 Aisha Yousaf
Aisha Yousaf Junfeng Liu
Junfeng Liu Sicheng Ye
Sicheng Ye Hua Chen
Hua Chen- 1CAS Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China
- 2China National Center for Bioinformation, Beijing, China
- 3University of Chinese Academy of Sciences, Beijing, China
- 4CAS Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming, China
The availability of high-quality genome sequences of great ape species provides unprecedented opportunities for genomic analyses. Herein, we reviewed the recent progress in evolutionary comparative genomic studies of the existing great ape species, including human, chimpanzee, bonobo, gorilla, and orangutan. We elaborate discovery on evolutionary history, natural selection, structural variations, and new genes of these species, which is informative for understanding the origin of human-specific phenotypes.
Introduction
The pronounced upsurge in modern sequencing technologies during the recent past has led to immense genomic data across a wide range of species. This spearheaded efforts to better understand the divergent species’ genome architecture, and unfold the potential adaptation mechanisms. Recently, interest has grown to provide enhanced insights into the genetic diversity of primates using comparative and population genomic data. Being Homo sapiens’ closely related cousins, non-human primates (NHPs) provide a stepping stone for better understanding the evolutionary origins of human-specific traits. Moreover, they can also serve as models for studying the genetic basis of human disease phenotypes. Humans markedly differ from the closely related NHPs on various grounds, including, but not limited to, brain size, cognitive capacities, social behavior, language, craniofacial features, bipedalism, hairless skin, and advanced tool usage (Gagneux and Varki, 2001; Carroll, 2003). Natural selection is deemed responsible for species’ adaptations to changing environments. During recent years, a plethora of studies focused on pinpointing the natural selection signatures in humans and their closely related cousins to understand the genetic basis of modern humans’ adaptation and their evolutionary uniqueness. Herein, we briefly review the recently conducted evolutionary comparative genomic studies of great apes regarding their evolutionary history, natural selection, new genes (originated in humans), and structural variation landscape. Figure 1 illustrates the phylogenetic relationship among great ape species.
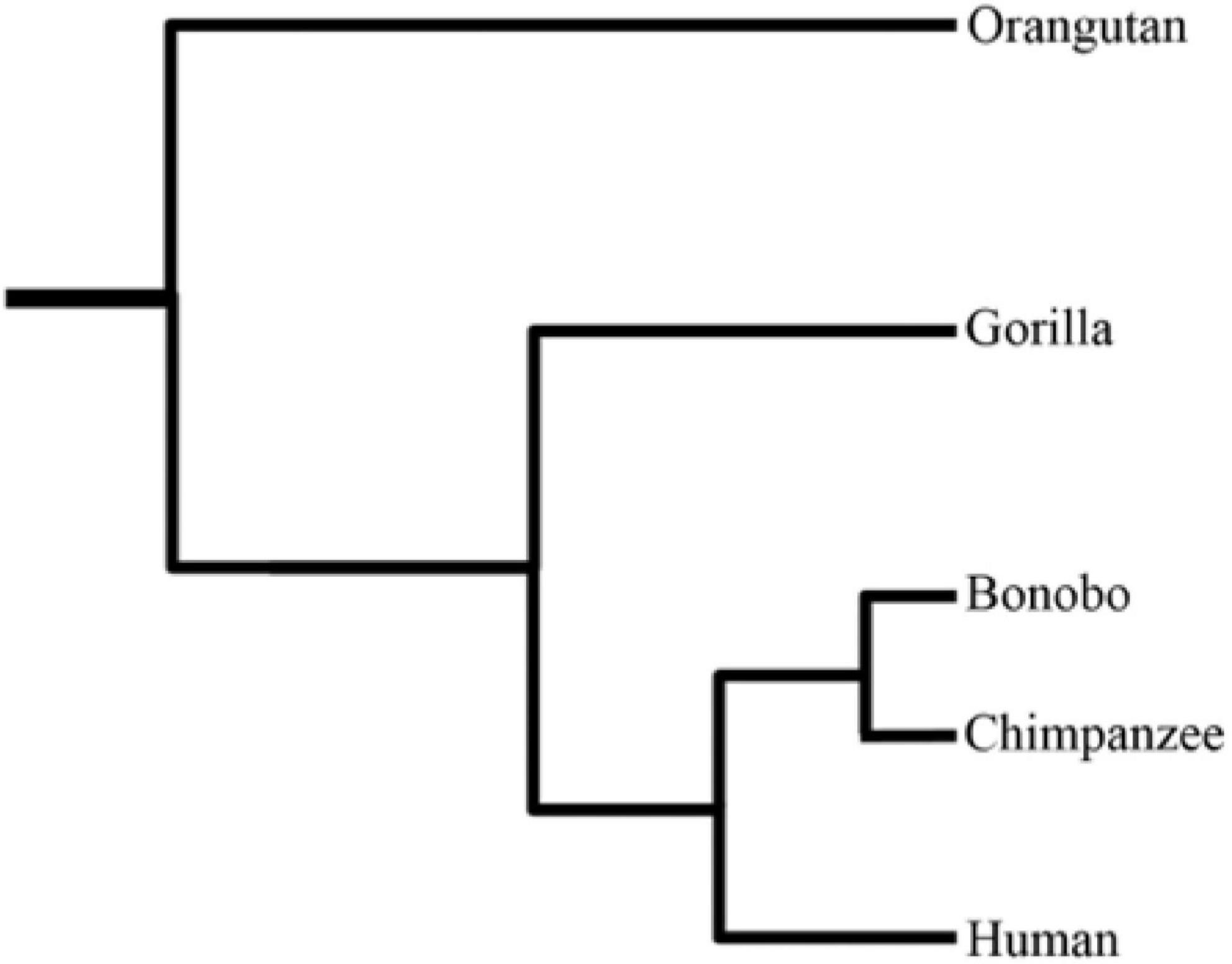
Figure 1. The phylogenetic tree of great ape species (Prado-Martinez et al., 2013).
