 Flore Ballaud1
Flore Ballaud1 Alexis Dufresne1
Alexis Dufresne1 André-Jean Francez1
André-Jean Francez1 Jonathan Colombet2,3
Jonathan Colombet2,3 Télesphore Sime-Ngando2,3
Télesphore Sime-Ngando2,3 Achim Quaiser1*
Achim Quaiser1*- 1UMR CNRS 6553 ECOBIO, Université de Rennes 1, Rennes, France
- 2Université Clermont Auvergne, Université Blaise Pascal, Clermont-Ferrand, France
- 3CNRS, UMR 6023, Laboratoire Microorganismes: Génome et Environnement, Aubière, France
Viruses impact microbial activity and carbon cycling in various environments, but their diversity and ecological importance in Sphagnum-peatlands are unknown. Abundances of viral particles and prokaryotes were monitored bi-monthly at a fen and a bog at two different layers of the peat surface. Viral particle abundance ranged from 1.7 x 106 to 5.6 x 108 particles mL-1, and did not differ between fen and bog but showed seasonal fluctuations. These fluctuations were positively correlated with prokaryote abundance and dissolved organic carbon, and negatively correlated with water-table height and dissolved oxygen. Using shotgun metagenomics we observed a shift in viral diversity between winter/spring and summer/autumn, indicating a seasonal succession of viral communities, mainly driven by weather-related environmental changes. Based on the seasonal asynchrony between viral and microbial diversity, we hypothesize a seasonal shift in the active microbial communities associated with a shift from lysogenic to lytic lifestyles. Our results suggest that temporal variations of environmental conditions rather than current habitat differences control the dynamics of virus-host interactions in Sphagnum-dominated peatlands.
Introduction
Viruses are present in virtually all ecosystems, and are considered to be the most abundant biological entities in the biosphere (Suttle, 2005). Through lytic and lysogenic life cycles, viruses affect the metabolism and abundance of their cellular hosts from all three domains of life (synthesis in) (Kirchman, 2012), impacting the diversity and the structure of microbial communities (Middelboe, 2000; Suttle, 2007; Sime-Ngando, 2014). Because viruses function as a top-down control on microbial production (Pradeep Ram et al., 2011; Chow et al., 2014), they affect biogeochemical cycles through the release of substantial amounts of organic carbon and nutrients in the environment (Middelboe and Lyck, 2002). While resources released by this viral shunt can be reused to sustain microbial biomass, it typically results in a reduction of the overall flow of organic matter and energy towards higher trophic levels (Fuhrman, 1999; Middelboe and Lyck, 2002; Ankrah et al., 2014). Besides the viral-shunt, a significant proportion of the released viruses represent a consequential amount of labile organic matter that can be readily decomposed (Dell’Anno et al., 2015). Viral lysis, along with bacterial grazing are important processes in microbial succession (Fierer et al., 2010), suggesting that coupled viral-host interactions can influence ecosystem-level carbon cycling, depending on the activity of cells and on the balance between lytic and lysogenic strategies. Consequently, considering viral ecology is critical to understanding ecosystem functioning.
Peatlands are a globally relevant component of the carbon cycle, storing a quarter of global soil carbon and more carbon than all vegetation (Yu, 2012; Turetsky et al., 2015). Boreal and temperate peatlands are often dominated by peat forming mosses, belonging to the Sphagnum genus. Many Sphagnum-dominated peatlands develop from a nutrient-rich, non-acidic fen (minerotrophic fen) during early stages, to a nutrient-poor acidic bog (rain-fed ombrotrophic bog) (Rydin and Jeglum, 2006). During this development process, peatlands accumulate large stocks of partly decomposed organic matter as peat. This accumulation is a consequence of the long-term imbalance between carbon uptake from photosynthesis and carbon losses via respiration, methanogenesis and DOC leaching, which results in the preservation of up to 15 % of the net primary production as peat (Clymo, 1984; Moore, 1987; Francez and Vasander, 1995; Roulet et al., 2007). The combination of abiotic conditions, such as low temperature, low pH, high soil water content and poor nutrient availability, as well as biotic factors (Sphagnum litter quality, allelopathy) constrains microbial activities, limiting decomposition and mineralization (Clymo, 1984; Rydin and Jeglum, 2006). Accumulation of organic matter over long time periods leads to stratification with a deep permanently anoxic catotelm in contact with underlying bedrock, an intermediate mesotelm layer that is periodically anoxic and oxic and the predominantly oxic acrotelm at the surface (Clymo and Bryant, 2008). The heterogeneity of peat, the variety of dynamic stages and sources of water and nutrients provide a large panel of habitats and niches for a broad diversity of microorganisms transforming different carbon substrates through aerobic and anaerobic pathways of decomposition (Juottonen et al., 2005; Artz, 2009; Andersen et al., 2010; Tveit et al., 2012).
Understanding of microbial diversity in peatlands, especially in regards to spatio-temporal patterns, is currently limited. The development of cultivation-independent approaches such as metagenomics provide a powerful tool to investigate microbial taxa and associated protein-coding genes across ecosystems (Lynch et al., 2004; Vandenkoornhuyse et al., 2010). More recently, sequencing of viral metagenomes (hereafter metaviromes) has dramatically expanded understanding of viral diversity, particularly in marine and freshwater environments (Comeau et al., 2008; Lopez-Bueno et al., 2009; Rohwer and Thurber, 2009; Roux et al., 2012; Smith et al., 2013; Labonté and Suttle, 2013; Wommack et al., 2015) compared to soil ecosystems (Kimura et al., 2008; Zablocki et al., 2014; Adriaenssens et al., 2015). Most analyses have focused on cross-biomes comparisons, while spatio-temporal dynamics remain poorly documented (Pagarete et al., 2013; Chow et al., 2014). Several viral marker genes, conserved within particular viral families, are considered to be reliable for taxonomic affiliation (Roux et al., 2011; Sakowski et al., 2014). Nevertheless, none of these marker genes are ubiquitous among viruses and they are far from the high level of conservation found in rRNA genes that are used for the classification of organisms from the three kingdoms. While several studies have investigated the diversity of microbial communities in peatlands (Dedysh et al., 2006; Artz et al., 2007; Peltoniemi et al., 2009; Dedysh, 2011; Mackelprang et al., 2011; Bragina et al., 2012; Serkebaeva et al., 2013; Mondav et al., 2014; Tveit et al., 2014; Hultman et al., 2015; Nunes et al., 2015) viral diversity in Sphagnum-dominated peatlands remains largely unknown and basic knowledge of virus ecology in these ecosystems is still lacking (Quaiser et al., 2015). In view of the importance of viruses in structuring and regulating prokaryotic communities and the implication of the latter in the carbon sink function of peatland; it is essential to understand the role of viruses in the dynamics of the microbial communities in this ecosystem.
In order to characterize virus ecology in a temperate Sphagnum-dominated peatland, we combined and integrated different approaches to study the spatio-temporal patterns of viral abundance and diversity. The goals of this work were: (1) to compare the seasonal abundance of viruses and prokaryotes at two different layers of the peat surface corresponding to a stratified analysis of acrotelm, the most active layer, of fen and bog; (2) to identify abiotic controls on abundance and distribution of these viruses and prokaryotes; and (3) to characterize the viral diversity over an annual cycle.
Materials and Methods
Site Description
Peat samples were collected at Les Pradeaux (3°55E; 45°32N), a Sphagnum-dominated peatland situated in the French Massif Central at an altitude of 1 350 m. The fen is dominated by Sphagnum fallax, Carex rostrata, Eriophorum angustifolium, and Menyanthes trifoliata, while the bog is mainly colonized by Sphagnum magellanicum, Sphagnum capillifolium, Andromeda polifolia, Vaccinium oxycoccos, and Eriophorum vaginatum.
Sampling Strategy and Experimental Design
Twelve field sessions were organized between May 2010 and November 2012 (Supplementary Table S1): One in May 2010, four in 2011 (June, August 12th, August 24th and October), and seven in 2012 (March, May, June, July, August, September, and November).
For each field session, three peat profiles (replicates) were analyzed in the fen and the bog. In order to avoid disturbance during the extraction of peat and water, sampling for viral counts and metagenomes followed a 3-step progressive cutting from surface to depth. The 0-5 cm layer corresponding to the living Sphagnum carpet (capitulum and “green” stems and leaves) was cut off and carefully removed. The 5-10 cm layer (called “upper surface layer” thereafter) was then cut off and a fraction of peat matrix was harvested and stored in a 50 mL tube for metagenome production. From the remaining undisturbed peat, pore-water was expressed for viral and prokaryote counts. The same method was applied to the 10-15 cm layer (called “lower surface layer” thereafter). For metaviromes production as well as for physico-chemical analysis pore-water was expressed from the remaining peat surface layers combined. At least 200 mL of pore-water were necessary for metavirome production.
Prokaryote and Viral-Like Particle Abundances
For each layer of each sampling point, at least 20 mL of peat water was extracted, prefiltered at 125 μm, and fixed in Glutaraldehyde (2%, Grade II, Sigma-Aldrich). Prokaryote abundance (PA) and viral particle abundance (VPA) were obtained using flow cytometry (BD Accuri C6) (Sime-Ngando et al., 2008).
Physico-Chemical Parameters
Peat temperature was measured directly in the peat profiles at 5, 10, and 15 cm under the Sphagnum capitula layer. Conductivity, pH and oxygen were measured in the field with filtered (125 μm) peat water. Dissolved organic carbon (DOC) and anion concentrations (nitrate, sulfate, and acetate) were measured at the “Biogeochemical Analysis” platform (ECOBIO and GEOSCIENCES - OSU Rennes), following water filtration at 0.2 μm (Whatman), using a Bioritech DOC Analyser and a Dionex Analyzer (Table 1).
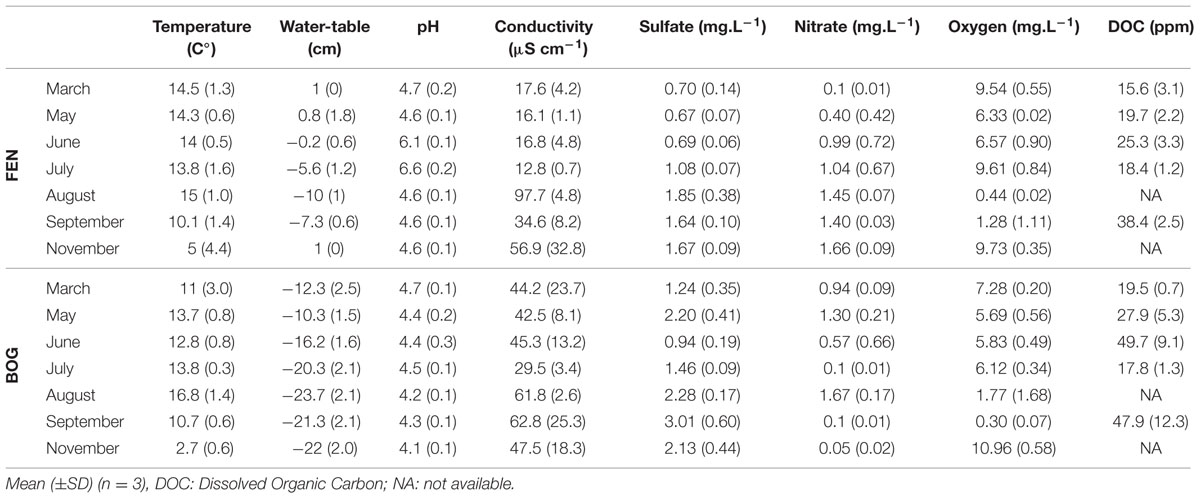
TABLE 1. Physico-chemical parameters measured in the peat samples from 2012.
