 Chongxu Han1†
Chongxu Han1† Dongsheng Han
Dongsheng Han- 1Clinical Medical Examination Center, Northern Jiangsu People's Hospital, Yangzhou, China
- 2Experimental Research Center, Northern Jiangsu People's Hospital, Yangzhou, China
Pandemic Vibrio parahaemolyticus is an emerging public health concern as it has caused numerous gastroenteritis outbreaks worldwide. Currently, the absence of a global overview of the phenotypic and molecular characteristics of pandemic strains restricts our overall understanding of these strains, especially for environmental strains. To generate a global picture of the sero-prevalence and genetic diversity of pandemic V. parahaemolyticus, pandemic isolates from worldwide collections were selected and analyzed in this study. After a thorough analysis, we found that the pandemic isolates represented 49 serotypes, which are widely distributed in 22 countries across four continents (Asia, Europe, America and Africa). All of these serotypes were detected in clinical isolates but only nine in environmental isolates. O3:K6 was the most widely disseminated serotype, followed by O3:KUT, while the others were largely restricted to certain countries. The countries with the most abundant pandemic serotypes were China (26 serotypes), India (24 serotypes), Thailand (15 serotypes) and Vietnam (10 serotypes). Based on MLST analysis, 14 sequence types (STs) were identified among the pandemic strains, nine of which fell within clonal complex (CC) 3. ST3 and ST305 were the only two STs that have been reported in environmental pandemic strains. Pandemic ST3 has caused a wide range of infections in as many as 16 countries. Substantial serotypic diversity was mainly observed among isolates within pandemic ST3, including as many as 12 combinations of O/K serotypes. At the allele level, the dtdS and pntA, two loci that perfectly conserved in CC3, displayed a degree of polymorphism in some pandemic strains. In conclusion, we provide a comprehensive understanding of sero-prevalence and genetic differentiation of clinical and environmental pandemic isolates collected from around the world. Although, further studies are needed to delineate the specific mechanisms by which the pandemic strains evolve and spread, the findings in this study are helpful when seeking countermeasures to reduce the spread of V. parahaemolyticus in endemic areas.
Introduction
Vibrio parahaemolyticus, an organism with a high genetic diversity, has emerged as a pathogen causing acute gastroenteritis with a worldwide distribution. Before 1996, V. parahaemolyticus infections usually exhibited a localized distribution and were linked to diverse serotypes (e.g., O2:K3, O3:K6, O4:K8) (Wong et al., 2000). In February 1996, the pandemic O3:K6 serotype emerged, resulting in an inexplicable increase of V. parahaemolyticus gastroenteritis in Kolkata city, India (Okuda et al., 1997). This unique serotype subsequently quickly spread into coastal regions of southern Asia (Bag et al., 1999), America (Martinez-Urtaza et al., 2004), Africa (Ansaruzzaman et al., 2005), and Europe (Martinez-Urtaza et al., 2005) and caused numerous outbreaks within a few years (Okuda et al., 1997; Chowdhury et al., 2000a; Nair et al., 2007). Such widespread occurrence of a single serotype of V. parahaemolyticus had not been previously reported.
All the pandemic O3:K6 strains share the following specific genetic markers: positivity for the thermostable direct hemolysin(tdh) gene, negativity for the TDH-related hemolysin(trh) gene and positivity for a toxRS/new gene, which can be amplified via a specific PCR method known as “GS-PCR” (Matsumoto et al., 2000; Chao et al., 2011; de Jesús Hernández-Díaz et al., 2015). To our surprise, in recent years, some new serotypes [e.g., O4:K68, O1:K25, O1:KUT(untypable)] have been detected that exhibit identical genotypes and molecular profiles to the pandemic O3:K6 serotype (Chang et al., 2000; Bhuiyan et al., 2002). These serotypes may diverge from the pandemic O3:K6 serotype in alteration of the O and/or K antigens and are referred to as “serovariants” of the pandemic O3:K6 serotype (Chowdhury et al., 2000b; Matsumoto et al., 2000). Currently, all of the pandemic serotypes are grouped as belonging to the “O3:K6 pandemic clone.” Through 2007, a total of 22 serotypes had been reported to belong to this clone (Nair et al., 2007).
Many surveys have shown that pandemic V. parahaemolyticus serovariants can be identified not only in clinical samples (Li et al., 2014; Pazhani et al., 2014; Ueno et al., 2016), but also in seafood and other environmental samples (Arakawa et al., 1999; Vuddhakul et al., 2000; Deepanjali et al., 2005; Quilici et al., 2005; Chao et al., 2009; Caburlotto et al., 2010), indicating that the pandemic strains have established ecological niches in many regions, resulting in a heightened perception of the threat to the public health of the local population. An accurate description of the distribution and spread of the pandemic strains is important for understanding the epidemiology of this pathogen and preventing outbreaks and sporadic illnesses. However, after G. Balakrish Nair and colleagues reviewed the global dissemination of pandemic V.parahaemolyticus serotype O3:K6 and its serovariants in 2007 (Nair et al., 2007), few studies have specifically for the pandemic V. parahaemolyticus on a global scale, especially concerning the worldwide dissemination of environmental strains. Therefore, it would be beneficial to integrate and update the available scientific data on pandemic V. parahaemolyticus.
The establishment of a multilocus sequence typing (MLST) scheme for V. parahaemolyticus has enhanced our knowledge of the population structure and genetic diversity of V. parahaemolyticus (Gonzalez-Escalona et al., 2008). Previous studies based on MLST assay have shown that the increasing prevalence of clonal complex 3 (CC3) has become an ongoing public health concern (Gonzalez-Escalona et al., 2008; Haendiges et al., 2014; Han et al., 2015), and most pandemic strains have been identified as belonging to CC3 (Chen et al., 2016). Thus, clarifying the genetic diversity among the pandemic strains will aid in the selection of preventative strategies targeting pandemic strain infections.
In this study, we collected data on pandemic strains mainly from the pubMLST database (http://pubmlst.org/vparahaemolyticus) and previous studies, in an effort to generate a comprehensive overview of the spread of clinical and environmental pandemic V. parahaemolyticus strains occurring over wide geographic areas since the emergence of this clone. Furthermore, through MLST phylogenetic analysis, we determined the genetic diversity of the pandemic clone to provide a holistic understanding of the microevolution of pandemic strains.
