Evolution and Diversity of the Antimicrobial Resistance Associated Mobilome in Streptococcus suis: A Probable Mobile Genetic Elements Reservoir for Other Streptococci
 Jinhu Huang1
Jinhu Huang1  Jiale Ma1
Jiale Ma1  Kexin Shang1
Kexin Shang1  Xiao Hu2 Yuan Liang1 Daiwei Li2,3
Xiao Hu2 Yuan Liang1 Daiwei Li2,3  Zuowei Wu2
Zuowei Wu2  Lei Dai2 Li Chen1
Lei Dai2 Li Chen1  Liping Wang1*
Liping Wang1*- 1Laboratory of Veterinary Pharmacology and Toxicology, College of Veterinary Medicine, Nanjing Agricultural University, Nanjing, China
- 2Department of Veterinary Microbiology and Preventive Medicine, Iowa State University, Ames, IA, USA
- 3Department of Pharmacy, The Second Hospital of Dalian Medical University, Dalian, China
Streptococcus suis is a previously neglected, newly emerging multidrug-resistant zoonotic pathogen. Mobile genetic elements (MGEs) play a key role in intra- and interspecies horizontal transfer of antimicrobial resistance (AMR) determinants. Although, previous studies showed the presence of several MGEs, a comprehensive analysis of AMR-associated mobilome as well as their interaction and evolution has not been performed. In this study, we presented the AMR-associated mobilome and their insertion hotspots in S. suis. Integrative conjugative elements (ICEs), prophages and tandem MGEs were located at different insertion sites, while 86% of the AMR-associated MGEs were inserted at rplL and rum loci. Comprehensive analysis of insertions at rplL and rum loci among four pathogenic Streptococcus species (Streptococcus agalactiae, Streptococcus pneumoniae, Streptococcus pyogenes, and S. suis) revealed the existence of different groups of MGEs, including Tn5252, ICESp1108, and TnGBS2 groups ICEs, Φm46.1 group prophage, ICE_ICE and ICE_prophage tandem MGEs. Comparative ICE genomics of ICESa2603 family revealed that module exchange and acquisition/deletion were the main mechanisms in MGEs' expansion and evolution. Furthermore, the observation of tandem MGEs reflected a novel mechanism for MGE diversity. Moreover, an in vitro competition assay showed no visible fitness cost was observed between different MGE-carrying isolates and a conjugation assay revealed the transferability of ICESa2603 family of ICEs. Our statistics further indicated that the prevalence and diversity of MGEs in S. suis is much greater than in other three species which prompted our hypothesis that S. suis is probably a MGEs reservoir for other streptococci. In conclusion, our results showed that acquisition of MGEs confers S. suis not only its capability as a multidrug resistance pathogen, but also represents a paradigm to study the modular evolution and matryoshkas of MGEs.
Introduction
There has recently been a worldwide rapid emergence of antibiotic-resistant streptococcal infection. In 2013, the US Centers for Disease Control and Prevention (CDC) declared the top 18 drug-resistant threats in the United States, including the “Serious” level drug-resistant Streptococcus pneumoniae, and the “Concerning” level erythromycin-resistant Streptococcus pyogenes and clindamycin-resistant Streptococcus agalactiae (CDC, 2013). Streptococcus suis is a previously neglected, newly emerging multidrug-resistant zoonotic pathogen that causes meningitis, septicemia, and arthritis in humans, and is one of the major pathogens that led to substantial economic losses in the intensive swine industry (Lun et al., 2007; Gottschalk et al., 2010). Outbreaks of human S. suis infections in China, of which strains were resistant to tetrecyclines and aminoglycosides, in 2005 posed a large public health challenge (Tang et al., 2006; Yu et al., 2006). Previous studies have suggested that S. suis is an important antimicrobial resistance (AMR) reservoir that can contribute to the spread of resistance genes to the above-mentioned streptococci (Palmieri et al., 2011; Huang et al., 2016).
Increasing resistance of streptococci to commonly used antimicrobials including tetracyclines (up to >90%) and macrolides (up to >70%) have been reported worldwide since the 1980s (Aarestrup et al., 1998; Zhang et al., 2008; Palmieri et al., 2011), and a number of AMR genes have been identified (http://faculty.washington.edu/marilynr/) (Roberts, 2005; Palmieri et al., 2011). Previous studies reported that the dissemination of these AMR genes in streptococci are associated with different mobile genetic elements (MGEs) (Roberts and Mullany, 2009; Varaldo et al., 2009). MGEs are ubiquitous among all prokaryotes and play a significant role in horizontal gene transfer (HGT) resulting in intra- and interspecies dissemination of AMR and virulence determinants (Dobrindt et al., 2004; Frost et al., 2005; Juhas et al., 2009; Bellanger et al., 2014). The pool of all genes within MGEs, such as integrative and conjugative elements (ICEs), plasmids, insertion sequences (IS), transposons, prophages, integrons and other genomic islands, are collectively referred to as “mobilome” (Frost et al., 2005).
Several MGEs, carrying AMR determinants for tetracyclines, macrolides, aminoglycosides, and chloramphenicol, have been identified in S. suis (Chen et al., 2007; Holden et al., 2009; Li et al., 2011; Palmieri et al., 2011). A tet(M)- and aadE-carrying 89-kb (89 K) pathogenicity island (PAI), which was found to be unusual in streptococcal toxic shock syndrome (STSS)-causing S. suis strains, was responsible for full bacterial virulence in two major outbreaks in China (Tang et al., 2006; Chen et al., 2007; Li et al., 2008). The 89 K shares similarity with conjugation modules of S. pneumoniae Tn5253, but its integrase belongs to the ICESa2603 family which site-specifically integrates at rplL site instead at rbgA site of Tn5253 (Ayoubi et al., 1991; Li et al., 2011). A tet(W)-carrying prophage ΦSsuD.1 was found in a S. suis strain isolated from a patient with meningitis and is highly similar with the main S. pyogenes Φm46.1 and S. agalactiae λSa04 (Brenciani et al., 2010; Palmieri et al., 2010). More recently, chimeric and tandem of ICEs in streptococci have been reported (Yao et al., 2015; Huang et al., 2016), which indicate interaction of MGEs occurs to extend MGE diversity and complexity.
Although, some MGEs have been identified in S. suis, the knowledge of the prevalence and diversity of MGEs remain largely underscored. Further, interaction of MGEs and their evolution in streptococci have not been identified. In this work, we present the AMR-associated mobilome and their insertion hotspots in S. suis to have a better understanding of S. suis resistome. Comparative and evolution analysis were conducted at rplL and rum loci in S. suis as well as S. pneumoniae, S. pyogenes, and S. agalactiae (in total 6491 genome sequences obtained from GenBank) to explore the diversity and evolution of MGEs. This is the first study that provides the landscape of the prevalence and diversity of MGEs at rplL and rum loci in S. suis, S. pneumoniae, S. pyogenes, and S. agalactiae to comprehend the underlying evolutionary mechanism of MGEs. Our work further confirms the hypothesis that S. suis is an important AMR reservoir and suggest that S. suis might be a MGEs reservoir for other streptococci which promoted the worldwide emergence of antibiotic-resistant streptococcal infection.
Materials and Methods
Bacterial Strains and Antimicrobial Susceptibility Tests
A total of seven S. suis isolates from our routine surveillance on antimicrobial resistance were chosen and sequenced in this study as they displayed different resistance genotype (Table S1). The complete genomes of five strains from GenBank database were also analyzed (Table S1). Isolated colonies were grown on Todd-Hewitt broth (Difco Laboratories) supplemented with 5% (v/v) sheep blood and incubated at 37°C. S. suis experiments were performed in a Biosafety level 2 laboratory. Erythromycin, tetracycline, streptomycin, and kanamycin were purchased from Sigma-Aldrich Co. LLC., Shanghai, China. Antimicrobial susceptibility tests (MICs) were determined by a standard broth micro dilution method.
Whole-Genome Sequencing and Amplification Experiments
The whole genomes of seven strains were sequenced and assembled on the Roche 454 GS Junior System at the Bioinformatics Center of Nanjing Agricultural University (Nanjing, China). These scaffolds of each genome were ordered according to the reference genome of the S. suis strain P1/7 (AM946016), using the Mauve v2.4.0 software (Darling et al., 2004). PCR assays were employed to close the gaps to obtain whole sequences of predicted MGEs.
Identification of Resistance Genes and Mobile Genetic Elements
MLST classification was derived directly from WGS data by the Web-based method (Larsen et al., 2012), and further confirmed with traditional PCR method (http://www.mlst.net/). Acquired antimicrobial resistance genes were identified using ResFinder 2.1 (Zankari et al., 2012).
MGEs candidates were roughly located when genomes were compared with S. suis reference strain P1/7 by MAUVE 2.4.1 (Darling et al., 2004). Prophages were predicted with Phage_Finder (Fouts, 2006). ICEs were predicted with the present of type IV secretion systems (T4SSs) and integrases (Int). Boundaries and insertion sites of both prophages and ICEs were manually checked. ISs were identified with ISfinder (Siguier et al., 2006). Transposons were identified by comparing candidate regions containing associated transposase genes against the NCBI nucleotide database. Putative insertion sites and att sequences were manually identified.