Metavirome and Metagenome Production
To get enough material fen and bog viral communities were sampled from pore water combining the upper and lower surface layers. Samples were recovered at four dates (Supplementary Table S1, Table 2): 07 June 2011 (vFen_June11, vBog_June11; along with peat for a study of the microbial communities), 12 August 2011 (vFen_Aug11, vBog_Aug11), 12 October 2011 (vFen_Oct11, vBog_Oct11), and 23 March 2012 (vFen_Mar12, vBog_Mar12). Due to low water content of the peat, only one sample was collected for each dynamic stage for the three first dates. In March 2012, the fen and bog were sampled in biological triplicates (vFen_Mar12_A, B, C; vBog_Mar12_A, B, C). In total we prepared six fen and six bog metaviromes. Sphagnum water was prefiltered at 125 μm. Viruses were concentrated using PEGylation (Colombet et al., 2007). Viral concentrates were filtered through a 0.20 μm filter (Minisart, Sartorius) and diluted 5x in H2O (Sigma) to a volume of 5 mL. Remaining non-viral DNA was digested with 10 U DNAse RQ1 RNAse free (Promega) at 37°C for 1 h. Viral DNA was extracted as described previously (Quaiser et al., 2015). DNA quality was checked with the High Sensitivity DNA kit on a 2100 Bioanalyzer (Agilent). Whole genome amplification (WGA) was applied in triplicate for each sample using GenomiPhi V2 (GE Healthcare) following manufacturers’ instructions and the triplicates were pooled.
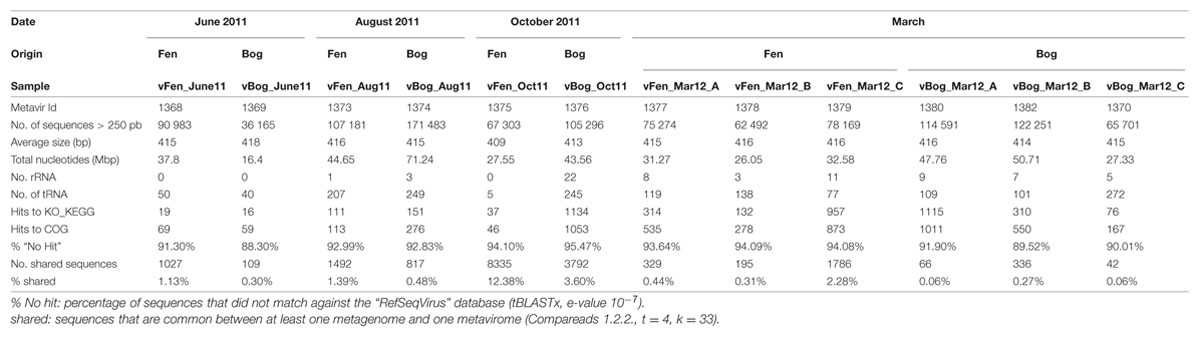
TABLE 2. Main characteristics of the 12 fen and bog metaviromes.
Subsequent pyrosequencing was performed on a GS FLX system (454 Life Sciences, Roche, Branford, CT, USA) at the “Functional and Environmental Genomics” platform (OSU, Rennes, France). Roche/454 filtering tools and stringent filters were used to ensure the highest sequence quality and to remove artificial replicates of sequences and sequences smaller than 250 bp as shown previously (Quaiser et al., 2014).
For the metagenomes, 12 un-pressed peat matrix samples, distinguishing bog/fen, upper and lower surface layers, each in triplicates, collected on 07 June 2011, underwent DNA extraction, pyrosequencing as well as size and quality trimming (Supplementary Table S2) as described previously (Quaiser et al., 2014). Briefly, 2 g of peat matrix were lysed in 15ml of extraction buffer containing 4% cetyltrimethylam- monium bromide (CTAB), 0.5% polyvinylpyrrolidone (PVP, Sigma-Aldrich), 0.7 M NaCl, 100 mM potassium phosphate (pH 6.8), 20 mM EDTA (pH 8.0), 1% beta-mercaptoethanol, 1 M guanidin thiocyanate and incubated at 65°C for 30 min. Homogenization was obtained by vigorous vortexing every 5 min during 1 min in the presence of glass beads. One volume of chloroform-isoamylalcohol (24:1) was added, vortexed for 1 min and incubated at room temperature for 5 min. The samples were centrifuged at 4000 g for 15 min at 4°C and the aqueous phases were transferred to new tubes. Binding conditions for silica-based RNA extraction were adjusted, applied on Nucleo Spin RNA II kit columns and subsequent purification was performed following the instructions of the manufacturer (Macherey-Nagel). DNA was nebulized to fragments of about 700 bp. The DNA was purified with Agencourt AMPur XP magnetic beads (Beckman-Coulter). DNA fragmentation quality was checked with the High sensitivity DNA kit on a 2100 Bioanalyzer (Agilent). Subsequent library construction and pyrosequencing was performed in technical duplicates on a GS FLX system (454/Roche) at the “Functional and Environmental Genomics” platform (OSU, Rennes, France). Roche/454 filtering tools and stringent filters developed locally were used to ensure the highest possible sequence quality and to suppress artificial replicates of sequences as well as sequences smaller than 250 bp.
The metaviromes are available under the Metavir IDs (http://metavir-meb.univ-bpclermont.fr/): 1368, 1369, 1370, 1373, 1374, 1375, 1376, 1377, 1378, 1379, 1380, and 1382 (project: VIRTOU). In addition, pyrosequencing reads reported in this publication have been deposited in the ENA Sequence Read Archive under the study accession number PRJEB11420 (metaviromes) and PRJEB11421 (metagenomes).
Metavirome Analysis
After pyrosequencing, sequence quality and size trimming, we obtained 481 402 and 615 487 sequences with an average length of 415 bp from fen and bog, respectively (Table 2). The quality of the virome extraction process was assessed by determining the amount of rRNA and tRNA sequences using Meta_RNA that identifies SSU and LSU rRNAs from the three kingdoms (Huang et al., 2009) and tRNAscan-SE (Lowe and Eddy, 1997). In total 69 rRNA (0.0063%) and 1681 tRNA (0.15%) sequences were identified indicating an insignificant level of potential contamination of microbial DNA. To avoid misinterpretation of the results, these sequences were excluded in subsequent analysis.
The viral diversity was analyzed using Metavir (Roux et al., 2014) and the sequences were subjected to tBLASTx (Altschul et al., 1997) against the NCBI RefSeqVirus database (e-value 10-7). Taxonomic assignment of the sequences was determined with MEGAN (Huson et al., 2007). Several accompanying tools were used on the Galaxy/Genouest bioinformatics platform (Le Bras et al., 2013).
To estimate the level of similarity between the viral communities, the proportion of similar sequences of each pair of metaviromes was computed with Compareads 1.2.2 (Maillet et al., 2012). Using this software, two sequences are considered to be similar if they present a defined number (t) of identical k-mers (k). To calibrate this analysis we tested 3 different numbers (t = 2; 4; 10) of identical 33mers (k = 33 nt). The most reliable results were obtained using four identical 33mers, parameters that were used for further analyses computed with Compareads. Compareads output is a percentage of similarity between a pair of metaviromes. These percentages were used to build a distance matrix, on which hierarchical clustering was performed using the R package pvclust package (Suzuki and Shimodaira, 2006) (distance = “correlation”, method = “average”).
Comparisons made with Compareads give a global estimate of the similarity between the metaviromes. In order to take into account the diversity of sequences within each metavirome, we analyzed the qualitative distribution (presence-absence) of clusters of highly similar sequences in the 12 metaviromes. This second analysis also allowed removal of potential bias due to the variation of the number of sequences obtained for each metavirome. Sequences from the 12 metaviromes were clustered using CD-HIT-EST (Huang et al., 2010) (c = 0.95; n = 8). Clustering results were used to compute Sørensen dissimilarity between pairs of metaviromes using MOTHUR (Schloss et al., 2009). Hierarchical clustering (pv-clust package) was used to represent compositional relatedness between metaviromes from the matrix of Sørensen dissimilarities. Clusters of sequences were split into different categories according to the amount of sequences they contained and the same analysis was performed for each size category.
Analysis of the Sequences Shared by a Metagenome and a Metavirome
In order to find viral sequences in the metagenomes, and to analyze the link between the viral and microbial communities, sequences shared by at least one metagenome and one metavirome were retrieved using Compareads (k = 33, t = 4), and clustered using CD-HIT-EST (see “Metaviromes Analysis”). To ensure that this selection of sequences did not alter the compositional patterns observed for the total metaviromes, Sørensen dissimilarity was calculated with these clusters for the “shared” sequences originating from the metaviromes. Correspondence Analysis (CA) was performed on the whole dataset of shared sequences and the sample dissimilarities carried on the two first axes was represented with a hierarchical clustering (ade4 package) (Dray and Dufour, 2007). Taxonomic assignment of the sequences was obtained using Metavir tBLASTx output (e-value 10-7) (Roux et al., 2014), and analyzed with MEGAN (Huson et al., 2007).
Statistical Analysis
Due to non-homogeneity of variance, one-factor Kruskal-Wallis tests were used on PA, VPA, and VPR in order to detect differences between sites (fen vs. bog), layers (upper vs. lower) and sampling dates. A principal component analysis (PCA) was performed on the physico-chemical dataset taking into account samples with available DOC (ade4 package) (Dray and Dufour, 2007). The first component was associated with the fluctuation of the physico-chemical variables through the habitats and seasons. We used the sample coordinates on the first component as a variable representing the spatio-temporal gradient (Legendre and Legendre, 1998; Ramette, 2007). Then potential relations between the gradient and log transformed PA, VPA, and VPR were tested using linear regression. All statistical analyses were performed using the open-source statistical software R (version 2.14) (R Development Core Team, 2013).
