 Katherine C. Goldfarb1 Ulas Karaoz1 China A. Hanson2 Clark A. Santee1 Mark A. Bradford3 Kathleen K. Treseder2 Matthew D. Wallenstein4 Eoin L. Brodie1*
Katherine C. Goldfarb1 Ulas Karaoz1 China A. Hanson2 Clark A. Santee1 Mark A. Bradford3 Kathleen K. Treseder2 Matthew D. Wallenstein4 Eoin L. Brodie1*- 1 Ecology Department, Earth Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
- 2 Ecology and Evolutionary Biology, University of California, Irvine, CA, USA
- 3 School of Forestry and Environmental Studies, Yale University, New Haven, CT, USA
- 4 Natural Resource Ecology Laboratory, Colorado State University, Fort Collins, CO, USA
Soils are immensely diverse microbial habitats with thousands of co-existing bacterial, archaeal, and fungal species. Across broad spatial scales, factors such as pH and soil moisture appear to determine the diversity and structure of soil bacterial communities. Within any one site however, bacterial taxon diversity is high and factors maintaining this diversity are poorly resolved. Candidate factors include organic substrate availability and chemical recalcitrance, and given that they appear to structure bacterial communities at the phylum level, we examine whether these factors might structure bacterial communities at finer levels of taxonomic resolution. Analyzing 16S rRNA gene composition of nucleotide analog-labeled DNA by PhyloChip microarrays, we compare relative growth rates on organic substrates of increasing chemical recalcitrance of >2,200 bacterial taxa across 43 divisions/phyla. Taxa that increase in relative abundance with labile organic substrates (i.e., glycine, sucrose) are numerous (>500), phylogenetically clustered, and occur predominantly in two phyla (Proteobacteria and Actinobacteria) including orders Actinomycetales, Enterobacteriales, Burkholderiales, Rhodocyclales, Alteromonadales, and Pseudomonadales. Taxa increasing in relative abundance with more chemically recalcitrant substrates (i.e., cellulose, lignin, or tannin–protein) are fewer (168) but more phylogenetically dispersed, occurring across eight phyla and including Clostridiales, Sphingomonadalaes, Desulfovibrionales. Just over 6% of detected taxa, including many Burkholderiales increase in relative abundance with both labile and chemically recalcitrant substrates. Estimates of median rRNA copy number per genome of responding taxa demonstrate that these patterns are broadly consistent with bacterial growth strategies. Taken together, these data suggest that changes in availability of intrinsically labile substrates may result in predictable shifts in soil bacterial composition.
Introduction
With over a billion individual cells and estimates of 104–105 distinct genomes per gram of soil (Gans et al., 2005; Tringe et al., 2005; Fierer et al., 2007b), bacteria in soil are the reservoirs for much of Earth’s genetic biodiversity. This vast phylogenetic and functional diversity can be attributed in part to the dynamic physical and chemical heterogeneity of soil, which results in spatial and temporal separation of microorganisms (Papke and Ward, 2004). Given the high diversity of carbon (C) – rich compounds in soils, the ability of each taxon to compete for only a subset of resources could also contribute to the high diversity of bacteria in soils through resource partitioning (Zhou et al., 2002). Indeed, Waldrop and Firestone (2004) have demonstrated distinct substrate preferences by broad microbial groups in grassland soils and C resource partitioning has been demonstrated to be a key contributor to patterns of bacterial co-existence in model communities on plant surfaces (Wilson and Lindow, 1994). Whether resource partitioning occurs in soil bacterial communities has not yet been addressed in a comprehensive manner, largely due to methodological limitations in sampling the enormous diversity and in measuring functional responses.
The development of high-throughput tools to assess the composition of soil bacterial communities is rapidly contributing to an improved understanding of bacterial diversity and biogeographical distribution (Drenovsky et al., 2009; Lauber et al., 2009; Chu et al., 2010). However, our ability to assess the functions of different bacterial taxa has not kept pace (Green et al., 2008). This limits our ability to interpret the functional consequences of shifts in community composition in response to environmental changes (Stein and Nicol, 2011). For example, most of our knowledge of the metabolic capabilities of bacterial taxonomic groups is derived from culture-based studies, which may not represent their activities in natural environments and is a small subset of the total diversity of bacteria in soils. Improving our understanding of the metabolic plasticity/rigidity of soil bacteria and determining their ability to utilize different types of C substrates, will enable us to better predict the consequences of changes in substrate availability on soil microbial community composition and diversity.
Phylogenetic approaches such as those based upon 16S rRNA genes are limited in their ability to extrapolate function since many functional traits are phylogenetically dispersed. For this reason, the use of tracer molecules such as stable-isotopes and the thymidine analog, 3-bromodeoxyuridine (BrdU), have been widely adopted in an effort to connect phylogeny to function. Stable-isotopes, particularly the heavy carbon isotope 13C, have been frequently used to identify microbial community members capable of catabolizing particular substrates (Radajewski et al., 2000; Griffiths et al., 2004; Buckley et al., 2007; Feth El Zahar et al., 2007; Schwartz, 2007). This technique requires separation of nucleic acids based on buoyant density, so high concentrations of isotopically labeled substrate are needed. Thus, this approach is costly and impractical for many complex organic compounds that are not commercially available. An alternative is the use of BrdU to monitor cell division following substrate addition. This approach was first applied to the study of bacterial populations over a decade ago (Urbach et al., 1999) and it has since been used to identify soil bacterial taxa that respond to various environmental stimuli (Borneman, 1999; Yin et al., 2000; Artursson and Jansson, 2003; Artursson et al., 2005). Recently, BrdU incorporation has been shown to detect a broad diversity of bacterial phyla in marine systems (Edlund et al., 2008) and fungal taxa in temperate (Hanson et al., 2008) and boreal forest soils (Allison et al., 2008).
In this study, we incubated soils from Harvard Forest (MA, USA) in the presence of BrdU and with a range of C substrates varying in chemical recalcitrance due to their molecular weight, structure, and the need for enzymatic digestion prior to microbial assimilation (glycine, sucrose, cellulose, lignin, tannin–protein). Next, we analyzed immunocaptured DNA by 16S rRNA PhyloChip (Brodie et al., 2006, 2007; DeSantis et al., 2007) to determine which members of the soil bacterial community increased their growth rates in response to substrate additions. We hypothesized that the soil bacterial community is comprised of relatively distinct phylogenetic subsets that are either metabolically versatile with regard to their C substrate preferences or display specific C substrate utilization profiles and that response to substrates can be related to rRNA copy number as a proxy for growth strategy (Klappenbach et al., 2000; Lee et al., 2009).
Materials and Methods
Soil Incubations, DNA Extraction, and BrdU Immunocapture
Soil cores were collected from the Harvard Forest Warming Experimental Site in Petersham, Massachusetts in September 2005 according to Hanson et al. (2008). Soil moisture was 16.1% (w/v) and mean water holding capacity was 60.9%, this is within the range of previous laboratory incubations (Paul et al., 2001; Fierer and Schimel, 2002; Bradford et al., 2010), therefore soil moisture was not adjusted prior to substrate addition. From each of four replicate soil samples 2 g were incubated for 48 h at room temperature in the dark with 260 μl of a solution containing 7.69 mM 3-bromodeoxyuridine (BrdU) and 20 mg of one of five substrates (glycine, sucrose, cellulose, lignin, and tannin–protein). A BrdU-only control incubation was also performed for each soil replicate. Soil DNA extraction and immunocapture of BrdU-labeled DNA was performed according to Hanson et al. (2008). Substrate concentrations were provided in excess of demand (at 1% w/w) across the 48-h, as demonstrated with preliminary trials (data not shown). Addition of substrate concentrations in excess of demand is a common approach for assessing both the potential respiration and microbial community responses of substrate utilization in soils (e.g., Bradford et al., 2010). However, soil microbial communities are typically considered substrate limited, and so the caveat of the approach is that substrate concentrations are above what would be observed in situ (e.g., van Hees et al., 2005). This should be considered when extrapolating patterns observed in this study to those observed in the field.
