Molecular Autopsy for Sudden Death in the Young: Is Data Aggregation the Key?
 Manuel Rueda1*
Manuel Rueda1*
 Jennifer L. Wagner1
Tierney C. Phillips1
Sarah E. Topol1
Jennifer L. Wagner1
Tierney C. Phillips1
Sarah E. Topol1
 Evan D. Muse1,2
Jonathan R. Lucas3
Glenn N. Wagner3
Evan D. Muse1,2
Jonathan R. Lucas3
Glenn N. Wagner3
 Eric J. Topol1,2
Eric J. Topol1,2  Ali Torkamani1*
Ali Torkamani1*
- 1The Scripps Translational Science Institute, Scripps Health, The Scripps Research Institute, La Jolla, CA, United States
- 2Division of Cardiology, Scripps Clinic, La Jolla, CA, United States
- 3Medical Examiner Department, San Diego County, San Diego, CA, United States
The Scripps molecular autopsy study seeks to incorporate genetic testing into the postmortem examination of cases of sudden death in the young (<45 years old). Here, we describe the results from the first 2 years of the study, which consisted of whole exome sequencing (WES) of a cohort of 50 cases predominantly from San Diego County. Apart from the individual description of cases, we analyzed the data at the cohort-level, which brought new perspectives on the genetic causes of sudden death. We investigated the advantages and disadvantages of using WES compared to a gene panel for cardiac disease (usually the first genetic test used by medical examiners). In an attempt to connect complex clinical phenotypes with genotypes, we classified samples by their genetic fingerprint. Finally, we studied the benefits of analyzing the mitochondrial DNA genome. In this regard, we found that half of the cases clinically diagnosed as sudden infant death syndrome had an increased ratio of heteroplasmic variants, and that the variants were also present in the mothers. We believe that community-based data aggregation and sharing will eventually lead to an improved classification of variants. Allele frequencies for the all cases can be accessed via our genomics browser at https://genomics.scripps.edu/browser.
Introduction
More than 10,000 individuals under the age of 45 years die suddenly and unexpectedly in the USA each year (1). While ischemic heart disease remains the predominant cause of cardiac arrest in the older population, most sudden deaths in the young (SDY) stem from cardiomyopathies (including channelopathies) (2, 3). SDY remains an elusive entity for the medical community, mostly because death is often the first manifestation of the disease. Postmortem forensic examination reveals a pre-existing structural abnormality on the heart (a.k.a., autopsy positive) in approximately two-thirds of the cases, however, for the remaining one-third of the cases no cause of death is identified by traditional postmortem examination (a.k.a., autopsy negative) (4–7). The lack of an identifiable diagnosis after autopsy leaves family members without an explanation for the cause of death of their relative and potentially at risk themselves despite having no manifestations of disease.
Many of the underlying causes of SDY are hereditary, thus, a postmortem genetic diagnosis [molecular autopsy (MA)] provides a landmark for both the identification of the cause of death and a potential resolution of the uncertainty for risk to living relatives (8–13). In this regard, minimally invasive molecular tests, enabled by recent technological advances in high-throughput DNA sequencing, have reduced the cost of genomic sequencing relative to the gene panel-based tests traditionally available to medical examiners (14–17). Thus, the MA represents an exciting opportunity to fill in or supplement the knowledge gaps from traditional clinical autopsies while potentially providing accurate genetic information that could facilitate prevention—especially in cases of SDY (6, 18–28).
In this light, we initiated the first systematic and prospective, family-based, MA study in 2014, jointly organized by the San Diego County medical examiner’s (ME) office and our academic medical center. In this study, whole exome sequencing (WES) of participants in addition to family members (preferably parents) was assessed for potential heritable causes of sudden death. Our preliminary analysis from 2016 (29) performed in 25 cases lead to a positive diagnosis—identification of a known or expected pathogenic in a gene related to sudden cardiac death (SCD)—in 20% of the cases. We also observed that variant interpretation and classification was complicated by the lack of a robust database of sudden death-associated genetic mutations. Since then, our efforts have been focused on recruiting other ME centers throughout the nation (e.g., Onondaga County, NY, USA; Las Vegas, NV, USA; Grand Rapids, MI, USA) to establish a repository of genetic variants that will allow for large-scale genetic studies and empower disease prevention.
Three years after initiating our program, we had sequenced 50 sudden death cases including autopsy negative (e.g., sudden infant death syndrome) and autopsy-positive [e.g., hypertrophic cardiomyopathy (HCM), ruptured aneurysm, etc.] cases and interpreted the results. Here, we describe major findings from the study, emphasizing data aggregation, with the objective of providing a broad view of the genetic causes of SDY.
Materials and Methods
Recruitment, Screening, and Consent
The Scripps Institutional Review Board approved the Scripps Molecular Autopsy Study (IRB-14-6386) in 2014. Informed consent was obtained from all living participants or from next of kin for deceased individuals. Individuals between birth and 45 years of age presenting with sudden unexpected death (autopsy-positive cases: where a previously undiagnosed structural abnormality of the heart or other viscera was reported by the ME) or sudden unexplained death (autopsy-negative cases: where no significant findings related to SDY were reported by the ME) etiology were eligible for enrollment. Deaths from an external cause or in persons with known comorbid chronic conditions are excluded. A full description of the inclusion and exclusion criteria can be found at http://clinicaltrials.gov (trial ID: NCT02168088). The San Diego ME’s Office and our research team screened each case for eligibility. In order to be accepted, the forensic autopsies needed to have external and internal examination of the body, as well as histological (including microscopic examination) and toxicological analysis (if requested). Additional cases from non-San Diego offices were screened upon request.
Sequence Data Generation, Analysis, and Interpretation
Once the cases were selected and informed consent was obtained, biological samples were to our lab at the Scripps Translational Science Institute for sequencing of the proband and biological family members. WES was performed on blood samples from the proband, whereas saliva samples were used for parents (when available) in cases where the proband was <18 years old. Full details of exome sequencing, analysis, and interpretation methodology have been described elsewhere (30). Briefly, exome sequence data generation was performed via standard Illumina HiSeq2500 using an Agilent SureSelect target enrichment exome capture that yielded a median coverage of 48× per sample (interquartile range: 43–52, min value: 43, max value: 52) Data analysis was performed according to the Genome Analysis Toolkit variant calling (GATK) best practices (31). After GATK stage, all samples were analyzed for internal quality control (coverage statistics, gender match, relatedness in trios, etc.) with state-of the-art tools (32, 33). Variants were then filtered, annotated, and categorized with our SG-ADVISER tools (34) in three categories: (a) likely causal (mutation previously reported or expected pathogenic in a SCD-related gene), (b) plausibly causal (mutation of unknown significance in an SCD gene), and (c) speculative (mutation previously reported in other disorders) cause of death. A MA research report was considered positive when at least one DNA variant was identified as a likely cause of sudden death. A report was considered negative when no DNA variants that definitively or likely explain the cause of sudden death were identified. Sanger sequencing was utilized to confirm candidate causal variants (35).
Results
Cohort Statistics
From August 2014 to March 2017, we compiled 50 cases, with ages ranging from 2 months to 44 years (Table 1; Table S1 in Supplementary Material), of which 19 were females and 31 were males (1:2 ratio). One of the cases (MA00024) consisted of two male sibling probands (01P and 02P), but, from the two, only the autopsy negative (02P) was included in the global analyses. When the proband was <18 years old we also performed WES on the biological parents (if available) to empower trio-based variant calling analysis that enabled identification of de novo variants. In the two cases where family segregation of potentially informative phenotypes such as palpitations or syncope existed (MA00024 and MA02002), we also sequenced other relatives. Due to the disparity of ages in the cases and the consequential effect of aging on the phenotypes, for analysis purposes we split the cohort in three overlapping groups (Table 1): (i) infants (n = 8; ages from 0 to 12 months), (ii) trios (n = 17; ages from 0 to 18 years), and (iii) full cohort (n = 50). Unless otherwise stated, only variants from the proband are displayed. The San Diego cohort (cases labeled MA0[01]xxx in Table 1) did not show any enrichment for any specific race and is representative of the demographic distribution of the County (31 cases: 52% White, 29% Hispanic, 13% African-American, and 6% Asian). Other states demographic trends were not analyzed due to the small sample size.
