 Somsubhra Nath
Somsubhra Nath Dishari Ghatak
Dishari Ghatak Pijush Das
Pijush Das Susanta Roychoudhury
Susanta Roychoudhury- Cancer Biology and Inflammatory Disorder Division, CSIR-Indian Institute of Chemical Biology, Kolkata, India
Research over the past few decades has well established the molecular functioning of mitosis. Deregulation of these functions has also been attributed to the generation of aneuploidy in different tumor types. Numerous studies have given insight into the regulation of mitosis by cell cycle specific proteins. Optimum abundance of these proteins is pivotal to timely execution of mitosis. Aberrant expressions of these mitotic proteins have been reported in different cancer types. Several post-transcriptional mechanisms and their interplay have subsequently been identified that control the level of mitotic proteins. However, to date, infrequent incidences of cancer-associated mutations have been reported for the genes expressing these proteins. Therefore, altered expression of these mitotic regulators in tumor samples can largely be attributed to transcriptional deregulation. This review discusses the biology of transcriptional control for mitosis and evaluates its role in the generation of aneuploidy and tumorigenesis.
Introduction
The propagation of eukaryotic life is orchestrated by the generation of descendent cells through the biological process of cell division. While mitosis controls the propagation of somatic cells, generation of germ cells is controlled by meiosis. The fidelity of mitosis determines the equal division of duplicated chromosomes to the two daughter cells. The first phase of mitosis, that is, nuclear division or karyokinesis is divided into four sub-phases. Prophase marks the initiation of mitosis bringing about chromosome condensation, separation of duplicated centrosomes, and recruitment of some mitotic checkpoint proteins to the kinetochores. Following this, disassembly of the nuclear envelope (NE) marks the entry into metaphase (prometaphase). Subsequently, the release of chromosomes into cytoplasm activates the spindle assembly checkpoint (SAC) at each unattached kinetochore. After microtubule capturing of each chromatid pair at their kinetochores and alignment at the midzone, silencing of the SAC occurs and cell overcomes the “wait anaphase” signal. During anaphase, sister chromatids are completely separated to the two opposite poles of the cell and the invagination of plasma membrane around the spindle midzone becomes visible. Telophase ends with chromosome decondensation and reassembly of the NE around polar chromosomes. Cytokinesis or cytoplasmic division giving rise to two daughter cells follows soon after. Intriguingly, each of these events is sequentially organized in a manner that minimizes any segregational errors. Therefore, defects in the operation of any mitotic event may lead to the generation of chromosomal instability (CIN), a hallmark of cancer (1–5). Having said this, precision and efficiency of the mitotic cell division depends on proper regulation of the expression and function of mitotic proteins. Indeed, most of these proteins show mitotic phase specific activity. This activity is chiefly regulated by post-translational modifications, namely, phosphorylation and ubiquitination, and some other mechanisms (6). Notably, transcription also plays a key role in the maintenance of cell cycle specific protein levels (7). However, little has been summarized about the transcriptional control of the mitotic phenomenon. In this review, we will discuss the role of transcription in mitotic regulation and provide evidence for transcriptional anomalies underlying abnormal mitotic events that lead to CIN and tumorigenesis.
Mitosis and Aneuploidy
Errors in chromosome partitioning often give rise to aneuploidy. There are several roads that lead to aneuploidy through mitotic errors (4, 8, 9). The first and foremost reason of mitotic aneuploidy is a faulty SAC. The SAC monitors bipolar segregation of duplicated chromosomes during metaphase to anaphase transition (10). Prior to anaphase, sister chromatids remain held together by the cohesin complex (11). At the onset of anaphase, securin gets ubiquitinated by the E3 ubiquitin ligase, anaphase promoting complex/cyclosome (APC/C). This degradation of securin, in turn, makes separase (a protease) free and active. The latter then cleaves cohesin and the chromosomes begin to separate (10, 12). In the presence of any unattached kinetochore or lack of amphitelic attachment SAC is activated. A number of proteins are involved in the tasks executed by the SAC (10). At the molecular level, the mitotic checkpoint complex (MCC) comprised of the Mad and Bub families of proteins, sequesters APC/C adapter protein Cdc20 (13), and APC/C remains inactive until the defects get corrected. After the completion of proper bipolar attachment Cdc20 is ubiquitinated by mitotic ubiquitin carrier protein UbcH10 and gets free from inhibitory MCC (14). Concordant with that, the SAC antagonist protein p31comet binds to the MCC component Mad2 and modulates extraction of Mad2 from MCC. This, in turn, causes disassembly of MCC and blocks further sequestration of Cdc20 (15, 16). Free Cdc20 activates APC/C, which then degrades anaphase inhibitors and cells progress through mitosis. The stepwise functioning of these events depends on the balanced level of the SAC proteins. While mutations in the SAC genes are infrequent in human cancers, their altered expressions are often reported in various cancers and have been associated with defective SAC-mediated aneuploidy (4). Hence, the balanced levels of different SAC proteins are important determinants of SAC behavior. The cell cycle specific transcriptional regulation of SAC proteins might be an elemental reason in maintaining this balance, deregulation of which might be involved in altered levels of the SAC molecules.
In search of other CIN inducing mitotic phenomena, genetic screens have identified cohesion defects as contributors to the onset of aneuploidy (3, 4, 8). Glitches in the machinery monitoring sister chromatid cohesion might promote aneuploidy. Consistent with this, a recent study identified mutations in STAG2 (which encodes a protein subunit of the cohesion complex) in a number of aneuploid primary tumors and cancer cell lines (17). Also, overexpression of securin and separase, two key regulators of cohesion, is reported to promote aneuploidy and tumorigenesis (18, 19). Chromosome missegregation may also occur in case of merotelic attachment where a single kinetochore attaches to microtubules emanating from both poles of the spindle (20, 21). Several molecular components, for example, Aurora kinase B, kinesin-13 proteins, MCAK, INCENP, Survivin, and Shugoshin are associated in this phenomenon and their overexpression are reported in cancers of various origins (21). A final source of aneuploidy is the prevalence of aberrant centromeres and multipolar mitosis (2–4). Centrosomes provide mitotic spindle poles and concurrently, presence of more than two centrosomes might produce multipolar spindles. Additionally, aberrant chromosome numbers and multipolarity are associated with CIN in various cancers (22). A number of cellular proteins, including Aurora kinase A, Plk1, Chk1, Chk2, Cyclin B1, and Cdk1, regulate centrosome duplication and the abnormal upstream regulation of these proteins is found in various cancers (2).
Molecular Control of Mitosis: Regulation of Mitotic Proteins
Mitosis is a complex event performed by multiple factors with distinct phase specific responsibilities. Regulation at the protein level plays a crucial role in the mitosis specific performances by these factors. These regulations can occur through several routes (Figure 1). First, ubiquitination-mediated protein degradation is believed to be pivotal. The mitotic ubiquitin ligase, APC/C promotes ubiquitination of various protein substrates in a spatial manner (23). By ubiquitinating and consequently targeting mitotic inhibitors for proteasomal degradation, this cellular phenomenon controls mitotic progression in a unidirectional manner. Second, phosphorylation controls functional activities of a number of mitotic proteins in a time-dependent manner. Mitotic cyclin dependent kinase Cdk1, in association with Cyclin A or B, phosphorylates more than 70 substrates involved in various steps of mitosis (24). Some other mitotic kinases like Aurora, Polo, and Nek families also participate in phosphorylation-mediated mitotic regulation (24). As a third mechanism, microRNA (miRNA)-mediated regulation of mitotic proteins is also currently emerging (25–30). In this list of regulatory pathways, the control of expression at the transcription level could be considered as momentous.
