 Hasan C. Tekedar
Hasan C. Tekedar Attila Karsi
Attila Karsi Joseph S. Reddy
Joseph S. Reddy Seong W. Nho
Seong W. Nho Safak Kalindamar
Safak Kalindamar Mark L. Lawrence
Mark L. Lawrence- 1College of Veterinary Medicine, Mississippi State University, Mississippi State, MS, USA
- 2Mayo Clinic, Department of Health Sciences Research, Jacksonville, FL, USA
Flavobacterium columnare is a Gram-negative fish pathogen causing columnaris disease in wild and cultured fish species. Although the pathogen is widespread in aquatic environments and fish worldwide, little is known about biology of F. columnare and mechanisms of columnaris disease pathogenesis. Previously we presented the complete genome sequence of F. columnare strain ATCC 49512. Here we present a comparison of the strain ATCC 49512 genome to four other Flavobacterium genomes. In this analysis, we identified predicted proteins whose functions indicate F. columnare is capable of denitrification, which would enable anaerobic growth in aquatic pond sediments. Anaerobic growth of F. columnare ATCC 49512 with nitrate supplementation was detected experimentally. F. columnare ATCC 49512 had a relatively high number of insertion sequences and genomic islands compared to the other Flavobacterium species, suggesting a larger degree of horizontal gene exchange and genome plasticity. A type VI subtype III secretion system was encoded in F. columnare along with F. johnsoniae and F. branchiophilum. RNA sequencing proved to be a valuable technique to improve annotation quality; 41 novel protein coding regions were identified, 16 of which had a non-traditional start site (TTG, GTG, and CTT). Candidate small noncoding RNAs were also identified. Our results improve our understanding of F. columnare ATCC 49512 biology, and our results support the use of RNA sequencing to improve annotation of bacterial genomes, particularly for type strains.
Introduction
Flavobacterium columnare is in the family Flavobacteriaceae, which is one as the main phyletic lines within the Bacteroidetes (Bernardet et al., 1996). This family has several important fish-pathogens, including Flavobacterium psychrophilum, Flavobacterium branchiophilum, and Flavobacterium columnare. F. columnare is the causative agent of columnaris disease, which is common in freshwater fish throughout the world, infecting populations of wild and cultured fish species. In the United States, channel catfish is the leading aquacultured fish species, and columnaris disease is the second leading cause of mortalities in commercial catfish aquaculture (Durborow et al., 1998).
F. columnare is a long Gram-negative rod (2–10 μm in length) forming yellow-pigmented and typically rhizoid colonies (Bernardet et al., 1996). Four distinct colony types have been reported for F. columnare strains (Kunttu et al., 2009), which are divided into three genomovars based on 16S rRNA gene-based RFLP (Wakabayashi and Wakabayashi, 1999), and F. columnare ATCC 49512 belongs to genomovar I (Michel et al., 2002). Strains from the different genomovars affect geographically distributed fish species at different virulence levels (Arias et al., 2004; Darwish and Ismaiel, 2005; Shoemaker et al., 2008), Genomovar II strains tend to be more virulent than genomovar I strains in most fish species (Olivares-Fuster et al., 2011; Lafrentz et al., 2012), but there is considerable strain variation in virulence within the genomovars. Recently, a standard and optimized protocol was developed to distinguish F. columnare isolates using expected restriction patterns for genomovars I, II, II-B, and III (Lafrentz et al., 2014, 2016).
Despite its importance, there is a lack of essential information concerning columnaris disease, which limits the implementation of methods to manage, treat, and prevent the disease. Moreover, mechanisms of F. columnare pathogenesis are not completely understood. The nucleotide sequence of a pathogen's genome is a major step for understanding the mechanisms of pathogenesis.
To date, several complete genomes from Flavobacterium species have been released, including F. psychrophilum (Duchaud et al., 2007), F. johnsoniae (McBride et al., 2009), F. branchiophilum (Touchon et al., 2011), F. indicum (Barbier et al., 2012), and F. columnare (Tekedar et al., 2012). Here we present comparative genome analysis of the F. columnare ATCC 49512 genome. For the purposes of this comparison, we chose representative closed genomes from five of the best characterized species to enable a more complete functional comparison. The analysis showed, for the first time, that F. columnare is capable of denitrification. Transcriptome analysis of strain ATCC 49512 confirmed predicted gene annotations and identified 41 new protein coding regions, 16 of which have a non-traditional start site (TTG, GTG, and CTT). Thirty-six of the newly identified proteins are conserved hypothetical proteins, of which 30 may be involved in virulence based on similarity to proteins in MvirDB (Zhou et al., 2007). Our results provide important new information about the physiology of F. columnare and yield strong evidence for the utility of RNA-Seq to improve annotation of bacterial genomes.
Materials and Methods
Library Preparation and Sequencing
F. columnare strain ATCC 49512 is the type strain in the genomovar I group; it was isolated in 1987 from a skin lesion of brown trout (Salmo trutta) fry in France (Bernardet, 1989). Bacteria were cultivated in Flavobacterium columnare growth medium (FCGM) broth or agar (Farmer, 2004) at 30°C with shaking at 200 rpm. Genomic DNA was isolated and short, medium, and large insert libraries were prepared and sequenced as described (Tekedar et al., 2012).
Anaerobic Growth
The ability of F. columnare ATCC 49512 to grow anaerobically using nitrate as an electron acceptor was determined. Bacteria were grown under aerobic conditions in FCGM broth [tryptone (8.00 g), yeast extract (0.80 g), MgSO4 7 H2O (1.00 g), CaCl2 2H2O (0.74 g), NaCl (5.00 g), and sodium citrate (1.50 g) per liter] to an OD600 of 1.0. The culture was diluted 1/10 in FCGM and FCGM with 5 mM sodium nitrate broth medium and incubated under anaerobic conditions at 30°C using a GasPak anaerobic system (Fisher Scientific, PA, USA) in a standard anaerobic jar. Bacterial growth was monitored using OD600 for 1 week.
Determination of Extracellular Nitrite
Nitrite is an intermediate product during denitrification and is reduced to nitric oxide by nitrite reductase. To determine nitrite production during anaerobic growth, a diazotization reaction was used as described (Griess, 1879) with some modifications. Supernatant was collected from cultured bacteria following centrifugation, and 100 μl was transferred to a 96-well plate (Corning, NY, USA). Serially diluted sodium nitrite was used for a standard curve. Fifty microliters of sulfanilamide solution (Sigma-Aldrich; 1% sulfanilamide in 5% phosphoric acid) was added to each sample, and the plate was incubated for 10 min without light. Finally, 50 μl of N-1-naphthylethylenediamine dihydrochloride (NED) solution (Sigma-Aldrich; 0.1% NED in water) was added to each followed by another 10-min incubation without light. Color development was measured at 540 nM using a microplate reader (Molecular Devices, CA, USA).
Comparative Analysis of Flavobacterium Genomes
Five publicly available annotated Flavobacterium genomes (Flavobacterium columnare ATCC 49512 [CP003222], Flavobacterium branchiophilum FL-15 [NC_016001], Flavobacterium indicum GPTSA100-9T [NC_017025], Flavobacterium johnsoniae UW101 [NC_009441], and Flavobacterium psychrophilum JIP02/86 [AM398681]) were used for genome comparisons. For pathway analyses and comparison of gliding motility components, Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Database (http://www.genome.jp/kegg/) was used (Ogata et al., 1999; Kanehisa and Goto, 2000). Rast annotation was used to conduct a sequence-based comparison of the Flavobacterium genomes (Overbeek et al., 2014).
All the annotated Flavobacterium genome proteins were categorized according to their function using BAsys annotation server (Van Domselaar et al., 2005). Mauve genome alignment software was used to identify potential horizontal transfer loci and genomic rearrangements (Darling et al., 2004). PSORTdb (http://db.psort.org) version 3.0 was used to predict protein localization (Rey et al., 2005; Yu et al., 2011). Orthology analysis was conducted using OrtholugeDB (Whiteside et al., 2013) and Rast annotation server (Aziz et al., 2008; Overbeek et al., 2014).
EDGAR was used to determine phylogenetic relationships between the Flavobacterium genomes and to calculate genome to genome distances (Blom et al., 2009). Pan-core genome and singleton analysis of the Flavobacterium genomes was conducted with EDGAR (Blom et al., 2009). EDGAR was also used to create a synteny plot for the Flavobacterium genomes, which helps to show colocalization of genes on a stretch of DNA (Blom et al., 2009).
Functional Systems Analysis of Flavobacterium Genomes
Genome-wide transcriptional regulatory networks (TRNs) were computationally predicted using P2TF (Predicted Prokaryotic Transcriptional Factors) and P2CS (Prokaryotic 2-Component System) (Ortet et al., 2012, 2015). In these analyses, bacterial transcription factors, one-component systems (OCS), DNA-binding response regulators (RR), sigma factors (SF), transcriptional regulators (TR), other DNA-binding proteins (ODP), two-component systems, histidine kinases (classic, hybrid, and unorthodox), and phosphotransferase proteins [histidine phosphotransferases (Hpt) and histidine kinase homodimeric domain proteins (HisKA)] were computationally predicted.
MacSyFinder and TXSScan were used to detect protein secretion systems and their appendages in the Flavobacterium genomes (Abby et al., 2014). Protein files were downloaded from NCBI database for each genome. “Ordered replicon” was used as the type of dataset, and “circular” was used as the topology setting. Default parameters were used for detection of all available secretion systems (maximal e-value: 1.0, maximal independent E-value: 0.001, minimal profile coverage: 0.5).
CRISPR elements were identified for the five Flavobacterium genomes using CRISPRfinder (Grissa et al., 2007). CAS elements were identified using MacSyFinder (Abby et al., 2014). Maximal E-value was set at 1.0, maximal independent E-value was 0.001, and minimal profile coverage parameters were set for CAS element identification.
To identify possible virulence factors encoded in F. columnare ATCC 49512 and other Flavobacterium strains, Microbial Virulence Database (MVirDB) was downloaded from http://mvirdb.llnl.gov/ (Zhou et al., 2007), and local BLAST was conducted with all the predicted ATCC 49512 and other Flavobacterium strains, proteins using CLC Genomic Workbench (version 6.5). To be considered a match, Flavobacterium proteins had to have BLAST result in the MvirDB with an E-value <1*10−10. For comparison with other Flavobacterium strains, the predicted F. columnare ATCC 49512 virulence factor proteins were uploaded to the NCBI website to identify the best BLAST hit results. Distribution of predicted F. columnare ATCC 49512 virulence factors in metabolic pathways was analyzed using Blast2GO (Conesa et al., 2005).
Genomics Islands, Insertion Elements, and Phage Analysis
IslandViewer was used to predict genomic islands in the completed Flavobacterium genomes (Langille and Brinkman, 2009; Dhillon et al., 2013). IslandViewer combines three different genomic island prediction methods: IslandPick (Langille et al., 2008), SIGI-HMM (Waack et al., 2006), and IslandPath-DIMOB (Hsiao et al., 2003). Issaga was used to identify individual insertion sequences (Varani et al., 2011).
To identify phage elements in Flavobacterium genomes, Genbank files were downloaded for all the evaluated Flavobacterium genomes and submitted to the PHASTER (PHAge Search Tool- Enhanced Release) server (Arndt et al., 2016).
Transcriptome Analysis by RNA-Seq
For RNA isolation, F. columnare strain ATCC 49512 was grown in 10 ml Shieh broth at 30°C to log phase (OD600 0.8) and harvested by centrifugation. RNAprotect Bacteria Reagent (Qiagen) was immediately added, and pellets were stored at −80°C. Total RNA was isolated using RNA Qiagen Mini Kit, and rRNA was removed with the Ribo-Zero™ Magnetic rRNA removal kit (Illumina, San Diego, CA). Sequencing libraries were prepared using Truseq RNA v2 (Illumina) at Global Biologics (Columbia, MO) and pooled. Single-end 50 bp reads were sequenced on two lanes using an Illumina HiSeq 2000.
To validate genome annotation generated by the NCBI Prokaryotic Genome Annotation Pipeline (08.13.2015) and to identify unannotated functional elements, mRNA expression profiling of F. columnare was performed using RNA-seq. Transcriptome profiling of F. columnare using Illumina Hi-Seq 2000 generated a total of 68,164,700 single-end reads across six samples. Reads were processed for adaptor removal and filtered for quality using FastX clipper and FastQC, respectively. Reads were aligned to the F. columnare ATCC 49512 genome (NC_016510.2) using Bowtie 2 (Langmead and Salzberg, 2012) at default settings.
Bacterial Intergenic Region Analysis Pipeline (BIRAP) (https://github.com/jsreddy82/birap) was used for analysis of expression and evaluation of annotation. BIRAP is a command-line Perl pipeline that integrates expression data with in silico annotation to identify expressed annotated as well as unannotated regions. The tool capitalizes on the single nucleotide resolution generated by RNA-seq when reads are initially aligned to the genome using tools such as Bowtie and its subsequent representation as reads per base (RPB) in the pileup output generated by SAMtools (Li et al., 2009). The pileup file generated by SAMtools contains all expressed nucleotides and the total number of reads aligned to each.