Materials and Methods
Datasets Utilized in the Present Study
A total of 267 representative clinical and environmental V. parahaemolyticus isolates with pandemic genetic marks (toxRS/new+, tdh+, and trh−) were selected as the research subject of this study, among which 263 isolates came from the literature and four from the pubMLST database (http://pubmlst.org/vparahaemolyticus/). To identify relevant publications, we conducted a comprehensive search of the US National Library of Medicine PubMed database and the Elsevier, Springer, and China National Knowledge Infrastructure databases for all relevant studies using combinations of the following terms: “Vibrio parahaemolyticus,” “pandemic clone,” “pandemic strains,” and “O3:K6 clone” (until July 1, 2015). Additional eligible studies were identified from references cited in the relevant articles. The full text of each potentially relevant paper was scrutinized and a total of 263 isolates with pandemic genetic marks (toxRS/new+, tdh+, and trh−) were finally extracted from 39 papers. Details on the individual isolates are summarized in Additional file 1: Table S1.
Multilocus Sequence Typing Analysis
To determine the genetic diversity of the pandemic strains through MLST analysis, another file containing 185 isolates were analyzed (see Additional file 2: Table S2). These isolates included 95 pandemic isolates from Table S1 and 90 strains that are non-pandemic but belong to CC3 from the pubMLST database. The sequence types (STs) of the 185 isolates were compared using eBURST V3 (http://eburst.mlst.net/) (Feil et al., 2004). Additionally, a “population snapshot” analysis of the entire V. parahaemolyticus population was also implemented based on the total pubMLST dataset, which illustrated the population differentiation of the whole population. Clonal complexes were defined conservatively as a cluster of STs in an eBURST diagram, in which all STs were linked as single-locus variants (SLVs, two STs differing from each other at a single locus) to at least one other ST (Feil et al., 2004). The singleton STs corresponded to STs differing from all the others by three or more of the seven loci (Esteves et al., 2015).
Genetic Diversity and Phylogenetic Analysis
The diversity of the seven loci in the pandemic isolates was revealed by DnaSP V5 (http://www.ub.edu/dnasp/) with respect to the following parameters: the number of alleles, number (%) of polymorphic sites, nucleotide diversity (per site) and Tajima's D value. The purpose of Tajima's D test is to distinguish housekeeping genes evolving randomly (“neutrally”) vs. those evolving under a non-random process (Tajima, 1989). A P > 0.05 indicates that the target gene is evolving randomly and that mutations in the gene have no effect on the fitness and survival of an organism (Tajima, 1989; Ferreira et al., 2008). A minimum-evolution (ME) tree for the concatenated sequences of each ST of the 185 isolates was generated using Mega 5 software with the Kimura two-parameter model to estimate genetic distances. The statistical support for the nodes in the ME tree was assessed through 1000 bootstrap resamplings.
Results
Global Spread of Pandemic Serovariants
According to a detailed review, a total of 49 pandemic serotypes from 22 countries across four continents (Asia, Europe, America, and Africa) were identified. All of these serotypes were detected in clinical isolates but only nine in environmental isolates. O3:K6 was the most widely disseminated serotype, and patients in all 22 countries had been infected with this subtype at some point in time. O3:KUT was the second most widely distributed serotype. Several serotypes, such as O1:K25, O1:KUT, and O4:K68, also exhibited multi-country distributions but were mainly restricted to Southeast Asia (Table 1). The sources of environmental pandemic isolates were diverse, mainly including shellfish, oyster, clam, and shrimp, sediment and seawater samples collected in nine countries (see in Additional file 1: Table S1).
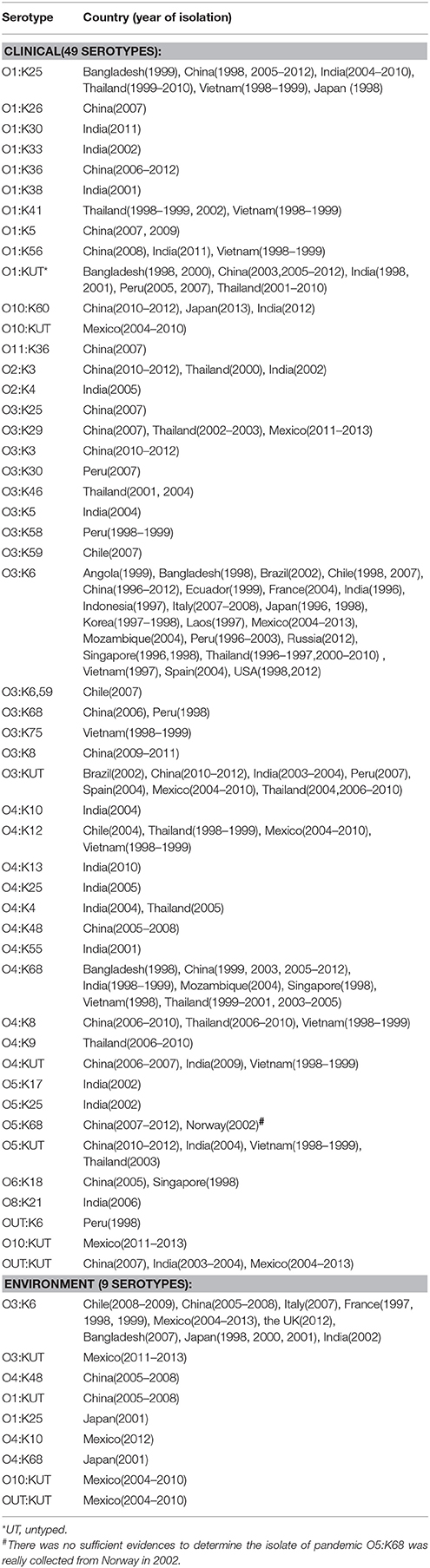
Table 1. Presence of clinical and environmental pandemic serovariants of V. parahaemolyticus occurring at a global scale.
A comprehensive map of the dissemination of the clinical and environmental pandemic serotypes on a global scale was generated (Figure 1). The serotypes of the pandemic clone were highly abundant and variable in coastal regions of China, India, Thailand and Vietnam. It was notable that most of the environmental pandemic serotypes present in a certain country were also detected in patients from that country. O3:K6 was the typical serotype. Four environmental serotypes (O3:K6, O3:KUT, O10:KUT, and OUT:KUT) in Mexico were also found spread in its local population.