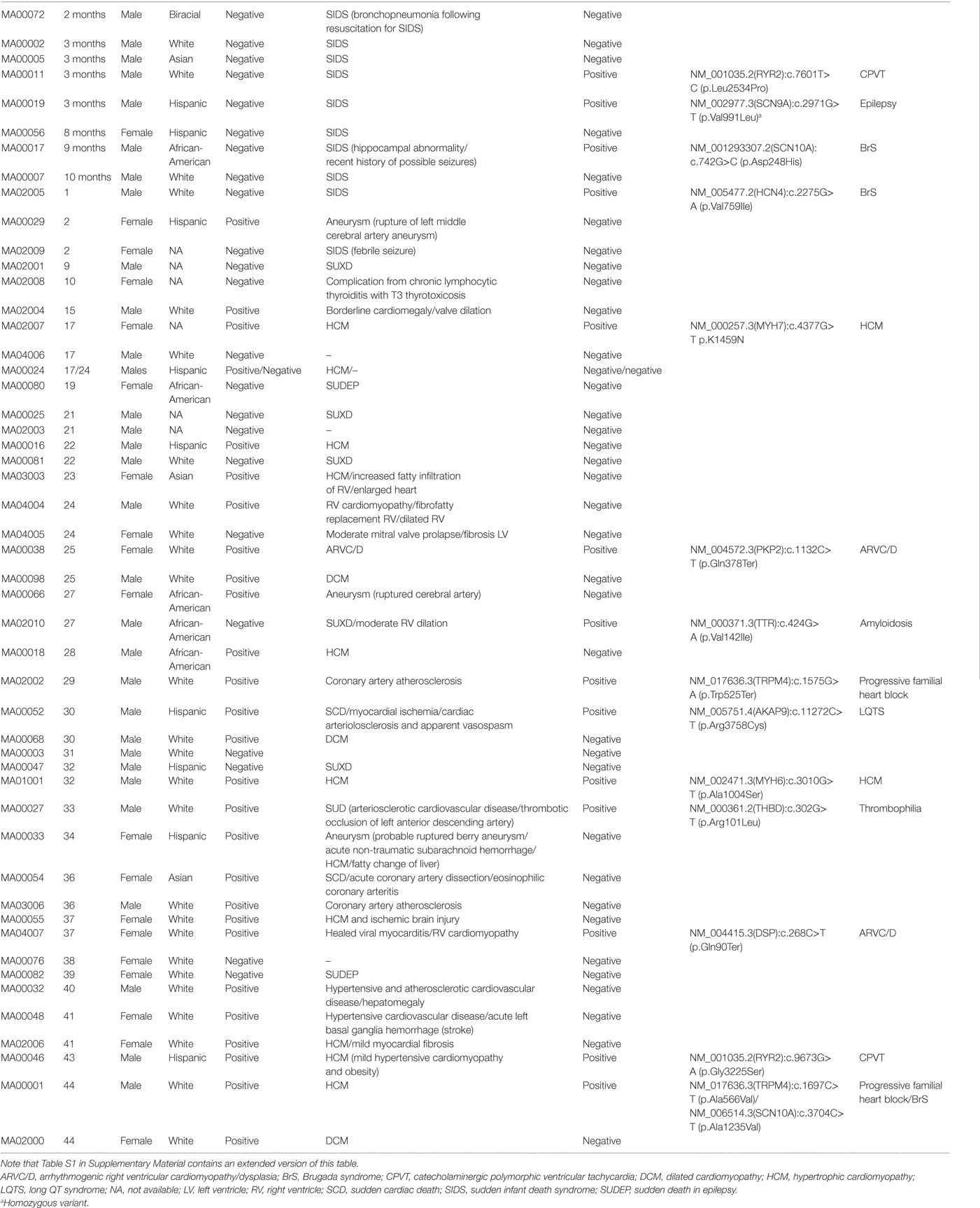
Table 1. Summary of the demographic characteristics and autopsy findings in the 50 cases of sudden death in the young from our study.
The cohort had a bimodal distribution of ages (Figure 1A). There was a peak for infant cases (eight cases) and then an exponential increase peaking with participants of 30–45 years of age. All forensic autopsies were negative for infants, transitioning to positive with increase in age. For the entire cohort, 52% (26 out of 50) of the forensic autopsies (FA) were positive. The FA+ consisted of 10 cases (38%) of HCM, 3 cases (12%) of aneurism, 3 (12%) cases associated with ischemic events due to coronary artery disease (not expected to be significant enough to cause sudden death), and the remaining 10 cases associated with other causes (Table 1; Table S1 in Supplementary Material). In terms of molecular autopsies, 28% (14 out of 50) cases were positive and 72% came out as negative. In this regard, a common concern is whether molecular autopsies improve the diagnostic yield over forensic ones (26). In Table 2, we compared the diagnostic yield from all four possibilities that appear when combining both types of autopsies (molecular/forensic). According to our data, 19 out of 50 cases (38%) were −/−, whereas 9 (18%) were +/+. Interestingly, the number of cases where MA could not help was around the same for FA+ and FA− cases (17 and 19, respectively, out of 50). Molecular autopsies found a likely causal variant in 9 FA+ cases and 5 FA− cases.
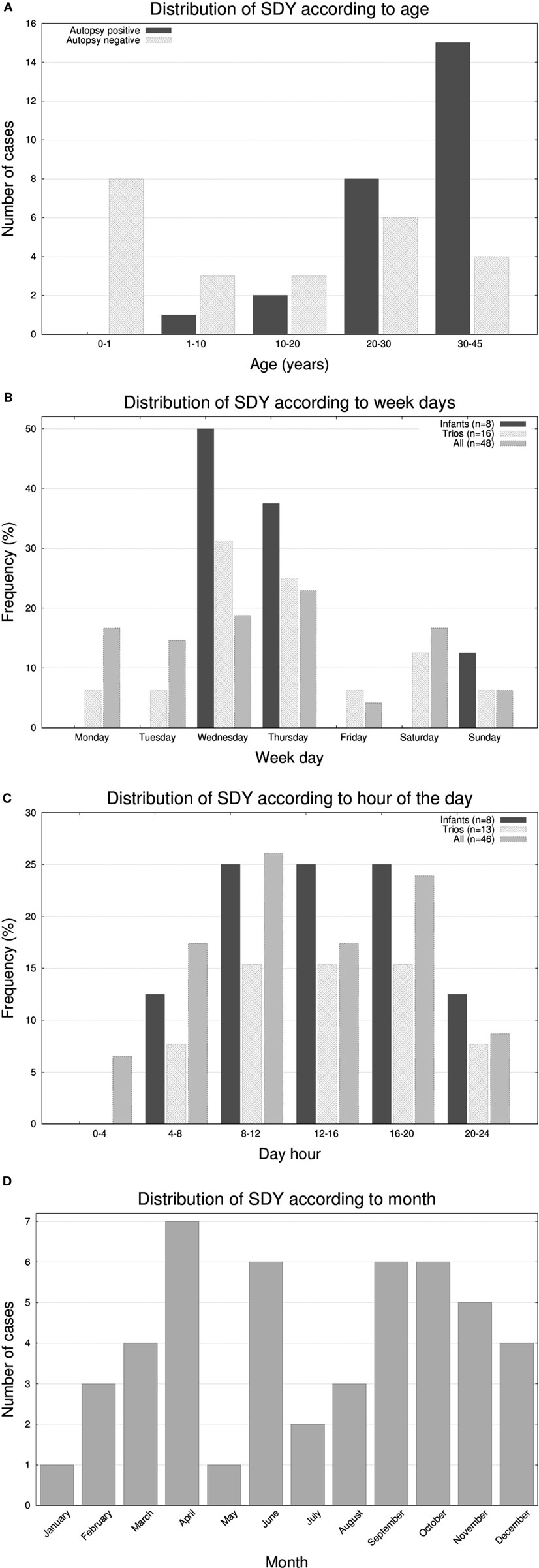
Figure 1. Histograms representing the number of molecular autopsy cases of sudden death in the young (SDY) according to (A) 6 group ages, (B) day of the week, (C) hour of the day, and (D) month of the year.