BIRAP establishes a basal expression cutoff for each sample as the 10th percentile RPB for all expressed bases in the genome (Wurtzel et al., 2010; Kumar et al., 2012; Reddy et al., 2012). After a baseline for expression is established, RPB is binary transformed to represent one of two states. A value of 1 represents expression above baseline while 0 indicates no expression. Once expression across all samples is binary transformed, a statistical model for binary expression is calculated to determine expression at each nucleotide in the genome.
Normalized and binary transformed RNA-seq data is used for evaluation of annotation. An annotated region is considered as expressed only if at least 60% of all bases spanning the length of the gene are above baseline expression. BIRAP then identifies all expressed intergenic regions (EIRs) and verifies their association with annotated regions. A region of at least 70 bp in length, identified as expressed, and located between two annotated regions is classified as an EIR. If an EIR is located within 10 bp up- or downstream of an annotated region, then the EIR is marked as associated with that annotated region in the result file, which helps evaluate the annotated region's start and stop sites. If EIRs are not in the vicinity of annotated regions, they are marked as unpredicted EIRs. BIRAP evaluates EIRs as potentially being putative non-coding RNAs when promoters and terminators are predicted in appropriate locations. Small non-coding RNAs are known to regulate gene expression and are generally under the influence of a promoter or a rho-independent terminator (Reddy et al., 2012).
EIRs identified by BIRAP were searched against the translated nucleotide non-redundant bacterial database using BlastX (Altschul et al., 1997). EIRs mapping along the full length of closely related orthologs was classified as novel protein coding regions. EIRs homologous to 5′ and 3′ ends of proteins in closely related species were used to evaluate start and stop sites of annotated regions in the F. columnare genome. Promoters and rho-independent terminators were predicted using Genome2D (Baerends et al., 2004) and TranstermHP (Kingsford et al., 2007), respectively. EIRs identified as putative non-coding RNAs by BIRAP were searched against BlastX to eliminate false positives, and those that remained were searched against Rfam (Gardner et al., 2011) for sequence conservation across other species.
To provide an overview of gene regulation and operon structures, Database of prOkaryotic OpeRons (DOOR2) (Mao et al., 2014) was used to predict operons using the most recent annotation of F. columnare ATCC 49512 generated by NCBI. Operons encoding two or more genes were evaluated for expression using the RNA-seq expression profile with BIRAP.
Accession Numbers
The genome sequence of F. columnare ATCC 49512 has been deposited in the DDBJ/EMBL/GenBank database under the following accession number: CP003222, version CP003222.2 (GI: 372863588). RNA-Seq accession numbers are: 116-SRR1122816, 117-SRR1122817, 118-SRR1122818, 119-SRR1122819, 120-SRR1122820, 121-SRR1122821.
Results
Genome Features and Comparative Genomics
F. columnare ATCC 49512 chromosome features compared against F. johnsoniae UW101, F. psychrophilum JIP02/86, F. branchiophilum FL-15, and F. indicum GPSTA100-9T are listed in Table 1. All of the Flavobacterium strains have low G+C content. Genome size varies from 2,861,988 to 6,096,872 bp, with the F. johnsoniae UW101 genome being twice the size of the other flavobacteria. Only F. psychrophilum JIP/86 and F. branchiophilum FL-15 carry plasmids. Amino acid composition was similar between all the Flavobacterium genomes (Figure 1A). Predicted protein lengths were similar for the Flavobacterium genomes, except F. branchiophilum FL-15 genome had shorter protein lengths than the other four (Figure 1B). Predicted subcellular localization of proteins encoded by the Flavobacterium genomes was also similar (Figure 1C). Genome to genome distance calculation showed a better correlation with 16s rRNA gene distances than other evaluation methods (Auch et al., 2010); therefore, we applied this calculation method. Results showed that the F. columnare and F. psychrophilum genomes are closer than the other Flavobacterium genomes (Figure 1D).
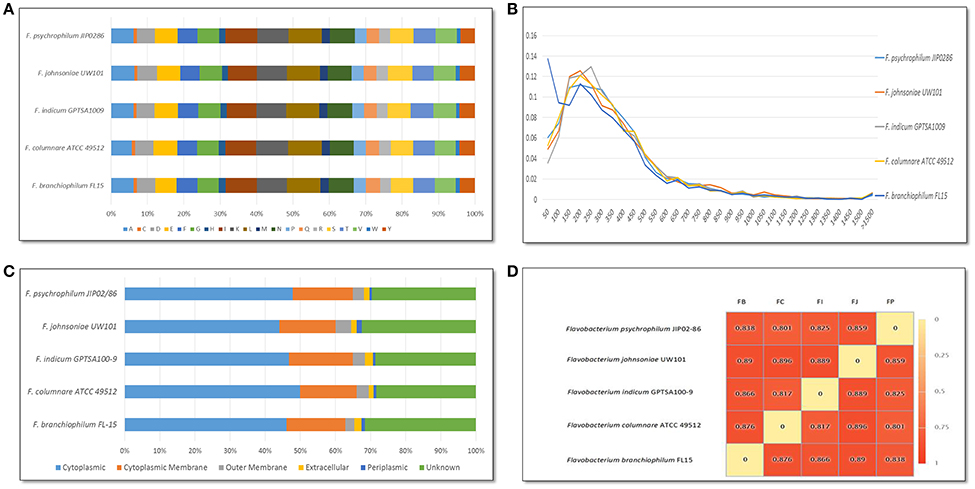
Figure 1. Comparison of Flavobacterium genome features. (A) Amino acid composition for Flavobacterium strains. Letter abbreviations: A, Alanine; C, Cysteine; D, Aspartic Acid; E, Glutamic Acid; F, Phenylalanine; G, Glycine; H, Histidine; I, Isoleucine; K, Lysine; L, Leucine; M, Methionine; N, Asparagine; P, Proline; Q, Glutamine; R, Arginine; S, Serine; T, Threonine; V, Valine; W, Tryptophan; Y, Tyrosine. (B) Predicted protein lengths for Flavobacterium strains. (C) Predicted subcellular localization of proteins encoded in Flavobacterium genomes using PSORTb. (D) Genome to genome distance calculation for Flavobacterium genomes. Species abbreviations: FB, F. branchiophilum FL15; FC, F. columnare ATCC 49512; FI, F. indicum GPTSA100-9; FJ, F. johnsoniae UW101; and FP, F. psychrophilum JIB02-86.
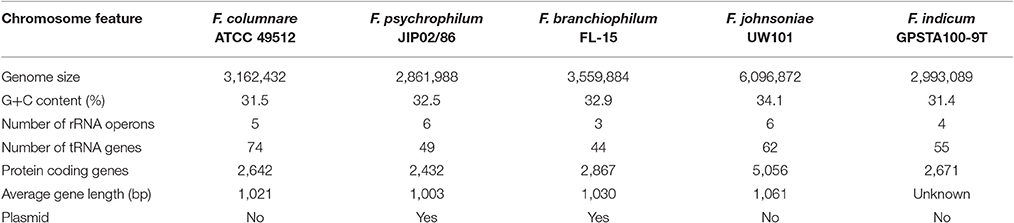
Table 1. Chromosome features of flavobacterium genomes.
Pan-Core and Singletons Genome Analysis
Bacterial pan-core and singletons analysis are used to identify unique proteins and to estimate the genomic diversity of evaluated genomes (Vernikos et al., 2015). Singletons are defined as genes encoding proteins unique to a particular species in the analysis. Our comparative pan-genome analyses of the five Flavobacterium genomes revealed that each Flavobacterium species' contributed unique proteins to the pan-genome (Figure 2A). The final core genome size was 1,252 genes, and the final pan-genome had 8,339 genes. An exponential decay model estimated that 930 new genes could be revealed for every new Flavobacterium genome added to the analysis (Figure 2B). A pan-core genome Venn diagram showed that F. columnare and F. psychrophilum had the least number of unique genes, possibly because these two species are the closest related (Figure 2C). F. branchiophilum and F. johnsoniae had the most shared genes because they are the two largest genomes in the comparison. As expected, F. johnsoniae had the highest number of unique genes, followed by F. branchiophilum.
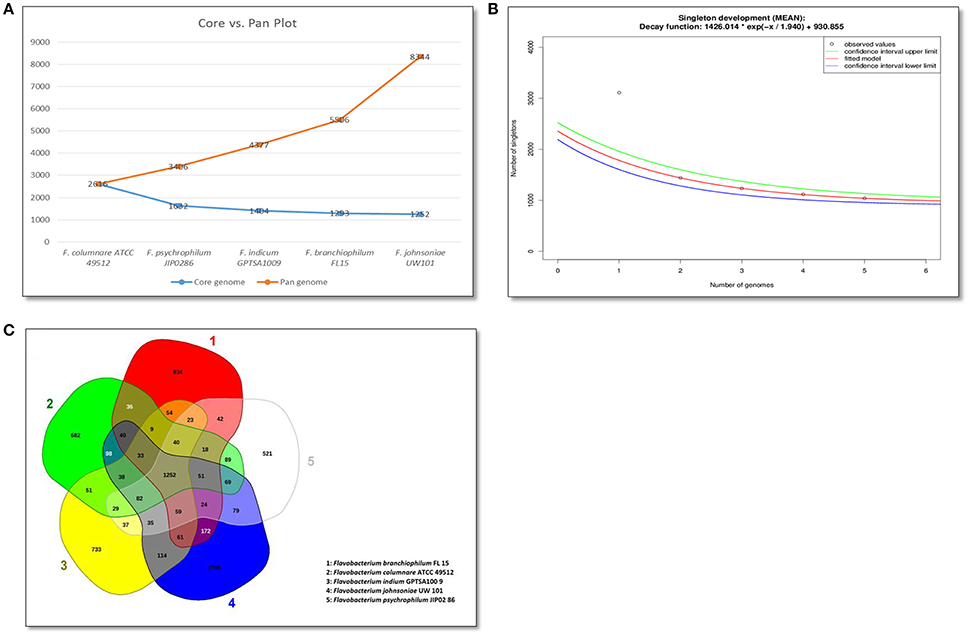
Figure 2. Flavobacterium pan core genomes analysis. (A) Core vs. pan-genome plot of the five evaluated Flavobacterium genomes. (B) Singleton (unique genes) analyses for Flavobacterium genomes. A decreasing number of singletons are observed with each sequential addition of a genome. (C) Venn diagram of Flavobacterium pan and core genomes based on orthology analysis.
Most of the unique genes for each species consisted of hypothetical proteins, insertion elements, phage components, and two component systems. More specifically, the F. columnare genome has unique nitrate/nitrite reductase components, IS3/IS911 insertion elements, cobalamin biosynthesis proteins, and CRISPR elements. The F. branchiophilum genome has unique IS595 insertion elements, bleomycin resistance proteins, and two-component system proteins. The F. psychrophilum genome has unique putative cell surface elements and IS3/IS982/IS256/IS1182 family insertion elements. The F. indicum genome has unique IS3/IS110 family proteins, and the F. johnsoniae genome has many unique hypothetical proteins and two component elements (Supplementary Table 1). Orthology analysis, described below, was used to better define the unique proteins for each species.
Orthology Analysis
Our orthology analysis clearly identified that the F. columnare ATCC 49512 genome has 495 genes encoding unique protein functions compared to the other Flavobacterium species analyzed. Of these, 319 encode hypothetical proteins. Many of the protein-coding genes unique to F. columnare and many of the conserved protein-coding genes were located in clusters (Figure 3). For example, highlighted region number one shows a unique F. columnare genomic island. Highlighted region number two is conserved in all the evaluated Flavobacterium genomes; these genes encode ribosomal subunit elements.
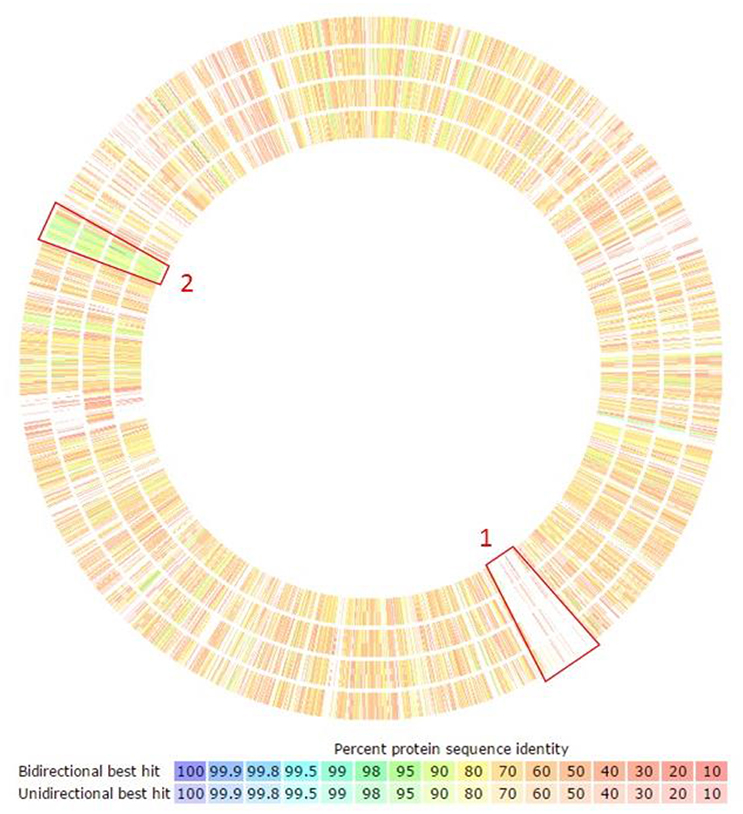
Figure 3. Orthology analysis of F. columnare compared against the other Flavobacterium species using RAST annotation. Circles display percent identity of proteins from each listed species compared to F. columnare. From outside to inside: Flavobacterium branchiophilum FL-15, Flavobacterium indicum GPTSA100-9, Flavobacterium johnsoniae UW101, and Flavobacterium psychrophilum JIP02/86. Boxed region 1 indicates a unique F. columnare genomic island. Boxed region 2 is conserved in all the evaluated Flavobacterium genomes; the genes in this region encode ribosomal subunit elements.