PCR Amplification of 16S rRNA Genes
The 0.5-μl of BrdU-labeled genomic DNA was used as template for PCR amplification of 16S rRNA genes. Eight replicate reactions containing 0.02 U/μL ExTaq (Takara Bio Inc., Japan), 1× ExTaq buffer, 0.2 mM dNTP mixture, 1 μg/μL bovine serum albumin (BSA), and 300 pM each of universal bacterial primers: 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) were performed for each sample. To minimize PCR bias due to variable template annealing efficiencies and random priming effects, PCR was performed with an eight temperature annealing gradient (48–58°C) and the following conditions: 95°C (3 min), followed by 30 cycles of 95°C (30 s), annealing (30 s), 72°C (2 min), and a final extension at 72°C (10 min). Reactions were combined for each sample and concentrated by precipitation with isopropanol, washed twice with ice-cold 70% ethanol and resuspended in 50 μL nuclease-free water.
Phylochip Microarray Analysis of 16S rRNA Gene Diversity
Five hundred nanogram of pooled PCR amplicons from each sample were spiked with known concentrations of control amplicons derived from yeast and bacterial metabolic genes. This mix was subject to fragmentation, biotin labeling, and hybridization to G2 PhyloChip microarrays according to manufacturer’s protocols and as described previously (Brodie et al., 2007).
Each PhyloChip was scanned and recorded as a pixel image, and initial data acquisition and intensity determination were performed using standard Affymetrix software (GeneChip microarray analysis suite, version 5.1). Background subtraction and probe-pair scoring were performed as reported previously (Brodie et al., 2006, 2007; DeSantis et al., 2007). The positive fraction (pf) was calculated for each probe set as the number of positive probe-pairs divided by the total number of probe-pairs in a probe set. Taxa were deemed present when the pf value met or exceeded 0.90. Intensities were summarized for each taxon/probe set using a trimmed mean (highest and lowest values removed before averaging) of the intensities of the perfect match (PM) probes minus their corresponding mismatch probes (MM).
Quantitative PCR
qPCR assays were performed on BrdU-captured DNA from each sample following Fierer et al. (2005, 2007a). Separate qPCR assays were conducted to quantify the abundance of 16S ribosomal RNA gene copies from total bacterial (“all bacteria” assay), in addition to rRNA gene copy numbers belonging to the following sub-groups of bacteria: Acidobacteria, Beta-proteobacteria, and Bacteroidetes. These assays were performed in triplicate for each sample in 96-well plates in a BioRad iCycler single-color real-time PCR detection system (BioRad, CA, USA) using SYBR Green I dye (BioRad, CA, USA). All primers, reaction conditions and reaction concentrations were identical to those used by Fierer et al. (2005, 2007a) with one modification: the annealing temperature for the Acidobacteria qPCR assay was increased to 52°C, to improve PCR specificity. Standard curves for each of the assays were generated using triplicate 10-fold dilutions of known plasmid standards containing PCR products from an appropriate positive control generated using the qPCR primers. After confirming the presence of target PCR product in each reaction using a melting curve analysis with the MyIQ software (BioRad), standard curves were used to calculate target copy numbers for each reaction. Relative abundances of each bacterial subgroup in each DNA sample were calculated as a ratio between the measured copy numbers of each group-specific assay and the “all bacteria” assay. Because amplification efficiencies can vary across DNA samples, copy numbers expressed as a fractional value more accurately reflect a relative index of the abundances of each of the targeted bacterial groups (Fierer et al., 2005).
Statistical Analysis of Phylochip Assays of Bacterial Community Composition
All statistical analyses were carried out in the R programming environment1. To correct for variation associated with quantification of amplicon target (quantification variation), and downstream variation associated with target fragmentation, labeling, hybridization, washing, staining, and scanning (microarray technical variation), hybridization intensities were subject to the following two-step normalization procedure. First, for each PhyloChip experiment, a scaling factor best explaining the intensities of the spiked control probes under a multiplicative error model was estimated using a maximum-likelihood procedure. The intensities in each experiment were multiplied by their corresponding optimal scaling factor. Second, the intensities for each experiment were corrected for the variation in total array intensity by dividing the intensities by their corresponding total array intensity for bacterial probe sets. Normalized intensities were then log10 transformed and only bacterial taxa present (pf ≥ 0.90) in at least one replicate soil incubation were considered. A distance matrix was calculated from the normalized transformed intensity values using the Bray–Curtis distance metric within the function “vegdist” in the R package “vegan.” The distance matrix was represented as a non-metric multidimensional scaling plot using the function “metaMDS” and variance partitioning was calculated with the function “adonis.” Bacterial taxa that were significantly enriched in abundance for a C substrate type relative to the BrdU-only control were determined by ANOVA (p < 0.05) after correction for multiple observations using the Benjamini and Hochberg (1995) procedure. Differences in distributions of pairwise distances between samples receiving substrate additions and BrdU-only controls were tested for significance using a Mann–Whitney test.
Phylogenetic Tree Construction
To display the phylogenetic breadth of organisms detected as capable of incorporating BrdU, a phylogenetic tree was generated using representative 16S rRNA sequences for those bacterial taxa detected in at least one sample following BrdU immunocapture. Representative sequences were exported aligned (7682 character NAST alignment; DeSantis et al., 2006b) from the greengenes database (DeSantis et al., 2006a) and local bootstrapped neighbor-joining trees constructed using FastTree2 with default settings (Price et al., 2009). Dendrogram branches were annotated according to phylum and according to substrate use range using the interactive tree of life (ITOL) web server3 (Letunic and Bork, 2007).
Analysis of Phylogenetic Clustering and Dispersion within Bacterial Communities Responding to Carbon Substrate Addition
To determine whether additions of a given substrate enriched bacterial taxa that were more or less phylogenetically related than expected by chance, we calculated the nearest taxon index (NTI) using the function “ses.mnnd” in the R package “picante”4. NTI measures the distance between each taxon and its nearest neighbor to determine terminal phylogenetic relatedness. Comparisons were made to a random community drawn from the entire pool of taxa observed to incorporate BrdU. Positive values indicate phylogenetic clustering while negative values demonstrate more even dispersion than would be expected by a random sampling across a phylogeny (Horner-Devine and Bohannan, 2006; Webb et al., 2008).
Relationship between Growth Response and rRNA Copy Number
To assess whether the growth response of bacteria following substrate addition was related to rRNA copy number as a proxy for ecological strategy we calculated median rRNA copy numbers for individual response groups which we have defined as follows: organisms that increase in relative abundance in response to any C substrate (1), organisms that decrease in relative abundance in response to any C substrate (2), organisms that increase relative abundance in response to labile C (3), or chemically recalcitrant C (4), and organisms increasing in relative abundance in response to each of the substrates glycine (5), sucrose (6), cellulose (7), lignin (8), tannin–protein (9). As rRNA copy numbers are typically similar across genera, rRNA copy numbers for taxa detected in this study were inferred from genome sequenced relatives found in the rrnDB5 as of April 12th 2010 (Lee et al., 2009).
Results
BrdU Was Incorporated into the Genomes of a Broad Diversity of Soil Bacteria
Across all BrdU-labeled incubations, we detected 2,233 bacterial taxa (1,938 taxa with sequence lengths >600 bp are displayed in the phylogenetic tree; Figure 1). These taxa were distributed across 43 of the 58 bacterial phyla detectable by the PhyloChip version G2 (available in Table S2 in Supplementary Material). The detected taxa represented the major lineages of soil bacteria (Proteobacteria, Firmicutes, Acidobacteria, Actinobacteria, Planctomycetes, Bacteroidetes, Verrucomicrobia, TM7, Spirochaetes, Gemmatimonadetes, Deinococcus, Nitrospira, Chloroflexi, and Cyanobacteria) and a strong phylogenetic signal for substrate utilization was apparent (Figures 1 and 3; Table S1 in Supplementary Material).