Table 2. Combined diagnostic yield for forensic autopsies and molecular ones.
It has been reported that SCDs follow circadian/diurnal/seasonal variation (1). To evaluate these trends in our cohort, we represented histograms of the frequency of deaths according to the day of the week (Figure 1B), hour (Figure 1C), and month (Figure 1D). Unexpectedly, we found a substantial accumulation of sudden unexplained deaths during the middle of the week in the very young population (seven out of eight infants passed away on Wednesdays or Thursdays; p-value <0.05). With respect the hour of the day, most deaths occurred in the 8 a.m. to 8 p.m. 12-h window, and, in terms of seasonality, we observed two peaks, one in the month April and another around early Fall.
Variant Distribution in Genes: The Case for Exome versus Gene Panel
Two of the most interesting observations that stem from WES of disease cohorts are the qualitative (location in the genome) and quantitative analysis of pathogenic variants. In our case, after applying GATK’s variant calling pipeline we obtained a median value of 98,667 variants per sample (see full statistics in Table S2 in Supplementary Material; aggregate data for all cases can be accessed at https://genomics.scripps.edu/browser). From these, a median of 80,445 variants per sample passed quality controls. The variant calling format (VCF) files generated were subsequently parsed with our additional in-house filter to discard common variants [variants with a minor allele frequency (MAF) ≥5% in our internal database]. This filter removed a very high percentage of the variants, ending up with a median of 4,684 variants per sample. The final step involved again filtering [keeping only variants with MAF < 1% in 1,000 genomes (36)], plus annotation and prioritization of variants according to the SG-ADVISER protocol (34) leading to a median of 354.5 variants per sample. After applying all these filters, only ~0.4% of the variants were retained from the original VCFs coming from GATK’s pipeline.
Exome sequencing was performed as a first pass in all 50 cases. However, many other laboratories performing varieties of molecular autopsies use more affordable methods such as gene panels specific for heart disease (23, 25, 26, 37). In that regard, a common topic of debate is whether or not “escalating” to exome helps in deciphering more cases, or if it simply adds unnecessary complexity to an already intricate procedure. To shed some light in this issue, here we analyzed the VCF files resulting from WES at both gene panel and exome level. In terms of a gene panel, we restricted the variant search to an ad hoc list of genes (labeled as “panel”) consisting of 233 relevant loci associated with heart disease (see full list of genes in Table S3 in Supplementary Material). For exome, we did a full search without restrictions to particular loci (labeled as “exome”).
In Figure 2, we present the abundance of variants per gene according to exome (Figure 2A) and gene panel (Figure 2B). The transition from exome to gene panel creates an important reduction in the number of variants obtained, going from a median of 354 per sample in exome (interquartile range: 325–396, min value: 235, max value: 739, total number of variants for the 50 cases was 19,505) to a median of 7 (interquartile range: 5–10, min value: 1, max value: 18, total number of variants for the 50 cases was 374). For exome, variants in TTN were the most abundant (56 variants), followed by MUC16 (53 variants) and SSPO (49 variants). TTN gene encodes titin, the largest protein of the human genome [305 kilobases (kb)] a protein involved in many cardiomyopathies (38). MUC16 (mucin 16) is a 132 kb gene encoding the mucin protein, a glycoprotein associated with cancers, whereas SSPO (subcommissural organ spondin) is a 58 kb gene that encodes a protein involved in the modulation of neuronal aggregation. Due to their large size, both TTN and MUC16 are frequently seen in exomes and caution is advised with the interpretation of variants (39). For TTN, the median number of variants per sample was 1 (interquartile range: 0–2, min value: 0, max value: 7), for MUC16 the median number was 1 (interquartile range: 0–1, min value: 0, max value: 13), and for SSPO the median number was 0 (interquartile range: 0–1, min value: 0, max value: 14). We found that MA00066, the case with the largest number of variants (739), carried 7 variants in TTN, 14 variants in SSPO, and 13 variants in MUC16. Case MA00066 is a 27-year-old African-American female for whom the cause of death was reported as a ruptured cerebral artery aneurysm. None of the variants in those three genes was annotated to be pathogenic, and actually our MA research report for this case came out as negative for nuclear variants. For the gene panel, we found again that TTN was the most abundant, with a 10-fold increase with respect the rest of the genes.
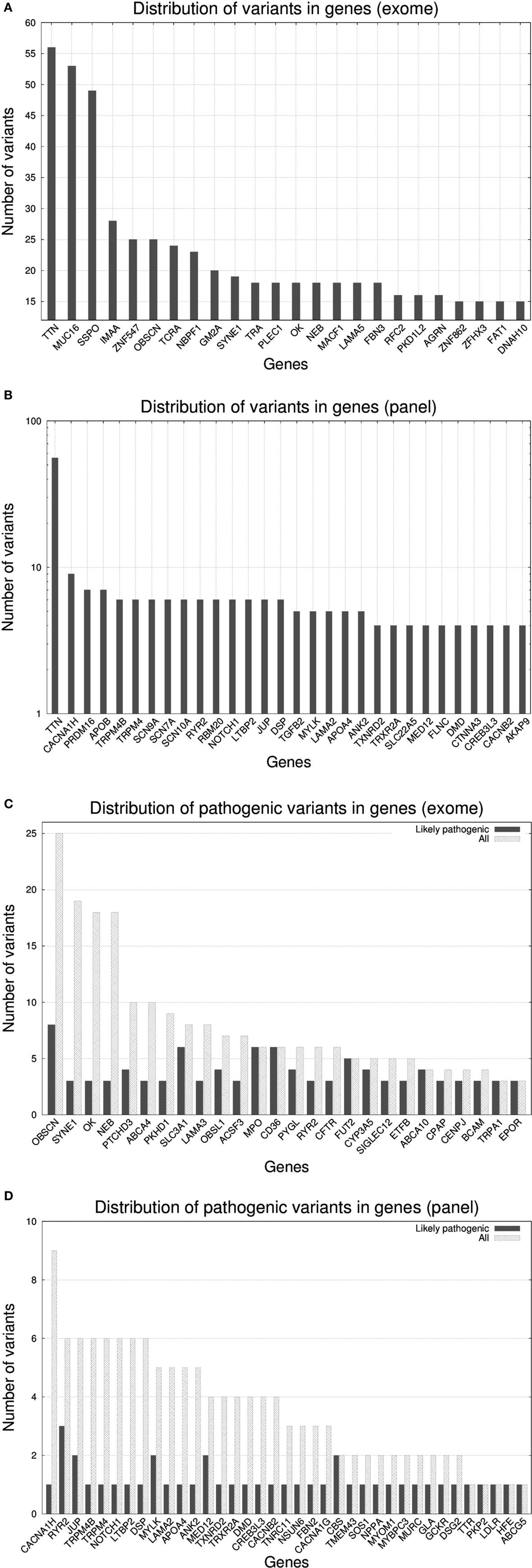
Figure 2. Histograms representing the number of variants per gene* for 50 molecular autopsy cases: (A) when taking into account all exome genes**, (B) when taking into account a gene panel consisting of 233 genes associated to sudden death (note log10 transformation in y-axis), (C) when taking into account exome genes with ≥3 likely pathogenic variants, (D) when taking into account panel genes with ≥1 likely pathogenic variant. *Non-coding RNA loci were excluded from the figures (e.g., AK093733). **Fourteen PCDHG[AB][1-9] loci (protocadherin gamma subfamily A and B) were excluded yet in total they carried 367 variants.