Genome Assemblies
The first genome sequence of chimpanzee was assembled in 2005, which was compared with the human genome for generating the catalog of genetic differences found among both species (The Chimpanzee Sequencing and Analysis Consortium, 2005). The Sumatran orangutan draft genome has been assembled in 2011 and compared to the other primates (Locke et al., 2011). Prüfer et al. (2012) sequenced and assembled the bonobo genome in 2012 for studying the evolutionary relationship with chimpanzee and human genomes. Although the short-read sequencing has been widely used for whole-genome assembly due to the decreasing cost and increasing throughput, the repetitive DNA sequences may make the assembly incomplete (Gordon et al., 2016). Recently developed long-read sequencing technologies, single-molecule real-time (SMRT) sequencing by PacBio sciences, and Oxford Nanopore Technologies’ (ONT) nanopore sequencing have proved promising in generating the highly contiguous genomes. Using SMRT sequencing, Gordon et al. (2016) presented the long-read sequence assembly of gorilla genome with a novel assembly algorithm, which can use long (>10 Kbp) sequence reads. Kronenberg et al. (2018) generated the genome assemblies of human, chimpanzee, and orangutan using SMRT long-read sequencing with over 65-fold coverage. Mao et al. (2021) reported a new high-quality bonobo genome assembly using the long-read PacBio platform with 74-fold sequence coverage. The details of recently assembled genomes of great apes can be found in Table 1. The availability of high-quality whole-genome sequences of extant great ape species provides an avenue to conduct highly refined comparative genomics research in great ape lineages.
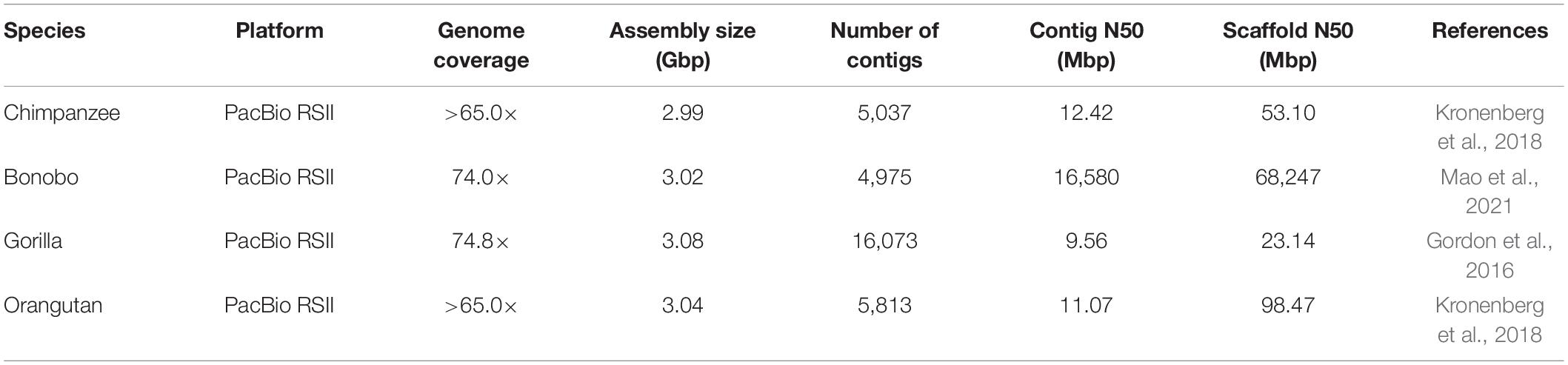
Table 1. A summary of whole-genome sequencing in great apes.
