Ca2+-permeable AMPA receptors in homeostatic synaptic plasticity
- The Solomon H. Snyder Department of Neuroscience, The Zanvyl Krieger Mind/Brain Institute, Johns Hopkins University, Baltimore, MD, USA
Neurons possess diverse mechanisms of homeostatic adaptation to overall changes in neural and synaptic activity, which are critical for proper brain functions. Homeostatic regulation of excitatory synapses has been studied in the context of synaptic scaling, which allows neurons to adjust their excitatory synaptic gain to maintain their activity within a dynamic range. Recent evidence suggests that one of the main mechanisms underlying synaptic scaling is by altering the function of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), including synaptic expression of Ca2+-permeable (CP-) AMPARs. CP-AMPARs endow synapses with unique properties, which may benefit adaptation of neurons to periods of inactivity as would occur when a major input is lost. This review will summarize how synaptic expression of CP-AMPARs is regulated during homeostatic synaptic plasticity in the context of synaptic scaling, and will address the potential functional consequences of altering synaptic CP-AMPAR content.
Introduction
Dynamic regulation of synaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) has emerged as a critical mechanism underlying multiple forms of synaptic plasticity across many brain areas. AMPARs are the main excitatory postsynaptic glutamate receptors in the central nervous system (CNS), and are comprised of 4 subunits (GluA1–4 or GluR1–4; see Table 1), which combine into tetrameric functional ligand-gated ion channels [reviewed in (Traynelis et al., 2010)]. The assembly of AMPARs occurs via a dimer-of-dimers, such that in most brain areas the predominant AMPARs are hetero-tetramers comprised of GluA1-dimers and GluA2-dimers (i.e., GluA1/GluA2 heteromers) [reviewed in (Traynelis et al., 2010)]. The majority of AMPARs in the mammalian CNS contains the GluA2 subunit, which has undergone RNA editing of the glutamine 607 at the pore loop to an arginine (Sommer et al., 1991; Burnashev et al., 1992). The positive charge under physiological pH and the bulkier side chain of the arginine confers GluA2-containing AMPARs with their generic properties, such as impermeability to Ca2+, linear current-voltage (I–V) relationship, and insensitivity to polyamines [reviewed in (Liu and Zukin, 2007)]. However, a subpopulation of AMPARs lacks the GluA2 subunit, and display Ca2+ permeability, sensitivity to polyamines, and inward rectification of current (Hollmann et al., 1991; Bowie and Mayer, 1995; Donevan and Rogawski, 1995). The latter two properties are due to the interaction between the positively charged polyamines to the negatively charged side chain of the glutamine 607 in the pore region. The binding of polyamines to the pore region predominantly occurs at depolarizing membrane potentials, and prevents outward current through the GluA2-lacking CP-AMPARs. Initially, GluA2 lacking CP-AMPARs were characterized only in a subsest of interneurons (McBain and Dingledine, 1993; Bochet et al., 1994; Otis et al., 1995; Isa et al., 1996; Washburn et al., 1997), but recent studies indicate that they are present at pyramidal cell synapses under specific conditions [reviewed in (Isaac et al., 2007; Liu and Zukin, 2007)]. These studies highlight that synaptic CP-AMPAR population is highly regulated, and depend on neuronal activity (Isaac et al., 2007; Liu and Zukin, 2007).
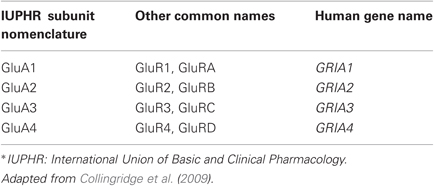
Table 1. Nomenclatures of AMPAR subunits.
Much of what we know about the activity-dependent regulation of AMPAR function has come from studies of synaptic plasticity involving long-term potentiation (LTP) and long-term depression (LTD), which are thought to be the cellular basis for memory formation and proper wiring of circuits during development. However, it was recognized early on that typical LTP- and LTD-type of synaptic modifications need to be adequately balanced to maintain homeostasis of the system. Specifically, many network models of LTP and LTD require normalization or other mechanisms to stabilize the overall activity of the neural network, otherwise the innate positive feedback of LTP and LTD destabilizes net neuronal activity and prevents meaningful synaptic modifications (Bienenstock et al., 1982; Bear et al., 1987; Miller and Mackay, 1994; Turrigiano and Nelson, 2004). There are diverse modes of homeostatic regulation of neural responses, ranging from adjustment of excitatory and inhibitory synaptic gain (Lissin et al., 1998; O'Brien et al., 1998; Rutherford et al., 1998; Turrigiano et al., 1998; Burrone et al., 2002; Chang et al., 2010; Gao et al., 2010; Kim and Alger, 2010), changes in intrinsic excitability (Desai et al., 1999; Aizenman et al., 2003; Pratt and Aizenman, 2007; Maffei and Turrigiano, 2008), and alterations in the induction mechanisms of LTP and LTD (Bienenstock et al., 1982; Bear et al., 1987; Quinlan et al., 1999a,b). While these require distinct mechanisms of neural regulation, they all share a common feature in that they allow neurons to maintain homeostasis despite on-going alterations in input activity and synaptic modifications. Therefore, homeostatic plasticity mechanisms are thought to be especially important during development where there is massive activity-dependent adjustment of neural circuits and synapses, and during periods of heightened plasticity, as would occur during regeneration and recovery following injury. Since the main focus of this review is to provide information on the role of CP-AMPARs in homeostatic synaptic plasticity, the discussion will be focused on the synaptic scaling mode of homeostatic regulation of excitatory synapses. In addition, known molecular mechanisms and potential functional consequences will be discussed.
Synaptic Scaling of Excitatory Synapses
According to the synaptic scaling model, postsynaptic neuronal activity is homeostatically adjusted by modifying the synaptic drive (Turrigiano et al., 1998, 2008; Turrigiano and Nelson, 2004). Synaptic scaling of excitatory synaptic transmission was initially demonstrated in cortical and spinal cord neuronal cultures via pharmacological manipulation of neuronal activity (O'Brien et al., 1998; Turrigiano et al., 1998). A key finding was that the strength of AMPAR-mediated excitatory synaptic transmission was enhanced as neuronal activity decreases, but increased after inhibiting neuronal activity. A fundamental feature of synaptic scaling is to allow neurons to maintain their average firing rate within a dynamic range for effective information transfer (Turrigiano et al., 1998; Turrigiano and Nelson, 2004). While it has not been explicitly stated, changes in synaptic drive can in principle alter the integration properties of synapses and potentially alter the induction threshold for LTP and LTD (see Section “Potential Functional Consequences of Regulating CP-AMPARs” for details).
While the initial studies of synaptic scaling described this phenomenon as a global change in synaptic strength across most of the synapses on a given neuron, it is now clear that synaptic scaling can also operate at specific synapses to individually tune to their own activity [reviewed in (Turrigiano, 2008)]. Global homeostatic changes are cell autonomous and hence can be induced by specifically blocking action potentials in the postsynaptic neuron by restricting tetrodotoxin (TTX) application to the postsynaptic soma (Ibata et al., 2008) or expressing inward-rectifying K+ channels in the postsynaptic neuron (Burrone et al., 2002). Similarly, increasing firing mainly in the postsynaptic neuron using optogenetics was sufficient to scale up synapses (Goold and Nicoll, 2010). On the other hand, synapse-specific homeostatic plasticity is observed following selective manipulation of presynaptic activity. For example, expressing inward-rectifying K+ channels (Beique et al., 2011) in a subset of presynaptic neurons specifically decreases synaptic transmission and triggers scaling up of those specific inputs. In addition, global blockade of neuronal activity by TTX can target specific sets of synapses in an intact network to produce input-specific homeostatic changes (Kim and Tsien, 2008). In these experiments, TTX led to homeostatic adjustment at synapses serving the feed-forward circuits, but opposite changes at feedback inputs. Collectively, these results suggest that there are largely two distinct modes of synaptic scaling: one that targets the majority of synapses, and the other being more selective to a restricted set of synapses. While the details of how these two forms of synaptic scaling interact need to be ironed out, there is some evidence that the underlying molecular mechanisms may differ [reviewed in (Turrigiano, 2008)]. For example, input-specific synaptic scaling has been shown to occur via local inhibition of NMDAR and dendritic synthesis of GluA1-containing CP-AMPARs, while regulation of somatic Ca2+ transients and transcriptional events have been linked to global synaptic scaling. However, there are exceptions to these cases. Also, in vivo sensory experience-induced homeostatic synaptic plasticity can be global or restricted to a subset of synapses depending on the age of the animals or the particular neurons studied in an intact circuit (Goel et al., 2006; Goel and Lee, 2007; Gao et al., 2010; Petrus et al., 2011).