Results
Viral and Prokaryotic Bundance
Viral particle abundance and PA were investigated for the two peatland development stages over 2 years (Figure 1) aiming for the detection of spatial trends in the abundance of biological entities. VPA ranged from 1.7 ± 0.9 × 106 (fen upper layer, July 2012) to 5.6 ± 2.1 x 108 particles mL-1 (bog lower layer, September 2012), and PA ranged from 2.8 ± 1.2 × 106 (fen upper layer, July 2012) to 6.3 ± 1.3 x 108 cells mL-1 (fen lower layer, May 2010). VPA and PA were significantly correlated (Spearman, r = 0.76; P < 10-15; N = 95). We did not observe significant differences in PA and VPA between fen and bog; however, we detected significant variations with time (PA: KW-test, P = 1.8 × 10-5, N = 95; VPA: KW-test, P = 1.1 × 10-8, N = 95). While PA was significantly higher at the lower layer (average abundance of 9.5 ± 7.1 × 107 cells mL-1 at the upper layer and 1.9 ± 1.6 × 108 cells mL-1 at the lower layer) (KW-test, P < 0.05, N = 95), VPA did not differ significantly with depth (KW-test, P = 0.056, N = 95). Nevertheless, VPA followed a similar trend, with higher average abundances at lower layer compared with the upper layer (1.8 ± 1.6 × 108 particles mL-1 vs0 1.2 ± 1.1 × 108 particles mL-1, respectively). The virus to prokaryote ratio (VPR) differed by sampling date (Figure 2) (KW-test, P < 0.01, N = 95). No significant differences were observed between development stages or sampling depths. The highest VPR, measured in June 2011 was due to low PA rather than to high VPA.
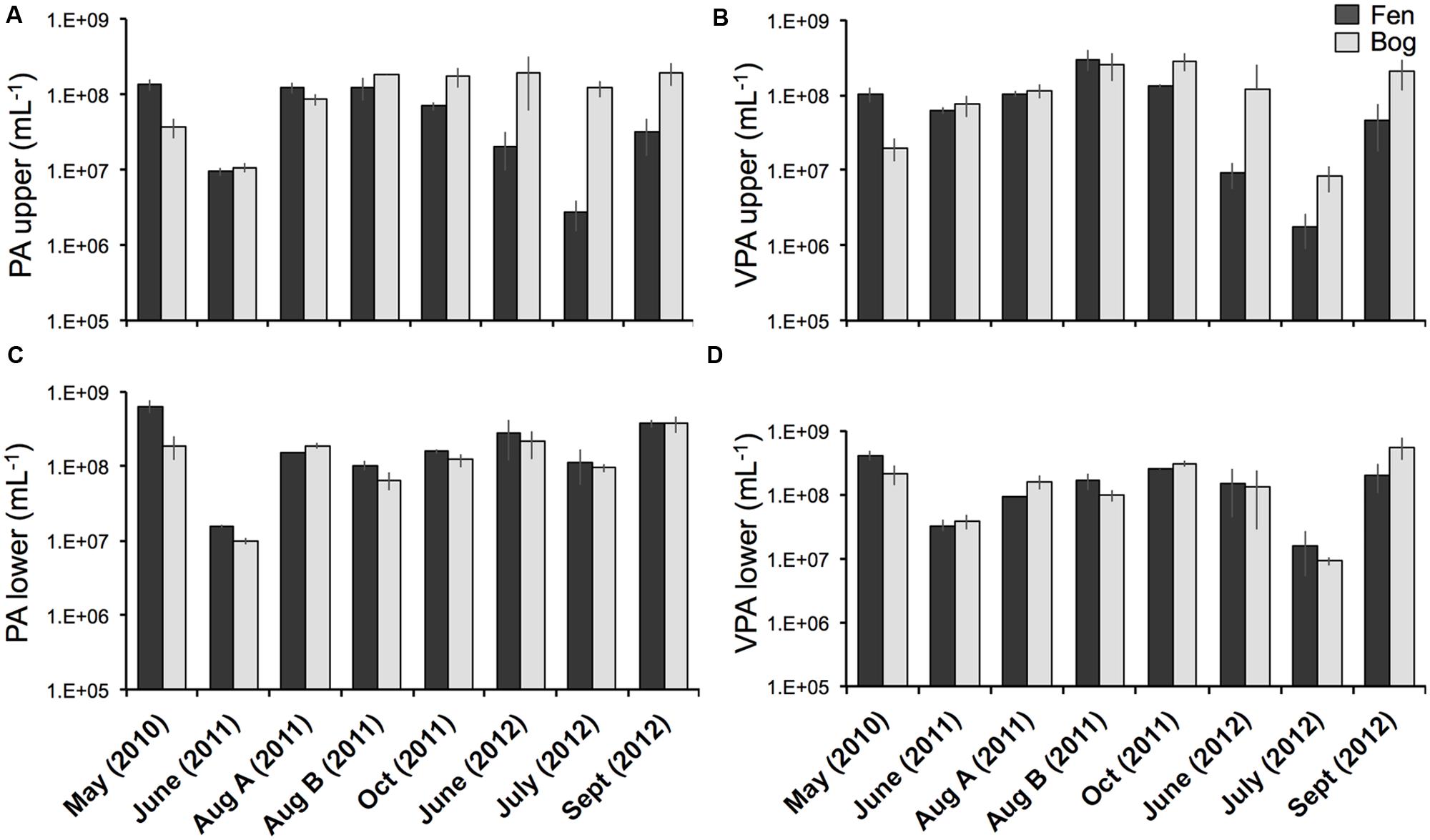
FIGURE 1. Prokaryote abundance (PA) and viral particle abundance (VPA) from the fen and bog of the Sphagnum-dominated peatland. (A) PA at the upper layer (5-10 cm), (B) PA at the lower layer (10-15 cm), (C) VPA at the upper layer, (D) VPA at the lower layer. Bars represent standard deviations (N = 3).
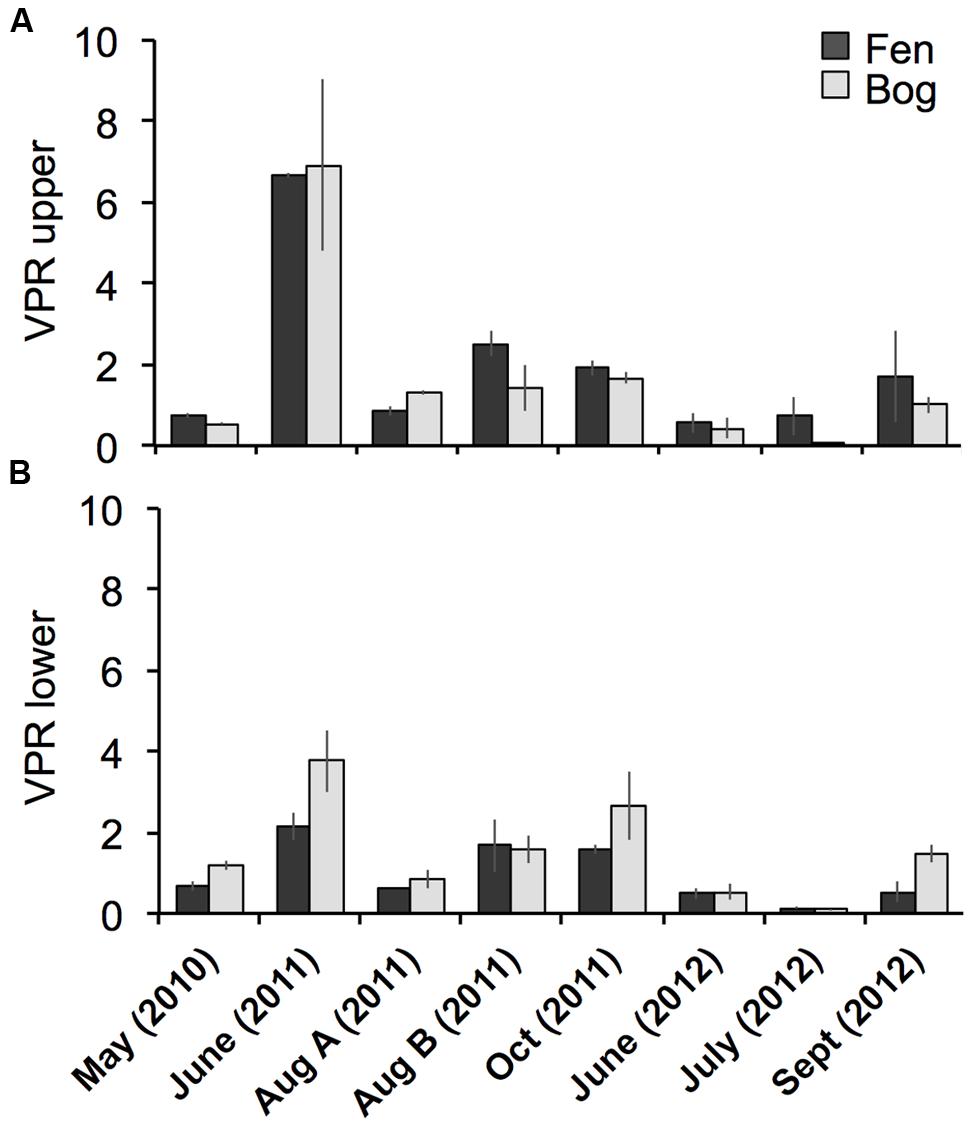
FIGURE 2. Comparison of the virus to prokaryote ratio (VPR) of the Sphagnum–dominated peatland. Bars represent standard deviations (N = 3).
Link Between Abiotic Variables, VPA and PA
The potential relationships between abiotic and biotic environment and virus communities were analyzed combining fluctuation of physico-chemical variables with viral and prokaryote abundance. The annual mean water-table level was -2.9 ± 4.5 cm in the fen and -18.0 ± 5.0 cm in the bog. Temperature varied more at the upper layer of the bog (annual mean: 11.6 ± 4.4°C) than at the upper layer of the fen (12.4 ± 3.8 °C), but was lower at the lower layer of both stages (fen: 9.3 ± 3.5°C; bog: 9.2 ± 3.5°C) (Table 1). We characterized the variation of abiotic physico-chemical parameters with a (PCA) (Figure 3). The first component of the PCA accounted for 49.8% of the variance, and was positively correlated with conductivity, sulfate (SO42-) and DOC and negatively correlated with water-table level and oxygen (O2). The second component accounted for 16.4% of the variance and was mainly correlated with nitrate concentration, which was higher in the fen in June, July, and September. The distribution of data points across the first two components emphasized the differences between fen and bog but also highlighted similar temporal trends within the two development stages, distinguishing the March and May samples from September. The seasonal fluctuations of the water-table were closely linked with variation in water-chemistry, suggesting that water sources and flowpaths affect nutrient concentrations, potentially due to dilution of a limited solute stock (for example, Spearman’s rank correlations; water-table to conductivity: R = -0.67, P = 6 × 10-5, N = 29; water-table to sulfate: R = -0.66, P = 9 × 10-5, N = 29) (Table 1; Figure 3). Because the first component represents an integrated variable of spatio-temporal variations of the physico-chemical and hydrological parameters, we used sample coordinates on this axis to model the seasonal abiotic fluctuations for both fen and bog. Log-transformed PA and VPA were both positively, linearly correlated with this abiotic gradient (N = 29, PA: r2= 0.53, P < 10-5; VPA: r2= 0.41, P < 10-3; Supplementary Figure S1), whereas VPR was unrelated to this gradient (r2= -0.03, P = 0.68).