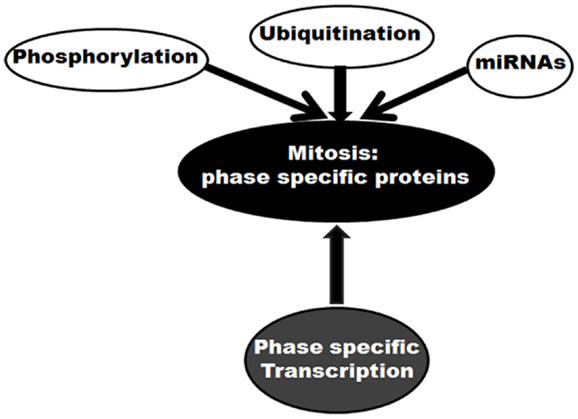
Figure 1. Molecular control of mitosis: regulation of mitotic proteins. Different regulatory mechanisms are shown.
Roads to Chromosomal Instability: Contribution of Mutation Versus Transcription of Mitotic Genes
Most of the tumors are reported to acquire a number of mutations in proto-oncogenes and tumor suppressor genes. Mutation of a gene may alter its product, qualitatively or quantitatively. Extensive search has shown mutations in >1% of candidate genes causally related to oncogenesis, termed as cancer genes (31). Given the fact that mitotic protein levels are pivotal in proper execution of mitosis, the mutational defects can be assumed prime factors in deregulation of mitosis. Simultaneously, a few reports have identified mutations in SAC as well as other mitotic genes in cancers of different origins (32–34). For example, a biallelic germline mutation of the SAC gene BUB1B has been diagnosed with mosaic variegated aneuploidy, a rare recessive condition of childhood cancer (35). The genetic alterations, such as gene amplification or depletion, also play a key role in the regulation of many mitotic genes. For example, the genes expressing Aurora-A and Ect2 are amplified in several types of tumors (36–38). Interestingly, despite these reports, mutations directly affecting a mitotic gene are not frequent among cancer types. In an in silico approach, we analyzed the mutation status of 526 genes from a list of 572 validated mitotic genes (39) using COSMIC v67 database1 (Table S1 in Supplementary Material). The percent mutation for each of these genes was obtained from the percentage of unique mutated samples out of total samples studied. The extracted dataset showed <1% of mutations in 84% (441) of the genes. On the other hand, only 5% of the genes showed mutations in >3% of the samples (Figure 2). In a separate approach, we tried to find out the expression status of validated mitotic hits (39). Using ONCOMINE 4.4 research edition database2 cancer versus normal expression patterns were obtained for 557 mitotic genes in 7 head and neck squamous cell carcinoma (HNSCC) datasets (Table S2 in Supplementary Material). Among these, 15% (82) of the genes were found to be overexpressed and 1% was found to be down-regulated in >60% of datasets (Figure 2). To find out whether mutations are responsible for the altered expression of these genes we correlated these two analyses (Table S3 in Supplementary Material). The analysis revealed that 73% of the overexpressed mitotic genes have mutations in <1% of the samples. Only 19% of the overexpressed genes were detected with mutations in 1–2% samples and 8% of the genes were detected with mutations in >3% of the samples. Among the down-regulated genes, 87% of the genes showed mutations in <1% of the samples while 13% of the genes showed mutations in 1–2% of the samples (Figure 2). This data clearly negate the involvement of mutations in regulating the expression of mitotic genes. The probable reasons behind these findings could be (a) mutation in any one of the mitotic genes including SAC regulators may weaken the checkpoint or other mitotic regulations; (b) mutation leading to complete inactivation of any crucial mitosis regulatory gene would be fatal and be eliminated by death of the affected cell(s). For instance, germline deletion of the SAC gene MAD2 is associated with the loss of pregnancy (40). Indeed, negligible cancer-associated mutations are reported for Aurora kinase B, Cdk1, Cyclin B, Nek2, and Pin1, proteins involved in initial events of mitosis (Table S1 in Supplementary Material) (6). Also, mutations in mitotic checkpoint genes themselves are not found responsible for abnormal checkpoint in cancer cells (8) and infrequently reported for core SAC proteins like Cdc20, Bub3, and Mad2, and SAC-associated proteins like Borealin, Zwint, Hec1, and Aurora kinase B (6).
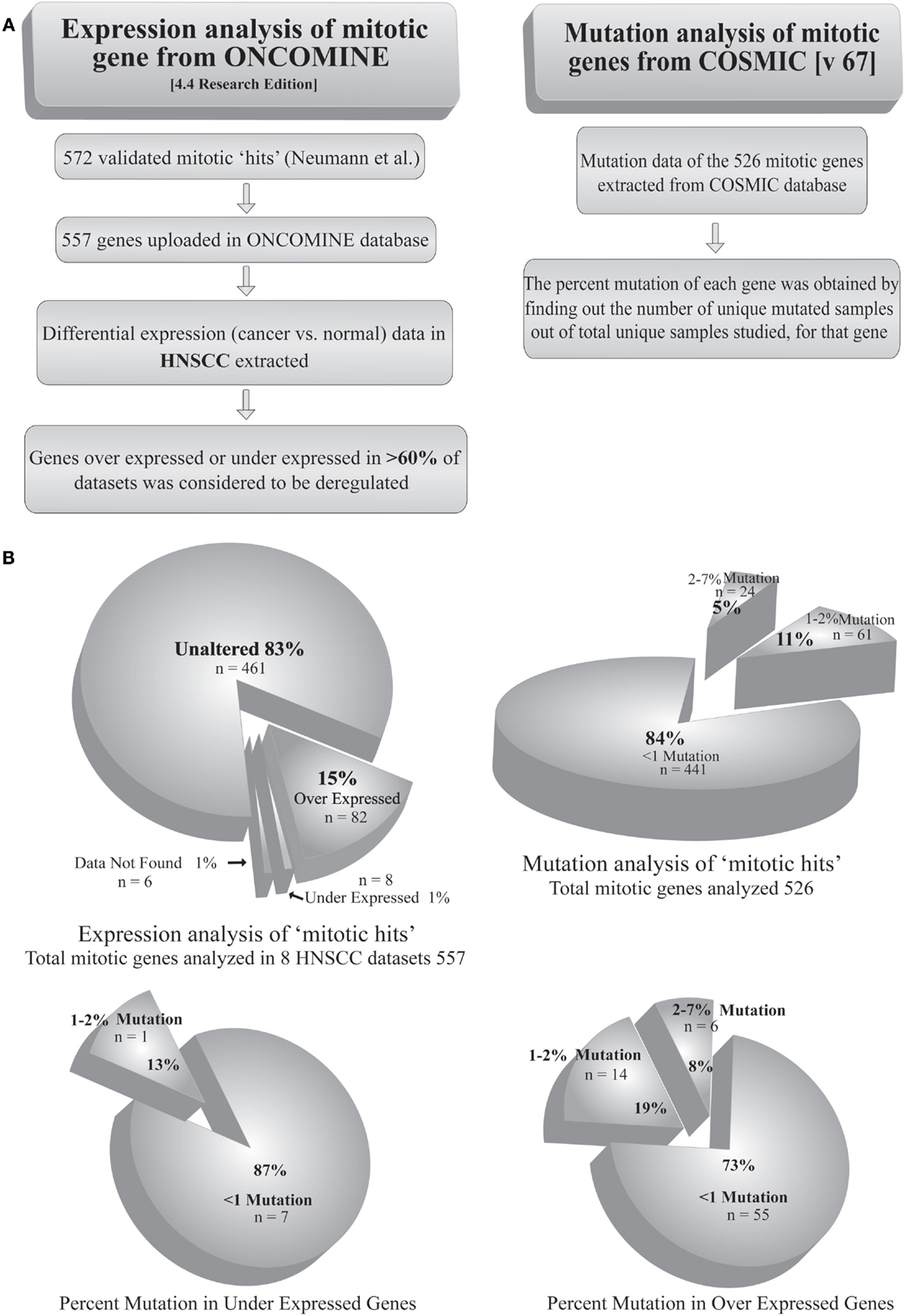
Figure 2. Analysis of mutation and transcriptional alteration in mitotic genes. (A) Expression analysis of mitotic genes was done using ONCOMINE (4.4 research edition) database and mutation analysis was done using COSMIC (v67) according to the given workflow. (B) The two analyses were correlated to obtain the percentage of mutations in overexpressed and downregulated genes.