Analysis of Insertions At rplL and rum Loci
To obtain the insertion landscape at rplL and rum loci, 523 genome sequence of S. suis were obtained from GenBank (December, 2015). For rplL and rum loci, sequences between rplL and hdy genes as well as rum and glf genes were analyzed, respectively. Three conditions will be considered: (1) no insertion, sequence length will be ~128 or ~561 bp; (2) with insertion and gap, no sequence will be acquired; (3) with full length insertion, sequence length will be insertion length plus ~128 or ~561 bp. The obtained sequences with insertion were further annotated by an online PATRIC server (Wattam et al., 2014). ISs, prophages, transposons, and ICEs were identified as above mentioned.
Comparative Analysis of ICEs Groups
For ICE identification, signature proteins of integrase, relaxase and VirB4 were searched by BLASTp comparison. Filters were defined by the coverage (>25% and cover function domain), E-value (1e-5) and length (>300 aa for Int, >150 aa for relaxases, and >320 aa for VirB4). If all three proteins were present, the elements were considered ICEs, elements that only harbored integrase and partially or no relaxase and VirB4 were considered defective ICEs (dICEs). Similar analysis of insertions was also done in 637 S. agalactiae, 5063 S. pneumoniae, and 268 S. pyogenes.
Phylogenetic tree of integrases, relaxases and VirB4 proteins were further analyzed. Protein identity of less than 60% was considered as different clade. ICEs were classified by the presence of signature integrases, relaxase, VirB4, and AMR profiles.
Two-hundred and ten ICESa2603 families of ICEs were retrieved from insertions at rplL and rum sites. BLAST atlas maps were built by comparing ICEs with reference sequences of 89 K (CP000407) using BLASTn with a >80% identity threshold within the CGView Comparison Tool (Grant et al., 2012).
Conjugation Assay
In mating experiments, two derivative strains S. suis P1/7RF (The same as BAA-853RF) and SS-1RF (tetracycline- and erythromycin-susceptible but rifampin- and fusidic acid-resistant) were used as recipient and S. suis strains (tetracycline- and erythromycin-resistant but rifampin- and fusidic acid- susceptible) which harbored different MGEs were utilized as donors. Filter mating assays were performed as previously described (Li et al., 2011). The transconjugant was further confirmed by PCR, sequencing, and MLST typing.
Fitness Measurements
The fitness difference between the MGEs-carrying clinical isolates and the reference strain P1/7 were calculated by in vitro growth and competition assay. In vitro competition, culture of each competitor was adjusted to OD600 = 0.2, mixed in a 1:1 ratio, and diluted 1:100 in 5 mL for 10 days. The cfu of the competitors was counted by plating onto medium with or without erythromycin and tetracycline. The relative fitness (w) was determined in competition experiments in triplicate and repeated at least twice, as previously described (Starikova et al., 2013).
Data Analysis
Statistical analysis was performed and illustrated with GraphPad Prism 5 (GraphPad Software, Inc.). Insertions analysis of the prevalence of each MGEs groups in these four species and statistics of growth rate and relative fitness (w) in fitness assay were analyzed using student t-test. A P < 0.05 was considered statistically significant.
Nucleotide Sequence Accession Number
The Whole Genome Shotgun (WGS) sequence of YY060816 has been deposited at GenBank under the accession number AYSB00000000. Sequences of 17 MGEs identified have been deposited at GenBank (Accessions: KX077882-KX077898).
Result
AMR-Associated Mobilome in S. suis Isolates
To gain insight into the role of MGEs in dissemination of antimicrobial resistance, seven representative isolates were sequenced. The draft genomes were ordered and compared with reference strain P1/7. Finally, we identified 17 intact MGEs in seven strains including six ICEs, eight prophages, two ICE_prophage type tandem composite mobile genetic elements (here assigned as CMGEs), and one ICE_ICE type tandem ICEs (Figure S1 and Table S1). MLST type, serotype, and acquired AMR genes of the isolates are listed in Table S1. AMR genes mediating resistance to macrolides [erm(B), mef (A), and msr(D)], tetracyclines [tet(O), tet(40), tet(L), tet(S), and tet(W)], and aminoglycosides [aph(3′)-III, sat4, aac(6′)-aph(2″), and aadE] were carried by corresponding MGEs (Table S1).
The size, GC content, insertion site, and insertion sequence of each MGEs were further analyzed (Table S1). ICEs were usually integrated at 3′ terminal of the rplL gene with the exception of ICESsuJH1308-1 (in SSU0468) and ICESsuZJ20091101-1 (in SSU1262) (Figure 1A). Conserve CDs of ICEs including the modules of integration/excision (Int and Xis) and conjugation (relaxase and T4SS proteins) are presented in ICESsu05SC260, ICESsuJH1308-2, ICESsuLP081102, and ICESsuJH1301 (Figure 1A), which can be classified into ICESa2603 family according to the ICE core structure (conjugation modules; Bi et al., 2012). Three integrases of ICESsuJH1308-1 and ICESsuZJ20091101-1 belonged to the serine family of recombinase instead of the tyrosine family recombinase of IntICESa2603 (Figure 1A and Table S1).
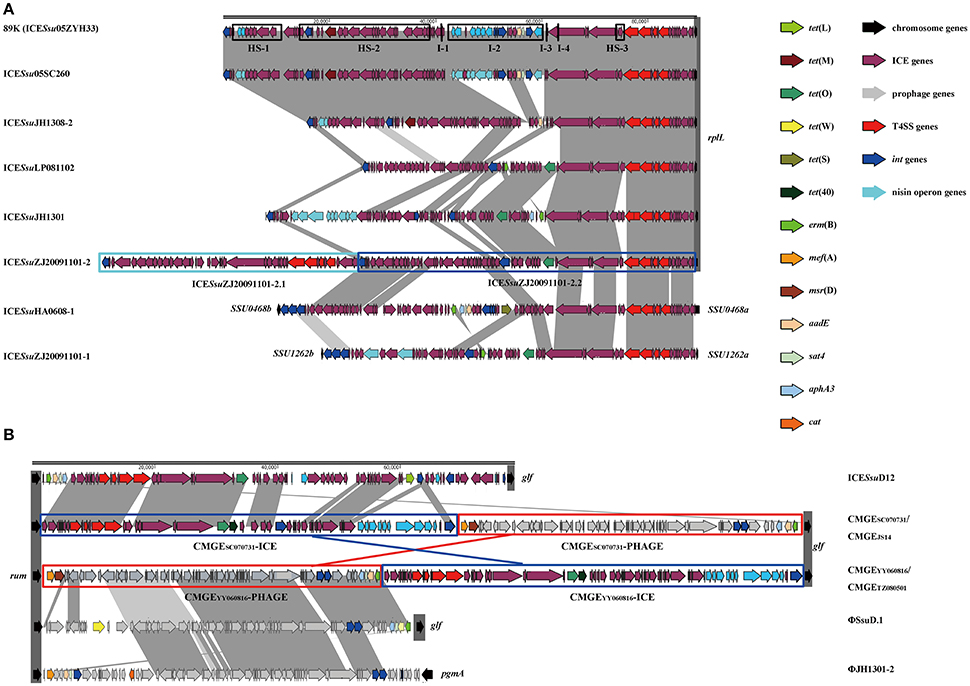
Figure 1. Genetic context of MGEs at rplL (A) and rum (B) loci in S. suis. ORFs in different color were listed representing different functions. Each part of tandem MGEs were highlighted in color boxes. (A) The 89 K PAI were used as references. Accession genes were highlighted in black box with Hotspot (HS) and Insertion (I) as previously described (Huang et al., 2016). ICESsuJH1308-1 and ICESsuZJ20091101-1 inserted in SSU0468 (lysS) and SSU1262 (mutT) were also indicated. (B) Multiple MGEs were inserted in 3′ of rum gene.
Prophages are known to have increased diversity, thus we chose to only focus on AMR-associated prophages in this study. The mef (A)-carrying prophages ΦJH1301-2 and two CMGEs (CMGEYY060816 and CMGETZ080501) were inserted in the 3′ terminal of rum gene (Figure 1B). CMGESC070731, CMGEJS14, ICESsuD12, and ΦSsuD.1 (Palmieri et al., 2010) identified from publicly-available genomes were also found inserted in rum gene (Table S1). A scheme of insertion site of the MGEs to the genome of reference strain P1/7 is shown in Figure 2. These results suggested that rplL and rum loci are major insertion hotspots for integration of MGEs in S. suis.