One of the postsynaptic changes associated with synaptic scaling is the regulation of AMPA receptor function. Synaptic scaling is observed as an increase in AMPAR-mediated miniature excitatory postsynaptic current (mEPSC) amplitude following inhibition of neuronal activity, and a decrease in AMPAR-mEPSC amplitude after a period of enhanced neuronal activity (O'Brien et al., 1998; Turrigiano et al., 1998). These functional changes are often accompanied by the regulation of synaptic AMPAR content (O'Brien et al., 1998; Ju et al., 2004; Thiagarajan et al., 2005; Wierenga et al., 2005). Incubating neuronal cultures in TTX or AMPAR antagonist (e.g., CNQX) for a period (hours to days) to block neuronal activity causes an accumulation of AMPARs at synapses that correlates with increases in AMPAR-mEPSC amplitude. On the other hand, increasing neuronal activity by incubating neurons in antagonists of GABAA receptors (e.g., bicucullin or picrotoxin), decreases synaptic AMPAR content and AMPAR-mEPSC amplitude. It is imperative to point out that synaptic scaling also has been shown to alter the frequency of AMPAR-mEPSCs (Thiagarajan et al., 2002, 2005), which may be restricted to certain synapses (Kim and Tsien, 2008) and/or dependent on the developmental stage of the neurons (Wierenga et al., 2006). The alteration in AMPAR-mEPSC frequency is often interpreted as changes in the number of functional synapses, and can be mediated by either altering presynaptic function or postsynaptic silencing/unsilencing of synapses. Whether synaptic scaling is observed as changes in AMPAR-mEPSC amplitude or frequency, it is often accompanied by alterations in synaptic CP-AMPAR content (Thiagarajan et al., 2002, 2005; Ju et al., 2004; Sutton et al., 2006; Aoto et al., 2008; Beique et al., 2011) [but see (Gainey et al., 2009; Anggono et al., 2011)].
Synaptic scaling type of homeostatic plasticity has been observed in vivo in different preparations. In line with the in vitro pharmacological activity manipulations, in vivo application of TTX to CA1 increases neuronal excitability, which correlated with an increased in AMPAR-mEPSC frequency or both frequency and amplitude depending on the developmental age of the animal (Echegoyen et al., 2007). More importantly, manipulation of in vivo sensory experience has been shown to produce homeostatic changes at cortical synapses. In particular, depriving vision scales up excitatory synapses in the visual cortex (Desai et al., 2002; Maffei et al., 2004; Goel et al., 2006, 2011; Goel and Lee, 2007; Maffei and Turrigiano, 2008; Petrus et al., 2011), while re-exposing the visually deprived animals to light scales down the synapses (Goel et al., 2006, 2011; Goel and Lee, 2007; Gao et al., 2010; Petrus et al., 2011). Interestingly, the sensory experience-induced homeostatic synaptic changes are mainly observed as regulation of the amplitude of AMPAR-mEPSCs with little effect on the frequency. This suggests that the in vivo sensory experience-dependent changes are mainly postsynaptic in nature at the level of AMPAR regulation. Furthermore, under certain conditions of sensory deprivation, the homeostatic regulation of AMPAR function with sensory experience is accompanied by changes in the level of synaptic CP-AMPARs (Goel et al., 2006, 2011).
CP-AMPAR Regulation and Homeostatic Synaptic Plasticity
Studies of synaptic scaling highlight that the one of the main molecular mechanisms is the regulation of postsynaptic AMPARs. Hours to days of neuronal inactivity or decreased sensory input to neurons results in synaptic accumulation of AMPAR subunits (O'Brien et al., 1998; Ju et al., 2004; Thiagarajan et al., 2005; Wierenga et al., 2005; Goel et al., 2006, 2011; Sutton et al., 2006; Aoto et al., 2008). While most studies agree on the accumulation of the GluA1 (or GluR1) subunit of AMPARs, there is varying degree of observations regarding the GluA2 (or GluR2) subunit. For instance, several studies reported that pharmacologically inhibiting neural activity increases synaptic GluA1 in cultured neurons often without much change in the GluA2 subunit levels (Lissin et al., 1998; Ju et al., 2004; Thiagarajan et al., 2005; Aoto et al., 2008). The increase in synaptic GluA1 requires local translation of GluA1 mRNA in the dendrites (Ju et al., 2004; Sutton et al., 2006; Maghsoodi et al., 2008), and is regulated by retinoic acid (RA) signaling (Aoto et al., 2008; Maghsoodi et al., 2008; Poon and Chen, 2008). This inactivity-induced increase in GluA1 results in accumulation of CP-AMPARs at synapses, which is detected as the appearance of polyamine sensitive AMPAR-mEPSCs (Thiagarajan et al., 2005; Sutton et al., 2006). Similarly, experience-dependent homeostatic synaptic plasticity observed in visual cortex is also associated with a rather selective regulation of GluA1 and CP-AMPAR at synapses as measured biochemically in isolated postsynaptic density (PSD) preparations and appearance of inward rectifying AMPAR currents (Goel et al., 2006, 2011). In this case, phosphorylation of the GluA1 subunit of AMPAR on the major cAMP-dependent protein kinase (PKA) site, serine-845 (S845) (Roche et al., 1996), correlated with and was necessary for scaling up AMPAR-mEPSCs with visual deprivation (Goel et al., 2006, 2011).
While many studies suggest regulation of GluA1 and CP-AMPARs during synaptic scaling, this is not universally observed. There are reports that pharmacologically blocking neuronal activity in dissociated neuronal cultures up-regulates both GluA1 and GluA2 subunits at synapses (O'Brien et al., 1998; Wierenga et al., 2005; Anggono et al., 2011), and is dependent on the GluA2 regulatory mechanisms (Gainey et al., 2009; Anggono et al., 2011). The discrepancy in results needs to be clarified, but one proposal is that it may depend on the means of blocking neuronal activity. It was been suggested that blocking neuronal firing alone may trigger a global synaptic scaling that regulates both GluA1 and GluA2 subunits, while blocking NMDAR in conjunction with activity blockade may specifically target a subset of synapses and recruit CP-AMPARs (Turrigiano, 2008). Indeed, studies reporting concurrent GluA1 and GluA2 regulation with synaptic scaling have used either TTX to block action potentials or CNQX to block AMPAR driven synaptic activity (O'Brien et al., 1998; Wierenga et al., 2005; Gainey et al., 2009; Anggono et al., 2011). On the other hand, combining TTX with NMDAR antagonist APV results in GluA1 regulation with minimal effect on the synaptic levels of GluA2 (Ju et al., 2004; Sutton et al., 2006; Aoto et al., 2008; Maghsoodi et al., 2008). Furthermore, synapse-specific inhibition of presynaptic activity also leads to the appearance of CP-AMPARs at synapses (Beique et al., 2011). However, there are cases where activity blockade alone without specific NMDAR inhibition can regulate GluA1 (Thiagarajan et al., 2005), hence there may be other variables at work. In vivo scaling triggered by sensory manipulations can cause preferential regulation of GluA1 and synaptic trafficking of CP-AMPARs (Goel et al., 2006, 2011), or lead to synaptic increase in Ca2+-impermeable (CI)-AMPARs dependent on GluA2 regulatory mechanisms (Gainey et al., 2009). As will be discussed later, this discrepancy may lie in the pattern of sensory loss (see Section “Potential Functional Consequences of Regulating CP-AMPARs”).
Molecular Mechanisms of CP-AMPAR Regulation
The regulation of CP-AMPARs at synapses is widely utilized under various synaptic plasticity mechanisms [reviewed in (Isaac et al., 2007; Liu and Zukin, 2007)]. Synaptic expression of CP-AMPARs is presumably very tightly regulated, especially in pyramidal neurons where there is very little basal expression. This is quite distinct from inhibitory interneurons with a larger subpopulation expressing GluA2 lacking CP-AMPARs at synapses (McBain and Dingledine, 1993; Bochet et al., 1994; Otis et al., 1995; Isa et al., 1996; Washburn et al., 1997). Synaptic CP-AMPARs display properties distinct from CI-AMPARs in that they are Ca2+ permeable, have larger single channel conductance and faster kinetics, and display voltage-dependent blockade by intracellular polyamines (Hollmann et al., 1991; Bowie and Mayer, 1995; Donevan and Rogawski, 1995). The faster kinetics of CP-AMPAR-mediated synaptic responses would benefit synapses that require precise temporal signaling, and the larger conductance together with the voltage-dependent block would allow more efficient synaptic integration. Furthermore, the polyamine block can be relieved by a period of high frequency activity (Rozov et al., 1998; Rozov and Burnashev, 1999), which will allow CP-AMPAR containing synapses to function differently depending on the recent history of synaptic activity.
A more thorough understanding of activity-dependent CP-AMPAR regulation has come from studies of the stellate cells in the cerebellum. These interneurons express CP-AMPARs at synapses under basal conditions, but high frequency activity replaces CP-AMPARs to GluA2-containing CI-AMPARs (Liu and Cull-Candy, 2000, 2002). This process is termed CP-AMPAR plasticity (CARP), and involves GluA2 interaction with Protein Interacting with C-Kinase 1 (PICK1) and N-ethylmaleimide-sensitive fusion protein (NSF) (Gardner et al., 2005; Liu and Cull-Candy, 2005). In addition, activation of both CP-AMPARs and Group 1 mGluRs are necessary to support CARP (Kelly et al., 2009). Recently, it was reported that CARP at the parallel fiber input to the stellate cells can be induced by emotional stress and require transcription of GluA2 mRNA (Liu et al., 2010). These results suggest that increased neuronal activity triggers GluA2 synthesis, which then traffic to synapses via PICK1 and NSF-dependent mechanisms. The activity-dependent switch from CP-AMPAR to CI-AMPAR would dampen the excitatory drive, hence will in a way act to maintain neuronal homeostasis.