Transcriptional Control of Mitotic Genes
Mitosis, like any other pathway, is essentially interplay among various protein molecules with tightly regulated phase specific functional activities. A number of mitotic genes show peak level of transcription when the cell passes through the G2 phase (Figure 3) (41). Promoters of these genes remain repressed during G0 and G1 phase. The relief from repression starts at the S-phase and peaks after reaching the G2 phase. The transcription factor, NF-Y is crucial in this timely expression (41). A number of mitotic genes contain two or three CCAAT boxes. These sites are recruited by hetero-trimeric NF-Y in association with histone acetyltransferase p300. This dynamic recruitment brings upon transcriptional activation of mitotic genes at the late phase of cell cycle (42). Two consensus sites, namely, cell cycle dependent element (CDE) and cell cycle genes homology region (CHR), have been extensively described in the global regulation of genes having mitosis specific expression (41). Transcriptional repression remains maintained during G0 and G1 phase through the binding of repressor proteins in CDE and/or CHR elements. The release from repression starts at the S-phase. Following this, activation of these promoters mostly occurs through CCAAT boxes after binding of NF-Y in combination with the co-activator, p300. Promoters of mitotic genes, namely, CCNA (Cyclin A), CCNB1 (Cyclin B1), CCNB2 (Cyclin B2), CDC2/CDK1, CDC25C, CKS1, MKLP1, PLK1, and TOME-1, with well documented mitotic phase specific regulation by CDE/CHR elements, are found to be activated through their CCAAT consensus elements (41). On the other hand, p53 has been associated with the repression of several mitotic genes through CDE/CHR elements (41). A number of mitotic genes, like CDC20, CKS1, CCNB1, CCNB2, and CDC2/CDK1, are repressed by p53 (43–46). However, cell cycle specific repression of some other genes without CDE/CHR has also been documented (41). Toward that, besides CDE/CHR site driven effect, a direct p53 binding element has been identified to regulate Cdc20 expression (47).
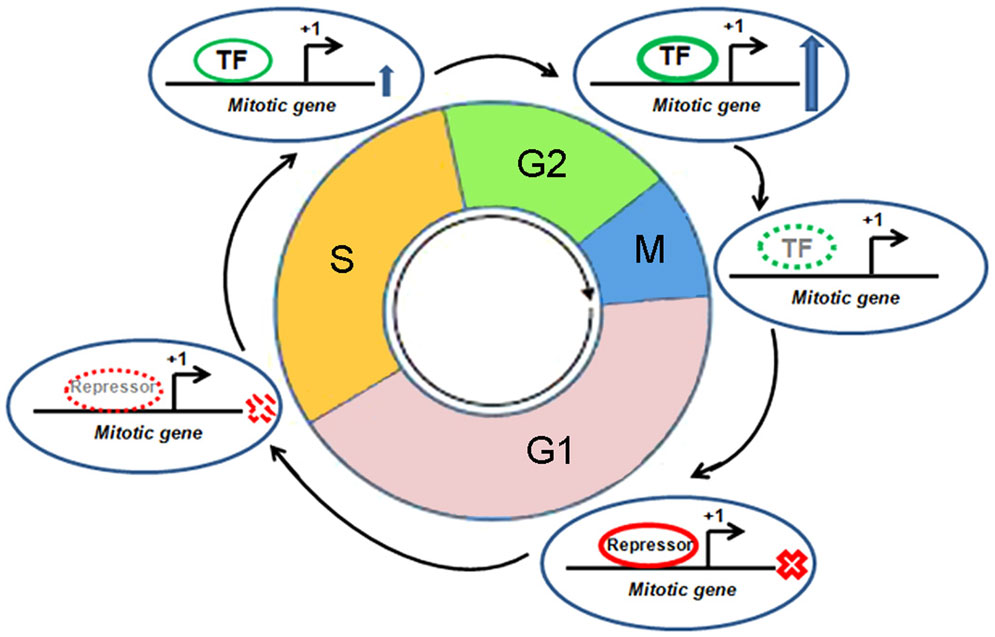
Figure 3. Cell cycle specific transcriptional regulation of a mitotic gene. Different cell cycle phases, mitotic gene, transcription start site, transcription factor (TF), and repressor are shown. “X” mark indicates “transcription off” condition and up arrow indicates “transcription on” condition. Dotted appearance indicates gradual reduction of recruitment of TF or repressor.
Beside this, several other transcription factors have also been reported to control the expression of genes in mitosis specific manner. As, for example, Forkbox M1 (FoxM1) has been identified as a master regulator of mitosis. Laoukili et al. have elegantly shown a transcriptional cluster to be regulated by FoxM1 (48). Another study conducted by Wang et al. also ended up with similar observation for FoxM1 as a master regulator of mitotic genes, like CDC25A, AURKA, AURKB, Survivin, CENPA, CENPB, CKS1, SKP2, and PLK1 (49). Subsequently, Fu et al. have shown that mitotic serine/threonine kinase protein, Polo like kinase 1 (PLK1), a target of FoxM1 itself, interacts with and phosphorylates FoxM1. This phosphorylation, indeed, regulates the transcriptional program driven by FoxM1 (50), thereby indicating a positive feedback loop as a driving force in mitotic transcriptional regulation.
In the last decade, E2F transcription factor family, well-known regulator of S-phase specific trans-regulation, has also been identified in transcriptional control of mitotic genes (51–57). The initial finding of E2F targets from microarray analysis was validated in more than one way and was followed up with identification of target genes involved in chromosome condensation and segregation, SAC functioning, centrosome organization and duplication, and cytokinesis. For instance, core SAC protein Mad2, mitotic ubiquitin carrier protein UbcH10 and PTTG1, a subunit of chromosome cohesion regulator Securin are shown in extensive detail to be G2/M specific E2F targets (58–60). Zhu et al. further showed that recruitment of an activator E2F to the promoter of mitotic cyclin-dependent kinase gene CDC2/CDK1 requires an adjacent CCAAT consensus site pre-occupied by NF-Y. Furthermore, the authors reported that the association of Myb family transcription factor, b-Myb to the promoter of CDC2 and CCNB1 depends on an intact E2F binding site, suggesting a co-operative nature of trans-factor binding in determining mitotic gene activation. Interestingly, b-Myb, itself being an early phase E2F target, links the E2F driven early phase (G1/S) and late phase (G2/M) transcription cascade. Cdc2, Cyclin A2, and Cyclin B1, three important regulators of mitotic entry and progression, were found to be under control of b-Myb-E2F mediated transcription (61).
