 Chloé Ambroset1,2†
Chloé Ambroset1,2† Charles Coluzzi1,2†
Charles Coluzzi1,2† Gérard Guédon1,2
Gérard Guédon1,2 Marie-Dominique Devignes3,4
Marie-Dominique Devignes3,4 Valentin Loux5
Valentin Loux5 Thomas Lacroix5
Thomas Lacroix5 Sophie Payot1,2
Sophie Payot1,2 Nathalie Leblond-Bourget1,2*
Nathalie Leblond-Bourget1,2*- 1DynAMic, Faculté des Sciences et Technologies, Université de Lorraine, UMR 1128, Vandœuvre-lès-Nancy, France
- 2DynAMic, Institut National de la Recherche Agronomique, UMR 1128, Vandœuvre-lès-Nancy, France
- 3Laboratoire Lorrain de Recherche en Informatique et ses Applications, Faculté des Sciences et Technologies, Université de Lorraine, UMR 7503, Vandœuvre-lès-Nancy, France
- 4CNRS, Laboratoire Lorrain de Recherche en Informatique et ses Applications, UMR 7503, Vandśuvre-lès-Nancy, France
- 5UR 1404 Mathématiques et Informatique Appliquées du Génome à l’Environnement, Institut National de la Recherche Agronomique, Jouy-en-Josas, France
Recent genome analyses suggest that integrative and conjugative elements (ICEs) are widespread in bacterial genomes and therefore play an essential role in horizontal transfer. However, only a few of these elements are precisely characterized and correctly delineated within sequenced bacterial genomes. Even though previous analysis showed the presence of ICEs in some species of Streptococci, the global prevalence and diversity of ICEs was not analyzed in this genus. In this study, we searched for ICEs in the completely sequenced genomes of 124 strains belonging to 27 streptococcal species. These exhaustive analyses revealed 105 putative ICEs and 26 slightly decayed elements whose limits were assessed and whose insertion site was identified. These ICEs were grouped in seven distinct unrelated or distantly related families, according to their conjugation modules. Integration of these streptococcal ICEs is catalyzed either by a site-specific tyrosine integrase, a low-specificity tyrosine integrase, a site-specific single serine integrase, a triplet of site-specific serine integrases or a DDE transposase. Analysis of their integration site led to the detection of 18 target-genes for streptococcal ICE insertion including eight that had not been identified previously (ftsK, guaA, lysS, mutT, rpmG, rpsI, traG, and ebfC). It also suggests that all specificities have evolved to minimize the impact of the insertion on the host. This overall analysis of streptococcal ICEs emphasizes their prevalence and diversity and demonstrates that exchanges or acquisitions of conjugation and recombination modules are frequent.
Introduction
Streptococci are Gram positive bacteria belonging to the phylum of Firmicutes. This genus comprises 110 recognized species (1July 24, 2015). Almost all streptococci are commensal or pathogen of humans and/or animals. Numerous streptococci, such as Streptococcus pneumoniae, Streptococcus pyogenes, Streptococcus mutans or Streptococcus agalactiae, are responsible for a wide variety of diseases worldwide, ranging from mild to invasive infections that have a severe impact on human and animal health and lead to significant morbidity and mortality (Mitchell, 2003; Kohler, 2007). Streptococci are also ubiquitously present as commensal inhabitants of the gastro-intestinal tracts of healthy adults and/or newborns. Streptococcus salivarius, in particular, is one of the first colonizers of the human oral cavity (Park et al., 2005; Nakajima et al., 2013) and is also a dominant part of the early life human intestinal microbiota (Arrieta et al., 2014). At last, two species deriving from commensal streptococci, S. thermophilus and S. macedonicus are used as starters in dairy industry to transform milk in yogurt and/or cheese (Franciosi et al., 2009).
During the last 20 years, it has become increasingly apparent that horizontal gene transfer (HGT) of genomic islands plays a key role in bacterial evolution and adaptation (Hacker and Kaper, 2000; Hacker and Carniel, 2001; Dobrindt et al., 2004; Juhas et al., 2009). In essence, genomic islands are chromosomal segments acquired by HGT that carry gene sets enhancing the fitness of their hosts. Recent data suggest that numerous genomic islands correspond to non-canonical classes of mobile genetic elements (MGEs) that can transfer by conjugation or are non-mobile elements deriving from such MGEs (Bellanger et al., 2014). Among them, the integrative and conjugative elements (ICEs) are mobile elements integrated in bacterial chromosomes or plasmids which encode their own excision, their transfer by conjugation, and their integration (Burrus et al., 2002b; Bellanger et al., 2014).
Integration of ICEs is catalyzed by three phylogenetically and structurally unrelated families of enzymes: tyrosine integrases, serine integrases, and DDE transposases (Wozniak and Waldor, 2010; Bellanger et al., 2014). Both tyrosine and serine integrases usually catalyze site-specific recombination between small (2–60 bp) similar or identical sequences included in the attI site of the circular form of the ICE and the attB site of the bacterial genome. This leads to the formation of attL and attR sites flanking the integrated elements; as a consequence, the integrated ICE is flanked by direct repeats (DR). Usually, tyrosine integrases catalyze ICE integration in a large array of specific sites, including the 3′ end of tRNA genes and the 3′ or 5′ end of genes encoding various housekeeping proteins (Bellanger et al., 2014). One exception is the tyrosine integrase of Tn916 that shows low integration specificity; as a consequence, Tn916 and its relatives are not flanked by DRs. Knowledge of the integration specificity of serine integrases from ICE is scarce. The third family of enzymes, DDE transposases, catalyzes transposition of DNA segments. Binding of the enzyme to terminal inverted repeats (IRs) at the extremities of elements enables strand cleavage required for the transposition reaction. DDE transposases have a low specificity of integration and catalyze the duplication of the target sequence (2–13 bp). Up to now, only one subfamily of DDE transposases was described for streptococcal ICEs. These DDE integrases catalyze the integration 15–16 bp upstream of the -35 box of the promoter region of various genes (Brochet et al., 2009; Guérillot et al., 2013).
The first step of the conjugative transfer is the excision of the ICE as a circular form that is ensured by the same enzyme as for integration. In general, tyrosine integrases need additional co-factors to carry out the reverse excision reaction (Groth and Calos, 2004); these are encoded by the element. So far, most ICEs (including all ICEs from Firmicutes) transfer as single-strand DNA. The transfer of the excised ICE would be similar to the transfer of conjugative plasmids that is well-known in Gram negative bacteria (Smillie et al., 2010; Low et al., 2014). The circularized ICE DNA is taken over by a relaxosome, a complex that includes a relaxase. A relaxase is a trans-esterase, acting as a dimer, that catalyzes a site and strand-specific cleavage at the nic site of the origin of transfer (oriT) of its cognate ICE. The relaxase, covalently bound to the 5′ end of the single-stranded DNA, is then recognized by the membrane-associated coupling protein (CP) that interacts with a type IV secretion system (T4SS). T4SSs are ATP-powered and multi-protein complexes that span the cellular envelopes of the donor cell in Gram negative bacteria. CP and T4SS translocate the DNA-relaxase complex through membranes and cell walls into the recipient cell. A rolling-circle replication of the element is likely concomitant to its transfer so that the ICE is not lost in the donor cell. Finally, the relaxase achieves the transfer by recircularizing the ICE (for a review see (Bellanger et al., 2014)). Although the conjugative transfer of DNA in Firmicutes is poorly known, it relies on relaxase, CP and T4SS (Goessweiner-Mohr et al., 2013; Guglielmini et al., 2014). Recent analyses suggest that T4SSs of Firmicutes include an homolog or analog of most T4SS membrane-spanning proteins found in inner membranes of Gram negative bacteria including the ATPase VirB4, VirB3, VirB6, VirB8 and the cell-wall degrading enzyme VirB1 (Goessweiner-Mohr et al., 2013; Guglielmini et al., 2014; Leonetti et al., 2015).
Like all other bacterial MGEs (Toussaint and Merlin, 2002), ICEs have a modular structure, i.e., the genes involved in the same biological function (such as conjugation or integration/excision) are physically linked. In addition to the integrase and recombination directionality factor genes, the integration/excision module includes the recombination site attI. It is thus generally located at one end of the integrated element. The conjugation module includes genes coding for the T4SS, the CP, the relaxase (and eventually accessory proteins of the relaxase) and oriT. The regulation module encodes all the genes involved in the regulation of ICE dissemination and maintenance. In addition to modules dedicated to gene transfer, all ICEs also carry at least one adaptation module that encodes adaptive traits that might be beneficial for bacteria under certain growth or environmental conditions. Adaptation modules are highly variable and include genes involved in antimicrobial resistance, virulence or alternative metabolic pathways (Burrus et al., 2002a,b; van der Meer and Sentchilo, 2003; Schubert et al., 2004; Heather et al., 2008; Croucher et al., 2009; Chuzeville et al., 2012).
Although ICEs have a major impact on gene flow and genome dynamics in bacteria, their prevalence and diversity remain largely underscored (Bellanger et al., 2014).
In this work, we took advantage of the increase of publicly available genomic sequences (124 complete genomes of Streptococcus available at the beginning of this work) to search for ICEs using the combined presence of signature proteins (from conjugation and integration/excision modules). Coding sequences (CDSs) encoding these signature proteins were localized on the chromosomes and a strategy was developed to search for ICEs boundaries and to identify their integration site. This work (i) gives a general overview of the high prevalence and diversity of ICEs within Streptococcus species, (ii) identifies their numerous sites of insertion, and (iii) sheds light on their phylogenetic relationships and on their modular evolution.
Materials and Methods
Genomes Examined and Database of Reference Proteins
The dataset of the 124 complete chromosomes from Streptococcus species available at the beginning of this work was taken from GenBank (2last accessed December 2013).
The initial database of reference proteins contains signature proteins from ICEs reported for Firmicutes in the literature at the beginning of this study. It includes protein sequences from 50 tyrosine integrases, 13 serine integrases, two DDE transposases, 50 relaxases, 37 CP, and 26 VirB4 proteins.
Search Strategy
The overall workflow of our search strategy to detect and characterize ICEs in streptococcal chromosomes is depicted in Figure 1.
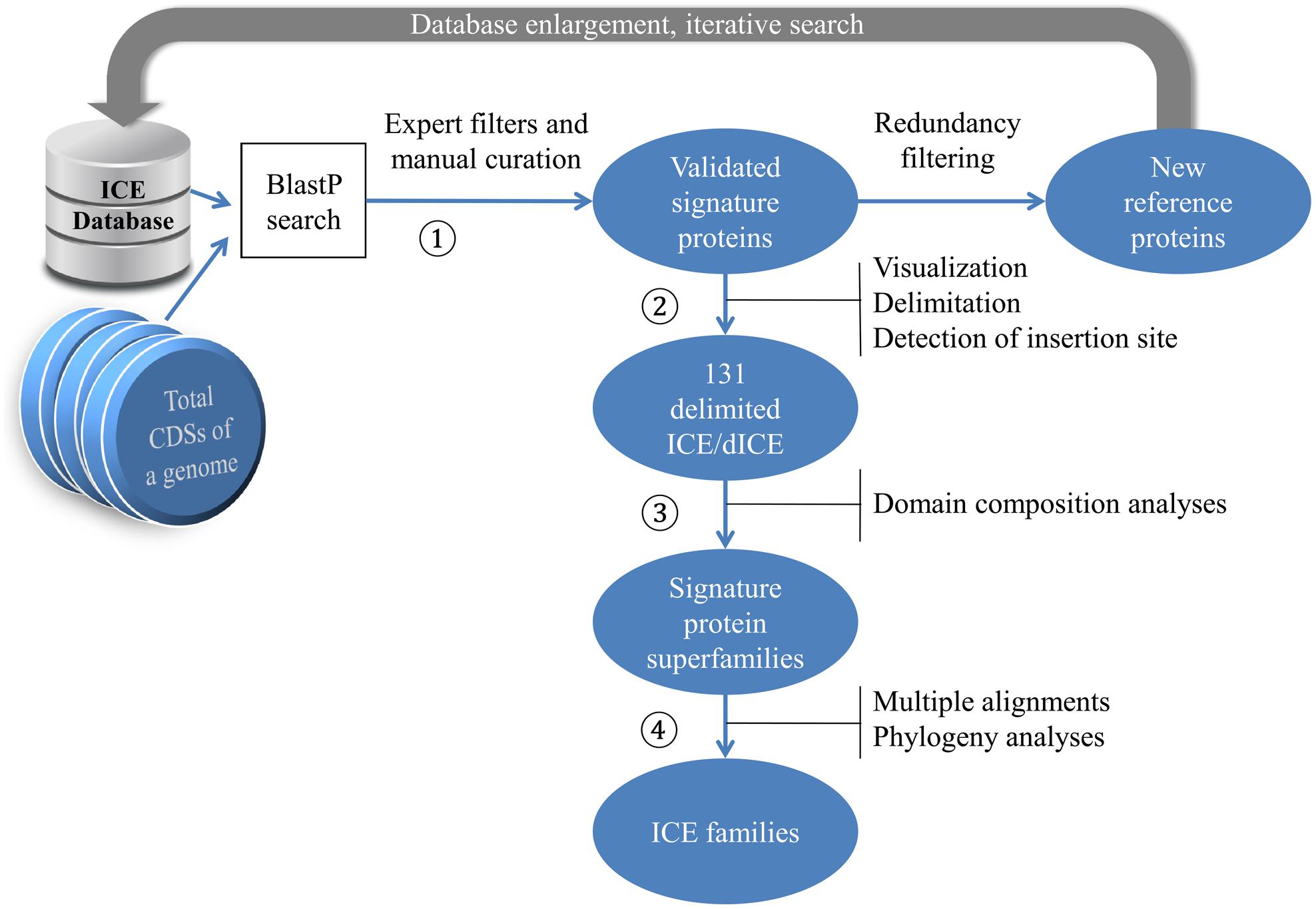
FIGURE 1. Procedure for identifying candidate ICEs in sequenced genomes. The amino-acid sequences encoded by a chromosome are collected as multifasta files and processed as follows (see Materials and Methods). AAA172 Signature proteins are identified by BlastP search using our reference sequences as query and the set of multifasta files as search database. Resulting hits are filtered and validated. AAA173 The location of genes encoding the validated signature proteins are visualized using Artemis and ICEs are delimited. AAA174 Domain composition of signature proteins are searched using Biomart and the signature proteins are grouped into classes. AAA175 Multiple alignments of signature proteins in each class are performed using Clustal Omega and their phylogeny is analyzed using maximum likelihood (ML) methods and BioNJ.