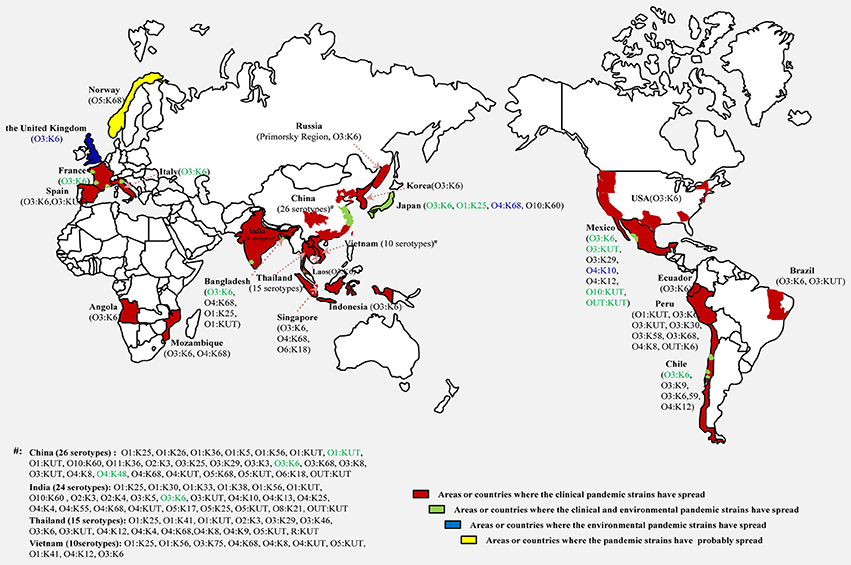
Figure 1. Map showing the dissemination of clinical and environmental pandemic serovariants of V. parahaemolyticus occurring at a global scale. Serotypes identified in clinical isolates (black), environmental isolates (blue) and both in clinical and environmental isolates (green) are marked respectively.
Widely Dispersed Clones of V. parahaemolyticus and Genetic Differentiation of the Pandemic Isolates
Until August 2015, a total of 954 STs had been identified in the V. parahaemolyticus pubMLST database, approximately two-thirds of which were detected in environmental isolates, while less than one-third came from clinical isolates, and only 26 were present both in environmental and clinical isolates. The total population displayed 19 CCs as well as some doublets and numerous singletons (Figure 2). CC3 was the most prevalent CC, being comprised of 18 STs with no less than 15 serotypes (Table 2).
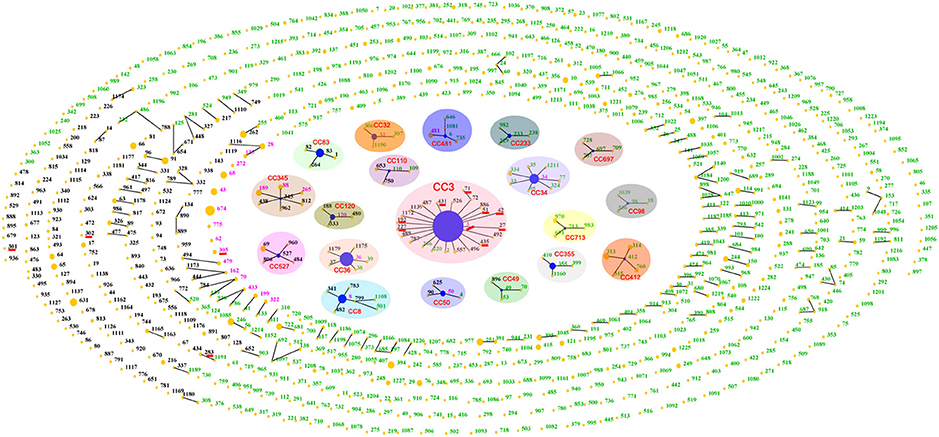
Figure 2. “Population snapshot” showing the clonal diversity of V. parahaemolyticus based on data archived in public database. Green numbers indicate STs typed in environmental isolates; black numbers represent clinical STs; and pink numbers are STs that found in both clinical and environmental collections. Nineteen clonal complexes are indicated with separate shades. STs that are SLVs of each other are connected with black lines. STs associated with pandemic spreading are underlined in red.
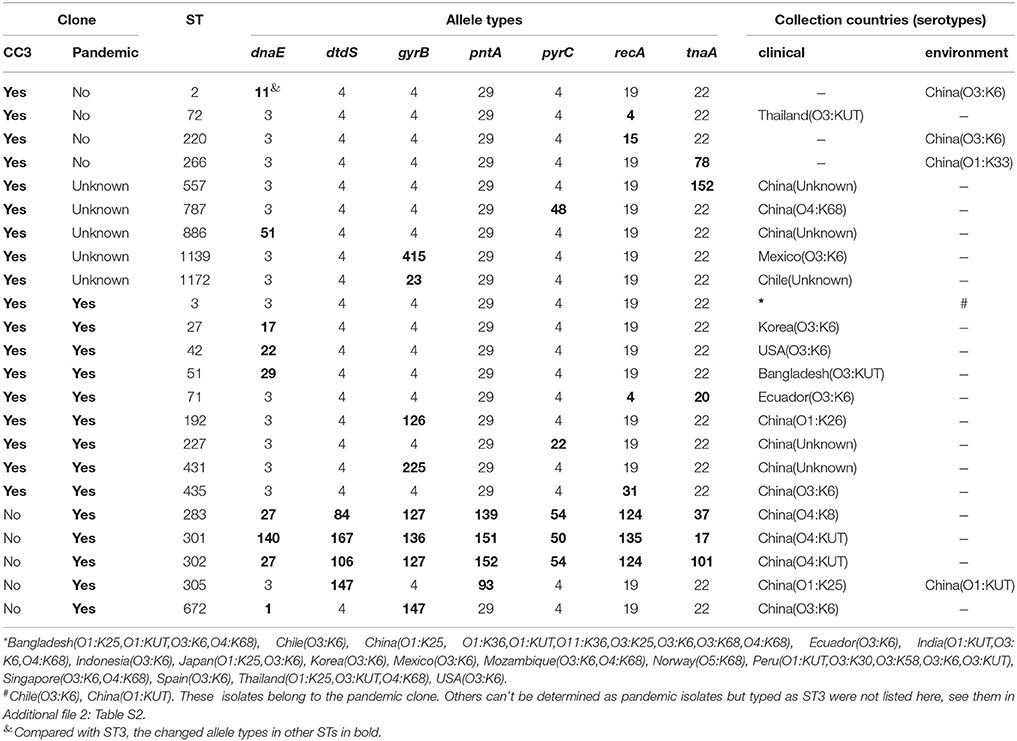
Table 2. Sequence types, allele profiles, and geographic locations of CC3 and pandemic V. parahaemolyticus clone.