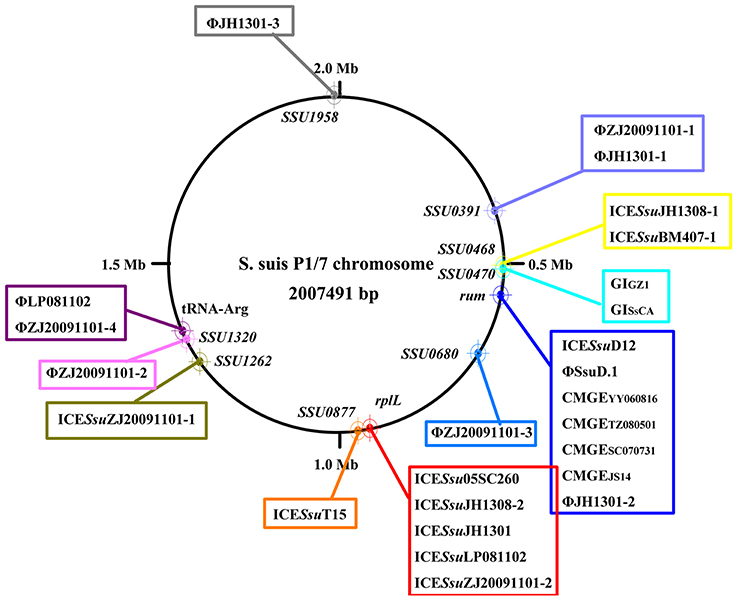
Figure 2. Schematic map of insertion sites of MGEs identified in this study and previously reported (Palmieri et al., 2011). Insertion sites were mapped to reference strain P1/7.
Insertion Analysis at rplL and rum Loci
As the data thus far had shown that MGEs inserted at rplL and rum loci were the main MGEs responsible for antimicrobial resistance in S. suis, to further obtain the prevalence and diversity of MGEs and their interaction, 523 genome sequences of S. suis strains were retrieved from GenBank and analyzed to identify MGEs. At rplL site, 326 (62.32%) of the strains contained insertions. Among them, 194 (59.51%) were fully sequenced with no gap between rplL and hdy genes (Table S2A). However, at rum site, the number of strains containing insertions and fully sequenced with no gap were 201 (38.43%) and 73 (36.32%), respectively (Table S2B). The isolation data, insertion length, MGEs profiles, and AMR genes are summarized in Table S2. ICEs, prophages, and tandem MGEs were identified at rplL and rum loci, but ICEs were the majority of MGEs in both loci.
Classification of ICE Groups
ICEs display a modular structure which had previously been analyzed as the modules of integration/excision, conjugation, and adaption (Bellanger et al., 2014). Herein, signature proteins of integration/excision (integrase) and conjugation (relaxase and VirB4) modules were searched by BLASTp comparison. In total, 265 serine or tyrosine family integrases, 252 relaxases, and 253 VirB4 proteins were detected. Phylogenetic analysis showed that integrases, relaxases, and VirB4 proteins could be classified into five, four, and three distinct clades, respectively (Figure 3). Further domain feathers of each clade/sub-clade are shown in Table S3. Clade I-III integrases had specific integrates in rum site belonging to serine family recombinase. Clade IV and V integrases belonged to tyrosine family integrases and specifically integrated at rplL site. According to the CONJscan-T4SSscan (Guglielmini et al., 2011), all relaxases and VirB4 proteins belonged to MOBp and VirB4 family, respectively. However, the 20 Clade I relaxases that contain a “Relaxase” domain (Pfam03432) were more distant than other groups which also contained an additional C-terminal “Streptin-Immun” domain (Pfam11083). All VirB4 proteins contained a unique “AAA_10” domain (Pfam12846) but belong to three distantly related Clades (Figure 3).
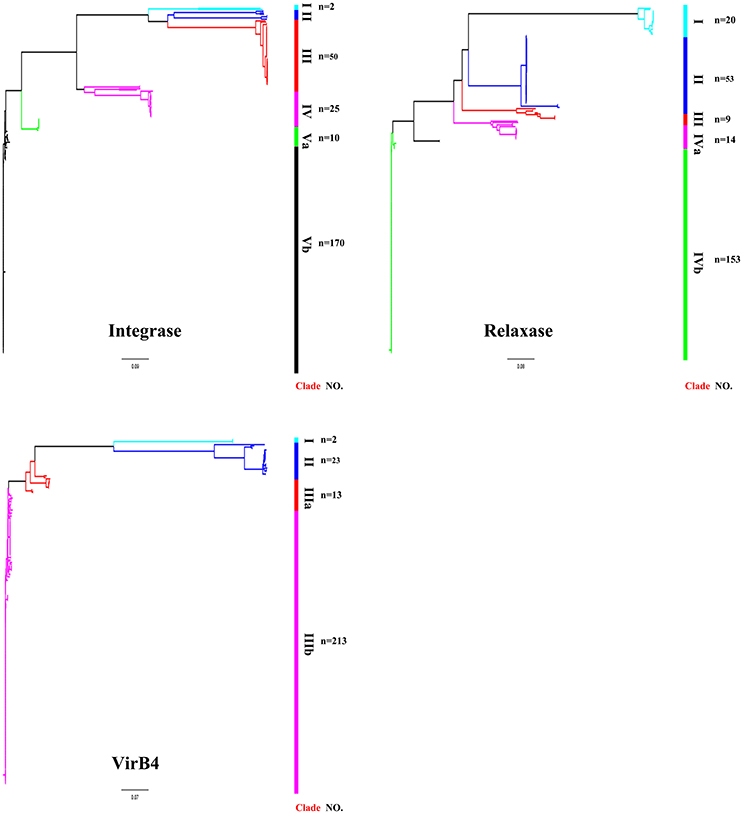
Figure 3. Phylogenetic tree of integrases, relaxases, and VirB4 proteins. Protein sequences were aligned by Clustal Omega and Neighbor-Joining tree was constructed. Sequence identity less than 60% were classified into different Clades. The Clade and number of isolates were shown in right of the tree. For detail information, see Tables S2, S3.
VirB4, the most conserved protein in conjugation module, is used in the classification of ICE groups (Guglielmini et al., 2011), which we also performed in this study. In addition to ICESa2603 family, we identified two novel types of ICEs, designated ICESsuTYPE2 and ICESsuTYPE3, based on phylogenetic trees of VirB4 proteins and followed by analysis of the core conjugation module (Figure 4). We also performed the occurrence of integrases and relaxases within each types of ICEs to evaluate the modular evolution of ICEs. Table 1 summarizes the co-occurrence of integrases, relaxases, VirB4 proteins, and AMR profiles in ICE groups. Further comparison of ICEs to other known streptococcal ICEs allowed us to reclassify ICEs into three groups named by their prototype: Tn5252, ICESp1108, and TnGBS2, respectively (Figure 4).
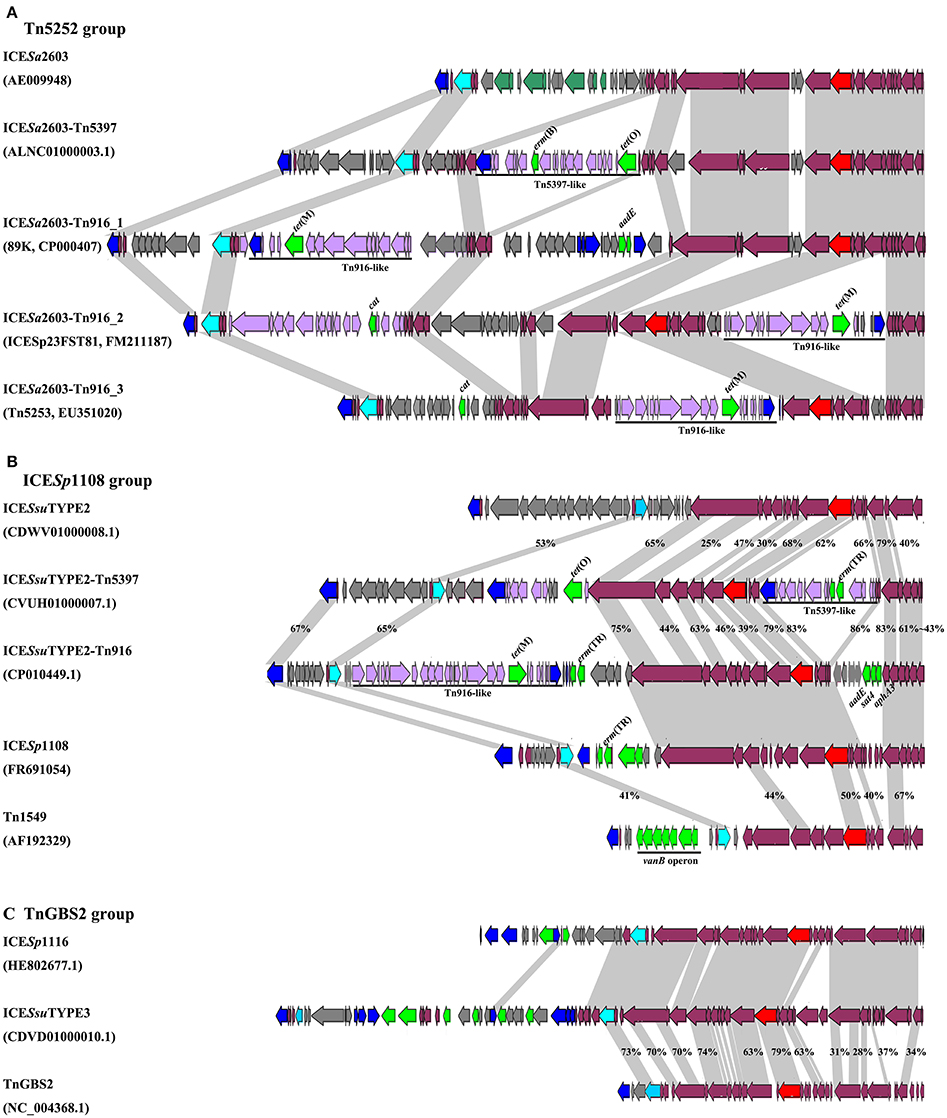
Figure 4. Schematic diagrams of three groups of ICEs inserted at rplL and rum loci. Int, relaxase, VirB4, and AMR determinants were indicated in blue, light blue, red, and green, respectively. MGEs integrated were highlighted in black straight line. ORFs identity was shown in light gray shadow and identity less than 90% were indicated. (A) Comparisons of Tn5252 groups. The prototype ICE Tn5253 sequence was retrieved from NCBI under accession EU351020. Tn5253 integrated at rbgA site. (B) Comparisons of ICESp1108 group. This group of ICEs was previously classified into Tn1549 group, but this study showed different genetic context thus was classified as ICESp1108 group. (C) Comparisons of TnGBS2 group. ICESsuTYPE3 is highly identical with ICESp1116 in core structure, a TnGBS2 family ICEs.