Due to the inherent difficulty of analyzing all exomic variants in a given sample, researches usually resort to methods that prioritize variants according to predicted pathogenicity. According to this, we restricted the search to variants assessed as likely pathogenic according SG-ADVISER (34) clinical categories 1 (variant is previously reported and is a recognized cause of the disorder), and 2 (variant is previously unreported and is of the type that is expected to cause the disorder). SG-ADVISER categories 1–2 overlap with the American College of Medical Genetics scoring guidelines [see discussion in Ref. (34)]. For exomes, the median number of likely pathogenic variants was 10.5 (interquartile range: 8–13, min value: 5, max value: 20) whereas for the gene panel we found that 23 out 50 samples (46%) had zero variants, 18 samples had 1 (36%), 6 samples had 2 (12%), and 3 samples had 3 (6%). According to these numbers, in the context of coding regions, “de-escalating” from exome to gene panel imposes an upper limit in the yield of positive molecular autopsies, as only 54% of cases analyzed with the gene panel carried a likely pathogenic variant. Conversely, when working at exome level, even the individual with the lowest number of likely pathogenic variants carried five of them (yet most variants were in genes a priori not associated with sudden death). With respect the effect of the mutations, for exome we identified 382 missense variants, 49 nonsense variants, and 74 frameshift variants, whereas for the gene panel, 32 were missense variants, 5 nonsense variants, and 2 were frameshift variants.
As seen previously (29), many variants that did not fall under the likely pathogenic category, ended being classified as variants of unknown significance (VUS). A high percentage of them were previously unreported in online variant databases such as ClinVar (40); thus, any source of information that facilitates their classification becomes fundamental. In that sense, estimation of the ratio of pathogenic variants per gene versus the total can be used a reference (41–43). In Figure 2C (exome) and Figure 2D (gene panel), we reported this ratio with a histogram and we observed that there existed genes (e.g., OBSCN, SYNE1, OK, and NEB in exome; CACNA1AH, TRPM4[B], NOTCH1, LTBP2, DSP, LAMA2, APO4, and ANK2 in gene panel) that carried many variants, but only a small fraction were annotated as likely pathogenic. Conversely, genes that did not tolerate mutations had a similar number of total and likely pathogenic variants. The list of genes displayed in Figures 2C,D only overlaps partially because many of the exome-only loci carried more variants than gene panel ones (note the different scale in y-axis).
Do Likely Pathogenic Mutations Cooccur?
After studying the distribution of variants in genes we investigated whether there existed cooccurrence of pathogenic variants in the same genes across individuals. In the case of gene panel, only a few cases carried >1 likely pathogenic variants being the affected genes dissimilar across individuals. In case of the exome, in Figure 3A, we represented the percentage of samples that shared up to three genes (with likely pathogenic variants) with at least another sample. Only 7 out of 50 samples shared three genes, 3 shared two, and all 50 samples shared at least one gene with another sample. Trios and infants had lower percentages due to the smaller group size, which limited the probability of two samples carrying mutations on the same genes. With regard to which were the genes were shared, in Figure 3B, we show the distribution of genes according to the number of samples that carried likely pathogenic mutations in them. The five most abundantly mutated loci were: OBSCN (obscurin, cytoskeletal calmodulin, and titin-interacting RhoGEF) that plays a role in the organization of myofibrils during sarcomere assembly and has been associated with HCM, SLC3A1 (neutral and basic amino acid transport protein rBAT) that is associated with cystinuria, MPO (myeloperoxidase) that has been associated with coronary artery disease, CD36 (cluster of differentiation 36) a class B scavenger receptor implicated in cardiovascular disease, and ABCA4 [ATP-binding cassette, subfamily A (ABC1), member 4] that has been associated with macular degeneration (see Figure 3C). None of the five protein products of these genes are part of the same biological network.
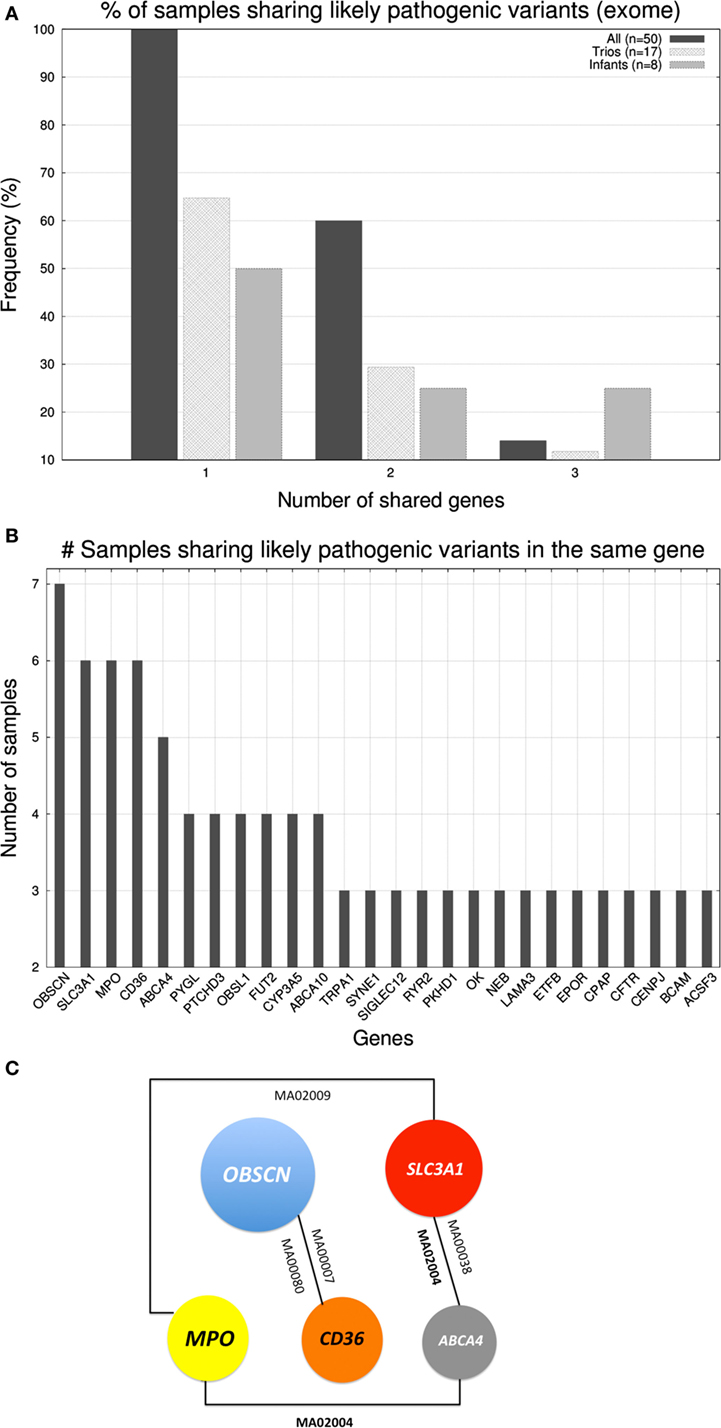
Figure 3. (A) Histogram displaying the percentage of samples that shared up to three genes (with likely pathogenic variants) with at least another sample. (B) Number of samples sharing genes with likely pathogenic variants (exome level). (C) Schematic representation of the cooccurrence of the five most shared genes (shared by ≥5 samples).
Is There a Genetic Fingerprint for Sudden Death?
The cardiovascular forensic examinations of each case often reveal complex phenotypes. For instance, a given individual could have a mixture of HCM, dilated cardiomyopathy, and the fibrofatty replacement typical from arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D). In terms of genetics variants, it is not uncommon to identify variants in genes associated with malignant arrhythmias along with mutations in genes associated with heart muscle disease (25). Under the assumption that similar genotypes will yield similar phenotypes, we investigated if the combination of affected genes could be used to classify samples.
In order to compare the samples we used a genetic fingerprint, created as follows: for each sample, we collapsed all variants (per gene) that fell into SG-ADVISER categories 1–3. We included SG-ADVISER category number 3 (variant is previously unreported and is of the type which may or may not be causative of the disorder) to increase the number of variants, as categories 1–2 yielded only a maximum overlap of three genes between two individuals (see above). Then, we used that gene list to carry out a pairwise (sample-based) comparison using an intersection over union index (Jaccard index), standardized with a Z-score. The resulting matrix of pairwise Z-scores was transformed to a heatmap with R software (44) on which we applied hierarchical clustering (Figure 4). After standardization, we obtained eight pairwise comparisons having Z-scores >3 resulting from the intersection of 6–10 genes over the union of 88–116. The highest Z-score = 5.44 was obtained between cases MA00007 and MA00025 (both forensic autopsy negatives) with an intersection of 10 genes (ASPM, CCDC88C, IDUA, KCNK18, MAST4, MYO15A, PKHD1, SPTBN2, SPTBN5, and SYNE1) over a union of 96.