Detection of Signature Sequences in the Genomes of Streptococci
The first step of our workflow consists in the search for signature proteins (tyrosine integrase, serine recombinase or DDE transposase, CP, relaxase, and VirB4 proteins). It was performed by BlastP comparison, using the accelerated BlastP version implemented in the ngKlast software (Nguyen and Lavenier, 2009; with default parameters except for disabled low-complexity filter). The queries were the sequences of all reference ICE proteins and the target was the set of multifasta files (one per genome) representing all translated CDSs of the studied genomes. Expert filters were designed in the ngKlast system to remove hits corresponding to the translation of pseudogenes and to related proteins not involved in transfer or integration of conjugative elements (i.e., recombinases involved in inversion of DNA segments or in resolution of DNA molecule multimers, transcriptional regulators carrying a HTH DNA binding domain, DNA translocase FtsK involved in cell division, some toxins, etc). These filters include a percentage cover threshold (>25%), an E-value threshold (<1.10-04 for relaxases and integrases and <1.10-05 for CPs and VirB4) and a length threshold (>320 amino acids for integrases, >180 amino acids for relaxases, between 180 and 700 aa and between 1000 and 1200 aa for CPs and >500 amino acids for VirB4). Related proteins with biological function other than conjugation (i.e., XerS) passing through filters were manually removed. An iterative search was performed with the reference protein database enlarged with the newly found proteins and the same parameters until no new hit was found. Thus, only elements lacking significant sequence similarity with all signature proteins of ICE or carried by a plasmid could be missed. Sequence redundancy was eliminated using the BlastClust program with an identity threshold of 90%. The final reference database for ICE signature proteins contains non-redundant sequences from 106 tyrosine integrases, 43 serine integrases, 10 DDE transposases, 72 relaxases, 45 CPs, and 36 VirB4 proteins.
The relative positions of the CDSs corresponding to the detected signature proteins were visualized using Artemis (Rutherford et al., 2000). This step allowed checking if the CDSs co-localize in the genome and can thus be part of the same ICE. The detection of VirB4 guaranties the retrieval of ICEs rather than IMEs (integrative and mobilizable elements) that never encode this protein.
Detection of Insertion Sites and Delimitation of Putative ICEs
In the second step of the workflow (Figure 1), CDSs known to be potential insertion sites for ICE encoding site-specific integrases were searched and retained as potential candidates for insertion sites if located close to the CDSs of signature proteins. In their absence, insertion site was examined by comparing synteny with other genomes.
Putative ICEs were delimited by searching DRs on both sides of the putative ICEs by BLASTn analysis with either the 3′ end or the 5′ end of the potential insertion CDS or tRNA genes as a “query.” If no such potential insertion CDS was detected, the intergenic sequence downstream from the integrase gene was used as a query. ICEs closely related to Tn916 were delimited by BLASTn with the ends of Tn916 from Enterococcus faecalis DS16 as queries.
ICE/Decayed ICE (dICE) Counting
All the elements delimited with DRs and containing CDSs for the four complete signature proteins (integrase, relaxase, CP, and VirB4) were considered as ICEs. When some signature CDSs were missing or were incomplete, the complete CDS encoded by the closest ICE was compared to the putative defective one by tBlastN in order to detect possible genome annotation errors (e.g., mis-identification of an authentic gene as a pseudogene most frequently due to the presence of a type II intron within the gene or mis-identification of START codon suggesting truncated genes). Elements that carry one or two defective signature CDSs, or that lack one of its extremity were considered as decayed ICEs (dICEs).
Domain Composition Analysis
The third step of our workflow (Figure 1) involved retrieving domain composition of all ICE signature proteins from Uniprot annotations using the BioMart Central Portal3. De novo CD-search for conserved domains4 was performed when no data was available through BioMart.
Tree Construction
In this step (Step 4 on Figure 1), signature proteins were aligned using Clustal Omega with default parameters (Sievers et al., 2011). Trees of ICE signature proteins were built with MEGA (Tamura et al., 2013) using both maximum likelihood (ML; tree shown) based on JTT with Freqs (+F) model (partial deletion of gaps and missing data (80% cutoff), Gamma distributed with Invariant sites G+I (five categories)) and BioNJ methods with the Poisson model (Gouy et al., 2010). Branch support of the groupings was estimated using bootstrap (100 replicates for the ML method and 1,000 replicates for BioNJ).
ICE Annotation and Comparative Analysis
The comparative analysis of the conserved CDSs within a given ICE family was performed only for those that were non- or mis-characterized and displayed a significant number of ICEs. Functional annotation of ICEs was performed using Agmial (Bryson et al., 2006). Protein product, gene name and EC_number were assigned using similarity with Uniprot databank.
Data mining of the orthologs and the conserved syntenies was performed using Insyght. Sequence alignments were carried out at the protein level (using BLASTp) to achieve the pairwise comparisons of all the CDSs of ICEs belonging to the same family. Two genes were considered orthologous if they gave rise to a bi-directional best hit (BDBH) of the corresponding ICE genomic regions and if the sequence alignment included more than 50% of the total proteins with an e-value less than 0.01. Two CDSs for which the E-value of the sequence alignment was less than 0.01 were considered homologous but were not analyzed unless they belong to a synteny. Syntenies were computed with a dynamic programming algorithm that determines the highest scoring paths amongst the chains of colinear homologs. The scores and penalties used were as follows: minimum synteny score: 8; ortholog BBDH: 4; homolog non BDBH: 2; mismatch: -3; gap creation: -4; gap extension: -2. This setting allows the insertion of small gaps within the conserved synteny. The “Orthologs table” view in Insyght was used to identify the conserved and idiosyncratic loci within ICEs.
Results
Prevalence of ICEs and dICEs within Streptococcal Chromosomal Genomes
A total of 105 ICEs and 26 dICEs were identified among the 124 streptococcal genomes analyzed in this work (Supplementary Table S1). About half (63/124) of the examined strains contain at least one element and among those strains 61% (39/63) harbor several ICEs or dICEs. ICE denomination indicates whether the element is an ICE or a dICE, followed by letters and numbers allowing species and strain identification. When ICE/dICE encodes a site-specific integrase, its denomination also specifies the name of the target-gene. Otherwise, it indicates the integrase family (Tn916 or DDE). For elements already well-characterized and named, the correspondence between names is indicated in Supplementary Table S1.
Some streptococcal species show relatively few ICEs and dICEs even if a significant number of genomes were analyzed (Table 1). In particular, in the salivarius group, only one element was found in the six analyzed genomes of S. thermophilus analyzed and no element in the three genomes of S. salivarius. By contrast, in the anginosus and bovis groups, there is an average of more than two ICEs or dICEs per genome, with extreme situations such as 13 and 11 elements found in the three genomes studied in S. anginosus and S. gallolyticus, respectively. The occurrence of ICEs and dICEs per species or strain can vary within a group. For example in the pyogenic group, there were only six elements found in the 19 S. pyogenes genomes analyzed but as many as 10 elements in the five S. dysgalactiae genomes analyzed.
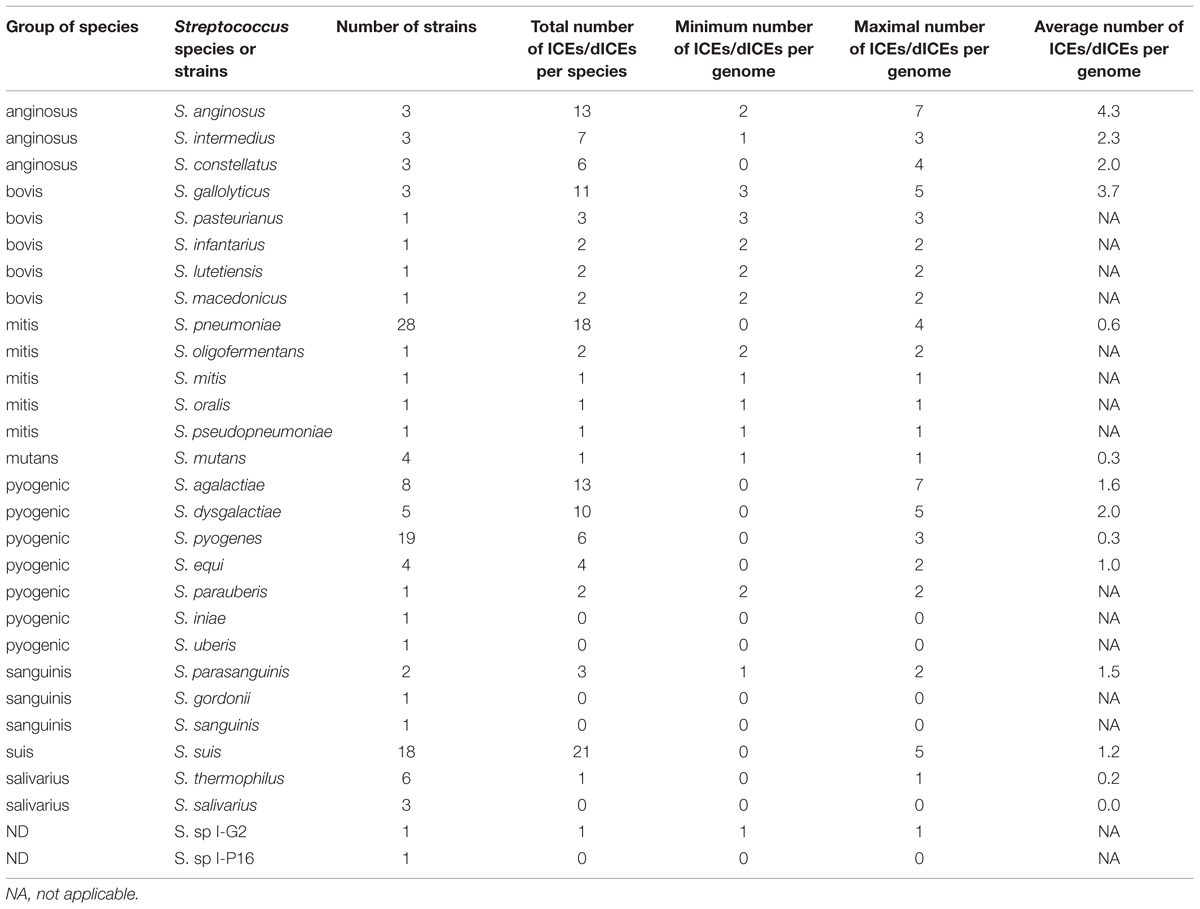
TABLE 1. Prevalence of ICEs and dICEs within streptococcal species.
Diversity of ICE and dICE Relaxases, and VirB4 in Streptococci
A total of 121 relaxases, encoded by ICEs or dICEs, were detected. They can be classified in three distinct classes on the basis of their domains (Table 2). The ‘Rel-I’ regroups 52 relaxases that contain a C-terminal catalytic “Rep_trans” domain (PF02486) associated with an N-terminal Helix-Turn-Helix (PF01381) DNA binding domain. According to the CONJscan-T4SSscan server (5Guglielmini et al., 2011), these 52 relaxases belong to the MOBT family that is related to initiators of rolling-circle replication of some plasmids and prophages (Guglielmini et al., 2014). The ‘Rel-II’ class regroups 62 relaxases sharing a common N terminal “relaxase” PF03432 catalytic domain and belonging to the MOBP family. Among them, 20 relaxases also carry a C terminal “Lantibiotic streptin immunity” PF11083 domain of unknown function. The ‘Rel-III’ class of relaxases contains seven proteins with no identified domains according to CD-search. These proteins are classified by the CONJscan-T4SSscan server in the MOBP family of relaxases.
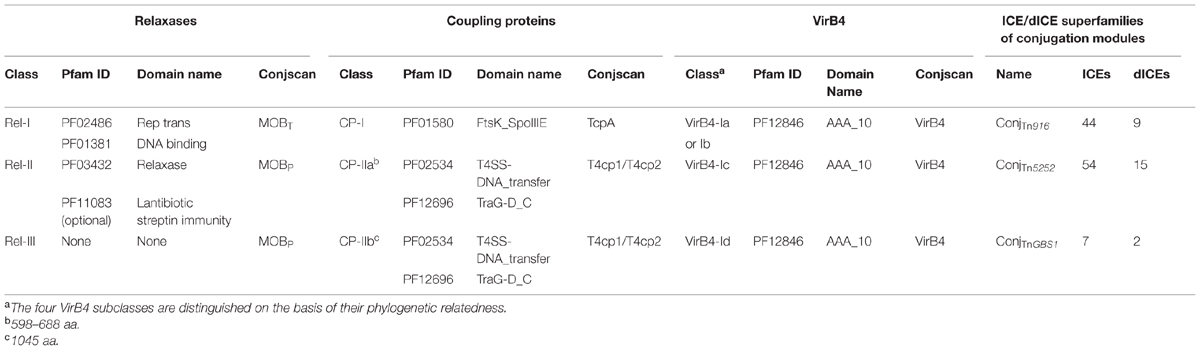
TABLE 2. Characterization of the ConjTn916, ConjTnGBS2, and ConjTnGBS1 superfamilies of conjugation modules.
The 126 CPs encoded by ICEs or dICEs of streptococci are divided into two classes. The CP-I class groups 50 CPs sharing a unique central FtsK_SpoIIIE catalytic domain (PF01580; Table 2). According to the CONJscan-T4SSscan server, they belong to a particular class of CPs named TcpA, unrelated to all CPs of Gram-negative bacteria (Guglielmini et al., 2014). The ‘CP-II’ class contains 76 CPs with a central catalytic TraG/TraD domain (PF02534) and an additional C-terminal ‘TraM recognition site of TraD and TraG’ PF12696 domain. According to the CONJscan-T4SSscan server, these proteins belong to the main family of CPs named VirD4 (Guglielmini et al., 2014). Among them, 67 CPs, constituting the ‘CP-IIa’ class, are 598 aa to 688 aa-long. The nine remaining CPs are composed of about 1045 aa and are representatives of the ‘CP-IIb’ class (Table 2).
All 124 VirB4 proteins from ICEs and dICEs show a unique C-terminal PF12846 ‘AAA-10’ catalytic domain. Reconstruction of their phylogenetic relatedness (data not shown) suggested that they can be grouped in four classes designated ‘VirB4-Ia’ (30 proteins), ‘VirB4-Ib’ (20 proteins), ‘VirB4-Ic’ (65 proteins), and ‘VirB4-Id’ (nine proteins; Table 2). These proteins are all identified as VirB4 using the CONJscan-T4SSscan server.
Classification of Conjugative Modules in Superfamilies and Families
Table 2 summarizes the co-occurrence of the different classes of signature proteins within the conjugation modules of ICEs and dICEs. This allowed classification of the conjugation modules into three distinct superfamilies, named according to the first characterized element of each superfamily: ConjTn916, ConjTn5252, and ConjTnGBS1.
The ConjTn916 Superfamily of Conjugation Modules
The ConjTn916 superfamily groups together 44 ICEs and nine dICEs. All encode a ‘Rel-I’ relaxase associated with a ‘CP-I’ and a ‘VirB4-Ia’ or ‘-Ib’ VirB4 protein.