Evolutionary History
Genomic Introgression
Previous studies based on molecular data report that humans and chimpanzees diverged ∼5–7 million years ago (Mya) (Chen and Li, 2001; Brunet et al., 2002). However, this estimate is incompatible with the divergence estimates drawn based on fossil data, which is ∼6–7 Mya. The study led by Langergraber indicates that human–chimpanzee split likely occurred at least 7–8 Mya (Langergraber et al., 2012). Recently, Besenbacher et al. (2019) yielded divergence estimates by extrapolating the non-human mutation rates over the great apes’ phylogenetic tree and dated humans and chimpanzees split to 10.6 Mya. The difference between the two genomes is ∼1%. By analyzing the divergence among hundreds of DNA sequences between the two genomes, Osada and Wu (2005) inferred that the speciation history between human and chimpanzee could not be the same for coding and intergenic regions. The results suggest that the speciation of human and chimpanzee may not be allopatric speciation. The divergence time between sequences, which belong to two closely related species, shows variation throughout the genome because of the polymorphism and the stochastic fluctuation. The exponential distribution can be used to describe the variation. However, gene flow occurring between the species elicits increased disparity in the divergence time between sequences. By comparing neutral autosomal loci between human and chimpanzee, the data unveiled gene flow in ancestral lineages leading to humans and chimpanzees millions of years ago (Yang, 2010; Liu et al., 2015).
Genetic admixture events among chimpanzee subspecies and bonobos were also investigated based on the genome-wide statistics and site frequency spectrum (SFS)-based modeling (de Manuel et al., 2016). A demographic model based on SFS supports multiple events of genetic admixture between chimpanzees and bonobos. According to this model, gene flow from bonobos into the central and eastern chimpanzees occurred approximately 200,000 and 550,000 years ago. This subsequently spread into Nigeria–Cameroon as a result of admixture between Nigeria–Cameroon and central and eastern chimpanzees. A more recent gene flow event occurred between bonobos and central chimpanzees approximately >200,000 years ago. Furthermore, Kuhlwilm et al. (2019) also detected the signatures of archaic gene flow between bonobos and extinct great ape populations and found the evidence for archaic admixture between bonobos and divergent great ape lineage. They compared the landscapes of introgression in humans and bonobos and showed that SERPINA11 and SERPINA9, which are related to adaptive immunity, are in the longest introgression regions.
Demographic History
Chimpanzees (Pan troglodytes) and bonobos (Pan paniscus), which together form the Pan genus, are the closest extant relatives of the humans, which experienced a bottleneck out of Africa (Li and Durbin, 2011). They have an estimated divergence time of 1–2 Mya (Prüfer et al., 2012). Despite the very short split time, both species markedly differ in terms of their demographic histories and also exhibit lineage-specific differences for several behavioral and phenotypic traits, which make them an interesting subject for comparative genomic studies. Chimpanzees are distributed across Africa ranging from central Africa to eastern and western regions of Africa. Radiation among chimpanzee subspecies began approximately 600,000 years ago (Hoelzel, 2016). Although substantial differences exist in the effective population sizes (Ne) of chimpanzee subspecies, all chimpanzee subspecies have greater Ne except western chimpanzees and harbor higher genetic diversity than their sister species bonobo (de Manuel et al., 2016). Bonobos are found in the Democratic Republic of the Congo (central Africa); central chimpanzees (Pan troglodytes troglodytes) bound their distribution to the west, and the eastern chimpanzee (Pan troglodytes schweinfurthii) to the north and south. Bonobos have experienced population bottlenecks and have a smaller long-term Ne (11,900–23,800) (Prado-Martinez et al., 2013; de Manuel et al., 2016). The rapid decline in the bonobo population during the recent past has rendered them among the endangered species.
In gorillas, there are four sub-species: eastern gorillas, western gorillas, cross-river gorillas, and mountain gorillas. The four gorilla species have different demographic histories over the past 200,000 years (Prado-Martinez et al., 2013). The western gorillas have the largest historical Ne while the historical Ne of the eastern gorillas is the smallest. Additionally, the mountain gorillas experienced severe population bottleneck and are a critically endangered subspecies in central Africa (Xue et al., 2015). Orangutans are the most distant relatives of humans among the extant great apes and have two sub-species: Sumatran orangutans and Bornean orangutans, which were designated as distinct species in 2001 (Xu and Arnason, 1996; Groves, 2001). They have an estimated divergence time of 1 Mya and also experienced severe population bottleneck, which makes it urgent to protect these endangered species (Prado-Martinez et al., 2013). However, Nater et al. (2017) show a new orangutan sub-species, which is an isolated population from Batang Toru and is distinct from Sumatran and Bornean populations. Their analyses revealed that the divergence time between Batang Toru populations and the ancestral populations of Sumatran orangutans and Bornean orangutans is ∼3.38 Mya.