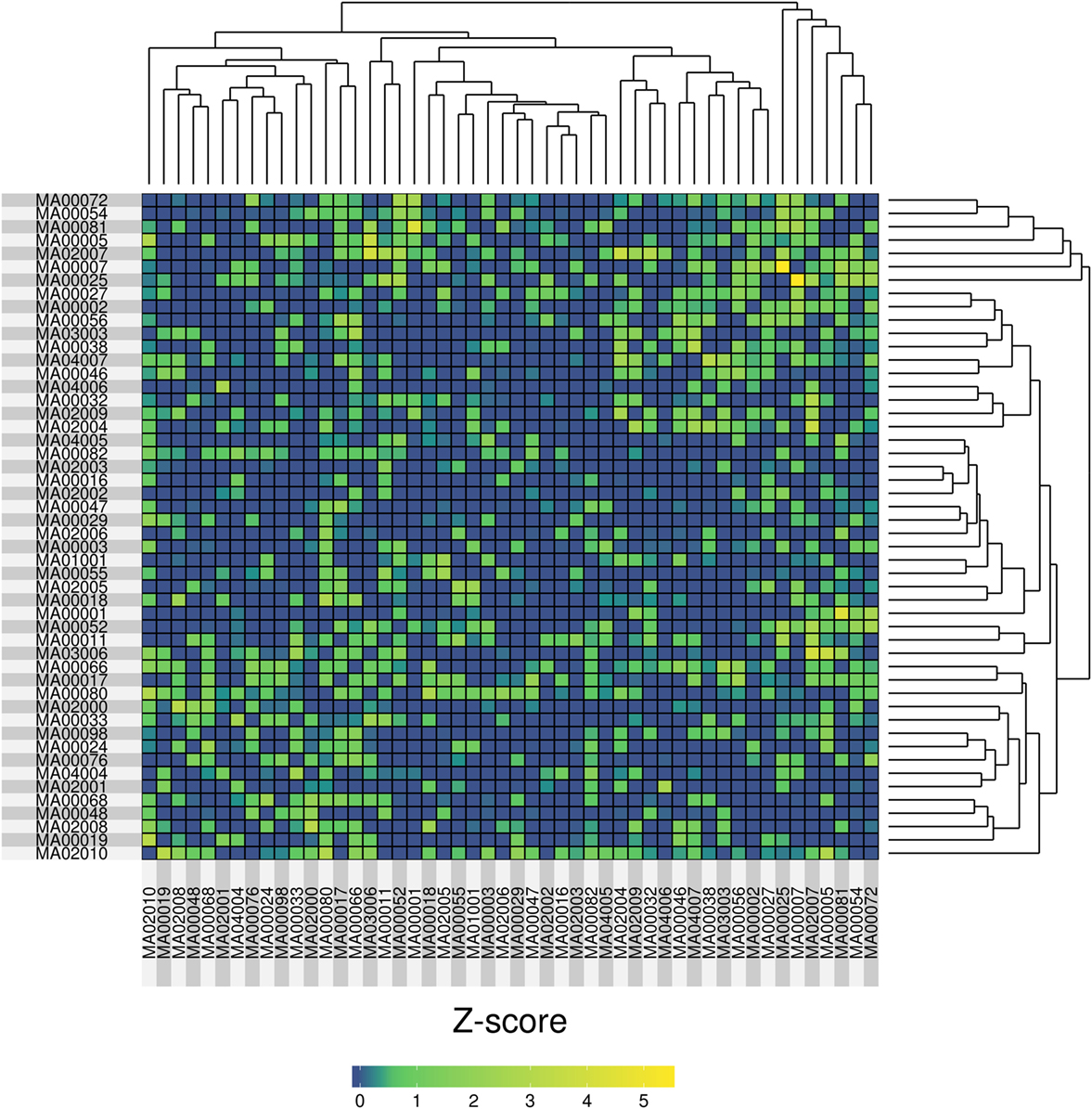
Figure 4. Heatmap representation of the pairwise similarity (measured by a Jaccard index, standardized with a Z-score) among 50 molecular autopsy cases. The Jaccard index was computed using exome genes that carried variants in SG-ADVISER categories 1–3.
Mitochondrial DNA (mtDNA) Variants
Due to the fact that cardiomyocytes are in constant need for energy, all the steps involved in ATP generation are of crucial importance. It is well established that mitochondria play roles in many cardiomyopathies (45), and it has been suggested that depletion of ATP (caused by an external stressor) in defective mitochondria might play a role in some forms of sudden infant death syndrome (10, 46). Despite that, downstream analysis of mtDNA variants is rarely performed in exome-based data (47). Here, we used our bioinformatics expertise in analysis of mtDNA data [methods described elsewhere (47)] to add this information to each case. After the variant calling, we only considered mtDNA variants that possessed a heteroplasmic fraction (HF) >0.2 and a MAF <0.05 in 1,000 Genomes (36).
The median number of heteroplasmic variants per case was 4 (interquartile range: 2–7, min value: 0, max value: 17). It is worth mentioning that the number of variants was not associated with differences in depth of coverage relative to the sequencing step (Figure S1 in Supplementary Material). We compared the number of heteroplasmic variants per sample with respect to age (Figure 5) and we observed that there was no relation between the two parameters, and that 8 (out of the 50) individuals carried ≥10 heteroplasmic variants. From those eight cases, four were infants (half of the infant group). A high percentage of the variants were in the MT-DLOOP region (Table 3), a locus recently associated with total levels of heteroplasmy (47). The heteroplasmic variants were not de novo, as they were also present in the mothers with equally high levels of HFs.
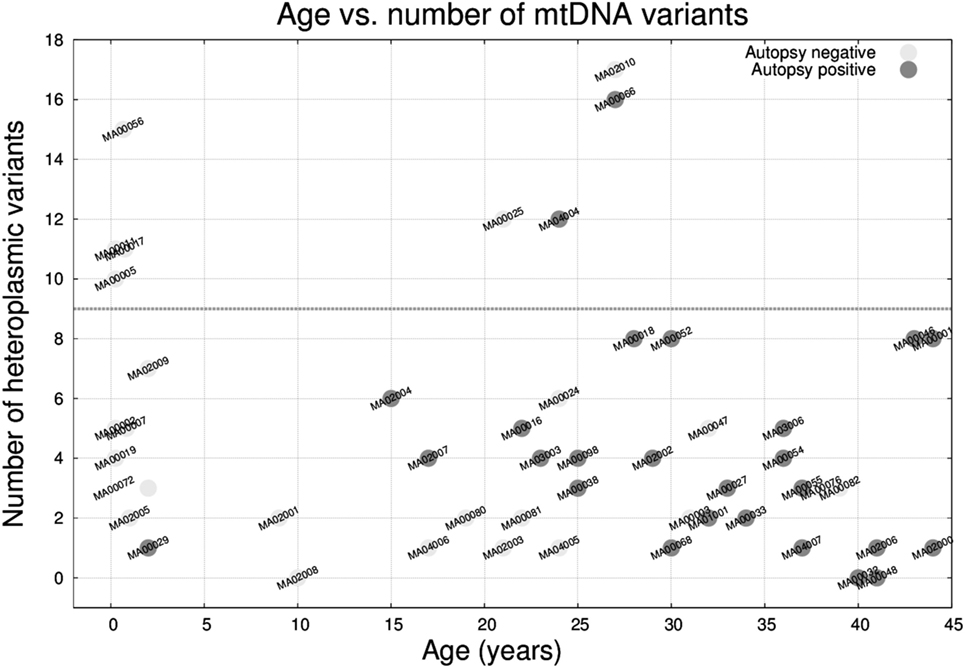
Figure 5. Age versus number of mitochondrial DNA heteroplasmic variants in the 50 molecular autopsy cases colored according to being forensic autopsy positive or negative. Note that 4 out 8 infants plus 4 other cases carried ≥10 variants.