The phylogenetic tree of relaxases (Figure 2A) of the ConjTn916 superfamily indicates three well-supported groups: relaxases related to the one of Tn916, to that of ICE_SmuA159_tRNAleu and to that of ICESt3. However, the latter two groups are not supported by the phylogenetic trees of CPs and VirB4 (Figures 2B,C). Therefore, two families of conjugative modules can be distinguished in the ConjTn916 superfamily: the ConjTn916 family and the ConjICESt3 family.
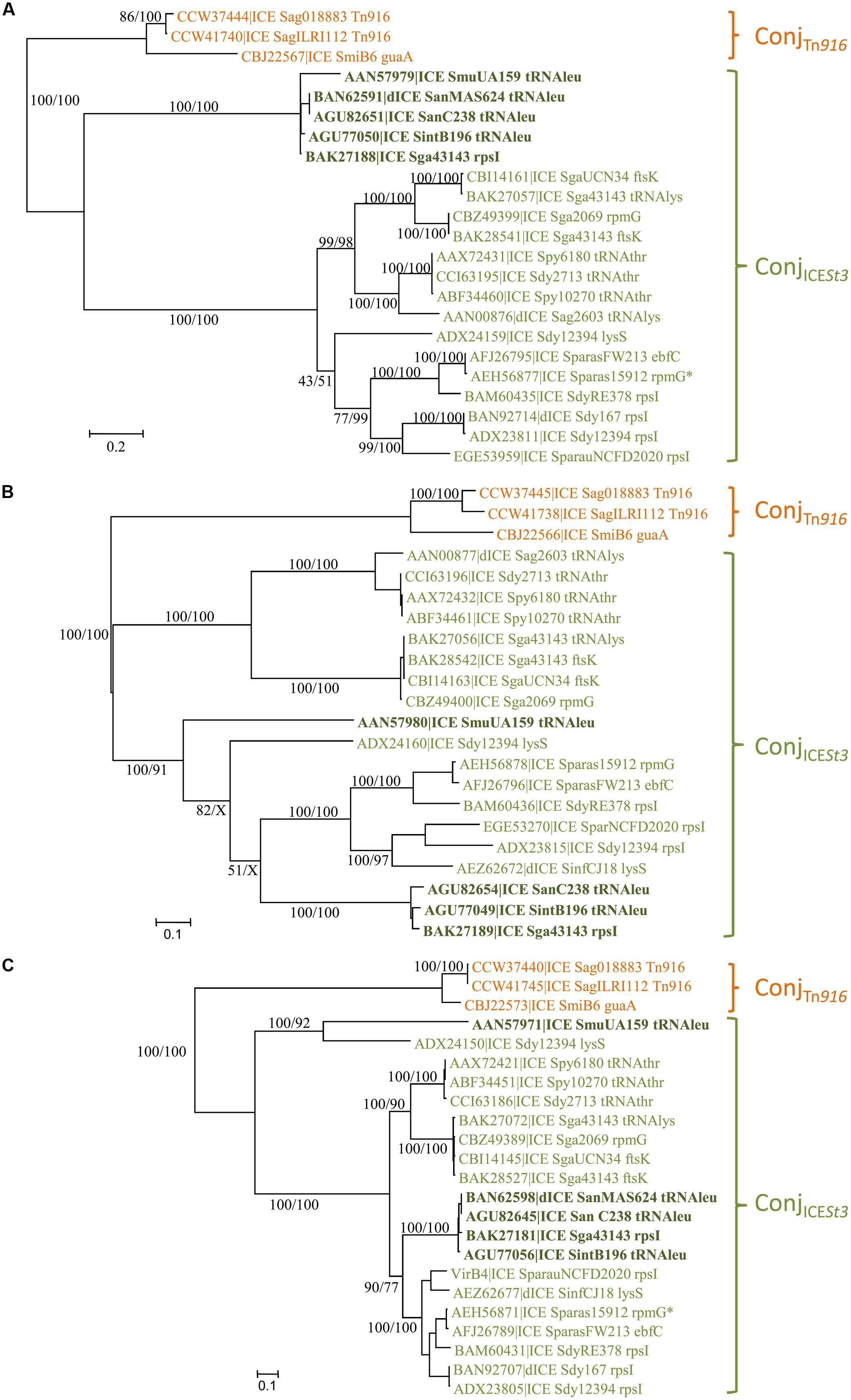
FIGURE 2. Phylogenetic analysis of ICEs and dICE belonging to the ConjTn916 superfamily. (A) Relaxases; (B) coupling proteins; (C) VirB4 proteins. Bootstrap supports are given as followed: ML/BioNJ. X marks the nodes that are not validated with BioNJ. ICE names are colored according to the family of conjugation module they encode: orange = ConjTn916, green = ConjICESt3 (including elements encoding relaxases related to the one of ICE_SmuA159_tRNAleu in bold dark green). Because of their very close phylogenetic relationships, relaxases (A), CP (B), and VirB4 (C) of only three ICEs representative of the ConjTn916 family are shown. Refer to Supplementary Table S1 for ICE/dICE and strain details.
The ConjTn916 family is well-supported by phylogenetic trees of the relaxase, CP and VirB4 protein sequences (Figures 2A–C). It clusters 27 ICEs (and five dICEs) whose signature proteins are almost identical to those of the well-described Tn916 element originally isolated from the Firmicute E. faecalis (Franke and Clewell, 1981). It also includes the more distant ICE_SmiB6_guaA whose signature proteins shared only 67, 66, and 80% identity with the E. faecalis Tn916 relaxase, CP and VirB4 protein, respectively. All the 12 genes of the conjugation module of the prototype Tn916 from E. faecalis DS16 were found with a similar organization in the vast majority of ICEs of the ConjTn916 family (data not shown) thus confirming their relationships. Others only differ by one or few deletions, pseudogenizations or insertions.
The ConjICESt3 family gathers together 21 elements whose signature proteins are much more variable in sequence than those of the ConjTn916 family. The sequence of relaxases, CPs and VirB4 proteins of the most distantly related elements displayed around 20, 30, and 60% of identity, respectively.
For a better characterization of the ConjICESt3 family, a search for conserved CDSs in all members of this family was undertaken. Orthologous CDSs were identified on the basis of the identity of their product using Insyght. The integration module of all elements of the ConjICESt3 family is composed of both a tyrosine integrase and an excisionase (Figure 3A). In addition to the relaxase, CP, VirB4 CDSs, tyrosine integrase and excisionase CDSs, they share seven other CDSs including CDSs involved in the T4SS formation (VirB1, VirB3, VirB6, and VirB8) and a CDS encoding a regulation protein (HTH regulator).

FIGURE 3. Identification of conserved CDSs within the ConjICESt3, ConjTn1549 and ConjTn5252 families of ICEs. (A) ConjICESt3 family; (B) ConjTn1549 family; (C) ConjTn5252 family. Genes encoding conserved proteins in all ICEs of a given family are indicated by arrows with below their putative function (or known homologs).
The ConjTn5252 Superfamily of Conjugation Modules
The ConjTn5252 superfamily gathers together 54 ICEs and 15 dICEs encoding a ‘Rel-II’ relaxase associated with a ‘CP-IIa’ CP and a ‘VirB4-IIc’ VirB4 (Table 2). The phylogenetic trees obtained independently for ‘Rel-II’ relaxases, ‘CP-IIa’ CPs and ‘VirB4-Ic’ proteins (Figure 4) are congruent and therefore only the CP one is shown. These data are consistent with the splitting of the ConjTn5252 superfamily into four distinct families: ConjvanG, ConjTn1549, ConjTnGBS2, ConjTn5252 that cluster 2, 21, 17, and 29 elements, respectively.
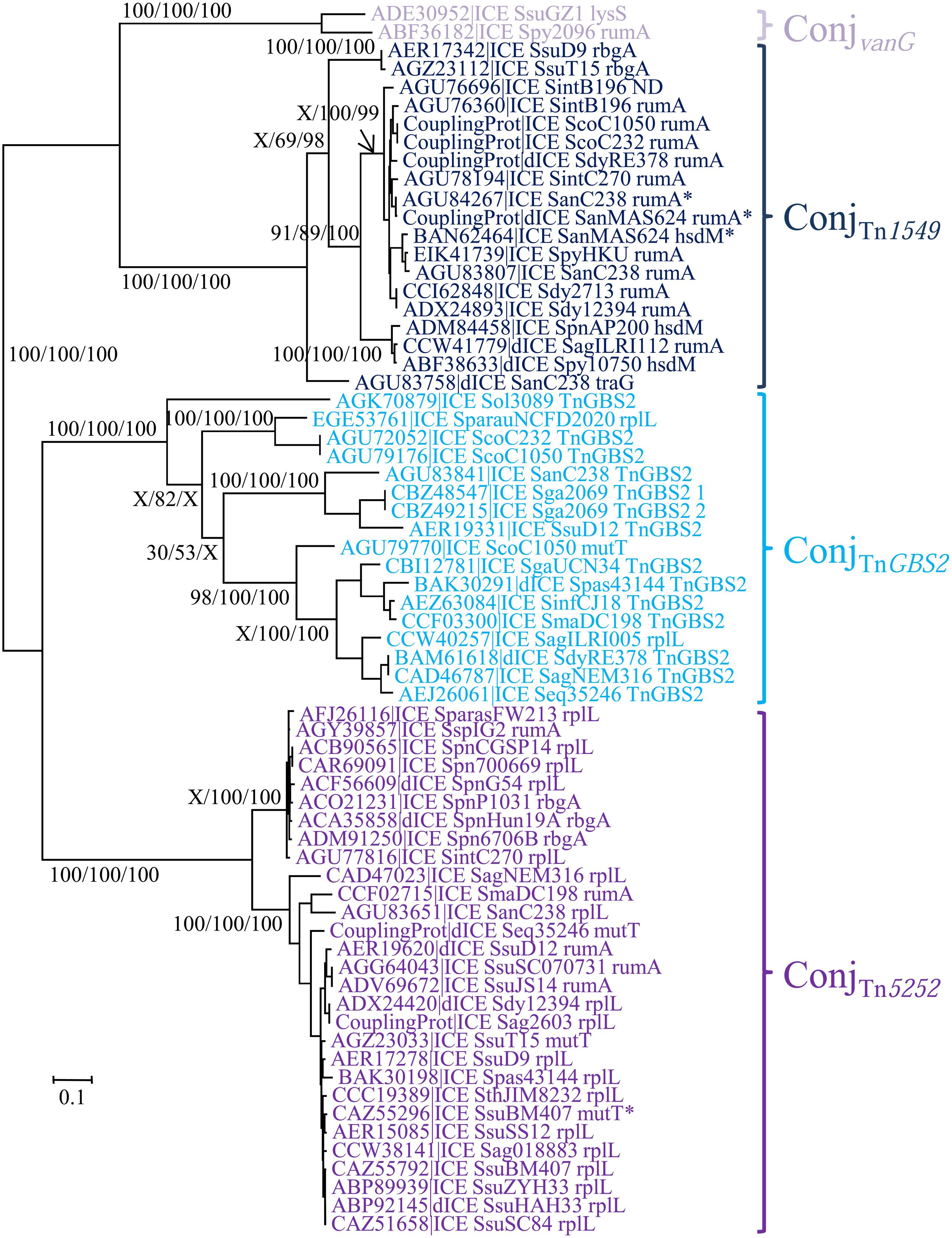
FIGURE 4. Phylogenetic analysis of ICEs and dICEs belonging to the ConjTn5252 superfamily. The trees of CPs are shown in this figure. ML boostrap values for relaxases/CPs/VirB4 (in this order) are given. X marks the nodes that are not validated with other proteins. Families are also supported by BioNJ analysis (data not shown). In mauve are ICEs/dICEs belonging to the ConjvanG family, in dark blue those belonging to the ConjTn1549 family, in light-blue those of the ConjTnGBS2 family and in purple that of the ConjTn5252 family. Elements marked with an asterisk are not integrated in their primary sites but in secondary ones. Refer to Table 1 for ICE/dICE and strain details.
As expected from Guérillot et al. (2013), sequence comparison of the 15 ICEs encoding a ConjTnGBS2 module identified 14 CDSs shared by all these TnGBS2-related elements (data not shown).
Using Insyght, CDSs comparison of the 14 ICEs of the ConjTn1549 family identified 14 CDSs shared by all of them, in addition to the relaxase, CP and VirB4 CDSs (Figure 3B). One of these CDSs encodes a sigma factor that may be involved in the regulation of ICE transfer; three others being probably involved in the T4SS formation (VirB1, VirB3, VirB6) and one in maintenance of the ICE after excision (segregation ATPase). Many of these ICEs also encode a protein carrying a repA_N domain that could be involved in maintenance of excised ICEs.
As for ICEs of the ConjTn1549, the 23 ICEs of the ConjTn5252 family do not share the same integration module. However, they display 24 conserved CDSs. In addition to the relaxase, and the CP VirB4 CDSs, they share three other CDSs probably involved in the T4SS formation (VirB1, VirB3, VirB6), and one encoding a replication initiator (repA_N; Figure 3C).
The ConjTnGBS1 Superfamily of Conjugation Module
The ConjTnGBS1 superfamily of conjugation modules is the least represented in streptococcal genomes with only seven ICEs and two dICEs exhibiting such a module. It is characterized by the presence of a ‘Rel-III’ relaxase co-occurring with a ‘CP-IIb’ CP and a ‘VirB4-Id’ protein. This superfamily was not identified by Guglielmini et al. (2014).
As for the ConjTn5252 superfamily, whatever the signature protein used, all phylogenetic trees are congruent and therefore only the CP one is shown (Figure 5). Sequence comparison of the signature proteins of the most divergent ICEs of this superfamily indicated that they shared more than 56% of identity. Thus, within streptococcal genomes, the ConjTnGBS1 superfamily is represented by the unique ConjTnGBS1 family. As expected from Guérillot et al. (2013), systematic comparisons of the protein sequence of the members of the ConjTnGBS1 family with TnGBS1 CDSs confirmed that they are related to TnGBS1 (data not shown).