Human MuvB core complex, comprising of Lin9, Lin37, Lin52, Lin54, and RBBP4, was also identified to regulate transcription of the genes required for the progression into mitosis. Knockdown of the members of this complex led to downregulation of mRNA levels of mitotic proteins including Plk1, Aurora kinase A, Bub1, CENP-E, Lap2, Cyclin A2, Cyclin B1, Cep55, Survivin, and Cdc2 (62, 63). Following this, Sadasivam et al. (64) explains the association and interplay among these master regulators of transcription during the course of cell cycle. They showed that DREAM complex (comprising of DP1, Rb-related protein p130, E2F4, and MuvB core complex) functions as a global repressor of mitotic genes during quiescence or G0 phase. Following entry of a cell in G1 phase after quiescence, this DREAM complex dissociates from MuvB core complex. The MuvB core complex then associates with b-Myb and gets recruited to the promoters of late phase mitotic genes (64). Subsequently, MuvB and b-Myb together facilitate the binding of FoxM1 to these promoters during G2 phase to promote the transcription of mitotic proteins like, Cyclin B1, Plk1, Cdc6, Aurora kinase A, and RacGAP1. The cell cycle regulated expression of three other mitotic genes, namely ECT2, MgcRacGAP, and MKLP1, also showed CHR dependent repression throughout G1 phase (65). These genes code for three important regulators of Rho GTPases, critical for mitotic progression, and cytokinesis. The cut homeobox 1 (Cux1) transcription factor coordinately induces the expression of these three genes from S-phase. Moreover, E2F1 was shown to be required in this Cux1 dependent trans-activation process (65).
Besides these well-known consensus elements and master regulators of transcription, some gene specific regulations are also documented in influencing the expression of several mitotic genes. For instance, the transcription of Cdc20 is reported to be regulated by E2F through a new element called Cell cycle Site Regulating p55Cdc/Fizzy transcription (SIRF) (66). Surprisingly, a few mitotic proteins are also identified with transcription regulatory activities. A report showed that WD40 domain containing mitotic checkpoint proteins could act as co-repressors during interphase. The WD40 domain containing SAC proteins, Cdc20 and Bub3, form a complex with histone deacetylases (HDAC1 and HDAC2) during the course of repression (67). On the other hand, we showed that Cdc20, in combination with APC/C and CBP/p300, transcriptionally activates the expression of UbcH10 (68). Furthermore, recruitment of this Cdc20 trans-complex showed dependence on E2F consensus element on the UBCH10 promoter (60). The mitotic kinase Plk1 was reported to regulate mitotic gene transcription program by phosphorylating FoxM1 (50). Together, these findings clearly indicate a co-ordination of several master regulators of transcription among themselves and with some gene specific co-activators in controlling cell cycle specific expression of mitotic players. This, in turn, points out the importance of transcription in maintenance of mitotic progression.
Transcriptional Alterations of Mitotic Genes and Association with Cancer
On a cellular level, cancer cells are associated with the loss-of-function mutations of tumor suppressors and the gain-of-function mutations of proto-oncogenes. As many of the mitotic genes are transcriptionally regulated by tumor suppressor or proto-oncoprotein trans-factors, the above-mentioned mutational incidences frequently deregulate the transcriptional outcome of the mitotic genes in tumor cells (Figure 4). This, in turn, results in the abnormal execution of mitosis and defects in the chromosomal segregation leading to aneuploidy. Concordant with that, abnormal expressions of many mitotic genes are often associated with the occurrence of oncogenesis.
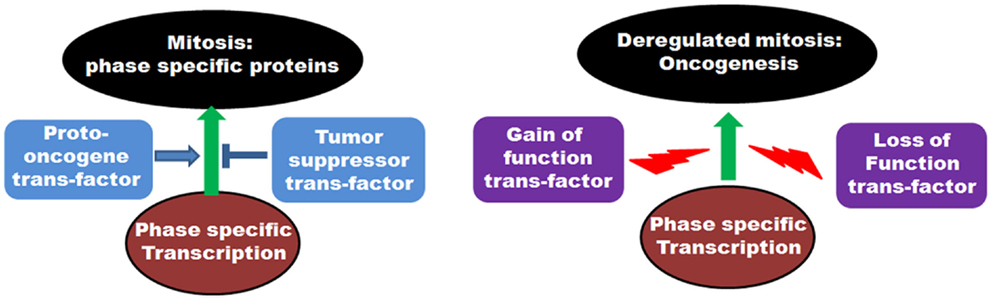
Figure 4. Role of transcription factors in regulation of mitosis: The left panel shows the involvement of various proto-oncogenic trans-factors as well as tumor suppressor transcription factors in regulation of phase specific expression of mitotic proteins. The right panel depicts the deregulation of transcription by gain of function mutations of proto-oncogene trans-factors as well as loss of function mutations of tumor suppressor trans-factors and onset of oncogenesis.
At the initial stages of mitosis (centrosome maturation, chromosome condensation, NE breakdown, and spindle formation), a number of proteins participate in an orchestrated fashion. Among them, expression of Cyclin B, a Cdk1 activator involved in G2/M progression, has been found to be regulated by the tumor suppressor p53 (69). The direct interaction of p53 to the promoter response element downregulates Cyclin B expression upon DNA damage-mediated checkpoint arrest (69). With alteration of p53 pathway, overexpression of Cyclin B has been shown to contribute to the alteration of SAC and occurrence of CIN in cancer samples (70–72). The Ser–Thr kinase, Plk1 (involved in mitotic initiation in more ways than one) showed elevated mRNA levels in a variety of tumors (73). This protein is transcriptionally coordinated during cell cycle, its level being low during interphase and maximum in mitosis (74). The cell cycle-dependent repression of Plk1 is mediated by Rb pathway. During DNA damage-mediated checkpoint activation, tumor suppressors like p53 and BRCA1 are found to influence levels of Plk1 (74, 75). Correlated with the loss of functional tumor suppressors, transcriptional deregulation of Plk1 is reported in various cancers and associated with CIN and oncogenic transformation (76). Furthermore, tumor suppressors like BRCA1 and Rb are reported to regulate the levels of another mitotic kinase Nek2. In co-ordination with the loss-of-function of tumor suppressors, the overexpression of this protein is associated with CIN and cancer (77–80). Cell cycle specific expression of mitotic Aurora kinases (Aurora kinase A and Aurora kinase B) are CDE-CHR element regulated (81, 82). Oncoproteins like EWS-Fli1 and Myc upregulate expression of aurora family proteins through binding on promoter response elements. On the other hand, tumor suppressors like p53, Brd4 also influence expression of Aurora kinases (83–86). In fact, the altered expression of Aurora kinases are potential markers of cancer progression (87). Regulated expression of kinetochore protein Hec1 is directly related to phosphorylation-mediated inactivation of Rb during the course of cell cycle. Beside this, the CREB family of oncoprotein transcription factors has been shown to upregulate the levels of kinetochore protein Hec1 (88). Disrupted pRb function is associated with transcriptional upregulation of Hec1, which may cause aneuploidy and tumor formation (89, 90).