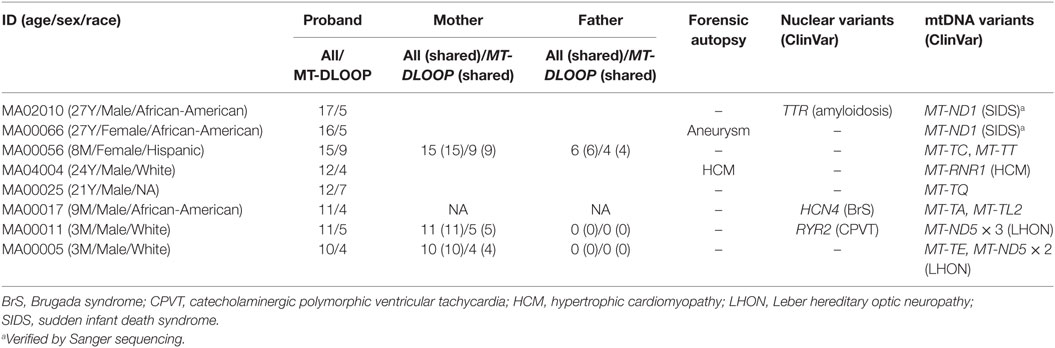
Table 3. Values for the eight molecular autopsy cases who carried ≥10 mtDNA heteroplasmic variants.
Discussion
Here, we analyzed 50 molecular autopsies of sudden unexpected and unexplained cases of cardiac death in the young (<45 years old) predominantly from the San Diego County. In keeping with previous studies of SDY, we observed a 1:2 female to men ratio (5, 22, 25, 27). The number of cases had a peak in infants, and exponentially increased up to the ages studied (2, 25). A surprisingly large percentage of sudden infant death syndromes happened during the middle of the week (Wednesdays and Thursdays), whereas for older individuals, there was a peak on Mondays, a day known for being enriched in sudden deaths due to ischemic events (1). Friday was the day less prone to deaths. While a relation between SCD and variation in weekly stress levels can be speculated in young adults with social life, such a connection seems less intuitive for infants. In that regard, it is known that heart rate and ventilation tends to synchronize in individuals sharing space (48–50), and it is also known that maternal environment (e.g., cortisol in breast milk) can influence infant development (51, 52). Given the limited sample size, we could not establish a legitimate explanation for the excess of infant deaths during the middle of the week. However, we cannot discount the possible contribution of environmental factors. We found that for the entire cohort (n = 50), the majority of deaths occurred between 8 a.m. and 8 p.m., a window that overlaps with previous findings [6 a.m. to 12 p.m. and late afternoon (1, 53)]. This circadian relationship has been associated with sympathetic activation (54) and protein regeneration (55). The deaths were not equally spread during the year, as we found a peak in April, followed by months of June, October, and September. Only two cases of SDY happened in January and May. With the exception of the lower mortality found in late summer, our seasonal findings do not overlap with previous studies on SCD (56), but it is important to remember that our cohort consisted of young individuals and did not include deaths by ischemic events per se. Molecular autopsies overall gave a modest yield in terms of likely pathogenic variants, irrespective of being performed in forensic autopsy positive or negative cases. A high percentage of the variants found fell under the category of plausible or VUS (15, 57). Still, combined use of forensic and molecular autopsies provided the maximum diagnostic yield.
We compared an exome-based approach to a simulated gene panel for cardiac disease. In that regard, we found that exome-based analysis led to many likely pathogenic variants that could potentially explain sudden death. However, due to the lack of robust databases for variants associated with SDY, we could not make conclusive statements since the genes themselves have not been associated with sudden death and we only have one off cases in these instances. On the other end of the spectrum, gene panel-based analysis provided a manageable number of likely pathogenic mutations, but the overall diagnostic yield was low. An obvious advantage of using an exome-based capture is that it allows for searches using lists of genes and opens the possibility of expanding the lists without repeating the sequencing experiments, but at the expense of losing depth of coverage (37, 58–61). The effect of other variants not studied here, such as somatic mutations, variants in non-coding regions, and copy number variations, is currently an active field of cardiovascular research (6, 62–65).
In terms of where the variants fall in the nuclear exome, the tendency was that larger genes (e.g., TTN) accumulated more variants. Regardless of size, some genes are less tolerant to mutations and this information can be useful to assess pathogenicity likelihood when an unseen variant is found (41–43). Even for recognized Mendelian diseases such as long QT syndrome, Brugada syndrome, or catecholaminergic polymorphic ventricular tachycardia, we often observed that multiple variants could potentially be contributing to the observed phenotype. In that sense, the separation of VUS with respect to true pathogenic variants is probably the most challenging task that the field of genetics will face during the upcoming years. In terms of sample classification, the small sample size and the lack of consistency on genes carrying pathogenic variants caused that a comparison based on a genetic fingerprint did not perform satisfactorily. Still, and under the assumption that similar genotypes should lead to similar phenotypes (10, 66), we believe that in the future, when larger cohorts with clinical ontologies become accessible (67), this departure from a “n-of-1” might help us see the forest through the trees and will facilitate the diagnostic of disease. In the case of cardiomyopathy, the cohort does not necessarily need to be restricted to deceased individuals (25), as it is possible to perform early screening in at risk populations (68).
Finally, we analyzed mtDNA variants from all 50 samples (and trios when available) and observed that 50% of the infants (4 out of 8) had a substantial increase in the number of heteroplasmic variants (≥10 variants) with respect to the rest of the cohort (median: 4; interquartile range: 2–7). The variants were not de novo (mothers were also carriers yet apparently asymptomatic) and had high heteroplasmic levels. Among those variants, only a subset (as annotated by external databases) was capable of producing mitochondrial disease. In that sense, we contemplate the possibility that an excess of heteroplasmic mtDNA variants might confer susceptibility for a mitochondrial “system-failure” under conditions of metabolic stress, especially when an immature metabolism is at play. Parenthetically, we recently found that people over 100 years free from disease tend to have low number of heteroplasmic variants (47), which supports the idea that heteroplasmic levels might be associated with metabolic efficiency. This is just a hypothesis since we cannot assess to what extent mtDNA variants contributed to the sudden death. We acknowledge that our cohort is small and that our findings need to be replicated. If heteroplasmy turns out to be associated with metabolic disadvantage, implementing a preconceptual/prenatal genetic test for risk for sudden infant death syndrome based on mtDNA should be straightforward.
We believe a comprehensive effort to collect and share genetic and phenotypic data is needed in order to define pathogenic variants more precisely, provide quantifiable risks to living relatives, and unravel the incomplete penetrance, variable expressivity, and gene–environment interactions evident in previous findings. The aggregate genomic data for all cases can be accessed at https://genomics.scripps.edu/browser.
Conclusion
We present here a systematic analysis of the first 50 cases from our MA for sudden death in the young study. We found that males were affected twice as often as females, and that deaths followed circadian, weekly and seasonal patterns. Molecular autopsies identified a likely causal variant in 14 cases, yielding maximum value when combined with forensic ones. Almost all the reported likely causal variants were in genes associated with cardiac disease, thus, “escalating” to exome did not improve the diagnostic yield. In that regard, most of the unreported variants were VUS. At the coding level, our cohort did not have many individuals sharing genes with pathogenic variants and thus a genotype-based classification was unsatisfactory. Adding mtDNA variants to molecular autopsies provided new insights as well as new uncertainties. We found that half of the infants carried out an unusually high number in heteroplasmic variants. Overall, the analysis of MA cases at the cohort-level adds a new dimension to the understanding of the genetic causes of SDY. We believe that a global effort to share genomic data in a centralized knowledge resource is needed to succeed in transitioning MA negatives to positives.
Ethics Statement
This study was carried out in accordance with the recommendations of Scripps Office for the Protection of Research Subjects, Protocol number IRB-14-6386 written or verbal consent was obtained from each subject or their authorized representative.