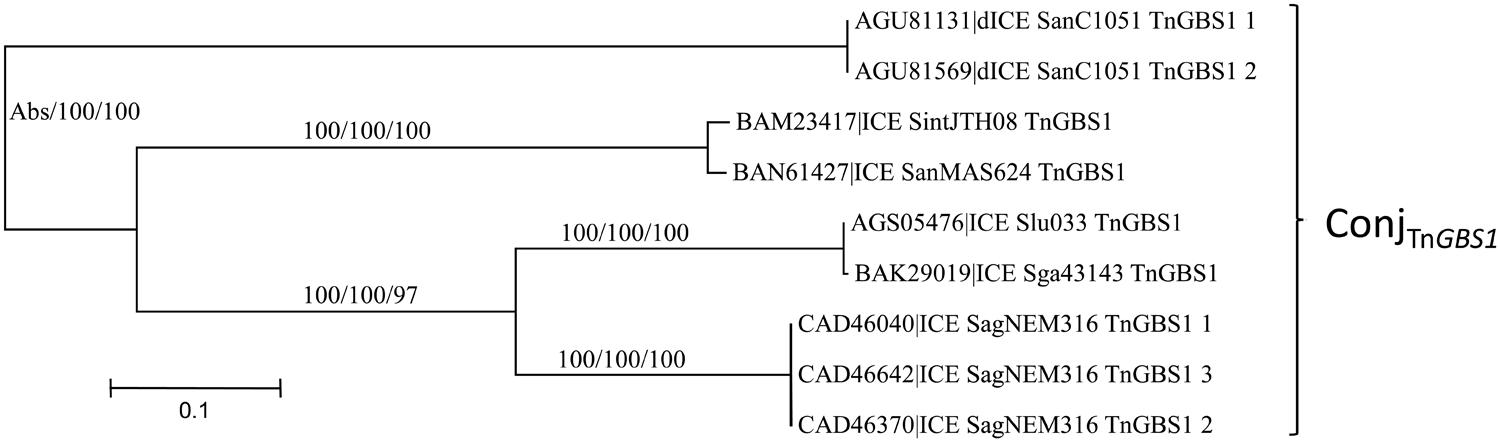
FIGURE 5. Phylogenetic analysis of ICEs and dICEs belonging to the ConjTnGBS1 superfamily. The trees of CPs are shown in this figure. ML boostrap values for relaxases/CPs/VirB4 is given in this order. Families are also supported by BioNJ analysis (data not shown). Abs means that one sequence was missing. Refer to Supplementary Table S1 for ICE/dICE and strain details.
Prevalence of the Different Families of Conjugation Modules within Streptococcal Species
In summary, seven families of conjugation modules belonging to three superfamilies were identified in streptococcal ICEs/dICEs. Most ICEs of the ConjTn916 family are found in S. suis and S. pneumoniae, while S. dysgalactiae and S. gallolyticus mainly harbor ICEs of the ConjICESt3 family. ICEs of the ConjTn5252 family are frequently found in S. suis, and ICEs of the ConjTnGBS1 family are only found in S. agalactiae, S. anginosus, S. intermedius, and S. lutetiensis genomes.
Diversity of Integration Modules and Integration Sites of Streptococcal ICEs and dICEs
Three unrelated families of integrases (tyrosine integrase, serine integrase, and DDE transposase) are encoded by streptococcal ICEs/dICEs. Most of these integrase genes are located at one extremity of the ICE and adjacent to the integration site.
Prevalence and Integration Site of ICEs/dICEs Encoding a Tyr Integrase
Tyrosine integrases are the most prevalent integrases detected as they are found in 53% of the elements. In total, 73 tyrosine integrases were identified (66 for ICEs and 7 for dICEs). Most of them are ∼400 aa long, except for five that are composed of 502 aa. Despite a variable degree of identity between these proteins, all tyrosine recombinases share a “phage-integrase” PF00589 domain in their N-terminal region. Most of them carry an additional N-terminal binding domain being either: (i) a pfam02920 “DNA binding domain characteristic of Tn916 integrase” or (ii) a PF14659 “Phage integrase, N-terminal SAM-like domain” (iii) and/or more rarely a PF14657 “AP2-like DNA-binding integrase domain.”
The phylogenetic tree of the tyrosine integrases reveals 11 well-supported groups (Figure 6). The overall finding is that almost all tyrosine integrases are grouped according to their insertion loci. Exceptions are the two non-grouped tyrosine integrases catalyzing integration in the 3′ end of the rpmG gene that display two distinct orientations and locations relative to the rpmG gene (Figure 6) and share only 21% of identity. Interestingly, all tyrosine recombinases catalyzing integration in a tRNA gene are grouped together. Half of the elements encoding a tyrosine integrase are inserted in the 3′ end of either tRNA CDSs (9/73) or a well-conserved housekeeping genes (30/73) such as rplL (L7/L12 ribosomal protein), rpsI (S9 ribosomal protein), lysS (lysyl-tRNA synthetase), rpmG (L33 ribosomal protein), or guaA (GMP synthase). In rare cases (7/73), the integration sites are found in the 5′ end of genes such as ftsK (DNA translocase involved in cell division), rbgA (ribosomal biogenesis GTPase) and ebfC (nucleoid associated protein). The remaining tyrosine integrases (27/73) catalyze low-specificity integration as previously shown for the very closely related integrase of Tn916 (Scott et al., 1994).
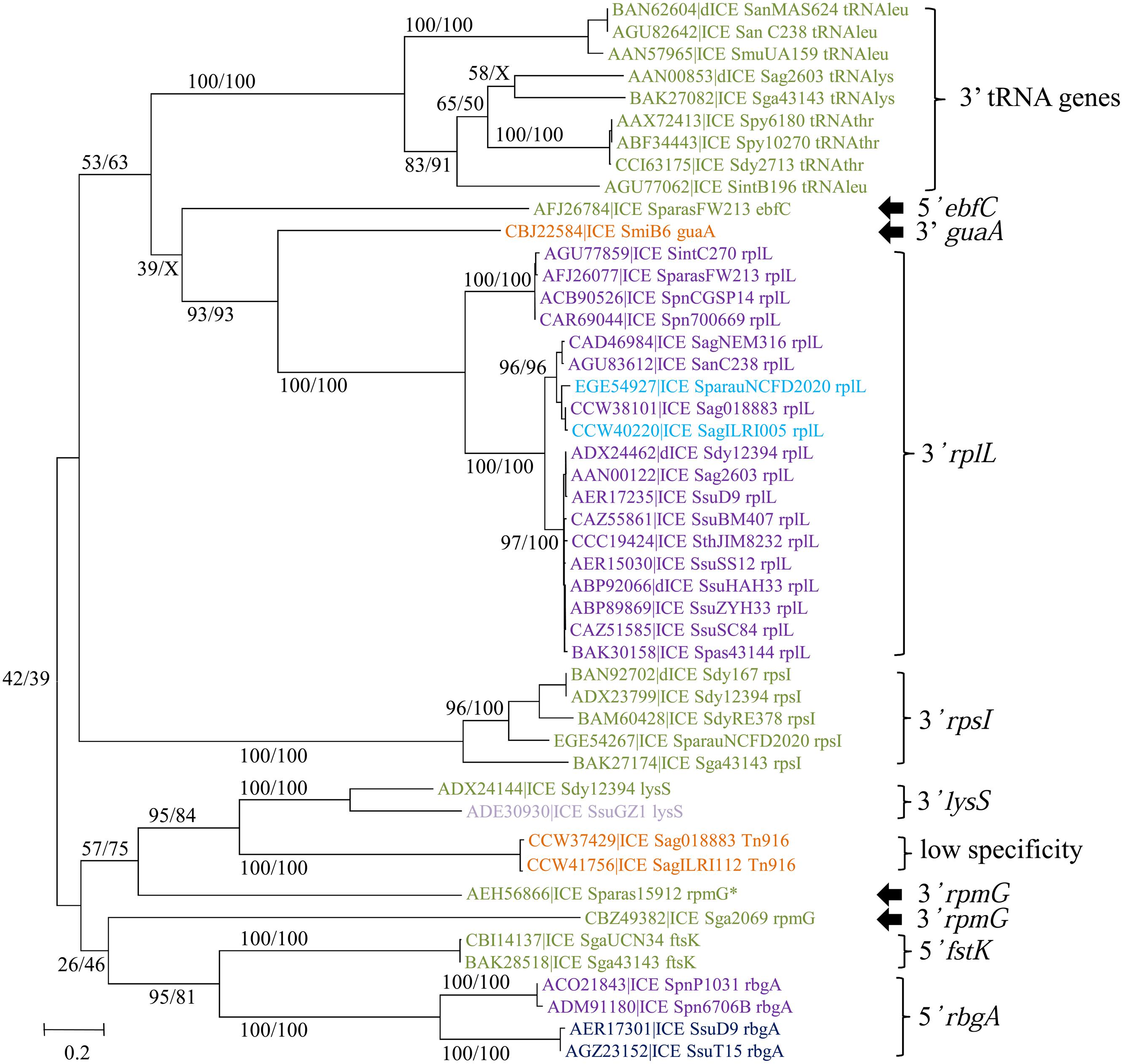
FIGURE 6. Phylogenetic trees of tyrosine integrases from streptococcal ICEs/dICEs. Bootstrap values are given as followed: ML/BioNJ. X marks the nodes that are not validated with BioNJ. ICE names are colored according to the family of conjugation module they encode: orange = ConjTn916, green = ConjICESt3, mauve = ConjvanG, dark-blue = ConjTn1549, purple = ConjTn5252, and in light-blue those of the ConjTnGBS2. Brackets gather together closely related integrases sharing the same specificity of integration. Integration specificity of the ICEs/dICEs is indicated. Elements marked with an asterisk are not integrated in their primary sites but in secondary ones. Refer to Supplementary Table S1 for ICE/dICE and strain details.
All the ICEs/dICEs encoding a site-specific tyrosine integrase are flanked by DRs whose size ranges from 12 to 54 bp. DRs in rplL, guaA or in genes encoding tRNAthr only contain the exact 3′ end of the target genes. By contrast, DRs of elements integrated in the 5′ end of ftsK and rbgA, and in the 3′ end of genes of tRNAleu and tRNAlys, overlap the flanking intergenic regions (Figure 7). Three elements are integrated in the lysS gene and are flanked by DRs with similar sequences. However, these DR sequences have distinct locations within lysS resulting from a slight difference in the length of this gene: one corresponds to the last 17 bp of the lysS gene while the others contain 13 bp and are more internal (Figure 7).
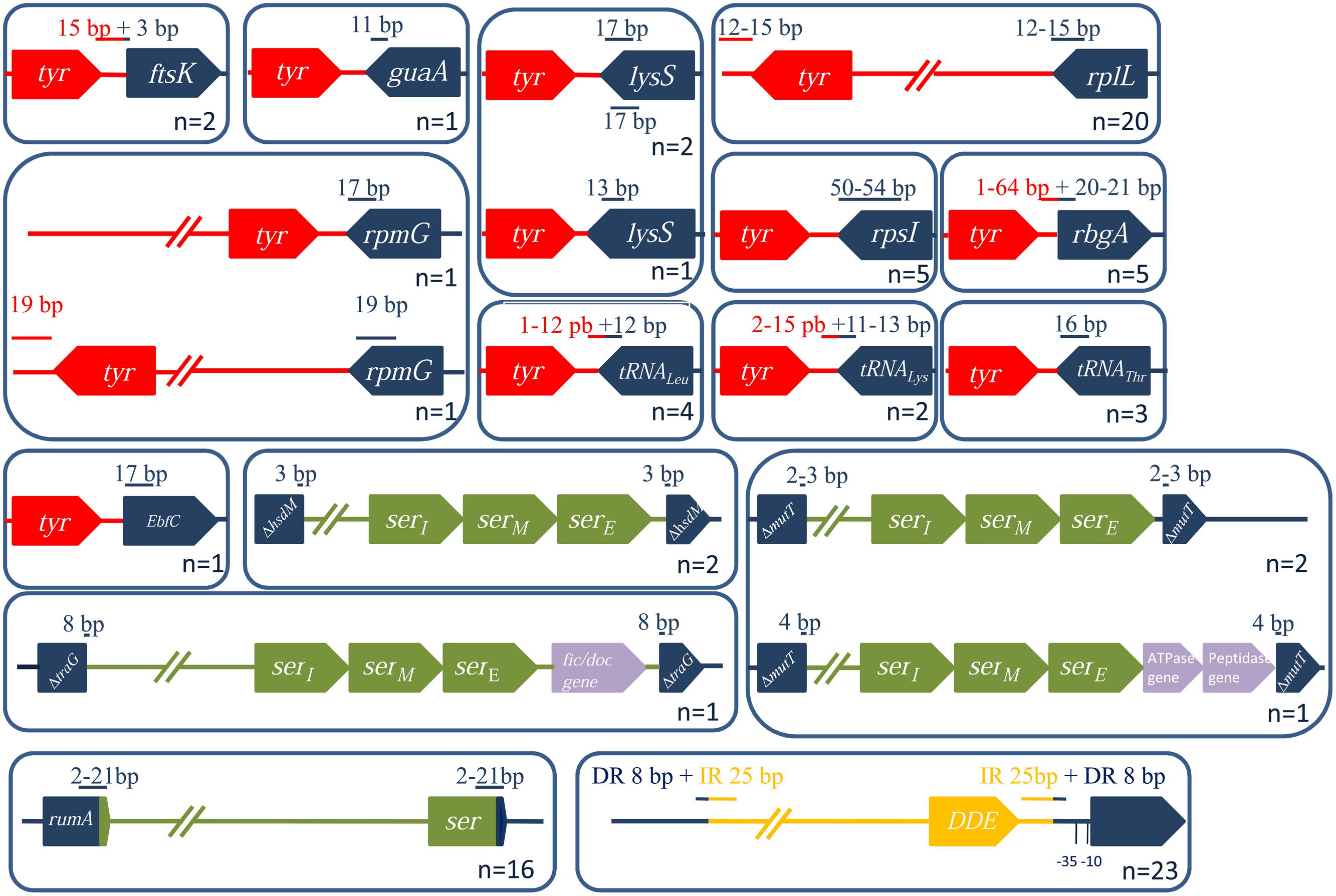
FIGURE 7. Characterization of ICE/dICE integration loci and their position relative to the integrase CDSs. The genes within (or next to) which an ICE is inserted are in blue. Tyrosine integrases are in red, serine recombinases in green and DDE transposases in yellow. The sizes (in bp) of the DR (or of IRs when specified) are indicated in red when the sequence is inside of the conjugative element (in blue, outside). Numbers represent the number of ICEs integrated in a given target gene.
Prevalence and Integration Sites of ICEs and dICEs Encoding a Serine Integrase
A total of 42 serine integrases were identified: 32 from ICEs and 10 from dICEs. They all displayed an N-terminal catalytic and dimerization ‘Resolvase’ domain (PF00239) that contains a conserved serine residue. This domain is always associated with a ‘Recombinase’ PF07508 domain. All of them but 10 also contain a pfam13408 ‘Zinc ribbon recombinase’ domain that is likely to play a DNA-binding role.
Streptococcal conjugative elements encode either a single serine integrase (14 ICEs and four dICEs) or a triplet of serine integrase genes (6 ICEs and 3 dICEs). The phylogenetic tree of the serine integrases is compatible with the existence of two groups of integrases (Figure 8). One of the groups clustered all the single serine integrases that target integration in rumA [23S rRNA (uracil-5-)methyltransferase]. ICEs/dICEs encoding such serine integrase are flanked by 2–21 bp-DRs localized at one end in the rumA gene and at the other end in the serine integrase gene (Figure 7). Integration leads to a reciprocal exchange of the 3′ part of the rumA and serine integrase genes leading to a modification of sequence and length of the C-terminal end of the corresponding proteins. It should be noticed that the replacement changes the translation frame of both proteins.