The initial events of mitosis are followed by chromosomal alignment at the equatorial plane of the cell during metaphase. The amphitelic metaphase alignment precedes SAC release, chromosomal segregation, and entry into anaphase. Consistent with their mitotic roles, a number of core SAC and SAC-associated proteins (Mad1, Mad2, Bub1, Cdc20, UbcH10 to name a few) accumulate gradually through the G2 phase with peak levels at mitosis (13, 68, 91, 92). Different transcription factors and chromatin modifiers regulate cell cycle specific promoter activities of these genes (68, 92, 93). The well-known tumor suppressor p53 is reported to control the transcription of CDC20 and BUB1B (46, 47, 94). Upon genotoxic stress, Cdc20 expression is indirectly suppressed by p53 through p21-dependent mechanism (46). On the other hand, a direct p53 binding element has been identified on the CDC20 promoter and shown to bring about repression of transcription through chromatin remodeling (47). Similarly, direct recruitment of p53 on the promoter consensus element brings about chromatin remodeling and the repression of Mad1 expression (95). Expression of Mad2 is regulated by E2F in a cell cycle-dependent manner. Rb inactivation leads to aberrant Mad2 expression by deregulating E2F activity and contributes to mitotic defects and aneuploidy (58). The tumor suppressor BRCA1 was also reported to regulate Mad2 expression (96). Cancer-associated defects in these tumor suppressors contribute to the abnormal expression of these proteins and a flawed SAC. Indeed, transcriptional abnormalities including differential promoter methylation of these SAC proteins are potential markers of cancers of various origins (97, 98). Their deregulated expressions are associated with CIN phenotype and incidence of cancer (6).
The final stages of mitosis involve cytokinesis and mitotic exit. Along with mitotic kinases like Aurora, Polo, and related families, some other molecular components also regulate this stage of the cell cycle. Protein regulator of cytokinesis 1 (PRC1) and the guanine nucleotide exchange factor, Ect2, the two major molecules of cytokinesis have been related with cancer-associated altered expressions and CIN (6). In conclusion, we have summarized a number of reports from the ever-growing lists of proto-onco gene as well as tumor suppressor trans-factors in regulation of mitosis and their deregulation in tumor background (Table 1).
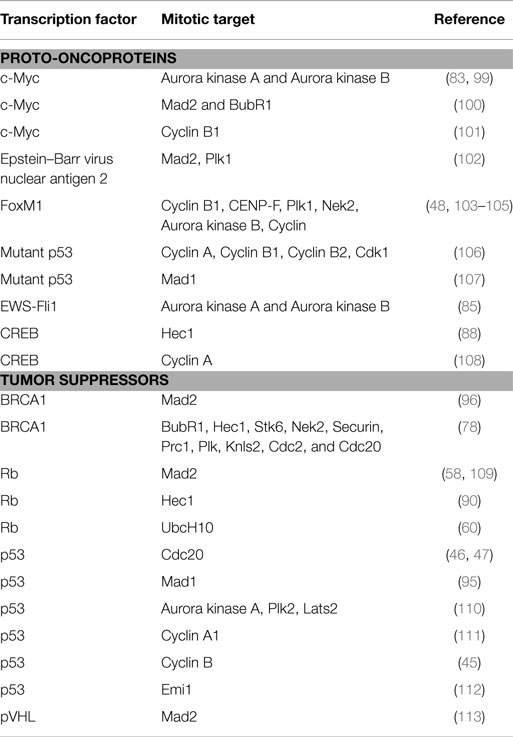
Table 1. Role of proto-oncoprotein and tumor suppressor transcription factors in mitosis and involvement in oncogenesis.
Conclusion
The role of transcriptional regulatory pathways behind the incidence of tumorigenesis remains an enigma. For a number of key cell cycle regulators, the transcriptional control represents an evolutionarily conserved mechanism to precisely maintain their abundance, working in conjunction with miRNA mediated silencing, translational control, and ubiquitin-mediated degradation (23, 26, 114, 115). Among these cell cycle regulators, a defined set of factors stringently control mitotic entry, progression, and exit. The interplay among these factors is naturally adjusted by their abundance. Abnormality in this abundance is associated with the occurrence of aneuploidy, a hallmark of cancer (2, 8, 116). In this review, we have discussed the maintenance of protein levels of the mitotic players whose transcription is regulated in a cell cycle specific manner. We further discussed the deregulation of their transcriptional control, working in concert with cancer onset. In this regard, mutations in various tumor suppressors and proto-oncogenes acting as co-factors of transcription are found to disharmonize the relative protein levels, rather than mutations in the mitotic genes themselves. Besides this, a few mitotic genes are reported to participate in transcriptional control. Furthermore, the list of transcripts whose transcription is affected by certain cell cycle or developmental transitions is being expanded owing to new genome-wide approaches. Answer to many open questions regarding the interplay between transcriptional regulation and mitotic progression will make an important contribution to the understanding of cell cycle control. This, in turn, will help to dissect the involvement of cell cycle progression in the onset of tumorigenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by CSIR-Mayo Clinic collaboration for Innovation and Translational Research Grant CMPP-08 and CSIR-NMITLI project TLP 0007 to Dr. SR. Pre-doctoral fellowship to SN and DG is supported by CSIR.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fendo.2015.00060
Footnotes
References
1. Jallepalli PV, Lengauer C. Chromosome segregation and cancer: cutting xthrough the mystery. Nat Rev Cancer (2001) 1:109–17. doi: 10.1038/35101065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Chi Y-H, Jeang K-T. Aneuploidy and cancer. J Cell Biochem (2007) 102:531–8. doi:10.1002/jcb.21484
3. Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol (2009) 10:478–87. doi:10.1038/nrm2718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep (2012) 13:501–14. doi:10.1038/embor.2012.55
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Pfau SJ, Amon A. Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep (2012) 13:515–27. doi:10.1038/embor.2012.65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. De Castro IP, de Cárcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis (2007) 28:899–912. doi:10.1093/carcin/bgm019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell (2002) 13:1977–2000. doi:10.1091/mbc.02-02-0030
8. Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer (2005) 5:773–85. doi:10.1038/nrc1714
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Siegel JJ, Amon A. New insights into the troubles of aneuploidy. Annu Rev Cell Dev Biol (2012) 28:189. doi:10.1146/annurev-cellbio-101011-155807
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol (2007) 8:379–93. doi:10.1038/nrm2163
11. Nasmyth K. Segregating sister genomes: the molecular biology of chromosome separation. Science (2002) 297:559–65. doi:10.1126/science.1074757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Bharadwaj R, Yu H. The spindle checkpoint, aneuploidy, and cancer. Oncogene (2004) 23:2016–27. doi:10.1038/sj.onc.1207374
