = 8; Figure 2C). The highest number of transfected CGNs per well was obtained using 1 pulse (190 ± 44 transfected CGNs; values represent mean ± SD, n = 8; Figure 2C) with a transfection efficiency of 25% relative to the total number of surviving neurons. Applying more than 1 pulse did not result in a significant increase in transfection efficiency compared to the non-electroporated control (Figure 2C), which was probably a result of extensive cell death. The mean number of CGNs was significantly affected by pulse number [df = 3(28), F = 314.16, P < 0.001, one way ANOVA, n = 8]. Dunnett’s post hoc test revealed that the mean number of CGNs was significantly decreased at all pulse numbers compared with the non-electroporated control (***P < 0.001, n = 8; Figure 2C). Approximately 800 neurons per well were obtained with one pulse and percentage viability at 1 pulse was 53%. One pulse was chosen as the optimal parameter as it gave the highest transfection efficiency and viability. Taken together these results show that the optimal parameters for electroporation of CGNs is 1 pulse at 300 V, for 1 ms and all further electroporation studies were carried out using these parameters.
Approximately 700–800 neurons per well of a 96-well plate is an ideal density for our assay: ~190 transduced neurons per well are nicely spaced, with room to spare when a RAG or drug promotes additional growth (Figures 1A and 3). This spacing also allows the IN Cell Analyser to accurately measure neurite lengths without high frequency obstructions from crossing or fasciculated neurites.
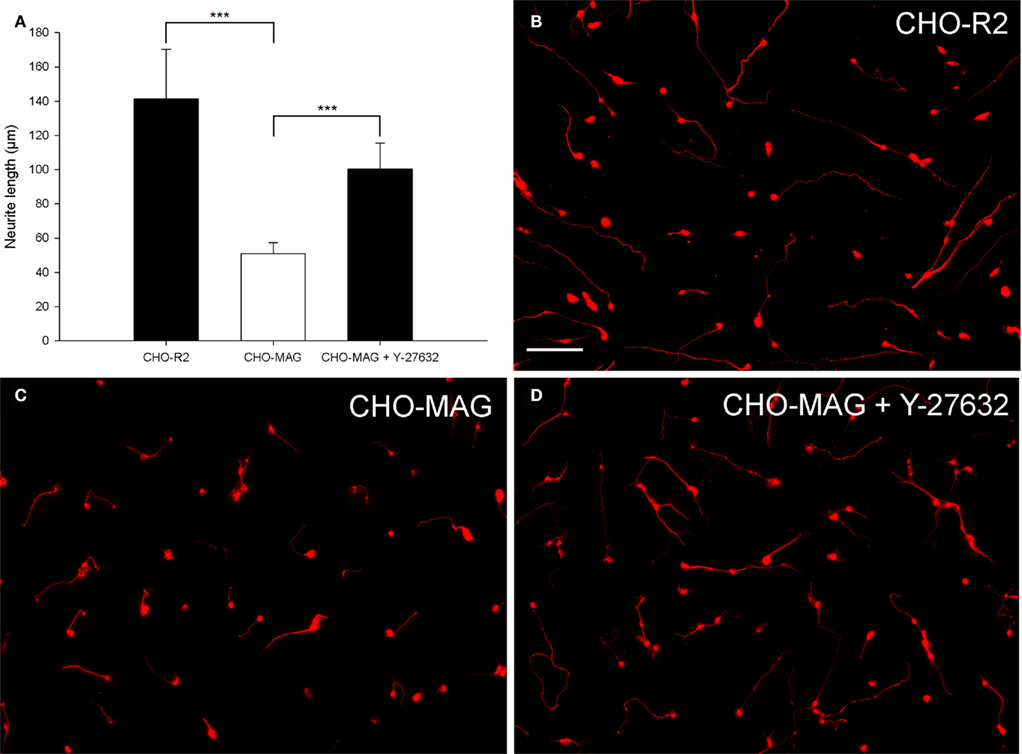
Figure 3. Chinese hamster ovary-MAG cells inhibit CGN neurite outgrowth, which can be partially reversed by the ROCK inhibitor Y-27632. (A) Quantification of the mean neurite length per transfected neuron of CGNs cultured on CHO-MAG or CHO-R2 cells. The mean neurite length per transfected CGN was significantly longer when cultured on CHO-R2 cells compared to CHO-MAG cells. The CHO-MAG inhibition could be partially reversed with 20 μM Y-27632. The mean neurite length per transfected CGN was significantly longer when cultured on CHO-MAG cells in the presence of Y-27632 compared to CGNs cultured on CHO-MAG cells without Y-37632. Values represent mean and SD; analysis was performed using one way ANOVA with Dunnett’s post hoc tests comparing to CHO-MAG cells (***P < 0.001), n = 8. (B) Non-inhibitory CHO-R2 cell substrate. (C) CGN neurite outgrowth is inhibited by the CHO-MAG cell substrate. (D) The ROCK inhibitor Y-27632 reverses the MAG inhibition and promotes neurite outgrowth. Scale bar: 100 μm.