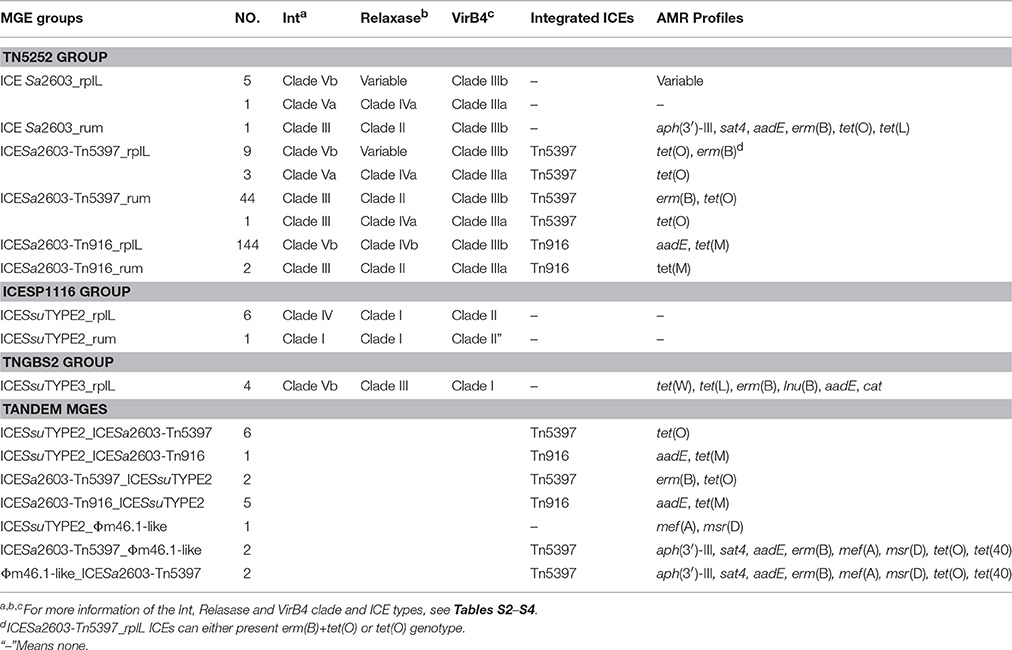
Table 1. Characterization of the ICEs and tandem MGEs at rplL and rum loci in S. suis.
The ICESa2603 family (Tn5252 group) consisted of the largest number of ICEs in both sites. Three subgroups were divided by the chimeric of Tn916 or Tn5397. The ICESa2603 subgroup contained six ICEs at rplL site and one ICE in rum site. Five ICEs at rplL site encode a Clade Vb Int and a Clade IIIb VirB4, and one ICE encoded a Clade Va Int, a Clade IVa relaxase, and a Clade IIIa VirB4. However, the ICESa2603_rum contained a Clade III Int, a Clade II relaxase, and a Clade IIIb VirB4. The ICESa2603-Tn5397 subgroup consisted of 12 ICEs at rplL site and 45 ICEs in rum site. This group ICEs resulted from the integration of Tn5397 in I-2 site of ICESa2603 (Figure 1A). The ICESa2603-Tn916 subgroup contained 144 ICEs at rplL site and two ICEs at the rum site. Similar to ICESa2603-Tn5397, ICESa2603-Tn916 was generated by the integration of Tn916 in I-2 or I-4 site of ICESa2603 (Figure 4A).
The ICESsuTYPE2 family (ICESp1108 group) included six ICEs at rplL site and one ICE in rum site. ICEs of this group encoded a Clade I relaxase and a Clade II VirB4 associated with a Clade IV (rplL site) or Clade I (rum site) Int (Table 1). However, the ICESsuTYPE3 family (TnGBS2 group) was only identified at rplL site. ICEs of this group encoded a Clade Vb Int which was identical to IntICESa2603, but the conjugation module, a Clade IV relaxase and a Clade I VirB4, was distinct from those of ICESa2603 family (Table 1). Notably, the ICESp1108 group encoded multiple AMR genes including tet(W), tet(L), erm(B), lnu(B), aadE, and cat. The schematic of the genetic organization of the three groups of ICEs are shown in Figure 4.
Genetic Variability and Evolution of ICESa2603 Family—Role of Acquisition/Deletion and Module Exchange
The 210 identified ICESa2603 family ICEs were compared to 89 K reference ICE using BLAST to detect genetic variability and rearrangements within core and variable regions. Figure 5 shows the BLAST atlas for genetic variability of all 210 ICEs against the 89 K.
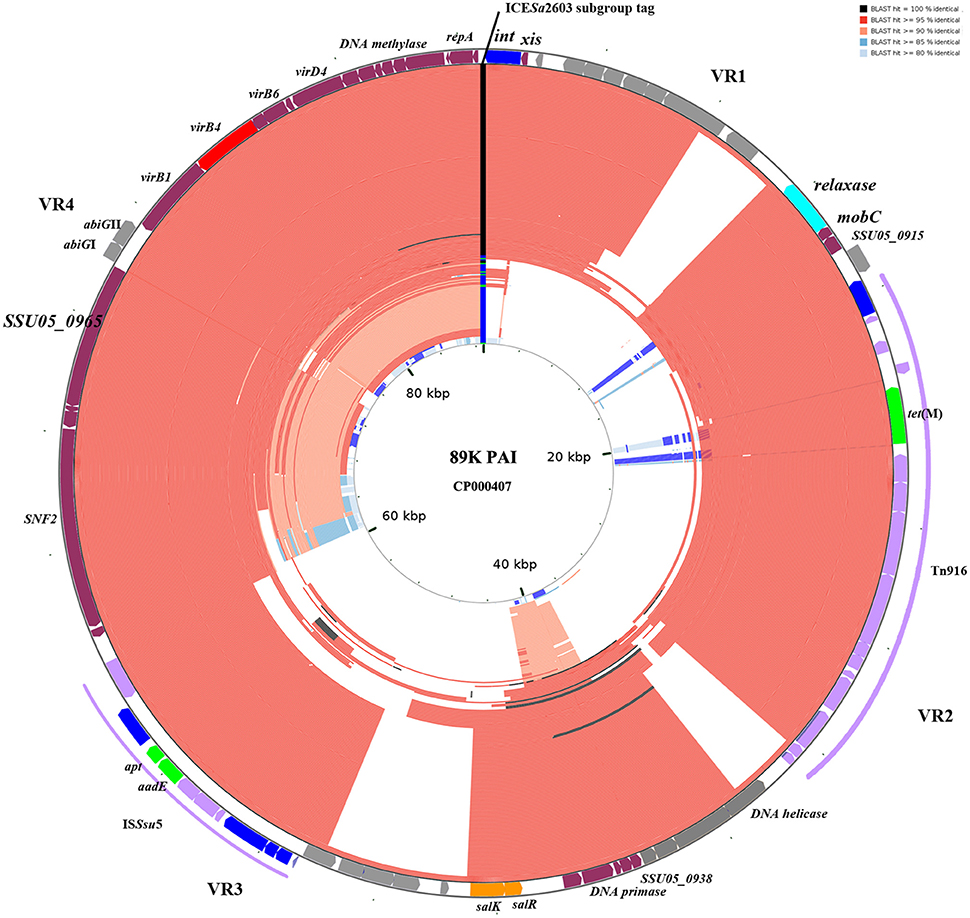
Figure 5. BLAST atlas of Tn5252 group ICEs. A total of 210 Tn5252 groups of ICEs were mapped against with the reference sequence of 89 K. ORFs of 89 K are mapped on the most outer circle. Four variable region (VR) are indicated. Different color in ICESa2603 subgroup tag represent ICESa2603 (green), ICESa2603-Tn5397 (blue), and ICESa2603-Tn916 (black). The 146 ICESa2603-Tn916 subgroup of ICEs are shown to have alignments at Tn916 region. Five ICEs with Clade IIIa VirB4 were shown in most inner circle. Color gradients are proportional to the BLAST percent identity (80–100%).