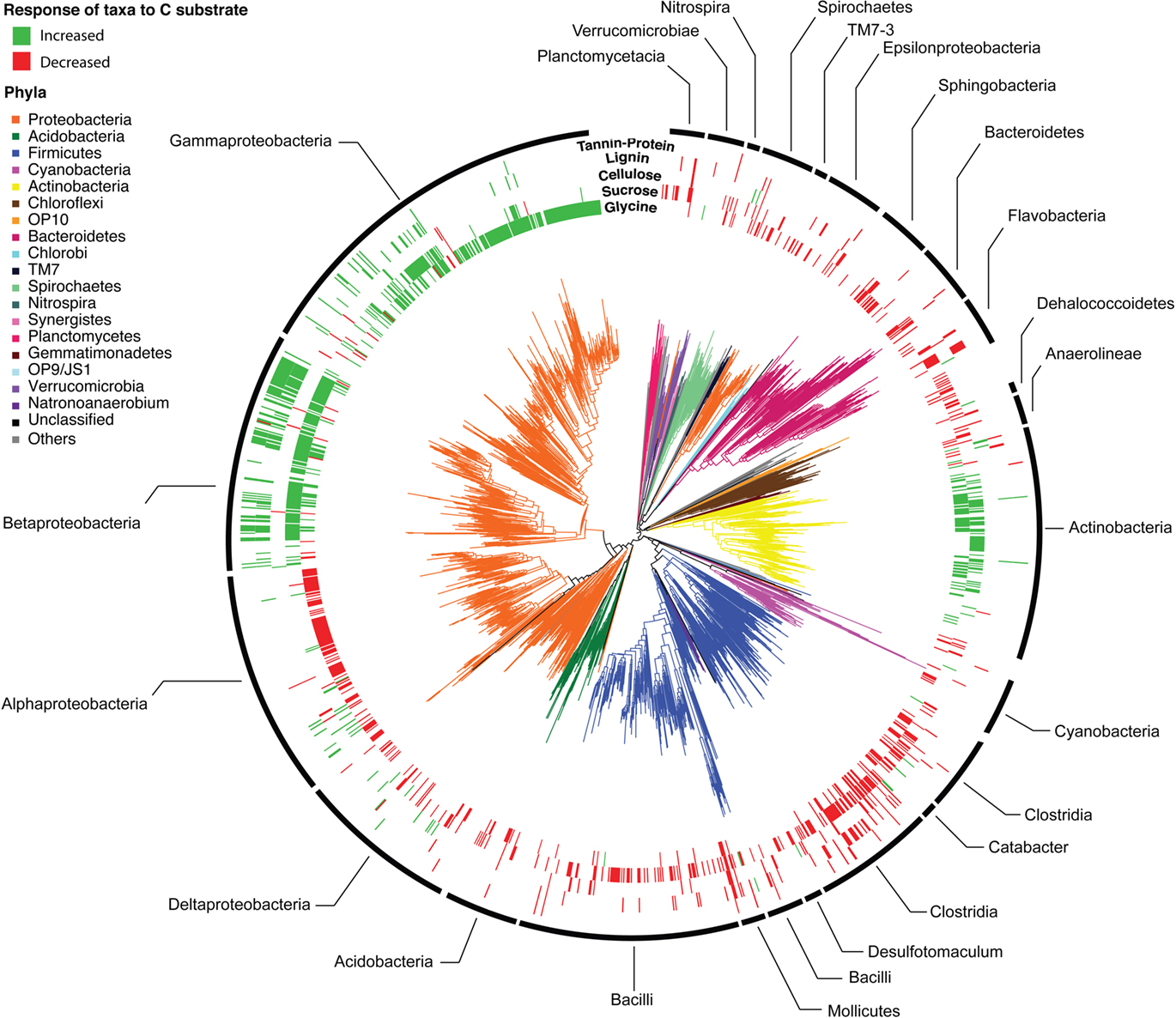
Figure 1. Phylogeny of bacterial taxa detected following BrdU incorporation. Phyla are designated by branch colors and selected sub-phyla/classes are annotated. The outer rings display the taxa whose relative abundance increased (green) or decreased (red) significantly (p < 0.05 following BH correction) in response to C substrate addition relative to the BrdU-only controls. Outer rings are arranged from the interior in order of expected substrate recalcitrance (glycine, sucrose, cellulose, lignin, tannin–protein).
The Active Bacterial Community is Impacted to a Greater Degree by Labile Organic C
Non-metric Multidimensional Scaling of inter-sample Bray–Curtis distances, based on PhyloChip probe set intensities, demonstrated that more labile substrates such as glycine and sucrose resulted in a greater divergence of the active bacterial population from the BrdU-only controls than did more chemically recalcitrant substrates (Figure 2). Many more bacterial taxa (>300) were significantly enriched by the addition of a labile substrate, whereas the addition of more chemically recalcitrant substrates stimulated 27 to 129 taxa (Figures 1 and 3).
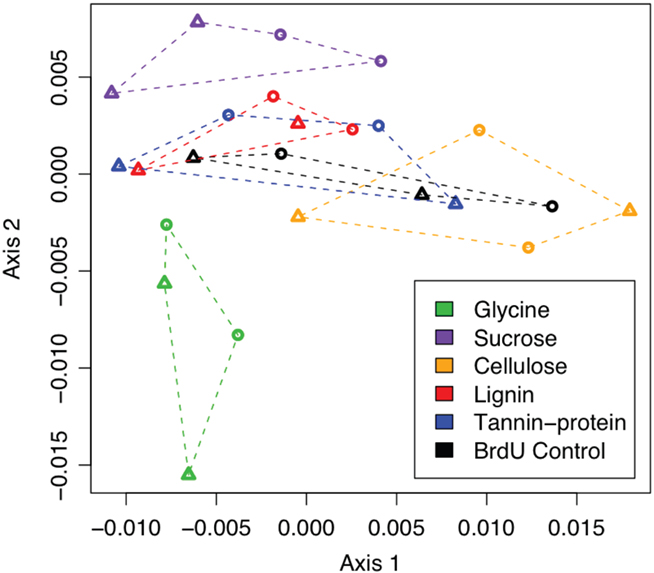
Figure 2. Non-metric Multidimensional Scaling (NMDS) projection of a Bray–Curtis distance matrix showing the response of actively replicating bacterial communities to C substrate addition. Substrate incubations are depicted with colors and grouped by dotted lines.
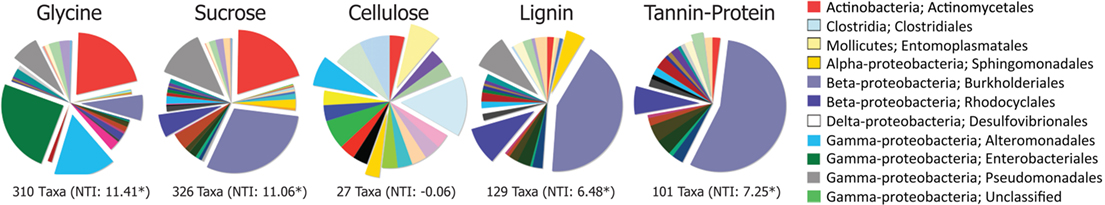
Figure 3. Order-level distributions of bacteria significantly (p < 0.05) enriched in response to substrate addition relative to the BrdU-only control. Numbers of enriched taxa are given under each pie and Nearest Taxon Index (NTI) values are in parentheses. *Denotes NTI value significantly different from the null community distribution (p < 0.05).
The greater effect of labile substrates was apparent at taxonomic levels ranging from order to OTU/taxon (Figure 1) and the order-level distribution of bacterial taxa responding to particular substrates varied substantially (Figure 3). Nevertheless, strong patterns were apparent. Taxa that became enriched were primarily restricted to a limited number of bacterial orders primarily Enterobacteriales, Actinomycetales, Burkholderiales, Alteromonadales, Pseudomonadales, Rhodocyclales, and Clostridiales and the NTI (Horner-Devine and Bohannan, 2006; Webb et al., 2008; Swenson, 2009) confirmed a phylogenetic clustering of the enriched taxa (Figure 3). All substrates, with the exception of cellulose, resulted in enrichment of phylogenetically clustered groups of bacteria. In other words, these substrates stimulated the growth of bacterial taxa that are more similar to each other than would be expected in a random sampling of taxa detected in Figure 1.