Unlike the interneurons, excitatory synapses of the principal neurons primarily express CI-AMPARs. However, CP-AMPARs can be recruited to these synapses in an activity-dependent manner in various preparations [reviewed in (Liu and Zukin, 2007)]. Furthermore, in some brain areas, CP-AMPARs are present on pyramidal neurons during early development. For instance, CP-AMPARs are present at postnatal ages 5–8 days (P5–P8) in a subset of synapses on CA1 neurons (Stubblefield and Benke, 2010), and similarly present at synapses of layer 5 pyramidal neurons until around P15 (Kumar et al., 2002; Shin et al., 2005). Collectively, these observations suggest that synaptic expression of CP-AMPARs in principal neurons is tightly regulated.
Initial evidence that hinted of CP-AMPAR recruitment to synapses of pyramidal neurons was from experiments where GluA1 homomers were exogenously expressed into CA1 pyramidal neurons in organotypic hippocampal slice cultures. In one of the initial studies, over-expression of GluA1 resulted in assembly of Ca2+-permeable GluA1 homomers, which were recruited for synaptic transmission following LTP induction (Hayashi et al., 2000). It is noteworthy that even in the over-expression system, synaptic trafficking of GluA1 homomers is strictly dependent on high frequency activity, NMDAR activation, and recruitment of CaMKII signaling (Hayashi et al., 2000). This suggests a high degree of regulation of GluA1 CP-AMPAR synaptic localization. Subsequent studies have reported that native CP-AMPARs can be transiently recruited to synapses following LTP and subsequently replaced by CI-AMPARs in hippocampal slices (Plant et al., 2006), albeit this is not universally observed (Adesnik and Nicoll, 2007; Gray et al., 2007; Volk et al., 2010). There are reports that this discrepancy may stem from specific requirement of CaMKI signaling (Guire et al., 2008; Fortin et al., 2010), the transient nature of the synaptic CP-AMPARs (Plant et al., 2006), or the specific developmental age of the animal (Lu et al., 2007). Alternatively, the changes may only happen at a subset of synapses. Furthermore, it was recently suggested that CP-AMPAR synaptic trafficking may differ in distinct brain areas. For instance, there is evidence that hippocampal CA1 synapses are more resilient to CP-AMPAR expression than synapses in the primary visual cortex (He et al., 2011; Lee and Kirkwood, 2011).
GluA1-Dependent Mechanisms
In several preparations, homeostatic synaptic scaling has been shown to regulate CP-AMPARs (see Section “CP-AMPAR Regulation and Homeostatic Synaptic Plasticity”), and the main mechanism depends on the regulation of synaptic GluA1 content (Figure 1). Inactivity results in synaptic accumulation of GluA1 subunits without much change in the GluA2 levels in dissociated neuronal cultures (Thiagarajan et al., 2005; Sutton et al., 2006; Maghsoodi et al., 2008), which correlates with synaptic appearance of polyamine sensitive CP-AMPAR current (Thiagarajan et al., 2005; Sutton et al., 2006). This effect is largely accelerated by inhibition of NMDAR-mediated mEPSCs (NMDAR-mEPSCs) (Sutton et al., 2006), and involves local dendritic synthesis of GluA1 subunits (Ju et al., 2004; Sutton et al., 2006; Maghsoodi et al., 2008; Poon and Chen, 2008). The local synthesis of GluA1 could in principle result in accumulation of CP-AMPARs at a specific set of synapses. Hence, concurrent blockade of action potentials and NMDAR-mEPSCs has been suggested to mediate synapse-specific scaling with CP-AMPAR expression, which is distinct from global cell-wide synaptic scaling (Turrigiano, 2008). However, blocking neuronal activity without an NMDAR antagonist can also up-regulate CP-AMPARs (Thiagarajan et al., 2005; Lindskog et al., 2010; Beique et al., 2011; Groth et al., 2011), which suggests that NMDAR blockade is not a requirement.
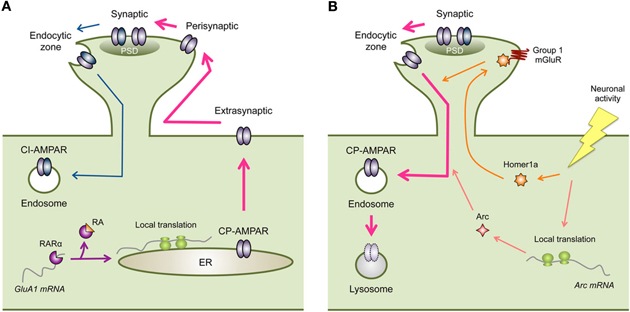
Figure 1. Regulation of CP-AMPARs in homeostatic synaptic plasticity. (A) Inactivity-induced synaptic expression of CP-AMPARs. Inactivity leads to local synthesis of GluA1. One mechanism is via RA signaling, where RA binds to RARα to relieve translational inhibition of GluA1 mRNA. Newly synthesized GluA1 containing CP-AMPARs are then eventually trafficked to the synapse (PSD, postsynaptic density). Cell surface expression of GluA1 containing CP-AMPARs is likely tied to phosphorylation of GluA1-S845. In visual cortex, the increase in extrasynaptic GluA1 leads to synaptic accumulation, however, in hippocampus an extra regulatory step from perisynaptic to synaptic trafficking may be needed. Inactivity driven synaptic localization of CP-AMPARs also depend on CaMKIIβ activity, which may act on GluA1-S831 phosphorylation. Synaptic expression of CP-AMPARs may occur in conjunction with removal of CI-AMPARs via GluA2-dependent mechanisms. (B) Removal of synaptic CP-AMPARs following an increase in neural activity. The increase in neuronal activity following a period of inactivity results in up-regulation of immediate early genes such as Arc and Homer1a. Both of these proteins lead to endocytosis of AMPARs. However, this process is not likely specific to CP-AMPARs and would result in removal of CI-AMPARs as well (not depicted in the figure). Homer1a acts to produce agonist-independent activation of Group 1 mGluRs by displacing long forms of Homer1. Homer1a signaling is specific for activity-dependent homeostatic scaling down of excitatory synapses. On the other hand, Arc is also involved in AMPAR endocytosis following mGluR-LTD.
Inactivity-driven local synthesis of GluA1 and synaptic scaling are mimicked by application of RA (Aoto et al., 2008). Retinoic acid receptor alpha (RARα) has been shown to bind to the 5′-untranslated region (UTR) of GluA1 mRNAs, which represses its basal translation. Inactivity leads to dendritic accumulation of RARα to dendritic RNA granules (Maghsoodi et al., 2008) and synthesis of RA (Aoto et al., 2008) in dissociated hippocampal neuronal cultures. Activation of RARα by RA relieves the translational repression leading to local synthesis of GluA1 (Maghsoodi et al., 2008; Poon and Chen, 2008). These results provide a mechanism for regulating dendritic synthesis of GluA1 following inactivity (Ju et al., 2004; Sutton et al., 2006; Aoto et al., 2008) (Figure 1A). While cell surface levels of AMPARs correlate with synaptic content (Kessels et al., 2009), simply over-expressing GluA1 does not normally lead to synaptic accumulation of functional GluA1 homomers in organotypic hippocampal slice cultures (Hayashi et al., 2000). Therefore, regulated synaptic trafficking of GluA1 containing CP-AMPARs is likely needed during synaptic scaling.
Synaptic trafficking of GluA1 homomers during LTP depends critically on CaMKII activation (Hayashi et al., 2000) and actin binding motor proteins myosin Va (Correia et al., 2008) and myosin Vb (Wang et al., 2008). While CaMKII activity is necessary for LTP (Malinow et al., 1989; Silva et al., 1992) and synaptic insertion of AMPARs (Hayashi et al., 2000), these are both independent of the phosphorylation on the GluA1-S831 site (Hayashi et al., 2000; Lee et al., 2010). Actin based motor proteins are involved in mobilizing AMPAR containing vesicles, and in particular myosin Vb is activated by Ca2+ and suggested to mobilize recycling endosomes for synaptic insertion of AMPARs (Wang et al., 2008). There is evidence that CaMKII function is also involved in synaptic scaling (Thiagarajan et al., 2002). Distinct isoforms of CaMKII have been suggested to respond to inactivity, such that CaMKIIα is down-regulated mainly by inhibiting NMDAR activity and CaMKIIβ is up-regulated by antagonizing AMPAR function in dissociated hippocampal neuronal cultures (Thiagarajan et al., 2002). Hence the balance of CaMKIIα and CaMKIIβ subunits has been suggested to regulate the polarity of synaptic scaling (Thiagarajan et al., 2002). In line with this, knocking down CaMKIIβ expression prevented the inactivity-induced increase in GluA1, in addition to preventing scaling up of mEPSC amplitude and frequency (Groth et al., 2011). This suggests that GluA1 synaptic localization depends on CaMKIIβ. Interestingly, the increase in mEPSC frequency following inactivity was triggered by Ca2+ influx through the synaptic CP-AMPARs and retrograde signaling to enhance presynaptic vesicle turnover (Lindskog et al., 2010). This suggests that synaptic incorporation of CP-AMPARs alters not only postsynaptic function, but also coordinates an increase in presynaptic release when activity blockade is removed.