Author Contributions
MR designed, analyzed, interpreted the data, and wrote the manuscript. JW and ST participated as clinical trials coordinators. TP performed laboratory experiments. EM provided case review and revised the manuscript. JL and GW conceived the study, oversaw forensic autopsies, and provided case review. ET conceived the study, obtained funding, and organized the collaboration with the medical examiner. AT obtained funding, interpreted the data and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the included families for their participation, as well as all the external medical examiners and their team members. We also thank Joe Davis, Stephen Campman, Alex Lippman, Emily G. Spencer, Sydney Running and Lifesharing for their help.
Funding
This work is supported by Scripps Genomic Medicine, an NIH-NCATS Clinical and Translational Science Award (CTSA; 5 UL1 RR025774) to ET. Further support is from NIH-U01 HG006476.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fcvm.2017.00072/full#supplementary-material.
References
1. Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation (2012) 125:620–37. doi:10.1161/CIRCULATIONAHA.111.023838
2. Faragli A, Underwood K, Priori SG, Mazzanti A. Is there a role for genetics in the prevention of sudden cardiac death? J Cardiovasc Electrophysiol (2016) 27:1124–32. doi:10.1111/jce.13028
3. Chugh SS. Sudden cardiac death in 2017: spotlight on prediction and prevention. Int J Cardiol (2017) 237:2–5. doi:10.1016/j.ijcard.2017.03.086
4. Erck Lambert AB, Parks SE, Camperlengo L, Cottengim C, Anderson RL, Covington TM, et al. Death scene investigation and autopsy practices in sudden unexpected infant deaths. J Pediatr (2016) 174:84–90.e81. doi:10.1016/j.jpeds.2016.03.057
5. Hefti MM, Kinney HC, Cryan JB, Haas EA, Chadwick AE, Crandall LA, et al. Sudden unexpected death in early childhood: general observations in a series of 151 cases: part 1 of the investigations of the San Diego SUDC Research Project. Forensic Sci Med Pathol (2016) 12:4–13. doi:10.1007/s12024-015-9724-2
6. Priest JR, Gawad C, Kahlig KM, Yu JK, O’Hara T, Boyle PM, et al. Early somatic mosaicism is a rare cause of long-QT syndrome. Proc Natl Acad Sci U S A (2016) 113:11555–60. doi:10.1073/pnas.1607187113
7. Parks SE, Erck Lambert AB, Shapiro-Mendoza CK. Racial and ethnic trends in sudden unexpected infant deaths: United States, 1995-2013. Pediatrics (2017) 139:e20163844. doi:10.1542/peds.2016-3844
8. Noseworthy PA, Newton-Cheh C. Genetic determinants of sudden cardiac death. Circulation (2008) 118:1854–63. doi:10.1161/CIRCULATIONAHA.108.783654
9. Campuzano O, Beltran-Alvarez P, Iglesias A, Scornik F, Perez G, Brugada R. Genetics and cardiac channelopathies. Genet Med (2010) 12:260–7. doi:10.1097/GIM.0b013e3181d81636
10. Van Norstrand DW, Ackerman MJ. Genomic risk factors in sudden infant death syndrome. Genome Med (2010) 2:86. doi:10.1186/gm207
11. Wilde AA, Behr ER. Genetic testing for inherited cardiac disease. Nat Rev Cardiol (2013) 10:571–83. doi:10.1038/nrcardio.2013.108
12. Spears DA, Gollob MH. Genetics of inherited primary arrhythmia disorders. Appl Clin Genet (2015) 8:215–33. doi:10.2147/TACG.S55762
13. Mcguire AL, Moore Q, Majumder M, Walkiewicz M, Eng CM, Belmont JW, et al. The ethics of conducting molecular autopsies in cases of sudden death in the young. Genome Res (2016) 26:1165–9. doi:10.1101/gr.192401.115
14. Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet (2014) 59:5–15. doi:10.1038/jhg.2013.114
15. Novelli V, Gambelli P, Memmi M, Napolitano C. Challenges in molecular diagnostics of channelopathies in the next-generation sequencing era: less is more? Front Cardiovasc Med (2016) 3:29. doi:10.3389/fcvm.2016.00029
16. Harper AR, Parikh VN, Goldfeder RL, Caleshu C, Ashley EA. Delivering clinical grade sequencing and genetic test interpretation for cardiovascular medicine. Circ Cardiovasc Genet (2017) 10:e001221. doi:10.1161/CIRCGENETICS.116.001221
17. Parikh VN, Ashley EA. Next-generation sequencing in cardiovascular disease: present clinical applications and the horizon of precision medicine. Circulation (2017) 135:406–9. doi:10.1161/CIRCULATIONAHA.116.024258
18. Tester DJ, Ackerman MJ. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation (2011) 123:1021–37. doi:10.1161/CIRCULATIONAHA.109.914838
19. Brion M, Sobrino B, Martinez M, Blanco-Verea A, Carracedo A. Massive parallel sequencing applied to the molecular autopsy in sudden cardiac death in the young. Forensic Sci Int Genet (2015) 18:160–70. doi:10.1016/j.fsigen.2015.07.010
20. Semsarian C, Ingles J, Wilde AA. Sudden cardiac death in the young: the molecular autopsy and a practical approach to surviving relatives. Eur Heart J (2015) 36:1290–6. doi:10.1093/eurheartj/ehv063
21. Ackerman JP, Bartos DC, Kapplinger JD, Tester DJ, Delisle BP, Ackerman MJ. The promise and peril of precision medicine: phenotyping still matters most. Mayo Clin Proc (2016) 91(11):1606–16. doi:10.1016/j.mayocp.2016.08.008
22. Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, Lam L, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med (2016) 374:2441–52. doi:10.1056/NEJMoa1510687
23. Methner DN, Scherer SE, Welch K, Walkiewicz M, Eng CM, Belmont JW, et al. Postmortem genetic screening for the identification, verification, and reporting of genetic variants contributing to the sudden death of the young. Genome Res (2016) 26:1170–7. doi:10.1101/gr.195800.115
24. Neubauer J, Haas C, Bartsch C, Medeiros-Domingo A, Berger W. Post-mortem whole-exome sequencing (WES) with a focus on cardiac disease-associated genes in five young sudden unexplained death (SUD) cases. Int J Legal Med (2016) 130:1011–21. doi:10.1007/s00414-016-1317-4
25. Sanchez O, Campuzano O, Fernandez-Falgueras A, Sarquella-Brugada G, Cesar S, Mademont I, et al. Natural and undetermined sudden death: value of post-mortem genetic investigation. PLoS One (2016) 11:e0167358. doi:10.1371/journal.pone.0167358
26. Lahrouchi N, Raju H, Lodder EM, Papatheodorou E, Ware JS, Papadakis M, et al. Utility of post-mortem genetic testing in cases of sudden arrhythmic death syndrome. J Am Coll Cardiol (2017) 69:2134–45. doi:10.1016/j.jacc.2017.02.046
27. Neubauer J, Lecca MR, Russo G, Bartsch C, Medeiros-Domingo A, Berger W, et al. Post-mortem whole-exome analysis in a large sudden infant death syndrome cohort with a focus on cardiovascular and metabolic genetic diseases. Eur J Hum Genet (2017) 25:404–9. doi:10.1038/ejhg.2016.199
28. Suktitipat B, Sathirareuangchai S, Roothumnong E, Thongnoppakhun W, Wangkiratikant P, Vorasan N, et al. Molecular investigation by whole exome sequencing revealed a high proportion of pathogenic variants among Thai victims of sudden unexpected death syndrome. PLoS One (2017) 12:e0180056. doi:10.1371/journal.pone.0180056
29. Torkamani A, Muse ED, Spencer EG, Rueda M, Wagner GN, Lucas JR, et al. Molecular autopsy for sudden unexpected death. JAMA (2016) 316:1492–4. doi:10.1001/jama.2016.11445
30. Bloss CS, Zeeland ASV, Topol SE, Darst BF, Boeldt DL, Erikson GA, et al. A genome sequencing program for novel undiagnosed diseases. Genet Med (2015) 17:995–1001. doi:10.1038/gim.2015.21
31. Mckenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res (2010) 20:1297–303. doi:10.1101/gr.107524.110
32. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics (2009) 25:2078–9. doi:10.1093/bioinformatics/btp352
33. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (2010) 26:841–2. doi:10.1093/bioinformatics/btq033
34. Pham PH, Shipman WJ, Erikson GA, Schork NJ, Torkamani A. Scripps genome ADVISER: annotation and distributed variant interpretation SERver. PLoS One (2015) 10:e0116815. doi:10.1371/journal.pone.0116815
35. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A (1977) 74:5463–7. doi:10.1073/pnas.74.12.5463
36. Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature (2015) 526:68–74. doi:10.1038/nature15393
37. Nunn LM, Lopes LR, Syrris P, Murphy C, Plagnol V, Firman E, et al. Diagnostic yield of molecular autopsy in patients with sudden arrhythmic death syndrome using targeted exome sequencing. Europace (2016) 18:888–96. doi:10.1093/europace/euv285
38. Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MR, et al. A review of the giant protein titin in clinical molecular diagnostics of cardiomyopathies. Front Cardiovasc Med (2016) 3:21. doi:10.3389/fcvm.2016.00021
39. Shyr C, Tarailo-Graovac M, Gottlieb M, Lee JJ, Van Karnebeek C, Wasserman WW. FLAGS, frequently mutated genes in public exomes. BMC Med Genomics (2014) 7:64. doi:10.1186/s12920-014-0064-y
40. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res (2014) 42:D980–5. doi:10.1093/nar/gkt1113
41. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet (2013) 9:e1003709. doi:10.1371/journal.pgen.1003709
42. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet (2014) 46:310–5. doi:10.1038/ng.2892
43. Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, Mcguire KJ, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med (2017) 19:192–203. doi:10.1038/gim.2016.90