Natural Selection
Many tests designed to detect the natural selection signals identify the genomic loci that show departure from a standard neutral model as probable targets of natural selection. Usually, dN/dS (the non-synonymous/synonymous rate) ratios provide a simplistic measure of selective pressures experienced by a genomic loci, with dN/dS >1 as an indicator of positive selection at the protein level (McDonald and Kreitman, 1991; Yang and Nielsen, 2002, 2008). In comparison, it is difficult to detect the adaptive evolution of non-coding DNA because of the lack of natural benchmark. Siepel et al. introduced methods to test natural selection on non-coding DNA (Gronau et al., 2013; Huang et al., 2017). The method analyzes polymorphism of a single population with sequences of one or several outgroup species, and can distinguish the effects of strong positive, strong negative, and weak negative selection on the basis of their influence on polymorphism and divergence patterns. The aforementioned methods provide an opportunity to detect the natural selection signal across the whole genome.
In earlier studies of Pan genus, the effectiveness of purifying selection has been analyzed and the results showed the correlation between past Ne and the efficacy of natural selection (Bataillon et al., 2015; Cagan et al., 2016). Cagan et al. (2016) presented the first global map of natural selection in great apes based on genome-wide information by combining several neutrality tests. They found that most signatures of positive selection are species-specific, such as the signature driven by the gene AMY2B related to diet. Daub et al. (2017) focused on four branches of the primate tree and identified the biological pathway signals of adaptation in the primate phylogeny. They found that the selection signals in the candidate pathways are elicited by different genes in the different branches during the course of primate evolution. Bertranpetit et al. tested signatures of adaptive introgression in chimpanzees based on SFS (Nye et al., 2018). They found the evidence of subspecies-specific adaptations in introgressed regions, which are involved in processes such as the male reproduction in central chimpanzees, the immune system in eastern chimpanzees, and the nervous system in Nigeria–Cameroon chimpanzees. Han et al. (2019) analyzed the largest available dataset of Pan populations and reported the demographic history and purifying selection as the underlying factors for existing genetic variation in Pan species. They also found that the small past Ne correlates with a larger number of deleterious alleles and the genes enriched with bonobo-specific non-synonymous changes are related to age at menarche in humans. Based on a new bonobo genome assembly, Mao et al. (2021) identified some novel genes and found that most of the novel genes showed selective sweeps in bonobo, such as DIRC1, GULP1, and ERC2.
Recently, Zhao et al. (2019) developed a method, HDMKPRF (aka high-dimensional MKPRF), which is built in the Poisson random field framework developed by Sawyer and Hartl (1992), and is an extension of their MKPRF method from two species to multiple species (Bustamante et al., 2001). They constructed a spatial–temporal landscape of natural selection signatures that occurred across the species’ evolutionary history (Zhao et al., 2019). The method pools information over multiple gene loci and gains power over the traditional single-gene-based MK test to detect positive selection signals (Zhao et al., 2019). Given the high efficiency of the method, genome-wide selection scans across great apes except of bonobos using the HDMKPRF method, which jointly analyzes the within-species polymorphism and cross-species divergence data, provide comprehensive insights into the lineage-specific selection across multiple species. The authors found that the positively selected genes identified in the human lineage are enriched in gene expression regulation pathways, immune system, and metabolic pathways.
Structural Variations
Structural variants (SVs) are an important class of variants that are at least 50 bp in size. SVs have increased capacity to rearrange genomic content than small-scale insertions/deletions (indels; <50 bp) or single-nucleotide variants (SNVs). Owing to their size and abundance, SVs potentially have increased capacity to affect gene expression, shape up genome evolution, and impact phenotypes (Perry et al., 2008; Weischenfeldt et al., 2013). A large number of studies aimed at detecting structural variations in humans and NHPs have contributed a great deal to our understanding of SV abundance and their functional impact. These studies used a wide array of technologies to discover SVs (Gokcumen et al., 2013; Kuderna et al., 2017; Catacchio et al., 2018; Kronenberg et al., 2018).
Catacchio et al. (2018) performed genome-wide comparisons between human, great ape, and macaque genomes and detected 156 putative inversions belonging to 136 human gene loci. These inversions showed considerable variation in their size ranging from 103 Kbp to 91 Mbp. They also found 67 inversions in either one or multiple primates, with 36 inversion breakpoints overlapping with 81 human genes. In addition, they also unveiled the evolutionary history of these genomic inversions to investigate functional differences among primate genomes. The genes impacted by these inversions showed enrichment in several functional categories, including transport proteins, DNA-binding proteins, receptors (G protein-coupled receptors and olfactory receptors), and drug metabolism (cytochrome P450). However, the importance of these genes in bringing about phenotypic differences among humans and other primates remains unknown. Kronenberg et al. (2018) employed long-read sequencing technology to generate high-quality assemblies of human, chimpanzee, and orangutan genomes and identified all SVs >50 bp in size within great ape genomes. They identified a total of 17,789 SVs specifically fixed in human lineage (fhSVs) that impact protein-coding regions as well as regulatory regions with some of these deletions related to human-specific phenotypes. For instance, a large human-specific deletion SV (65 Kbp) was detected in two genes, FADS1 and FASD2, which are also positive selection targets in modern humans and are implicated in fatty-acid biosynthesis (Ameur et al., 2012). This fhSV may have functionally contributed to differential dietary habits of great apes (from herbivores to omnivores) during the course of their evolution (Ye et al., 2017). Notably, two fhSVs were also found in WEE1 and CDC25C genes, which are cell cycle regulators and act as ultrasensitive antagonists. These genes are expressed in radial glia and, as a result of increased cell division therein, are speculated to underlie neocortical expansions in human lineage.