After thoroughly analyzing the sequence data for the 185 isolates (in Additional file 2: Table S2), we found that the pandemic strains exhibited 14 STs, only two of which (ST3 and ST305) had ever been identified in environmental samples (Figure 3C). China was the country with the most pandemic STs (10 STs). Nine of these 14 pandemic STs could be classified into CC3, among which, ST3 was the only pandemic ST that had caused a wide range of infections in as many as 16 countries (Table 2, Figure 3A). ST305 and ST672 were DLVs of ST3 but were not members of CC3 because there was no ST in CC3 could act as their SLV. The other three STs (ST283, ST301, and ST302) originating from coastal areas of China were identified as singletons with no relationship to CC3.
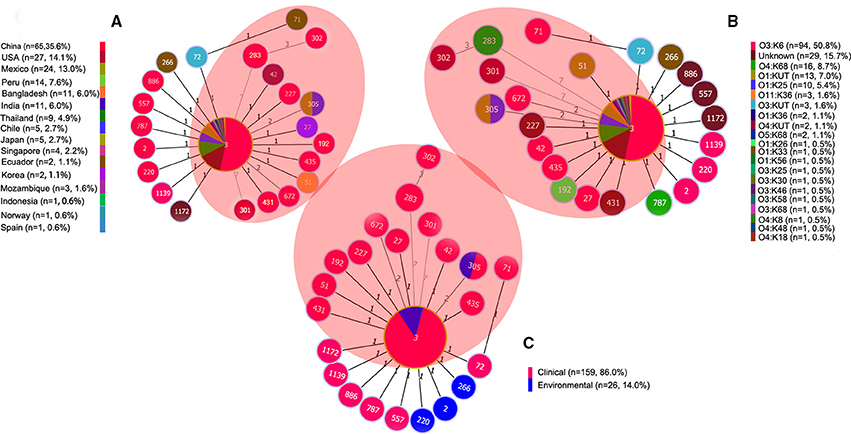
Figure 3. goeBURST full MST of the STs consisting of 185 representative isolates associated with CC3 and/or the pandemic clone. (A) Geographical distribution, (B) Serotype distribution, (C) Source distribution. The sizes of the circles vary according to the frequency of the ST. The number of different alleles is presented between STs connected via a line. ST3 and its SLVs (one allele diversity) form the CC3. STs in the shaded area are associated with the pandemic clone. “n” in the figure legend indicates the number of isolates.
The Association between Pandemic STs and Serotypes
The Minimum spanning tree of the 14 pandemic STs resulting from the MLST analysis showed a substantial serotypic diversity among isolates within ST3, but not among isolates of the other 13 STs (Figure 3B). Specifically, pandemic ST3 comprised isolates of 12 serotypes (O1:K25, O1:K36, O1:KUT, O3:K6, O3:K25, O3:K30, O3:K58, O3:K68, O3:KUT, O4:K68, O5:K68, and O11:K36). ST305 included isolates belonging to O1:K25 and O1:KUT serotypes. The remaining STs were consisted of a single serotype, respectively (Table 2, Figure 3B). From another perspective, the pandemic O3:K6 serotype was shared by six different STs (ST3, ST27, ST42, ST71, ST435, and ST672). Other serotypes were clustered in no more than two different pandemic STs, respectively (Table 2).
Genetic Diversity of the Pandemic Isolates
The data on the nucleotide and allelic diversity of the pandemic isolates are summarized in Table 3. The highest percentage of polymorphic sites was detected in dtdS (5.46%). Nucleotide diversity ranged from 0.01082 (pyrC) to 0.02926 (dtdS). dtdS and pntA were perfectly conserved in CC3 (the allele types were dtdS4 and pntA29), but in the pandemic isolates, five different alleles were detected for each of the two genes; the number of SNPs was 25(5.46%) for dtdS and 10(2.33%) for pntA (Table 3).
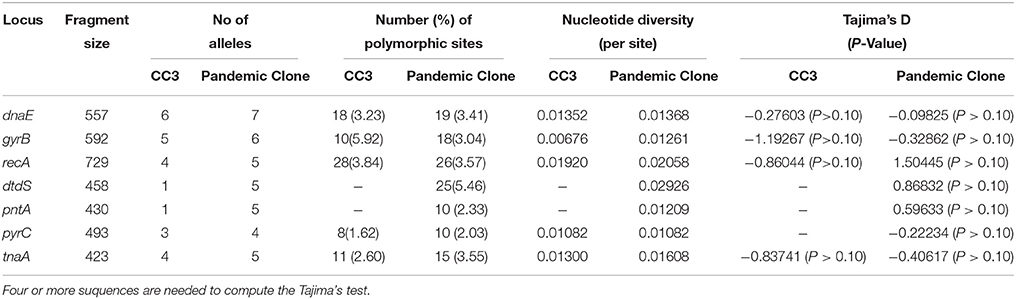
Table 3. Sequence analysis of the seven loci studies for the isolates of CC3 and pandemic clone.
Phylogenetic Analysis of Pandemic Isolates
Phylogenetic analysis may provide a better resolution and elucidate some phylogenetic relationships among CCs or singletons that are not observed or resolved using goeBURST. Therefore, an ME tree representing the concatenated sequences of the seven housekeeping gene fragments in the 185 isolates is shown in Figure 4. In the goeBURST analysis, five pandemic STs (ST305, S672, ST301, ST302, and ST283) were not grouped into CC3. However, in the ME tree analysis, ST305 and ST672 were clustered together with STs of CC3, and only ST301, ST302, and ST283 exhibited relatively greater evolutionary distances from STs in CC3. In fact, the number of SNPs in the seven alleles of these last STs was greater than in ST305 and ST672 when compared with the STs of CC3.