Comparison of the Carbon Substrate Range of Soil Bacterial Taxa
Many taxa (390) were enriched in response to labile substrates only, with 195 taxa only responding to glycine and 131 only to sucrose (Figure 4). Sixty-four taxa responded to both glycine and sucrose but not to any chemically recalcitrant substrate. Fewer taxa responded only to chemically recalcitrant substrates. Notably, of the 27 taxa that became enriched in response to cellulose, 24 responded only to its addition. In contrast, 123 of the 129 lignin-enriched taxa were also enriched by the addition of labile substrates. Only one taxon (an unclassified γ-Proteobacterium) was enriched only by tannin–protein additions.
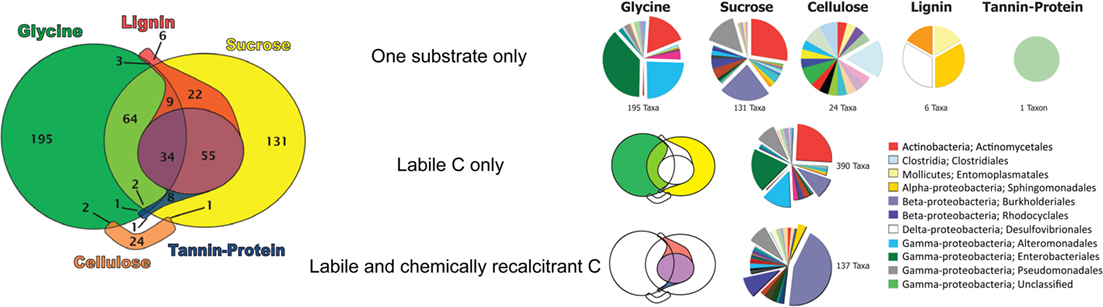
Figure 4. Euler diagram showing substrate use range of bacterial taxa significantly enriched in response to substrate addition. Pies display the order level taxonomic composition of bacterial taxa enriched only by single substrates, by labile substrates only, and by both labile and chemically recalcitrant substrates. Only expanded pie wedges are included in the legend, a full color legend can be found as supplementary material (Figure S1 in Supplementary Material).
Most of the taxa responding only to labile C were Actinobacteria (Actinomycetales), γ-Proteobacteria (Enterobacteriales, Alteromonadales, Pseudomonadales), and β-Proteobacteria (Burkholderiales). Organisms enriched by glycine only were mostly γ-Proteobacteria (Enterobacteriales, Alteromonadalaes) and Actinobacteria (Actinomycetales); those enriched by sucrose only were mostly Actinobacteria (Actinomycetales), β-Proteobacteria (Burkholderiales, Rhodocyclales), and γ-Proteobacteria (Pseudomonadales; Figure 1; Table S1 in Supplementary Material). Although certain Burkholderiales taxa were enriched only by labile substrates, many more displayed broader substrate preferences as did many members of the Rhodocyclales and the Pseudomonadales. Some taxa within the Sphingomonadalaes responded positively to both sucrose and lignin, but declined in response to glycine (Table S1 in Supplementary Material).
Very few taxa responded only to chemically recalcitrant substrates, with only 24 for cellulose (Clostridiales; termite gut clones, Deinococcus, Chloroflexi, Acetobacter, Desulfobacteraceae, Geobacteraceae, Syntrophobacteraceae, Treponema), 6 for lignin (Xanthomonadaceae, Desulfovibrionaceae, Entomoplastamaceae, Sphingomonadaceae), and 1 for tannin–protein (an unclassified γ-Proteobacterium; Table S1 in Supplementary Material). Organisms responding only to a specific substrate displayed a distinct taxonomic composition according to the C substrate applied (Figure 4).
Substrate additions also elicited negative effects on certain taxa (Figure 1; Table S1 in Supplementary Material). Specifically, glycine and sucrose additions reduced the relative abundance of approximately 300 taxa. Moreover, cellulose, lignin, and tannin–protein decreased 69, 81, and 21 taxa, respectively. Although not universal, C substrates generally impacted a number of Clostridia and Bacilli families negatively. Glycine addition also negatively affected Acidobacteria and Bacteroidetes which was confirmed by qPCR (Table S3 in Supplementary Material) as well as many α-Proteobacteria and other taxa. Sucrose addition also negatively affected Acidobacteria and Bacteroidetes (Figure 1; Tables S1 and S3 in Supplementary Material) in addition to many δ-Proteobacteria, Planctomycetes, and Gemmatimonadetes amongst others. Overall, qPCR estimates of changes in fractional abundance of specific bacterial groups agreed well with PhyloChip estimates (r = 0.77, Table S3 in Supplementary Material).
Table 1 shows the estimated rRNA copy numbers/genome for bacterial taxa responding to C substrate addition. Overall, taxa that increased in relative abundance in response to C addition had greater median rRNA copy numbers than those that decreased. Bacteria increasing in response to labile C had higher median rRNA copy numbers than those increasing in response to more chemically recalcitrant C. This latter trend is clear when comparing the median rRNA copy numbers by substrate applied.
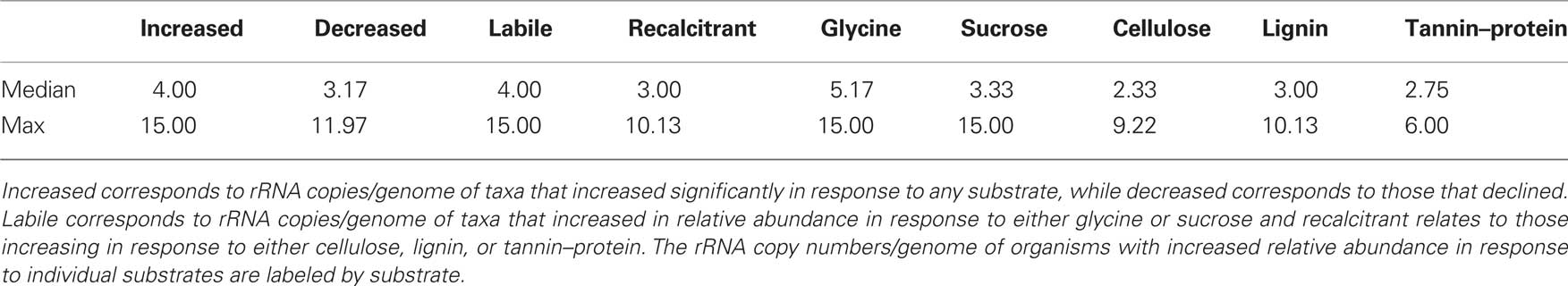
Table 1. Estimated rRNA copy numbers/genome of bacterial taxa responding significantly to substrate addition.
Discussion
The microbial diversity of soils is vast, but the mechanisms maintaining this diversity are not fully understood. A major contributor to this diversity may be the development of distinct resource niches whereby many organisms can co-inhabit the same location by utilizing different substrates. BrdU labeling allowed us to characterize some of these niches by identifying the bacterial members who responded (through DNA replication) to the presence of specific C substrates.
Soil microbes, with the exception of those inhabiting the rhizosphere, are typically considered to be C-limited (Zak et al., 1993; Curtis et al., 1994; Alden et al., 2001; Garbeva and De Boer, 2009). This together with the fact that soil C pools are heterogeneous and vary in chemical recalcitrance and turnover times (Dixon et al., 1994; Lichtfouse et al., 1995; Knorr et al., 2005) suggests that specialization on C substrates may contribute to the high phylogenetic diversity observed in soils. Indeed, it has been suggested that one of the main factors controlling bacterial species diversity is the diversity of C substrates (Zhou et al., 2002). Here, we identified hundreds of bacterial taxa that responded only to labile C additions (Figure 4). In fact, addition of labile C (glycine or sucrose) resulted in a greater divergence of bacterial communities from controls than did chemically recalcitrant C (Figure 2). This finding is supported by our estimates of rRNA copy numbers/genome of taxa responding to labile substrates, where we see a large number of fast-growing (copiotrophs) responding quickly to a pulsed resource. Conversely, slow-growing (oligotrophs) with lower rRNA copy numbers, such as some alpha-proteobacteria and Acidobacteria decline following substrate addition.