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Yu H. Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell (2007) 27:3–16. doi:10.1016/j.molcel.2007.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Reddy SK, Rape M, Margansky WA, Kirschner MW. Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature (2007) 446:921–5. doi:10.1038/nature05734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Mapelli M, Filipp FV, Rancati G, Massimiliano L, Nezi L, Stier G, et al. Determinants of conformational dimerization of Mad2 and its inhibition by p31comet. EMBO J (2006) 25:1273–84. doi:10.1038/sj.emboj.7601033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Westhorpe FG, Tighe A, Lara-Gonzalez P, Taylor SS. p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J Cell Sci (2011) 124:3905–16. doi:10.1242/jcs.093286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science (2011) 333:1039–43. doi:10.1126/science.1203619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Yu R, Lu W, Chen J, McCabe CJ, Melmed S. Overexpressed pituitary tumor-transforming gene causes aneuploidy in live human cells. Endocrinology (2003) 144:4991–8. doi:10.1210/en.2003-0305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Zhang N, Ge G, Meyer R, Sethi S, Basu D, Pradhan S, et al. Overexpression of separase induces aneuploidy and mammary tumorigenesis. Proc Natl Acad Sci U S A (2008) 105:13033–8. doi:10.1073/pnas.0801610105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, Salmon ED. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol (2001) 153:517–28. doi:10.1083/jcb.153.3.517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Cimini D. Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim Biophys Acta (2008) 1786:32–40. doi:10.1016/j.bbcan.2008.05.003
22. Saunders W. Centrosomal amplification and spindle multipolarity in cancer cells. Semin Cancer Biol (2005) 15:25–32. doi:10.1016/j.semcancer.2004.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Peters J-M. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol (2006) 7:644–56. doi:10.1038/nrm1988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Malumbres M, Barbacid M. Cell cycle kinases in cancer. Curr Opin Genet Dev (2007) 17:60–5. doi:10.1016/j.gde.2006.12.008
25. Lal A, Navarro F, Maher CA, Maliszewski LE, Yan N, O’Day E, et al. . miR-24 inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3’ UTR microRNA recognition elements. Mol Cell (2009) 35:610–25. doi:10.1016/j.molcel.2009.08.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Bueno MJ, Malumbres M. MicroRNAs and the cell cycle. Biochim Biophys Acta (2011) 1812:592–601. doi:10.1016/j.bbadis.2011.02.002
27. Shi W, Alajez NM, Bastianutto C, Hui AB, Mocanu JD, Ito E, et al. Significance of Plk1 regulation by miR-100 in human nasopharyngeal cancer. Int J Cancer (2010) 126:2036–48. doi:10.1002/ijc.24880
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Huang V, Place RF, Portnoy V, Wang J, Qi Z, Jia Z, et al. Upregulation of cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res (2012) 40:1695–707. doi:10.1093/nar/gkr934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Bhattacharjya S, Nath S, Ghose J, Maiti GP, Biswas N, Bandyopadhyay S, et al. miR-125b promotes cell death by targeting spindle assembly checkpoint gene MAD1 and modulating mitotic progression. Cell Death Differ (2013) 20:430–42. doi:10.1038/cdd.2012.135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Bhattacharjya S, Roy KS, Ganguly A, Sarkar S, Panda CK, Bhattacharyya D, et al. Inhibition of nucleoporin member Nup214 expression by miR-133b perturbs mitotic timing and leads to cell death. Mol Cancer (2015) 14:42. doi:10.1186/s12943-015-0299-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer (2004) 4:177–83. doi:10.1038/nrc1299
32. Yuen KW, Montpetit B, Hieter P. The kinetochore and cancer: what’s the connection? Curr Opin Cell Biol (2005) 17:576–82. doi:10.1016/j.ceb.2005.09.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Weaver BA, Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol (2006) 18:658–67. doi:10.1016/j.ceb.2006.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. De Carcer G, Perez de Castro I, Malumbres M. Targeting cell cycle kinases for cancer therapy. Curr Med Chem (2007) 14:969–85. doi:10.2174/092986707780362925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Hanks S, Coleman K, Reid S, Plaja A, Firth H, FitzPatrick D, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet (2004) 36:1159–61. doi:10.1038/ng1449
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell (2003) 3:51–62. doi:10.1016/S1535-6108(02)00235-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Staff S, Isola J, Jumppanen M, Tanner M. Aurora-A gene is frequently amplified in basal-like breast cancer. Oncol Rep (2010) 23:307–12. doi:10.3892/or_00000637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Murata Y, Minami Y, Iwakawa R, Yokota J, Usui S, Tsuta K, et al. ECT2 amplification and overexpression as a new prognostic biomarker for early-stage lung adenocarcinoma. Cancer Sci (2014) 105:490–7. doi:10.1111/cas.12363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Neumann B, Walter T, Heriche J-K, Bulkescher J, Erfle H, Conrad C, et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature (2010) 464:721–7. doi:10.1038/nature08869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Nath S, Moghe M, Chowdhury A, Godbole K, Godbole G, Doiphode M, et al. Is germline transmission of MAD2 gene deletion associated with human fetal loss? Mol Hum Reprod (2012) 18:554–62. doi:10.1093/molehr/gas031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Müller GA, Engeland K. The central role of CDE/CHR promoter elements in the regulation of cell cycle-dependent gene transcription. FEBS J (2010) 277:877–93. doi:10.1111/j.1742-4658.2009.07508.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Caretti G, Salsi V, Vecchi C, Imbriano C, Mantovani R. Dynamic recruitment of NF-Y and histone acetyltransferases on cell-cycle promoters. J Biol Chem (2003) 278:30435–40. doi:10.1074/jbc.M304606200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Rother K, Li Y-Y, Tschöp K, Kirschner R, Müller GA, Mössner J, et al. Expression of cyclin-dependent kinase subunit 1 (Cks1) is regulated during the cell cycle by a CDE/CHR tandem element and is downregulated by p53 but not by p63 or p73. Cell Cycle (2007) 6:853–62. doi:10.4161/cc.6.7.4017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. De Toledo SM, Azzam EI, Keng P, Laffrenier S, Little JB. Regulation by ionizing radiation of CDC2, cyclin A, cyclin B, thymidine kinase, topoisomerase IIalpha, and RAD51 expression in normal human diploid fibroblasts is dependent on p53/p21Waf1. Cell Growth Differ (1998) 9:887–96.