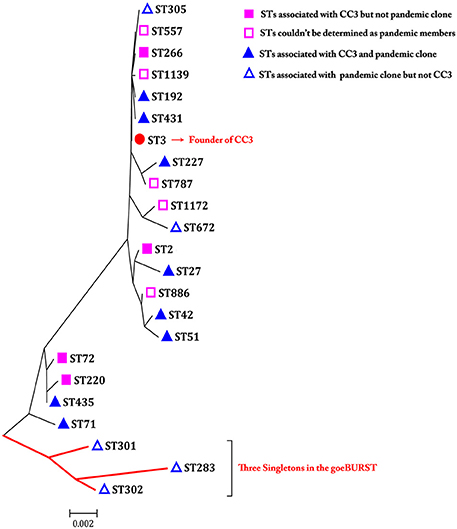
Figure 4. A minimum evolutionary tree built using the concatenated sequences of the seven loci of each ST associated with pandemic clone and/or CC3. The scale bar represents the evolutionary distance.
Discussion
In previous studies, we successfully made extensive descriptions of strains from a global clinical collection and from Chinese patients, respectively, exhibiting a highly degree of genetic diversity and a complicated population structure of V. parahaemolyticus in general (Han et al., 2014, 2015). In this study, we elucidated the sero-prevalence and genetic differentiation of the pandemic clone, which has becoming an emerging public health concern (Martinez-Urtaza et al., 2010; Velazquez-Roman et al., 2012, 2014; Powell et al., 2013; Li et al., 2014; Pazhani et al., 2014). The results will be useful in uncovering the microevolution relationships among pandemic V. parahaemolyticus strains. Serotyping is the primary basis of the classification of V. parahaemolyticus strains. Pandemic strains exhibit rapidly changing their serotypes (Nair et al., 2007). From1996 to 2007, 22 pandemic serotypes were identified (Nair et al., 2007). In the present study, as many as 49 serotypes identified to date in investigations conducted by different laboratory groups around the world could be confirmed as being associated with the pandemic clone.
Several lines of evidence have been presented in support of the hypothesis that these new serotypes might have emerged from the pandemic O3:K6 strains through replacement of the putative O and K antigen gene clusters (Okura et al., 2008; Harth et al., 2009). In the present study, as many as 12 combinations of O/K serotypes were grouped in pandemic ST3, demonstrating a remarkably high degree of serotypic diversity among the pandemic isolates and suggesting that the O- and K-antigen encoding loci are subject to exceptionally high rates of recombination in isolates with the same genotype (Gavilan et al., 2013; Theethakaew et al., 2013). Herein, we agree that the high frequency of alterations in the O and/or K antigens is a significant biological characteristic of pandemic V. parahaemolyticus strains, which might be an important means of survival in the face of changing external environments and host immunological resistance.
Regional persistence of the clinical pandemic O3:K6 serotype has been identified in coastal areas of many countries, such as Mexico(2004–2015) (Velazquez-Roman et al., 2012; de Jesús Hernández-Díaz et al., 2015), Peru(2007) (Gil et al., 2007), Chile(2007–2009) (Cabello et al., 2007; Garcia et al., 2009), China(2007–2012) (Zhang et al., 2013; Li et al., 2014), India(2001–2012) (Pazhani et al., 2014), and Thailand(2006–2010) (Thongjun et al., 2013). However, it is not obvious what specific factors conferred upon this serotype the ability to disseminate around world. Some environmental conditions (e.g., seawater temperature, PH or salinity effects) affecting survival and the unique pathogenic potential of pandemic O3:K6 strains vs. other strains have been compared, but the specific advantage of pandemic O3:K6 strains over other strains remains unclear (Wong et al., 2000; Yeung et al., 2002). Further, investigations should focus on revealing the routes and mechanisms of the rapid spread of the pandemic clone.
In addition to the O3:K6 serotype, other pandemic serotypes have been isolated in both clinical and environmental samples from some certain countries, such as O1:KUT and O4:K48 in China (Chao et al., 2009), O1:K25 in Japan (Hara-Kudo et al., 2003) and O3:KUT, O10:KUT, and OUT:OUT in Mexico (Velazquez-Roman et al., 2012; de Jesús Hernández-Díaz et al., 2015). Although, the specific relationships between environmental serotypes and those leading to illnesses have not been determined, it is important to first understand epidemic situation of these serotypes through active surveillance.
In the present study, we showed that the population structure of V. parahaemolyticus was extremely genetically diverse based on the successful identification of 19 CCs and a large number of singletons, in agreement with previous findings (Han et al., 2015). Over half of the pandemic STs belonged to CC3 according to goeBURST analysis. The dtdS and pntA genes were found to be perfectly conserved throughout the evolution of CC3, whereas they presented some degree of polymorphism in pandemic strains. In our analysis, none of the values of Tajima's D was significantly different from zero (P > 0.10), suggesting that the housekeeping genes of the pandemic strains evolve under a random process (“neutrally”) and are subject to low selective pressure. The similar conclusion was obtained in studies based on the entire V. parahaemolyticus population (Theethakaew et al., 2013).
According to the available data, 64.3% of the STs (9/14) of the pandemic clones were isolated from China, suggesting that this country represents an important reservoir for the emergence of novel pandemic strains. If a global network for the prevention and control of V. parahaemolyticus infection is established in the future, the coastal regions of China should be recognized as important monitoring points. Three special STs (ST283, ST301, and ST302) typed in pandemic isolates originating from China were identified as singletons presenting distant relationships with other STs of the pandemic clone in this study. However, in a study by Chen et al. (2012), the corresponding strains were clustered together with other pandemic strains based on other molecular typing methods, such as enterobacterial repetitive intergenic consensus sequence PCR (ERIC-PCR) and sequence analysis of the gyrB gene. Thus, it can be observed that current molecular typing methods, including MLST, could lead to controversial results, making it difficult to draw conclusions, although such methods have been confirmed to provide a high level of resolution and information for elucidating the evolution of the V. parahaemolyticus clonal complex (Chen et al., 2012). Therefore, to accurately portray the relationships among strains at the molecular level, combined use of different molecular typing techniques with better discrimination could be considered in epidemiological investigations of V. parahaemolyticus. Whole genome sequencing (WGS), a powerful typing method with a robust differentiation ability for characterizing related isolates, is another outstanding alternative for analyzing the evolution and population structure of V. parahaemolyticus (Haendiges et al., 2015).