CHO-MAG Cell Inhibition
To evaluate the axon growth potential of electroporated neurons, two different growth inhibitory substrates (MAG and CSPGs) were optimized. CHO cells expressing MAG on their cell surface (CHO-MAG) or CHO cells expressing the reverse peptide sequence of MAG (CHO-R2) were cultured to a confluent monolayer. P7–9 CGNs were electroporated and cultured on top of the CHO cells for 48 h. To determine whether the MAG inhibition could be overcome, the electroporated neurons were treated with Y-27632, a selective ROCK inhibitor, which has previously been shown to reverse growth inhibition on MAG (Niederost et al., 2002; Chivatakarn et al., 2007). The cultures were then fixed, stained, and the mean neurite length per transfected neuron was measured. There was a significant difference in the neurite length between groups [df = 2(21), F = 35.95, P < 0.001, one way ANOVA, n = 8]. Dunnett’s post hoc test revealed that the CGNs cultured on the CHO-MAG cells had significantly reduced neurite outgrowth compared to CGNs cultured on the CHO-R2 cells (CHO-R2 141 ± 29 μm; CHO-MAG, 51 ± 6 μm; values represent mean ± SD, n = 8, ***P < 0.001; Figures 3A–C). Addition of 20 μM of the ROCK inhibitor Y-27632 to CGNs cultured on the CHO-MAG cells resulted in a significant increase in the mean neurite length (CHO-MAG + Y-27632 100 ± 15 μm; CHO-MAG, 51 ± 6 μm; values represent mean ± SD, n = 8, ***P < 0.001; Figures 3A,D).
Sample size calculations showed that only four electroporation replicates per gene would be required to identify a RAG that promoted a 40% or greater increase in neurite length on CHO-MAG cells (comparing two groups: RAG vs. mCherry). Power calculations showed that a “true hit” would be detected on 87% of occasions and “false hits” would be rejected on 95% of occasions. This proves the assay has very high “hit” rates and acceptably low false positive rates.
CSPG Inhibition
To evaluate an alternative inhibitory substrate, a combination of CSPGs and laminin was used with laminin alone as a control. These were coated onto the surface of a 96-well plate and then electroporated CGNs were cultured on top for 48 h. To establish whether the CSPG inhibition could be overcome, neurons cultured on the CSPG substrate were treated with Y-27632 a selective ROCK inhibitor which is known to reverse growth inhibition on CSPGs (Monnier et al., 2003; Sivasankaran et al., 2004; Lingor et al., 2007). The mean neurite length per transfected CGN was measured after 48 h. After analysis the substrate was shown to have a significant effect on CGN neurite length [df = 2(21), F = 17.83, P < 0.001, one way ANOVA, n = 8]. Dunnett’s post hoc tests revealed that CGNs plated on CSPG + laminin had significantly shorter neurites than CGNs plated on Laminin alone (Laminin 203 ± 54 μm; CSPG + laminin, 62 ± 28 μm; values represent mean ± SD, n = 8, ***P < 0.001; Figures 4A–C). Addition of 20 μM ROCK inhibitor Y-27632 to the electroporated CGNs cultured on the CSPG + laminin resulted in a significant increase in neurite length (CSPG + Y-27632 137 ± 55 μm; CSPG + laminin, 62 ± 28 μm; values represent mean ± SD, n = 8, **P < 0.01; Figures 4A,D).