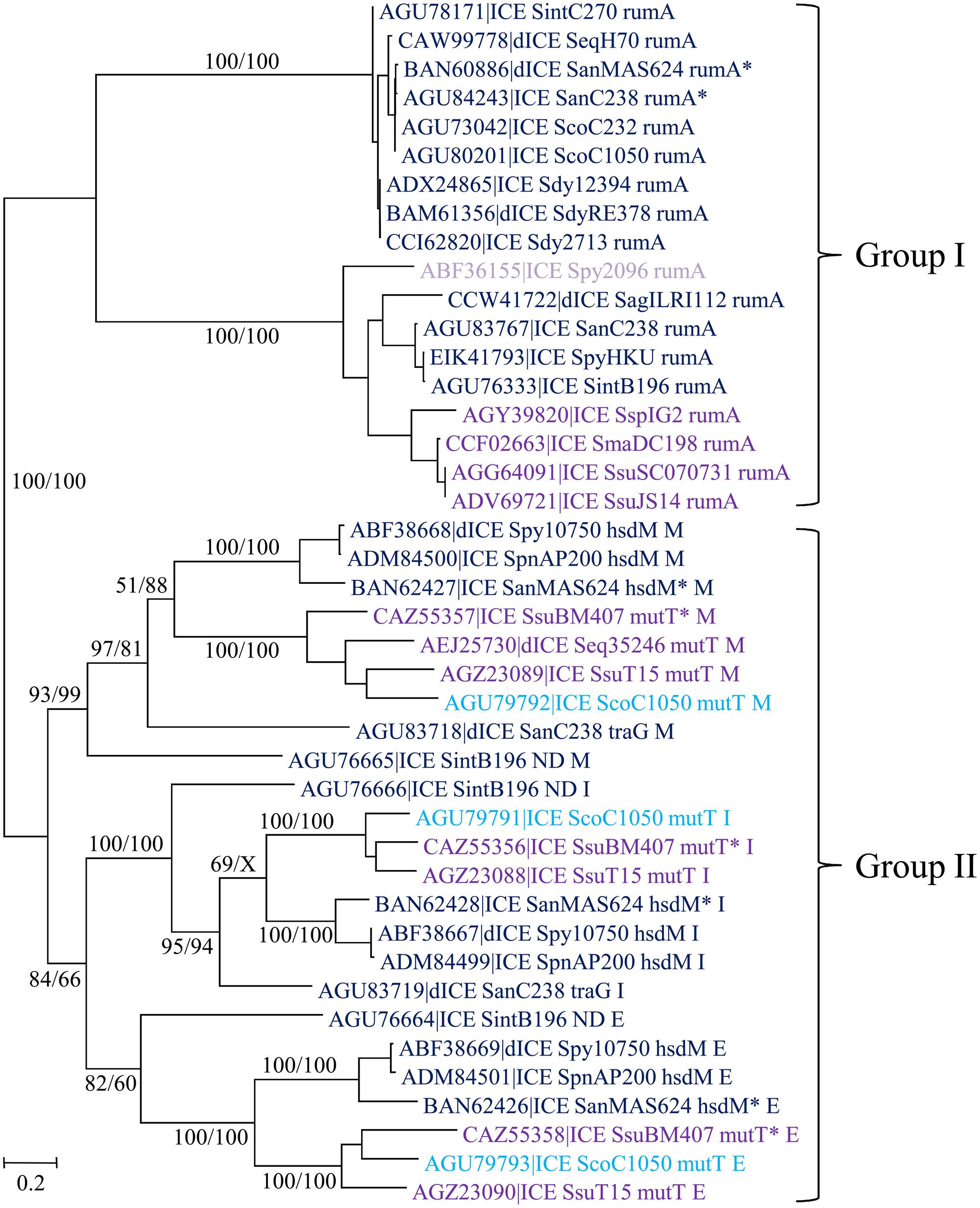
FIGURE 8. Phylogenetic tree of serine integrases from streptococcal ICE/dICE. Bootstrap values are given as followed: ML/BioNJ. X marks the nodes that are not validated with MP. ICE name are colored according to the family of conjugation module they encode (dark-blue = ConjTn1549, purple = ConjTn5252, mauve = ConjvanG), and in light-blue those of the ConjTnGBS2. Brackets gather together closely related integrases sharing the same specificity of integration. Genes in which the ICEs/dICEs are integrated are indicated. Refer to Supplementary Table S1 for ICE/dICE and strain details. Elements marked with an asterisk are not integrated in their primary sites but in secondary ones.
The second group of serine integrases is composed of the 24 integrases that are organized in triplets within the elements (Figure 8). Interestingly, these serine integrases are clustered according to their position within the triplets: external (E), middle (M), or internal (I) with respect to the ICE/dICE extremity (Figure 8). This suggests that all these modules derive from an ancestral module that already encoded three serine recombinases and that the presence of triplet results from two successive duplications. Triplets of serine integrases catalyze site-specific integration within several genes thus leading to their disruption: mutT (Nudix hydrolase), traG (CP of another ICE, dICE, or ICE remnant) and hsdM (methyltransferase subunit of type I restriction-modification systems). Insertion of ICEs/dICEs encoding triplets of serine integrases leads to very small 2–8 bp DRs (Figure 7). In some cases, one or several CDSs separated the triplets of serine integrases from the target gene (Figure 7). All serine integrases have the same orientation with respect to the target gene.
In several cases (element names marked with an asterisk in Figure 8), the comparison of the observed integration sites with the ones of elements with closely related integrases strongly suggests that these elements are not integrated in their primary sites but in secondary ones. ICE_SanMAS624_hsdM∗ disrupts a gene encoding a protein carrying the domain PF0267 (“Adenine nucleotide alpha hydrolase”). ICE_SsuBM407_mutT∗ disrupts a gene encoding a luciferase-like protein. dICE_SanMAS624_rumA∗ and ICE_SanC238_rumA∗ disrupts sstT, a gene encoding a serine/threonine transporter. The relaxase, CP and VirB4 of these last two elements are very closely related (Figure 4) as well as their integrases (Figure 8). This suggests that they were both inherited from the last common ancestor of their hosts.
Prevalence and Integration Sites of ICEs/dICEs Encoding a DDE Transposase
A total of 23 DDE transposases related to those encoded by TnGBS1 and TnGBS2 elements (Guérillot et al., 2013) were detected. All show an “Uncharacterized protein family” PF06782 conserved domain with unknown function. Alignment of the amino acid sequences of the most divergent DDE transposases revealed that they share 42% identity showing that all these transposases belong to the same family. However, DDE transposases encoded by ICEs carrying a ConjTnGBS1 module are clustered together and are distinct from those encoded by ICEs with a CONJTnGBS2 module (Figure 9).
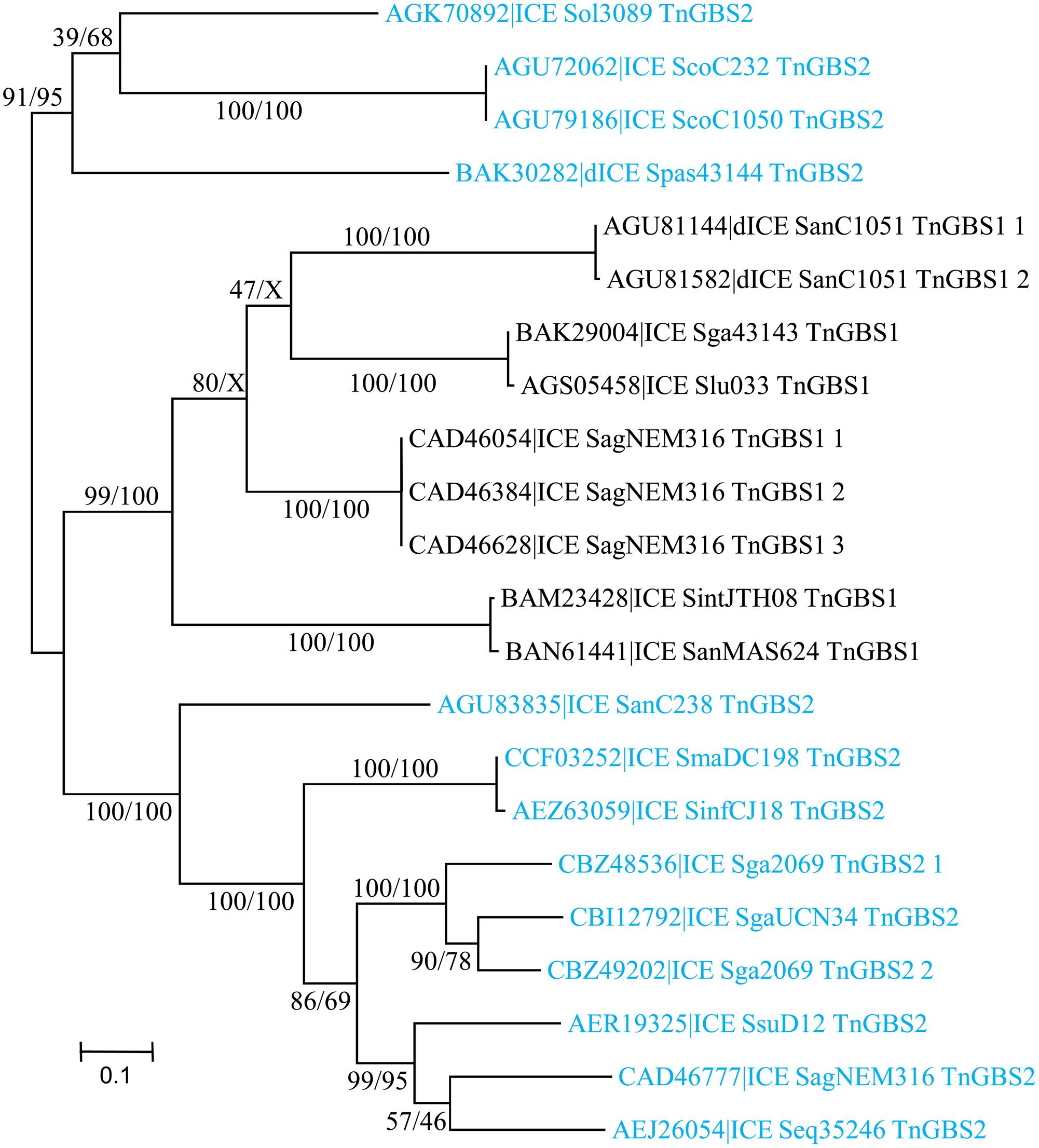
FIGURE 9. Phylogenetic tree of DDE transposases from streptococcal ICE/dICE. Bootstrap values are given as followed: ML/BioNJ. ICE name are colored according to the family of the conjugation module they encode (black = ConjTnGBS1, light-blue = ConjTnGBS2. Refer to Table 1 for ICE/dICE and strain details.
Analysis of the junction sequence of ICEs encoding DDE transposases shows an 8-bp DR sequence that results from the duplication of the target sequence. Comparison of the insertion sites between all ICEs and dICEs of this group did not reveal any significant sequence similarity among the duplicated sequences but they are all located 15–16 pb upstream from -35 boxes of sigma A promoters as previously reported (Brochet et al., 2009).
Diversity and Evolution of ICEs/dICEs
The determination of the limits of each element allows the comparison of their size. If one excludes the ConjvanG ICE family (only two elements), the ConjTn916 and ConjTnGBS1 families are the most homogeneous in size with elements from 18 to 26 kb and from 40 to 53 kb, respectively. By contrast, the size of the ConjICESt3 elements is much more variable: most of them are 19- to 37-kb long and one exceeds 60 kb. The size of the ConjTn1549 elements can double (from 36 to 72 kb) and that of ConjTnGBS2 elements shows a very large disparity (from 25 to 82 kb). When ICEs/dICEs are present within Streptococcus genomes, they contribute to 1–13% of chromosomal DNA.
Analysis of streptococcal ICEs and dICES also allowed determining their typical combination of conjugation and integration/excision modules (Table 3). Almost all conjugation modules of the ConjTn916 family are associated with a tyrosine integrase identical, or almost identical, to the one of Tn916, that is known to have a low specificity of integration (Scott et al., 1994). The only exception is ICE_SmiB6_guaA that encodes a conjugation module of the ConjTn916 family but is site-specifically integrated in the 3′ end of guaA. Elements with a ConjICESt3 module are associated with site-specific tyrosine integrases catalyzing integration in eight distinct integration sites (3′ end of three types of tRNA encoding genes, rpsI, rpmG, and lysS as well as the 5′ end of ftsK or ebfC). The conjugation modules of ConjTn5252 superfamily can be associated with a tyrosine recombinase, a serine site specific recombinase or a DDE transposase. Thus, ICEs and dICEs carrying a ConjTn1549 module are associated with: (i) a single serine integrase catalyzing the insertion in rumA; (ii) a triplet of serine integrases catalyzing the insertion in hsdM or traG or (iii) a tyrosine integrase catalyzing the insertion in the 5′ end of rbgA. Those carrying a ConjTn5252 module can encode: (i) a tyrosine integrase catalyzing the integration in the 3′ end of rplL or the 5′ end of rbgA; (ii) a single serine integrase catalyzing the integration in rumA; (iii) or a triplet of serine integrases catalyzing the insertion in mutT. The two ICE (ICE_SsuGZ1_lysS and ICE_Spy2096_rumA) displaying a ConjvanG conjugation module are associated with a tyrosine or a serine integrase, respectively. All the ConjTnGBS1 and 14 of the 17 ConjTnGBS2 conjugation modules are associated with a DDE transposase. However, three ICEs carrying a ConjTnGBS2 module are not associated with a DDE transposase: ICE_Sco1050_mutT encodes a triplet of serine recombinases and both ICE_SparauNCFD2020_rplL and ICE_SagILRI005_rplL encode a tyrosine integrase. Sequence comparison suggests that ICE_SagILRI005_rplL is composite (Figure 10). Its ConjTnGBS2 conjugation module is closely related to the one of ICE_SgaUCN34_TnGBS2. However, its left part is closely related to the left part of ICE_Sag018883_rplL, an ICE carrying a conjugation module belonging to CONJTn5252 family. It includes not only the recombination module but also a lactose utilization module and a pseudogene of relaxase typical of the ConjTn5252 family. The right end of ICE_SagILRI005_rplL carries a gene encoding a repA_N domain closely related to a gene located in the right of ICE_Sga018883_rplL and additionally a pseudogen, whose product also carry a repA_N domain.
