
- 1 Institute for Advanced Biosciences, Keio University, Tokyo, Japan
- 2 Systems Biology Program, Graduate School of Media and Governance, Keio University, Tokyo, Japan
- 3 Department of Molecular and Cellular Bioanalysis, Graduate School of Pharmaceutical Sciences, Kyoto University, Kyoto, Japan
Our recently developed rice proteogenomics database (OryzaPG-DB) is the first sustainable resource for rice shotgun-based proteogenomics, providing information on peptides identified in rice protein digested peptides measured by means of liquid chromatography–tandem mass spectrometry (LC–MS/MS), and mapping of the peptides to their genomic origins and the genomic novelty of each peptide. The sequences of the peptides, proteins, cDNAs and genes, and the gene annotations are available for download in FASTA and GFF3 formats, respectively. Further, an annotated visualization of the gene models, corresponding peptides, and genomic novelty is available for each gene, and MS/MS spectra are available for each peptide. In this article, we discuss the utilization of OryzaPG-DB and report on its development, recent content expansions, and newly added features in the current version (OryzaPG-DB v1.1).
Introduction
The rapid improvement of analytical instruments for biological research has resulted in the accumulation of enormous amounts of both raw and analyzed data (Helmy et al., 2012). Such huge amounts of data contain valuable biological information, which can only be uncovered by performing appropriate computational analysis using proper bioinformatics tools. Since this data is very rich, applying different kinds of computational analysis or using different bioinformatics tools can lead to different discoveries according to the goal of the research. Therefore, such biological data are generally deposited in biological databases, which are collections or libraries of biological data sorted and organized in a way that allows easy access, management, and updating in a sustainable format so that interested scientists can have consistent access over a prolonged period (Birney and Clamp, 2004). Data sustainability and ease of use are the main benefits of storing biological data in databases. Further, long-term projects, e.g., the human genome project, require extendable and sustainable databases that can store data from different phases of the projects (McGourty, 1989; Hyman, 2011). Almost all kinds of biological data are stored in databases including, for example, sequence data, structural data, interaction data, and proteogenomic data (Birney and Clamp, 2004).
Proteogenomics is an alliance between proteomics and genomics, and aims mainly to use proteomics data and technologies for identifying novel genomic features, such as novel un-annotated genes, and for improving, correcting, or confirming the structural and functional genome annotation (Ansong et al., 2008). However, it is also applicable to deeper and wider goals, such as understanding the mechanisms of environmental adaptation between different species, discovery of biomarkers, and identification of the targets of antibodies (Huang et al., 2004; Sigdel and Sarwal, 2008; de Groot et al., 2009). In plants, particularly, Arabidopsis received most of the proteogenomic efforts (Baerenfaller et al., 2008; Castellana et al., 2008). However, proteogenomic analysis was performed for several other plants, such as rice, and several major plant pathogens (Bindschedler et al., 2009, 2011; Bringans et al., 2009; Helmy et al., 2011).
Proteogenomic analyses usually rely on high-throughput genomic and proteomic data, such as genome sequencing and liquid chromatography–tandem mass spectrometry (LC–MS/MS) data. The scheme of proteogenomic analysis can differ from project to project depending on the available genomic and proteomic data, the status of the genome annotation of the organism in question (annotated or newly sequenced) and the availability of sufficient informatics tools and computational power to deal with such large amounts of data (Helmy et al., 2012). However, three steps are always shared among different proteogenomic projects: (1) Mapping proteomic data (peptide sequences derived from the MS/MS analysis) to the genome. (2) Evaluating the genomic novelty of the proteomic data. (3) Updating or confirming the current genome annotation by integrating the newly discovered genomic information into the current genome annotation (Ansong et al., 2008; Helmy et al., 2012). These considerations distinguish proteogenomics databases from other types of biological databases.
Thus, a proteogenomic database is a biological database that holds genomic and proteomic sequence data, together with the mapping of the proteomic data to the genome and the genome annotation. It can also store other information concerning the organism’s genes and proteins, such as mRNA and cDNA sequences and biological or biochemical functions. Therefore, proteogenomic databases require special design and implantation (Helmy et al., 2011).