Many studies have shown that the synaptic localization of GluA1 is dependent on phosphorylation of key residues, one of which is the serine-845 (S845) [reviewed in (Lee, 2006)]. S845 is a major PKA phosphorylation site on the GluA1 subunit (Roche et al., 1996), and is targeted by various neuromodulators linked to cAMP signaling (Chao et al., 2002; Hu et al., 2007; Seol et al., 2007). Manipulations that increase GluA1-S845 phosphorylation have been shown to enhance cell surface expression of GluA1-containing AMPARs (Sun et al., 2005; Oh et al., 2006; Man et al., 2007; Goel et al., 2011; He et al., 2011) via promoting receptor recycling to the plasma membrane (Ehlers, 2000). And the GluA1-S845 site is considered a necessary pre-requisite step in synaptic trafficking of GluA1-containing AMPARs during LTP (Esteban et al., 2003). On the flip side, dephosphorylation of GluA1-S845 is correlated with endocytosis of AMPARs (Ehlers, 2000; Lee et al., 2003; Man et al., 2007), and is necessary for LTD in hippocampus and visual cortex (Lee et al., 1998, 2003, 2010; Seol et al., 2007). These results highlight that a reversible regulation of GluA1-S845 phosphorylation is key in bidirectional synaptic plasticity [reviewed in (Lee and Huganir, 2008)]. Furthermore, visual deprivation-induces scaling up of mEPSCs and the appearance of synaptic CP-AMPARs is correlated with an increase in GluA1-S845 phosphorylation in visual cortex (Goel et al., 2006, 2011), and is abolished in mice specifically lacking this phosphorylation site (GluA1-S845A mice) (Goel et al., 2011). This suggests a pivotal role of S845 phosphorylation in cell surface and synaptic targeting of GluA1 containing CP-AMPARs, likely GluA1 homomers. Furthermore, the GluA1-S845 site is necessary for maintaining a perisynaptic pool of CP-AMPARs, which could be recruited to participate in synaptic transmission upon Group 1 mGluR activation (He et al., 2009). This suggests that GluA1-S845 phosphorylation is likely a pre-requisite for synaptic recruitment of CP-AMPARs to synapses. Consistent with this idea, enhancing GluA1-S845 phosphorylation alone increases mEPSC amplitude at visual cortex synapses, but it is not sufficient for producing multiplicative synaptic scaling as seen with visual deprivation (Goel et al., 2011). Therefore, there are likely additional signals needed, besides GluA1-S845 phosphorylation, which allows normal multiplicative synaptic scaling. This is reminiscent of the role of GluA1-S845 in LTP, where it “primes” LTP but the ultimate induction is dependent on NMDAR activation (Oh et al., 2006; Seol et al., 2007). In any case, collectively these results suggest that synaptic regulation of GluA1 during LTP/LTD and homeostatic synaptic plasticity is surprisingly conserved. This would suggest that despite different neural activity required for induction, the neurons utilize similar downstream AMPAR regulatory mechanisms to postsynaptically alter the strength of excitatory synaptic transmission.
In addition to GluA1-S845, CaMKII phosphorylation of GluA1-S831 site has been implicated in LTP (Barria et al., 1997; Lee et al., 2000). However, the role of this site in homeostatic synaptic plasticity is not as clear. This may be because unlike LTP, which requires CaMKIIα (Silva et al., 1992), scaling up of mEPSCs depends more on CaMKIIβ (Thiagarajan et al., 2002; Groth et al., 2011). Interestingly, mice lacking the GluA1-S831 site display abnormal synaptic scaling of visual cortex synapses, such that visual deprivation scales down mEPSCs, which is opposite from what is observed in wild-type mice (Goel et al., 2011). Similarly, knocking down CaMKIIβ in dissociated neuronal cultures results in scaling down of mEPSCs with inactivity (Groth et al., 2011). Therefore, it is plausible that phosphorylation of GluA1-S831, perhaps via CaMKIIβ signaling, may play a yet to be determined function in homeostatic up-regulation of excitatory synaptic strength. However, phosphorylation of this site does not always correlate with scaling up of mEPSCs, at least in the in vivo models of synaptic scaling of cortical synapses (Goel et al., 2006, 2011). Therefore, further research is needed to clarify the role of GluA1-S831 phosphorylation in homeostatic synaptic plasticity.
GluA2-Dependent Mechanisms
In addition to GluA1-dependent mechanisms, there is evidence that regulation of GluA2 is involved in homeostatic regulation of CP-AMPARs. Most of the studies on the role of GluA2 in homeostatic synaptic plasticity come from experimental conditions where CP-AMPAR regulation is not observed (Gainey et al., 2009; Anggono et al., 2011). For example, expressing GluA2 carboxy-terminal (ct), but not GluA1-ct, prevents scaling up of mEPSCs with inactivity (Gainey et al., 2009). Also, knocking out GluA2 interacting protein PICK1 occludes scaling up of mEPSCs with inactivity (Anggono et al., 2011). In these two studies done in dissociated cortical cultures, scaling up of mEPSCs was triggered by TTX and was not accompanied by synaptic localization of CP-AMPARs (Gainey et al., 2009; Anggono et al., 2011). These results would suggest that GluA2-dependent mechanisms may dominate TTX-induced homeostatic synaptic scaling involving CI-AMPARs. However, as will be discussed later, there is some indication that GluA2-dependent mechanisms may contribute to CP-AMPAR regulation during homeostatic synaptic plasticity. The GluA2-ct contains numerous binding regions for various interacting molecules, such as NSF, PICK1, and glutamate receptor interacting protein (GRIP)/AMPA receptor binding protein (ABP) [reviewed in (Song and Huganir, 2002)]. The interaction of GluA2 with these binding partners regulates its cell-surface expression and synaptic targeting. In particular, binding of PICK1 and GRIP/ABP to the c-terminal extreme PDZ ligand region of GluA2 (-SVKICOOH), is tightly regulated by phosphorylation of S880 present at the -3 site (Matsuda et al., 1999; Chung et al., 2000). Phosphorylation of GluA2-S880 shifts the preference of binding from GRIP to PICK1, and this change in interaction is critical for the endocytosis of AMPARs and expression of LTD (Chung et al., 2000; Matsuda et al., 2000; Seidenman et al., 2003). At cerebellar stellate cell synapses, activity-dependent removal of CP-AMPARs requires NSF, PICK1, and GRIP interactions (Gardner et al., 2005; Liu and Cull-Candy, 2005). Therefore, it is possible that GluA2 interaction with its binding partners is involved in scaling down of synapses, which is when the removal of CP-AMPARs occurs. The idea is that CP-AMPAR removal may be coordinated with replacement by CI-AMPARs, but it is possible that both CP-AMPARs and CI-AMPARs are removed together to scale down synapses (Figure 1B). Moreover, it is also likely that GluA2-binding molecules play a role in CP-AMPAR expression at synapses with inactivity (Figure 1A). It was observed that PICK1 over-expression increases AMPAR-EPSCs and appearance of synaptic CP-AMPARs in acute cultured hippocampal slices (Terashima et al., 2004), which depends on NMDAR activation (Terashima et al., 2008). Furthermore, sensory experience-induced synaptic CP-AMPAR expression in barrel cortex also depends on PICK1 (Clem et al., 2010). Therefore, PICK1-dependent endocytosis of GluA2-containing AMPARs may occur in conjunction with GluA1-dependent CP-AMPAR synaptic targeting. Whether GluA2-dependent mechanisms play a role in homeostatic synaptic plasticity under conditions that recruit CP-AMPARs needs to be verified experimentally.
Potential Interaction Between GluA1- and GluA2-Dependent Mechanisms
Most of our understanding on the mechanisms of GluA1- and GluA2-dependent regulation of AMPAR derives from studies of homomeric receptors. However, it is clear that the majority of native AMPARs in the principal neurons are heteromeric complexes of GluA1 and GluA2 (Wenthold et al., 1996; Lu et al., 2009). Therefore, the native AMPARs have the potential for regulation via both subunits. However, there are only few studies addressing how the different subunits dependent mechanisms may interact to regulate synaptic AMPARs by activity. Most of the evidence suggests that synaptic trafficking of AMPAR is dominated by specific subunits depending on the subunit unit composition of the AMPAR and the direction of trafficking. For example, synaptic trafficking of GluA1/GluA2 heteromers in organotypic hippocampal slices follows the pattern of GluA1 homomers in that it requires neuronal activity, which differs from constitutive synaptic accumulation of GluA2/GluA3 heteromers (Shi et al., 2001). On the other hand, agonist-induced internalization of GluA1/GluA2 heteromers follow a similar pattern of sorting as GluA2 homomers and are targeted to lysosomes rather than like GluA1 homomers which remain in recycling endosomes (Lee et al., 2004).