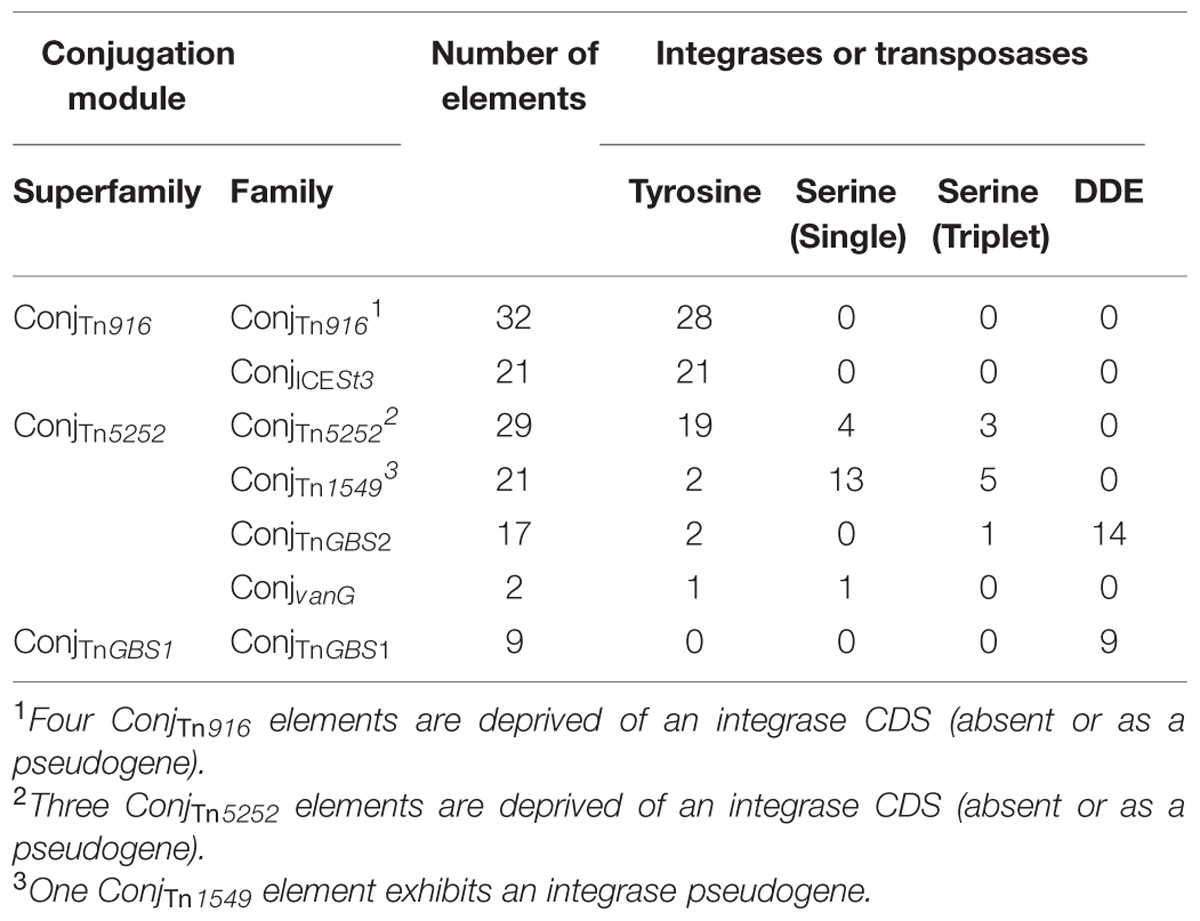
TABLE 3. Various combinations of conjugation and integration modules in ICEs and dICEs.
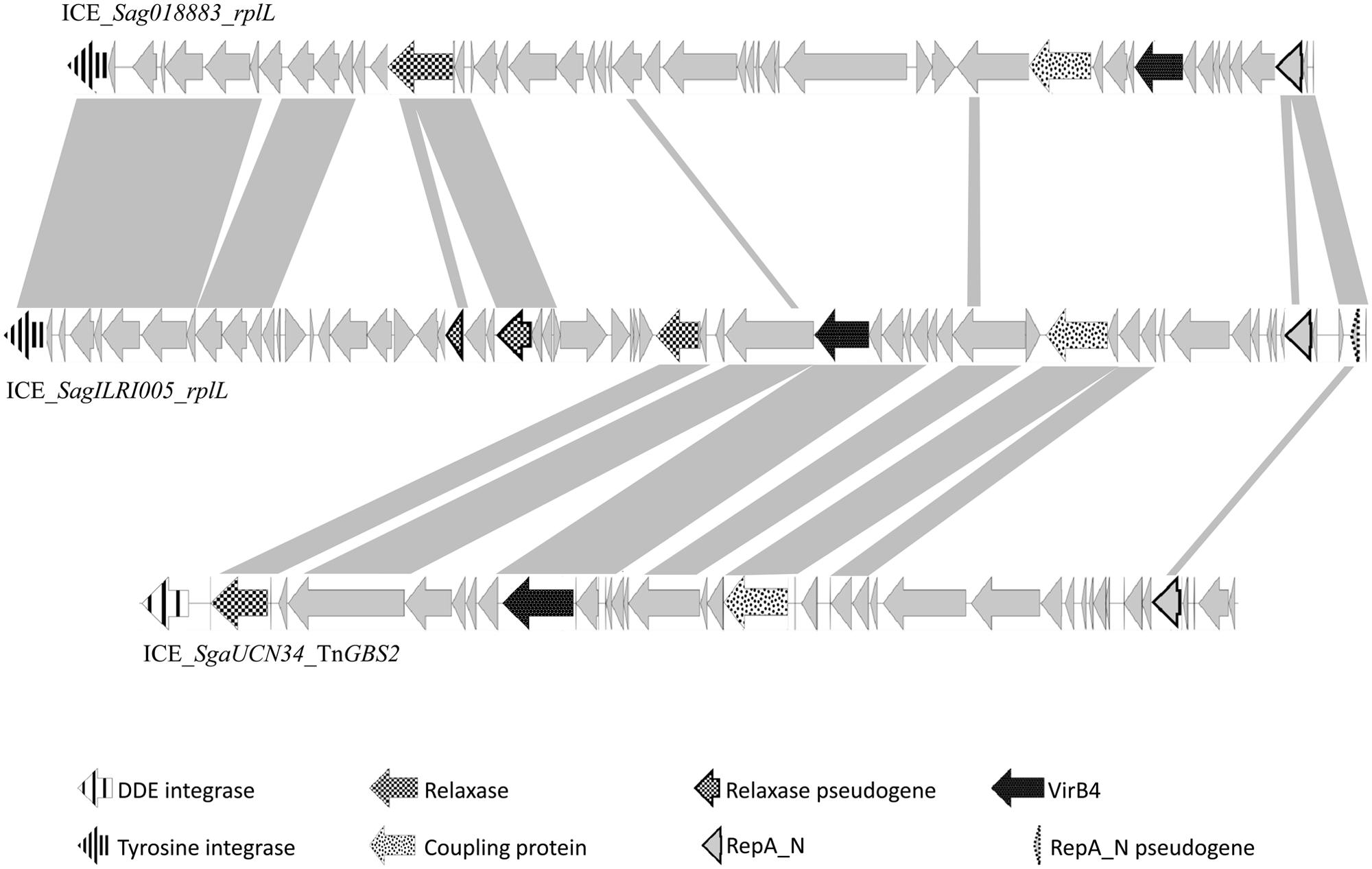
FIGURE 10. Genetic organization of ICE_SagILRI005_rplL and comparison against ICE_SgaUCN34_TnGBS2 and ICE_ Sag018883_rplL. For the comparison, gray shading between the genetic elements represents regions with >50% amino acid sequence identity. The arrows represent the individual ORFs. Putative functions of the conserved genes are those deduced from the functional annotation of the ICE using Agmial.
Discussion
Detection of ICEs and dICEs in Streptococcal Genomes
Burrus et al. (2002a), a precursor analysis of 24 genomes from various Firmicutes, led to the identification of 17 putative ICEs and suggested that these elements are widespread at least in this division of bacteria. Over the last decade, with the revolution of sequencing technology, the number of fully sequenced bacterial genomes greatly increased. By the same time, efforts were made to improve in silico analysis of the data sets and in particular those allowing the detection of genomic islands including ICEs. Almost all searches of ICEs in bacterial genomes were only performed with a strain-centric or an ICE family centric point of view. However, few recent studies also reported extensive characterization of these conjugative elements. The first extensive study identified 335 chomosomal conjugative modules by scanning 1124 genomes of prokaryotes for conjugative genes (using HMM profiles of conjugative proteins of essentially proteobacterial plasmids; Guglielmini et al., 2011). Soon after, Ghinet et al. (2011) reported the characterization of 161 ICEs within 275 genomes of Actinobacteria but did not identify their limits. More recently, Puymège et al. (2015) searched for genetic elements integrated in the tRNAlys CTT gene in 303 genomes of S. agalactiae, leading to the identification and delimitation of 108 putative ICEs or derivatives. It should be noticed that in 2012, Bi et al. (2012) developed a web database6, compiling information on ICEs from both Gram+ and Gram- bacteria. However, if this database has the merit to list a large number and a great diversity of elements, it was not updated since November 2012 and some studies describing novel streptococcal ICEs published before this date escaped to the attention of the authors (for example, Brochet et al., 2008, reporting 10 novel ICEs). ICEberg have limitations considering ICEs from Streptococcus since (i) about half of ICE/dICE boundaries are incorrectly delimitated in ICEberg and (ii) the insertion site of many of them, although published, is not registered in this database. More widely, information on numerous elements from Firmicutes is inconsistent or wrong. Thus, while the authors indicated that a family should include only elements that carry both related integration and conjugation modules, they also included in the Tn916 family ICEs that carry Tn916-unrelated integration modules (such as Tn5397 or ICELm1), Tn916-unrelated conjugation modules (such as Tn1549) or elements completely unrelated to Tn916 but carrying conjugation modules related to the one of Tn1549 (such as CTn2 or CTn5). Furthermore, although ICELm1 and Tn5801 carry almost identical integration and conjugation modules (Burrus, plasmid 2002, cited in ICEberg for ICELm1), they were included in different families, Tn916 and Tn5801, respectively. Even more problematically, the Tn1207.3 family and 10750-RD.1 family only contain elements unrelated to ICEs, prophages for the first one and highly decayed derivates of integrative mobilizable elements for the second one. In general, these failures (and many others not mentioned here) make this database unreliable and very difficult to use for ICEs from Streptococci and other Firmicutes.
Here, we present the results of ICE detection within 124 complete streptococcal genomes. Our search is based on an iterative search for genes encoding signature proteins from ICEs. The co-occurrence of an integrase and three proteins of the conjugation module guaranties the retrieval of ICEs or dICEs. When one or two signature CDSs appeared to be a pseudogene or to be absent, an analysis of the whole element was undertaken to confirm its nature.
This work led to the identification and characterization in 63 Streptococcus genomes of 131 ICEs/dICEs whose extremities were precisely mapped on the genome. Elements that were already precisely identified are marked by a reference in Supplementary Table S1.
Distribution of ICEs and dICEs in the Different Streptococcal Species
Among the 27 streptococcal species analyzed, all, except 5 (S. iniae, S. gordonii, S. uberis, S. sanguinis, for which only one complete genome is available and S. salivarius for which three genome sequences exist), contain ICEs/dICEs showing the ubiquity of these elements in Streptococcus. S. suis appears as the species containing the highest number of ICEs/dICEs since 61% of the strains carry at least one ICE/dICE. However, the high prevalence of ICEs/dICEs in this species might be due to strain sampling since many strains carrying an ICE are related. This is also the case for S. pneumoniae genomes of which 40% (11/28) encode at least one ICE. Among them, seven are derived from clinical isolates known to be resistant to one or multiple antibiotics. All of them carry at least one ICE/dICE suggesting a possible correlation between the presence of ICEs and resistance to antibiotics. Indeed among the families detected, Tn916 (Roberts and Mullany, 2011), Tn5252 (Korona-Glowniak et al., 2015), and Tn1549 (Garnier et al., 2000) are known vectors of antibiotic resistance genes.
Definition of Different Families of Elements on the Basis of their Conjugative Module
These ICEs were classified into seven distinct families belonging to three superfamilies on the basis of their conjugation modules: (i) ConjTn916 and ConjICESt3 belonging to the ConjTn916 superfamily, (ii) ConjvanG, ConjTn5252, ConjTn1549, and ConjTnGBS2 belonging to the ConjTn5252 superfamily and the ConjTnGBS1 family. The ConjTn916 and the ConjTn5252 superfamilies of conjugation modules belong respectively to the MPFFA and to the MPFFATA classes of T4SS involved in bacterial conjugation as defined by Guglielmini et al. (2014). No match was found for the ConjTnGBS1 family.
Modular Evolution
The phylogenetic trees obtained for relaxase, CP and VirB4 encoded by elements belonging to the ConjTn916 superfamily (Figure 2), the ConjTn5252 superfamily (Figure 4), and the ConjTnGBS1 family (Figure 5) are highly similar, suggesting that genes exchanges or replacements within the conjugation modules of these families have not occurred or are rare. However, incongruences were found for relaxases, CPs and VirB4 proteins of some elements belonging to ICESt3 family, suggesting that some gene exchanges or replacements have occurred within the conjugation modules belonging to this family.
Unrelated or very distantly related integrases were found to be encoded by at least some of the ICEs belonging to the same family (except for TnGBS1 family) and frequently by closely or very related elements (for example in the Tn5252 family). Furthermore, related site-specific integrases were found in unrelated or distantly related ICEs. Such incongruences are due to multiple exchanges of integration/excision and/or conjugation modules between and/or within ICE families. For most cases, the data do not allow to determine what precisely happened. However, it was previously reported that the last common ancestor of TnGBS2 family acquired its DDE transposase from an insertion sequence and that the last common ancestor of TnGBS1 family acquired its DDE transposase from an ICE belonging to TnGBS2 family (Guérillot et al., 2013). Here, the comparison of phylogenetic trees obtained for CONJTn5252 superfamily, serine integrases and tyrosine integrases clearly shows three independent replacements of the DDE transposases by unrelated integrases. One of these ICEs, ICE_SagILRI005_rplL, probably results from: (i) the integration of an ICE belonging to TnGBS2 family (encoding a low specificity/site-preferential DDE transposase and a RepA_N protein) into an ICE belonging to Tn5252 family (encoding a tyrosine integrase specific of rplL and another RepA_N protein), and (ii) the loss of the conjugation and replication modules of the Tn5252-related element and the deletion of the DDE transposase gene of the TnGBS2-related ICE.