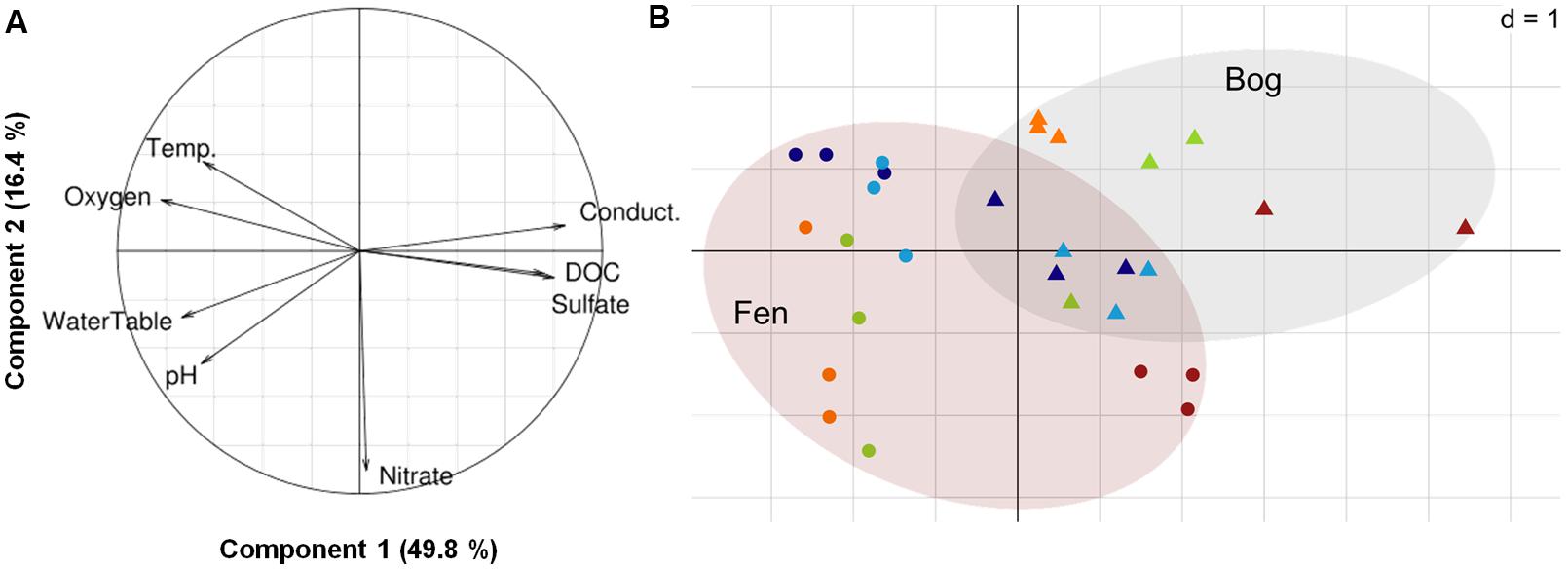
FIGURE 3. Principal Component Analysis on the physico-chemical dataset. (A) Variance explained per component, and correlation circle. (B) Projection of the samples. Dot shape represents the habitat (circle: fen, triangle: bog). Dot colors represent the sampling date (dark blue: March 2012, light blue: May 2012, light green: June 2012, orange: July 2012, red: September 2012).
Viral Community Composition and Diversity
Pyrosequencing of the 12 metaviromes yielded 1 096 889 sequences with an average length of 415 bp (Table 2). Analysis revealed that sequences of ribosomal RNA genes accounted for 0.0063% (69 rRNA sequences), which were excluded from subsequent analysis. In addition, the predicted protein coding sequences that matched the functional category databases (KEGG) and the cluster of orthologous genes database (COG) accounted for only 0.38 and 0.45%, respectively. This corresponds to 10-50 times fewer matches than are typically found in conventional short read metagenomes (Table 2; Quaiser et al., 2011), indicating a very low level of contamination by genomic DNA from microorganisms (Roux et al., 2013b). This allows the precise characterization of viral diversity and variation in fen and bog through the year.
Taxonomic Composition
Sequences were compared against viral genomes from the NCBI RefSeqVirus database. Only a small proportion of sequences, ranging from 4.2% (vBog_Oct11, v = virus/Fen or Bog/sampling date) to 10.9% (vBog_June11) matched the available viral genomes indicating the presence of currently undetected viruses (Table 2; Figure 4). Matches associated with ssDNA viruses were most common, accounting for a mean of 4.5% of the total number of sequences, with primary assignment to the bacteriophage family Microviridae (1.7 ± 1.3%) and to the eukaryal ssDNA family Circoviridae (0.9 ± 0.7%). Matches with dsDNA viruses appeared mostly affiliated with the order of Caudovirales (1.3 ± 0.8%), which can be hosted by both Bacteria and Archaea. While the protocol was not designed to preserve RNA viruses, we detected sequences matching to ssRNA viruses affiliated with Tombusviridae and Sclerophtora macrospora virus A representing likely the recently identified so called “chimeric viruses” (Diemer and Stedman, 2012; Roux et al., 2013a). They were present in all samples and accounted for 0.1% (vFen_Aug11) to up to 2.8% (vBog_Aug11) of the total metavirome sequences (Figure 4). Nevertheless, the interpretation of these results must be considered with respect to the applied multiple displacement amplification, that was shown to be bias prone towards ssDNA viruses (Kim and Bae, 2011). Due to potential biases no statistical analyses were performed on the proportions of the viral types.
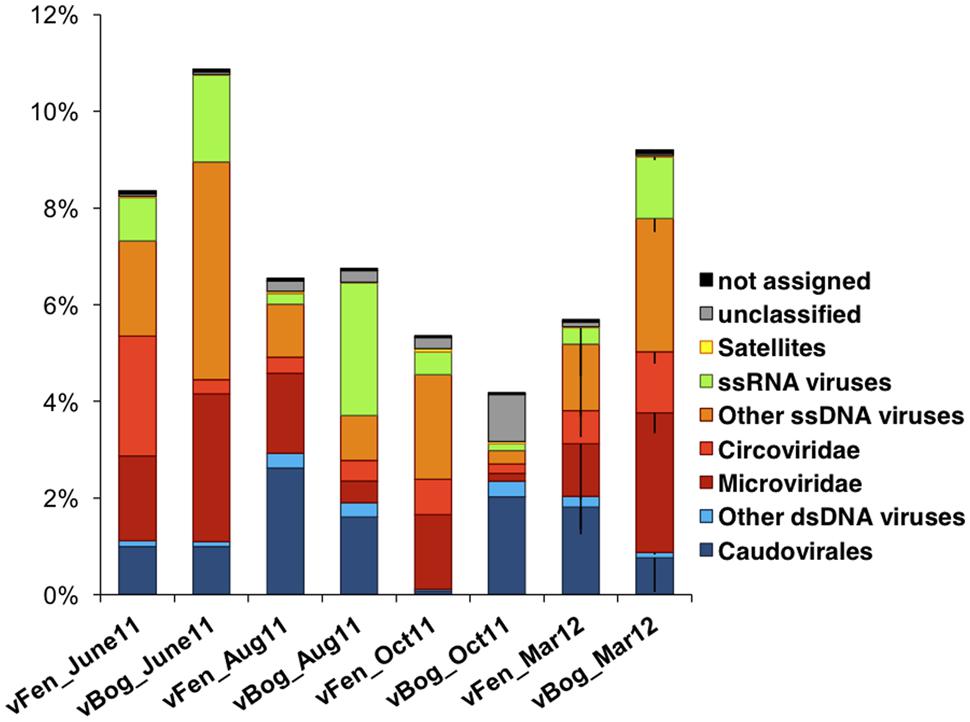
FIGURE 4. Relative abundances of the different viral types in the six fen and six bog metaviromes. Taxonomic affiliations were obtained by tBLASTx against RefSeqVirus (e-value 10-7). Relative abundances were averaged for March fen and bog metavirome triplicates. Error bars are shown only for negative values and represent standard deviation (N = 3).
Genomic Diversity Based Comparisons of the Metaviromes
Due to the lack of virus reference sequences in the databases the majority of sequences (88.3 to 95.4%) remained unaffiliated to known viral taxa. To characterize the remaining, unidentified metaviromes sequences, we analyzed the proportion of similar sequences (four identical kmers of 33 bp) between each pair of metaviromes with Compareads (Maillet et al., 2012). The dendrogram built from the similarity matrix showed two well-separated clusters (Supplementary Figure S2A). One group was composed of metaviromes collected during summer and autumn 2011 (vBog_Aug2011, vFen_Oct11, vBog_Oct2011), while the second group included the communities sampled in winter 2012 (March 2012) and spring 2011 (June 2011) regardless of the peatland development stage.
Sequence comparisons with Compareads provide a global estimate of the proportion of similar sequences without taking into account the internal structure of the sequence sets. Therefore we clustered sequences with a 95% identity threshold to assess the diversity of protein-coding gene sequences and to determine which groups of sequences drive the similarity between metaviromes in the Compareads analysis. Sørensen dissimilarity was calculated for every pair of metaviromes. This distance is based on the distribution (presence-absence) of sequences from the different clusters in the 12 metaviromes. Sørensen dissimilarities between pairwise metaviromes were uncorrelated with the amount of sequences in pairwise comparisons (Spearman’s rank correlation, R = 0.08, P = 0.495, N = 66). The pattern of similarity between the metaviromes supported the first analysis done with Compareads (Supplementary Figure S2B), with nearly identical grouping of summer and autumn communities and winter and spring communities.
Unique sequences represented 28% of the total metaviromes and 49% of the sequences belonged to clusters consisting of 10 to more than 1 000 sequences. We split clusters of highly similar sequences into different size categories, depending on the number of sequences they included (Figure 5) and computed hierarchical clustering based on Sørensen dissimilarities for each size category. Dendrograms indicated the same contrasted pattern between March viral communities and the summer and autumn group of metaviromes to the exception of vFen_Aug11. This distinction was not significant for clusters smaller than five sequences. This suggests a fundamental change in viral community between the two main groups of metaviromes with low intergroup Sørensen similarity (Supplementary Figure S3). Resemblance between winter and spring metaviromes (Fen and Bog from June 2011 and March 2012) was only significant for clusters larger than 250 sequences. Thus, the resemblance between June and March metaviromes appears to be due to a small number of large clusters.