Genetic variations were mainly observed in four variable regions (VR1-VR4, Figure 5). Gaps of VR1-VR4 in BLAST atlas were due to the different variable-region content of each ICE or absence of a sequence (i.e., the abiGI/abiGII genes presented in VR4 of some ICEs but absent in other ICEs; Figure 5). The SalR/SalK, which is essential for full virulence of highly invasive Streptococcus suis serotype 2 (Li et al., 2008), was only presented in VR3 of ICESa2603-Tn916 subgroups. Further, acquisition of other MGEs into the ICEs was also observed. The ICESa2603-Tn916 was composed of the Tn916 insertion in VR2 of the ICESa2603 structure. An extensive analysis showed that Tn916 could be inserted in different sites which greatly expanded ICE diversity (Figure 4A). Some ICEs of this subgroup even acquired ISSsu5 composite transposon (Figure 5). Deletions were also observed in some genes near variable region probably due to the destruction from recombination/insertion. These results revealed the important role of acquisition of additional genes in ICE diversity and evolution.
The role of module exchange in ICE diversity was evaluated. Most genes in the conjugation module were conserved except three DNA processing genes (relaxase, mobC, and SSU05_0915). Phylogenetic analysis of relaxases and VirB4 proteins showed different combination in subgroups, which suggested that exchange of DNA processing modules may occur although the evolution of relaxases into different Clade cannot be ruled out (Table 1). Integrases of the 210 ICEs analyzed belonged to tyrosine (at rplL site) or serine recombinase (at rum site) families but harbor a common conjugation module (Figure 5 and Table S3). The above ICESsuJH1308-1 and ICESsuZJ20091101-1 contained the same conjugation module of ICESa2603 but encoded three serine recombinases and integrated into different sites (Figure 1A). ICEs encoding the same integration/excision module but with unrelated conjugation modules were also observed in this study (Figure S2). Overall, exchanges of integration/excision module frequently occurred which enhanced ICE diversity and broadened the insertion hotspot for ICE integration. A schematic model for module exchange and sites for integrating of MGEs or other adaptation genes are presented in Figure 6A.
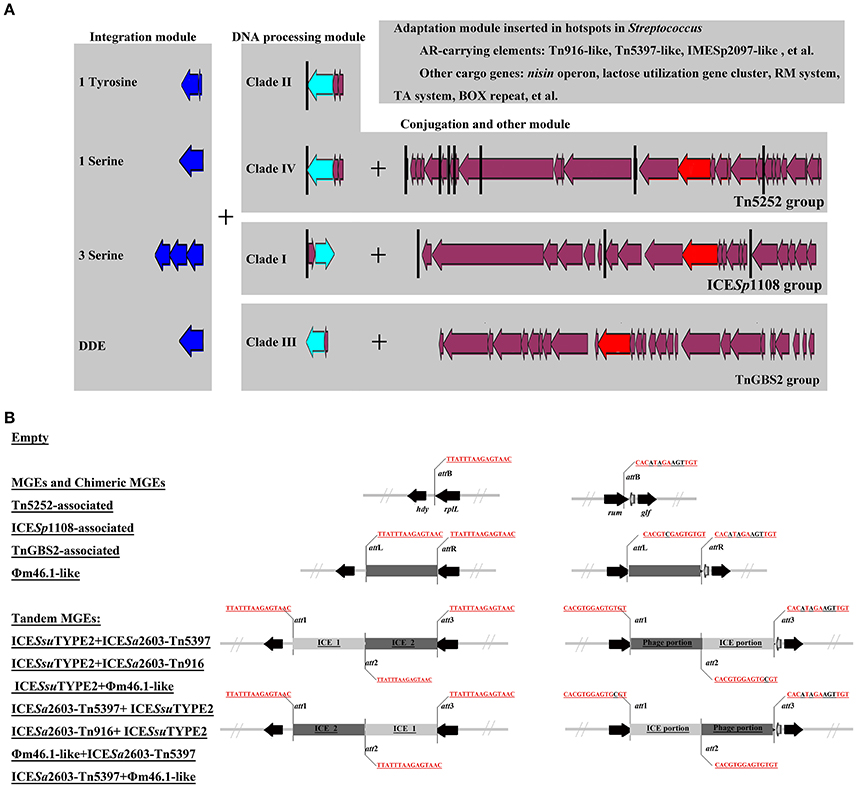
Figure 6. A schematic model for module exchange and acquisition/deletion (A), and tandem accretion (B). (A) Int, relaxase, VirB4, and AMR determinants were indicated in blue, light blue, red, and green, respectively. Core conjugation genes were shown in purple. Black vertical line indicates the insertion sites for variable DNA. Exchange of integration module may occur to form different combination of ICEs. (B) Schematic map of different types of MGEs and empty form of MGEs at rplL and rum sites.
Tandem Accretion—Novel Mechanism for MGE Diversity
An MGE encoding site-specific integrase can integrate into either att site of a cell already harboring an MGE thus can form a tandem MGE. By mobilome analysis of clinical S. suis isolates above, we identified two types of tandem MGEs, ICESsuZJ20091101-2 (ICE_ICE) and CMGEYY060816/CMGETZ080501 (ICE_Prophage). Further insertion analysis at rplL and rum sites identified 17 tandem MGEs displaying various tandem combinations (Table 1). These tandem MGEs typically contained three att sites flanking MGEs (Figure 6B). Notably, the phage_ICE (or ICE_phage) CMGE-carrying isolates were identified in different regions of China from the southwest (SC070731, Sichuan) to southeast (TZ081102, Zhejiang). A clinical screen of the coexistence of the CMGEs' core ICE and phage genes showed the percentage as ~11.11% (data not shown), which may underscore a potential dissemination of this novel CMGEs-carrying S. suis.
Transferability and Fitness of MGEs
In conjugation assay, four clinical MGEs-carrying strains 05SC260, YY060816, LP081102, and ZJ20091101 were used as donors and strains S. suis P1/7RF and SS-1RF were utilized as recipients. However, we only obtained clones of transconjugant carrying ICESsu05SC260 in both P1/7RF and SS-1RF at a low frequency of ~4.25 × 10−8 and 6.1 × 10−8, respectively.
We then detected the fitness of these isolates by in vitro growth and competition. Figure S3 shows the growth curve of four isolates (05SC260, YY060816, LP081102, and ZJ20091101) and a reference strain P1/7. The growth of YY060816 exhibited ~3.5 h detention and the maximum density was 14.7% lower than P1/7 although there were no differences in growth rate at logarithmic phase (Figure S3A). No significant differences were observed between 05SC260, LP081102, ZJ20091101, and P1/7 (Figure S3A). In vitro competition was also performed to determine whether there were fitness differences between those isolates to reference strain P1/7 (Figure S3B); however, no visible fitness cost was observed between different MGE-carrying isolates.
The Prevalence and Diversity of MGEs in S. suis, S. agalactiae, S. pneumoniae, and s. pyogenes
Since rplL and rum loci are also the insertion hotspots in other Streptococcal species, we further analyzed the insertions in three clinically important species, S. agalactiae, S. pneumoniae, and S. pyogenes. Tables S4A,B summarizes the isolation data, insertion length, MGEs profiles, and AMR genes of S. agalactiae, S. pneumoniae, and S. pyogenes isolates obtained from GenBank database. Statistics of the percentage of isolates with insertions within each species is shown in Figure 7A. Among the isolates with insertions, the prevalence of MGEs groups is further shown in Figure 7B. The above-mentioned three groups of ICEs in S. suis were also identified in S. agalactiae, S. pneumoniae, and S. pyogenes which indicated interspecies transfer of MGEs in Streptococcus species. The Tn5252 group presented at rplL site in S. suis (83.51%), S. agalactiae (6.78%) and S. pneumoniae (2.34%), while at rum site the percentage was 65.75% (S. suis), 2.06% (S. agalactiae), and 0.28% (S. pneumoniae), respectively. The ICESp1108 group presented at rum site of S. pyogenes (68.18%), S. agalactiae (18.56%), S. suis (1.37%), and S. pneumoniae (0.62%), but at rplL site only presented in S. suis (3.09%) (Figure 7B and Table S4). The TnGBS2 group only presented in rplL site of S. suis (2.06%) and S. agalactiae (0.85%). Collectively, the prevalence and diversity of MGEs (three groups of ICEs, prophages, and tandem MGEs) is much greater than other three species (Figure 7B).
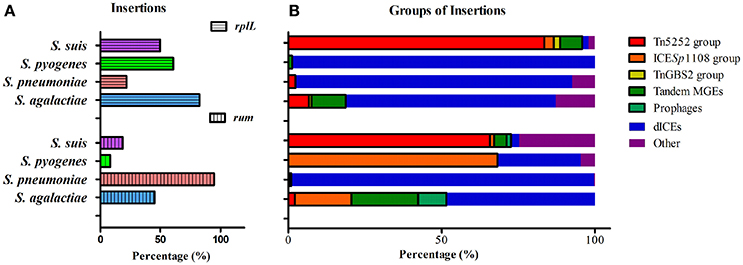
Figure 7. Prevalence of insertions (A) and percentage of MGEs groups in isolates with insertion (B) in four species at rplL and rum sites.