45. Krause K, Wasner M, Reinhard W, Haugwitz U, Lange-zu Dohna C, Mössner J, et al. The tumour suppressor protein p53 can repress transcription of cyclin B. Nucleic Acids Res (2000) 28:4410–8. doi:10.1093/nar/28.22.4410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Kidokoro T, Tanikawa C, Furukawa Y, Katagiri T, Nakamura Y, Matsuda K. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene (2008) 27:1562–71. doi:10.1038/sj.onc.1210799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Banerjee T, Nath S, Roychoudhury S. DNA damage induced p53 downregulates Cdc20 by direct binding to its promoter causing chromatin remodeling. Nucleic Acids Res (2009) 37:2688–98. doi:10.1093/nar/gkp110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Laoukili J, Kooistra MR, Brás A, Kauw J, Kerkhoven RM, Morrison A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol (2005) 7:126–36. doi:10.1038/ncb1217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Wang I-C, Chen Y-J, Hughes D, Petrovic V, Major ML, Park HJ, et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol (2005) 25:10875–94. doi:10.1128/MCB.25.24.10875-10894.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Fu Z, Malureanu L, Huang J, Wang W, Li H, Van Deursen JM, et al. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol (2008) 10:1076–82. doi:10.1038/ncb1767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, et al. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol (2001) 21:4684–99. doi:10.1128/MCB.21.14.4684-4699.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, et al. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev (2002) 16:245–56. doi:10.1101/gad.949802
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Polager S, Kalma Y, Berkovich E, Ginsberg D. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene (2002) 21:437–46. doi:10.1038/sj.onc.1205102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Stevaux O, Dyson NJ. A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol (2002) 14:684–91. doi:10.1016/S0955-0674(02)00388-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. DeGregori J. The genetics of the E2F family of transcription factors: shared functions and unique roles. Biochim Biophys Acta (2002) 1602:131–50. doi:10.1016/S0304-419X(02)00051-3
56. Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene (2005) 24:2810–26. doi:10.1038/sj.onc.1208612
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Cam H, Dynlacht BD. Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell (2003) 3:311–6. doi:10.1016/S1535-6108(03)00080-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Hernando E, Nahlé Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature (2004) 430:797–802. doi:10.1038/nature02820
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Zhou C, Wawrowsky K, Bannykh S, Gutman S, Melmed S. E2F1 induces pituitary tumor transforming gene (PTTG1) expression in human pituitary tumors. Mol Endocrinol (2009) 23:2000–12. doi:10.1210/me.2009-0161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Nath S, Chowdhury A, Dey S, Roychoudhury A, Ganguly A, Bhattacharyya D, et al. Deregulation of Rb-E2F1 axis causes chromosomal instability by engaging the transactivation function of Cdc20–anaphase-promoting complex/cyclosome. Mol Cell Biol (2015) 35:356–69. doi:10.1128/MCB.00868-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Zhu W, Giangrande PH, Nevins JR. E2Fs link the control of G1/S and G2/M transcription. EMBO J (2004) 23:4615–26. doi:10.1038/sj.emboj.7600459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Osterloh L, von Eyss B, Schmit F, Rein L, Hübner D, Samans B, et al. The human synMuv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. EMBO J (2007) 26:144–57. doi:10.1038/sj.emboj.7601478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Kittler R, Pelletier L, Heninger A-K, Slabicki M, Theis M, Miroslaw L, et al. Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat Cell Biol (2007) 9:1401–12. doi:10.1038/ncb1659
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev (2012) 26:474–89. doi:10.1101/gad.181933.111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Seguin L, Liot C, Mzali R, Harada R, Siret A, Nepveu A, et al. CUX1 and E2F1 regulate coordinated expression of the mitotic complex genes Ect2, MgcRacGAP, and MKLP1 in S phase. Mol Cell Biol (2009) 29:570–81. doi:10.1128/MCB.01275-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Haugwitz U, Tschöp K, Engeland K. SIRF – a novel regulator element controlling transcription from the p55Cdc/Fizzy promoter during the cell cycle. Biochem Biophys Res Commun (2004) 320:951–60. doi:10.1016/j.bbrc.2004.06.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Yoon Y-M, Baek K-H, Jeong S-J, Shin H-J, Ha G-H, Jeon A-H, et al. WD repeat-containing mitotic checkpoint proteins act as transcriptional repressors during interphase. FEBS Lett (2004) 575:23–9. doi:10.1016/j.febslet.2004.07.089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Nath S, Banerjee T, Sen D, Das T, Roychoudhury S. Spindle assembly checkpoint protein Cdc20 transcriptionally activates expression of ubiquitin carrier protein UbcH10. J Biol Chem (2011) 286:15666–77. doi:10.1074/jbc.M110.160671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Lipski R, Lippincott DJ, Durden BC, Kaplan AR, Keiser HE, Park J-H, et al. p53 Dimers associate with a head-to-tail response element to repress cyclin B transcription. PLoS One (2012) 7:e42615. doi:10.1371/journal.pone.0042615
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Sarafan-Vasseur N, Lamy A, Bourguignon J, Le Pessot F, Hieter P, Sesbouee R, et al. Overexpression of B-type cyclins alters chromosomal segregation. Oncogene (2002) 21:2051–7. doi:10.1038/sj.onc.1205257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Soria J-C, Jang SJ, Khuri FR, Hassan K, Liu D, Hong WK, et al. Overexpression of cyclin B1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res (2000) 60:4000–4.
72. Takeno S, Noguchi T, Kikuchi R, Uchida Y, Yokoyama S, Müller W. Prognostic value of cyclin B1 in patients with esophageal squamous cell carcinoma. Cancer (2002) 94:2874–81. doi:10.1002/cncr.10542
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer (2006) 6:321–30. doi:10.1038/nrc1841
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Martin BT, Strebhardt K. Polo-like kinase 1: target and regulator of transcriptional control. Cell Cycle (2006) 5:2881–5. doi:10.4161/cc.5.24.3538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Tategu M, Nakagawa H, Sasaki K, Yamauchi R, Sekimachi S, Suita Y, et al. Transcriptional regulation of human polo-like kinases and early mitotic inhibitor. J Genet Genomics (2008) 35:215–24. doi:10.1016/S1673-8527(08)60030-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene (2005) 24:267–76. doi:10.1038/sj.onc.1208273
77. Patel D, Incassati A, Wang N, McCance DJ. Human papillomavirus type 16 E6 and E7 cause polyploidy in human keratinocytes and up-regulation of G2-M-phase proteins. Cancer Res (2004) 64:1299–306. doi:10.1158/0008-5472.CAN-03-2917
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Bae I, Rih JK, Kim HJ, Kang HJ, Hassan B, Kirilyuk A, et al. BRCA1 regulates gene expression for orderly mitotic progression. Cell Cycle (2005) 4:1641–66. doi:10.4161/cc.4.11.2152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Hayward DG, Fry AM. Nek2 kinase in chromosome instability and cancer. Cancer Lett (2006) 237:155–66. doi:10.1016/j.canlet.2005.06.017
80. Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res (2004) 64:7370–6. doi:10.1158/0008-5472.CAN-04-0960