In the case of homeostatic synaptic scaling, GluA1-dependent mechanism may dominate for inactivity-induced scaling up of synapses, because most studies are done on native AMPARs, which are mainly GluA1/GluA2 heteromers, and involve synaptic expression of CP-AMPARs, likely GluA1-homomers (Figure 1A). While this is the case for most studies (see Section “GluA1-Dependent Mechanisms” for Details), there are studies suggesting dependence on GluA2 (Section “GluA2-Dependent Mechanisms”). Whether the discrepancy is due to differences in how neuronal inactivity was achieved (e.g., TTX only or in combination with APV) or involves other experimental parameters is unclear at this point. However, considering that both GluA1 and GluA2 dependent mechanisms have been suggested to play a role in sensory experience-induced homeostatic synaptic plasticity (Goel et al., 2006, 2011; Gainey et al., 2009), there are likely interactions between the two modes of regulation. On the other hand, activity-dependent scaling down of synapses may utilize both GluA1- and GluA2-dependent mechanisms, because synaptic removal of CP-AMPARs (Figure 1B) may occur together with down-regulation of GluA1/GluA2 heteromers. In any case, further studies are needed to determine the exact nature of the interaction between GluA1- and GluA2-dependent mechanisms, and how each mechanism is recruited under different circumstances during homeostatic adaptation.
Possible Induction Mechanisms
So far the studies reviewed above suggest a rather surprising conservation of AMPAR regulatory mechanisms between LTP/LTD type of synaptic plasticity and homeostatic synaptic plasticity. This was initially unexpected, since these two types of synaptic plasticity mechanisms are triggered by almost opposite changes in neuronal activity. For example, LTP is induced by a brief increase in patterned neural activity, while homeostatic scaling up of synapses is induced by a rather prolonged duration of inactivity. Then, how do these two distinct patterns of neuronal activity recruit similar downstream AMPAR regulatory mechanisms to strengthen synapses? The answer probably lies in the distinct induction mechanisms and initial signaling pathways that converge downstream to tap into AMPAR regulatory mechanisms. One signaling candidate that may distinguish LTP and LTD from homeostatic synaptic plasticity may be Group 1 mGluRs. While Group 1 mGluRs have also been implicated to play a role in various forms of LTP and LTD, a recent study suggests that different modes of activating this receptor may be a key component. A recent study suggested that switching Group 1 mGluR from an agonist-dependent to agonist-independent mode by an immediate early gene Homer1a is critical for homeostatic synaptic scaling in cultured cortical neurons (Hu et al., 2010). Group 1 mGluRs undergo two distinct mode of activation: one that is agonist-dependent and the other that is agonist-independent (Ango et al., 2001). Agonist-independent activation requires the expression of Homer1a, which then displaces mGluR interaction with the long forms of Homer1. Because Homer1a expression is triggered by neuronal activity (Brakeman et al., 1997), it allows increased neuronal activity to cause constitutive activation of mGluRs (Ango et al., 2001). Preventing agonist-independent activity of both mGluR1 and mGluR5 scales up mEPSCs and prevents activity-induced scaling down (Hu et al., 2010). Furthermore, Homer1a KO shows larger basal mEPSCs and more synaptic AMPARs, and lack scaling up and down of mEPSCs (Hu et al., 2010). Homer1a seems specific for homeostatic synaptic plasticity, especially because it is not required for mGluR-LTD (Hu et al., 2010), which also is a form of activity-dependent synaptic weakening. These results collectively suggest that one of the key differences between AMPAR down-regulation during homeostatic synaptic plasticity and LTD may be at the level of recruiting Homer1a signaling (Figure 1B). However, it is important to note that in addition to Homer1a mediated scaling down of mEPSCs, there is an independent parallel pathway mediated by another immediate early gene Arc (Shepherd et al., 2006; Gao et al., 2010; Beique et al., 2011), which is independent of Group 1 mGluR signaling (Hu et al., 2010) (Figure 1B). Further studies are needed to determine how these two signaling pathways interact or are recruited differentially to mediate scaling down of synapses.
In the case of inactivity-driven homeostatic scaling up of mEPSCs, a decrease in somatic Ca2+ signal and CaMKIV mediated somatic transcriptional regulation has been implicated (Ibata et al., 2008). However, this is likely applicable for global cell-wide synaptic scaling. For scaling up synapses locally, a synapse-specific decrease in Ca2+ via inactivation of NMDAR may be responsible. And as discussed previously, a local synthesis of GluA1 and trafficking of these receptors to synapses is required (see Section “GluA1-Dependent Mechanisms”).
Potential Functional Consequences of Regulating CP-AMPARS
Synaptic recruitment of CP-AMPARs during homeostatic synaptic plasticity suggests that the postsynaptic signaling during synaptic transmission under basal conditions and after a period of inactivity is distinct. Due to their faster kinetics and activity-dependent polyamine block, CP-AMPARs will alter the pattern of synaptic summation. Faster decay kinetics (Hollmann et al., 1991) predict a diminished summation at low input frequencies, and the activity-dependent relief from polyamine block will enhance the summation of synaptic responses following a period of high input activity (Rozov et al., 1998; Rozov and Burnashev, 1999). In this sense, synaptic CP-AMPARs are expected to endow the synapses to act in accordance with prior input activity. In addition, because of their Ca2+ permeability, they can potentially trigger postsynaptic signaling cascades that depend on Ca2+. Induction of postsynaptic forms of LTP/LTD is critically dependent on the magnitude and temporal profile of postsynaptic Ca2+ transients, such that larger transients induce LTP and smaller/prolonged Ca2+ signals produce LTD (Malenka et al., 1992; Yang et al., 1999). Furthermore, manipulation of postsynaptic Ca2+ transients has been shown to slide the LTP/LTD induction threshold (Cummings et al., 1996). While the “sliding threshold” theory is mainly considered to act on NMDAR regulations (Bear, 1996), the theory itself does not specify the mechanism as to how the LTP/LTD induction threshold slides (Bienenstock et al., 1982; Bear et al., 1987). Therefore, in principle, the trafficking of Ca2+-permeable AMPA receptors (CP-AMPARs) to synapses could alter the Ca2+ signature and affect LTP/LTD induction. In support of this, mice expressing CP-AMPARs at synapses (i.e., GluR2 knockouts) produce LTP (Jia et al., 1996; Meng et al., 2003; Wiltgen et al., 2010) solely mediated by Ca2+ through CP-AMPARs (Asrar et al., 2009) and even at hyperpolarizing membrane potentials (Wiltgen et al., 2010). However, how CP-AMPARs contribute to synapses with predominantly CI-AMPARs is unknown. In principle, the extra Ca2+ influx through CP-AMPARs could add to the traditional means of Ca2+ elevation at synapses, such as through NMDARs or voltage-gated Ca2+ channels. Further studies are needed to determine whether the addition of CP-AMPARs to synapses can alter the synaptic modification threshold.
Homeostatic regulation of CP-AMPARs is observed in vivo following change in sensory experience. For instance, several days of visual deprivation causes synaptic incorporation of CP-AMPARs (Goel et al., 2006, 2011). This suggests that homeostatic regulation of CP-AMPARs may play an adaptive role for the cortex deprived of its primary inputs. However, the synaptic incorporation of CP-AMPARs seems specific to the mode of visual deprivation and is observed when rearing the animals in the dark (e.g., dark-rearing or dark-exposure) (Goel et al., 2006, 2011), but not when vision is deprived by intraocular TTX injections (Gainey et al., 2009). These two modes of visual deprivation affect neural activity differently. For instance, dark-rearing/dark-exposure type of manipulation only silences visually-driven activity but leaves spontaneous retinal activity intact, while intraocular TTX injections will silence them both. How these differences contribute to synaptic regulation of CP-AMPARs is unknown, but considering the differential requirement of neural activity in GluA1- and GluA2-dependent AMPAR trafficking, it is perhaps not a surprise. Synaptic trafficking of GluA1-containing AMPARs critically depends on neuronal activity, but GluA1-lacking AMPARs (i.e., GluA2/GluR3 heteromers) can get to synapses constitutively (Shi et al., 2001). Perhaps coordinated action of both GluA1- and GluA2-dependent mechanisms support synaptic CP-AMPAR incorporation (Figure 1A), and GluA2-only mechanisms may cause homeostatic synaptic scaling without CP-AMPAR expression. In line with this interpretation, dark-rearing/dark-exposure causes appearance of CP-AMPAR and scaling up of mEPSCs in visual cortex synapses, which depend on GluA1 phosphorylation (Goel et al., 2006, 2011). On the other hand, intraocular TTX injection scales up mEPSCs without CP-AMPARs at synapses, and is dependent on GluA2-ct but not GluA1-ct (Gainey et al., 2009). In any case, the homeostatic regulation of CP-AMPARs is expected to alter synaptic function so as to decrease the threshold for LTP induction. This will allow the cortex deprived of its primary inputs to strengthen connections that were previously subthreshold for LTP. This may provide a way for recruiting the deprive cortex for processing previously weak inputs, such as those arising from other sensory systems. In line with this, it was proposed that homeostatic synaptic plasticity induced in primary sensory cortices following visual deprivation may be cellular substrates of cross-modal plasticity (Goel et al., 2006). For instance, scaling up of excitatory synapses and synaptic recruitment of CP-AMPARs in primary visual cortex by visual deprivation could allow recruitment of visual cortical neurons for processing previously subthreshold auditory or tactile inputs, which could explain the recruitment of primary visual cortex when blind individuals read Braille (Sadato et al., 1996, 2002; Buchel et al., 1998). A similar case scenario may apply to cortical areas denervated by injury, where the deprived cortical area may use homeostatic recruitment of CP-AMPARs for enhancing plasticity to recover function. Whether this is the case is unknown, but many neuronal injury and insult models report up-regulation of CP-AMPARs [e.g., (Liu et al., 2006; Spaethling et al., 2008)].