Invalid data in the pubMLST database were one problem restricting our analysis in this study. As of 15th July 2015, a total of 1844 records of isolates had been deposited, but definite STs were only available for 1700. Moreover, information on the corresponding biological characteristics of many uploaded isolates, such as sample sources, regions, drug sensitivity, serotypes and virulence genes was deficient. This lack of information is not conducive to conducting further epidemiologic and etiologic analyses of V. parahaemolyticus at a global scale. In this study, for five STs belonging to CC3 (ST557, ST787, ST886, ST1139, and ST1172), it could not be determined whether they were associated with pandemic clone, because of missing of toxRS/new gene and/or tdh gene sequences. As MLST assays play an important role in studies on the molecular epidemiology of V. parahaemolyticus, we recommend that the researchers uploaded their data on isolates as accurately and completely as possible.
In summary, the present study provides novel information on the abundance and prevalence of pandemic V. parahaemolyticus based on the analysis of clinical and environmental isolates from a worldwide collection. We showed that the regional persistence of pandemic O3:K6 has been established in coastal areas of many countries. The presence and persistence of pandemic V. parahaemolyticus strains, and especially the continuous appearance of environmental pandemic strains, is a matter of concern for public health authorities. We analyzed the genetic diversity of the pandemic clone to provide a comprehensive understanding of the microevolutionary relationships between pandemic strains. The answers to some unresolved questions about the pandemic clone, such as the advantage of pandemic O3:K6 over other strains and the mechanisms underlying the spread of strains with pandemic genetic marks, remain speculative, and require further investigations.
Author Contributions
Conceived and designed the experiments: DH, HT, CH. Performed the experiments: DH, CR. Analyzed the data: DH, HT, XZ. Contributed reagents/materials/analysis tools: DH, CH. Wrote the paper: DH.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was jointly supported by grant NO.81400899 from the National Natural Science Foundation of China, grant NO.YZ2014061 from the Science & Technology Foundation Program of Yangzhou city, and two research projects (NO.yzucms201425 and NO.yzucms201403) of Northern Jiangsu People's Hospital, Yangzhou, China.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00567
References
Ansaruzzaman, M., Lucas, M., Deen, J. L., Bhuiyan, N. A., Wang, X. Y., Safa, A., et al. (2005). Pandemic serovars (O3:K6 and O4:K68) of Vibrio parahaemolyticus associated with diarrhea in Mozambique: spread of the pandemic into the African continent. J. Clin. Microbiol. 43, 2559–2562. doi: 10.1128/JCM.43.6.2559-2562.2005
Arakawa, E., Murase, T., Shimada, T., Okitsu, T., Yamai, S., and Watanabe, H. (1999). Emergence and prevalence of a novel Vibrio parahaemolyticus O3:K6 clone in Japan. Jpn. J. Infect. Dis. 52, 246–247.
Bag, P. K., Nandi, S., Bhadra, R. K., Ramamurthy, T., Bhattacharya, S. K., Nishibuchi, M., et al. (1999). Clonal diversity among recently emerged strains of Vibrio parahaemolyticus O3:K6 associated with pandemic spread. J. Clin. Microbiol. 37, 2354–2357.
Bhuiyan, N. A., Ansaruzzaman, M., Kamruzzaman, M., Alam, K., Chowdhury, N. R., Nishibuchi, M., et al. (2002). Prevalence of the pandemic genotype of Vibrio parahaemolyticus in Dhaka, Bangladesh, and significance of its distribution across different serotypes. J. Clin. Microbiol. 40, 284–286. doi: 10.1128/JCM.40.1.284-286.2002
Cabello, F. C., Espejo, R. T., Hernandez, M. C., Rioseco, M. L., Ulloa, J., and Vergara, J. A. (2007). Vibrio parahaemolyticus O3:K6 epidemic diarrhea, Chile, 2005. Emerg. Infect. Dis. 13, 655–656. doi: 10.3201/eid1304.061152
Caburlotto, G., Gennari, M., Ghidini, V., Tafi, M., and Lleo, M. M. (2010). Serological and molecular characterization of Vibrio parahaemolyticus marine strains carrying pandemic genetic markers. ISME J. 4, 1071–1074. doi: 10.1038/ismej.2010.34
Chang, B., Yoshida, S., Miyamoto, H., Ogawa, M., Horikawa, K., Ogata, K., et al. (2000). A unique and common restriction fragment pattern of the nucleotide sequences homologous to the genome of vf33, a filamentous bacteriophage, in pandemic strains of Vibrio parahaemolyticus O3:K6 O4:K68, and O1:K untypeable. FEMS Microbiol. Lett. 192, 231–236. doi: 10.1111/j.1574-6968.2000.tb09387.x
Chao, G., Jiao, X., Zhou, X., Yang, Z., Huang, J., Pan, Z., et al. (2009). Serodiversity, pandemic O3:K6 clone, molecular typing, and antibiotic susceptibility of foodborne and clinical Vibrio parahaemolyticus isolates in Jiangsu, China. Foodborne Pathog. Dis. 6, 1021–1028. doi: 10.1089/fpd.2009.0295
Chao, G., Wang, F., Zhou, X., Jiao, X., Huang, J., Pan, Z., et al. (2011). Origin of Vibrio parahaemolyticus O3:K6 pandemic clone. Int. J. Food Microbiol. 145, 459–463. doi: 10.1016/j.ijfoodmicro.2011.01.022
Chen, W., Xie, Y., Xu, J., Wang, Q., Gu, M., Yang, J., et al. (2012). Molecular typing of Vibrio parahaemolyticus isolates from the middle-east coastline of China. Int. J. Food Microbiol. 153, 402–412. doi: 10.1016/j.ijfoodmicro.2011.12.001
Chen, Y., Chen, X., Yu, F., Wu, M., Wang, R., Zheng, S., et al. (2016). Serology, virulence, antimicrobial susceptibility and molecular characteristics of clinical Vibrio parahaemolyticus strains circulating in southeastern China from 2009 to 2013. Clin. Microbiol. Infec. 22, 258.e9–258.e16. doi: 10.1016/j.cmi.2015.11.003