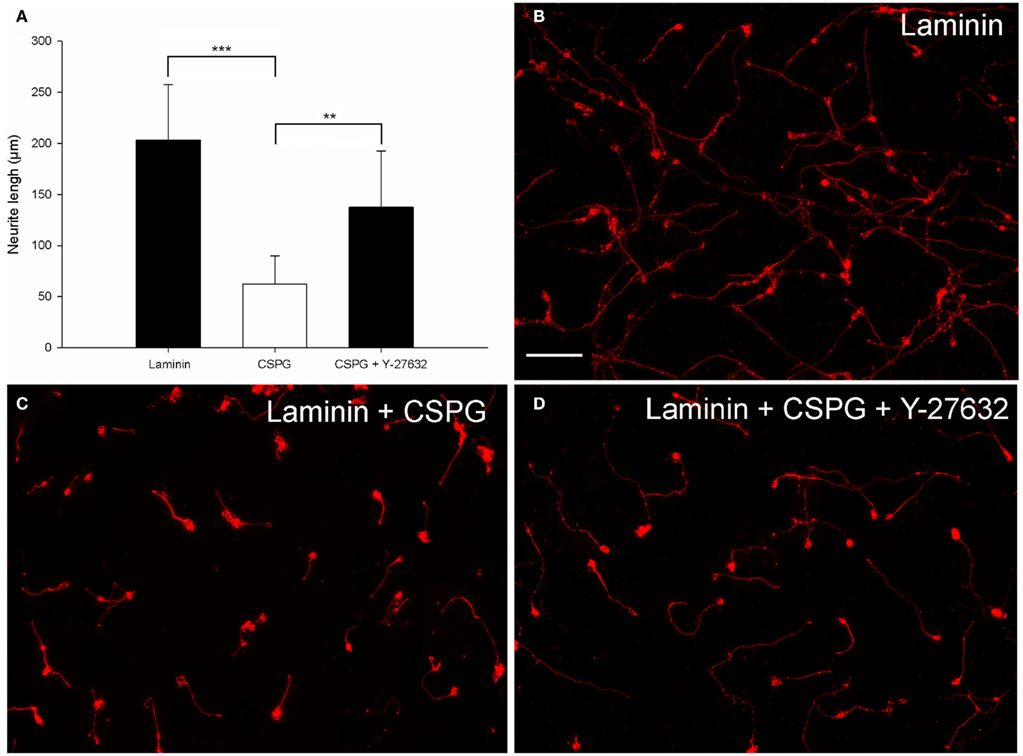
Figure 4. CSPG inhibits postnatal CGN neurite outgrowth, which can be partially reversed by the ROCK inhibitor Y-27632. (A) Quantification of the mean neurite length per transfected neuron of CGNs cultured on Laminin and CSPG substrates. CGNs cultured on laminin had significantly longer neurites than CGNs cultured on a CSPG substrate. The CSPG inhibition could be partially reversed using 20 μM Y-27632. The mean neurite length per transfected CGN was significantly longer when cultured on the CSPG substrate in the presence of Y-27632 compared to CGNs cultured on the CSPG substrate without Y-37632. Values represent mean and SD; analysis was performed using one way ANOVA with Dunnett’s post hoc tests comparing to CSPG substrate (***P < 0.001, **P < 0.01), n = 8. (B) Non-inhibitory laminin substrate. (C) Neurite outgrowth is inhibited by the CSPG substrate. (D) The ROCK inhibitor Y-27632 can promote CGN neurite outgrowth and overcome the CSPG inhibition. Scale bar: 100 μm.
Dual Transfection
The pCMVSPORT6 plasmid encoding the RAGs was purchased for cost–effective screening (Mammalian Gene Collection) but it does not contain a reporter gene. To identify transfected neurons a second plasmid expressing GFP was co-transfected. An optimal ratio of the two plasmids was determined so that the majority of cells expressing GFP also expressed the RAG. Plasmid ratios were tested using a red fluorescent protein, mCherry, which was cloned in place of the RAG. Co-transfection of this plasmid with the GFP plasmid at different ratios resulted in different levels of co-expression that could be optimized. The percentage of cells co-expressing both fluorescent proteins increased with the ratio of mCherry to GFP, peaking at a ratio of 4:1 where 84% of the GFP expressing cells also expressed mCherry (Figures 5A,B).