In a recent study, Soto et al. (2020) performed long-range and -read sequencing (using optical nanopore sequencing) of two chimpanzee individuals (belonging to the Pan. troglodytes verus subspecies) in order to characterize their SV landscape. A total of 124 novel SVs with size ≥10 kb (including 88 deletions and 36 inversions) were identified in chimpanzees. The study highlighted that 56 genes impacted by putative chimpanzee-specific SVs could lead to chimpanzee-specific phenotypic traits. Deletion SVs showed overrepresentation in “sensory perception of smell” and “G-protein coupled receptor signaling pathway.” Furthermore, some of the genes impacted by SVs appeared likely targets of natural selection, which is suggestive of the significance of SVs in impacting the chimpanzee adaptation during the course of evolution. Comparing the bonobo genome to the other great apes’ genomes, Mao et al. (2021) identified 22,868 bonobo-specific SVs with size >50 bp (including 7,082 deletions and 15,786 insertions), among which there are 1,965 fixed deletions and 3,604 fixed insertions.
New Genes
New genes are often implicated in the emergence of lineage-specific traits and serve as an important resource for evolutionary innovation. For decades, the birth of new genes was attributed to the modification of preexisting genes (Ohno, 1970). In the recent past, studies also reported the birth of new genes from scratch, by DNA- or RNA-mediated mechanisms (Kaessmann, 2010; Betrán, 2015). These mechanisms include gene fusion, horizontal gene transfer, virus domestication, retroduplication, and de novo gene origination. De novo genes also known as “motherless” or “orphan” genes may constitute a significant proportion (up to 10%) of all new genes (Zhang et al., 2010). Interestingly, de novo protein-coding genes have also been found in humans similar to other species (Wu et al., 2011). Wu et al. (2011) identified 60 protein-coding genes of de novo origin in human lineage implicating a higher-than-expected rate of de novo gene birth. Some of these genes were highly expressed in the cerebral cortex pertaining to their involvement in cognitive capacities.
Cai and Petrov (2010) classified human and chimpanzee orthologous protein-coding genes into distinct age classes (i.e., young and old genes) based on their distribution breadth across the phylogenetic tree. Old genes are broadly distributed compared with young genes, which exhibit restricted phylogenetic distribution and appear in closely related species. Notably, the study unveiled frequent non-synonymous polymorphism experienced by young genes contrary to old genes. These findings suggested that stronger purifying selection acts upon old genes while young genes experience weaker purifying selection and hence evolve faster (Cai and Petrov, 2010). Furthermore, the study also highlighted disparities in the deleterious mutation burden of two gene classes with older genes being abundant in slightly deleterious mutations than younger genes.
Xie et al. (2012) conducted genome-wide identification of de novo protein-coding genes that were specific to hominoids and unveiled the precise origin timing of these genes in vertebrate phylogeny. Furthermore, comparative transcriptomic profiling was also performed in human, chimpanzee, and rhesus macaque to address how these de novo protein-coding genes came into being. Notably, most of these protein-coding genes were found to be originated from non-coding RNAs (Xie et al., 2012). The study presented a “semi-product” model of de novo gene birth and evolution.
Focusing on new genes, Shao et al. (2019) created an integrated online database, named GenTree. GenTree gleans age estimations from multiple gene-dating methods and incorporates the functional genomic data, gathered from Human Protein Atlas, for new genes. By performing genome-wide comparison of the existing age estimation methods, the study unveiled synteny-based pipeline (SBP) as the most suitable method for dating recently duplicated genes. However, for dating ancient genes, protein-family-based methods proved promising. Shao et al. (2019) also curated a list of 254 SBP-dated primate-specific protein-coding genes (PSGs) with different levels of protein evidence. Classifying these PSGs into co-expressing modules spotlighted the functional bias of these PSGs. Notably, PSGs were predominantly involved in male reproduction, defense response, mother–fetus interaction, and brain development. These findings highlighted that PSGs are recruited to processes under strong selection pressure or show biased recruitment in organs with rapidly evolving pathways, for instance, an expanded brain and placenta. Taken together, PSGs were contemplated as a group of genes potentially contributing to primate-specific phenotypic evolution.
Conclusion
Over the past years, the successful completion of the human genome project and the release of whole-genome sequences of NHPs led to a plethora of studies highlighting the most conspicuous differences between humans and their closely related phylogenetic cousins. Natural selection is deemed responsible for conferring unique characteristics to a species or within species trait divergence that includes behavioral, cognitive, dietary, and phenotypic differences, among others. Furthermore, the natural selection signatures identified in humans and their closely related cousins help reveal the mechanism of human evolution. In addition, the systematic discovery of SVs by comparing improved great ape genome assemblies provides enhanced insights into the evolution of structural variations and their contribution to lineage-specific phenotypes during the course of great apes’ evolution.