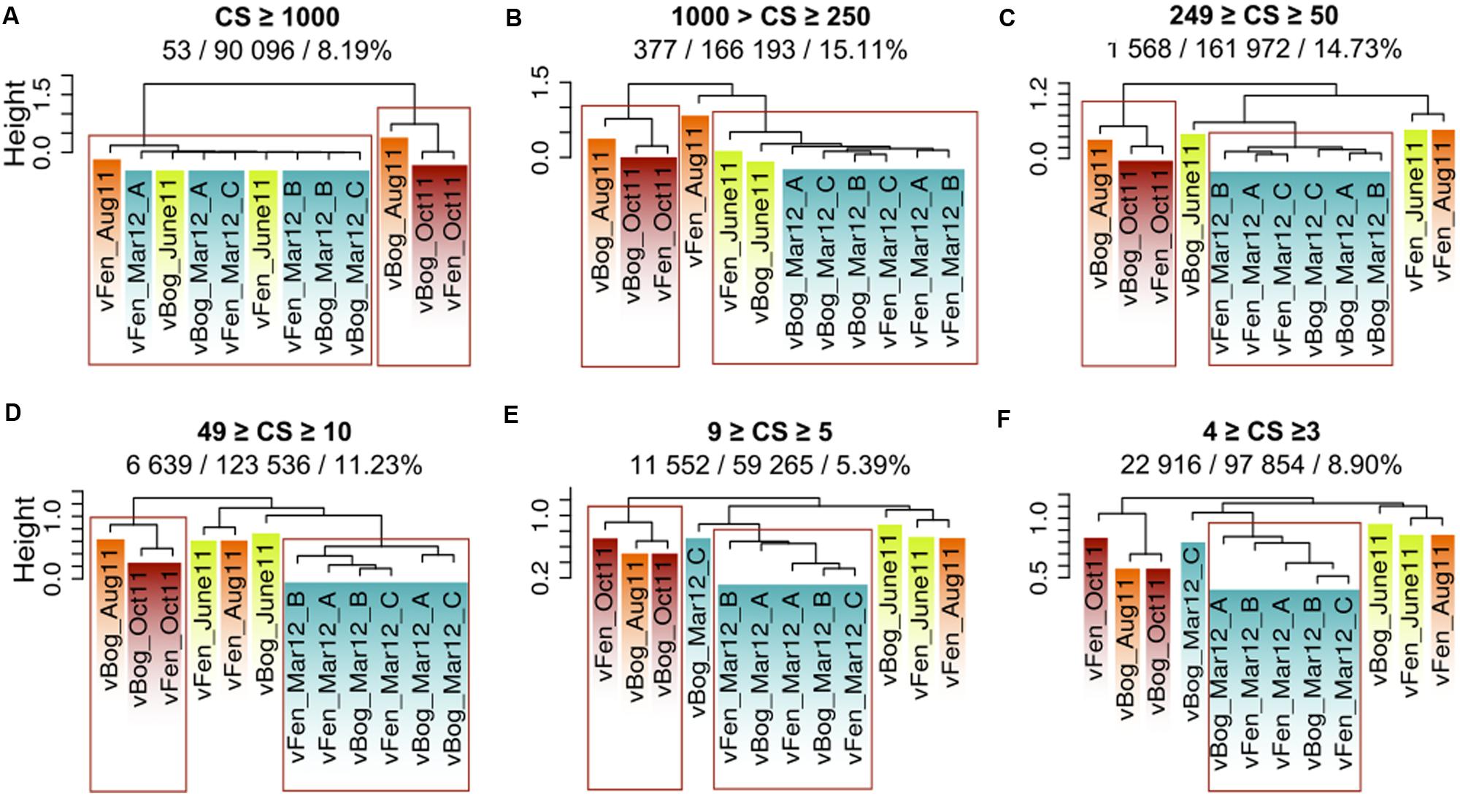
FIGURE 5. Metaviromes similarities based on Sørensen Index and on the number of sequences gathered in the clusters. (A-F) represent different size categories, ranging from the category of clusters that gathered more than 1000 sequences (A) to the category of clusters represented by three or four sequences (F). Abbreviations: CS: Cluster Size. Numbers below the class size: No. of clusters represented in the size category / No. of sequences represented in the size category/% of the total metaviromes sequences. Red rectangles represent the most robust clusters (pvclust. au = 0.05).
To analyze the genetic similarities with other metaviromes, we compared the 12 peatland metaviromes with 49 available metaviromes from eight different ecosystem types by hierarchical clustering and tBLASTx (Supplementary Figure S4, see Materials and Methods). Peatland viruses formed a distinct group, clearly separated even from geographically close viral communities originating from freshwater lakes, indicating that these metaviromes represent a unique community characteristic of and structured by its ecosystem (Roux et al., 2012).
Link Between Viral and Microbial Communities
To investigate the interactions between viruses and the microbial communities in Sphagnum-dominated peatlands, we sequenced 12 metagenomes from the fen and bog prokaryotic communities (Supplementary Table S2) from the same day and site as the metaviromes vFen_June11 and vBog_June11 (Supplementary Table S1). Metagenome DNA was extracted from the peat matrix allowing finer spatial sampling. In addition, the peat matrix contained the peat pore-water from which the viral particles were sampled. Based on taxonomic affiliations fen and bog prokaryotic communities (hereafter called pFen and pBog) appeared to be predominantly composed of the same main phyla (Supplementary Figure S5). However, regardless of depth, fen and bog appeared to harbor prokaryotic communities with distinct structures as shown by non-metric multidimensional scaling ordination of euclidean distances between metagenomes (Supplementary Figure S6) and with the analysis of similarity (ANOSIM, Euclidean distance, R = 0.68, P < 0.01). In order to identify viral signatures in these metagenomes, we identified sequences shared by metagenomes and metaviromes using Compareads (four identical kmers of 33 bp). We obtained 18,676 “shared” sequences, of which 30 were from bog metagenomes (pBog), 320 were from fen metagenomes (pFen) (Supplementary Table S2), and 18,326 from the 12 metaviromes (Table 2). In most metaviromes, the number of “shared” sequences represented less than 1% of the total sequences but reached up to 12% in vFen_Oct11 (Table 2). We clustered sequences with a 95% identity threshold, built a contingency matrix based on the number of sequences from each sample in the different clusters and performed a CA on this contingency matrix. The first two axes of the CA contained 21% of the total information. Hierarchical clustering based on the first two components revealed three groups of samples (Figure 6). Two groups included metagenomes sampled in June 2011 from fen and bog, respectively (pFen and pBog_ samples) as well as metaviromes from August and October 2011. The third group contained the metaviromes from June 2011 and March 2012. Thus metaviromes from June 2011 did not cluster with the metagenomes sampled on the same day. Clustering and Sørensen dissimilarity analysis based solely on the subset of metavirome sequences shared with metagenomes revealed the same summer and autumn and winter and spring groupings as obtained for complete metaviromes (Supplementary Figure S7). Among these 18 676 “shared” sequences, a total of 774 sequences (4%) were assigned to references in RefSeqVirus (tBLASTx, e-value 10-7) indicating that the vast majority (96%) originate from currently unidentified viruses. Most hits were associated with the Microviridae subfamily Gokushovirinae (ssDNA viruses, 409 hits) and to a lesser extend with the ssDNA viruses Circoviridae (173 hits), Caudovirales (dsDNA viruses, 134 hits), and Sclerophtora macrospora virus A (ssRNA viruses, 19 hits).
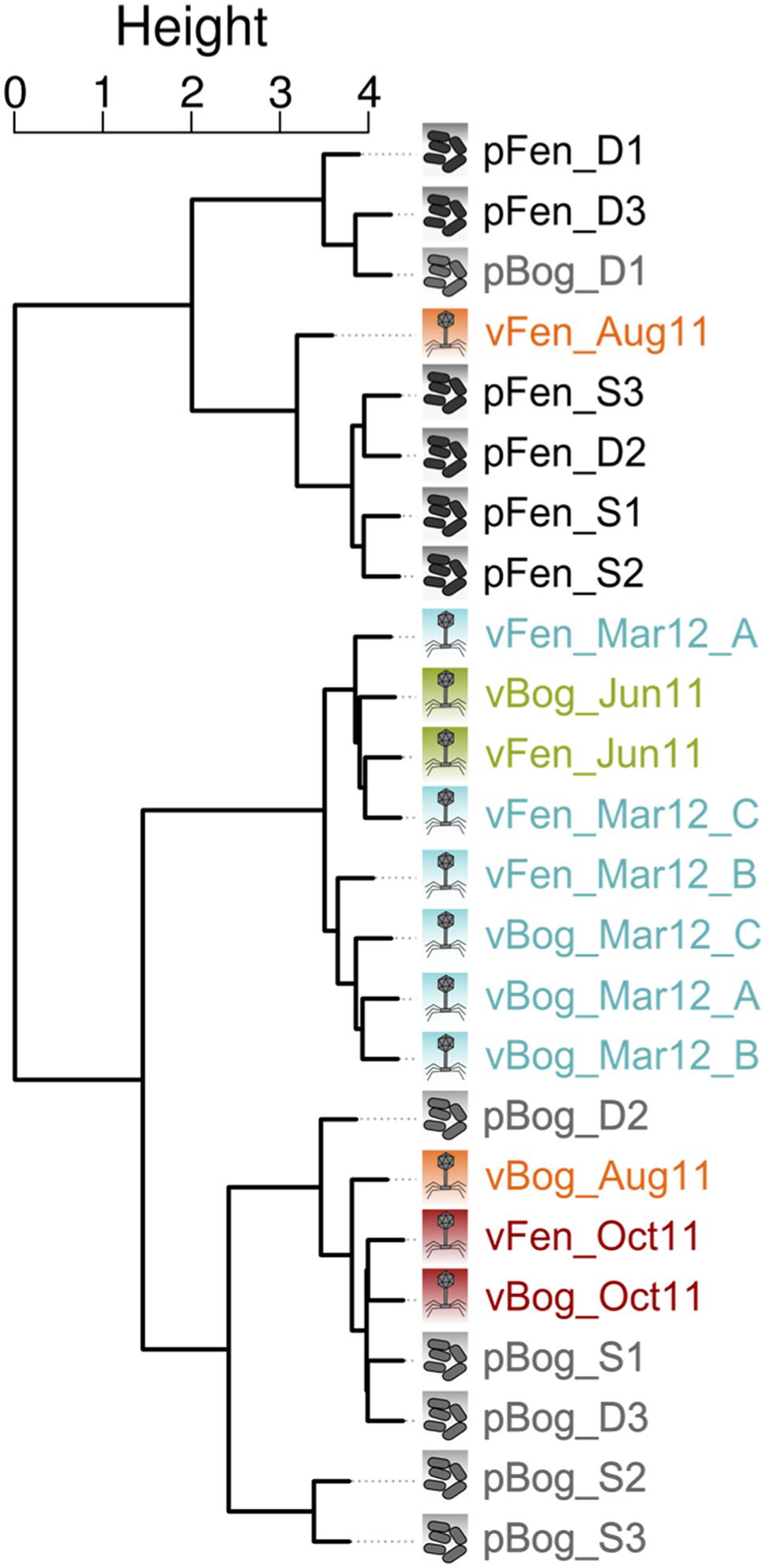
FIGURE 6. Hierarchical clustering representing metagenome and metavirome similarity based on sequences shared by at least one metagenome and one metavirome. Similarities were obtained using Compareads (k = 33; t = 4). Shared sequences have been clustered using CD-HIT-EST, and a Correspondence Analysis (CA) was performed on the clusters. Hierarchical clustering is based on the two first axes of the CA. Abbreviations: pFen/pBog: metagenome from fen/bog sampled in June 2011; S/D: upper/lower; 1,2,3: metagenome replicates; vFen/vBog: metavirome from fen/bog; (A,B,C): metavirome replicates.