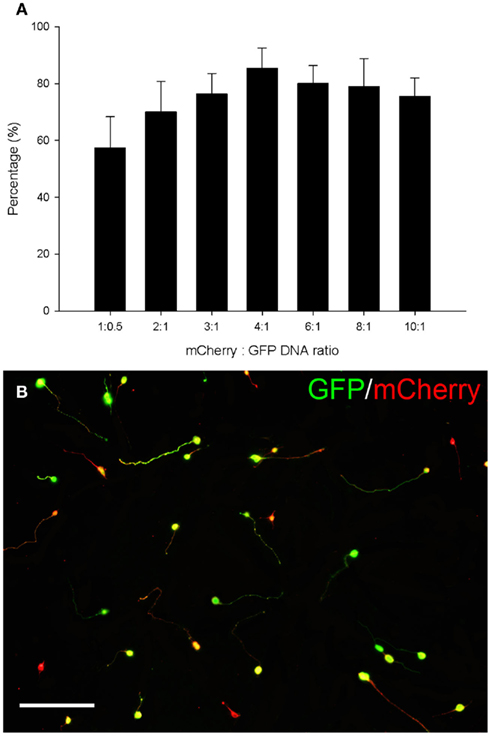
Figure 5. Eighty four percent co-expression of mCherry and GFP was obtained using a 4:1 DNA ratio. (A) Quantification of the percentage of cells co-expressing GFP and mCherry. The percentage of cells co-expressing both fluorescent proteins increased with the ratio of mCherry to GFP, peaking at a ratio of 4:1 where 84% of the GFP expressing cells also expressed mCherry. Values represent mean and SD, n = 8. (B) CGNs co-transfected with mCherry and GFP using a ratio of 4:1. CGNs co-expressing GFP (green) and mCherry (red) appear yellow. Scale bar: 100 μm.
Evaluating RAG over-expression
Over-expression of the RAG ATF-3 was confirmed in transfected CGNs. CGNs were co-transfected with the pCMVSPORT6 plasmid encoding either the RAG ATF-3 or the control gene mCherry and the pmaxGFP reporter plasmid at a ratio of 4:1. ATF-3 expression was assessed using qRT-PCR, measuring the relative level of ATF-3 mRNA using a standard curve and normalizing to GAPDH. ATF-3 mRNA levels were significantly increased (2700-fold) in cells transfected with the plasmid expressing ATF-3 compared to CGNs transfected with the plasmid expressing mCherry (df = 4, **P < 0.01, Student’s t-test for independent samples, n = 4; Figure 6A).
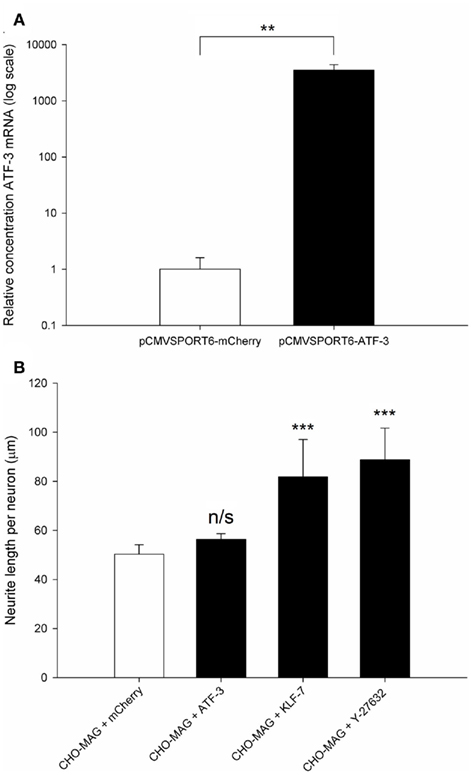
Figure 6. Regeneration-associated genes are successfully over-expressed following electroporation, and over-expression of KLF-7 but not ATF-3 was able to enhance neurite outgrowth on an inhibitory MAG substrate. (A) Quantification of the relative level of ATF-3 mRNA in electroporated CGNs. Significantly higher levels (2700-fold) of ATF-3 were detected in CGNs transfected with the ATF-3 plasmid compared to CGNs transfected with the mCherry plasmid. Values represent mean and SD; analysis was performed using an independent samples Student’s t-test (**P < 0.01), n = 4. (B) Quantification of the mean neurite length per transfected neuron of CGNs cultured on CHO-MAG cells. The CHO-MAG inhibition could be partially reversed by over-expressing KLF-7 or addition of 20 μM Y-27632 but not by over-expressing ATF-3. Values represent mean and SD; analysis was performed using one way ANOVA with Dunnett’s post hoc tests comparing to CHO-MAG cells (***P < 0.001), n = 8.