Although a large number of fossils of humans have been found, they are mainly used to calibrate the divergence time in the studies of evolutionary comparative genomics of great apes. However, ancient DNA that can be extracted from the fossil has great potential advantages in the evolutionary comparative genomics research (Pickrell and Reich, 2014; Hofreiter et al., 2015). Firstly, ancient DNA can be used to track migration and natural selection directly. Secondly, ancient DNA makes it possible to observe genetic variation patterns of extinct species directly. Finally, ancient DNA allows us to study history interactions between extinct species and modern species. Therefore, ancient DNA from archaic hominins, such as Neanderthals and Denisovans, can serve as more closely related outgroups and provide novel insights into the evolution of humans (Slatkin and Racimo, 2016; Yang and Fu, 2018; Zhang and Fu, 2020). Furthermore, how to take advantage of ancient DNA in the studies of evolutionary comparative genomics of great apes will be a promising direction for future research.
Author Contributions
HC designed the manuscript. AY, JL, SY, and HC wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key R&D Program of China (2018YFC1406902), the National Natural Science Foundation of China (Grant Nos. 31571370, 91631106, and 91731302), and CAS-TWAS President’s fellowship to AY.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DG declared a past co-authorship with one of the authors SY to the handling editor.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ameur, A., Enroth, S., Johansson, A., Zaboli, G., Igl, W., Johansson, A. C. V., et al. (2012). Genetic adaptation of fatty-acid metabolism: a human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am. J. Hum. Genet. 90, 809–820. doi: 10.1016/j.ajhg.2012.03.014
Bataillon, T., Duan, J., Hvilsom, C., Jin, X., Li, Y., Laurits, S., et al. (2015). Inference of purifying and positive selection in three subspecies of chimpanzees (Pan troglodytes) from exome sequencing. Genome Biol. Evol. 7, 1122–1132. doi: 10.1093/gbe/evv058
Besenbacher, S., Hvilsom, C., Marques-Bonet, T., Mailund, T., and Schierup, M. H. (2019). Direct estimation of mutations in great apes reconciles phylogenetic dating. Nat. Ecol. Evol. 3, 286–292. doi: 10.1038/s41559-018-0778-x
Betrán, E. (2015). The “life histories” of genes. J. Mol. Evol. 80, 186–188. doi: 10.1007/s00239-015-9668-x
Brunet, M., Guy, F., Pilbeam, D., Mackaye, H. T., Likius, A., Ahounta, D., et al. (2002). A new hominid from the upper miocene of chad, Central Africa. Nature 418, 145–151. doi: 10.1038/nature00879
Bustamante, C. D., Wakeley, J., Sawyer, S., and Hartl, D. L. (2001). Directional selection and the site-frequency spectrum. Genetics 159, 1779–1788. doi: 10.1093/genetics/159.4.1779
Cagan, A., Theunert, C., Laayouni, H., Santpere, G., Pybus, M., Casals, F., et al. (2016). Natural selection in the great apes. Mol. Biol. Evol. 33, 3268–3283. doi: 10.1093/molbev/msw215
Cai, J. J., and Petrov, D. A. (2010). Relaxed purifying selection and possibly high rate of adaptation in primate lineage-specific genes. Genome Biol. Evol. 2, 393–409. doi: 10.1093/gbe/evq019
Carroll, S. B. (2003). Genetics and the making of Homo sapiens. Nature 422, 849–857. doi: 10.1038/nature01495
Catacchio, C. R., Maggiolini, F. A. M., D’Addabbo, P., Bitonto, M., Capozzi, O., Signorile, M. L., et al. (2018). Inversion variants in human and primate genomes. Genome Res. 28, 910–920. doi: 10.1101/gr.234831.118
Chen, F. C., and Li, W. H. (2001). Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 68, 444–456. doi: 10.1086/318206
Daub, J. T., Moretti, S., Davydov, I. I., Excoffier, M., and Robinson-Rechavi, M. (2017). Detection of pathways affected by positive selection in primate lineages ancestral to humans. Mol. Biol. Evol. 34, 1391–1402. doi: 10.1093/molbev/msx083
de Manuel, M., Kuhlwilm, M., Frandsen, P., Sousa, V. C., Desai, T., Prado-Martinez, J., et al. (2016). Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 354, 477–481. doi: 10.1126/science.aag2602
Gagneux, P., and Varki, A. (2001). Genetic differences between humans and great apes. Mol. Phylogenet. Evol. 18, 2–13. doi: 10.1006/mpev.2000.0799
Gokcumen, O., Tischler, V., Tica, J., Zhu, Q., Iskow, R. C., Lee, E., et al. (2013). Primate genome architecture influences structural variation mechanisms and functional consequences. Proc. Natl. Acad. Sci. U.S.A. 110, 15764–15769. doi: 10.1073/pnas.1305904110