Discussion
Quality of the Metaviromes
To explore the diversity and potential ecological role of viruses in Sphagnum-dominated peatland, we analyzed and compared six fen and six bog metaviromes covering the seasonal periods. The quality of the viromes is essential for comprehensive analysis, since contaminations with microbial genomic DNA would falsify the results. To assure that microbial DNA contamination was satisfactorily low, we applied the pegylation procedure to enrich viral particles (Colombet et al., 2007), DNAse treatment to degrade “free” DNA not protected by capsids, and triplicate whole genome amplification to balance potential amplification bias. The high quality of the virome sequences was shown by the very low abundance of rRNA sequences as well as the low number of matches to functional databases (i.e., KEGG_KO and COG). This is in accordance with the high diversity present in the viral genomic pool and with the high rates of evolutionary changes in viral genomes that are much less conserved than microbial genes (Duffy et al., 2008). Whole genome amplification, as applied here, is known to amplify preferentially circular ssDNA viruses (Kim and Bae, 2011), a bias that cannot be prevented when pooling separate triplicate amplification (Marine et al., 2014). Therefore, the interpretation of the results must be considered with caution. Nevertheless, since all viromes were generated the same way, the inevitably introduced biases should be the same for all allowing reliable comparative analysis.
It has been hypothesized that viruses infecting eukaryotes might be more important in terrestrial ecosystems and wetlands, where protozoan and fungal biomass is higher, while bacteriophages dominate viral consortia in marine and freshwater ecosystems (Farnell-Jackson and Ward, 2003; Jackson and Jackson, 2008; Kimura et al., 2008). Concerning the peatland metaviromes, among the sequences that matched viral genome databases, similar proportions of sequences were assigned to eukaryotic viruses, such as Circoviridae and Sclerophtora macrospora virus A-like viruses, and to prokaryotic viruses, such as Caudovirales or Microviridae. However, due to the vast majority of sequences being unassigned and to the high variability of viral genes, it remains impossible to determine whether viral communities in peatlands are dominated by prokaryote or eukaryote infecting viruses.
Successional Patterns of Viral and Microbial Communities
Ecological integration of the viral compartment into ecosystem functioning is mainly obtained through approaches combining virome sequences and viral abundance analysis (Wommack et al., 2015). In order to characterize viral ecology of Sphagnum-dominated peatlands, we monitored seasonal abundance and diversity of viruses and prokaryotes at two different depths of fen and bog and attempted to identify whether these were correlated to abiotic factors. Fens and bogs are development stages of peatlands that differ fundamentally in vegetation (Sphagnum and vascular plants) and associated physico-chemistry (Rydin and Jeglum, 2006). Thus, as already observed for microbial diversity (Opelt et al., 2007; Bragina et al., 2012), we hypothesized that peatland development stage and associated Sphagnum habitat would be the major driver in the distribution of virus and prokaryote abundance and diversity. Our results confirm that the physico-chemical conditions and the structure of the prokaryotic communities differ between the 2 dynamic stages, but, surprisingly, we did not observe any significant difference in VPA, PA, and viral diversity between fen and bog. The viral communities showed no systematic spatial trend and high variability even within replicates.
While we did not detect any significant spatial differences, we observed a significant seasonal fluctuation of virus diversity and abundance. For both fen and bog, VPA and PA (log-transformed data) were strongly correlated with the seasonal fluctuations of water-table, DOC, conductivity and sulfate: i.e., VPA and PA were higher when water-table was low and DOC and sulfate were high (Supplementary Figure S1). DOC has been recognized as a key factor in the C-balance of Sphagnum-dominated peatlands (Billett et al., 2004) and its patterns are driven by both biological activity (microbial production and consumption, plant exudation) and abiotic variables such temperature, water-table level or acidity (Clark et al., 2009) with seasonal fluctuations as a consequence (Moore, 1987). While the pH is recognized as an integrated physico-chemical variable, we did not register strong influence on PA and VPA. The increased pH observed during June and July in the fen is potentially due to photosynthetic activity with strong assimilation of dissolved inorganic carbon by microalgae, which significantly develop in submerged Sphagnum-fen at the beginning of summer (Gilbert et al., 1998). As viral activity is dependent on bacterial production (Middelboe, 2000), the correlations between DOC and VPA and PA suggest a net production of DOC with increasing microbial activity including viral lysis. DOC concentrations also depend on temperature and water flows, which play a key role in the production and redistribution of carbon in the peat (Waddington and Roulet, 1997; Clark et al., 2009), the two factors interacting during drawdown and flooding periods. However, we did not evidence a clear combination of DOC and temperature. The fluctuations of the water-table depend largely on hydrologic inputs that are rainfall in the bog and mainly runoff and groundwater inflow in the fen, which occur at the yearly scale (seasons) and at a short-time scale (episodic events). Sulfate also interacts with DOC concentrations, especially during drought periods when significant production of sulfate may increase peat acidification and ionic strength (Clark et al., 2009). In our study, sulfate concentrations were significantly higher during water-table drawdowns and were positively linked to DOC. In addition, we could not show whether the production of sulfate was linked to nitrate through microbial sulfur oxidation as already demonstrated in peatlands (Burgin and Hamilton, 2008). Nitrate seems to play a key role only during summer in the fen (see component 2 of the ACP), a dynamic stage in which nitrogen mineralization significantly occurs at this season (Francez and Loiseau, 1999). Our results suggest that viruses and prokaryotes are more abundant at the lower surface layer regardless of peatland development stage. This is in accordance with a previous study concerning prokaryote abundance (Dedysh et al., 2006) and potentially due to more buffered temperature and water-table fluctuations at the lower layer providing more stable conditions. Altogether, these results indicate that seasonal changes in temperature and precipitations (allogenic variables) influence PA and VPA via water-table fluctuations and consequently nutrient concentrations with larger effects at the upper than at the lower layer.
Comparisons of metaviromes with metagenomes showed that temporal variations are more influential than differences in peatland habitat in structuring viral communities. There was a particularly substantial shift in sequence composition from spring to autumn with distinct patterns of composition and abundance, suggesting the existence of ecological succession of viral communities at the seasonal scale. This pattern appears to be consistent inter-annually, with metaviromes sampled about a year apart (June 2011 and March 2012) clustering together. These findings suggest, for the first time, that a cyclic succession in peatlands affects free-occurring viruses at the community level. Recent studies on marine ecosystems also described seasonal fluctuations of viral communities at the ocean surface (Chow and Fuhrman, 2012; Pagarete et al., 2013). In these studies, which focused on the diversity of a viral gene marker, seasonality was mainly characterized by fluctuations of dominant viral types while in our study, seasonality was associated with a general change in the composition of viral communities.
Viruses and Carbon Cycling in Sphagnum-Dominated Peatlands
Viruses are believed to be key components of the carbon cycle in many ecosystems, both altering carbon fluxes and contributing to C-redistribution through bacterial lysis (Fuhrman, 1999; Middelboe and Lyck, 2002; Ankrah et al., 2014). Despite recent analyses of peatland microbial food-webs (Lamentowicz et al., 2013), the significance of viruses in the functioning of Sphagnum-dominated peatlands remains unknown.
In viral ecology, VPR is generally considered as an indicator of the bacterial hosts metabolic state (Williamson et al., 2005; Kimura et al., 2008) because viral burst size, and thus viral abundance is positively correlated with microbial growth rate (Middelboe, 2000). In the studied peatland, the VPR was low and did not differ between fen and bog, despite the differences between dead organic matter produced in the two dynamic stages, in relation to the dominant Sphagnum species (Francez, 1995; Thormann et al., 2003). The low VPR compared with other ecosystems is likely due to lower metabolic activity of microorganisms, that is in accordance with the functioning of Sphagnum-dominated peatlands where decomposition is slowed down due to constraining conditions (Rydin and Jeglum, 2006; Artz, 2009) and the presence of a significant proportion of dormant cells in the community (Dedysh et al., 2006; Pankratov et al., 2011).
We detected a VPR peak in June 2011, just before a broad modification of viral community composition in fen and bog. Viruses interact with their hosts through at least two main strategies: the lytic and the lysogenic life cycles, the latter is believed to be favored when microbial activity is low (Danovaro et al., 2002; Payet and Suttle, 2013; Sime-Ngando, 2014). The change in the viral community composition in summer could result from a seasonal shift in the active part of the microbial community and related C-cycling processes via decomposition that show seasonal patterns (Basiliko et al., 2005; Sun et al., 2012). This illustrates a transition from lysogenic to lytic strategies of the viruses infecting the newly active prokaryotes. This hypothesis is supported by the low PA associated with the VPR peak in June 2011, which could result from virus-mediated bacterial lysis, and by the similarities between spring metagenomes (June) and summer and autumn viromes, suggesting the presence of prophages in the microbial genomes in June, that were later released and detected in the metaviromes in August and October.
Conclusion
We applied an integrated approach linking virome sequence analysis, viral particle and prokaryote abundance, physico-chemical parameters and metagenome-virome comparison to get insights into the ecological functioning of the viral community in peatlands. We found that viral community abundance and diversity in Sphagnum-dominated peatlands express an ecological succession, and that viruses, as well as their hosts, are strongly influenced by the temporal fluctuations at the peatland surface. The observed low VPR, compared with other ecosystems, is in accordance with slowed down decomposition processes in Sphagnum-dominated peatlands. The observed shift in viral diversity suggests a seasonal change of the microbial community and the associated switch of viral life-cycle strategies during summer and autumn highlighting the importance of virus-host interactions as they control the dynamics of microbial communities. These patterns may be related to changes in C-cycling processes but further studies are needed to strictly link microbial and viral diversity with C-transformations in the peat. These should focus on the seasonality of viruses that infect different kind of hosts (prokaryotes/eukaryotes) in order to identify the main factors driving this succession and changes in functioning processes, as already suggested for seasonal fluctuations of plankton (Sommer et al., 2012).
To assess the ecological relevance of the diversity of viral communities, analysis of the spatio-temporal dynamics of ecosystem-specific metaviromes as applied here, rather than cross-biomes comparisons, represent a powerful approach to overcome the lack of viral genomes in the databases, and to take advantage of the whole diversity carried in the sequenced viral communities.
Funding
This work was supported by funding from the INSU-CNRS EC2CO program to A-JF (“Virtou”) (Program MICROBIEN, 2011–2012) and grants from the University of Rennes/CNRS and from the “Observatoire des Sciences de l’Univers de Rennes”(OSUR) to AQ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the ‘Environmental and Functional Genomics Platform’ (Biogenouest Genomics) for sequencing and the molecular ecology facility (UMR 6553 ECOBIO) for support. We thank Benjamin W. Abbott for manuscript editing. We thank Patricia Madec, Nathalie Le Bris, and Sophie Coudouel (OSU Rennes) for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01494
References
Adriaenssens, E. M., Van Zyl, L., De Maayer, P., Rubagotti, E., Rybicki, E., Tuffin, M., et al. (2015). Metagenomic analysis of the viral community in Namib Desert hypoliths. Environ. Microbiol. 17, 480–495. doi: 10.1111/1462-2920.12528
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Andersen, R., Grasset, L., Thormann, M. N., Rochefort, L., and Francez, A. J. (2010). Changes in microbial community structure and function following Sphagnum peatland restoration. Soil Biol. Biochem. 42, 291–301. doi: 10.1016/j.soilbio.2009.11.006
Ankrah, N. Y. D., May, A. L., Middleton, J. L., Jones, D. R., Hadden, M. K., Gooding, J. R., et al. (2014). Phage infection of an environmentally relevant marine bacterium alters host metabolism and lysate composition. ISME J. 8, 1089–100. doi: 10.1038/ismej.2013.216
Artz, R. (2009). “Microbial community structure and carbon substrate use in northern peatlands,” in Carbon Cycling in Northern Peatlands, eds A. J. Baird, L. R. Belyea, X. Comas, A. S. Reeve, L. D. Slater (Washington, DC: American Geophysical Union), 111–129. doi: 10.1029/2008GM000806.