Labile C also significantly alters fungal community structure in this site, as does cellulose which Hanson et al. (2008) observed to be mineralized rapidly. In contrast, cellulose did not induce a large change in bacterial community structure (Figure 2), although we did not directly compare the relative abundances of bacteria and fungi in these data, suggesting that fungi – not bacteria – may be the main decomposers of cellulose in these soils. Numerous bacterial taxa were enriched by labile C (Figure 3) but they were phylogenetically clustered and represented few bacterial orders suggesting that labile C (such as that found in rhizosphere exudates) promotes outgrowth of low diversity assemblages, which results in a heavily skewed distribution of taxa abundance at the community level. In this study we detected significant increases in the relative abundances of many β-Proteobacteria (Burkholderiales and Rhodocyclales), γ-Proteobacteria (Enterobacteriales and Pseudomonadales), and Actinobacteria (Actinomycetales) in response to labile substrates. This does not necessarily indicate that other taxa are not capable of utilizing labile substrates, but does indicate that they were not competitive for these resources under these conditions. The rapid response of fast-growing bacteria (such as those that responded to glycine and/or sucrose) to labile C may partly explain the rhizosphere effect. Smalla et al. (2001) reported an enrichment of Actinobacteria in the rhizosphere of several plant species and DeAngelis et al. (2009) recently documented a higher abundance of both β-Proteobacteria and Actinobacteria in the rhizosphere of wild oats. In the latter study, of the 44 phyla detected in the grassland soil, root movement through soil only significantly affected ~7% of taxa within 8 days. Drigo et al. (2009) found that increased rhizodeposition of sugars due to elevated CO2 increased the abundance of Burkholderia and Pseudomonads. In our study, almost 50% of actively replicating bacterial taxa (1115 from 2233) were significantly altered by C substrate addition in 2 days, but as in the study by DeAngelis et al. (2009) they represented few phyla. Collectively, these findings suggest that in complex communities, labile substrates are primarily utilized by fast-growing copiotrophs, depleting these resources in time and space before oligotrophs can assimilate them.
Even though a limited number of phyla responded to labile C addition, substrate preference was still apparent. For example, glycine addition stimulated the growth of many γ-Proteobacteria (Enterobacteriales, Alteromonadalaes), whereas sucrose addition stimulated growth of many Actinobacteria (Actinomycetales), β-Proteobacteria (Burkholderiales, Rhodocyclales), and other γ-Proteobacteria (Pseudomonadales; Figures 1 and 3B). This substrate preference may allow co-existence between these functional groups, although our results also suggest that many organisms within these groups can compete for either form of labile C, which may be advantageous in a relatively C-limited environment such as mineral soil.
In contrast to the phylogenetic clustering observed among labile C users, chemically recalcitrant C users – primarily cellulose users – were less phylogenetically related. Since relatively few bacterial taxa responded positively to chemically recalcitrant C, it appears that less functional redundancy exists in the turnover of these C pools. On the other hand, cellulose users were relatively phylogenetically diverse (eight phyla), which may broaden the range of environmental conditions under which this metabolism may operate. Many of the taxa responding positively to cellulose have previously been shown to be capable of cellulose hydrolysis (e.g., bacteria from the families Clostridiaceae and Lachnospiraceae (Schwarz, 2001), Sphingomonas spp. (Kurakake et al., 2007), and Spirochetes (Warnecke et al., 2007), while Acetobacter aceti (Moonmangmee et al., 2002) is typically considered a cellulose producer rather than a consumer. In contrast, other cellulose-stimulated taxa from our study, such as Desulfobacterium sp., Syntrophobacteraceae, Geobacteraceae, Coriobacteriaceae, and Anaerolineae have not been previously implicated in cellulose hydrolysis directly.
Only six taxa were stimulated by lignin addition but no other substrate, these included two from the order Desulfovibrionales (δ-Proteobacteria), two from Sphingomonadalaes (α-Proteobacteria) and a Xanthomonadales (γ-Proteobacteria). Lignin hydrolysis has been documented under sulfate–reducing conditions (Dittmar and Lara, 2001) and Desulfovibrio desulfuricans has been shown previously to oxidize lignin under anaerobic conditions, a process that impacts both the polyphenolic backbone and functional side groups on the compound (Ziomek and Williams, 1989). Sphingomonas species have been reported to grow on several dimeric model compounds of lignin as a C and energy source using O demethylation systems (Sonoki et al., 2000) and Xanthomonas species have previously been demonstrated to use lignin as a C source for growth (Kern and Kirk, 1987; Kirk and Farrell, 1987). Only a single taxon was stimulated by tannin–protein alone; this probe set represented an unclassified γ-Proteobacterium.
Almost 140 taxa responded to both labile and chemically recalcitrant C particularly from within the Burkholderiales families Alcaligenaceae (including Alcaligenes spp.), Comamonadaceae (including Acidovorax and Variovorax spp.), and Burkholderiaceae (mostly Burkholderia spp.; Table S1 in Supplementary Material). These versatile taxa responded mostly to sucrose, lignin, and tannin–protein and this agrees with previous studies that demonstrated either an ability to metabolize lignin/lignin monomers (Krishna and Sunil, 1993; Nigel et al., 1995; Kato et al., 1998; Mitsui et al., 2003) or presence of lignin peroxidase gene homologs (in the case of Acidovorax avenae). Many Rhodocyclales (including Azoarcus and Thauera spp.) and Pseudomonadales (mostly Pseudomonas spp.) were also stimulated by multiple forms of C. Bacterial taxa such as Burkholderia and Pseudomonas species are known to be metabolically versatile (Yoder-Himes et al., 2009), a trait probably related to a capacity for genome re-arrangement (Lin et al., 2008; Silby et al., 2009) and maintenance of accessory genomes (Wolfgang et al., 2003; Mahenthiralingam and Drevenik, 2007; Mathee et al., 2008; Sim et al., 2008). The ability of such organisms to catabolize labile and chemically recalcitrant C may make them efficient competitors in a frequently C-limited environment. They may also serve as contributors to rhizosphere priming, in which plant roots, through exudation, stimulate the mineralization of soil organic matter (Fu and Cheng, 2002; Bader and Cheng, 2007).
Changes in the abundance of taxa were relative within a community, so increases in relative abundance of one taxon were matched by decreases in relative abundance of others. Although a change in relative abundance does not necessarily imply a change in absolute number, it represents a shift in the rank order of an organism within a community. This shift could lead to a change in the ability of that organism to compete for resources and influence ecosystem processes. Specifically, we found a consistent decrease in relative abundance of more than 16 families of α-Proteobacteria in response to glycine (Figure 1; Table S1 in Supplementary Material). Similarly, DeAngelis et al. (2009) found that α-Proteobacteria were significantly altered by root movement through the soil. Moreover, many taxa within this sub-phylum decrease in relative abundance at the root tips, where exudation peaks (Egeraat, 1975; Personeni et al., 2007). Conversely, these bacteria are enriched near root hairs and mature roots compared to bulk soil populations. Glycine has been shown to have a high rate of efflux from roots of multiple plant species (Lesuffleur et al., 2007) which may explain lower relative abundances of α-Proteobacteria at the root tip. The mechanism for this observation is unclear. Glycine may inhibit these bacteria directly or through competitive exclusion by glycine users (e.g., the Enterobacteriales). Alternatively, it may simply be that the relative decline in α-Proteobacteria is not a product of an absolute decline. Future work is required to discern whether C substrate additions to soil provoke absolute declines in the abundance of specific taxa and, if so, the mechanisms underlying these declines.