The Rice Proteogenomic Database (OryzaPG-DB)
The Rice Proteogenomic Database (OryzaPG-DB) incorporates the genomic features of experimental shotgun proteomics data for rice (Oryza sativa; Helmy et al., 2011). It was developed using whole proteome data of rice undifferentiated cultured cells, generated from 27 nanoLC–MS/MS runs on a hybrid ion trap–orbitrap mass spectrometer, and the rice genome annotation database at Michigan State University (MSU), which offers rice protein, cDNA, transcript and genome databases, and rice genome annotation (Ouyang et al., 2007). The MS/MS spectra are filtered using our previously described method (Ravichandran et al., 2009) then searched against the above-mentioned databases in the order mentioned, using Mascot (v.2.3; Perkins et al., 1999) with peptide acceptance criteria ≥99.9%. The lists of identified peptides are merged and filtered to remove all peptides shorter than seven amino acids (Choudhary et al., 2001). Then, the list of accepted peptides is filtered again to remove redundancy. Therefore, the final list contains only accurate and unique peptides (here, unique means a unique combination of sequence and modifications).
The identified peptides were used to perform proteogenomic analysis of the rice genome by mapping the identified peptides to their genomic origins. Mapping was performed through alignment of peptides identified from the protein, cDNA, and transcript databases to the un-spliced-genomic mRNA of the corresponding genes. Peptides identified from the genome database were aligned to the corresponding chromosome then mapped to the genes using the alignment coordinates. Next, the alignment results were converted to GFF3 format and the peptide’s GFF3 line was appending to the annotation of the corresponding gene and new GFF3 files were created. The new files are submitted to the ProteoGenomic Features Evaluator (PGFeval) software tool to evaluate the genomic novelty of each peptide (Helmy et al., 2011). PGFeval analysis provided 51 novel genomic features in 40 rice genes. Further, PGFeval exported a visualized annotation file for each gene in PNG image format. Finally, a tailored proteogenomic database was designed and implemented to host all this data, with the capability of expansion to host data from future phases of rice proteogenomics analysis, and to make the data publicly available for interested scientists in the rice biology community.
OryzaPG-DB Design
Since proteogenomics data has a complex structure, as described above, a tailored database design is required for a proteogenomic database. Further, when we designed OryzaPG-DB, we decided to make the design as generic as possible, in order to help other researchers working on the design of databases suitable for proteogenomic data (Helmy et al., 2011). In the original design (Figure 1A), the main entity of the database is the gene. Then, information on the corresponding protein(s), LC–MS/MS measured peptide(s), cDNA(s), mRNA(s), mapping information, and updated gene annotation are attached to this entity. In addition, several files are attached to each gene, including protein, peptide, cDNA and mRNA sequences, gene annotation in GFF3 format, and visualization of the gene annotation (PGFeval output).
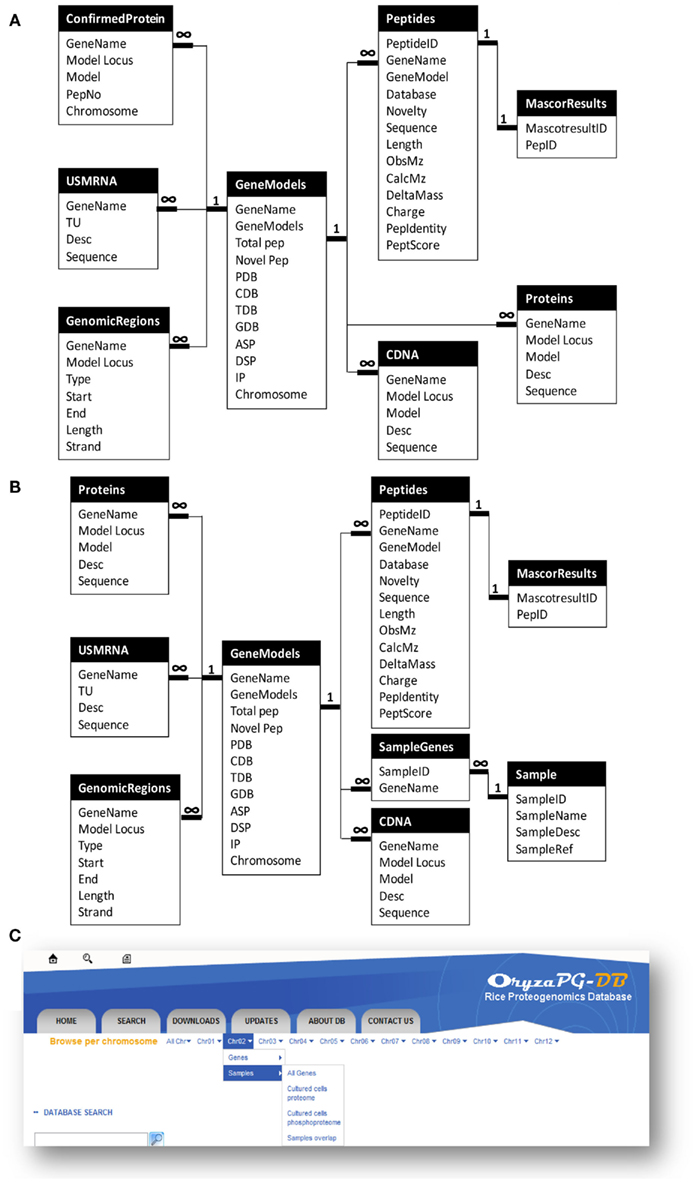
Figure 1. Updated design of OryzaPG-DB and browsing options. (A) OryzaPG-DB original database design adopted from Helmy et al. (2011). (B) OryzaPG-DB (v1.1) database design. (C) Updated database browsing options.
Updates on OryzaPG-DB Design and Development