Artz, R., Anderson, I. C., Chapman, S. J., Hagn, A., Schloter, M., Potts, J. M., et al. (2007). Changes in fungal community composition in response to vegetational succession during the natural regeneration of cutover peatlands. Microb. Ecol. 54, 508–22. doi: 10.1007/s00248-007-9220-7
Basiliko, N., Moore, T. R., Lafleur, P. M., and Roulet, N. T. (2005). Seasonal and inter-annual decomposition, microbial biomass, and nitrogen dynamics in a canadian bog. Soil Sci. 170, 902–912. doi: 10.1097/01.ss.0000196765.59412.14
Billett, M. F., Palmer, S. M., Hope, D., Deacon, C., Storeton-West, R., Hargreaves, K. J., et al. (2004). Linking land-atmosphere-stream carbon fluxes in a lowland peatland system. Global Biogeochem. Cycles 18, GB1024. doi: 10.1029/2003GB002058
Bragina, A., Berg, C., Cardinale, M., Shcherbakov, A., Chebotar, V., and Berg, G. (2012). Sphagnum mosses harbour highly specific bacterial diversity during their whole lifecycle. ISME J. 6, 802–813. doi: 10.1038/ismej.2011.151
Burgin, A. J., and Hamilton, S. K. (2008). NO3 -Driven SO4 2- production in freshwater ecosystems: implications for N and S cycling. Ecosystems 11, 908–922. doi: 10.1007/s10021-008-9169-5
Chow, C.-E. T., and Fuhrman, J. A. (2012). Seasonality and monthly dynamics of marine myovirus communities. Environ. Microbiol. 14, 2171–2183. doi: 10.1111/j.1462-2920.2012.02744.x
Chow, C.-E. T., Kim, D. Y., Sachdeva, R., Caron, D. A., and Fuhrman, J. A. (2014). Top-down controls on bacterial community structure: microbial network analysis of bacteria, T4-like viruses and protists. ISME J. 8, 816–29. doi: 10.1038/ismej.2013.199
Clark, J. M., Ashley, D., Wagner, M., Chapman, P. J., Lane, S. N., Evans, C. D., et al. (2009). Increased temperature sensitivity of net DOC production from ombrotrophic peat due to water table draw-down. Glob. Chang. Biol. 15, 794–807. doi: 10.1111/j.1365-2486.2008.01683.x
Clymo, R. S. (1984). The limits to peat bog growth. Philos. Trans. R. Soc. B Biol. Sci. 303, 605–654. doi: 10.1098/rstb.1984.0002
Clymo, R. S., and Bryant, C. L. (2008). Diffusion and mass flow of dissolved carbon dioxide, methane, and dissolved organic carbon in a 7-m deep raised peat bog. Geochim. Cosmochim. Acta 72, 2048–2066. doi: 10.1016/j.gca.2008.01.032
Colombet, J., Robin, A., Lavie, L., Bettarel, Y., Cauchie, H. M. M., and Sime-Ngando, T. (2007). Virioplankton “pegylation”: use of PEG (polyethylene glycol) to concentrate and purify viruses in pelagic ecosystems. J. Microbiol. Methods 71, 212–219. doi: 10.1016/j.mimet.2007.08.012
Comeau, A. M., Hatfull, G. F., Krisch, H. M., Lindell, D., Mann, N. H., and Prangishvili, D. (2008). Exploring the prokaryotic virosphere. Res. Microbiol. 159, 306–313. doi: 10.1016/j.resmic.2008.05.001
Danovaro, R., Manini, E., and Dell’Anno, A. (2002). Higher abundance of bacteria than of viruses in deep Mediterranean sediments. Proc. Natl. Acad. Sci. U.S.A. 68, 1468–1472. doi: 10.1128/aem.68.3.1468-1472.2002
Dedysh, S. N. (2011). Cultivating uncultured bacteria from northern wetlands: knowledge gained and remaining gaps. Front. Microbiol. 2:184. doi: 10.3389/fmicb.2011.00184
Dedysh, S. N., Pankratov, T. A., Belova, S. E., Kulichevskaya, I. S., and Liesack, W. (2006). Phylogenetic analysis and in situ identification of bacteria community composition in an acidic sphagnum peat bog. Appl. Environ. Microbiol. 72, 2110–2117. doi: 10.1128/AEM.72.3.2110-2117.2006
Dell’Anno, A., Corinaldesi, C., and Danovaro, R. (2015). Virus decomposition provides an important contribution to benthic deep-sea ecosystem functioning. Proc. Natl. Acad. Sci. U.S.A. 112, E2014–E2019. doi: 10.1073/pnas.1422234112
Diemer, G. S., and Stedman, K. M. (2012). A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol. Dir. 7:13. doi: 10.1186/1745-6150-7-13
Dray, S., and Dufour, A. B. (2007). The ade4 Package: implementing the Duality Diagram for Ecologists. J. Stat. Softw. 22, 1–20.
Duffy, S., Shackelton, L. A., and Holmes, E. C. (2008). Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 9, 267–276. doi: 10.1038/nrg2323
Farnell-Jackson, E. A., and Ward, A. K. (2003). Seasonal patterns of viruses, bacteria and dissolved organic carbon in a riverine wetland. Freshw. Biol. 48, 841–851. doi: 10.1046/j.1365-2427.2003.01052.x
Fierer, N., Nemergut, D., Knight, R., and Craine, J. M. (2010). Changes through time: integrating microorganisms into the study of succession. Res. Microbiol. 161, 635–42. doi: 10.1016/j.resmic.2010.06.002
Francez, A.-J. (1995). Dynamique du carbone et de l’azote chez le Carex rostrata, l’ Eriophorum vaginatumet le Calluna vulgarisdans une tourbière à sphaignes des monts du Forez (France). Can. J. Bot. 73, 121–129. doi: 10.1139/b95-014
Francez, A.-J., and Loiseau, P. (1999). Devenir de l’azote mineral dans une tourbiere a Sphagnum fallax Klinggr. et Carex rostrata Stokes du Massif central (France). Can. J. Bot. 77, 1136–1143. doi: 10.1139/b99-100
Francez, A.-J., and Vasander, H. (1995). Peat accumulation and peat decomposition after human disturbance in French and finnish mires. Acta Oecol. 16, 599–608.
Fuhrman, J. A. (1999). Marine viruses and their biogeochemical and ecological effects. Nature 399, 541–548. doi: 10.1038/21119
Gilbert, D., Amblard, C., Bourdier, G., and Francez, A.-J. (1998). The microbial loop at the surface of a peatland:structure, function, and impact of nutrient input. Microb. Ecol. 35, 83–93. doi: 10.1007/s002489900062
Huang, Y., Gilna, P., and Li, W. (2009). Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25, 1338–1340. doi: 10.1093/bioinformatics/btp161.
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. doi: 10.1093/bioinformatics/btq003
Hultman, J., Waldrop, M. P., Mackelprang, R., David, M. M., McFarland, J., Blazewicz, S. J., et al. (2015). Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 521, 208–212. doi: 10.1038/nature14238
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Jackson, E. F., and Jackson, C. R. (2008). Viruses in wetland ecosystems. Freshw. Biol. 53, 1214–1227. doi: 10.1111/j.1365-2427.2007.01929.x
Juottonen, H., Galand, P. E., Tuittila, E.-S., Laine, J., Fritze, H., and Yrjälä, K. (2005). Methanogen communities and Bacteria along an ecohydrological gradient in a northern raised bog complex. Environ. Microbiol. 7, 1547–1557. doi: 10.1111/j.1462-2920.2005.00838.x
Kim, K.-H., and Bae, J.-W. (2011). Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 77, 7663–7668. doi: 10.1128/AEM.00289-11
Kimura, M., Jia, Z. J., Nakayama, N., and Asakawa, S. (2008). Ecology of viruses in soils: past, present and future perspectives. Soil Sci. Plant Nutr. 54, 1–32. doi: 10.1111/j.1747-0765.2007.00197.x
Labonté, J. M., and Suttle, C. A. (2013). Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 7, 2169–2177. doi: 10.1038/ismej.2013.110
Lamentowicz, M., Bragazza, L., Buttler, A., Jassey, V. E. J., and Mitchell, E. A. D. (2013). Seasonal patterns of testate amoeba diversity, community structure and species-environment relationships in four Sphagnum-dominated peatlands along a 1300m altitudinal gradient in Switzerland. Soil Biol. Biochem. 67, 1–11. doi: 10.1016/j.soilbio.2013.08.002
Le Bras, Y., Roult, A., Monjeaud, C., Bahin, M., and Quénez, O. (2013). Towards a Life Sciences Virtual Research Environment. e-biogenouest.org. Available at: https://www.e-biogenouest.org/resources/129/download/jobim_YLeBras_2013.pdf
Legendre, P., and Legendre, L. (1998). Numerical ecology. Numer. Ecol. Second English Ed. 20:870. doi: 10.1021/ic050220j
Lopez-Bueno, A., Tamames, J., Velazquez, D., Moya, A., Quesada, A., and Alcami, A. (2009). High diversity of the viral community from an antarctic lake. Science 8, 858–861. doi: 10.1126/science.1179287
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Lynch, J. M., Benedetti, A., Insam, H., Nuti, M. P., Smalla, K., Torsvik, V., et al. (2004). Microbial diversity in soil: ecological theories, the contribution of molecular techniques and the impact of transgenic plants and transgenic microorganisms. Biol. Fertil. Soils 40, 363–385. doi: 10.1007/s00374-004-k0784–789
Mackelprang, R., Waldrop, M. P., DeAngelis, K. M., David, M. M., Chavarria, K. L., Blazewicz, S. J., et al. (2011). Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480, 368–371. doi: 10.1038/nature10576
Maillet, N., Lemaitre, C., Chikhi, R., Lavenier, D., and Peterlongo, P. (2012). Compareads: comparing huge metagenomic experiments. BMC Bioinformatics 13:S10. doi: 10.1186/1471-2105-13-S19-S10
Marine, R., McCarren, C., Vorrasane, V., Nasko, D., Crowgey, E., Polson, S. W., et al. (2014). Caught in the middle with multiple displacement amplification: the myth of pooling for avoiding multiple displacement amplification bias in a metagenome. Microbiome 2:3. doi: 10.1186/2049-2618-2-3
Middelboe, M. (2000). Bacterial growth rate and marine virus-host dynamics. Microb. Ecol. 40, 114–124.