One of the protein functions unique to F. columnare ATCC 49512 is vitamin B12 synthesis. B12 biosynthesis occurs strictly in some bacteria and archaea; animals and protists require B12 but are not capable of synthesizing it (Martens et al., 2002). In the F. columnare ATCC 49512 genome, cobalamin biosynthesis elements, including CbiX, CbiD, CobD, CobW, and CobN subunit were identified. Also, two additional elements were identified that contribute to cobalamin biosynthesis: cobyric acid synthase and a,c-diamide synthase.
The F. columnare ATCC 49512 genome also encodes unique nickel permease function, which could play an important role in several cellular processes. Nickel can be essential for energy metabolism, nitrogen metabolism, detoxification, and virulence (Schauer et al., 2007). Helicobacter pylori is an example of a species where nickel metabolism contributes to virulence (Atherton, 2006). F. columnare ATCC 49512 is also unique in encoding neprilysin, which is considered to have a nutritional role in bacteria. It is a membrane-bound protein that is responsible for the degradation of biologically active peptides (Rawlings and Salvesen, 2013).
F. branchiophilum FL-15 encodes 722 unique proteins. Of these, 529 of them are hypothetical proteins. Some of the other genes encode bleomycin resistance proteins, glycoside family proteins, LytTR family two-component system response regulatory proteins, and transposases. Two of the unique proteins in the F. branchiophilum genome, TerZ/TerD, are widely present in prokaryotes. They can mediate resistance to tellurite and may be activated by signal transduction pathways triggered by oxidizing agents such as tellurite or hydrogen peroxide. It has been proposed that TerZ/TerD are responsible for intracellular Ca2+ signaling (Pan et al., 2011).
A total of 534 proteins is uniquely encoded by F. indicum GPTSA100-9. Of these, 424 are hypothetical proteins. Some of the other proteins include a glycosyltransferase, four OmpA outer membrane protein precursors, IS3 and IS110 family transposases, and two-component system response regulatory proteins.
The F. psychrophilum JIP0286 genome encodes 360 unique proteins; 293 are annotated as hypothetical proteins. Three two-component system response regulator proteins and a universal stress protein are unique to F. psychrophilum. A unique protein encoded by the bmgB gene was studied in Bacteroides fragilis and shown to play a role in DNA mobilization/horizontal gene transfer (Bass and Hecht, 2002).
As expected, the F. johnsoniae UW101 genome has the highest number of unique proteins: 2297. Of these, 1,199 are annotated as hypothetical. Some of these proteins include a ferric citrate transport system: FecR anti-Fecl sigma factor and TonB dependent receptors/siderophores. In this systejom, ferric citrate initiates the signal transduction mechanism by interacting with FecA protein using a TonB-dependent element. The signal is transmitted to FecR, which activates transcription of transport genes (Angerer et al., 1995).
Transcriptional Factors and Two-Component Systems
The percentage of TFs (transcriptional factors) and TCs (two-component systems) in the fish pathogens' genomes [F. branchiophilum FL-15 (3.04% TFs, 1.51% TCs), F. columnare ATCC49512 (2.49% TFs, 1.17% TCs), and F. psychrophilum JIP0286 (2.96% TFs, 1.32% TCs)], is slightly less than the percentage in environmental isolates F. indicum GPTSA100-9 (3.11% TFs, 2.28% TCs) and F. johnsoniae UW101 (5.59% TFs, 2.89% TCs) (Table 2) (Supplementary Table 2). F. johnsoniae UW101 has more TFs and TCs than any other Flavobacterium genome, reflecting its more complex environmental niche. Notably, TC response regulator PglZ is conserved among all the Flavobacterium genomes. Histidine phosphotransferases (HPT), a part of the TCs, are present only in the F. branchiophilum FL-15 and F. columnare ATCC49512 genomes. F. branchiophilum FL-15 encodes two putative unorthodox histidine kinases. F. branchiophilum FL-15 encodes more TFs than the other two fish pathogenic species. However, a similar percentage of histidine kinases and response regulators are encoded in all fish pathogenic Flavobacterium genomes.
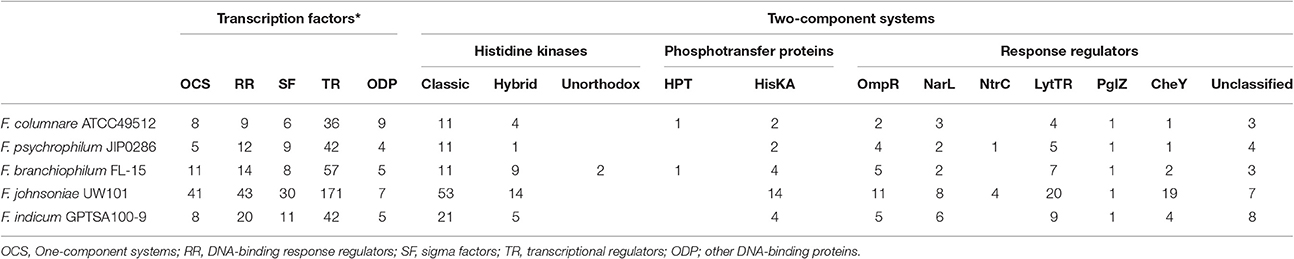
Table 2. Transcriptional factors and two-component systems in Flavobacterium genomes.
Secretion Systems
Gram-negative bacteria have several types of secretion systems (Koster et al., 2000; Johnson et al., 2006). Flavobacterium secretion systems are shown in Table 3. The T9SS system is unique in functioning as both a secretion system and contributing to gliding motility (McBride and Nakane, 2015). All five of the Flavobacterium genomes encode one whole T9SS operon, but the porQ gene is present in all Flavobacterium genomes twice.
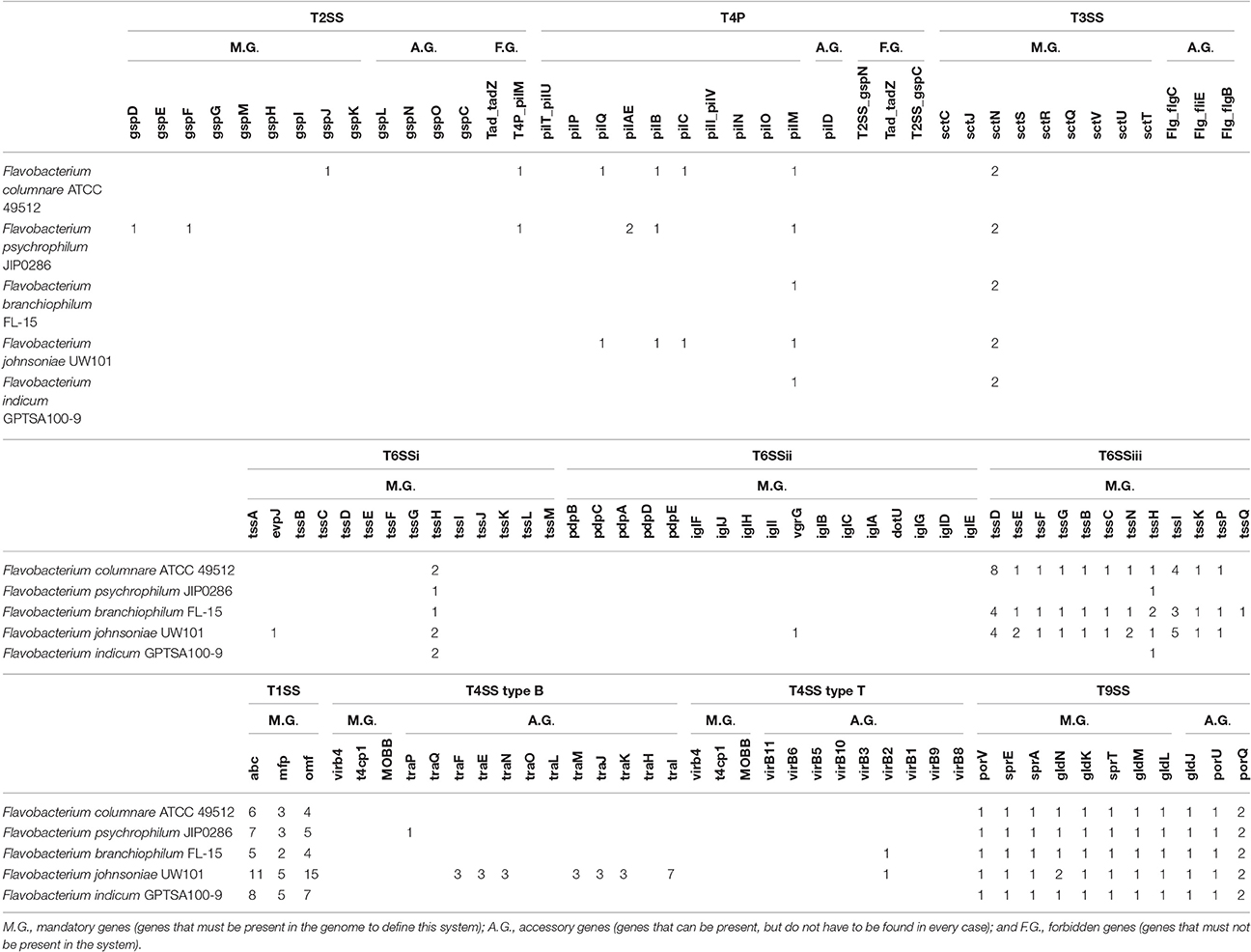
Table 3. Secretion systems distribution in Flavobacterium genomes.
The type VI secretion-related pathway (designated subtype III or T6SSiii) is restricted to the Bacteroidetes phylum (Russell et al., 2014; Abby et al., 2016), but our comparative analysis showed that F. psychrophilum JIP0286 and F. indicum GPTSA100-9 do not carry T6SSiii. On the other hand, F. branchiophilum FL-15 encodes the entire operon of the T6SSiii, whereas F. columnare ATCC 49512 and F. johnsoniae UW101 are missing the tssQ gene. Interestingly, one of the components of T6SSiii is TssD, which has eight gene copies in F. columnare and four in F. branchiophilum and F. johnsoniae. TssD is one of the core elements of the T6SSiii injection mechanism, participating in the assembly of the secretion system and export of its effectors (Silverman et al., 2013).
All five of the Flavobacterium genomes carry a complete Type I secretion system, which consists of three essential components: ABC-transporter (abc), porin (omf) and the inner-membrane-anchored adaptor protein (mfp). The T1SS is encoded multiple times in the five flavobacterial genomes. T1SS is responsible for secretion of toxins and various other proteins ranging in size from 5.8 to 900 kDa (Kanonenberg et al., 2013).
Interestingly, only the F. johnsoniae UW101 genome carries T4SS elements. However, although it carries multiple copies of some T4SS type B accessory elements, F. johnsoniae has none of the mandatory T4SS genes, indicating a functional T4SS is not present in any of the Flavobacterium genomes. This system is used by some pathogenic bacteria to translocate virulence genes and mediate horizontal gene transfer (Wallden et al., 2010). T3SS, which is important in many Gram-negative pathogens, is not encoded by any of the Flavobacterium species analyzed. T2SS is also not encoded by these five species; this system can perform a wide range of functions such as adhesion, signaling, and motility (Nivaskumar and Francetic, 2014). Some of the components of type IV pili (T4P) are encoded by F. columnare, F. psychrophilum, and F. johnsoniae. However, other critical elements of a T4P system are not encoded by these species. T4P is very similar to T2SS.
CRISPR-Cas Systems Analysis
All of the evaluated Flavobacterium genomes either carry conserved or possible CRISPR (Clustered regularly interspaced short palindromic repeats)-Cas regions and direct repeat sequences. All the identified CRISPR elements and their components are listed in Table 4. The Flavobacterium genomes have CRISPR spacers, and such repeat regions could be the result of the horizontal transfer and contribute to flavobacterial genome plasticity (Lopatina et al., 2016). Some bacteria collected from Antarctic surfaces in a metagenomics study carry more spacers with a high level of diversity, which may result from a need for mobile genetic elements to adapt to extreme Antarctic conditions (Lopatina et al., 2016). CRISPR regions in the F. columnare ATCC 49512, F. psychrophilum JIP0286, and F. branchiophilum FL-15 genomes carry multiple spacer elements ranging in size from 29 to 78 bp.