The original version of OryzaPG-DB has been publicly available online since 2010 and was officially opened in 2011. Since that time, several developments have been added to the original version. Here, we review the recent updates and newly added features in the current version (OryzaPG-DB v1.1).
OryzaPG-DB Updated Database Design
The above design of OryzaPG-DB was suitable for hosting the rice proteogenomic data available at the database launch. However, we were aware that this design would need to be updated to accommodate new proteomes from other samples/organs or other types of analysis, such as phosphoproteome analysis, as mentioned in the future work section of the original publication on OryzaPG-DB (Helmy et al., 2011). Thus, the database design of the current version of OryzaPG-DB (v1.1) has been updated to include information about the experimental sample (Figure 1B).
Adding the ability to include sample information makes the design sufficiently generic to host all proteogenomic data of an organism while retaining information about the source of each peptide and the expression organ of each gene/protein, as well as providing the basis for several other grouping features that can be useful in performing functional analysis, organ-specific analysis and/or inter-organ comparisons. It is now possible, with the updated design, to select peptides identified from a certain sample and/or organ and/or analysis and/or database, with the corresponding genes. For instance, in a proteogenomic study with extensive sampling, such as the Arabidopsis proteogenomic study (Baerenfaller et al., 2008), it is important to have an easy way to create lists of genes/peptides identified from various combinations of sample, organ, and condition for comparison in order to find organ or life stage specific biomarkers. Such a task is now straightforward with the updated design of OryzaPG-DB v1.1.
OryzaPG-DB New Browsing Options
The original version of OryzaPG-DB had the “browse per chromosome” feature that allows fetching data of all genes, updated genes, proteins, cDNAs, or transcripts of all chromosomes or a single chromosome. However, updating the database design consequently requires updating the “browse per chromosome” options to add sample level options. In the current version, we added the sample level to the “browse per chromosome” menu. This allows fetching data of all genes, genes identified in a particular sample/organ/analysis, or genes overlapping samples for all chromosomes or single chromosome (Figure 1C). With the new “Save to File” option (see below), it is possible to save the browsing/search results to a CSV file for download, so that creating lists of genes, proteins, cDNAs, transcripts or peptides per sample, or those overlapping samples for all chromosomes or a single chromosome, has become a single-click task.
OryzaPG-DB New and Updated Application Programming Interfaces
An application programming interface (API) is an implemented interface that allows other programs or the operating system to interact with the whole program or particular parts or functions of the program (Tulach, 2008). To increase the usability of the data stored in OryzaPG-DB, we provided six URL APIs to help researchers fetch OryzaPG-DB dynamically with scripts or integrate OryzaPG-DB data into their applications. The URL APIs of OryzaPG-DBv1.1 contains seven APIs (Table 1), which are updated versions of the six APIs of the original OryzaPG-DB plus a new API for samples. The seven APIs are briefly described.