Chowdhury, N. R., Chakraborty, S., Eampokalap, B., Chaicumpa, W., Chongsa-Nguan, M., Moolasart, P., et al. (2000a). Clonal dissemination of Vibrio parahaemolyticus displaying similar DNA fingerprint but belonging to two different serovars (O3:K6 and O4:K68) in Thailand and India. Epidemiol. Infect. 125, 17–25. doi: 10.1017/S0950268899004070
Chowdhury, N. R., Chakraborty, S., Ramamurthy, T., Nishibuchi, M., Yamasaki, S., Takeda, Y., et al. (2000b). Molecular evidence of clonal Vibrio parahaemolyticus pandemic strains. Emerg. Infect. Dis. 6, 631–636. doi: 10.3201/eid0606.000612
Deepanjali, A., Kumar, H. S., Karunasagar, I., and Karunasagar, I. (2005). Seasonal variation in abundance of total and pathogenic Vibrio parahaemolyticus bacteria in oysters along the southwest coast of India. Appl Environ. Microbiol. 71, 3575–3580. doi: 10.1128/AEM.71.7.3575-3580.2005
de Jesús Hernández-Díaz, L., Leon-Sicairos, N., Velazquez-Roman, J., Flores-Villaseñor, H., Guadron-Llanos, A. M., Martinez-Garcia, J. J., et al. (2015). A pandemic Vibrio parahaemolyticus O3:K6 clone causing most associated diarrhea cases in the Pacific Northwest coast of Mexico. Front. Microbiol. 6:221. doi: 10.3389/fmicb.2015.00221
Esteves, K., Mosser, T., Aujoulat, F., Hervio-Heath, D., Monfort, P., and Jumas-Bilak, E. (2015). Highly diverse recombining populations of Vibrio cholerae and Vibrio parahaemolyticus in French Mediterranean coastal lagoons. Front. Microbiol. 6:708. doi: 10.3389/fmicb.2015.00708
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Ferreira, J. M., Oliveira, L. R. D., Wynen, L., Bester, M. N., Guinet, C., Moraes-Barros, N., et al. (2008). Multiple origins of vagrant Subantarctic fur seals: a long journey to the Brazilian coast detected by molecular markers. Polar Biol. 31, 303–308. doi: 10.1007/s00300-007-0358-z
Garcia, K., Torres, R., Uribe, P., Hernandez, C., Rioseco, M. L., Romero, J., et al. (2009). Dynamics of clinical and environmental Vibrio parahaemolyticus strains during seafood-related summer diarrhea outbreaks in southern Chile. Appl. Environ. Microbiol. 75, 7482–7487. doi: 10.1128/AEM.01662-09
Gavilan, R. G., Zamudio, M. L., and Martinez-Urtaza, J. (2013). Molecular epidemiology and genetic variation of pathogenic Vibrio parahaemolyticus in Peru. PLoS Negl. Trop. Dis. 7:e2210. doi: 10.1371/journal.pntd.0002210
Gil, A. I., Miranda, H., Lanata, C. F., Prada, A., Hall, E. R., Barreno, C. M., et al. (2007). O3:K6 serotype of Vibrio parahaemolyticus identical to the global pandemic clone associated with diarrhea in Peru. Int. J. Infect. Dis. 11, 324–328. doi: 10.1016/j.ijid.2006.08.003
Gonzalez-Escalona, N., Martinez-Urtaza, J., Romero, J., Espejo, R. T., Jaykus, L. A., and DePaola, A. (2008). Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 190, 2831–2840. doi: 10.1128/JB.01808-07
Haendiges, J., Rock, M., Myers, R. A., Brown, E. W., Evans, P., and Gonzalez-Escalona, N. (2014). Pandemic Vibrio parahaemolyticus, Maryland, USA, 2012. Emerg. Infect. Dis. 20, 718–720. doi: 10.3201/eid2004.130818
Haendiges, J., Timme, R., Allard, M. W., Myers, R. A., Brown, E. W., and Gonzalez-Escalona, N. (2015). Characterization of Vibrio parahaemolyticus clinical strains from Maryland (2012-2013) and comparisons to a locally and globally diverse V. parahaemolyticus strains by whole-genome sequence analysis. Front. Microbiol. 6:125. doi: 10.3389/fmicb.2015.00125
Han, D., Tang, H., Lu, J., Wang, G., Zhou, L., Min, L., et al. (2014). Population structure of clinical Vibrio parahaemolyticus from 17 coastal countries, determined through multilocus sequence analysis. PLoS ONE 9:e107371. doi: 10.1371/journal.pone.0107371
Han, D., Tang, H., Ren, C., Wang, G., Zhou, L., and Han, C. (2015). Prevalence and genetic diversity of clinical Vibrio parahaemolyticus isolates from China, revealed by multilocus sequence typing scheme. Front. Microbiol. 6:291. doi: 10.3389/fmicb.2015.00291
Hara-Kudo, Y., Sugiyama, K., Nishibuchi, M., Chowdhury, A., Yatsuyanagi, J., Ohtomo, Y., et al. (2003). Prevalence of pandemic thermostable direct hemolysin-producing Vibrio parahaemolyticus O3:K6 in seafood and the coastal environment in Japan. Appl. Environ. Microbiol. 69, 3883–3891. doi: 10.1128/AEM.69.7.3883-3891.2003
Harth, E., Matsuda, L., Hernandez, C., Rioseco, M. L., Romero, J., Gonzalez-Escalona, N., et al. (2009). Epidemiology of Vibrio parahaemolyticus outbreaks, southern Chile. Emerg. Infect. Dis. 15, 163–168. doi: 10.3201/eid1502.071269
Li, Y., Xie, X., Shi, X., Lin, Y., Qiu, Y., Mou, J., et al. (2014). Vibrio parahaemolyticus, Southern Coastal Region of China, 2007-2012. Emerg. Infect. Dis. 20, 685–688. doi: 10.3201/eid2004.130744
Martinez-Urtaza, J., Bowers, J. C., Trinanes, J., and DePaola, A. (2010). Climate anomalies and the increasing risk of Vibrio parahaemolyticus and Vibrio vulnificus illnesses. Food Res. Int. 43, 1780–1790. doi: 10.1016/j.foodres.2010.04.001
Martinez-Urtaza, J., Lozano-Leon, A., DePaola, A., Ishibashi, M., Shimada, K., Nishibuchi, M., et al. (2004). Characterization of pathogenic Vibrio parahaemolyticus isolates from clinical sources in Spain and comparison with Asian and North American pandemic isolates. J. Clin. Microbiol. 42, 4672–4678. doi: 10.1128/JCM.42.10.4672-4678.2004
Martinez-Urtaza, J., Simental, L., Velasco, D., DePaola, A., Ishibashi, M., Nakaguchi, Y., et al. (2005). Pandemic Vibrio parahaemolyticus O3:K6, Europe. Emerg. Infect. Dis. 11, 1319–1320. doi: 10.3201/eid1108.050322
Matsumoto, C., Okuda, J., Ishibashi, M., Iwanaga, M., Garg, P., Rammamurthy, T., et al. (2000). Pandemic spread of an O3:K6 clone of Vibrio parahaemolyticus and emergence of related strains evidenced by arbitrarily primed PCR and toxRS sequence analyses. J. Clin. Microbiol. 38, 578–585.