Integration Specificity of ICEs in Streptococcal Genomes: Impact on Host Fitness and on the Evolution of Elements
In this work, efforts were made to identify the boundaries of the ICEs and therefore to identify the insertion site of each of them. This analysis of integration/excision modules and site specificity is the first one carried for a large array of ICEs encoding their transfer as single-stranded DNA. The ICEs and dICEs of Streptococci carry diverse integration/excision modules (75 encoding a tyrosine recombinase, 20 encoding a unique serine recombinase, nine encoding three serine recombinases, and 23 a DDE transposase) and have a large array of integration specificity (low or preferential integration, 18 different site-specific integrations). This work led to the detection of eight new target-genes for streptococcal ICE insertion that have not been identified previously (ftsK, guaA, lysS, mutT, rpmG, rpsI, traG, and ebfC).
It should be noticed that, among the 131 ICEs/dICEs identified, only nine (restricted to one family) were found to be integrated into the 3′ end of genes encoding tRNAs. This contrasts with the results of the analysis of actinobacterial ICEs (most of these ICEs carry conjugation modules unrelated to the ones of streptococcal ICEs and transfer as double-stranded DNA). Among the 144 actinobacterial ICEs analyzed, 100 were found integrated in the 3′ end of a tRNA gene (Ghinet et al., 2011).
In most streptococcal ICEs, as for almost all other known ICEs, the attI site is located in the vicinity of the integrase gene. However, it should also be noticed that for all streptococcal ICEs/dICEs integrated in rumA, the attR site is located within the serine integrase gene and consequently the integrase gene carried by the excised ICE has a different length and C terminus. att sites are found within the genes of their cognate integrase in very few integrative elements, such as the prophage Mx8 from Myxococcus xanthus for which the phage attachment site, attP, is located within the tyrosine integrase gene (Magrini et al., 1999). Site-specific integration of Mx8 leads to the replacement of the 112-residue C-terminal sequence by a 13-residue C terminus. This modified integrase is less active that the integrase encoded by the excised element. Therefore, it seems probable that the differences between the integrases encoded by the ICEs integrated in rumA and the integrases encoded by the excised elements can lead to differences in the function of the two forms of the integrase.
Besides mechanistic constraints leading to integration in conserved palindromic sequences for many tyrosine integrases (Williams, 2002), one would expect that selection criteria for ICE integration in evolution would be (i) to have the least effect on host fitness and, (ii) since many have a large host range (Bellanger et al., 2014), to allow integration into a wide range of strains and species. Numerous ICEs encoding a tyrosine integrase were found to be site-specifically integrated in the conserved 3′ end of essential conserved genes that are isolated or are the last gene of an operon. The insertion does not modify the gene product (tRNA, ribosomal proteins) or leads to very little modification of the 3′ end of the protein (lysyl-tRNA synthetase). Hence, such integrations would have no effect on host fitness and can occur in a large array of species.
All (except one) the streptococcal ICEs belonging to the Tn916 family detected in this study encode a tyrosine integrase identical or almost identical to the Tn916 integrase. Analyses of a high number of insertion sites in various hosts showed that Tn916, despite having a low specificity of integration, still has a preference for AT rich regions (consensus TTTTTnnnnnnAAAAA; Hosking et al., 1998; Cookson et al., 2011). Furthermore, the analyses of insertion sites after conjugal transfer to Butyrivibrio proteoclasticus B316T, whose genome has a similar GC percent as the ones of Streptococci (39%), showed that only 34% of the 123 analyzed insertions disrupt annotated ORFs even if 90% of this genome is made of ORFs (Cookson et al., 2011). This may be due to lower GC ratio (34.7%) of intergenic regions. Therefore, the AT-rich region preference of Tn916 probably leads to a null or low impact of most Tn916 insertion events on host fitness. Since MGEs have generally a lower G+C percent than their host genome (Rocha and Danchin, 2002), this preference could also explain the frequent presence of Tn916 or Tn916-related elements in plasmids or Tn5252-related elements (Clewell and Gawron-Burke, 1986; Ayoubi et al., 1991; Ding et al., 2009; Mingoia et al., 2011; Chancey et al., 2015). This putative preference for MGEs would also lead to a lower impact on the fitness of the bacterial host. It was previously shown that a Tn916 element carried by a Tn5252-related element can be transferred alone (Santoro et al., 2010) or as a part of the Tn5252 element (Ayoubi et al., 1991). Therefore, besides a low impact on host fitness, this A+T rich preference could increase the transfer ability of Tn916 (either autonomously or by mobilization in cis).
Some ICEs integrate in the conserved 5′ end of the first gene of an operon or of an isolated gene that encodes an essential protein (the DNA translocase FtsK that coordinates cell division and chromosome segregation; the nucleoid-associated protein EbfC; the ribosome assembly GTPase RbgA). Importantly, the insertion does not modify the N-terminus of the protein encoded by the target gene. Moreover, for three ICEs integrated in rbgA and two ICEs integrated in ftsK, the integration does not change the 15–64 bp sequence located upstream from the START codon. However, in the two other ICEs integrated in rbgA and in the one integrated in ebfC, the sequence upstream from the gene, including its promoter, is completely different, suggesting that the expression of the gene is impacted by the integration of the element. This situation is reminiscent of the integration of the putative satellite prophage SpyCI from S. pyogenes. In stationary phase, SpyCI is integrated into the 5′ end of the DNA mismatch repair gene mutL, disrupting its expression and that of three other genes located downstream (Nguyen and McShan, 2014). During early exponential growth, SpyCI excises from the bacterial chromosome and replicates as an episome, thus allowing the expression of mutL and of downstream genes. Concerning the ebfC gene, it is known that in the spirochaete Borrelia burgdorferi, it is highly expressed in rapidly growing bacteria but mRNA is undetectable in stationary phase (Jutras et al., 2012). Thus, ICE integration in the promoter of this gene in S. parasanguinis FW213 may not alter host fitness at all: expression of ebfC gene would not be required in the stationary phase when the element is integrated and excision of the element in the exponential phase restores the expression of this gene.
TnGBS1 and TnGBS2 are two known elements from S. agalactiae encoding DDE transposases that integrate in various intergenic regions located 15 or 16 bp upstream from the 35 box of sigma A promoters (Brochet et al., 2009; Guérillot et al., 2013). In this study, all elements belonging to TnGBS1 and TnGBS2 families (except three TnGBS2 that encode serine or tyrosine recombinases) are also integrated in such location. Insertion into intergenic regions is expected to minimize the effects caused by the transposon insertion on host fitness. However, such insertions may interfere with the transcription level of the downstream gene. However, it should be noticed that the insertion of TnGBS2 does not seem to affect significantly the transcription level of the gene located downstream from the preferred insertion site.
All insertions of the 29 ICEs encoding serine integrase disrupt genes encoding proteins. A large majority are site-specific. Most of these genes are widespread but are not essential for the strain (rumA, hsdM, mutT). The rumA gene encodes a widespread rRNA methyltransferase. In Escherichia coli, the deletion of this gene has little effect on growth or on the fidelity of translation, but alters the sensitivity of the ribosomes to fusidic acid and capreomycin (Persaud et al., 2010). The hdsM gene encodes the methyltransferase subunit of type I restriction-modification systems (Murray, 2000). The mutT gene encodes a Nudix hydrolase that removes oxidized nucleotide precursors so that they cannot be incorporated in DNA during replication (Lu et al., 2001). One element, dICE_SanC238_traG is site-specifically integrated into the traG gene that encodes the CP of an ICE remnant (not detailed in this report because this remnant is too much decayed) belonging to Tn1549 family.
The consequences of the integration/excision balance of ICE encoding serine recombinases on the expression of the target genes encoding proteins have never been studied. However, several examples can be cited for prophages or prophage-related elements (Stragier et al., 1989; Kunkel et al., 1990; Rabinovich et al., 2012). Thus, the DNA uptake competence system of the intracellular bacterial pathogen Listeria monocytogenes serovar 1/2 was considered non-functional because the competence master activator gene, comK, is disrupted by the insertion of the temperate prophage A118 encoding a serine recombinase (Rabinovich et al., 2012). However, the prophage excises not only during the activation of lytic phase but also during intracellular growth, primarily within phagosomes of macrophages, without any production of progeny virions, thus allowing expression of the comK gene (Rabinovich et al., 2012). In the same way, ICEs integrated within specific genes and disrupting them may excise when these genes are useful for the host cell (rumA, hsdM, mutT) or for the host ICE (traG) to reduce the impact on host fitness and guarantee their maintenance in the cell. Four elements encoding serine integrases are integrated in secondary sites within protein-encoding genes. As for primary integration sites, if the elements are still able to excise, expression of the target gene might not be impacted. The integration of CTn5, an ICE belonging to the Tn1549 family and encoding a serine integrase occurs in an adhesin gene in Clostridium difficile 630 (Sebaihia et al., 2006). However, comparison of this genome with that of the derived strain 630Δerm showed that CTn5 has excised from its original location and has inserted in rumA (CD3393) of 630Δerm (van Eijk et al., 2015). This suggests that an ICE integrated in a secondary site is able to excise and reintegrate in its primary site and conversely.
Globally, the impact of the integration upstream from promoters, in the 5′ end of CDSs or within CDSs could be reduced if the ICE excises when the targeted genes are expressed. It was initially thought that ICEs do not replicate autonomously in the cell, although conjugative transfer can be seen as an intercellular replication (Burrus et al., 2002b). Therefore, although the excision could be advantageous for the host, if the cell divides when the ICE is excised, the ICE would be lost in one of the daughter cell. Nevertheless, several recent studies showed or strongly suggested that various single-strand DNA transferring ICEs are capable of extrachromosomal replication in both Gram-positive and Gram-negative bacteria (Carraro et al., 2015 and references therein). In particular, replication was found to be involved in maintenance of TnGBS1 and TnGBS2 in transconjugants before their integration (Guérillot et al., 2013). These elements encode a protein that carries a repA_N domain and is related to the protein controlling the θ replication of various plasmids and a protein related to ParA, a protein involved in maintenance of some plasmids (Guérillot et al., 2013). We found genes encoding a protein with a RepA_N domain in all ICE belonging to the Tn5252 family and numerous ICEs belonging to Tn1549 family. We also found ParA segregation ATPase CDSs in all ICEs belonging to Tn1549 family and vanG family suggesting that all these elements can replicate as episomes. Evidence of intracellular extrachromosomal replication was also recently obtained for ICESt3 (Carraro et al., 2011) and another element belonging to ICESt3 family, RD2 (i.e., ICE_Spy6180_tRNAthr) of S. pyogenes (Sitkiewicz et al., 2011) although these elements do not carry any replication module. Moreover, extrachromosomal replication of ICEBs1 from Bacillus subtilis, an element belonging to the Tn916 superfamily (Burrus et al., 2002a) was found to be involved in the stability of the element. This intracellular replication is initiated from the ICEBs1 oriT and required the ICEBs1-encoded relaxase (Lee et al., 2010). At last, all ICEs belonging to the Tn916 superfamily encode a peculiar relaxase (MOBT) related to rolling circle replication initiators involved in maintenance of various plasmids (Guglielmini et al., 2014), suggesting that all these elements are also able to replicate as episomes.
Conclusion
This study greatly enriches our understanding of the classification and integration sites of ICEs/dICEs in streptococci genomes. In the future, it will be updated and further extended to take into account newly sequenced genomes and to confirm all the trends proposed here. An automated bioinformatics procedure will be developed to keep pace with the constantly growing number of available genomes. Extension to other species of Firmicutes and to the search for IMEs is also envisaged.
Author Contributions
GG and SP conceived the reference database of signature proteins. GG, SP, NL-B, and M-DD contributed to the conception of the work. CA, CC, GG, NL-B, M-DD, SP, VL, and TL were involved in the acquisition and/or the analysis of the data. NL-B, GG, CC, and SP contribute to the drafting of the manuscript. NL-B and CC elaborated the figures and tables. All authors criticized and finally approved this final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
CC is recipient of a scholarship of the Ministère de l’Enseignement Supérieur et de la Recherche. This work received financial support from the Région Lorraine and Université de Lorraine.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01483
Footnotes
- ^http://www.bacterio.net/streptococcus.html
- ^http://www.ncbi.nlm.nih.gov/genome/browse/
- ^http://central.biomart.org/
- ^http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
- ^http://mobyle.pasteur.fr/cgi-bin/portal.py#forms::CONJscan-T4SSscan
- ^http://db-mml.sjtu.edu.cn/ICEberg/index.php
References
Arrieta, M. C., Stiemsma, L. T., Amenyogbe, N., Brown, E. M., and Finlay, B. (2014). The intestinal microbiome in early life: health and disease. Front. Immunol. 5:427. doi: 10.3389/fimmu.2014.00427
Ayoubi, P., Kilic, A. O., and Vijayakumar, M. N. (1991). Tn5253, the pneumococcal omega (cat tet) BM6001 element, is a composite structure of two conjugative transposons, Tn5251 and Tn5252. J. Bacteriol. 173, 1617–1622.