Table 1. OryzaPG-DB v1.1 APIs.
1. Genes API: allows users to retrieve the gene information for a particular gene, all genes in a particular chromosome or all chromosomes.
2. Updated genes API: allows users to retrieve the gene(s) with updated annotations and novel genomic features; per gene, all updated genes in one chromosome or all updated genes.
3. Proteins API: allows users to retrieve the protein information for a particular gene, all genes in a particular chromosome or all genes. The result can be shown in tabular view or in FASTA format.
4. cDNAs API: allows users to retrieve the cDNA information for a particular gene, all genes in a particular chromosome or all genes. The result can be shown in tabular view or in FASTA format.
5. Transcripts API: allows users to retrieve the transcript (un-spliced-genomic mRNA) information for a particular gene, all genes in a particular chromosome or all genes. The result can be shown in tabular view or in FASTA format.
6. Peptides API: allows users to retrieve peptides information for peptides identified from a particular gene or gene products (protein, cDNA, or mRNA), all genes in a particular chromosome or all genes covered by our analysis. The result can be shown in tabular view or in FASTA format. Also, a special parameter can be used to select novel peptides only instead of all peptides.
7. Samples API (new): allows users to retrieve the genes identified in a particular sample, all samples or overlapping samples for a particular chromosome or all chromosomes.
For all APIs, a new option was added that allows saving the API execution result to a CSV file by setting the to_file parameter to true (Table 1).
OryzaPG-DB Data Expansion
A key feature of biological databases is their expandability, so that the database can expand to host more data related to the original content when such data becomes available. The OryzaPG-DB original and updated database designs both aim to host rice proteogenomics data in an expandable and sustainable way, as described above. In the current version of OryzaPG-DB v1.1, the rice proteogenomic data derived from the proteogenomic analysis of the original 27 nanoLC–MS/MS runs of cultured rice cells were recently expanded to 61 runs in total, demonstrating the sustainability of OryzaPG-DB (Table 2). The updated design of the database, by adding sample information, is able to distinguish peptides and genes identified in each sample or those identified in both samples (Figure 1C). The proteogenomic analysis of the newly added sample covers 845 new genes which were not present in the original OryzaPG-DB coverage, and adds new peptides and/or novel genomic features to 914 of the originally existing genes, expanding the database coverage to 3973 genes. The numbers of genes with novel peptides and genes with novel genomic features are increased from 119 and 40 to 160 and 62, respectively.
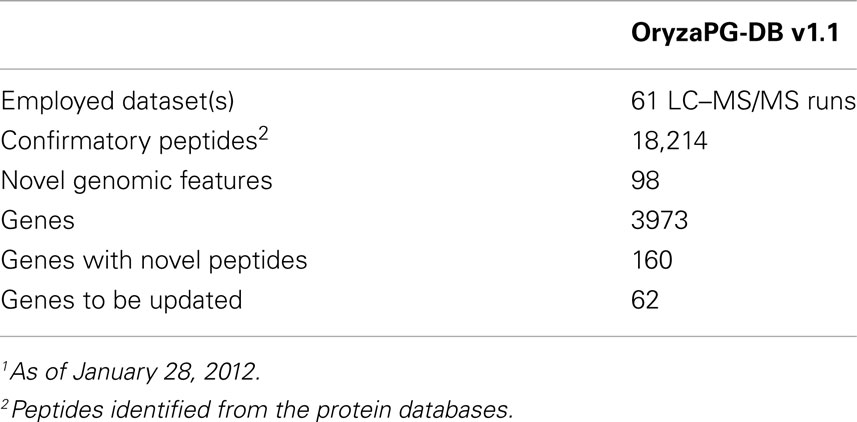
Table 2. OryzaPG-DB current status1.
OryzaPG-DB New and Updated Features
The updated database design and the recent data expansion required the development of several new features that take advantage of the new developments and improve data fetching. In this section we present a brief description of some of the new features, which are mainly related to database search and content download.
1. Adding sample to the advanced search: this option makes use of the new database design, in which one can limit the search to be within the genes/peptides of a certain sample or within the genes/peptides identified across all samples.