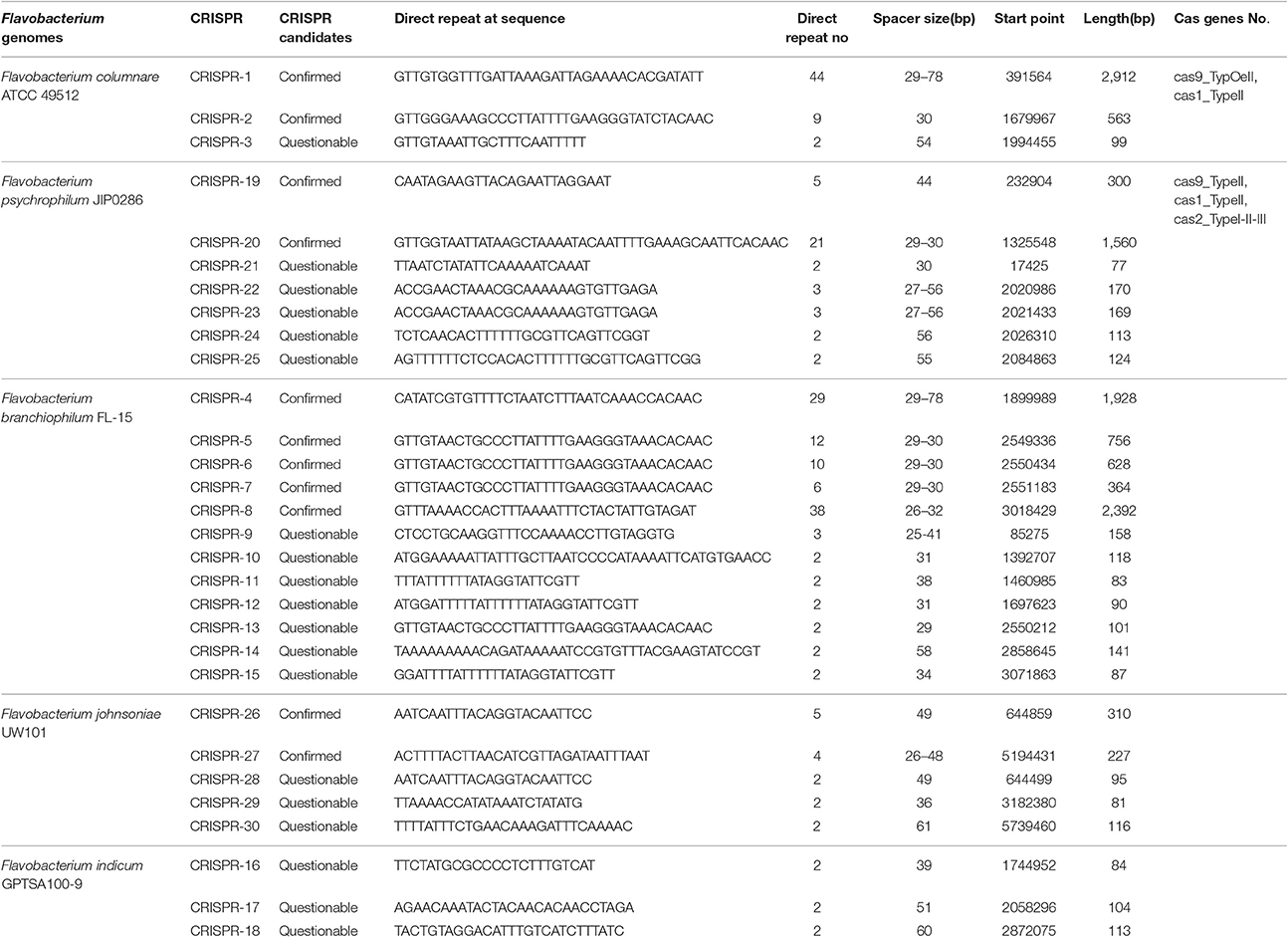
Table 4. CRISPR-Cas systems analysis of Flavobacterium genomes.
Interestingly, only F. columnare ATCC 49512 and F. psychrophilum JIP0286 genomes carry cas genes flanking the CRISPR regions. The F. columnare genome carries two different types of cas genes; one of them is cas9, and the other one is cas1. The F. psychrophilum genome carries three different types of cas genes: cas1, cas2, and cas9. Bacteria use the CRISPR-Cas system to provide resistance against foreign elements such as plasmids or phages. Cas1 and Cas2 catalyze spacer acquisition from foreign DNA, and Cas9 (also called Csn1) participates in blocking transcription (Nunez et al., 2015).
Genomic Islands
Genomic islands are clusters of genes that often are associated with microbial adaptations of medical or environmental interest (Langille et al., 2008). Often the G+C content of pathogenicity islands differs from that of the rest of the host genome, clearly indicating that those regions originated or adapted from other bacteria by horizontal gene transfer (Dobrindt and Reidl, 2000). Interestingly, G+C content of genomic islands in the flavobacterial genomes did not differ dramatically from the overall genome G+C content (for example, F. columnare ATCC 49512 genome G+C content is 31.5% compared to 32.3% for the genomic islands), but genomic islands are still identifiable using other features.
Fourteen genomic islands were identified in F. columnare ATCC 49512 ranging from 5 to 30 kb in length. By comparison, F. johnsoniae UW101 has 29 predicted genomic islands, F. psychrophilum JIP02/86 has five, F. branchiophilum FL-15 has seven, and F. indicum GPSTA100-9T has eight. All of the genomic islands appear unique; none are shared between species. Genomic islands often carry transmissible genomic elements such as transposons, bacteriophages, or plasmids (Hacker et al., 1997). Three of the F. columnare islands include genes encoding integrase, integrase catalytic subunit, or integrase family protein, suggesting horizontal gene transfer was the source of these genomic islands. These genes are commonly found in pathogenicity islands and can be responsible for the formation, integration, deletion, and mobility of pathogenicity islands (Hacker and Kaper, 2000). Putative proteins encoded in the predicted F. columnare ATCC 49512 and other Flavobacterium genomes' genomic islands are provided in Supplementary Table 3.
Insertion Elements
Insertion elements are widely distributed in bacteria and play an important role in prokaryotic genome evolution and organization due to horizontal gene transfer (Kusumoto et al., 2011). Insertion element expansion can also contribute to genome rearrangement or genome reduction. Eleven different insertion element families in the F. columnare ATCC 49512 genome and 73 total insertion elements were identified (Varani et al., 2011). In comparison, F. psychrophilum JIP02/86 had 12 families and 50 insertion elements, F. branchiophilum FL-15 had 12 families and 46 insertion elements, F. indicum GPTSA100-9 had 5 families and 14 insertion elements, and F. johnsoniae UW101 had 8 families and 22 insertion elements (Supplementary Table 4) (Figure 4). F. columnare ATCC 49512 had the largest number of insertion elements, but F. branchiophilum FL-15 and F. psychrophilum JIP02/86 had slightly more variety of insertion families. Interestingly, F. johnsoniae had fewer insertion elements. Therefore, it appears that the number of flavobacterial insertion elements is not directly related to genome size.
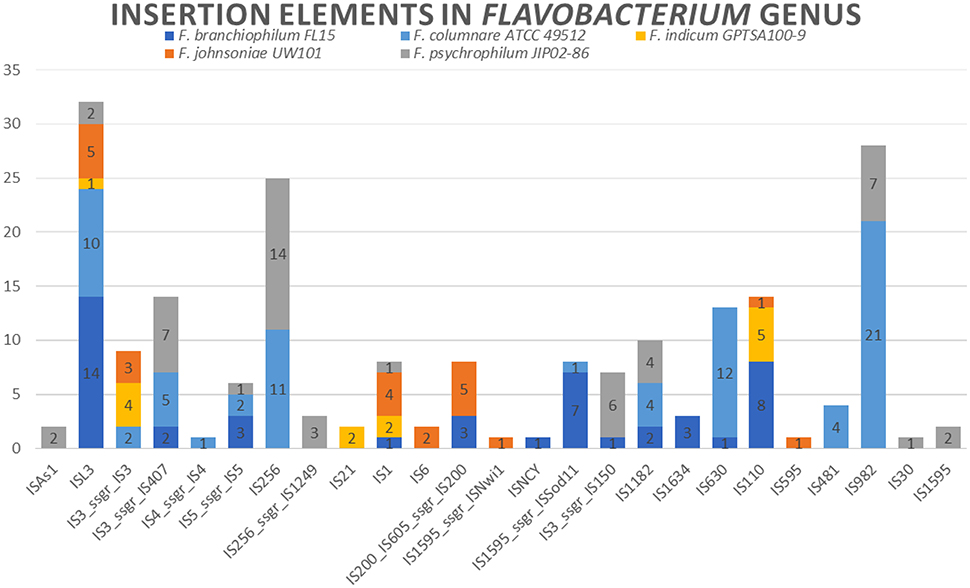
Figure 4. Insertion elements distribution in Flavobacterium genomes.
All the Flavobacterium genomes carry ISL3 family insertion elements. More than 80 bacterial species carry insertion elements in this family. Genes encoding these insertion elements range in size from 1,186 to 1,553 bp. The typically have imperfect repeats ranging in size from 15 to 39 bp along with 8 bp direct repeats. Additionally, the protein usually includes an α-helical domain (Siguier et al., 2015). In Pseudomonas stutzeri, ISL3 elements' translocation can be induced by zygotic induction after conjugative interaction (Christie-Oleza et al., 2010). There is evidence that insertion elements do not always insert randomly but often target preferred sequence elements (Siguier et al., 2014). Target preference for ISL3 elements is not known, but our comparative analysis and general Flavobacterium low G+C genome content suggests that ISL3 members could target AT-rich regions.
IS982 and IS256 elements are present only in F. columnare ATCC 49512 and F. psychrophilum JIP02/86 genomes. In particular, IS256 elements contribute to genome flexibility, genome evolution, and adaptation (Hennig and Ziebuhr, 2010).
Prophages
Prophages can induce virulence of opportunistic bacteria by activating virulence factors (such as in Vibrio cholerae) (Waldor and Mekalanos, 1996). Some bacteria can gain resistance against prophages, which can have the effect of reduced virulence (such as in Salmonella enterica) (Rychlik et al., 2001). F. columnare ATCC 49512 has one incomplete bacteriophage region (Supplementary Table 5). Three incomplete regions are in the F. branchiophilum FL-15 genome, and one incomplete and one questionable region are in the F. johnsoniae UW101 genome. Only the F. psychrophilum JIP02/86 genome carries a complete phage region, which has been previously described (Castillo et al., 2014) and is absent in other Flavobacterium genomes (Wiens et al., 2014). F. indicum GPTSA100-9 genome does not carry any predicted bacteriophage regions.
The incomplete F. columnare phage region size is ~19 kb, and it contains 18 ORFs. Most encode hypothetical proteins, but some of the ORFs encode proteins with predicted functions, including AttL, phage tail tape measure protein, and phage lysin proteins. The incomplete phage regions in F. branchiophilum FL-15 are 10.5 kb (12 ORFs), 6.8 kb (7 ORFs), and 7.5 kb (8 ORFs), and they include ORFs encoding chemical-damaging agent resistance protein and toxic ion resistance protein, which are unique to this genome. The incomplete phage region in F. johnsoniae UW101 has 21 ORFs (15.6 kb), and one of its ORFs encodes arsenate reductase. F. columnare, F. psychrophilum, and F. johnsoniae phage regions all include attL gene, and F. psychrophilum and F. johnsoniae also carry the attR gene. AttL and AttR proteins are responsible for creating recombinant junctions with the help of phage integrase gene.
The phage region in F. psychrophilum JIP02/86 is classified as bacteriophage type 6H, which is an abundant type of temperate phage that is lysogenized in a large number of F. psychrophilum strains (Castillo et al., 2014). It is a 47-kb phage region that has 64 ORFs (Supplementary Table 5). One of the ORFs encodes LuxR family protein, which is present only in the F. psychrophilum genome. The majority of the ORFs in the F. psychrophilum phage region encode hypothetical proteins.
Predicted Virulence Factors
The Flavobacterium genomes are valuable tools for identification of potential virulence mechanisms. To be considered candidate virulence factors in the current study, Flavobacterium proteins had to have BLAST result in MvirDB with an E-value <1*10−10. Using these criteria, F. columnare ATCC 49512 encodes 567 potential virulence proteins, F. branchiophilum FL-15 has 604, F. indicum GPTSA100-9 encodes 560, F. johnsoniae UW101 has 1141, and F. psychrophilum JIP02/86 has 557 (Supplementary Table 6). However, it should be noted that sequence similarities to proteins in MvirDB, which is a repository of microbial virulence proteins, does not necessarily indicate that the identified flavobacterial proteins have the same virulence functions. In particular, F. indicum and F. johnsoniae are not known to be pathogenic, so it is likely that many of these protein matches from these species do not function in virulence.
An example of a putative virulence factor in this list is KatG (catalase-peroxidase), which is present in F. columnare ATCC 49512 and other Flavobacterium genomes. KatG can enable resisting the oxidative burst in phagocytes. In the fish pathogen Edwardsiella tarda, KatG provides protection against endogenous and exogenous H2O2,and it is involved in pathogenesis (Xiao et al., 2012). Another example is GdhA (glutamate dehydrogenase), which is encoded in the F. columnare ATCC 49512 genome and other Flavobacterium genomes. GdhA is important in bacterial nitrogen metabolism; in Streptococcus pneumoniae, GdhA is expressed in nitrogen limitation and is tightly regulated by virulence proteins GlnR and CodY (Hendriksen et al., 2008). Another potential virulence gene identified encodes ClpP protease, which plays an important role in stress response and biofilm formation in pathogenic bacteria. Mutation of this gene in Actinobacillus pleuropneumoniae showed that it mediates tolerance to several stressors and regulates virulence and iron utilization (Xie et al., 2013).