Postnatal day 7–9 CGNs were co-transfected with ATF-3 and pmaxGFP, KLF-7 and pmaxGFP or mCherry and pmaxGFP and then cultured on an inhibitory CHO-MAG substrate for 48 h. The cultures were then fixed, stained for beta III tubulin and the mean neurite length per transfected neuron measured. Statistical analysis showed a significant difference between the groups [df = 3(28), F = 27.05, P < 0.001, one way ANOVA, n = 8]. Dunnett’s post hoc tests revealed that over-expressing ATF-3 did not significantly increase neurite outgrowth on an inhibitory MAG substrate (CHO-MAG + ATF-3 56 ± 2 μm; CHO-MAG + mCherry, 50 ± 4 μm; values represent mean ± SD, n = 8, P > 0.05; Figure 6B). This is consistent with what has been previously been reported on an inhibitory myelin substrate (Seijffers et al., 2007). However, over-expressing KLF-7 or addition of 20 μM Y-27632 to electroporated CGNs resulted in a significant increase in neurite outgrowth (CHO-MAG + KLF-7 82 ± 15 μm; CHO-MAG + Y-27632 89 ± 13 μm; CHO-MAG + mCherry, 50 ± 4 μm; values represent mean ± SD, n = 8, ***P < 0.001; Figure 6B). These results demonstrate that we have developed a sensitive and powerful in vitro assay that will allow us to identify novel pro-regenerative genes that can enhance neurite outgrowth on inhibitory substrates.
Discussion
Following electroporation the transfection efficiency and viability of mammalian cells has previously been shown to be dependent on the cell type, voltage, pulse duration, and pulse number (Wolf et al., 1994; Gabriel and Teissie, 1995, 1997). These parameters were therefore optimized for postnatal rat CGNs and established as 1 pulse at 300 V for 1 ms. These gave an average transfection efficiency of 28% and viability of 51%, which are similar those obtained by previous studies (Buchser et al., 2006; Gartner et al., 2006).
Overall the results from the electroporation optimization experiments demonstrate that increasing the voltage or pulse length enhanced transfection efficiency. This is probably a result of increasing the area or degree of cell permeabilization thereby facilitating DNA entry into the cell (Wolf et al., 1994; Gabriel and Teissie, 1997). However this also reduces cell viability via increased loss of vital intracellular molecules or increased intracellular levels of extracellular ions (e.g., Ca2+), which can lead to the induction of excitotoxicity and apoptosis (Gabriel and Teissie, 1994; Meldrum et al., 1999).
The optimal electroporation parameters identified in this study were chosen as a compromise between transfection efficiency and cell viability. The optimal voltage (300 V) was similar to what had previously been reported using CGNs from postnatal mice (Buchser et al., 2006). The optimal pulse length (1 ms) and pulse number (1 pulse) were consistent with a previous study which demonstrated that the highest yield of viable permeabilized cells was obtained using a single pulse, 1 ms in duration (Gabriel and Teissie, 1995).
Successful transfection of a range of primary neurons has been reported using several different electroporation systems. Using a BTX 96-well electroporation system identical to the one in the present study, identical buffers and similar parameters (340 V, 900 μs, 1 pulse), postnatal mouse CGNs have been previously transfected with an average efficiency of 26.8% and a viability of 37% (Buchser et al., 2006). These values are consistent with our results although a slightly higher cell viability (50%) was observed in the present study, which is probably a result of the lower voltage used.
CGNs have also previously been transfected using the Bio-Rad electroporation system, which is not a medium-throughput system and requires larger numbers of neurons (Mertz et al., 2002). This study reported a transfection efficiency of 10% and cell viability of 44%. The transfection efficiency is considerably lower than what was observed in the present study. Possible reasons for this include the electroporation buffer: Mertz et al. (2002) used complete culture medium, which has been shown to result in low transfection efficiencies (Buchser et al., 2006). Instead a low Ca2+ containing buffer such as the INB buffer used in the present study and optimized by Buchser et al. (2006) should increase transfection efficiency. In addition, Mertz et al. (2002) left the cells on ice for 10 min following electroporation and it has been reported that immediate warming following electroporation increases transfection efficiency and cell viability (Rols et al., 1994).