Concluding Remarks
In many brain regions, the regulation of CP-AMPARs is rather tightly controlled, perhaps due to the fact that excess Ca2+ through these receptors can lead to neurotoxicity [reviewed in (Tanaka et al., 2000; Liu and Zukin, 2007)]. Consistent with this, many interneurons, which normally express CP-AMPARs at synapses, also express a variety of Ca2+ binding proteins. Despite the potentially negative impacts CP-AMPARs may have on neuronal function, they possess unique properties, which may benefit synaptic function, especially under conditions that trigger homeostatic adaptation to inactivity. However, even under homeostatic synaptic plasticity conditions, synaptic expression of CP-AMPARs is not universally observed, which underscores the high degree of regulation and specific circumstances required for CP-AMPARs to function at synapses. Despite the rather specific requirements, homeostatic CP-AMPAR regulation is readily observed (Thiagarajan et al., 2002, 2005; Ju et al., 2004; Goel et al., 2006; Sutton et al., 2006; Aoto et al., 2008; Beique et al., 2011), and depend on specific signals that tap into local protein synthesis in the dendrites (Sutton et al., 2006; Maghsoodi et al., 2008) as well as synaptic trafficking dependent on phosphorylation of the GluA1 subunit (Goel et al., 2011). Furthermore, CP-AMPAR regulation during homeostatic synaptic plasticity is not an isolated observation in vitro, but is also observed in vivo following certain paradigms of sensory deprivation (Goel et al., 2006, 2011). While there are predicted functional consequences of having CP-AMPARs at synapses, how these receptors actually contribute to altering brain function following inactivity requires further studies.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author would like to thank Dr. A. Kirkwood for helpful comments on this manuscript and Ms. E. Petrus for help on the manuscript. This work was support by a NIH grant (R01-EY014882) to Hey-Kyoung Lee.
References
Adesnik, H., and Nicoll, R. A. (2007). Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J. Neurosci. 27, 4598–4602.
Aizenman, C. D., Akerman, C. J., Jensen, K. R., and Cline, H. T. (2003). Visually driven regulation of intrinsic neuronal excitability improves stimulus detection in vivo. Neuron 39, 831–842.
Anggono, V., Clem, R. L., and Huganir, R. L. (2011). PICK1 loss of function occludes homeostatic synaptic scaling. J. Neurosci. 31, 2188–2196.
Ango, F., Prezeau, L., Muller, T., Tu, J. C., Xiao, B., Worley, P. F., Pin, J. P., Bockaert, J., and Fagni, L. (2001). Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein Homer. Nature 411, 962–965.
Aoto, J., Nam, C. I., Poon, M. M., Ting, P., and Chen, L. (2008). Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60, 308–320.
Asrar, S., Zhou, Z., Ren, W., and Jia, Z. (2009). Ca(2+) permeable AMPA receptor induced long-term potentiation requires PI3/MAP kinases but not Ca/CaM-dependent kinase II. PLoS One 4, e4339. doi: 10.1371/journal.pone.0004339
Barria, A., Muller, D., Derkach, V., Griffith, L. C., and Soderling, T. R. (1997). Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276, 2042–2045.
Bear, M. F. (1996). A synaptic basis for memory storage in the cerebral cortex. Proc. Natl. Acad. Sci. U.S.A. 93, 13453–13459.
Bear, M. F., Cooper, L. N., and Ebner, F. F. (1987). A physiological basis for a theory of synapse modification. Science 237, 42–48.
Beique, J. C., Na, Y., Kuhl, D., Worley, P. F., and Huganir, R. L. (2011). Arc-dependent synapse-specific homeostatic plasticity. Proc. Natl. Acad. Sci. U.S.A. 108, 816–821.
Bienenstock, E. L., Cooper, L. N., and Munro, P. W. (1982). Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J. Neurosci. 2, 32–48.
Bochet, P., Audinat, E., Lambolez, B., Crepel, F., Rossier, J., Iino, M., Tsuzuki, K., and Ozawa, S. (1994). Subunit composition at the single-cell level explains functional properties of a glutamate-gated channel. Neuron 12, 383–388.
Bowie, D., and Mayer, M. L. (1995). Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron 15, 453–462.
Brakeman, P. R., Lanahan, A. A., O'Brien, R., Roche, K., Barnes, C. A., Huganir, R. L., and Worley, P. F. (1997). Homer: a protein that selectively binds metabotropic glutamate receptors. Nature 386, 284–288.
Buchel, C., Price, C., Frackowiak, R. S., and Friston, K. (1998). Different activation patterns in the visual cortex of late and congenitally blind subjects. Brain 121 (Pt 3), 409–419.
Burnashev, N., Monyer, H., Seeburg, P. H., and Sakmann, B. (1992). Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8, 189–198.
Burrone, J., O'Byrne, M., and Murthy, V. N. (2002). Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420, 414–418.
Chang, M. C., Park, J. M., Pelkey, K. A., Grabenstatter, H. L., Xu, D., Linden, D. J., Sutula, T. P., McBain, C. J., and Worley, P. F. (2010). Narp regulates homeostatic scaling of excitatory synapses on parvalbumin-expressing interneurons. Nat. Neurosci. 13, 1090–1097.
Chao, S. Z., Lu, W., Lee, H. K., Huganir, R. L., and Wolf, M. E. (2002). D(1) dopamine receptor stimulation increases GluR1 phosphorylation in postnatal nucleus accumbens cultures. J. Neurochem. 81, 984–992.
Chung, H. J., Xia, J., Scannevin, R. H., Zhang, X., and Huganir, R. L. (2000). Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J. Neurosci. 20, 7258–7267.
Clem, R. L., Anggono, V., and Huganir, R. L. (2010). PICK1 regulates incorporation of calcium-permeable AMPA receptors during cortical synaptic strengthening. J. Neurosci. 30, 6360–6366.
Collingridge, G. L., Olsen, R. W., Peters, J., and Spedding, M. (2009). A nomenclature for ligand-gated ion channels. Neuropharmacology 56, 2–5.
Correia, S. S., Bassani, S., Brown, T. C., Lise, M. F., Backos, D. S., El-Husseini, A., Passafaro, M., and Esteban, J. A. (2008). Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat. Neurosci. 11, 457–466.
Cummings, J. A., Mulkey, R. M., Nicoll, R. A., and Malenka, R. C. (1996). Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron 16, 825–833.
Desai, N. S., Cudmore, R. H., Nelson, S. B., and Turrigiano, G. G. (2002). Critical periods for experience-dependent synaptic scaling in visual cortex. Nat. Neurosci. 5, 783–789.
Desai, N. S., Rutherford, L. C., and Turrigiano, G. G. (1999). Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat. Neurosci. 2, 515–520.
Donevan, S. D., and Rogawski, M. A. (1995). Intracellular polyamines mediate inward rectification of Ca(2+)-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Proc. Natl. Acad. Sci. U.S.A. 92, 9298–9302.
Echegoyen, J., Neu, A., Graber, K. D., and Soltesz, I. (2007). Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence. PLoS One 2, e700. doi: 10.1371/journal.pone.0000700
Ehlers, M. D. (2000). Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28, 511–525.
Esteban, J. A., Shi, S. H., Wilson, C., Nuriya, M., Huganir, R. L., and Malinow, R. (2003). PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 6, 136–143.
Fortin, D. A., Davare, M. A., Srivastava, T., Brady, J. D., Nygaard, S., Derkach, V. A., and Soderling, T. R. (2010). Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J. Neurosci. 30, 11565–11575.
Gainey, M. A., Hurvitz-Wolff, J. R., Lambo, M. E., and Turrigiano, G. G. (2009). Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J. Neurosci. 29, 6479–6489.
Gao, M., Sossa, K., Song, L., Errington, L., Cummings, L., Hwang, H., Kuhl, D., Worley, P., and Lee, H. K. (2010). A specific requirement of Arc/Arg3.1 for visual experience-induced homeostatic synaptic plasticity in mouse primary visual cortex. J. Neurosci. 30, 7168–7178.