Nair, G. B., Ramamurthy, T., Bhattacharya, S. K., Dutta, B., Takeda, Y., and Sack, D. A. (2007). Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin. Microbiol. Rev. 20, 39–48. doi: 10.1128/CMR.00025-06
Okuda, J., Ishibashi, M., Hayakawa, E., Nishino, T., Takeda, Y., Mukhopadhyay, A. K., et al. (1997). Emergence of a unique O3:K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from Southeast Asian travelers arriving in Japan. J. Clin. Microbiol. 35, 3150–3155.
Okura, M., Osawa, R., Tokunaga, A., Morita, M., Arakawa, E., and Watanabe, H. (2008). Genetic analyses of the putative O and K antigen gene clusters of pandemic Vibrio parahaemolyticus. Microbiol. Immunol. 52, 251–264. doi: 10.1111/j.1348-0421.2008.00027.x
Pazhani, G. P., Bhowmik, S. K., Ghosh, S., Guin, S., Dutta, S., Rajendran, K., et al. (2014). Trends in the epidemiology of pandemic and non-pandemic strains of Vibrio parahaemolyticus isolated from diarrheal patients in Kolkata, India. PLoS Neglect. Trop. D. 8:e2815. doi: 10.1371/journal.pntd.0002815
Powell, A., Baker-Austin, C., Wagley, S., Bayley, A., and Hartnell, R. (2013). Isolation of pandemic Vibrio parahaemolyticus from UK water and shellfish produce. Microb. Ecol. 65, 924–927. doi: 10.1007/s00248-013-0201-8
Quilici, M. L., Robert-Pillot, A., Picart, J., and Fournier, J. M. (2005). Pandemic Vibrio parahaemolyticus O3:K6 spread, France. Emerg. Infect. Dis. 11, 1148–1149. doi: 10.3201/eid1107.041008
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595.
Theethakaew, C., Feil, E. J., Castillo-Ramirez, S., Aanensen, D. M., Suthienkul, O., Neil, D. M., et al. (2013). Genetic relationships of Vibrio parahaemolyticus isolates from clinical, human carrier, and environmental sources in Thailand, determined by multilocus sequence analysis. Appl. Environ. Microb. 79, 2358–2370. doi: 10.1128/AEM.03067-12
Thongjun, J., Mittraparp-arthorn, P., Yingkajorn, M., Kongreung, J., Nishibuchi, M., and Vuddhakul, V. (2013). The trend of Vibrio parahaemolyticus infections in Southern Thailand from 2006 to 2010. Trop. Med. Health 41, 151–156. doi: 10.2149/tmh.2013-06
Ueno, H., Tomari, K., Kikuchi, K., Kobori, S., and Miyazaki, M. (2016). The first report of Vibrio parahaemolyticus O10:K60 in Japan, a new combination of O and K serotypes strain isolated from a gastroenteritis patient. Jpn. J. Infect. Dis. 69, 28–32. doi: 10.7883/yoken.JJID.2014.538
Velazquez-Roman, J., León-Sicairos, N., de Jesus Hernández-Díaz, L., and Canizalez-Roman, A. (2014). Pandemic Vibrio parahaemolyticus O3:K6 on the American continent. Front. Cell. Infect. Mi. 3:110. doi: 10.3389/fcimb.2013.00110
Velazquez-Roman, J., Leon-Sicairos, N., Flores-Villasenor, H., Villafana-Rauda, S., and Canizalez-Roman, A. (2012). Association of Pandemic Vibrio parahaemolyticus O3:K6 present in the coastal environment of Northwest Mexico with cases of recurrent Diarrhea between 2004 and 2010. Appl. Environ. Microb. 78, 1794–1803. doi: 10.1128/AEM.06953-11
Vuddhakul, V., Chowdhury, A., Laohaprertthisan, V., Pungrasamee, P., Patararungrong, N., Thianmontri, P., et al. (2000). Isolation of a pandemic O3:K6 clone of a Vibrio parahaemolyticus strain from environmental and clinical sources in Thailand. Appl. Environ. Microbiol. 66, 2685–2689. doi: 10.1128/AEM.66.6.2685-2689.2000
Wong, H. C., Liu, S. H., Wang, T. K., Lee, C. L., Chiou, C. S., Liu, D. P., et al. (2000). Characteristics of Vibrio parahaemolyticus O3:K6 from Asia. Appl. Environ. Microbiol. 66, 3981–3986. doi: 10.1128/AEM.66.9.3981-3986.2000
Yeung, P. S., Hayes, M. C., DePaola, A., Kaysner, C. A., Kornstein, L., and Boor, K. J. (2002). Comparative phenotypic, molecular, and virulence characterization of Vibrio parahaemolyticus O3:K6 isolates. Appl. Environ. Microbiol. 68, 2901–2909. doi: 10.1128/AEM.68.6.2901-2909.2002
Keywords: Vibrio parahaemolyticus, multilocus sequence typing, pandemic clone, gastroenteritis, phylogenetic analysis
Citation: Han C, Tang H, Ren C, Zhu X and Han D (2016) Sero-Prevalence and Genetic Diversity of Pandemic V. parahaemolyticus Strains Occurring at a Global Scale. Front. Microbiol. 7:567. doi: 10.3389/fmicb.2016.00567
Received: 18 October 2015; Accepted: 05 April 2016;
Published: 22 April 2016.
Edited by:
Julio Alvarez, University of Minnesota, USAReviewed by:
Biao Kan, National Institute for Communicable Disease Control and Prevention, ChinaKai Zhou, University Medical Center Groningen, Netherlands
Copyright © 2016 Han, Tang, Ren, Zhu and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongsheng Han, hands1103@163.com
†These authors have contributed equally to this work.