44. R_Core_Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing (2015).
45. Meyers DE, Basha HI, Koenig MK. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Tex Heart Inst J (2013) 40:385–94.
46. Hofmann S, Jaksch M, Bezold R, Mertens S, Aholt S, Paprotta A, et al. Population genetics and disease susceptibility: characterization of central European haplogroups by mtDNA gene mutations, correlation with D loop variants and association with disease. Hum Mol Genet (1997) 6:1835–46. doi:10.1093/hmg/6.11.1835
47. Rueda M, Torkamani A. SG-ADVISER mtDNA: a web server for mitochondrial DNA annotation with data from 200 samples of a healthy aging cohort. BMC Bioinformatics (2017) 18:373. doi:10.1186/s12859-017-1778-6
48. Richard CA, Mosko SS. Mother-infant bedsharing is associated with an increase in infant heart rate. Sleep (2004) 27:507–11. doi:10.1093/sleep/27.3.507
49. Lucas-Thompson R, Golbderg WA, Germo GR, Keller MA, Davis EP, Sandman CA. Sleep arrangements and night waking at 6 and 12 months in relation to infants’ stress-induced cortisol responses. Infant Child Dev (2009) 18:521–44. doi:10.1002/icd.636
50. Helm JL, Sbarra D, Ferrer E. Assessing cross-partner associations in physiological responses via coupled oscillator models. Emotion (2012) 12:748–62. doi:10.1037/a0025036
51. Grey KR, Davis EP, Sandman CA, Glynn LM. Human milk cortisol is associated with infant temperament. Psychoneuroendocrinology (2013) 38:1178–85. doi:10.1016/j.psyneuen.2012.11.002
52. Hinde K, Skibiel AL, Foster AB, Del Rosso L, Mendoza SP, Capitanio JP. Cortisol in mother’s milk across lactation reflects maternal life history and predicts infant temperament. Behav Ecol (2015) 26:269–81. doi:10.1093/beheco/aru186
53. Cohen MC, Rohtla KM, Lavery CE, Muller JE, Mittleman MA. Meta-analysis of the morning excess of acute myocardial infarction and sudden cardiac death. Am J Cardiol (1997) 79:1512–6. doi:10.1016/S0002-9149(97)00181-1
54. Peters RW. Propranolol and the morning increase in sudden cardiac death: the beta-blocker heart attack trial experience. Am J Cardiol (1990) 66:57G–9G. doi:10.1016/0002-9149(90)90398-K
55. Jeyaraj D, Haldar SM, Wan X, Mccauley MD, Ripperger JA, Hu K, et al. Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature (2012) 483:96–9. doi:10.1038/nature10852
56. Arntz HR, Willich SN, Schreiber C, Bruggemann T, Stern R, Schultheiss HP. Diurnal, weekly and seasonal variation of sudden death. Population-based analysis of 24,061 consecutive cases. Eur Heart J (2000) 21:315–20. doi:10.1053/euhj.1999.1739
57. Ackerman MJ. Genetic purgatory and the cardiac channelopathies: exposing the variants of uncertain/unknown significance issue. Heart Rhythm (2015) 12:2325–31. doi:10.1016/j.hrthm.2015.07.002
58. Waldmuller S, Schroeder C, Sturm M, Scheffold T, Imbrich K, Junker S, et al. Targeted 46-gene and clinical exome sequencing for mutations causing cardiomyopathies. Mol Cell Probes (2015) 29:308–14. doi:10.1016/j.mcp.2015.05.004
59. Anderson JH, Tester DJ, Will ML, Ackerman MJ. Whole-exome molecular autopsy after exertion-related sudden unexplained death in the young. Circ Cardiovasc Genet (2016) 9:259–65. doi:10.1161/CIRCGENETICS.115.001370
60. Pua CJ, Bhalshankar J, Miao K, Walsh R, John S, Lim SQ, et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J Cardiovasc Transl Res (2016) 9:3–11. doi:10.1007/s12265-016-9673-5
61. Seidelmann SB, Smith E, Subrahmanyan L, Dykas D, Abou Ziki MD, Azari B, et al. Application of whole exome sequencing in the clinical diagnosis and management of inherited cardiovascular diseases in adults. Circ Cardiovasc Genet (2017) 10:e001573. doi:10.1161/CIRCGENETICS.116.001573
62. Barc J, Briec F, Schmitt S, Kyndt F, Le Cunff M, Baron E, et al. Screening for copy number variation in genes associated with the long QT syndrome: clinical relevance. J Am Coll Cardiol (2011) 57:40–7. doi:10.1016/j.jacc.2010.08.621
63. Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet (2011) 88:273–82. doi:10.1016/j.ajhg.2011.01.016
64. Lara-Pezzi E, Dopazo A, Manzanares M. Understanding cardiovascular disease: a journey through the genome (and what we found there). Dis Model Mech (2012) 5:434–43. doi:10.1242/dmm.009787
65. Mademont-Soler I, Pinsach-Abuin ML, Riuro H, Mates J, Perez-Serra A, Coll M, et al. Large genomic imbalances in Brugada syndrome. PLoS One (2016) 11:e0163514. doi:10.1371/journal.pone.0163514
66. Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med (2017) 376:21–31. doi:10.1056/NEJMoa1516767
67. Kohler S, Vasilevsky NA, Engelstad M, Foster E, Mcmurry J, Ayme S, et al. The human phenotype ontology in 2017. Nucleic Acids Res (2017) 45:D865–76. doi:10.1093/nar/gkw1039
Keywords: molecular autopsy, sudden cardiac death, whole exome sequencing, gene panel, mitochondrial DNA
Citation: Rueda M, Wagner JL, Phillips TC, Topol SE, Muse ED, Lucas JR, Wagner GN, Topol EJ and Torkamani A (2017) Molecular Autopsy for Sudden Death in the Young: Is Data Aggregation the Key? Front. Cardiovasc. Med. 4:72. doi: 10.3389/fcvm.2017.00072
Received: 29 July 2017; Accepted: 27 October 2017;
Published: 09 November 2017
Edited by:
Valeria Novelli, Fondazione Policlinico Universitario A. Gemelli; Università Cattolica del Sacro Cuore, ItalyReviewed by:
Najim Lahrouchi, University of Amsterdam, NetherlandsCostantino Smaldone, Ospedale San Carlo, Italy
Copyright: © 2017 Rueda, Wagner, Phillips, Topol, Muse, Lucas, Wagner, Topol and Torkamani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel Rueda, mrueda@scripps.edu;
Ali Torkamani, atorkama@scripps.edu