2. Adding new peptide novelty categories to the advanced search: the advanced search form in the original version of OryzaPG-DB allows the user to limit the search results to show genes with particular type of peptide novelty, e.g., showing genes with intronic peptides only. Since we have adopted the newer version of PGFeval (Helmy et al., 2011), the new form includes two new peptide novelty categories, 3′UTR and, 5′UTR, indicating peptides identified from the 3′UTR and 5′UTR, respectively.
3. Adding Save to File option to the database browsing results: the database browsing feature allow fetching data by sample or genes for all chromosomes or a single chromosome (see above). The retrieved data is displayed per gene with links to all details related to this gene such as protein, cDNA, mRNA, peptides, and annotation. Consequently, the data is usually huge, so it is displayed in pages of 50 genes per page. In the original version of OryzaPG-DB, there was no way to display or save all retrieved data through database browsing. Therefore, we developed the Save to File option that appears in all database browsing results, and allows saving all the data to a downloadable file in CSV file format.
4. Adding Save Search Results option to the database searching results: similar to the database browsing feature, the database searching feature can display huge amounts of data and there was no way to display all this data or save it. Therefore, we added the Save Search Results option that allows saving all database searching results to a downloadable file in CSV file format in a similar way and format to the Save to File option described above.
OryzaPG-DB Utility and Availability
OryzaPG-DB is the first database that provides a sustainable resource for proteogenomic analysis of an economically important crop that includes genes, gene products (mRNA, cDNA, and protein), experimental expression evidence (MS/MS peptide spectra), and mapping of the peptides to their genomic origin. Further, the sequences of each gene and its products and the gene annotation are available in GFF3 format and can be graphically visualized. Such data can be of great value for plant biologists in general and rice biologists in particular. Furthermore, the generic OryzaPG-DB database design provides a template that should be applicable to data from other similar projects/analyses. The new or updated features are discussed in detail above.
OryzaPG-DB is freely available online at the servers of the Institute for Advanced Biosciences (IAB), Keio University, Japan at http://oryzapg.iab.keio.ac.jp/.
OryzaPG-DB Future Work
The current version of OryzaPG-DB includes several developments and features that were foreshadowed in the future work section of our original article describing OryzaPG-DB (Helmy et al., 2011), such as data expansion, adding sample level information, and updating the advanced search parameters, together with features that not mentioned then, but which we thought would improve the utility of the database such as save to file and save search results options. Future developments are expected to focus mainly on data expansion and proteogenomic analysis of newly added data. In addition, more informatics updates will be included, such as offering downloadable Perl script that will be useful for automation of OryzaPG-DB data acquisition through the available URL APIs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank members of the proteomics group in our institute for their contributions. This study is funded by Yamagata Prefecture and Tsuruoka City grants to Keio University, as well as by the Egyptian Ministry of Higher Education, the Egyptian Bureau of Culture, Science and Education – Tokyo, and JSPS Grants-in-Aid for Scientific Research (No. 236172) to Mohamed Helmy, Science and Technology Incubation Program in Advanced Regions from Japan Science and Technology Agency and JSPS Grants-in-Aid for Scientific Research (No. 21310129) to Yasushi Ishihama.
References
Ansong, C., Purvine, S. O., Adkins, J. N., Lipton, M. S., and Smith, R. D. (2008). Proteogenomics: needs and roles to be filled by proteomics in genome annotation. Brief. Funct. Genomic. Proteomic. 7, 50–62.
Baerenfaller, K., Grossmann, J., Grobei, M. A., Hull, R., Hirsch-Hoffmann, M., Yalovsky, S., Zimmermann, P., Grossniklaus, U., Gruissem, W., and Baginsky, S. (2008). Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320, 938–941.
Bindschedler, L. V., Burgis, T. A., Mills, D. J., Ho, J. T., Cramer, R., and Spanu, P. D. (2009). In planta proteomics and proteogenomics of the biotrophic barley fungal pathogen Blumeria graminis f. sp. hordei. Mol. Cell Proteomics 8, 2368–2381.
Bindschedler, L. V., Mcguffin, L. J., Burgis, T. A., Spanu, P. D., and Cramer, R. (2011). Proteogenomics and in silico structural and functional annotation of the barley powdery mildew Blumeria graminis f. sp. hordei. Methods 54, 432–441.