Several proteins with matches in MvirDB are encoded in all the evaluated Flavobacterium genomes. These include alginate o-acetyltransferase (AlgI), ABC transporter ATP-binding proteins, aspartate alpha-decarboxylase, bacteriophage components, capsular polysaccharide synthesis enzymes, cold shock-like proteins, CTP synthetize, ferric uptake regulators, glutamate dehydrogenase, invasion regulatory proteins, and thioredoxin reductase (TrxB).
F. columnare ATCC 49512 encodes 89 unique potential virulence proteins that are not in the other four flavobacterial genomes. Among these are a catalase and peroxiredoxin, which are both involved in bacterial resistance to toxic peroxides. A TonB-dependent siderophore receptor is also unique to F. columnare, as well as a thiol-activated cytolysin that is similar to listeriolysin O and streptolysin O. Several polysaccharide envelope synthesis enzymes are unique to F. columnare, including an N-acetyl neuramic acid synthase, a UDP-N-acetylglucosamine-2-epimerase, glycosyl transferases, and capsular polysaccharide synthesis enzymes. Several transcriptional regulators and sensor histidine kinases are also unique to F. columnare ATCC 49512.
Metabolism
Metabolic pathways of F. psychrophilum have been extensively described (Duchaud et al., 2007). Pathway analyses using the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Database (Ogata et al., 1999; Kanehisa and Goto, 2000) revealed that F. columnare ATCC 49512 genome has a complete tricarboxylic acid (TCA) cycle and glycolysis pathway. All of the other Flavobacterium strains in our comparative analysis also have complete TCA and glycolysis pathways. However, some carbohydrate metabolism components are unique to F. johnsoniae such as pentose phosphate pathway and pentose/glucuronate interconversion elements. Moreover, pathway comparison showed that starch and sucrose utilization pathways are present only in F. johnsoniae and F. branchiophilum.
F. columnare ATCC 49512 genome has predicted protein functions in oxidative phosphorylation, which is driven by the electron transport chain and TCA cycle (McNeil and Fineran, 2013). Oxidative phosphorylation consists of five different complexes, and our comparative analysis showed that all Flavobacterium strains encode oxidative phosphorylation components except for two missing genes. One of the missing genes encodes a protein in complex II, which is an important respiratory enzyme participating in both electron transport chain and TCA cycle. Complex II consists of four subunits: SdhA, SdhB, SdhC, and SdhD. SdhD is a hydrophobic membrane anchor and is missing in all the Flavobacterium strains (McNeil and Fineran, 2013). F. johnsoniae is also missing one gene encoding a protein from complex IV, cytochrome c oxidase cbb3-type subunit III. F. johnsoniae and F. branchiophilum also encode two cytochrome bd complex proteins (CydA and CydB) that are missing in the other Flavobacterium species.
F. columnare and F. johnsoniae are unique among the sequenced Flavobacterium species in that they encode more proteins in nitrogen metabolism. The other sequenced Flavobacterium strains do not carry 80% of the nitrogen metabolism genes carried by F. columnare and F. johnsoniae. In particular, F. columnare ATCC 49512 encodes protein functions suggesting it is capable of denitrification, which results in a reduction of nitrate and nitrite to molecular nitrogen (N2). It has predicted protein functions indicating that it is capable of nitrous oxide reduction, nitric oxide reduction, nitrite reduction, nitrate reduction, and nitrite/nitrate transport. Nitrate and nitrite are abundant in eutrophic freshwater environments where F. columnare is commonly found, particularly in warm water aquaculture systems where it is a common fish pathogen. Its ability to utilize nitrate and nitrite as electron acceptors for the electron transport system suggest that it is capable of actively metabolizing and multiplying in pond sediments that are commonly oxygen depleted or anaerobic.
Interestingly, F. columnare has a complete urea cycle, but it does not encode a urease. Ammonia is typically abundant in eutrophic freshwater environments, but the advantage to F. columnare of converting ammonia to urea is not currently clear. In Helicobacter pylori, it appears the urea cycle is used to maintain intrabacterial nitrogen balance (Mendz and Hazell, 1996). In marine diatoms growing in nutrient-rich environments, the urea cycle is important for the response of diatoms to episodic changes in nitrogen availability (Allen et al., 2011).
Fatty acids are crucial components of membranes and play an important role in energy metabolism. To maintain membrane lipid homeostasis, fatty acid biosynthesis and degradation pathways must be controlled coordinately (Fujita et al., 2007). Our metabolic pathway analysis showed that all the sequenced Flavobacterium species have fatty acid biosynthesis, whereas fatty acid degradation pathway is present only in the F. johnsoniae genome.
Bacteria require iron as an essential nutrient. Iron acquisition is particularly important for pathogenic bacteria because of limited availability inside the host (Beaz-Hidalgo and Figueras, 2013). Therefore, the iron acquisition is linked to bacterial virulence (Guan et al., 2013). Gram-negative bacteria iron uptake mechanisms have been reviewed (Brown and Holden, 2002). Recently, two iron acquisition elements were described from F. columnare (Guan et al., 2013). One of them was TonB-dependent ferrichrome-iron receptor precursor FhuA, and the second was putative ferric uptake regulator Fur. We identified a predicted gene encoding FhuA in F. columnare ATCC 49512, and the genome also has three iron ABC transporter genes encoding FhuD, FhuB, and FhuC. Also, a gene encoding ferric iron uptake transcriptional regulator (Fur) is encoded in the F. columnare genome. F. johnsoniae, F. psychrophilum, F. branchiophilum, and F. indicum have the same iron acquisition elements.
Gliding Motility
Bacterial gliding motility is an energy requiring process of bacterial translocation over a surface. In this process, flagella are not required, and bacterial movement is along the long axis of the bacterium. Gliding motility results in thin spreading edges on colonies (McBride, 2001), and it is considered one of the characteristics of some species in the phylum Bacteroidetes. Many of the motility genes from this phylum are novel and are not found outside the Bacteroidetes. There are two major protein systems responsible for gliding motility. First, Gld proteins are components of the “motor” that moves the cell. Proteins encoded by gldA, gldB, gldD, gldF, gldG, gldH, gldI, gldJ, gldK, gldL, gldM, and gldN are required for gliding (Nelson et al., 2008). Second, Spr proteins are cell surface proteins responsible for adhesion or protein secretion (McBride et al., 2009). Deletion or disruption of genes encoding Gld proteins results in loss of motility, but spr deletion causes partial loss of gliding motility (Nelson et al., 2008).
Gliding motility of flavobacteria is well described (Bernardet et al., 1996; Decostere et al., 1998), particularly in F. johnsoniae. However, it has not been characterized in F. columnare. In the F. columnare ATCC 49512 genome, 13 gliding motility genes were identified (including a gene encoding GldL, which is currently annotated as a hypothetical protein). GldE gene is missing in F. columnare ATCC 49512, F. psychrophilum JIB02/86, and F. indicum GPSTA100-9T. In F. johnsoniae, overexpression of gldE gene partly suppressed the motility defects of a gldB mutant, suggesting that gldB and gldE might have overlapping functions (Hunnicutt and McBride, 2001). Interestingly, GldO is present only in F. johnsoniae UW101; this protein is similar to GldN, and the two proteins can partially replace each other for function (Rhodes et al., 2010). Genes encoding SprA, SprB, SprC, SprD, SprE, SprF, and SprT were identified in all the evaluated Flavobacterium genomes. Moreover, like most Flavobacterium species, they encode multiple paralogs of some spr genes in their genomes.
F. columnare Anaerobic Growth
Denitrification occurs in the absence of O2.Nitrate serves as an electron acceptor for anaerobic respiration. Although not as efficient as aerobic respiration, it provides microbes with a relatively high amount of energy. In bacteria with denitrification capability, nitrate reduction is mediated by one of two enzymes; one is a membrane-bound nitrate reductase, and the other is a soluble cytoplasmic nitrate reductase (Gennis and Stewart, 1996). The latter is encoded in the genome of F. columnare ATCC 49512. Subsequently, the reduction of nitrite to dinitrogen through nitric oxide and nitrous oxide is catalyzed by reductases encoded by nirK, norB, and nosZ.
F. columnare ATCC 49512 has the genetic capability to completely reduce nitrate into nitrogen due to the presence of genes encoding all the denitrification reductases (Tekedar et al., 2012), suggesting that F. columnare ATCC 49512 can grow anaerobically using nitrogenous compounds as electron acceptors for anaerobic respiration. In the present study, we compared F. columnare ATCC 49512 growth under aerobic and anaerobic conditions in presence and absence of nitrate supplementation (Figure 5). Our results indicate that nitrate did not improve aerobic growth, but it did improve anaerobic growth. As expected, anaerobic growth with nitrate supplementation was not as efficient as aerobic growth. However, our results suggest for the first time that F. columnare is capable of anaerobic growth, and that anaerobic growth is improved in the presence of 5 mM sodium nitrate. Extracellular nitrite production was not detected, indicating this intermediate product was likely subsequently reduced to nitric oxide without accumulation.
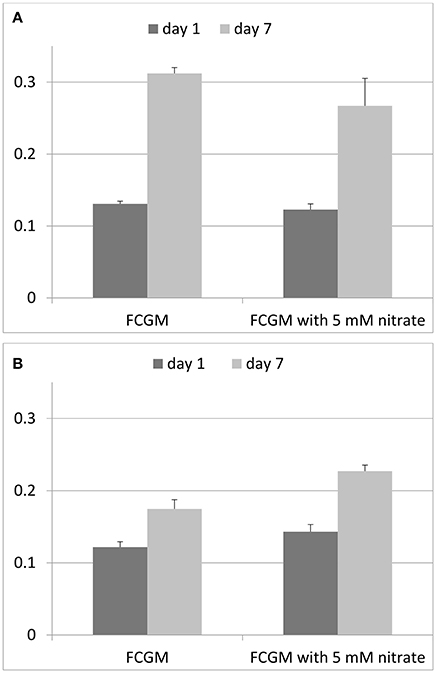
Figure 5. Growth of F. columnare ATCC 49512 in FCGM and FCGM supplemented with 5 mM nitrate. (A) Growth under aerobic conditions and (B) growth under anaerobic conditions.
Transcriptome Analysis
A total of 63,390,073 reads were aligned to the genome using Bowtie 2 with an average 98% alignment per sample. Genome annotation for F. columnare was downloaded from NCBI FTP and used for analysis of expression with BIRAP. From the expression profile generated by RNA-seq, a basal expression cutoff was calculated per million reads for each sample. Of the 2,772 annotated genes, BIRAP identified 2,635 as expressed above baseline. Of the 137 that were not expressed, 64 were annotated as hypothetical proteins (~47%).
A total of 1,132 EIRs of size 70 bp or more were identified by BIRAP and were analyzed. BlastX search against the non-redundant bacterial database identified 41 that had matches with annotated bacterial proteins in other species (Table 5), 16 of which had a non-traditional start site (TTG, GTG, and CTT). Thirty-six of these proteins matched with conserved hypothetical proteins, while a few proteins matched with proteins that had annotated functions defined. Identified protein functions include an ATPase, a DUF4295 domain-containing protein, molybdopterin molybdenum transferase MoeA, rod shape-determining protein MreD, and a Teichoic-acid-transporting ATPase. All 41 proteins were searched against virulence database MvirDB (Zhou et al., 2007), and 30 of them had matches, suggesting potential roles in virulence (Table 5).