The Lonza single cuvette electroporation system has been demonstrated to successfully electroporate several neuronal types. Postnatal CGNs have been electroporated with an average transfection efficiency of 20% (Gartner et al., 2006), which is consistent with the results presented in this study. Embryonic hippocampal and cortical neurons have been successfully electroporated, obtaining an average transfection efficiency of 50% (Dityateva et al., 2003; Gartner et al., 2006). RGCs and DRG neurons have also been electroporated with reported transfection efficiencies of 28 and 20% respectively (Leclere et al., 2005). Lonza also produce a 96-well medium-throughput electroporation system, which has been reported to provide transfection efficiencies of 40% using embryonic hippocampal neurons (Zeitelhofer et al., 2007).
These studies demonstrate that different electroporation systems, buffers and protocols can be used to successfully transfect a range of neurons to study gene function. However, the number of cells required for the single cuvette electroporation systems is inappropriate for medium-throughput screening. In the present study a 96-well medium-throughput electroporation system from BTX was chosen as it allows complete control of the electroporation parameters delivered to the cells, unlike the predetermined programs of the 96-well Lonza system. In addition, the recent optimization of an effective electroporation buffer (INB) composed of inexpensive benchtop reagents removed the requirement to use the expensive proprietary electroporation buffers (Buchser et al., 2006).
In the present study two growth inhibitory substrates were characterized. CHO cells expressing the inhibitory protein MAG on their cell surface were shown to consistently inhibit the neurite outgrowth of electroporated postnatal CGNs, which could be partially reversed using the synthetic ROCK inhibitor Y-27632. This is consistent with several previous studies that have demonstrated a similar inhibitory affect of CHO-MAG cells on the neurite outgrowth of primary neurons (Mukhopadhyay et al., 1994; DeBellard et al., 1996; Niederost et al., 2002; Domeniconi et al., 2005; Venkatesh et al., 2005; Chivatakarn et al., 2007).
A combination of CSPG and laminin was also successfully used to inhibit the neurite outgrowth of electroporated postnatal CGNs, which could again be partially reversed using the ROCK inhibitor Y-27632. This was consistent with previous studies, which have used similar inhibitory CSPG substrates to inhibit neurite outgrowth (Snow et al., 1990, 2002; Snow and Letourneau, 1992; Monnier et al., 2003; Sivasankaran et al., 2004; Lingor et al., 2007).
With 25–30% transfection efficiency it is important to be able to identify and analyze the transfected neurons. Ideally the DNA plasmid encoding a gene of interest would also encode a reporter gene but this dramatically increases the cost of the commercially available pre-cloned plasmid making screening a large dataset of genes uneconomical and unfeasible. We therefore used a co-transfection method where an optimal ratio of two plasmids (one expressing the RAG and the other a GFP reporter) was determined so that the vast majority of cells expressing GFP also expressed the RAG. This was achieved using a red fluorescent protein (mCherry) that was cloned in place of the RAG in the pCMVSPORT6 plasmid. Co-transfection of this plasmid with the pmaxGFP plasmid at different ratios resulted in different levels of co-expression that could be optimized. Using a ratio of 4:1 mCherry:GFP 84% co-transfection was observed, which is similar to reports from previous studies (Buchser et al., 2006, 2010; Moore et al., 2009; Blackmore et al., 2010).
It was also essential to confirm that following electroporation, the RAG being over-expressed was significantly up-regulated. We demonstrated that ATF-3 mRNA was significantly up-regulated in neurons transfected with a plasmid over-expressing ATF-3. This demonstrates that transfection with plasmids over-expressing RAGs provides high and consistent levels of expression, resulting in RAG up-regulation. We also demonstrated that over-expression of KLF-7 but not ATF-3 was able to enhance neurite outgrowth on an inhibitory MAG substrate, which is consistent with what has previously been reported (Seijffers et al., 2007; Moore et al., 2009).
In conclusion this study has established and characterized a 96-well electroporation and neurite outgrowth assay. This assay should facilitate the screening and discovery of novel gene targets that augment the intrinsic growth state of CNS neurons and enhance neurite outgrowth in vitro.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Marie Filbin for the CHO-MAG and CHO-R2 cell lines. This study was supported by a Research Councils UK Academic Fellowship (Lawrence D. F. Moon), the British Pharmacological Society’s Integrative Pharmacology Fund (Lawrence D. F. Moon), Friends of Guy’s Hospital Research Grant (Lawrence D. F. Moon), a Biotechnology and Biological Sciences Research Council’s Doctoral Training Grant (Thomas H. Hutson) and a Grant from The Henry Smith Charity (Lawrence D.F. Moon and Thomas H. Hutson).