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Tanaka M, Ueda A, Kanamori H, Ideguchi H, Yang J, Kitajima S, et al. Cell-cycle-dependent regulation of human aurora A transcription is mediated by periodic repression of E4TF1. J Biol Chem (2002) 277:10719–26. doi:10.1074/jbc.M108252200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Kimura M, Uchida C, Takano Y, Kitagawa M, Okano Y. Cell cycle-dependent regulation of the human aurora B promoter. Biochem Biophys Res Commun (2004) 316:930–6. doi:10.1016/j.bbrc.2004.01.178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood (2010) 116:1498–505. doi:10.1182/blood-2009-11-251074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Mao J-H, Wu D, Perez-Losada J, Jiang T, Li Q, Neve RM, et al. Crosstalk between Aurora-A and p53: frequent deletion or downregulation of Aurora-A in tumors from p53 null mice. Cancer Cell (2007) 11:161–73. doi:10.1016/j.ccr.2006.11.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Wakahara K, Ohno T, Kimura M, Masuda T, Nozawa S, Dohjima T, et al. EWS-Fli1 up-regulates expression of the Aurora A and Aurora B kinases. Mol Cancer Res (2008) 6:1937–45. doi:10.1158/1541-7786.MCR-08-0054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. You J, Li Q, Wu C, Kim J, Ottinger M, Howley PM. Regulation of aurora B expression by the bromodomain protein Brd4. Mol Cell Biol (2009) 29:5094–103. doi:10.1128/MCB.00299-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Giet R, Petretti C, Prigent C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link? Trends Cell Biol (2005) 15:241–50. doi:10.1016/j.tcb.2005.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Cheng L, Li L, Qiao X, Liu J, Yao X. Functional characterization of the promoter of human kinetochore protein HEC1: novel link between regulation of the cell cycle protein and CREB family transcription factors. Biochim Biophys Acta (2007) 1769:593–602. doi:10.1016/j.bbaexp.2007.07.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Diaz-Rodríguez E, Sotillo R, Schvartzman J-M, Benezra R. Hec1 overexpression hyperactivates the mitotic checkpoint and induces tumor formation in vivo. Proc Natl Acad Sci U S A (2008) 105:16719–24. doi:10.1073/pnas.0803504105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Ferretti C, Totta P, Fiore M, Mattiuzzo M, Schillaci T, Ricordye R, et al. Expression of the kinetochore protein Hec1 during the cell cycle in normal and cancer cells and its regulation by the pRb pathway. Cell Cycle (2010) 9:4174–82. doi:10.4161/cc.9.20.13457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Rape M, Kirschner MW. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature (2004) 432:588–95. doi:10.1038/nature03023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Myslinski E, Gérard M-A, Krol A, Carbon P. Transcription of the human cell cycle regulated BUB1B gene requires hStaf/ZNF143. Nucleic Acids Res (2007) 35:3453–64. doi:10.1093/nar/gkm239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Li H, Cuenin C, Murr R, Wang Z-Q, Herceg Z. HAT cofactor Trrap regulates the mitotic checkpoint by modulation of Mad1 and Mad2 expression. EMBO J (2004) 23:4824–34. doi:10.1038/sj.emboj.7600479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Oikawa T, Okuda M, Ma Z, Goorha R, Tsujimoto H, Inokuma H, et al. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol Cell Biol (2005) 25:4046–61. doi:10.1128/MCB.25.10.4046-4061.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Chun AC, Jin D-Y. Transcriptional regulation of mitotic checkpoint gene MAD1 by p53. J Biol Chem (2003) 278:37439–50. doi:10.1074/jbc.M307185200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Wang R-H, Yu H, Deng C-X. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. Proc Natl Acad Sci U S A (2004) 101:17108–13. doi:10.1073/pnas.0407585101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Jeong S-J, Shin H-J, Kim S-J, Ha G-H, Cho B-I, Baek K-H, et al. Transcriptional abnormality of the hsMAD2 mitotic checkpoint gene is a potential link to hepatocellular carcinogenesis. Cancer Res (2004) 64:8666–73. doi:10.1158/0008-5472.CAN-03-3455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Park H, Jeon Y, Shin H, Kim I, Kang H, Jeong S, et al. Differential promoter methylation may be a key molecular mechanism in regulating BubR1 expression in cancer cells. Exp Mol Med (2007) 39:195. doi:10.1038/emm.2007.22
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Courapied S, Cherier J, Vigneron A, Troadec M-B, Giraud S, Valo I, et al. Regulation of the Aurora-A gene following topoisomerase I inhibition: implication of the Myc transcription factor. Mol Cancer (2010) 9:205. doi:10.1186/1476-4598-9-205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Menssen A, Epanchintsev A, Lodygin D, Rezaei N, Jung P, Verdoodt B, et al. c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle (2007) 6:339–52. doi:10.4161/cc.6.3.3808
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci U S A (2002) 99:6274–9. doi:10.1073/pnas.082005599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Pan S-H, Tai C-C, Lin C-S, Hsu W-B, Chou S-F, Lai C-C, et al. Epstein-Barr virus nuclear antigen 2 disrupts mitotic checkpoint and causes chromosomal instability. Carcinogenesis (2009) 30:366–75. doi:10.1093/carcin/bgn291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Wierstra I, Alves J. FOXM1, a typical proliferation-associated transcription factor. Biol Chem (2007) 388:1257–74. doi:10.1515/BC.2007.159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Wierstra I. FOXM1 (Forkhead box M1) in tumorigenesis: overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor-suppressive properties, and target of anticancer therapy. Adv Cancer Res (2012) 119:191–419. doi:10.1016/B978-0-12-407190-2.00016-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell (2006) 10:191–202. doi:10.1016/j.ccr.2006.08.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Iwanaga Y, Jeang K-T. Expression of mitotic spindle checkpoint protein hsMAD1 correlates with cellular proliferation and is activated by a gain-of-function p53 mutant. Cancer Res (2002) 62:2618–24.
108. Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A, et al. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell (2005) 7:351–62. doi:10.1016/j.ccr.2005.02.018
109. Orr B, Compton DA. A double-edged sword: how oncogenes and tumor suppressor genes can contribute to chromosomal instability. Front Oncol (2013) 3:164. doi:10.3389/fonc.2013.00164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Kurinna S, Stratton SA, Coban Z, Schumacher JM, Grompe M, Duncan AW, et al. p53 regulates a mitotic transcription program and determines ploidy in normal mouse liver. Hepatology (2013) 57:2004–13. doi:10.1002/hep.26233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Rivera A, Mavila A, Bayless KJ, Davis GE, Maxwell SA. Cyclin A1 is a p53-induced gene that mediates apoptosis, G2/M arrest, and mitotic catastrophe in renal, ovarian, and lung carcinoma cells. Cell Mol Life Sci (2006):1425–39. doi:10.1007/s00018-006-5521-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Lehman NL, Verschuren EW, Hsu JY, Cherry AM, Jackson PK. Overexpression of the anaphase promoting complex/cyclosome inhibitor Emi1 leads to tetraploidy and genomic instability of p53-deficient cells. Cell Cycle (2006) 5:1569–73. doi:10.4161/cc.5.14.2925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Thoma CR, Toso A, Gutbrodt KL, Reggi SP, Frew IJ, Schraml P, et al. VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol (2009) 11:994–1001. doi:10.1038/ncb1912
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Kronja I, Orr-Weaver TL. Translational regulation of the cell cycle: when, where, how and why? Philos Trans R Soc Lond B Biol Sci (2011) 366:3638–52. doi:10.1098/rstb.2011.0084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Lindqvist A, Rodríguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol (2009) 185:193–202. doi:10.1083/jcb.200812045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Suijkerbuijk SJ, Kops GJ. Preventing aneuploidy: the contribution of mitotic checkpoint proteins. Biochim Biophys Acta (2008) 1786:24–31. doi:10.1016/j.bbcan.2008.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: mitosis, aneuploidy, cancer, transcription, mutation
Citation: Nath S, Ghatak D, Das P and Roychoudhury S (2015) Transcriptional control of mitosis: deregulation and cancer. Front. Endocrinol. 6:60. doi: 10.3389/fendo.2015.00060
Received: 25 February 2015; Paper pending published: 16 March 2015;
Accepted: 08 April 2015; Published: 05 May 2015
Edited by:
Wen Zhou, Columbia University, USAReviewed by:
Thomas E. Adrian, United Arab Emirates University, United Arab EmiratesHua Chen, University of Miami, USA
Shanshan Duan, NYU Langone Medical Center, USA
Copyright: © 2015 Nath, Ghatak, Das and Roychoudhury. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susanta Roychoudhury, Cancer Biology and Inflammatory Disorder Division, CSIR-Indian Institute of Chemical Biology, 4, Raja S.C. Mullick Road, Kolkata 700 032, India, susanta@iicb.res.in; susantarc@gmail.com
†Present address: Somsubhra Nath, Genetics, Cell Biology and Anatomy Division, University of Nebraska Medical Center, Omaha, NE, USA