Gardner, S. M., Takamiya, K., Xia, J., Suh, J. G., Johnson, R., Yu, S., and Huganir, R. L. (2005). Calcium-permeable AMPA receptor plasticity is mediated by subunit-specific interactions with PICK1 and NSF. Neuron 45, 903–915.
Goel, A., Jiang, B., Xu, L. W., Song, L., Kirkwood, A., and Lee, H. K. (2006). Cross-modal regulation of synaptic AMPA receptors in primary sensory cortices by visual experience. Nat. Neurosci. 9, 1001–1003.
Goel, A., and Lee, H. K. (2007). Persistence of experience-induced homeostatic synaptic plasticity through adulthood in superficial layers of mouse visual cortex. J. Neurosci. 27, 6692–6700.
Goel, A., Xu, L. W., Snyder, K. P., Song, L., Goenaga-Vazquez, Y., Megill, A., Takamiya, K., Huganir, R. L., and Lee, H. K. (2011). Phosphorylation of AMPA receptors is required for sensory deprivation-induced homeostatic synaptic plasticity. PLoS One 6, e18264. doi: 10.1371/journal.pone.0018264
Goold, C. P., and Nicoll, R. A. (2010). Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron 68, 512–528.
Gray, E. E., Fink, A. E., Sarinana, J., Vissel, B., and O'Dell, T. J. (2007). Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. J. Neurophysiol. 98, 2488–2492.
Groth, R. D., Lindskog, M., Thiagarajan, T. C., Li, L., and Tsien, R. W. (2011). Beta Ca2+/CaM-dependent kinase type II triggers upregulation of GluA1 to coordinate adaptation to synaptic inactivity in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 108, 828–833.
Guire, E. S., Oh, M. C., Soderling, T. R., and Derkach, V. A. (2008). Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J. Neurosci. 28, 6000–6009.
Hayashi, Y., Shi, S. H., Esteban, J. A., Piccini, A., Poncer, J. C., and Malinow, R. (2000). Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287, 2262–2267.
He, K., Goel, A., Ciarkowski, C. E., Song, L., and Lee, H.-K. (2011). Brain area specific regulation of synaptic AMPA receptors by phosphorylation. Commun. Integr. Biol. 4, 569–572.
He, K., Song, L., Cummings, L. W., Goldman, J., Huganir, R. L., and Lee, H. K. (2009). Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1-S845 phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106, 20033–20038.
Hollmann, M., Hartley, M., and Heinemann, S. (1991). Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science 252, 851–853.
Hu, H., Real, E., Takamiya, K., Kang, M. G., Ledoux, J., Huganir, R. L., and Malinow, R. (2007). Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell 131, 160–173.
Hu, J. H., Park, J. M., Park, S., Xiao, B., Dehoff, M. H., Kim, S., Hayashi, T., Schwarz, M. K., Huganir, R. L., Seeburg, P. H., Linden, D. J., and Worley, P. F. (2010). Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron 68, 1128–1142.
Ibata, K., Sun, Q., and Turrigiano, G. G. (2008). Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 57, 819–826.
Isa, T., Iino, M., and Ozawa, S. (1996). Spermine blocks synaptic transmission mediated by Ca(2+)-permeable AMPA receptors. Neuroreport 7, 689–692.
Isaac, J. T., Ashby, M., and McBain, C. J. (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871.
Jia, Z., Agopyan, N., Miu, P., Xiong, Z., Henderson, J., Gerlai, R., Taverna, F. A., Velumian, A., Macdonald, J., Carlen, P., Abramow-Newerly, W., and Roder, J. (1996). Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 17, 945–956.
Ju, W., Morishita, W., Tsui, J., Gaietta, G., Deerinck, T. J., Adams, S. R., Garner, C. C., Tsien, R. Y., Ellisman, M. H., and Malenka, R. C. (2004). Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat. Neurosci. 7, 244–253.
Kelly, L., Farrant, M., and Cull-Candy, S. G. (2009). Synaptic mGluR activation drives plasticity of calcium-permeable AMPA receptors. Nat. Neurosci. 12, 593–601.
Kessels, H. W., Kopec, C. D., Klein, M. E., and Malinow, R. (2009). Roles of stargazin and phosphorylation in the control of AMPA receptor subcellular distribution. Nat. Neurosci. 12, 888–896.
Kim, J., and Alger, B. E. (2010). Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat. Neurosci. 13, 592–600.
Kim, J., and Tsien, R. W. (2008). Synapse-specific adaptations to inactivity in hippocampal circuits achieve homeostatic gain control while dampening network reverberation. Neuron 58, 925–937.
Kumar, S. S., Bacci, A., Kharazia, V., and Huguenard, J. R. (2002). A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J. Neurosci. 22, 3005–3015.
Lee, H. K., Barbarosie, M., Kameyama, K., Bear, M. F., and Huganir, R. L. (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959.
Lee, H. K., and Huganir, R. L. (2008). “AMPA receptor regulation and the reversal of synaptic plasticity – LTP, LTD, depotentiation, and dedepression,” in Learning and Memory: A Comprehensive Reference, ed J. D. Sweatt (Elsevier Press).
Lee, H. K., Kameyama, K., Huganir, R. L., and Bear, M. F. (1998). NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21, 1151–1162.
Lee, H. K., and Kirkwood, A. (2011). AMPA receptor regulation during synaptic plasticity in hippocampus and neocortex. Semin. Cell Dev. Biol. 22, 514–520.
Lee, H. K., Takamiya, K., Han, J. S., Man, H., Kim, C. H., Rumbaugh, G., Yu, S., Ding, L., He, C., Petralia, R. S., Wenthold, R. J., Gallagher, M., and Huganir, R. L. (2003). Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112, 631–643.
Lee, H. K., Takamiya, K., He, K., Song, L., and Huganir, R. L. (2010). Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J. Neurophysiol. 103, 479–489.
Lee, S. H., Simonetta, A., and Sheng, M. (2004). Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron 43, 221–236.
Lindskog, M., Li, L., Groth, R. D., Poburko, D., Thiagarajan, T. C., Han, X., and Tsien, R. W. (2010). Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc. Natl. Acad. Sci. U.S.A. 107, 21806–21811.
Lissin, D. V., Gomperts, S. N., Carroll, R. C., Christine, C. W., Kalman, D., Kitamura, M., Hardy, S., Nicoll, R. A., Malenka, R. C., and Von Zastrow, M. (1998). Activity differentially regulates the surface expression of synaptic AMPA and NMDA glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 95, 7097–7102.
Liu, B., Liao, M., Mielke, J. G., Ning, K., Chen, Y., Li, L., El-Hayek, Y. H., Gomez, E., Zukin, R. S., Fehlings, M. G., and Wan, Q. (2006). Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J. Neurosci. 26, 5309–5319.
Liu, S. J., and Cull-Candy, S. G. (2002). Activity-dependent change in AMPA receptor properties in cerebellar stellate cells. J. Neurosci. 22, 3881–3889.
Liu, S. J., and Cull-Candy, S. G. (2005). Subunit interaction with PICK and GRIP controls Ca2+ permeability of AMPARs at cerebellar synapses. Nat. Neurosci. 8, 768–775.
Liu, S. J., and Zukin, R. S. (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134.
Liu, S. Q., and Cull-Candy, S. G. (2000). Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature 405, 454–458.
Lu, Y., Allen, M., Halt, A. R., Weisenhaus, M., Dallapiazza, R. F., Hall, D. D., Usachev, Y. M., Mcknight, G. S., and Hell, J. W. (2007). Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 26, 4879–4890.
Liu, Y., Formisano, L., Savtchouk, I., Takayasu, Y., Szabo, G., Zukin, R. S., and Liu, S. J. (2010). A single fear-inducing stimulus induces a transcription-dependent switch in synaptic AMPAR phenotype. Nat. Neurosci. 13, 223–231.
Lu, W., Shi, Y., Jackson, A. C., Bjorgan, K., During, M. J., Sprengel, R., Seeburg, P. H., and Nicoll, R. A. (2009). Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62, 254–268.
Maffei, A., Nelson, S. B., and Turrigiano, G. G. (2004). Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat. Neurosci. 7, 1353–1359.
Maffei, A., and Turrigiano, G. G. (2008). Multiple modes of network homeostasis in visual cortical layer 2/3. J. Neurosci. 28, 4377–4384.
Maghsoodi, B., Poon, M. M., Nam, C. I., Aoto, J., Ting, P., and Chen, L. (2008). Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 105, 16015–16020.
Malenka, R. C., Lancaster, B., and Zucker, R. S. (1992). Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron 9, 121–128.
Malinow, R., Schulman, H., and Tsien, R. W. (1989). Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245, 862–866.
Man, H. Y., Sekine-Aizawa, Y., and Huganir, R. L. (2007). Regulation of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc. Natl. Acad. Sci. U.S.A. 104, 3579–3584.
Matsuda, S., Launey, T., Mikawa, S., and Hirai, H. (2000). Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J. 19, 2765–2774.