Unlike ICEs in S. suis, in which exchange of integration/excision module and tandem accretion of MGEs play important roles in ICE diversity and evolution, deletion of modules was predominant in ICEs of S. agalactiae, S. pneumoniae, and S. pyogenes at rplL and rum sites, which, as a result, generated defective ICEs (dICEs) probably with no function (Table S4). With the exception of S. pyogenes at the rum site which only had 8.33% (22/268) of the isolates harbor insertions (Figure 7A), the most prevalent MGEs in other species or at other site were dICEs (Figure 7B), including the S. pyogenes 10270-RD.1 (Beres et al., 2006; Beres and Musser, 2007), an ICE-like region of difference which contained relaxase and site-specific recombinase but no conjugation module.
Discussion
We combined our sequenced genomes and publicly-available genomes to identify AMR-associated mobilome and their insertion hotspots in S. suis, an emerging drug-resistant zoonotic pathogen that most recently caused two human outbreaks in China (Tang et al., 2006; Ye et al., 2008). ICEs, prophages and tandem MGEs were detected at different insertion sites (Figure 2); but 86% of the AMR-associated MGEs were inserted at rplL and rum loci (Table S1), two cross-species conserved insertion hotspots in Streptococcus and other Gram-positive bacteria (Depardieu et al., 2003; Srinivasan et al., 2014; Marini et al., 2015; Huang et al., 2016). To date, many AMR-associated MGEs have been reported at rplL and rum sites. At rplL site, these include the ICESa2603 of S. agalactiae (Tettelin et al., 2002), the ICESp23FST81 of S. pneumoniae (Croucher et al., 2009), the 10750-RD.1 (dICEs) of S. pyogenes (Beres and Musser, 2007), and the 89 K and ICESsu32457 of S. suis (Li et al., 2011; Palmieri et al., 2012). At rum site, λSa04 in S. agalactiae (Tettelin et al., 2005), Φm46.1 in S. pyogenes (Brenciani et al., 2010), and ΦSsuD.1 in S. suis (Palmieri et al., 2010) have been identified. However, the prevalence and diversity of MGEs in these species have not been precisely characterized. Moreover, systemic evolutionary analysis involving interaction of MGEs has been lacking in Streptococcus species. By analysis of insertions at rplL and rum loci in 6491 genome of four Streptococcus species (Tables S2, S4), we provide the landscape of the prevalence and diversity of MGEs which deepens our comprehension of the underlying evolutionary mechanism of MGEs in Streptococci.
Among the identified MGEs, ICEs play a key role in bacterial evolution and adaptation. We classified ICEs among the pathogenic Streptococcus species (S. agalactiae, S. pneumoniae, S. pyogenes, and S. suis) into at least three groups on the basis of their conjugation modules: Tn5252, ICESp1108, and TnGBS2 (Figure 4). S. suis appeared to be the species with the highest rates of ICEs since 88.66% (at rplL site) and 67.12% (at rum site) of the strains with insertions harbored ICEs, while dICEs were more prevalent at both sites in S. agalactiae, S. pneumoniae, and S. pyogenes (Figure 7). The Tn5252 group, although its prototype was originally identified in S. pneumoniae (Ayoubi et al., 1991), was widely distributed in S. suis at both sites, 83.51% and 65.75%, respectively, but a lower rate (< 7%) was shown in S. agalactiae, S. pneumoniae, and S. pyogenes. The ICESp1108 group, first reported at rum site of S. pyogenes (Brenciani et al., 2011), was also present in the other three species at rum site but only in S. suis at rplL sites. Further, TnGBS2 group, tandem MGEs, and prophages are presented in S. suis and S. agalactiae but not in S. pneumoniae, and S. pyogenes at both sites (Figure 7). As such, the prevalence and diversity of MGEs in S. suis was much greater than other species. Similar results were also observed in a recent study for identification of ICEs in complete genome of Streptococcus species (Ambroset et al., 2015). Previously, conjugation transfer of Tn5252 group of ICEs between Streptococcus species was reported (Davies et al., 2009; Haenni et al., 2010). Recently, the S. suis Tn5252 group of ICE (ICESsu32457) was shown to have the transferability to S. agalactiae, S. pneumoniae, and S. pyogenes (Palmieri et al., 2012). Recombination of ICESsu32457 and S. agalactiae ICESa2603 generated a tandem ICE, which is transferable to S. pyogenes strains (Marini et al., 2015). All these observations strengthen the notion that S. suis is an important antibiotic resistance reservoir that can contribute to the spread of resistance genes to the above-mentioned streptococci, and is tempting to speculate that S. suis is probably a MGEs reservoir for other streptococci.
Our analysis revealed the role of module exchange in shaping ICEs structure and driving its evolution. Exchange of integration module has been reported in several ICEs like SXT/R391 family (Wozniak et al., 2009) and ICESa2603 family (Huang et al., 2016). However, these analyses only used very few integrases encoded by ICEs and therefore may not be significant. In this study, we showed that Tn5252 groups of ICEs can hijack different types of integrating modules including distinct tyrosine integrases and different number of serine integrases. At least two clades of tyrosine integrases, IntICESa2603 and IntTn5253, were shown in Tn5252 group of ICEs integrating at rplL and rbgA sites, respectively. Tn5252 group of ICEs with a unique integrase or three serine integrases were observed at rum or SSU1262 (mutT) and SSU0468 (lysS) in this study. Although, Tn5252 group of ICEs encodes a DDE transposase was not found in S. suis, this type of ICEs may occur during interaction of the ICEs. Further, exchange of integration module also occurred in ICESp1108 group at least in S. suis if not other streptococci (Tables S2, S4). TnGBS2, that originally encodes a DDE transposase was identified in S. agalactiae (Brochet et al., 2009), was also recently identified in other streptococci (Guerillot et al., 2013). Our study further confirms an exchange of DDE transposase by IntICESa2603 in TnGBS2 group (ICESsuTYPE3) in S. suis (Figure S2), which also suggests the potential occurrence of Tn5252 group of ICEs with a DDE transposase. Exchange of integration module may also occur between ICEs and prophages since the serine integrase of Φm46.1 phylogenetic resides between that of ICESp1108 and Tn5252 (Figure 3).
As module exchange plays key role in forming new types of ICEs and expanding the insertion sites for integration, acquisition/deletion of modules play important roles in ICE diversity and adaption. Deletion of conjugation module rarely occurred in S. suis, while it was frequently identified in S. agalactiae, S. pneumoniae, and S. pyogenes (Figure 7). Acquisition/deletion of adaption module frequently occurred in the MGEs studied. Here, we further discussed the ICESa2603 family (Tn5252 group) in depth to speculate their evolution mechanism. The original host might be S. suis since the most majority of ICEs have been identified in this species, including those within other insertion sites (Ambroset et al., 2015). ICESa2603 at rplL site interacts with other MGEs and exchanges the integration module to potentially form new subgroups of ICEs, for example, ICESa2603_rum, ICESa2603_mutT, ICESa2603_lysS. ICESa2603 can also acquire other MGEs or elements including the tet(M)-carrying Tn916, erm(B)- and/or tet(O)-carrying Tn5397, and aadE-carrying ISSsu5 composite transposon generating the “chimeric” or “composite” MGEs: ICESa2603-Tn5397 and ICESa2603-Tn916. ICESa2603, ICESa2603-Tn5397, and ICESa2603-Tn916 can undertake exchanging of modules between with MGEs or conjugating transfer to other streptococci. ICEs in their host continue to acquire AMR and virulence genes in order to adapt to the host or degrade into dICEs by deletion of core modules of ICEs. Figure 4A shows the different combination of Tn916/Tn5397 integration in different regions of ICESa2603 core structure. Notably, by acquiring of SalK-SalR constituent, the tet(M)- and aadE-carrying 89 K PAI caused two major human outbreaks in China (Li et al., 2008), underscoring the potential spread of ICEs that combination of AMR and virulence determinants.
In addition to module evolution, tandem accretion of MGEs have been reported in Streptococcus thermophiles which forms “tandem MGE.” Pavlovic et al. (2004) reported the ICESt1 from S. thermophiles evolved by deletion and tandem accretion of ICEs and CIMEs resulting from site-specific recombination. A recent study showed that conjugation of S. suis ICESsu32457 and S. agalactiae ICESa2603 generated a tandem ICE, which was transferable between S. pyogenes strains under laboratory conditions (Marini et al., 2015). In the present study, our data indicated that tandem accretion with different combination of ICE_ICE and ICE_prophage occurred in S. suis (~7%) and S. agalactiae (~16%). The prevalence of tandem MGEs in S. suis as well as other streptococci indicating that tandem accretion plays a role in evolution of MGEs (Pavlovic et al., 2004; Bellanger et al., 2014).