References
Blackmore, M. G., Moore, D. L., Smith, R. P., Goldberg, J. L., Bixby, J. L., and Lemmon, V. P. (2010). High content screening of cortical neurons identifies novel regulators of axon growth. Mol. Cell. Neurosci. 44, 43–54.
Buchser, W. J., Pardinas, J. R., Shi, Y., Bixby, J. L., and Lemmon, V. P. (2006). 96-Well electroporation method for transfection of mammalian central neurons. Biotechniques 41, 619–624.
Buchser, W. J., Slepak, T. I., Gutierrez-Arenas, O., Bixby, J. L., and Lemmon, V. P. (2010). Kinase/phosphatase overexpression reveals pathways regulating hippocampal neuron morphology. Mol. Syst. Biol. 6, 391.
Chivatakarn, O., Kaneko, S., He, Z., Tessier-Lavigne, M., and Giger, R. J. (2007). The Nogo-66 receptor NgR1 is required only for the acute growth cone-collapsing but not the chronic growth-inhibitory actions of myelin inhibitors. J. Neurosci. 27, 7117–7124.
DeBellard, M. E., Tang, S., Mukhopadhyay, G., Shen, Y. J., and Filbin, M. T. (1996). Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Mol. Cell. Neurosci. 7, 89–101.
Dityateva, G., Hammond, M., Thiel, C., Ruonala, M. O., Delling, M., Siebenkotten, G., Nix, M., and Dityatev, A. (2003). Rapid and efficient electroporation-based gene transfer into primary dissociated neurons. J. Neurosci. Methods 130, 65–73.
Domeniconi, M., Zampieri, N., Spencer, T., Hilaire, M., Mellado, W., Chao, M. V., and Filbin, M. T. (2005). MAG induces regulated intramembrane proteolysis of the p75 neurotrophin receptor to inhibit neurite outgrowth. Neuron 46, 849–855.
Faul, F., Erdfelder, E., Lang, A. G., and Buchner, A. (2007). G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 39, 175–191.
Gabriel, B., and Teissie, J. (1994). Generation of reactive-oxygen species induced by electropermeabilization of Chinese hamster ovary cells and their consequence on cell viability. Eur. J. Biochem. 223, 25–33.
Gabriel, B., and Teissie, J. (1995). Control by electrical parameters of short- and long-term cell death resulting from electropermeabilization of Chinese hamster ovary cells. Biochim. Biophys. Acta 1266, 171–178.
Gabriel, B., and Teissie, J. (1997). Direct observation in the millisecond time range of fluorescent molecule asymmetrical interaction with the electropermeabilized cell membrane. Biophys. J. 73, 2630–2637.
Gartner, A., Collin, L., and Lalli, G. (2006). Nucleofection of primary neurons. Meth. Enzymol. 406, 374–388.
Leclere, P. G., Panjwani, A., Docherty, R., Berry, M., Pizzey, J., and Tonge, D. A. (2005). Effective gene delivery to adult neurons by a modified form of electroporation. J. Neurosci. Methods 142, 137–143.
Lingor, P., Teusch, N., Schwarz, K., Mueller, R., Mack, H., Bahr, M., and Mueller, B. K. (2007). Inhibition of Rho kinase (ROCK) increases neurite outgrowth on chondroitin sulphate proteoglycan in vitro and axonal regeneration in the adult optic nerve in vivo. J. Neurochem. 103, 181–189.
Malo, N., Hanley, J. A., Cerquozzi, S., Pelletier, J., and Nadon, R. (2006). Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 24, 167–175.
Meldrum, R. A., Bowl, M., Ong, S. B., and Richardson, S. (1999). Optimisation of electroporation for biochemical experiments in live cells. Biochem. Biophys. Res. Commun. 256, 235–239.
Mertz, K. D., Weisheit, G., Schilling, K., and Luers, G. H. (2002). Electroporation of primary neural cultures: a simple method for directed gene transfer in vitro. Histochem. Cell Biol. 118, 501–506.
Mitchell, P. J., Hanson, J. C., Quets-Nguyen, A. T., Bergeron, M., and Smith, R. C. (2007). A quantitative method for analysis of in vitro neurite outgrowth. J. Neurosci. Methods 164, 350–362.