Gordon, D., Huddleston, J., Chaisson, M. J., Hill, C. M., Kronenberg, Z. N., Munson, K. M., et al. (2016). Long-read sequence assembly of the gorilla genome. Science 352:aae0344. doi: 10.1126/science.aae0344
Gronau, I., Arbiza, L., Mohammed, J., and Siepel, A. (2013). Inference of natural selection from interspersed genomic elements based on polymorphism and divergence. Mol. Biol. Evol. 30, 1159–1171. doi: 10.1093/molbev/mst019
Han, S., Andrés, A. M., Marques-Bonet, T., and Kuhlwilm, M. (2019). Genetic variation in pan species is shaped by demographic history and harbors lineage-specific functions. Genome Biol. Evol. 11, 1178–1191. doi: 10.1093/gbe/evz047
Hoelzel, A. R. (2016). The road to speciation runs both ways. Science 354, 414–415. doi: 10.1126/science.aaj2007
Hofreiter, M., Paijmans, J. L. A., Googchild, H., Speller, C. F., Barlow, A., Fortes, G. G., et al. (2015). The future of ancient DNA: technical advances and conceptual shifts. Bioessays 37, 284–293. doi: 10.1002/bies.201400160
Huang, Y.-F., Gulko, B., and Siepel, A. (2017). Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet. 49, 618–624. doi: 10.1038/ng.3810
Kaessmann, H. (2010). Origins, evolution, and phenotypic impact of new genes. Genome Res. 20, 1313–1326. doi: 10.1101/gr.101386.109
Kronenberg, Z. N., Fiddes, I. T., Gordon, D., Murali, S., Cantsilieris, S., Meyerson, O. S., et al. (2018). High-resolution comparative analysis of great ape genomes. Science 360:eaar6343. doi: 10.1126/science.aar6343
Kuderna, L. F. K., Tomlinson, C., Hillier, L. W., Tran, A., Fiddes, I. T., Armstrong, J., et al. (2017). A 3-way hybrid approach to generate a new high-quality chimpanzee reference genome (Pan_tro_3.0). Gigascience 6, 1–6. doi: 10.1093/gigascience/gix098
Kuhlwilm, M., Han, S., Sousa, V. C., Excoffier, L., and Marues-Bonet, T. (2019). Ancient admixture from an extinct ape lineage into bonobos. Nat. Ecol. Evol. 3, 957–965. doi: 10.1038/s41559-019-0881-7
Langergraber, K. E., Prüfer, K., Rowney, C., Boesch, C., Crockford, C., Fawcett, K., et al. (2012). Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proc. Natl. Acad. Sci. U.S.A. 109, 15716–15721. doi: 10.1073/pnas.1211740109
Li, H., and Durbin, R. (2011). Inference of human population history from individual whole-genome sequences. Nature 475, 493–U84. doi: 10.1038/nature10231
Liu, J., Zhang, D.-X., and Yang, Z. (2015). A discrete-beta model for testing gene flow after speciation. Methods Ecol. Evol. 6, 715–724. doi: 10.1111/2041-210X.12356
Locke, D. P., Hillier, L. W., Warren, W. C., Worley, K. C., Nazareth, L. V., Muzny, D. M., et al. (2011). Comparative and demographic analysis of orang-utan genomes. Nature 469, 529–533. doi: 10.1038/nature09687
Mao, Y., Catacchio, C. R., Hillier, L. W., Porubsky, D., Li, R., Sulovari, A., et al. (2021). A high-quality bonobo genome refines the analysis of hominoid evolution. Nature 594, 77–81. doi: 10.1038/s41586-021-03519-x
McDonald, J. H., and Kreitman, M. (1991). Adaptive protein evolution at the adh locus in Drosophila. Nature 351, 652–654. doi: 10.1038/351652a0
Nater, A., Mattle-Greminger, M. P., Nurcahyo, A., Nowak, M. G., de Manuel, M., Desai, T., et al. (2017). Morphometric, behavioral, and genomic evidence for a new orangutan species. Curr. Biol. 27, 3487–3498. doi: 10.1016/j.cub.2017.09.047
Nye, J., Laayouni, H., Kuhlwilm, M., Mondal, M., Marques-Bonet, T., and Bertranpetit, J. (2018). Selection in the Introgressed Regions of the Chimpanzee Genome. Genome Biol. Evol. 10, 1132–1138. doi: 10.1093/gbe/evy077
Osada, N., and Wu, C. I. (2005). Inferring the mode of speciation from genomic data: a study of the great apes. Genetics 169, 259–264. doi: 10.1534/genetics.104.029231
Perry, G. H., Ben-Dor, A., Tsalenko, A., Sampas, N., Rodriguez-Revenga, L., Tran, C. W., et al. (2008). The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 82, 685–695. doi: 10.1016/j.ajhg.2007.12.010
Pickrell, J. K., and Reich, D. (2014). Toward a new history and geography of human genes informed by ancient DNA. Trends Genet. 30, 377–389. doi: 10.1016/j.tig.2014.07.007
Prado-Martinez, J., Sudmant, P., Kidd, J., Li, H., Kelley, J. H., Lorente-Galdos, B., et al. (2013). Great ape genetic diversity and population history. Nature 499, 471–475. doi: 10.1038/nature12228
Prüfer, K., Munch, K., Hellmann, I., Akagi, K., Miller, J. R., Walenz, B., et al. (2012). The bonobo genome compared with the chimpanzee and human genomes. Nature 486, 527–531. doi: 10.1038/nature11128
Sawyer, S. A., and Hartl, D. L. (1992). Population genetics of polymorphism and divergence. Genetics 32, 1161–1176.