Birney, E., and Clamp, M. (2004). Biological database design and implementation. Brief. Bioinformatics 5, 31–38.
Bringans, S., Hane, J. K., Casey, T., Tan, K. C., Lipscombe, R., Solomon, P. S., and Oliver, R. P. (2009). Deep proteogenomics; high throughput gene validation by multidimensional liquid chromatography and mass spectrometry of proteins from the fungal wheat pathogen Stagonospora nodorum. BMC Bioinformatics 10, 301. doi:10.1186/1471-2105-10-301
Castellana, N. E., Payne, S. H., Shen, Z., Stanke, M., Bafna, V., and Briggs, S. P. (2008). Discovery and revision of Arabidopsis genes by proteogenomics. Proc. Natl. Acad. Sci. U.S.A. 105, 21034–21038.
Choudhary, J. S., Blackstock, W. P., Creasy, D. M., and Cottrell, J. S. (2001). Matching peptide mass spectra to EST and genomic DNA databases. Trends Biotechnol. 19, S17–S22.
de Groot, A., Dulermo, R., Ortet, P., Blanchard, L., Guerin, P., Fernandez, B., Vacherie, B., Dossat, C., Jolivet, E., Siguier, P., Chandler, M., Barakat, M., Dedieu, A., Barbe, V., Heulin, T., Sommer, S., Achouak, W., and Armengaud, J. (2009). Alliance of proteomics and genomics to unravel the specificities of Sahara bacterium Deinococcus deserti. PLoS Genet. 5, e1000434. doi:10.1371/journal.pgen.1000434
Helmy, M., Tomita, M., and Ishihama, Y. (2011). OryzaPG-DB: rice proteome database based on shotgun proteogenomics. BMC Plant Biol. 11, 63. doi:10.1186/1471-2229-11-63
Helmy, M., Tomita, M., and Ishihama, Y. (2012). Peptide identification by searching large-scale tandem mass spectra against large databases: bioinformatics methods in proteogenomics. Genes Genome Genomics 6, 76–85.
Huang, Y., Franklin, J., Gifford, K., Roberts, B. L., and Nicolette, C. A. (2004). A high-throughput proteo-genomics method to identify antibody targets associated with malignant disease. Clin. Immunol. 111, 202–209.
Hyman, S. E. (2011). Genome-sequencing anniversary. The meaning of the human genome project for neuropsychiatric disorders. Science 331, 1026.
Ouyang, S., Zhu, W., Hamilton, J., Lin, H., Campbell, M., Childs, K., Thibaud-Nissen, F., Malek, R. L., Lee, Y., Zheng, L., Orvis, J., Haas, B., Wortman, J., and Buell, C. R. (2007). The TIGR rice genome annotation resource: improvements and new features. Nucleic Acids Res. 35, D883–D887.
Perkins, D. N., Pappin, D. J., Creasy, D. M., and Cottrell, J. S. (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567.
Ravichandran, A., Sugiyama, N., Tomita, M., Swarup, S., and Ishihama, Y. (2009). Ser/Thr/Tyr phosphoproteome analysis of pathogenic and non-pathogenic Pseudomonas species. Proteomics 9, 2764–2775.
Keywords: proteomics, proteogenomics, bioinformatics, rice, LC–MS/MS, database
Citation: Helmy M, Sugiyama N, Tomita M and Ishihama Y (2012) The rice proteogenomics database OryzaPG-DB: development, expansion, and new features. Front. Plant Sci. 3:65. doi: 10.3389/fpls.2012.00065
Received: 29 January 2012; Accepted: 18 March 2012;
Published online: 02 April 2012.
Edited by:
Joshua L. Heazlewood, Lawrence Berkeley National Laboratory, USAReviewed by:
Ning Li, University of Washington, USASubhra Chakraborty, National Institute of Plant Genome Research, India
Copyright: © 2012 Helmy, Sugiyama, Tomita and Ishihama. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Yasushi Ishihama, Department of Molecular and Cellular Bioanalysis, Graduate School of Pharmaceutical Sciences, Kyoto University, Kyoto 606-8501, Japan. e-mail: yishiham@pharm.kyoto-u.ac.jp