The quality and quantity of C substrates is expected to be one of the primary drivers of microbial community composition in soils, at least at the phylum level (Fierer et al., 2007a). For example, Acidobacteria have been reported to show the highest relative abundance in soils where C mineralization rates are low and so classified as oligotrophs (Fierer et al., 2007a), while β-Proteobacteria and Bacteriodetes are more abundant in soils with high C mineralization rates, and therefore classified as copiotrophs. Consistent with these global-scale patterns, ß-Proteobacteria increased in abundance following sucrose additions in our study. On the one hand, our results confirm that increases in the availability of specific substrates can stimulate the growth of the taxa that can best compete for those resources, which may quickly lead to changes in microbial community composition. On the other hand, the addition of substrates that are resistant to degradation (e.g., lignin and cellulose) did not change the overall composition of the active microbial community, although a small number of specialist taxa did show a growth response. Even though the abundance of these specialist taxa remained rare, they likely still play an important ecological role in these soils. Thus, we suggest that the function of microbial communities cannot be revealed by broad characterizations of microbial community structure, but rather must also consider the functional contributions of the rare biosphere.
Here we demonstrated that by combining the BrdU labeling with a high-resolution microbial community analytical tool (16S rRNA PhyloChip), we could achieve a deep and broad coverage of soil bacterial diversity and its response to C substrates. Overall, 2,233 taxa from 43 phyla were detected following BrdU incorporation and these phyla include the predominant soil lineages (Janssen, 2006; Hawkes et al., 2007; Lauber et al., 2009). Rare phyla (Urich et al., 2008) such as Chlorobi, Dictoglomi, SPAM, TM6, and Termite group 1 were also detected (Figure 1; Table S1 in Supplementary Material). Although a previous study has suggested that BrdU incorporation may not be as efficient in Gram-positive bacteria (Urbach et al., 1999). As our approach compares the response of individual taxa relative to a control, BrdU incorporation by Gram-positive bacteria, even if it is inefficient, should be constant per taxon. Our data suggests this does not preclude the use of BrdU as an effective tool to monitor changes in bacterial replication.
The combination of BrdU incorporation and sensitive methods of community analysis such as high-density microarrays provides a powerful tool to investigate the response of soil microbes to changing resource availability. This technique could also have broad application for determining the effects of environmental perturbations, such as altered precipitation patterns and elevated temperature or CO2. Indeed, almost 50% of the actively replicating bacterial taxa in the soil samples we investigated were significantly altered by C substrate addition, with labile C inducing the greatest effect on community structure. Our findings provide insight into the factors responsible for maintaining the high diversity of bacteria observed in soils and represent a platform for future analysis of the mechanisms underlying environmental-driven alterations in soil bacterial communities, and the potential implications for biogeochemical cycling.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Steven Allison for critical review of this manuscript. Part of this work was performed under the auspices of the U.S. Department of Energy by the University of California, Lawrence Berkeley National Laboratory, under Contract DE-AC02-05CH11231 and was supported in part by the Laboratory Directed Research and Development Program of Lawrence Berkeley National Laboratory (to Eoin L. Brodie). Further support was provided by U.S. Department of Energy Program for Ecosystem Research grant number DE-FG02-04ER63893 (to Mark A. Bradford, Kathleen K. Treseder, and Matthew D. Wallenstein), by National Science Foundation grant number DEB-0445458 (to Kathleen K. Treseder), and a grant from the Warner College of Natural Resources at Colorado State University (to Matthew D. Wallenstein).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Terrestrial_Microbiology/10.3389/fmicb.2011.00094/abstract/
Figure S1|Full color legend for pie charts in Figures 3 and 4 that display the order-level taxonomic composition of bacterial taxa enriched only by single substrates, by labile substrates only, and by both labile and chemically recalcitrant substrates.
Table S1|Bacterial taxa displaying significantly different relative abundance relative to the BrdU-only control following C substrate addition.
Table S2|Excel spreadsheet containing taxon presence/absence data (probe-positive fraction; pf) and taxon intensity data (natural log transformed normalized array intensities) for all 8432 bacterial taxa represented on the PhyloChip version G2.
Table S3|Comparison of the relative impact addition of C substrates of varying chemical recalcitrance on three bacterial phyla/sub-phyla by microarray analysis and qPCR. For each substrate, values shown for microarray analysis represent the sum of mean differences in log10 fluorescence intensity between BrdU-substrate incubations and BrdU-controls for those that were statistically significant in Table S1. For qPCR data, values represent mean differences in fractional abundance of groups within BrdU-substrate incubations relative to the BrdU-control. The correlation (R) between the PhyloChip measure of changes in relative abundance and qPCR measures of changes in fractional abundance is 0.77.
Footnotes
References
Alden, L., Demoling, F., and Baath, E. (2001). Rapid method of determining factors limiting bacterial growth in soil. Appl. Environ. Microbiol. 67, 1830–1838.
Allison, S. D., Czimczik, C. I., and Treseder, K. K. (2008). Microbial activity and soil respiration under nitrogen addition in alaskan boreal forest. Glob. Change Biol. 14, 1156–1168.
Artursson, V., Finlay, R. D., and Jansson, J. K. (2005). Combined bromodeoxyuridine immunocapture and terminal-restriction fragment length polymorphism analysis highlights differences in the active soil bacterial metagenome due to glomus mosseae inoculation or plant species. Environ. Microbiol. 7, 1952–1966.
Artursson, V., and Jansson, J. K. (2003). Use of bromodeoxyuridine immunocapture to identify active bacteria associated with arbuscular mycorrhizal hyphae. Appl. Environ. Microbiol. 69, 6208–6215.
Bader, N. E., and Cheng, W. (2007). Rhizosphere priming effect of populus fremontii obscures the temperature sensitivity of soil organic carbon respiration. Soil Biol. Biochem. 39, 600–606.
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300.
Borneman, J. (1999). Culture-independent identification of microorganisms that respond to specified stimuli. Appl. Environ. Microbiol. 65, 3398–3400.
Bradford, M. A., Watts, B. W., and Davies, C. A. (2010). Thermal adaptation of heterotrophic soil respiration in laboratory microcosms. Glob. Change Biol. 16, 1576–1588.
Brodie, E. L., DeSantis, T. Z., Joyner, D. C., Baek, S. M., Larsen, J. T., Andersen, G. L., Hazen, T. C., Richardson, P. M., Herman, D. J., Tokunaga, T. K., Wan, J. M. M., and Firestone, M. K. (2006). Application of a high-density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl. Environ. Microbiol. 72, 6288–6298.
Brodie, E. L., DeSantis, T. Z., Parker, J. P., Zubietta, I. X., Piceno, Y. M., and Andersen, G. L. (2007). Urban aerosols harbor diverse and dynamic bacterial populations. Proc. Natl. Acad. Sci. U.S.A. 104, 299–304.
Buckley, D. H., Huangyutitham, V., Hsu, S. F., and Nelson, T. A. (2007). Stable isotope probing with 15N2 reveals novel noncultivated diazotrophs in soil. Appl. Environ. Microbiol. 73, 3196–3204.
Chu, H., Fierer, N., Lauber, C. L., Caporaso, J. G., Knight, R., and Grogan, P. (2010). Soil bacterial diversity in the arctic is not fundamentally different from that found in other biomes. Environ. Microbiol. 12, 2998–3006.
Curtis, P., O’neill, E., Teeri, J., Zak, D., and Pregitzer, K. (1994). Belowground responses to rising atmospheric CO2: implications for plants, soil biota and ecosystem processes. Plant Soil 165, 1–6.
DeAngelis, K. M., Brodie, E. L., DeSantis, T. Z., Andersen, G. L., Lindow, S. E., and Firestone, M. K. (2009). Selective progressive response of soil microbial community to wild oat roots. ISME J. 3, 168–178.