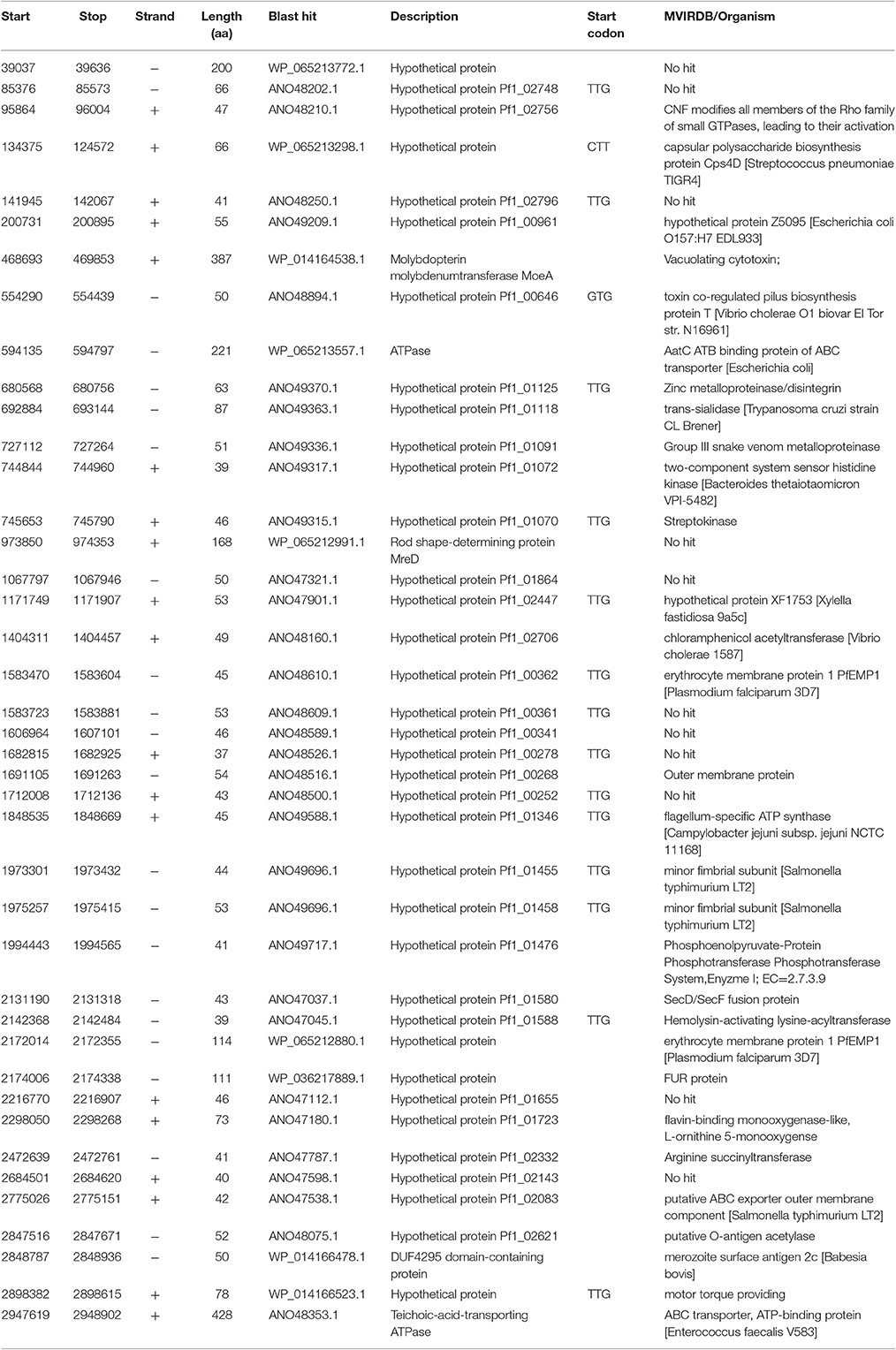
Table 5. Novel protein coding regions.
Some of the EIRs were adjacent to the 5′ end of annotated genes in the F. columnare ATCC 49512 genome, suggesting they could represent start codon corrections. BlastX searches of these EIRs resulted in updated start sites of 25 annotated protein coding regions (Supplementary Table 7), three of which (WP_041253481.1, WP_014166307.1, WP_041253585.1) used an alternate start codon (TTG). Of the 25 protein coding regions, 15 were hypothetical, and 10 had annotated functions.
EIRs with promoters or terminators associated with their loci and that did not have protein matches by BlastX were considered candidate putative non-coding RNAs (Supplementary Table 8). These were searched against Rfam database, but none of the putative non-coding RNAs matched conserved non-coding RNAs in Rfam.
Operons in the F. columnare ATCC 49512 genome were predicted using DOOR2. Our RNA-seq data enables experimental evaluation of these predictions. A total of 546 operons with two or more proteins were predicted by DOOR2, and RNA-seq confirmed expression of 526 operons. The complete operon structure determined by RNA-seq and comparison against DOOR2 predictions is presented in Supplementary Table 9.
Discussion
In this study, we aimed to compare the complete genome sequence of F. columnare ATCC 49512 against four other closed and annotated representative flavobacterial genomes to identify unique features and increase our understanding of this important fish pathogen. One of the novel capabilities we identified in the F. columnare ATCC 49512 genome is anaerobic respiration using nitrate, nitrite, nitric oxide, and nitrous oxide as electron acceptors. We confirmed this capability experimentally. We also aimed to confirm the predicted annotation using RNA-seq and identify expressed intergenic regions not predicted by our automated annotation method. Our results revealed that RNA-seq is an important tool to improve genome annotation by identification of unannotated intergenic expressed regions, including sRNAs.
Bacterial pan-core and singletons analysis have been used extensively to understand genomic variation between species. The F. johnsoniae genome had the greatest contribution of singletons in our analysis, increasing pan-genome size from 5,506 to 8,344 genes, which reflects its unique metabolism adapted to environmental growth. The pan-genome to core genome ratio of the flavobacterial genomes is relatively high compared to other pan-genome analyses such as Propionibacterium acnes (Tomida et al., 2013) and Erwinia amylovora (Mann et al., 2013). This likely reflects that our analysis was at the genus level, and it also reflects the variety of environmental niches for the flavobacterial species we analyzed. However, despite the variety of their lifestyles, the flavobacterial genomes also had a significant group of core functions (1,252 genes), reflecting similar adaptations to aquatic environments and conserved metabolic strategies for survival. The large pan-genome size of the Flavobacterium species may also reflect significant horizontal gene acquisition; in the aquatic and fish host environments, flavobacteria are expected to be exposed to a variety of bacterial species.
Analysis of the F. columnare ATCC 49512 genome revealed it has similar core metabolic functions as other flavobacterial genomes, including a complete TCA cycle and glycolysis pathway. Fatty acid biosynthesis pathways are encoded in the F. columnare ATCCC 49512 genome, whereas the fatty acid degradation pathway is missing. This indicates that F. columnare relies more on carbohydrates and proteins for energy than lipids. Like the other flavobacteria, F. columnare ATCC 49512 encodes oxidative phosphorylation functions. However, unlike the other flavobacteria, F. columnare is unique in having the capability of using alternate electron acceptors for respiration. Namely, it encodes proteins whose functions indicate F. columnare is capable of denitrification, which would allow it to utilize nitrate, nitrite, nitric oxide, and nitrous oxide as electron acceptors for the electron transport system. This suggests that F. columnare is capable of anaerobic growth in aquatic pond sediments, which are typically nitrogen rich. This provides important new information on the potential environmental reservoir for F. columnare in aquaculture ponds. Our study is the first to identify this predicted capability, and we have now confirmed experimentally that F. columnare can grow anaerobically. Furthermore, anaerobic growth is more efficient in the presence of nitrate supplementation, indicating that F. columnare is using nitrate as an electron acceptor for anaerobic respiration.
The F. columnare ATCC 49512 genome revealed that the species has several other unique metabolic capabilities. Besides denitrification, the F. columnare ATCC 49512 genome uniquely encodes nickel permease function, which could play an important role in several cellular processes such as energy metabolism, nitrogen metabolism, detoxification, and virulence (Schauer et al., 2007). F. columnare ATCC 49512 is also unique in encoding neprilysin, which is an endopeptidase considered to have a nutritional role in bacteria. Also, it is a membrane-bound protein that is responsible for degradation of biologically active peptides (Rawlings and Salvesen, 2013).
F. columnare ATCC 49512 has similar iron acquisition strategies as the other Flavobacterium species, and it also has a similar gliding motility system encoded in its genome. However, the flavobacteria vary in the spr genes they encode, which reflects variety in the types of surfaces they are required to use as substrates for motility (McBride et al., 2009).
Fish pathogenic strains F. branchiophilum FL-15, F. columnare ATCC49512, and F. psychrophilum JIP0286 carry fewer transcriptional factors and two-component systems elements than environmental isolates F. indicum GPTSA100-9 and F. johnsoniae UW10. Maintaining a large regulatory network has high genetic cost (Capra and Laub, 2012), and the number of regulatory systems correlates with the number of environmental niches bacteria are required to survive in. The F. johnsoniae UW101 transcriptional network is particularly noteworthy for being large relative to the size of its genome (Visweswariah and Busby, 2015), which likely reflects the need to adapt to a variety of environmental conditions. By contrast, the host environment is relatively constant compared to the aquatic environment, so F. columnare, F. psychrophilum, and F. branchiophilum appear to require fewer regulatory systems. However, the host environment is stressful and requires rapid adaptation to varying host defense systems, so the regulatory functions they carry are critical for survival. For example, F. psychrophilum senses temperature with a two-component system that activates virulence-related genes at its optimal growth temperature (Hesami et al., 2011).
All the evaluated Flavobacterium genomes carry T9SS and T1SS. The T9SS is important for gliding motility, which is a common feature of flavobacteria. Notably, the flavobacteria do not have a T2SS, which is the main terminal branch of the general secretory system. However, only some of the Flavobacterium species carry T6SSiii. F. branchiophilum FL-15 encodes an entire T6SS, and F. johnsoniae UW101 and F. columnare ATCC 49512 carry almost the entire T6SS. F. psychrophilum JIP0286 and F. indicum GPTSA100-9 genomes do not carry T6SSiii. The T6SS is particularly known for delivering anti-bacterial toxins, giving a competitive advantage in multi-species environments.
CRISPR-Cas systems are small RNA-based systems that protect bacteria from foreign DNA (bacteriophage and plasmids). A CRISPR segment consists of three elements: CRISPR array, flanking regions consisting of short direct repeats separated by short variable DNA sequences and cas genes, and short repeat regions (Makarova et al., 2015). CRISPR arrays store “memory” sequences using spacers (short DNA sequences), which originate from foreign DNA (Amitai and Sorek, 2016). Because of their unique ecological environments, it is expected that each species will carry different memory sequences depending on the types of foreign DNA they are exposed to. However, only the two pathogens F. columnare and F. psychrophilum carry genes encoding Cas enzymes that catalyze acquisition of spacer sequences from foreign DNA, possibly indicating more pressure from foreign DNA. The mechanism used by F. indicum GPTSA100-9, F. branchiophilum FL-15, and F. johnsoniae UW101 to obtain CRISPR regions and direct repeat elements is not known because they do not carry cas genes.
Despite carrying cas genes, F. columnare ATCC 49512 had the largest number of insertion elements and second highest number of genomic islands, indicating a relatively high degree of horizontal gene exchange and plasticity among the flavobacterial genomes. As expected, F. johnsoniae, the species with the largest genome, had the largest number of genomic islands, but it had surprisingly few insertion elements. F. branchiophilum FL-15 and F. psychrophilum JIP02/86 had fewer insertion elements than F. columnare ATCC 49512, but had a similar variety of insertion families. ISL3 elements are the most distributed family of insertion sequences in the Flavobacterium genomes we evaluated. Prophages are not as prevalent in flavobacterial genomes as other bacterial species, with only one (F. psychrophilum JIP02/86) carrying a complete phage region. The other four species only carry remnants or incomplete phage regions, with three incomplete regions in F. branchiophilum FL-15.
Few F. columnare virulence factors have been identified. An initial characterization of F. columnare iron acquisition proteins was described (Guan et al., 2013). Biofilm formation in the three F. columnare genomovars was compared (Cai et al., 2013). In addition, development of rifampicin-resistant mutants from a virulent F. columnare genomovar II strain caused loss of virulence, but the mechanism for attenuation was not identified (Mohammed et al., 2013). Genomic analysis revealed F. columnare potential virulence genes and possible virulence mechanisms that warrant further investigation. Some of these potential virulence genes are common to the Flavobacterium species, and many are unique to F. columnare ATCC 49512. In particular, peroxide resistance, iron metabolism, and stress response were revealed as potentially important virulence mechanisms. Exotoxins/cytotoxins were also identified that could contribute to virulence, and the role of polysaccharide coat in columnaris disease is not known.
Finally, we used RNA-Seq to improve the F. columnare ATCC 49512 genome annotation. Using this method, we identified 41 unannotated protein-coding genes, 16 of which had a non-traditional start site (TTG, GTG, and CTT). Also, RNA-seq provided experimental validation for predicted protein-coding ORFs that were identified using automated annotation for the F. columnare ATCC 49512 genome (particularly hypothetical proteins), and it allowed analysis of operon structure within the genome. RNA-seq also enables identification of small non-coding RNAs, which are not annotated using automated pipelines. We identified candidate sRNAs in the F. columnare ATCC 49512 genome, none of which had matches in the Rfam database. This is likely due to a paucity of data on sRNAs in the Bacteroides group, and we consider this to be the first experimental identification of sRNAs in the Flavobacterium genus. Based on our results, we advocate RNA-seq as a relatively low-cost method to improve annotation of reference genomes, particularly in identifying novel proteins and sRNAs that are not identified by automated annotation pipelines.
In summary, the F. columnare ATCC 49512 genome revealed several conserved functions the species shares with the other Flavobacterium species. Like the other flavobacteria, F. columnare likely derives energy primarily from carbohydrates and proteins rather than lipids, and it has similar gliding motility functions. However, unlike the other flavobacteria, F. columnare has the genetic capability of utilizing nitrate and other nitrogen molecules for anaerobic respiration. We present experimental evidence to phenotypically substantiate this capability. F. columnare ATCC 49512 also has evidence for more horizontal gene exchange than the other flavobacterial species, as evidenced by a high number of insertion sequences and genomic islands. RNA-seq improved the F. columnare ATCC 49512 annotation and added value by validating automated annotation and adding operon structure. We also report for the first-time candidate non-coding small RNAs in F. columnare.
Author Contributions
HT, AK, JR, and ML conceived and designed the experiments. HT, JR, SK, and SN performed the experiments and analyzed the data. HT wrote and ML and AK edited the manuscript. All authors approve submission of this manuscript to Frontiers in Microbiology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the United States Department of Agriculture, National Institute of Food and Agriculture (2006-35600-16571) and by the Mississippi State University College of Veterinary Medicine. The authors would like to thank Michelle Banes for technical assistance in RNA isolation, Amanda Cooksey for cDNA library construction, and Tony Arick for RNA-seq expression analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00588/full#supplementary-material
Supplementary Table 1. Unique and shared genes for all the Flavobacterium species.