Middelboe, M., and Lyck, P. G. (2002). Regeneration of dissolved organic matter by viral lysis in marine microbial communities. Aquat. Microb. Ecol. 27, 187–194. doi: 10.3354/ame027187
Mondav, R., Woodcroft, B. J., Kim, E.-H., McCalley, C. K., Hodgkins, S. B., Crill, P. M., et al. (2014). Discovery of a novel methanogen prevalent in thawing permafrost. Nat. Commun. 5:3212. doi: 10.1038/ncomms4212
Moore, T. R. (1987). Patterns of dissolved organic matter in subarctic peatlands. Earth Surf. Process. Landforms 12, 387–397. doi: 10.1002/esp.3290120405
Nunes, F. L. D., Aquilina, L., de Ridder, J., Francez, A.-J., Quaiser, A., Caudal, J.-P., et al. (2015). Time-Scales of hydrological forcing on the geochemistry and bacterial community structure of temperate peat soils. Sci. Rep. 5:14612. doi: 10.1038/srep14612
Opelt, K., Berg, C., Schönmann, S., Eberl, L., and Berg, G. (2007). High specificity but contrasting biodiversity of Sphagnum-associated bacterial and plant communities in bog ecosystems independent of the geographical region. ISME J. 1, 502–516. doi: 10.1038/ismej.2007.58
Pagarete, A., Chow, C., Johannessen, T., Fuhrman, J. A., Thingstad, T. F., and Sandaa, R. A. (2013). Strong seasonality and interannual recurrence in marine myovirus communities. Appl. Environ. Microbiol. 79, 6253–6259. doi: 10.1128/AEM.01075–1013
Pankratov, T. A., Ivanova, A. O., Dedysh, S. N., and Liesack, W. (2011). Bacterial populations and environmental factors controlling cellulose degradation in an acidic Sphagnum peat. Environ. Microbiol. 13, 1800–1814. doi: 10.1111/j.1462-2920.2011.02491.x
Payet, J., and Suttle, C. A. (2013). To kill or not to kill: the balance between lytic and lysogenic viral infection is driven by trophic status. Limnol. Oceanogr. 58, 465–474. doi: 10.4319/lo.2013.58.2.0465
Peltoniemi, K., Fritze, H., and Laiho, R. (2009). Response of fungal and actinobacterial communities to water-level drawdown in boreal peatland sites. Soil Biol. Biochem. 41, 1902–1914. doi: 10.1016/j.soilbio.2009.06.018
Pradeep Ram, A. S., Rasconi, S., Jobard, M., Palesse, S., Colombet, J., and Sime-Ngando, T. (2011). High lytic infection rates but low abundances of prokaryote viruses in a humic lake (Vassiviere, Massif Central, France). Appl. Environ. Microbiol. 77, 5610–5618. doi: 10.1128/AEM.01370-10
Quaiser, A., Bodi, X., Dufresne, A., Naquin, D., Francez, A. J., Dheilly, A., et al. (2014). Unraveling the stratification of an iron-oxidizing microbial mat by metatranscriptomics. PLoS ONE 9:e102561. doi: 10.1371/journal.pone.0102561
Quaiser, A., Dufresne, A., Ballaud, F., Roux, S., Zivanovic, Y., Colombet, J., et al. (2015). Diversity and comparative genomics of Microviridae in Sphagnum- dominated peatlands. Front. Microbiol. 6:375. doi: 10.3389/fmicb.2015.00375
Quaiser, A., Zivanovic, Y., Moreira, D., and López-García, P. (2011). Comparative metagenomics of bathypelagic plankton and bottom sediment from the Sea of Marmara. ISME J. 5, 285–304. doi: 10.1038/ismej.2010.113
Ramette, A. (2007). Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 62, 142–160. doi: 10.1111/j.1574-6941.2007.00375.x
R Development Core Team (2013). R Development Core Team. R A Language Environment Statics Computer. Available at: http://www.r-project.org/
Rohwer, F., and Thurber, R. V. (2009). Viruses manipulate the marine environment. Nature 459, 207–212. doi: 10.1038/nature08060
Roulet, N. T., Lafleur, P. M., Richard, P. J. H., Moore, T. R., Humphreys, E. R., and Bubier, J. (2007). Contemporary carbon balance and late Holocene carbon accumulation in a northern peatland. Glob. Chang. Biol. 13, 397–411. doi: 10.1111/j.1365-2486.2006.01292.x
Roux, S., Enault, F., Bronner, G., Vaulot, D., Forterre, P., and Krupovic, M. (2013a). Chimeric viruses blur the borders between the major groups of eukaryotic single-stranded DNA viruses. Nat. Commun. 4:2700. doi: 10.1038/ncomms3700
Roux, S., Krupovic, M., Debroas, D., Forterre, P., and Enault, F. (2013b). Assessment of viral community functional potential from viral metagenomes may be hampered by contamination with cellular sequences. Open Biol. 3:130160. doi: 10.1098/rsob.130160
Roux, S., Enault, F., Robin, A., Ravet, V., Personnic, S., Theil, S., et al. (2012). Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLoS ONE 7:e33641. doi: 10.1371/journal.pone.0033641
Roux, S., Faubladier, M., Mahul, A., Paulhe, N., Bernard, A., Debroas, D., et al. (2011). Metavir: a web server dedicated to virome analysis. Bioinformatics 27, 3074–3075. doi: 10.1093/bioinformatics/btr519
Roux, S., Tournayre, J., Mahul, A., Debroas, D., and Enault, F. (2014). Metavir 2: new tools for viral metagenome comparison and assembled virome analysis. BMC Bioinformatics 15:76. doi: 10.1186/1471-2105-15–76
Rydin, H., and Jeglum, J. (2006). “The biology of peatlands,” in The Biology of Habitats (New York, NY: Oxford University Press), 343.
Sakowski, E. G., Munsell, E. V., Hyatt, M., Kress, W., Williamson, S. J., Nasko, D. J., et al. (2014). Ribonucleotide reductases reveal novel viral diversity and predict biological and ecological features of unknown marine viruses. Proc. Natl. Acad. Sci. U.S.A. 111, 15786–15791. doi: 10.1073/pnas.1401322111
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-9
Serkebaeva, Y. M., Kim, Y., Liesack, W., and Dedysh, S. N. (2013). Pyrosequencing-based assessment of the bacteria diversity in surface and subsurface peat layers of a northern wetland, with focus on poorly studied phyla and candidate divisions. PLoS ONE 8:e63994. doi: 10.1371/journal.pone.0063994
Sime-Ngando, T. (2014). Environmental bacteriophages: viruses of microbes in aquatic ecosystems. Front. Microbiol. 5:355. doi: 10.3389/fmicb.2014.00355
Sime-Ngando, T., Colombet, J., Personnic, S., Domaizon, I., Dorigo, U., Perney, P., et al. (2008). Short-term variations in abundances and potential activities of viruses, bacteria and nanoprotists in Lake Bourget. Ecol. Res. 23, 851–861. doi: 10.1007/s11284-007-0448-y
Smith, R. J., Jeffries, T. C., Roudnew, B., Seymour, J. R., Fitch, A. J., Simons, K. L., et al. (2013). Confined aquifers as viral reservoirs. Environ. Microbiol. Rep. 5, 725–730. doi: 10.1111/1758-2229.12072
Sommer, U., Adrian, R., De Senerpont Domis, L., Elser, J. J., Gaedke, U., Ibelings, B., et al. (2012). Beyond the plankton ecology group (PEG) model: mechanisms driving plankton succession. Annu. Rev. Ecol. Evol. Syst. 43, 429–448. doi: 10.1146/annurev-ecolsys-110411-160251
Sun, C. L., Brauer, S. L., Cadillo-Quiroz, H., Zinder, S. H., and Yavitt, J. B. (2012). Seasonal changes in methanogenesis and methanogenic community in three peatlands, Newyork State. Front. Microbiol. 3:81. doi: 10.3389/fmicb.2012.00081
Suttle, C. A. (2007). Marine viruses-major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812. doi: 10.1038/nrmicro1750
Suzuki, R., and Shimodaira, H. (2006). Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542. doi: 10.1093/bioinformatics/btl117
Thormann, M. N., Currah, R. S., and Bayley, S. E. (2003). Succession of microfungal assemblages in decomposing peatland plants. Plant Soil 250, 323–333. doi: 10.1023/A:1022845604385
Turetsky, M. R., Benscoter, B., Page, S., Rein, G., van der Werf, G. R., and Watts, A. (2015). Global vulnerability of peatlands to fire and carbon loss. Nat. Geosci. 8, 11–14. doi: 10.1038/ngeo2325
Tveit, A., Schwacke, R., Svenning, M. M., and Urich, T. (2012). Organic carbon transformations in high-Arctic peat soils: key functions and microorganisms. ISME J. 7, 299–311. doi: 10.1038/ismej.2012.99
Tveit, A., Urich, T., and Svenning, M. M. (2014). Metatranscriptomic analysis of Arctic peat soil microbiota. Appl. Environ. Microbiol. 81, 2841–2851. doi: 10.1128/AEM.01030-1014
Vandenkoornhuyse, P., Dufresne, A., Quaiser, A., Gouesbet, G., Binet, F., Francez, A. J., et al. (2010). Integration of molecular functions at the ecosystemic level: breakthroughs and future goals of environmental genomics and post-genomics. Ecol. Lett. 13, 776–791. doi: 10.1111/j.1461-0248.2010.01464.x
Waddington, J. M., and Roulet, N. T. (1997). Groundwater flow and dissolved carbon movement in a boreal peatland. J. Hydrol. 191, 122–138. doi: 10.1016/S0022-1694(96)03075-3072
Williamson, K. E., Radosevich, M., and Wommack, K. E. (2005). Abundance and diversity of viruses in six Delaware soils. Appl. Environ. Microbiol. 71, 3119–3125. doi: 10.1128/AEM.71.6.3119-3125.2005
Wommack, K. E., Nasko, D. J., Chopyk, J., and Sakowski, E. G. (2015). Counts and sequences, observations that continue to change our understanding of viruses in nature. J. Microbiol. 53, 181–192. doi: 10.1007/s12275-015-5068-6
Yu, Z. C. (2012). Northern peatland carbon stocks and dynamics: a review. Biogeosciences 9, 4071–4085. doi: 10.5194/bg-9-4071-2012
Zablocki, O., van Zyl, L., Adriaenssens, E. M., Rubagotti, E., Tuffin, M., Cary, S. C., et al. (2014). High-level diversity of tailed phages, eukaryote-associated viruses, and virophage-like elements in the metaviromes of antarctic soils. Appl. Environ. Microbiol. 80, 6888–6897. doi: 10.1128/AEM.01525-1514
Keywords: fen, bog, metaviromes, prokaryotes, ecological succession, microbial interactions
Citation: Ballaud F, Dufresne A, Francez A-J, Colombet J, Sime-Ngando T and Quaiser A (2016) Dynamics of Viral Abundance and Diversity in a Sphagnum-Dominated Peatland: Temporal Fluctuations Prevail Over Habitat. Front. Microbiol. 6:1494. doi: 10.3389/fmicb.2015.01494
Received: 01 September 2015; Accepted: 11 December 2015;
Published: 06 January 2016.
Edited by:
Graeme W. Nicol, Université de Lyon, FranceReviewed by:
Kim Yrjälä, University of Helsinki, FinlandEvelien M. Adriaenssens, University of Pretoria, South Africa
Copyright © 2016 Ballaud, Dufresne, Francez, Colombet, Sime-Ngando and Quaiser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Achim Quaiser, achim.quaiser@univ-rennes1.fr