DeSantis, T. Z., Brodie, E. L., Moberg, J. P., Zubieta, I. X., Piceno, Y. M., and Andersen, G. L. (2007). High-density universal 16S rRNA microarray analysis reveals broader diversity than typical clone library when sampling the environment. Microb. Ecol. 53, 371–383.
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., Huber, T., Dalevi, D., Hu, P., and Andersen, G. L. (2006a). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with Arb. Appl. Environ. Microbiol. 72, 5069–5072.
DeSantis, T. Z. Jr., Hugenholtz, P., Keller, K., Brodie, E. L., Larsen, N., Piceno, Y. M., Phan, R., and Andersen, G. L. (2006b). NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 34, W394–W399.
Dittmar, T., and Lara, R. J. (2001). Molecular evidence for lignin degradation in sulfate-reducing mangrove sediments (Amazonia, Brazil). Geochim. Cosmochim. Acta 65, 1417–1428.
Dixon, R. K., Solomon, A. M., Brown, S., Houghton, R. A., Trexier, M. C., and Wisniewski, J. (1994). Carbon pools and flux of global forest ecosystems. Science 263, 185–190.
Drenovsky, R. E., Steenwerth, K. L., Jackson, L. E., and Scow, K. M. (2009). Land use and climatic factors structure regional patterns in soil microbial communities. Glob. Ecol. Biogeogr. 19, 27–39.
Drigo, B., Van Veen, J. A., and Kowalchuk, G. A. (2009). Specific rhizosphere bacterial and fungal groups respond differently to elevated atmospheric CO2. ISME J. 3, 1204–1217.
Edlund, A., Hardeman, F., Jansson, J. K., and Sjoling, S. (2008). Active bacterial community structure along vertical redox gradients in Baltic Sea sediment. Environ. Microbiol. 10, 2051–2063.
Egeraat, A. (1975). Exudation of ninhydrin-positive compounds by pea-seedling roots: a study of the sites of exudation and of the composition of the exudate. Plant Soil 42, 37–47.
Feth El Zahar, H., Wafa, A., Richard, C., Thierry, H., Christine, M., Marie-France, M., Christophe, M., Lionel, R., Jèrùme, B., and Odile, B. (2007). Identification of cellulolytic bacteria in soil by stable isotope probing. Environ. Microbiol. 9, 625–634.
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007a). Toward and ecological classification of soil bacteria. Ecology 88, 1354–1364.
Fierer, N., Breitbart, M., Nulton, J., Salamon, P., Lozupone, C., Jones, R., Robeson, M., Edwards, R. A., Felts, B., Rayhawk, S., Knight, R., Rohwer, F., and Jackson, R. B. (2007b). Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microbiol. 73, 7059–7066.
Fierer, N., Jackson, J. A., Vilgalys, R., and Jackson, R. B. (2005). Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl. Environ. Microbiol. 71, 4117–4120.
Fierer, N., and Schimel, J. P. (2002). Effects of drying-rewetting frequency on soil carbon and nitrogen transformations. Soil Biol. Biochem. 34, 777–787.
Fu, S., and Cheng, W. (2002). Rhizosphere priming effects on the decomposition of soil organic matter in C4 and C3 grassland soils. Plant Soil 238, 289–294.
Gans, J., Wolinsky, M., and Dunbar, J. (2005). Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309, 1387–1390.
Garbeva, P., and De Boer, W. (2009). Inter-specific interactions between carbon-limited soil bacteria affect behavior and gene expression. Microb. Ecol. 58, 36–46.
Green, J. L., Bohannan, B. J. M., and Whitaker, R. J. (2008). Microbial biogeography: from taxonomy to traits. Science 320, 1039–1043.
Griffiths, R. I., Manefield, M., Ostle, N., McNamara, N., O’Donnell, A. G., Bailey, M. J., and Whiteley, A. S. (2004). 13CO2 pulse labelling of plants in tandem with stable isotope probing: methodological considerations for examining microbial function in the rhizosphere. J. Microbiol. Methods 58, 119–129.
Hanson, C. A., Allison, S. D., Bradford, M. A., Wallenstein, M. D., and Treseder, K. K. (2008). Fungal taxa target different carbon sources in forest soil. Ecosystems 11, 1157–1167.
Hawkes, C. V., DeAngelis, K. M., and Firestone, M. K. (2007). “Root interactions with microbial communities and processes,” in The Rhizosphere: An Ecological Perspective, eds J. L. Whitbeck, and Z. G. Cardon (San Diego, CA: Academic Press), 1–31.
Horner-Devine, M. C., and Bohannan, B. J. (2006). Phylogenetic clustering and overdispersion in bacterial communities. Ecology 87, S100–S118.
Janssen, P. H. (2006). Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72, 1719–1728.
Kato, K., Kozaki, S., and Sakuranaga, M. (1998). Degradation of lignin compounds by bacteria from termite guts. Biotechnol. Lett. 20, 459–462.
Kern, H. W., and Kirk, T. K. (1987). Influence of molecular size and ligninase pretreatment on degradation of lignins by Xanthomonas sp. strain 99. Appl. Environ. Microbiol. 53, 2242–2246.
Kirk, T. K., and Farrell, R. L. (1987). Enzymatic “combustion”: the microbial degradation of lignin. Annu. Rev. Microbiol. 41, 465–501.
Klappenbach, J. A., Dunbar, J. M., and Schmidt, T. M. (2000). rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 66, 1328–1333.
Knorr, W., Prentice, I. C., House, J. I., and Holland, E. A. (2005). Long-term sensitivity of soil carbon turnover to warming. Nature 433, 298–301.
Krishna, M., and Sunil, K. (1993). Degradation of veratraldehyde by Alcaligenes paradoxus. FEMS Microbiol. Lett. 108, 361–366.
Kurakake, M., Ide, N., and Komaki, T. (2007). Biological pretreatment with two bacterial strains for enzymatic hydrolysis of office paper. Curr. Microbiol. 54, 424–428.
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120.
Lee, Z. M. P., Bussema, C., and Schmidt, T. M. (2009). rrndb: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acids Res. 37, D489–D493.
Lesuffleur, F., Paynel, F., Bataillé, M.-P., Le Deunff, E., and Cliquet, J.-B. (2007). Root amino acid exudation: measurement of high efflux rates of glycine and serine from six different plant species. Plant Soil 294, 235–246.
Letunic, I., and Bork, P. (2007). Interactive tree of life (itol): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128.
Lichtfouse, R., Dou, S., Houot, S., and Barriuso, E. (1995). Isotope evidence for soil organic carbon pools with distinct turnover rates – ii. Humic substances. Org. Geochem. 23, 845–847.
Lin, C. H., Bourque, G., and Tan, P. (2008). A comparative synteny map of Burkholderia species links large-scale genome rearrangements to fine-scale nucleotide variation in prokaryotes. Mol. Biol. Evol. 25, 549–558.
Mahenthiralingam, E., and Drevenik, P. (2007). “Comparative genomics of Burkholderia species,” in Burkholderia, eds T. Coeyne, and P. Vamdamme (Norwich: Horizon Scientific Press), 53–79.
Mathee, K., Narasimhan, G., Valdes, C., Qiu, X., Matewish, J. M., Koehrsen, M., Rokas, A., Yandava, C. N., Engels, R., Zeng, E., Olavarietta, R., Doud, M., Smith, R. S., Montgomery, P., White, J. R., Godfrey, P. A., Kodira, C., Birren, B., Galagan, J. E., and Lory, S. (2008). Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. U.S.A. 105, 3100–3105.
Mitsui, R., Kusano, Y., Yurimoto, H., Sakai, Y., Kato, N., and Tanaka, M. (2003). Formaldehyde fixation contributes to detoxification for growth of a nonmethylotroph, Burkholderia cepacia TM1, on vanillic acid. Appl. Environ. Microbiol. 69, 6128–6132.
Moonmangmee, S., Kawabata, K., Tanaka, S., Toyama, H., Adachi, O., and Matsushita, K. (2002). A novel polysaccharide involved in the pellicle formation of Acetobacter aceti. J. Biosci. Bioeng. 93, 192–200.