Bellanger, X., Payot, S., Leblond-Bourget, N., and Guedon, G. (2014). Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol. Rev. 38, 720–760. doi: 10.1111/1574-6976.12058
Bi, D., Xu, Z., Harrison, E. M., Tai, C., Wei, Y., He, X., et al. (2012). ICEberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Res. 40, D621–D626. doi: 10.1093/nar/gkr846
Brochet, M., Couve, E., Glaser, P., Guedon, G., and Payot, S. (2008). Integrative conjugative elements and related elements are major contributors to the genome diversity of Streptococcus agalactiae. J. Bacteriol. 190, 6913–6917. doi: 10.1128/JB.00824-08
Brochet, M., Da Cunha, V., Couve, E., Rusniok, C., Trieu-Cuot, P., and Glaser, P. (2009). Atypical association of DDE transposition with conjugation specifies a new family of mobile elements. Mol. Microbiol. 71, 948–959. doi: 10.1111/j.1365-2958.2008.06579.x
Bryson, K., Loux, V., Bossy, R., Nicolas, P., Chaillou, S., Van De Guchte, M., et al. (2006). AGMIAL: implementing an annotation strategy for prokaryote genomes as a distributed system. Nucleic Acids Res. 34, 3533–3545. doi: 10.1093/nar/gkl471
Burrus, V., Pavlovic, G., Decaris, B., and Guedon, G. (2002a). The ICESt1 element of Streptococcus thermophilus belongs to a large family of integrative and conjugative elements that exchange modules and change their specificity of integration. Plasmid 48, 77–97. doi: 10.1016/S0147-619X(02)00102-6
Burrus, V., Pavlovic, G., Decaris, B., and Guédon, G. (2002b). Conjugative transposons: the tip of the iceberg. Mol. Microbiol. 46, 601–610. doi: 10.1046/j.1365-2958.2002.03191.x
Carraro, N., Libante, V., Morel, C., Decaris, B., Charron-Bourgoin, F., Leblond, P., et al. (2011). Differential regulation of two closely related integrative and conjugative elements from Streptococcus thermophilus. BMC Microbiol. 11:238. doi: 10.1186/1471-2180-11-238
Carraro, N., Poulin, D., and Burrus, V. (2015). Replication and active partition of Integrative and Conjugative Elements (ICEs) of the SXT/R391 family: the line between ICEs and conjugative plasmids is getting thinner. PLoS Genet. 11:e1005298. doi: 10.1371/journal.pgen.1005298
Chancey, S. T., Agrawal, S., Schroeder, M. R., Farley, M. M., Tettelin, H., and Stephens, D. S. (2015). Composite mobile genetic elements disseminating macrolide resistance in Streptococcus pneumoniae. Front. Microbiol. 6:26. doi: 10.3389/fmicb.2015.00026
Chuzeville, S., Puymege, A., Madec, J. Y., Haenni, M., and Payot, S. (2012). Characterization of a new CAMP factor carried by an integrative and conjugative element in Streptococcus agalactiae and spreading in Streptococci. PLoS ONE 7:e48918. doi: 10.1371/journal.pone.0048918
Clewell, D. B., and Gawron-Burke, C. (1986). Conjugative transposons and the dissemination of antibiotic resistance in streptococci. Annu. Rev. Microbiol. 40, 635–659. doi: 10.1146/annurev.mi.40.100186.003223
Cookson, A. L., Noel, S., Hussein, H., Perry, R., Sang, C., Moon, C. D., et al. (2011). Transposition of Tn916 in the four replicons of the Butyrivibrio proteoclasticus B316(T) genome. FEMS Microbiol. Lett. 316, 144–151. doi: 10.1111/j.1574-6968.2010.02204.x
Croucher, N. J., Walker, D., Romero, P., Lennard, N., Paterson, G. K., Bason, N. C., et al. (2009). Role of conjugative elements in the evolution of the multidrug-resistant pandemic clone Streptococcus pneumoniae Spain23F ST81. J. Bacteriol. 191, 1480–1489. doi: 10.1128/JB.01343-08
Ding, F., Tang, P., Hsu, M. H., Cui, P., Hu, S., Yu, J., et al. (2009). Genome evolution driven by host adaptations results in a more virulent and antimicrobial-resistant Streptococcus pneumoniae serotype 14. BMC Genomics 10:158. doi: 10.1186/1471-2164-10-158
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Franciosi, E., Settanni, L., Cavazza, A., and Poznanski, E. (2009). Biodiversity and technological potential of wild lactic acid bacteria from raw cows’ milk. Int. Dairy J. 19, 3–11. doi: 10.1016/j.idairyj.2008.07.008
Franke, A. E., and Clewell, D. B. (1981). Evidence for a chromosome-borne resistance transposon (Tn916) in Streptococcus faecalis that is capable of “conjugal” transfer in the absence of a conjugative plasmid. J. Bacteriol. 145, 494–502.
Garnier, F., Taourit, S., Glaser, P., Courvalin, P., and Galimand, M. (2000). Characterization of transposon Tn1549, conferring VanB-type resistance in Enterococcus spp. Microbiology 146(Pt 6), 1481–1489. doi: 10.1099/00221287-146-6-1481
Ghinet, M. G., Bordeleau, E., Beaudin, J., Brzezinski, R., Roy, S., and Burrus, V. (2011). Uncovering the prevalence and diversity of integrating conjugative elements in actinobacteria. PLoS ONE 6:e27846. doi: 10.1371/journal.pone.0027846
Goessweiner-Mohr, N., Arends, K., Keller, W., and Grohmann, E. (2013). Conjugative type IV secretion systems in Gram-positive bacteria. Plasmid 70, 289–302. doi: 10.1016/j.plasmid.2013.09.005
Gouy, M., Guindon, S. P., and Gascuel, O. (2010). SeaView Version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Groth, A. C., and Calos, M. P. (2004). Phage integrases: biology and applications. J. Mol. Biol. 335, 667–678. doi: 10.1016/j.jmb.2003.09.082
Guérillot, R., Da Cunha, V., Sauvage, E., Bouchier, C., and Glaser, P. (2013). Modular evolution of TnGBSs, a new family of integrative and conjugative elements associating insertion sequence transposition, plasmid replication, and conjugation for their spreading. J. Bacteriol. 195, 1979–1990. doi: 10.1128/JB.01745-12
Guglielmini, J., Neron, B., Abby, S. S., Garcillan-Barcia, M. P., De La Cruz, F., and Rocha, E. P. (2014). Key components of the eight classes of type IV secretion systems involved in bacterial conjugation or protein secretion. Nucleic Acids Res. 42, 5715–5727. doi: 10.1093/nar/gku194
Guglielmini, J., Quintais, L., Garcillan-Barcia, M. P., De La Cruz, F., and Rocha, E. P. (2011). The repertoire of ICE in prokaryotes underscores the unity, diversity, and ubiquity of conjugation. PLoS Genet. 7:e1002222. doi: 10.1371/journal.pgen.1002222
Hacker, J., and Carniel, E. (2001). Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2, 376–381. doi: 10.1093/embo-reports/kve097
Hacker, J., and Kaper, J. B. (2000). Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54, 641–679. doi: 10.1146/annurev.micro.54.1.641
Heather, Z., Holden, M. T., Steward, K. F., Parkhill, J., Song, L., Challis, G. L., et al. (2008). A novel streptococcal integrative conjugative element involved in iron acquisition. Mol. Microbiol. 70, 1274–1292. doi: 10.1111/j.1365-2958.2008.06481.x
Hosking, S. L., Deadman, M. E., Moxon, E. R., Peden, J. F., Saunders, N. J., and High, N. J. (1998). An in silico evaluation of Tn916 as a tool for generalized mutagenesis in Haemophilus influenzae Rd. Microbiology 144(Pt 9), 2525–2530. doi: 10.1099/00221287-144-9-2525
Juhas, M., Van Der Meer, J. R., Gaillard, M., Harding, R. M., Hood, D. W., and Crook, D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Jutras, B. L., Chenail, A. M., and Stevenson, B. (2012). Changes in bacterial growth rate govern expression of the Borrelia burgdorferi OspC and Erp infection-associated surface proteins. J. Bacteriol. 195, 757–764. doi: 10.1128/JB.01956-12
Kohler, W. (2007). The present state of species within the genera Streptococcus and Enterococcus. Int. J. Med. Microbiol. 297, 133–150. doi: 10.1016/j.ijmm.2006.11.008
Korona-Glowniak, I., Siwiec, R., and Malm, A. (2015). Resistance determinants and their association with different transposons in the antibiotic-resistant Streptococcus pneumoniae. Biomed. Res. Int. 2015, 836496. doi: 10.1155/2015/836496
Kunkel, B., Losick, R., and Stragier, P. (1990). The Bacillus subtilis gene for the development transcription factor sigma K is generated by excision of a dispensable DNA element containing a sporulation recombinase gene. Genes Dev. 4, 525–535. doi: 10.1101/gad.4.4.525
Lee, C. A., Babic, A., and Grossman, A. D. (2010). Autonomous plasmid-like replication of a conjugative transposon. Mol. Microbiol. 75, 268–279. doi: 10.1111/j.1365-2958.2009.06985.x
Leonetti, C. T., Hamada, M. A., Laurer, S. J., Broulidakis, M. P., Swerdlow, K. J., Lee, C. A., et al. (2015). Critical components of the conjugation machinery of the Integrative and Conjugative Element ICEBs1 of Bacillus subtilis. J. Bacteriol. 197, 2558–2567. doi: 10.1128/JB.00142-15
Low, H. H., Gubellini, F., Rivera-Calzada, A., Braun, N., Connery, S., Dujeancourt, A., et al. (2014). Structure of a type IV secretion system. Nature 508, 550–553. doi: 10.1038/nature13081
Lu, A. L., Li, X., Gu, Y., Wright, P. M., and Chang, D. Y. (2001). Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem. Biophys. 35, 141–170. doi: 10.1385/CBB
Magrini, V., Creighton, C., and Youderian, P. (1999). Site-specific recombination of temperate Myxococcus xanthus phage Mx8: genetic elements required for integration. J. Bacteriol. 181, 4050–4061.
Mingoia, M., Tili, E., Manso, E., Varaldo, P. E., and Montanari, M. P. (2011). Heterogeneity of Tn5253-like composite elements in clinical Streptococcus pneumoniae isolates. Antimicrob. Agents Chemother. 55, 1453–1459. doi: 10.1128/AAC.01087-10
Mitchell, T. J. (2003). The pathogenesis of streptococcal infections: from tooth decay to meningitis. Nat. Rev. Microbiol. 1, 219–230. doi: 10.1038/nrmicro771
Murray, N. E. (2000). Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol. Mol. Biol. Rev. 64, 412–434. doi: 10.1128/MMBR.64.2.412-434.2000
Nakajima, T., Nakanishi, S., Mason, C., Montgomery, J., Leggett, P., Matsuda, M., et al. (2013). Population structure and characterization of viridans group streptococci (VGS) isolated from the upper respiratory tract of patients in the community. Ulster Med. J. 82, 164–168.
Nguyen, S. V., and McShan, W. M. (2014). Chromosomal islands of Streptococcus pyogenes and related streptococci: molecular switches for survival and virulence. Front. Cell. Infect. Microbiol. 4:109. doi: 10.3389/fcimb.2014.00109
Nguyen, V. H., and Lavenier, D. (2009). PLAST: parallel local alignment search tool for database comparison. BMC Bioinformatics 10:329. doi: 10.1186/1471-2105-10-329
Park, H. K., Shim, S. S., Kim, S. Y., Park, J. H., Park, S. E., Kim, H. J., et al. (2005). Molecular analysis of colonized bacteria in a human newborn infant gut. J. Microbiol. 43, 345–353.
Persaud, C., Lu, Y., Vila-Sanjurjo, A., Campbell, J. L., Finley, J., and O’connor, M. (2010). Mutagenesis of the modified bases, m(5)U1939 and psi2504, in Escherichia coli 23S rRNA. Biochem. Biophys. Res. Commun. 392, 223–227. doi: 10.1016/j.bbrc.2010.01.021
Puymège, A., Bertin, S., Guedon, G., and Payot, S. (2015). Analysis of Streptococcus agalactiae pan-genome for prevalence, diversity and functionality of integrative and conjugative or mobilizable elements integrated in the tRNA gene. Mol. Genet. Genomics 290, 1727–1740. doi: 10.1007/s00438-015-1031-9
Rabinovich, L., Sigal, N., Borovok, I., Nir-Paz, R., and Herskovits, A. A. (2012). Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 150, 792–802. doi: 10.1016/j.cell.2012.06.036
Roberts, A. P., and Mullany, P. (2011). Tn916-like genetic elements: a diverse group of modular mobile elements conferring antibiotic resistance. FEMS Microbiol. Rev. 35, 856–871. doi: 10.1111/j.1574-6976.2011.00283.x
Rocha, E. P., and Danchin, A. (2002). Base composition bias might result from competition for metabolic resources. Trends Genet. 18, 291–294. doi: 10.1016/S0168-9525(02)02690-2
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M. A., et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. doi: 10.1093/bioinformatics/16.10.944
Santoro, F., Oggioni, M. R., Pozzi, G., and Iannelli, F. (2010). Nucleotide sequence and functional analysis of the tet(M)-carrying conjugative transposon Tn5251 of Streptococcus pneumoniae. FEMS Microbiol. Lett. 308, 150–158. doi: 10.1111/j.1574-6968.2010.02002.x
Schubert, S., Dufke, S., Sorsa, J., and Heesemann, J. (2004). A novel integrative and conjugative element (ICE) of Escherichia coli: the putative progenitor of the Yersinia high-pathogenicity island. Mol. Microbiol. 51, 837–848. doi: 10.1046/j.1365-2958.2003.03870.x
Scott, J. R., Bringel, F., Marra, D., Van Alstine, G., and Rudy, C. K. (1994). Conjugative transposition of Tn916: preferred targets and evidence for conjugative transfer of a single strand and for a double-stranded circular intermediate. Mol. Microbiol. 11, 1099–1108. doi: 10.1111/j.1365-2958.1994.tb00386.x
Sebaihia, M., Wren, B. W., Mullany, P., Fairweather, N. F., Minton, N., Stabler, R., et al. (2006). The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38, 779–786. doi: 10.1038/ng1830
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi: 10.1038/msb.2011.75
Sitkiewicz, I., Green, N. M., Guo, N., Mereghetti, L., and Musser, J. M. (2011). Lateral gene transfer of streptococcal ICE element RD2 (region of difference 2) encoding secreted proteins. BMC Microbiol. 11:65. doi: 10.1186/1471-2180-11-65
Smillie, C., Garcillan-Barcia, M. P., Francia, M. V., Rocha, E. P., and De La Cruz, F. (2010). Mobility of plasmids. Microbiol. Mol. Biol. Rev. 74, 434–452. doi: 10.1128/MMBR.00020-10
Stragier, P., Kunkel, B., Kroos, L., and Losick, R. (1989). Chromosomal rearrangement generating a composite gene for a developmental transcription factor. Science 243, 507–512. doi: 10.1126/science.2536191
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Toussaint, A., and Merlin, C. (2002). Mobile elements as a combination of functional modules. Plasmid 47, 26–35. doi: 10.1006/plas.2001.1552
van der Meer, J. R., and Sentchilo, V. (2003). Genomic islands and the evolution of catabolic pathways in bacteria. Curr. Opin. Biotechnol. 14, 248–254. doi: 10.1016/S0958-1669(03)00058-2
van Eijk, E., Anvar, S. Y., Browne, H. P., Leung, W. Y., Frank, J., Schmitz, A. M., et al. (2015). Complete genome sequence of the Clostridium difficile laboratory strain 630Deltaerm reveals differences from strain 630, including translocation of the mobile element CTn5. BMC Genomics 16:31. doi: 10.1186/s12864-015-1252-7
Williams, K. P. (2002). Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Res. 30, 866–875. doi: 10.1093/nar/30.4.866
Keywords: integrative and conjugative elements, T4SS, integrase, integration site, Streptococcus
Citation: Ambroset C, Coluzzi C, Guédon G, Devignes M-D, Loux V, Lacroix T, Payot S and Leblond-Bourget N (2016) New Insights into the Classification and Integration Specificity of Streptococcus Integrative Conjugative Elements through Extensive Genome Exploration. Front. Microbiol. 6:1483. doi: 10.3389/fmicb.2015.01483
Received: 27 July 2015; Accepted: 08 December 2015;
Published: 06 January 2016.
Edited by:
John R. Battista, Louisiana State University and A & M College, USAReviewed by:
Awdhesh Kalia, University of Texas MD Anderson Cancer Center, USAWilliam John Kelly, AgResearch Ltd., New Zealand
Copyright © 2016 Ambroset, Coluzzi, Guédon, Devignes, Loux, Lacroix, Payot and Leblond-Bourget. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathalie Leblond-Bourget, nathalie.leblond@univ-lorraine.fr
†These authors have contributed equally to this work.