Matsuda, S., Mikawa, S., and Hirai, H. (1999). Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J. Neurochem. 73, 1765–1768.
McBain, C. J., and Dingledine, R. (1993). Heterogeneity of synaptic glutamate receptors on CA3 stratum radiatum interneurones of rat hippocampus. J. Physiol. 462, 373–392.
Meng, Y., Zhang, Y., and Jia, Z. (2003). Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron 39, 163–176.
Miller, K. D., and Mackay, D. J. C. (1994). The role of constraints in Hebbian learning. Neural Comput. 6, 100–126.
O'Brien, R. J., Kamboj, S., Ehlers, M. D., Rosen, K. R., Fischbach, G. D., and Huganir, R. L. (1998). Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21, 1067–1078.
Oh, M. C., Derkach, V. A., Guire, E. S., and Soderling, T. R. (2006). Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem. 281, 752–758.
Otis, T. S., Raman, I. M., and Trussell, L. O. (1995). AMPA receptors with high Ca2+ permeability mediate synaptic transmission in the avian auditory pathway. J. Physiol. 482 (Pt 2), 309–315.
Petrus, E., Anguh, T. T., Pho, H., Lee, A., Gammon, N., and Lee, H. K. (2011). Developmental switch in the polarity of experience-dependent synaptic changes in layer 6 of mouse visual cortex. J. Neurophysiol. 106, 2499–2505.
Plant, K., Pelkey, K. A., Bortolotto, Z. A., Morita, D., Terashima, A., McBain, C. J., Collingridge, G. L., and Isaac, J. T. (2006). Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604.
Poon, M. M., and Chen, L. (2008). Retinoic acid-gated sequence-specific translational control by RARalpha. Proc. Natl. Acad. Sci. U.S.A. 105, 20303–20308.
Pratt, K. G., and Aizenman, C. D. (2007). Homeostatic regulation of intrinsic excitability and synaptic transmission in a developing visual circuit. J. Neurosci. 27, 8268–8277.
Quinlan, E. M., Olstein, D. H., and Bear, M. F. (1999a). Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc. Natl. Acad. Sci. U.S.A. 96, 12876–12880.
Quinlan, E. M., Philpot, B. D., Huganir, R. L., and Bear, M. F. (1999b). Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat. Neurosci. 2, 352–357.
Roche, K. W., O'Brien, R. J., Mammen, A. L., Bernhardt, J., and Huganir, R. L. (1996). Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16, 1179–1188.
Rozov, A., and Burnashev, N. (1999). Polyamine-dependent facilitation of postsynaptic AMPA receptors counteracts paired-pulse depression. Nature 401, 594–598.
Rozov, A., Zilberter, Y., Wollmuth, L. P., and Burnashev, N. (1998). Facilitation of currents through rat Ca2+-permeable AMPA receptor channels by activity-dependent relief from polyamine block. J. Physiol. 511 (Pt 2), 361–377.
Rutherford, L. C., Nelson, S. B., and Turrigiano, G. G. (1998). BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron 21, 521–530.
Sadato, N., Okada, T., Honda, M., and Yonekura, Y. (2002). Critical period for cross-modal plasticity in blind humans: a functional MRI study. Neuroimage 16, 389–400.
Sadato, N., Pascual-Leone, A., Grafman, J., Ibanez, V., Deiber, M. P., Dold, G., and Hallett, M. (1996). Activation of the primary visual cortex by Braille reading in blind subjects. Nature 380, 526–528.
Seidenman, K. J., Steinberg, J. P., Huganir, R., and Malinow, R. (2003). Glutamate receptor subunit 2 Serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J. Neurosci. 23, 9220–9228.
Seol, G. H., Ziburkus, J., Huang, S., Song, L., Kim, I. T., Takamiya, K., Huganir, R. L., Lee, H. K., and Kirkwood, A. (2007). Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron 55, 919–929.
Shepherd, J. D., Rumbaugh, G., Wu, J., Chowdhury, S., Plath, N., Kuhl, D., Huganir, R. L., and Worley, P. F. (2006). Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52, 475–484.
Shi, S., Hayashi, Y., Esteban, J. A., and Malinow, R. (2001). Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105, 331–343.
Shin, J., Shen, F., and Huguenard, J. R. (2005). Polyamines modulate AMPA receptor-dependent synaptic responses in immature layer v pyramidal neurons. J. Neurophysiol. 93, 2634–2643.
Silva, A. J., Stevens, C. F., Tonegawa, S., and Wang, Y. (1992). Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science 257, 201–206.
Sommer, B., Kohler, M., Sprengel, R., and Seeburg, P. H. (1991). RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67, 11–19.
Song, I., and Huganir, R. L. (2002). Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 25, 578–588.
Spaethling, J. M., Klein, D. M., Singh, P., and Meaney, D. F. (2008). Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J. Neurotrauma 25, 1207–1216.
Stubblefield, E. A., and Benke, T. A. (2010). Distinct AMPA-type glutamatergic synapses in developing rat CA1 hippocampus. J. Neurophysiol. 104, 1899–1912.
Sun, X., Zhao, Y., and Wolf, M. E. (2005). Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J. Neurosci. 25, 7342–7351.
Sutton, M. A., Ito, H. T., Cressy, P., Kempf, C., Woo, J. C., and Schuman, E. M. (2006). Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125, 785–799.
Tanaka, H., Grooms, S. Y., Bennett, M. V., and Zukin, R. S. (2000). The AMPAR subunit GluR2: still front and center-stage. Brain Res. 886, 190–207.
Terashima, A., Cotton, L., Dev, K. K., Meyer, G., Zaman, S., Duprat, F., Henley, J. M., Collingridge, G. L., and Isaac, J. T. (2004). Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. J. Neurosci. 24, 5381–5390.
Terashima, A., Pelkey, K. A., Rah, J. C., Suh, Y. H., Roche, K. W., Collingridge, G. L., McBain, C. J., and Isaac, J. T. (2008). An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron 57, 872–882.
Thiagarajan, T. C., Lindskog, M., and Tsien, R. W. (2005). Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47, 725–737.
Thiagarajan, T. C., Piedras-Renteria, E. S., and Tsien, R. W. (2002). alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36, 1103–1114.
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., Hansen, K. B., Yuan, H., Myers, S. J., Dingledine, R., and Sibley, D. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496.
Turrigiano, G. G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435.
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896.
Turrigiano, G. G., and Nelson, S. B. (2004). Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 5, 97–107.
Volk, L., Kim, C. H., Takamiya, K., Yu, Y., and Huganir, R. L. (2010). Developmental regulation of protein interacting with C kinase 1 (PICK1) function in hippocampal synaptic plasticity and learning. Proc. Natl. Acad. Sci. U.S.A. 107, 21784–21789.
Wang, Z., Edwards, J. G., Riley, N., Provance, D. W. Jr., Karcher, R., Li, X. D., Davison, I. G., Ikebe, M., Mercer, J. A., Kauer, J. A., and Ehlers, M. D. (2008). Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell 135, 535–548.
Washburn, M. S., Numberger, M., Zhang, S., and Dingledine, R. (1997). Differential dependence on GluR2 expression of three characteristic features of AMPA receptors. J. Neurosci. 17, 9393–9406.
Wenthold, R. J., Petralia, R. S., Blahos, J. II, and Niedzielski, A. S. (1996). Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J. Neurosci. 16, 1982–1989.
Wierenga, C. J., Ibata, K., and Turrigiano, G. G. (2005). Postsynaptic expression of homeostatic plasticity at neocortical synapses. J. Neurosci. 25, 2895–2905.
Wierenga, C. J., Walsh, M. F., and Turrigiano, G. G. (2006). Temporal regulation of the expression locus of homeostatic plasticity. J. Neurophysiol. 96, 2127–2133.
Wiltgen, B. J., Royle, G. A., Gray, E. E., Abdipranoto, A., Thangthaeng, N., Jacobs, N., Saab, F., Tonegawa, S., Heinemann, S. F., O'Dell, T. J., Fanselow, M. S., and Vissel, B. (2010). A role for calcium-permeable AMPA receptors in synaptic plasticity and learning. |PLoS One| 5. doi: 10.1371/journal.pone.0012818
Keywords: homeostasis, synaptic scaling, CP-AMPAR, GluA1, activity-dependent, inactivity
Citation: Lee H (2012) Ca2+-permeable AMPA receptors in homeostatic synaptic plasticity. Front. Mol. Neurosci. 5:17. doi: 10.3389/fnmol.2012.00017
Received: 02 September 2011; Accepted: 02 February 2012;
Published online: 13 February 2012.
Edited by:
R. Suzanne Zukin, Albert Einstein College of Medicine, USAReviewed by:
Kei Cho, University of Bristol, UKMatthias Kneussel, Zentrum für Molekulare Neurobiologie Hamburg, Germany
Copyright: © 2012 Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hey-Kyoung Lee, The Solomon H. Snyder Department of Neuroscience, The Zanvyl Krieger Mind/Brain Institute, Johns Hopkins University, 3400 N. Charles Street, Dunning Hall Room 348, Baltimore, MD 21218, USA. e-mail: heykyounglee@jhu.edu