Shao, Y., Chen, C., Shen, H., He, B. Z., Yu, D., Jiang, S., et al. (2019). GenTree, an integrated resource for analyzing the evolution and function of primate-specific coding genes. Genome Res. 29, 682–696. doi: 10.1101/gr.238733.118
Slatkin, M., and Racimo, F. (2016). Ancient DNA and human history. Proc. Natl. Acad. Sci. U.S.A. 113, 6380–6387. doi: 10.1073/pnas.1524306113
Soto, D. C., Shew, C., Mastoras, M., Schmidt, J. M., Sahasrabudhe, R., Kaya, G., et al. (2020). Identification of structural variation in chimpanzees using optical mapping and nanopore sequencing. Genes 11:276. doi: 10.3390/genes11030276
The Chimpanzee Sequencing and Analysis Consortium. (2005). Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87. doi: 10.1038/nature04072
Weischenfeldt, J., Symmons, O., Spitz, F., and Korbel, J. O. (2013). Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14, 125–138. doi: 10.1038/nrg3373
Wu, D. D., Irwin, D. M., and Zhang, Y. P. (2011). De novo origin of human protein-coding genes. PLoS Genet. 8:e1002942. doi: 10.1371/journal.pgen.1002942
Xie, C., Zhang, Y. E., Chen, J. Y., Liu, C. J., Zhou, W. Z., Li, Y., et al. (2012). Hominoid-specific De Novo protein-coding genes originating from long non-coding RNAs. PLoS Genet. 8:e1002942.
Xu, X., and Arnason, U. (1996). The mitochondrial DNA molecule of Sumatran orangutan and a molecular proposal for two (Bornean and Sumatran) species of orangutan. J. Mol. Evol. 43, 431–437. doi: 10.1007/BF02337514
Xue, Y., Prado-Martinez, J., Sudmant, P. H., Narasimhan, V., Ayub, Q., Szpak, M., et al. (2015). Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 348, 242–245. doi: 10.1126/science.aaa3952
Yang, M. A., and Fu, Q. (2018). Insights into modern human prehistory using ancient genomes. Trends Genet. 34, 184–196. doi: 10.1016/j.tig.2017.11.008
Yang, Z. (2010). A likelihood ratio test of speciation with gene flow using genomic sequence data. Genome Biol. Evol. 2, 200–211. doi: 10.1093/gbe/evq011
Yang, Z., and Nielsen, R. (2002). Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 19, 908–917. doi: 10.1093/oxfordjournals.molbev.a004148
Yang, Z., and Nielsen, R. (2008). Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol. Biol. Evol. 25, 568–579. doi: 10.1093/molbev/msm284
Ye, K., Gao, F., Wang, D., Bar-Yosef, O., and Keinan, A. (2017). Dietary adaptation of FADS genes in Europe varied across time and geography. Nat. Ecol. Evol. 1:167. doi: 10.1038/s41559-017-0167
Zhang, M., and Fu, Q. (2020). Human evolutionary history in eastern Eurasia using insights from ancient DNA. Curr. Opin. Genet. Dev. 62, 78–84. doi: 10.1016/j.gbe.2020.06.009
Zhang, Y. E., Vibranovski, M. D., Krinsky, B. H., and Long, M. (2010). Age-dependent chromosomal distribution of male-biased genes in Drosophila. Genome Res. 20, 1526–1533. doi: 10.1101/gr.107334.110
Keywords: evolutionary comparative genomics, natural selection, structural variations, new genes, great apes
Citation: Yousaf A, Liu J, Ye S and Chen H (2021) Current Progress in Evolutionary Comparative Genomics of Great Apes. Front. Genet. 12:657468. doi: 10.3389/fgene.2021.657468
Received: 22 January 2021; Accepted: 15 July 2021;
Published: 11 August 2021.
Edited by:
Irina Ruf, Senckenberg Research Institute and Natural History Museum Frankfurt, GermanyReviewed by:
Martin Kuhlwilm, Pompeu Fabra University, SpainDeyan Ge, Institute of Zoology, Chinese Academy of Sciences (CAS), China
Copyright © 2021 Yousaf, Liu, Ye and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junfeng Liu, liujunfeng@big.ac.cn; Hua Chen, chenh@big.ac.cn