Monnier, P. P., Sierra, A., Schwab, J. M., Henke-Fahle, S., and Mueller, B. K. (2003). The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol. Cell. Neurosci. 22, 319–330.
Moore, D. L., Blackmore, M. G., Hu, Y., Kaestner, K. H., Bixby, J. L., Lemmon, V. P., and Goldberg, J. L. (2009). KLF family members regulate intrinsic axon regeneration ability. Science 326, 298–301.
Mukhopadhyay, G., Doherty, P., Walsh, F. S., Crocker, P. R., and Filbin, M. T. (1994). A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13, 757–767.
Neumann, E., Kakorin, S., and Toensing, K. (1999). Fundamentals of electroporative delivery of drugs and genes. Bioelectrochem. Bioenerg. 48, 3–16.
Niederost, B., Oertle, T., Fritsche, J., Mckinney, R. A., and Bandtlow, C. E. (2002). Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. J. Neurosci. 22, 10368–10376.
Rols, M. P., Delteil, C., Serin, G., and Teissie, J. (1994). Temperature effects on electrotransfection of mammalian cells. Nucleic Acids Res. 22, 540.
Rols, M. P., and Teissie, J. (1998). Electropermeabilization of mammalian cells to macromolecules: control by pulse duration. Biophys. J. 75, 1415–1423.
Seijffers, R., Mills, C. D., and Woolf, C. J. (2007). ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci. 27, 7911–7920.
Sivasankaran, R., Pei, J., Wang, K. C., Zhang, Y. P., Shields, C. B., Xu, X.-M., and He, Z. (2004). PKC mediates inhibitory effects of myelin and chondroitin sulfate proteoglycans on axonal regeneration. Nat. Neurosci. 7, 261–268.
Snow, D., Lemmon, V. A., Carrino, D. A., Caplan, A. I., and Silver, J. (1990). Sulfated proteoglycans in astroglial barriers inhibit neurite outgrowth in vitro. Exp. Neurol. 109, 111–130.
Snow, D., and Letourneau, P. C. (1992). Neurite outgrowth on a step gradient of chondroitin sulphate proteoglycan (CS-PG). J. Neurobiol. 23, 322–336.
Snow, D. M., Smith, J. D., and Gurwell, J. A. (2002). Binding characteristics of chondroitin sulfate proteoglycans and laminin-1, and correlative neurite outgrowth behaviors in a standard tissue culture choice assay. J. Neurobiol. 51, 285–301.
Teissie, J., Golzio, M., and Rols, M. P. (2005). Mechanisms of cell membrane electropermeabilization: a minireview of our present (lack of ?) knowledge. Biochim. Biophys. Acta 1724, 270–280.
Venkatesh, K., Chivatakarn, O., Lee, H., Joshi, P. S., Kantor, D. B., Newman, B. A., Mage, R., Rader, C., and Giger, R. J. (2005). The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J. Neurosci. 25, 808–822.
Washbourne, P., and McAllister, A. K. (2002). Techniques for gene transfer into neurons. Curr. Opin. Neurobiol. 12, 566–573.
Wolf, H., Rols, M. P., Boldt, E., Neumann, E., and Teissie, J. (1994). Control by pulse parameters of electric field-mediated gene transfer in mammalian cells. Biophys. J. 66, 524–531.
Keywords: electroporation, 96-well, neurite outgrowth, transfection, CNS neuron, neuronal regeneration, RAG, inhibitory substrate
Citation: Hutson TH, Buchser WJ, Bixby JL, Lemmon VP and Moon LDF (2011) Optimization of a 96-well electroporation assay for postnatal rat CNS neurons suitable for cost–effective medium-throughput screening of genes that promote neurite outgrowth. Front. Mol. Neurosci. 4:55. doi: 10.3389/fnmol.2011.00055
Received: 03 November 2011; Paper pending published: 24 November 2011;
Accepted: 04 December 2011; Published online: 23 December 2011.
Edited by:
Hans-Georg Breitinger, The German University in Cairo, EgyptReviewed by:
Mark R. Cookson, National Institutes of Health, USA;Guoku Hu, Creighton University, USA
Copyright: © 2011 Noori and Fornal. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Thomas H. Hutson, Neurorestoration Group, Wolfson Centre for Age-Related Diseases, King’s College London, Guy’s Campus, London, SE1 1UL, UK. e-mail: thomas.hutson@kcl.ac.uk