Acquisition of MGEs was thought to impose an immediate biological cost, the initial costs of MGE carriage may be mitigated during growth (Starikova et al., 2013). The fitness assays indicated no significant fitness cost between MGE-carrying and MGE-free isolates which may be mitigated during transmission. Furthermore, the ICESa2603-Tn916 group (ICESsu05SC260 in our study) was shown to be transferable, which was consistent with other studies (Li et al., 2011; Huang et al., 2016). Although, the transfer assay of tandem MGEs was not successful in our study, the transferability of tandem MGEs cannot be excluded because the horizontal transfer of tandem MGEs ICESt1 was observed (Bellanger et al., 2009). These results may explain the observation that ICESa2603-Tn916, ICESa2603-Tn5397, and tandem MGEs are widely distributed in S. suis.
In summary, the study provides an overview of AMR-associated mobilome and their insertion hotspots in S. suis, and illustrates the role of module exchange, acquisition/deletion, and tandem accretion in diversity and evolution of MGEs among four pathogenic Streptococcus species which suggests that S. suis is probably a MGEs reservoir for other streptococci.
Author Contributions
JH, LW developed the concept and designed experiments. JH, JM, KS, YL, and LC performed the experiments and collected the data. JH, XH, and ZW conducted all bioinformatics analyses. JH, DL, LD, and LW prepared the manuscript. All authors have contributed to, seen and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study was supported by the National Natural Science Foundation of China (No. 31572567), Qinlan project of Jiangsu Province (2014) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). JH was awarded a scholarship by the China Scholarship Council (CSC; No. 201406850028) and the Research Innovation Program for College Graduates of Jiangsu Province (KYLX_0595).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fcimb.2016.00118
Table S1. Identification of mobile genetic elements identified in the genome of S. suis strains analyzed in this study. aWhole genome sequences of MGEs were closed by PCR. bInsertion site genes were checked referring to P1/7. cPutative att sites were manually checked by compare of the junction sequences.
Table S2A. Identification of MGEs at rplL Locus of S. suis. 523 genome sequence were obtained from genbank. Of the 326 (62.32%) sequences which contain inserts, 194 (59.51%) can obtain non-gap insert sequences. The 194 MGE candidates were further annotated and analyzed for IS, Transposon, phage, and ICEs identification. Int, Relaxase, and VirB4 type and AMR profiles were also analyzed. Strain P1/7 were listed as no insert reference.
Table S2B. Identification of MGEs at rum Locus of S. suis. 523 genome sequence were obtained from genbank. Of the 395 (75.53%) sequences which contain inserts, 73 (18.48%) can obtain non-gap insert sequences. The 73 MGE candidates were further annotated and analyzed for IS, Transposon, phage, and ICEs identification. Int, Relaxase, and VirB4 type and AMR profiles were also analyzed. Strain P1/7 were listed as no insert reference.
Table S3. Classification of Int, Rlaxase, and VirB4. Sequence identity less than 60% were classified into different Clade. Domain information of each Clade were also indicated. For Int, Clade 1–3 belong to serine recombinase family, Clade 4 and 5 belong to tyrosine recombinase family. Identity between Clade may indicate.
Table S4A. Identification of MGEs at rplL Locus of S. agalactiae, S. pneumoniae, and S. pyogene.
Table S4B. Identification of MGEs at rum Locus of S. agalactiae, S. pneumoniae, and S. pyogene.
Figure S1. Comparative genomics of S.suis isolates and localization of MGEs in genomes. The scaffolds of each strains was ordered by MAUVE 2.4 with the whole genome sequenced reference strain including P1/7, 05ZYH33, BM407. The ordered genome sequences were further compared with P1/7. Black Box showed the MGEs after manually confirmed.
Figure S2. Schematic diagram of ICESa2603-Tn5397_rum, ICESa2603-Tn916_rplL, and ICESsuTYPE3_rplL. Int, relaxase, VirB4, and AMR determinants were indicated in blue, light blue, red, and green, respectively. ORFs identity was shown in light gray shadow and identity of amino acids (aa) was indicated. ICESa2603-Tn5397_rum and ICESa2603-Tn916_rplL showed encoding identical conjugation module but different integration module. ICESa2603-Tn916_rplL and ICESsuTYPE3_rplL had identical integration module but distinct conjugation module.
Figure S3. In vitro growth (A) and competition (B) of strain 05SC260, LP081102, YY060816, and ZJ20091101. Strain 05SC260 carry an ICESsu05SC260 which is nearly identical to 89K; LP081102 carry an ICE, ICESsuLP081102, and a prohage, ΦLP081102; YY060816 carry an ICE-phage tandem MGE, CMGEYY060816; and ZJ20091101 carry an ICE-ICE tandem MGE, ICESsuZJ20091101-2 and 5 other MGEs (See Table S1).
References
Aarestrup, F. M., Rasmussen, S. R., Artursson, K., and Jensen, N. E. (1998). Trends in the resistance to antimicrobial agents of Streptococcus suis isolates from Denmark and Sweden. Vet. Microbiol. 63, 71–80. doi: 10.1016/S0378-1135(98)00228-4
Ambroset, C., Coluzzi, C., Guedon, G., Devignes, M. D., Loux, V., Lacroix, T., et al. (2015). New insights into the classification and integration specificity of streptococcus integrative conjugative elements through extensive genome exploration. Front. Microbiol. 6:1483. doi: 10.3389/fmicb.2015.01483
Ayoubi, P., Kilic, A. O., and Vijayakumar, M. N. (1991). Tn5253, the pneumococcal omega (cat tet) BM6001 element, is a composite structure of two conjugative transposons, Tn5251 and Tn5252. J. Bacteriol. 173, 1617–1622.
Bellanger, X., Payot, S., Leblond-Bourget, N., and Guedon, G. (2014). Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol. Rev. 38, 720–760. doi: 10.1111/1574-6976.12058
Bellanger, X., Roberts, A. P., Morel, C., Choulet, F., Pavlovic, G., Mullany, P., et al. (2009). Conjugative transfer of the integrative conjugative elements ICESt1 and ICESt3 from Streptococcus thermophilus. J. Bacteriol. 191, 2764–2775. doi: 10.1128/JB.01412-08
Beres, S. B., and Musser, J. M. (2007). Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS ONE 2:e800. doi: 10.1371/journal.pone.0000800
Beres, S. B., Richter, E. W., Nagiec, M. J., Sumby, P., Porcella, S. F., Deleo, F. R., et al. (2006). Molecular genetic anatomy of inter- and intraserotype variation in the human bacterial pathogen group A Streptococcus. Proc. Natl. Acad. Sci. U.S.A. 103, 7059–7064. doi: 10.1073/pnas.0510279103
Bi, D., Xu, Z., Harrison, E. M., Tai, C., Wei, Y., He, X., et al. (2012). ICEberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Res. 40, D621–D626. doi: 10.1093/nar/gkr846
Brenciani, A., Bacciaglia, A., Vignaroli, C., Pugnaloni, A., Varaldo, P. E., and Giovanetti, E. (2010). Phim46.1, the main Streptococcus pyogenes element carrying mef(A) and tet(O) genes. Antimicrob. Agents Chemother. 54, 221–229. doi: 10.1128/AAC.00499-09
Brenciani, A., Tiberi, E., Bacciaglia, A., Petrelli, D., Varaldo, P. E., and Giovanetti, E. (2011). Two distinct genetic elements are responsible for erm(TR)-mediated erythromycin resistance in tetracycline-susceptible and tetracycline-resistant strains of Streptococcus pyogenes. Antimicrob. Agents Chemother. 55, 2106–2112. doi: 10.1128/AAC.01378-10
Brochet, M., Da Cunha, V., Couve, E., Rusniok, C., Trieu-Cuot, P., and Glaser, P. (2009). Atypical association of DDE transposition with conjugation specifies a new family of mobile elements. Mol. Microbiol. 71, 948–959. doi: 10.1111/j.1365-2958.2008.06579.x
CDC (2013). Antibiotic Resistance Threats in the United States, 2013. Available online at: http://www.cdc.gov/drugresistance/threat-report-2013 (Accessed January 12, 2016).