Nigel, A., Janet, E. T., and Robin, W. (1995). Degradation of homovanillate by a strain of Variovorax paradoxus via ring hydroxylation. FEMS Microbiol. Lett. 134, 213–219.
Papke, R. T., and Ward, D. M. (2004). The importance of physical isolation to microbial diversification. FEMS Microbiol. Ecol. 48, 293–303.
Paul, E. A., Morris, S. J., and Böhm, S. (2001). “The determination of soil C pool sizes and turnover rates: biophysical fractionation and tracers,” in Assessment Methods of Soil Carbon, eds R. Lal, J. M. Kimble, R. F. Follett, and B. A. Stewart (Boca Raton: CRC Press), 193–205.
Personeni, E., Nguyen, C., Marchal, P., and Pages, L. (2007). Experimental evaluation of an efflux-influx model of C exudation by individual apical root segments. J. Exp. Bot. 58, 2091–2099.
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650.
Radajewski, S., Ineson, P., Parekh, N. R., and Murrell, J. C. (2000). Stable-isotope probing as a tool in microbial ecology. Nature 403, 646–649.
Schwartz, E. (2007). Characterization of growing microorganisms in soil by stable isotope probing with H218O. Appl. Environ. Microbiol. 73, 2541–2546.
Schwarz, W. H. (2001). The cellulosome and cellulose degradation by anaerobic bacteria. Appl. Microbiol. Biotechnol. 56, 634–649.
Silby, M., Cerdeno-Tarraga, A., Vernikos, G., Giddens, S., Jackson, R., Preston, G., Zhang, X.-X., Moon, C., Gehrig, S., Godfrey, S., Knight, C., Malone, J., Robinson, Z., Spiers, A., Harris, S., Challis, G., Yaxley, A., Harris, D., Seeger, K., Murphy, L., Rutter, S., Squares, R., Quail, M., Saunders, E., Mavromatis, K., Brettin, T., Bentley, S., Hothersall, J., Stephens, E., Thomas, C., Parkhill, J., Levy, S., Rainey, P., and Thomson, N. (2009). Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol. 10, R51.
Sim, S. H., Yu, Y., Lin, C. H., Karuturi, R. K. M., Wuthiekanun, V., Tuanyok, A., Chua, H. H., Ong, C., Paramalingam, S. S., Tan, G., Tang, L., Lau, G., Ooi, E. E., Woods, D., Feil, E., Peacock, S. J., and Tan, P. (2008). The core and accessory genomes of Burkholderia pseudomallei: implications for human melioidosis. PLoS Pathog. 4, e1000178. doi: 10.1371/journal.ppat.1000178
Smalla, K., Wieland, G., Buchner, A., Zock, A., Parzy, J., Kaiser, S., Roskot, N., Heuer, H., and Berg, G. (2001). Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed. Appl. Environ. Microbiol. 67, 4742–4751.
Sonoki, T., Obi, T., Kubota, S., Higashi, M., Masai, E., and Katayama, Y. (2000). Coexistence of two different o demethylation systems in lignin metabolism by Sphingomonas paucimobilis SYK-6: cloning and sequencing of the lignin biphenyl-specific o-demethylase (ligx) gene. Appl. Environ. Microbiol. 66, 2125–2132.
Stein, L. Y., and Nicol, G. W. (2011). Grand challenges in terrestrial microbiology. Front. Microbiol. 2:6. doi: 10.3389/fmicb.2011.00006
Swenson, N. G. (2009). Phylogenetic resolution and quantifying the phylogenetic diversity and dispersion of communities. PLoS ONE 4, e4390. doi: 10.1371/journal.pone.0004390
Tringe, S. G., Von Mering, C., Kobayashi, A., Salamov, A. A., Chen, K., Chang, H. W., Podar, M., Short, J. M., Mathur, E. J., Detter, J. C., Bork, P., Hugenholtz, P., and Rubin, E. M. (2005). Comparative metagenomics of microbial communities. Science 308, 554–557.
Urbach, E., Vergin, K. L., and Giovannoni, S. J. (1999). Immunochemical detection and isolation of DNA from metabolically active bacteria. Appl. Environ. Microbiol. 65, 1207–1213.
Urich, T., Lanzen, A., Qi, J., Huson, D. H., Schleper, C., and Schuster, S. C. (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 3, e2527. doi: 10.1371/journal.pone.0002527
van Hees, P. A. W., Jones, D. L., Finlay, R., Godbold, D. L., and Lundström, U. S. (2005). The carbon we do not see-the impact of low molecular weight compounds on carbon dynamics and respiration in forest soils: a review. Soil Biol. Biochem. 37, 1–13.
Waldrop, M. P., and Firestone, M. K. (2004). Microbial community utilization of recalcitrant and simple carbon compounds: impact of oak-woodland plant communities. Oecologia 138, 275–284.
Warnecke, F., Luginbuhl, P., Ivanova, N., Ghassemian, M., Richardson, T. H., Stege, J. T., Cayouette, M., Mchardy, A. C., Djordjevic, G., Aboushadi, N., Sorek, R., Tringe, S. G., Podar, M., Martin, H. G., Kunin, V., Dalevi, D., Madejska, J., Kirton, E., Platt, D., Szeto, E., Salamov, A., Barry, K., Mikhailova, N., Kyrpides, N. C., Matson, E. G., Ottesen, E. A., Zhang, X., Hernandez, M., Murillo, C., Acosta, L. G., Rigoutsos, I., Tamayo, G., Green, B. D., Chang, C., Rubin, E. M., Mathur, E. J., Robertson, D. E., Hugenholtz, P., and Leadbetter, J. R. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565.
Webb, C. O., Ackerly, D. D., and Kembel, S. W. (2008). Phylocom: software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics 24, 2098–2100.
Wilson, M., and Lindow, S. E. (1994). Coexistence among epiphytic bacterial populations mediated through nutritional resource partitioning. Appl. Environ. Microbiol. 60, 4468–4477.
Wolfgang, M. C., Kulasekara, B. R., Liang, X., Boyd, D., Wu, K., Yang, Q., Miyada, C. G., and Lory, S. (2003). Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 100, 8484–8489.
Yin, B., Crowley, D., Sparovek, G., De Melo, W. J., and Borneman, J. (2000). Bacterial functional redundancy along a soil reclamation gradient. Appl. Environ. Microbiol. 66, 4361–4365.
Yoder-Himes, D. R., Chain, P. S., Zhu, Y., Wurtzel, O., Rubin, E. M., Tiedje, J. M., and Sorek, R. (2009). Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc. Natl. Acad. Sci. U.S.A. 106, 3976–3981.
Zak, D., Pregitzer, K., Curtis, P., Teeri, J., Fogel, R., and Randlett, D. (1993). Elevated atmospheric CO2 and feedback between carbon and nitrogen cycles. Plant Soil 151, 105–117.
Zhou, J., Xia, B., Treves, D. S., Wu, L. Y., Marsh, T. L., O’Neill, R. V., Palumbo, A. V., and Tiedje, J. M. (2002). Spatial and resource factors influencing high microbial diversity in soil. Appl. Environ. Microbiol. 68, 326–334.
Keywords: soil, bacteria, carbon, substrate quality, bromo-deoxyuridine, microarray, rRNA copy number
Citation: Goldfarb KC, Karaoz U Hanson CA, Santee CA, Bradford MA, Treseder KK, Wallenstein MD and Brodie EL (2011) Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front. Microbio. 2:94. doi: 10.3389/fmicb.2011.00094
Received: 01 February 2011;
Accepted: 18 April 2011;
Published online: 02 May 2011.
Edited by:
Jay Lennon, Michigan State University, USAReviewed by:
Marina Kalyuzhnaya, University of Washington, USABruce A. Hungate, Northern Arizona University, USA
Copyright: © 2011 Goldfarb, Karaoz, Hanson, Santee, Bradford, Treseder, Wallenstein and Brodie. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Eoin L. Brodie, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, MS70A-3317, Berkeley, CA 94720, USA. e-mail: elbrodie@lbl.gov