Supplementary Table 2. Flavobacterium transcriptional factors and two-component systems.
Supplementary Table 3. Genomic islands in Flavobacterium genomes.
Supplementary Table 4. Flavobacterium genus insertion elements.
Supplementary Table 5. Prophage elements in Flavobacterium genus.
Supplementary Table 6. Potential virulence factors in Flavobacterium genomes.
Supplementary Table 7. F. columnare ATCC 49512 proteins with updated start sites based on transcriptome analysis.
Supplementary Table 8. F. columnare ATCC 49512 putative non-coding RNAs based on transcriptome analysis.
Supplementary Table 9. F. columnare ATCC 49512 operons predicted by DOOR2 and evidence of expression.
References
Abby, S. S., Cury, J., Guglielmini, J., Neron, B., Touchon, M., and Rocha, E. P. (2016). Identification of protein secretion systems in bacterial genomes. Sci. Rep. 6:23080. doi: 10.1038/srep23080
Abby, S. S., Neron, B., Menager, H., Touchon, M., and Rocha, E. P. (2014). MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS ONE 9:e110726. doi: 10.1371/journal.pone.0110726
Allen, A. E., Dupont, C. L., Obornik, M., Horak, A., Nunes-Nesi, A., McCrow, J. P., et al. (2011). Evolution and metabolic significance of the urea cycle in photosynthetic diatoms. Nature 473, 203–207. doi: 10.1038/nature10074
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Amitai, G., and Sorek, R. (2016). CRISPR-Cas adaptation: insights into the mechanism of action. Nat. Rev. Microbiol. 14, 67–76. doi: 10.1038/nrmicro.2015.14
Angerer, A., Enz, S., Ochs, M., and Braun, V. (1995). Transcriptional regulation of ferric citrate transport in Escherichia coli K-12. Fecl belongs to a new subfamily of sigma 70-type factors that respond to extracytoplasmic stimuli. Mol. Microbiol. 18, 163–174. doi: 10.1111/j.1365-2958.1995.mmi_18010163.x
Arias, C. R., Welker, T. L., Shoemaker, C. A., Abernathy, J. W., and Klesius, P. H. (2004). Genetic fingerprinting of Flavobacterium columnare isolates from cultured fish. J. Appl. Microbiol. 97, 421–428. doi: 10.1111/j.1365-2672.2004.02314.x
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, 16–21. doi: 10.1093/nar/gkw387
Atherton, J. C. (2006). The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu. Rev. Pathol. 1, 63–96. doi: 10.1146/annurev.pathol.1.110304.100125
Auch, A. F., Von Jan, M., Klenk, H. P., and Goker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Baerends, R. J., Smits, W. K., De Jong, A., Hamoen, L. W., Kok, J., and Kuipers, O. P. (2004). Genome2D: a visualization tool for the rapid analysis of bacterial transcriptome data. Genome Biol. 5:R37. doi: 10.1186/gb-2004-5-5-r37
Barbier, P., Houel, A., Loux, V., Poulain, J., Bernardet, J. F., Touchon, M., et al. (2012). Complete genome sequence of Flavobacterium indicum GPSTA100-9T, isolated from warm spring water. J. Bacteriol. 194, 3024–3025. doi: 10.1128/JB.00420-12
Bass, K. A., and Hecht, D. W. (2002). Isolation and characterization of cLV25, a Bacteroides fragilis chromosomal transfer factor resembling multiple Bacteroides sp. mobilizable transposons. J. Bacteriol. 184, 1895–1904. doi: 10.1128/JB.184.7.1895-1904.2002
Beaz-Hidalgo, R., and Figueras, M. J. (2013). Aeromonas spp. whole genomes and virulence factors implicated in fish disease. J. Fish Dis. 36, 371–388. doi: 10.1111/jfd.12025
Bernardet, J. F. (1989). ‘Flexibacter columnaris’: first description in France and comparison with bacterial strains from other origins. Dis. Aquat. Organ. 6, 37–44. doi: 10.3354/dao006037
Bernardet, J. F., Segers, P., Vancanneyt, M., Berthe, F., Kersters, K., and Vandamme, P. (1996). Cutting a Gordian knot: emended classification and description of the genus Flavobacterium, emended description of the family Flavobacteriaceae, and proposal of Flavobacterium hydatis nom. nov. (basonym, Cytophaga aquatilis Strohl and Tait 1978). Int. J. Syst. Bacteriol. 46, 128–148. doi: 10.1099/00207713-46-1-128
Blom, J., Albaum, S. P., Doppmeier, D., Puhler, A., Vorholter, F. J., Zakrzewski, M., et al. (2009). EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics 10:154. doi: 10.1186/1471-2105-10-154
Brown, J. S., and Holden, D. W. (2002). Iron acquisition by Gram-positive bacterial pathogens. Microbes Infect. 4, 1149–1156. doi: 10.1016/S1286-4579(02)01640-4
Cai, W., De La Fuente, L., and Arias, C. R. (2013). Biofilm formation by the fish pathogen Flavobacterium columnare: development and parameters affecting surface attachment. Appl. Environ. Microbiol. 79, 5633–5642. doi: 10.1128/AEM.01192-13
Capra, E. J., and Laub, M. T. (2012). Evolution of two-component signal transduction systems. Annu. Rev. Microbiol. 66, 325–347. doi: 10.1146/annurev-micro-092611-150039
Castillo, D., Espejo, R., and Middelboe, M. (2014). Genomic structure of bacteriophage 6H and its distribution as prophage in Flavobacterium psychrophilum strains. FEMS Microbiol. Lett. 351, 51–58. doi: 10.1111/1574-6968.12342
Christie-Oleza, J. A., Nogales, B., Lalucat, J., and Bosch, R. (2010). TnpR encoded by an ISPpu12 isoform regulates transposition of two different ISL3-like insertion sequences in Pseudomonas stutzeri after conjugative interaction. J. Bacteriol. 192, 1423–1432. doi: 10.1128/JB.01336-09
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Darwish, A. M., and Ismaiel, A. A. (2005). Genetic diversity of Flavobacterium columnare examined by restriction fragment length polymorphism and sequencing of the 16S ribosomal RNA gene and the 16S-23S rDNA spacer. Mol. Cell. Probes 19, 267–274. doi: 10.1016/j.mcp.2005.04.003
Decostere, A., Haesebrouck, F., and Devriese, L. A. (1998). Characterization of four Flavobacterium columnare (Flexibacter columnaris) strains isolated from tropical fish. Vet. Microbiol. 62, 35–45. doi: 10.1016/S0378-1135(98)00196-5
Dhillon, B. K., Chiu, T. A., Laird, M. R., Langille, M. G., and Brinkman, F. S. (2013). IslandViewer update: improved genomic island discovery and visualization. Nucleic Acids Res. 41, W129–W132. doi: 10.1093/nar/gkt394
Dobrindt, U., and Reidl, J. (2000). Pathogenicity islands and phage conversion: evolutionary aspects of bacterial pathogenesis. Int. J. Med. Microbiol. 290, 519–527. doi: 10.1016/S1438-4221(00)80017-X
Duchaud, E., Boussaha, M., Loux, V., Bernardet, J. F., Michel, C., Kerouault, B., et al. (2007). Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat. Biotechnol. 25, 763–769. doi: 10.1038/nbt1313
Durborow, R. M., Thune, R. L., Hawke, J. P., and Camus, A. C. (1998). Columnaris Disease: A Bacterial Infection Caused by Flavobacterium columnare. Stoneville, MS: Southern Regional Aquaculture Center.
Farmer, B. (2004). Improved Methods for the Isolation and Characterization of Flavobacterium columnare. Baton Rouge, LA: Louisiana State University.
Fujita, Y., Matsuoka, H., and Hirooka, K. (2007). Regulation of fatty acid metabolism in bacteria. Mol. Microbiol. 66, 829–839. doi: 10.1111/j.1365-2958.2007.05947.x
Gardner, P. P., Daub, J., Tate, J., Moore, B. L., Osuch, I. H., Griffiths-Jones, S., et al. (2011). Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 39, D141–D145. doi: 10.1093/nar/gkq1129
Gennis, R. B., and Stewart, V. (1996). “Respiration,” in Escherichia coli and Salmonella: Cellular and Molecular Biology, eds F. C. Neidhardt, R. Curtiss, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (Washington, DC: ASM Press), 217–261.
Griess, P. (1879). Bemerkungen zu der Abhandlung der HH. Weselsky und Benedikt “Ueber einige Azoverbindungen.” Ber. Deutsch. Chem. Ges. 12, 426–428. doi: 10.1002/cber.187901201117
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Guan, L., Santander, J., Mellata, M., Zhang, Y., and Curtiss, R. III (2013). Identification of an iron acquisition machinery in Flavobacterium columnare. Dis. Aquat. Org. 106, 129–138. doi: 10.3354/dao02635
Hacker, J., Blum-Oehler, G., Muhldorfer, I., and Tschape, H. (1997). Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol. Microbiol. 23, 1089–1097. doi: 10.1046/j.1365-2958.1997.3101672.x
Hacker, J., and Kaper, J. B. (2000). Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54, 641–679. doi: 10.1146/annurev.micro.54.1.641
Hendriksen, W. T., Kloosterman, T. G., Bootsma, H. J., Estevao, S., De Groot, R., Kuipers, O. P., et al. (2008). Site-specific contributions of glutamine-dependent regulator GlnR and GlnR-regulated genes to virulence of Streptococcus pneumoniae. Infect. Immun. 76, 1230–1238. doi: 10.1128/IAI.01004-07
Hennig, S., and Ziebuhr, W. (2010). Characterization of the transposase encoded by IS256, the prototype of a major family of bacterial insertion sequence elements. J. Bacteriol. 192, 4153–4163. doi: 10.1128/JB.00226-10
Hesami, S., Metcalf, D. S., Lumsden, J. S., and Macinnes, J. I. (2011). Identification of cold-temperature-regulated genes in Flavobacterium psychrophilum. Appl. Environ. Microbiol. 77, 1593–1600. doi: 10.1128/AEM.01717-10
Hsiao, W., Wan, I., Jones, S. J., and Brinkman, F. S. (2003). IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19, 418–420. doi: 10.1093/bioinformatics/btg004
Hunnicutt, D. W., and McBride, M. J. (2001). Cloning and characterization of the Flavobacterium johnsoniae gliding motility genes gldD and gldE. J. Bacteriol. 183, 4167–4175. doi: 10.1128/JB.183.14.4167-4175.2001
Johnson, T. L., Abendroth, J., Hol, W. G., and Sandkvist, M. (2006). Type II secretion: from structure to function. FEMS Microbiol. Lett. 255, 175–186. doi: 10.1111/j.1574-6968.2006.00102.x
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kanonenberg, K., Schwarz, C. K., and Schmitt, L. (2013). Type I secretion systems - a story of appendices. Res. Microbiol. 164, 596–604. doi: 10.1016/j.resmic.2013.03.011
Kingsford, C. L., Ayanbule, K., and Salzberg, S. L. (2007). Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 8:R22. doi: 10.1186/gb-2007-8-2-r22
Koster, M., Bitter, W., and Tommassen, J. (2000). Protein secretion mechanisms in Gram-negative bacteria. Int. J. Med. Microbiol. 290, 325–331. doi: 10.1016/S1438-4221(00)80033-8
Kumar, R., Lawrence, M. L., Watt, J., Cooksey, A. M., Burgess, S. C., and Nanduri, B. (2012). RNA-seq based transcriptional map of bovine respiratory disease pathogen “Histophilus somni 2336”. PLoS ONE 7:e29435. doi: 10.1371/journal.pone.0029435
Kunttu, H. M., Suomalainen, L. R., Jokinen, E. I., and Valtonen, E. T. (2009). Flavobacterium columnare colony types: connection to adhesion and virulence? Microb. Pathog. 46, 21–27. doi: 10.1016/j.micpath.2008.10.001
Kusumoto, M., Ooka, T., Nishiya, Y., Ogura, Y., Saito, T., Sekine, Y., et al. (2011). Insertion sequence-excision enhancer removes transposable elements from bacterial genomes and induces various genomic deletions. Nat. Commun. 2:152. doi: 10.1038/ncomms1152
Lafrentz, B. R., Garcia, J. C., Dong, H. T., Waldbieser, G. C., Rodkhum, C., Wong, F. S., et al. (2016). Optimized reverse primer for 16S-RFLP analysis and genomovar assignment of Flavobacterium columnare. J. Fish Dis. doi: 10.1111/jfd.12583. [Epub ahead of print].