Chen, C., Tang, J. Q., Dong, W., Wang, C. J., Feng, Y. J., Wang, J., et al. (2007). A glimpse of streptococcal toxic shock syndrome from comparative genomics of S-suis 2 Chinese Isolates. PLoS ONE 2:e315. doi: 10.1371/journal.pone.0000315
Croucher, N. J., Walker, D., Romero, P., Lennard, N., Paterson, G. K., Bason, N. C., et al. (2009). Role of conjugative elements in the evolution of the multidrug-resistant pandemic clone Streptococcus pneumoniae Spain23F ST81. J. Bacteriol. 191, 1480–1489. doi: 10.1128/JB.01343-08
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Davies, M. R., Shera, J., Van Domselaar, G. H., Sriprakash, K. S., and Mcmillan, D. J. (2009). A novel integrative conjugative element mediates genetic transfer from group G Streptococcus to other {β}-hemolytic Streptococci. J. Bacteriol. 191, 2257–2265. doi: 10.1128/JB.01624-08
Depardieu, F., Bonora, M. G., Reynolds, P. E., and Courvalin, P. (2003). The vanG glycopeptide resistance operon from Enterococcus faecalis revisited. Mol. Microbiol. 50, 931–948. doi: 10.1046/j.1365-2958.2003.03737.x
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Fouts, D. E. (2006). Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 34, 5839–5851. doi: 10.1093/nar/gkl732
Frost, L. S., Leplae, R., Summers, A. O., and Toussaint, A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732. doi: 10.1038/nrmicro1235
Gottschalk, M., Xu, J., Calzas, C., and Segura, M. (2010). Streptococcus suis: a new emerging or an old neglected zoonotic pathogen? Future Microbiol. 5, 371–391. doi: 10.2217/fmb.10.2
Grant, J. R., Arantes, A. S., and Stothard, P. (2012). Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics 13:202. doi: 10.1186/1471-2164-13-202
Guerillot, R., Da Cunha, V., Sauvage, E., Bouchier, C., and Glaser, P. (2013). Modular evolution of TnGBSs, a new family of integrative and conjugative elements associating insertion sequence transposition, plasmid replication, and conjugation for their spreading. J. Bacteriol. 195, 1979–1990. doi: 10.1128/JB.01745-12
Guglielmini, J., Quintais, L., Garcillan-Barcia, M. P., De La Cruz, F., and Rocha, E. P. (2011). The repertoire of ICE in prokaryotes underscores the unity, diversity, and ubiquity of conjugation. PLoS Genet 7:e1002222. doi: 10.1371/journal.pgen.1002222
Haenni, M., Saras, E., Bertin, S., Leblond, P., Madec, J. Y., and Payot, S. (2010). Diversity and mobility of integrative and conjugative elements in bovine isolates of Streptococcus agalactiae, S. dysgalactiae subsp dysgalactiae, and S. uberis. Appl. Environ. Microbiol. 76, 7957–7965. doi: 10.1128/AEM.00805-10
Holden, M. T. G., Hauser, H., Sanders, M., Thi, H. N., Cherevach, I., Cronin, A., et al. (2009). Rapid evolution of virulence and drug resistance in the emerging zoonotic pathogen Streptococcus suis. PLoS ONE 4:e6072. doi: 10.1371/journal.pone.0006072
Huang, J., Liang, Y., Guo, D., Shang, K., Ge, L., Kashif, J., et al. (2016). Comparative genomic analysis of the ICESa2603 family ICEs and spread of erm(B)- and tet(O)-carrying transferable 89K-Subtype ICEs in swine and bovine Isolates in China. Front. Microbiol. 7:55. doi: 10.3389/fmicb.2016.00055
Juhas, M., Van Der Meer, J. R., Gaillard, M., Harding, R. M., Hood, D. W., and Crook, D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Li, M., Shen, X., Yan, J., Han, H., Zheng, B., Liu, D., et al. (2011). GI-type T4SS-mediated horizontal transfer of the 89K pathogenicity island in epidemic Streptococcus suis serotype 2. Mol. Microbiol. 79, 1670–1683. doi: 10.1111/j.1365-2958.2011.07553.x
Li, M., Wang, C., Feng, Y., Pan, X., Cheng, G., Wang, J., et al. (2008). SalK/SalR, a two-component signal transduction system, is essential for full virulence of highly invasive Streptococcus suis serotype 2. PLoS ONE 3:e2080. doi: 10.1371/journal.pone.0002080
Lun, Z. R., Wang, Q. P., Chen, X. G., Li, A. X., and Zhu, X. Q. (2007). Streptococcus suis: an emerging zoonotic pathogen. Lancet Infect. Dis. 7, 201–209. doi: 10.1016/S1473-3099(07)70001-4
Marini, E., Palmieri, C., Magi, G., and Facinelli, B. (2015). Recombination between Streptococcus suis ICESsu32457 and Streptococcus agalactiae ICESa2603 yields a hybrid ICE transferable to Streptococcus pyogenes. Vet. Microbiol. 178, 99–104. doi: 10.1016/j.vetmic.2015.04.013
Palmieri, C., Magi, G., Mingoia, M., Bagnarelli, P., Ripa, S., Varaldo, P. E., et al. (2012). Characterization of a Streptococcus suis tet(O/W/32/O)-carrying element transferable to major Streptococcal Pathogens. Antimicrob. Agents Chemother. 56, 4697–4702. doi: 10.1128/AAC.00629-12
Palmieri, C., Princivalli, M. S., Brenciani, A., Varaldo, P. E., and Facinelli, B. (2010). Different genetic elements carrying the tet(W) gene in two human clinical isolates of Streptococcus suis. Antimicrob. Agents Chemother. 55, 631–636. doi: 10.1128/AAC.00965-10
Palmieri, C., Varaldo, P. E., and Facinelli, B. (2011). Streptococcus suis, an emerging drug-resistant animal and human pathogen. Front. Microbiol. 2:235. doi: 10.3389/fmicb.2011.00235
Pavlovic, G., Burrus, V., Gintz, B., Decaris, B., and Guedon, G. (2004). Evolution of genomic islands by deletion and tandem accretion by site-specific recombination: ICESt1-related elements from Streptococcus thermophilus. Microbiology 150, 759–774. doi: 10.1099/mic.0.26883-0
Roberts, A. P., and Mullany, P. (2009). A modular master on the move: the Tn916 family of mobile genetic elements. Trends Microbiol. 17, 251–258. doi: 10.1016/j.tim.2009.03.002
Roberts, M. C. (2005). Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 245, 195–203. doi: 10.1016/j.femsle.2005.02.034
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Srinivasan, V., Metcalf, B. J., Knipe, K. M., Ouattara, M., Mcgee, L., Shewmaker, P. L., et al. (2014). vanG element insertions within a conserved chromosomal site conferring vancomycin resistance to Streptococcus agalactiae and Streptococcus anginosus. MBio 5, e01386–e01314. doi: 10.1128/mBio.01386-14
Starikova, I., Al-Haroni, M., Werner, G., Roberts, A. P., Sorum, V., Nielsen, K. M., et al. (2013). Fitness costs of various mobile genetic elements in Enterococcus faecium and Enterococcus faecalis. J. Antimicrob. Chemother. 68, 2755–2765. doi: 10.1093/jac/dkt270
Tang, J., Wang, C., Feng, Y., Yang, W., Song, H., Chen, Z., et al. (2006). Streptococcal toxic shock syndrome caused by Streptococcus suis serotype 2. PLoS Med. 3:e151. doi: 10.1371/journal.pmed.0030151
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tettelin, H., Masignani, V., Cieslewicz, M. J., Eisen, J. A., Peterson, S., Wessels, M. R., et al. (2002). Complete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiae. Proc. Natl. Acad. Sci. U.S.A. 99, 12391–12396. doi: 10.1073/pnas.182380799
Varaldo, P. E., Montanari, M. P., and Giovanetti, E. (2009). Genetic elements responsible for erythromycin resistance in streptococci. Antimicrob. Agents Chemother. 53, 343–353. doi: 10.1128/AAC.00781-08
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591. doi: 10.1093/nar/gkt1099
Wozniak, R. A., Fouts, D. E., Spagnoletti, M., Colombo, M. M., Ceccarelli, D., Garriss, G., et al. (2009). Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet. 5:e1000786. doi: 10.1371/journal.pgen.1000786
Yao, X., Li, M., Wang, J., Wang, C., Hu, D., Zheng, F., et al. (2015). Isolation and characterization of a native avirulent strain of Streptococcus suis serotype 2: a perspective for vaccine development. Sci. Rep. 5:9835. doi: 10.1038/srep09835
Ye, C., Bai, X., Zhang, J., Jing, H., Zheng, H., Du, H., et al. (2008). Spread of Streptococcus suis sequence type 7, China. Emerg. Infect. Dis. 14, 787–791. doi: 10.3201/eid1405.070437
Yu, H., Jing, H., Chen, Z., Zheng, H., Zhu, X., Wang, H., et al. (2006). Human Streptococcus suis Outbreak, Sichuan, China. Emerg. Infect. Dis. 12, 914–920. doi: 10.3201/eid1206.051194
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: antimicrobial resistance, mobilome, ICEs, MGEs, prophages, Streptococcus, evolution
Citation: Huang J, Ma J, Shang K, Hu X, Liang Y, Li D, Wu Z, Dai L, Chen L and Wang L (2016) Evolution and Diversity of the Antimicrobial Resistance Associated Mobilome in Streptococcus suis: A Probable Mobile Genetic Elements Reservoir for Other Streptococci. Front. Cell. Infect. Microbiol. 6:118. doi: 10.3389/fcimb.2016.00118
Received: 26 July 2016; Accepted: 21 September 2016;
Published: 07 October 2016.
Edited by:
Martin John McGavin, University of Western Ontario, CanadaReviewed by:
Hua Xie, Meharry Medical College, USAGena D. Tribble, University of Texas Health Science Center at Houston, USA
Copyright © 2016 Huang, Ma, Shang, Hu, Liang, Li, Wu, Dai, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liping Wang, wlp71@163.com