Lafrentz, B. R., Lapatra, S. E., Shoemaker, C. A., and Klesius, P. H. (2012). Reproducible challenge model to investigate the virulence of Flavobacterium columnare genomovars in rainbow trout Oncorhynchus mykiss. Dis. Aquat. Org. 101, 115–122. doi: 10.3354/dao02522
Lafrentz, B. R., Waldbieser, G. C., Welch, T. J., and Shoemaker, C. A. (2014). Intragenomic heterogeneity in the 16S rRNA genes of Flavobacterium columnare and standard protocol for genomovar assignment. J. Fish Dis. 37, 657–669. doi: 10.1111/jfd.12166
Langille, M. G., and Brinkman, F. S. (2009). IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25, 664–665. doi: 10.1093/bioinformatics/btp030
Langille, M. G., Hsiao, W. W., and Brinkman, F. S. (2008). Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 9:329. doi: 10.1186/1471-2105-9-329
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lopatina, A., Medvedeva, S., Shmakov, S., Logacheva, M. D., Krylenkov, V., and Severinov, K. (2016). Metagenomic analysis of bacterial communities of antarctic surface snow. Front. Microbiol. 7:398. doi: 10.3389/fmicb.2016.00398
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736. doi: 10.1038/nrmicro3569
Mann, R. A., Smits, T. H., Buhlmann, A., Blom, J., Goesmann, A., Frey, J. E., et al. (2013). Comparative genomics of 12 strains of Erwinia amylovora identifies a pan-genome with a large conserved core. PLoS ONE 8:e55644. doi: 10.1371/journal.pone.0055644
Mao, X., Ma, Q., Zhou, C., Chen, X., Zhang, H., Yang, J., et al. (2014). DOOR 2.0: presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 42, D654–D659. doi: 10.1093/nar/gkt1048
Martens, J. H., Barg, H., Warren, M. J., and Jahn, D. (2002). Microbial production of vitamin B12. Appl. Microbiol. Biotechnol. 58, 275–285. doi: 10.1007/s00253-001-0902-7
McBride, M. J. (2001). Bacterial gliding motility: multiple mechanisms for cell movement over surfaces. Annu. Rev. Microbiol. 55, 49–75. doi: 10.1146/annurev.micro.55.1.49
McBride, M. J., and Nakane, D. (2015). Flavobacterium gliding motility and the type IX secretion system. Curr. Opin. Microbiol. 28, 72–77. doi: 10.1016/j.mib.2015.07.016
McBride, M. J., Xie, G., Martens, E. C., Lapidus, A., Henrissat, B., Rhodes, R. G., et al. (2009). Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl. Environ. Microbiol. 75, 6864–6875. doi: 10.1128/AEM.01495-09
McNeil, M. B., and Fineran, P. C. (2013). Prokaryotic assembly factors for the attachment of flavin to complex II. Biochim. Biophys. Acta 1827, 637–647. doi: 10.1016/j.bbabio.2012.09.003
Mendz, G. L., and Hazell, S. L. (1996). The urea cycle of Helicobacter pylori. Microbiology 142(Pt 10), 2959–2967. doi: 10.1099/13500872-142-10-2959
Michel, C., Messiaen, S., and Bernardet, J. F. (2002). Muscle infections in imported neon tetra, Paracheirodon innesi Myers: limited occurrence of microsporidia and predominance of severe forms of columnaris disease caused by an Asian genomovar of Flavobacterium columnare. J. Fish Dis. 25, 253–263. doi: 10.1046/j.1365-2761.2002.00364.x
Mohammed, H., Olivares-Fuster, O., Lafrentz, S., and Arias, C. R. (2013). New attenuated vaccine against columnaris disease in fish: choosing the right parental strain is critical for vaccine efficacy. Vaccine 31, 5276–5280. doi: 10.1016/j.vaccine.2013.08.052
Nelson, S. S., Bollampalli, S., and McBride, M. J. (2008). SprB is a cell surface component of the Flavobacterium johnsoniae gliding motility machinery. J. Bacteriol. 190, 2851–2857. doi: 10.1128/JB.01904-07
Nivaskumar, M., and Francetic, O. (2014). Type II secretion system: a magic beanstalk or a protein escalator. Biochim. Biophys. Acta 1843, 1568–1577. doi: 10.1016/j.bbamcr.2013.12.020
Nunez, J. K., Harrington, L. B., Kranzusch, P. J., Engelman, A. N., and Doudna, J. A. (2015). Foreign DNA capture during CRISPR-Cas adaptive immunity. Nature 527, 535–538. doi: 10.1038/nature15760
Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., and Kanehisa, M. (1999). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34. doi: 10.1093/nar/27.1.29
Olivares-Fuster, O., Bullard, S. A., McElwain, A., Llosa, M. J., and Arias, C. R. (2011). Adhesion dynamics of Flavobacterium columnare to channel catfish Ictalurus punctatus and zebrafish Danio rerio after immersion challenge. Dis. Aquat. Org. 96, 221–227. doi: 10.3354/dao02371
Ortet, P., De Luca, G., Whitworth, D. E., and Barakat, M. (2012). P2TF: a comprehensive resource for analysis of prokaryotic transcription factors. BMC Genomics 13:628. doi: 10.1186/1471-2164-13-628
Ortet, P., Whitworth, D. E., Santaella, C., Achouak, W., and Barakat, M. (2015). P2CS: updates of the prokaryotic two-component systems database. Nucleic Acids Res. 43, D536–D541. doi: 10.1093/nar/gku968
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Pan, Y. R., Lou, Y. C., Seven, A. B., Rizo, J., and Chen, C. (2011). NMR structure and calcium-binding properties of the tellurite resistance protein TerD from Klebsiella pneumoniae. J. Mol. Biol. 405, 1188–1201. doi: 10.1016/j.jmb.2010.11.041
Reddy, J. S., Kumar, R., Watt, J. M., Lawrence, M. L., Burgess, S. C., and Nanduri, B. (2012). Transcriptome profile of a bovine respiratory disease pathogen: Mannheimia haemolytica PHL213. BMC Bioinformatics 13(Suppl. 15):S4. doi: 10.1186/1471-2105-13-S15-S4
Rey, S., Acab, M., Gardy, J. L., Laird, M. R., Defays, K., Lambert, C., et al. (2005). PSORTdb: a protein subcellular localization database for bacteria. Nucleic Acids Res. 33, D164–D168. doi: 10.1093/nar/gki027
Rhodes, R. G., Samarasam, M. N., Shrivastava, A., Van Baaren, J. M., Pochiraju, S., Bollampalli, S., et al. (2010). Flavobacterium johnsoniae gldN and gldO are partially redundant genes required for gliding motility and surface localization of SprB. J. Bacteriol. 192, 1201–1211. doi: 10.1128/JB.01495-09
Russell, A. B., Wexler, A. G., Harding, B. N., Whitney, J. C., Bohn, A. J., Goo, Y. A., et al. (2014). A type VI secretion-related pathway in Bacteroidetes mediates interbacterial antagonism. Cell Host Microbe 16, 227–236. doi: 10.1016/j.chom.2014.07.007
Rychlik, I., Sebkova, A., Gregorova, D., and Karpiskova, R. (2001). Low-molecular-weight plasmid of Salmonella enterica serovar Enteritidis codes for retron reverse transcriptase and influences phage resistance. J. Bacteriol. 183, 2852–2858. doi: 10.1128/JB.183.9.2852-2858.2001
Schauer, K., Gouget, B., Carriere, M., Labigne, A., and De Reuse, H. (2007). Novel nickel transport mechanism across the bacterial outer membrane energized by the TonB/ExbB/ExbD machinery. Mol. Microbiol. 63, 1054–1068. doi: 10.1111/j.1365-2958.2006.05578.x
Shoemaker, C. A., Olivares-Fuster, O., Arias, C. R., and Klesius, P. H. (2008). Flavobacterium columnare genomovar influences mortality in channel catfish (Ictalurus punctatus). Vet. Microbiol. 127, 353–359. doi: 10.1016/j.vetmic.2007.09.003
Siguier, P., Gourbeyre, E., and Chandler, M. (2014). Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol. Rev. 38, 865–891. doi: 10.1111/1574-6976.12067
Siguier, P., Gourbeyre, E., Varani, A., Ton-Hoang, B., and Chandler, M. (2015). Everyman's guide to bacterial insertion sequences. Microbiol. Spectr. 3:MDNA3-0030-2014. doi: 10.1128/microbiolspec.mdna3-0030-2014
Silverman, J. M., Agnello, D. M., Zheng, H., Andrews, B. T., Li, M., Catalano, C. E., et al. (2013). Haemolysin coregulated protein is an exported receptor and chaperone of type VI secretion substrates. Mol. Cell 51, 584–593. doi: 10.1016/j.molcel.2013.07.025
Tekedar, H. C., Karsi, A., Gillaspy, A. F., Dyer, D. W., Benton, N. R., Zaitshik, J., et al. (2012). Genome sequence of the fish pathogen Flavobacterium columnare ATCC 49512. J. Bacteriol. 194, 2763–2764. doi: 10.1128/JB.00281-12
Tomida, S., Nguyen, L., Chiu, B. H., Liu, J., Sodergren, E., Weinstock, G. M., et al. (2013). Pan-genome and comparative genome analyses of propionibacterium acnes reveal its genomic diversity in the healthy and diseased human skin microbiome. MBio 4, e00003–e00013. doi: 10.1128/mBio.00003-13
Touchon, M., Barbier, P., Bernardet, J. F., Loux, V., Vacherie, B., Barbe, V., et al. (2011). Complete genome sequence of the fish pathogen Flavobacterium branchiophilum. Appl. Environ. Microbiol. 77, 7656–7662. doi: 10.1128/AEM.05625-11
Van Domselaar, G. H., Stothard, P., Shrivastava, S., Cruz, J. A., Guo, A., Dong, X., et al. (2005). BASys: a web server for automated bacterial genome annotation. Nucleic Acids Res. 33, W455–W459. doi: 10.1093/nar/gki593
Varani, A. M., Siguier, P., Gourbeyre, E., Charneau, V., and Chandler, M. (2011). ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 12:R30. doi: 10.1186/gb-2011-12-3-r30
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. doi: 10.1016/j.mib.2014.11.016
Visweswariah, S. S., and Busby, S. J. (2015). Evolution of bacterial transcription factors: how proteins take on new tasks, but do not always stop doing the old ones. Trends Microbiol. 23, 463–467. doi: 10.1016/j.tim.2015.04.009
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142. doi: 10.1186/1471-2105-7-142
Wakabayashi, T., and Wakabayashi, H. (1999). Genotypic diversity of strains of Flavobacterium columnare from diseased fishes. Fish Pathol. 34, 65–71. doi: 10.3147/jsfp.34.65
Waldor, M. K., and Mekalanos, J. J. (1996). Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272, 1910–1914. doi: 10.1126/science.272.5270.1910
Wallden, K., Rivera-Calzada, A., and Waksman, G. (2010). Type IV secretion systems: versatility and diversity in function. Cell. Microbiol. 12, 1203–1212. doi: 10.1111/j.1462-5822.2010.01499.x
Whiteside, M. D., Winsor, G. L., Laird, M. R., and Brinkman, F. S. (2013). OrtholugeDB: a bacterial and archaeal orthology resource for improved comparative genomic analysis. Nucleic Acids Res. 41, D366–D376. doi: 10.1093/nar/gks1241
Wiens, G. D., Lapatra, S. E., Welch, T. J., Rexroad, C. III, Call, D. R., Cain, K. D., et al. (2014). Complete genome sequence of Flavobacterium psychrophilum strain CSF259-93, used to select rainbow trout for increased genetic resistance against bacterial cold water disease. Genome Announc. 2. doi: 10.1128/genomeA.00889-14
Wurtzel, O., Sapra, R., Chen, F., Zhu, Y., Simmons, B. A., and Sorek, R. (2010). A single-base resolution map of an archaeal transcriptome. Genome Res. 20, 133–141. doi: 10.1101/gr.100396.109
Xiao, J., Chen, T., Wang, Q., and Zhang, Y. (2012). Comparative analysis of the roles of catalases KatB and KatG in the physiological fitness and pathogenesis of fish pathogen Edwardsiella tarda. Lett. Appl. Microbiol. 54, 425–432. doi: 10.1111/j.1472-765X.2012.03225.x
Xie, F., Zhang, Y., Li, G., Zhou, L., Liu, S., and Wang, C. (2013). The ClpP protease is required for the stress tolerance and biofilm formation in Actinobacillus pleuropneumoniae. PLoS ONE 8:e53600. doi: 10.1371/journal.pone.0053600
Yu, N. Y., Laird, M. R., Spencer, C., and Brinkman, F. S. (2011). PSORTdb–an expanded, auto-updated, user-friendly protein subcellular localization database for Bacteria and Archaea. Nucleic Acids Res. 39, D241–D244. doi: 10.1093/nar/gkq1093
Keywords: Flavobacterium columnare, comparative genomics analysis, fish health, denitrification, RNA-Seq
Citation: Tekedar HC, Karsi A, Reddy JS, Nho SW, Kalindamar S and Lawrence ML (2017) Comparative Genomics and Transcriptional Analysis of Flavobacterium columnare Strain ATCC 49512. Front. Microbiol. 8:588. doi: 10.3389/fmicb.2017.00588
Received: 07 January 2017; Accepted: 21 March 2017;
Published: 19 April 2017.
Edited by:
Shicheng Chen, Michigan State University, USAReviewed by:
Mark J. McBride, University of Wisconsin–Milwaukee, USAGregory D. Wiens, Agricultural Research Service (USDA), USA
Copyright © 2017 Tekedar, Karsi, Reddy, Nho, Kalindamar and Lawrence. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark L. Lawrence, lawrence@cvm.msstate.edu