 Fuminori Mizukoshi1
Fuminori Mizukoshi1 Koo Nagasawa2
Koo Nagasawa2 Yen H. Doan3Kei Haga4Shima Yoshizumi5Yo Ueki6Michiyo Shinohara7Mariko Ishikawa8Naomi Sakon9Naoki Shigemoto10Reiko Okamoto-Nakagawa11Akie Ochi12Koichi Murakami2Akihide Ryo13
Yen H. Doan3Kei Haga4Shima Yoshizumi5Yo Ueki6Michiyo Shinohara7Mariko Ishikawa8Naomi Sakon9Naoki Shigemoto10Reiko Okamoto-Nakagawa11Akie Ochi12Koichi Murakami2Akihide Ryo13 Yoshiyuki Suzuki14Kazuhiko Katayama4*
Yoshiyuki Suzuki14Kazuhiko Katayama4* Hirokazu Kimura2,13*
Hirokazu Kimura2,13*- 1Department of Microbiology, Tochigi Prefectural Institute of Public Health and Environmental Science, Utsunomiya-shi, Japan
- 2Infectious Disease Surveillance Center, National Institute of Infectious Diseases, Musashimurayama-shi, Japan
- 3Department of Virology II, National Institute of Infectious Diseases, Musashimurayama-shi, Japan
- 4Laboratory of Viral infection I, Kitasato Institute for Life Sciences Graduate School of Infection Control Sciences, Kitasato University, Minato-ku, Japan
- 5Department of Infectious Diseases, Hokkaido Institute of Public Health, Sapporo-shi, Japan
- 6Department of Microbiology, Miyagi Prefectural Institute of Public Health and Environment, Sendai-shi, Japan
- 7Virus Division, Saitama Institute of Public Health, Hiki-gun, Japan
- 8Division of Virology, Kawasaki City Institute for Public Health, Kawasaki-shi, Japan
- 9Department of Microbiology, Osaka Prefectural Institute of Public Health, Osaka-shi, Japan
- 10Hiroshima Prefectural Technology Research Institute Public Health and Environment Center, Hiroshima-shi, Japan
- 11Department of Health Science, Yamaguchi Prefectural Institute of Public Health and Environment, Yamaguchi-shi, Japan
- 12Department of Microbiology, Ehime Prefectural Institute of Public Health and Environmental Science, Matsuyama-shi, Japan
- 13Department of Microbiology, Yokohama City University Graduate School of Medicine, Yokohama-shi, Japan
- 14Division of Biological Science, Nagoya City University, Nagoya-shi, Japan
The RNA-dependent RNA polymerase (RdRp) and capsid (VP1) genes of 51 GII.2 human norovirus (HuNoV) strains collected during the period of 2004–2015 in Japan were analyzed. Full-length analyses of the genes were performed using next-generation sequencing. Based on the gene sequences, we constructed the time-scale evolutionary trees by Bayesian Markov chain Monte Carlo methods. Time-scale phylogenies showed that the RdRp and VP1 genes evolved uniquely and independently. Four genotypes of GII.2 (major types: GII.P2-GII.2 and GII.P16-GII.2) were detected. A common ancestor of the GII.2 VP1 gene existed until about 1956. The evolutionary rates of the genes were high (over 10−3 substitutions/site/year). Moreover, the VP1 gene evolution may depend on the RdRp gene. Based on these results, we hypothesized that transfer of the RdRp gene accelerated the VP1 gene evolution of HuNoV genotype GII.2. Consequently, recombination between ORF1 (polymerase) and ORF2 (capsid) might promote changes of GII.2 antigenicity.
Introduction
Human norovirus (HuNoV) is a major causative agent of gastroenteritis in humans (Green, 2013). The HuNoV genogroup II (GII), in particular, is frequently detected in outbreaks. The HuNoV GII strains can be classified into 22 genotypes (Kroneman et al., 2013). Moreover, the most worldwide prevalent HuNoV GII genotypes belong to GII genotype 2 (GII.2), GII.3, GII.4, GII.6, and GII.17 (Centers for Disease Control and Prevention. CaliciNet Data [cited 2016])1. Since national surveillance began, nearly 3 million cases of NoV gastroenteritis have been recorded, and Japan was experiencing its second most serious norovirus outbreak during November 2016 to February 2017 (National Institute of Infectious Diseases. Japan. Infectious Gastroenteritis. [cited 4th April 2017, in Japanese])2 Importantly, HuNoV GII.2 emerged as a major cause of this outbreak in Japan, although the GII.4 strains were the most prevalent genotype during the past 10 years (National Institute of Infectious Diseases. Japan. Flash report of norovirus in Japan [cited 4th April 2017, in Japanese])3.
Very recent studies suggested that the evolutionary patterns of human and animal NoV genotypes are distinct (Kobayashi et al., 2015, 2016). Although all viral proteins may act as antigens, the HuNoV VP1 protein is also involved in viral infection. Furthermore, HuNoV frequently experiences recombination at the ORF1/ORF2 junction, resulting in new chimera viruses with different types of the RNA-dependent RNA polymerase (RdRp) genes and capsid (VP1) genes. Most studies have focused on the molecular evolution of HuNoV GII.4. Only a few examined that gene in other HuNoV genotypes, including GII.2. To gain insight into this process, we examined the molecular evolution of the GII.2 RdRp and VP1 genes, including chimera viruses, based on the full genome analyses of those detected in Japan over a period of 10 years (2004–2015 seasons).
Materials and Methods
To investigate the molecular evolution of the HuNoV VP1 and RdRp genes, 950 stool specimens were collected from various areas (13 prefectures) of Japan during the 2004–2015 seasons. These samples were obtained from patients with acute gastroenteritis due to HuNoV infections, in compliance with the Food Sanitation Law and the Law Concerning the Prevention of Infections and Medical Care for Patients of Infections of Japan. The personal data related to these samples were anonymized. RNA was extracted from 10% PBS suspensions of the specimens, and the HuNoV genomes were comprehensively analyzed by next-generation sequencing as described (Matsushima et al., 2015). Of 950 samples, the complete genome sequences of 538 strains were obtained (a success rate of 57%). Next, HuNoV genotypes were confirmed with the Norovirus Typing Tool (Version1.0), based on the nucleotide sequences of RdRp and VP1 genes as described by Kroneman et al. (2011). GII.2 strains were selected from these all genotyped strains, and then a few of strains having the undetermined base sequences (e.g., N, Y, R, and V) were omitted. Finally, 51 GII.2 strains were used for evolutionary analyses for the present study (Supplementary Table 1). The obtained nucleotide sequences for the GII.2 strains were deposited in GenBank under the accession numbers LC209431 to LC209481.
Time-scale evolutionary analyses were performed using the Bayesian Markov Chain Monte Carlo method (MCMC) with the BEAST package v1.8.3 (Drummond and Rambaut, 2007) and Tracer4 as a demographic model. Substitution models were calculated with Kakusan4 (Tanabe, 2011). The substitution model for the VP1 or the RdRp gene was the GTR-Γ or GTR-Γ invariant model, respectively. Based on Akaike's Information Criterion for MCMC values, we used the random local clock as a clock model, and used the logistic growth model (VP1 gene) or the constant size model (RdRp gene) as a tree model. Convergence was evaluated with an effective sample size (acceptable more than 200). The MCMC chain length was 3 × 108 steps with sampling every 1,000 steps for the MCMC tree of the VP1 gene. To exactly estimate the evolutionary rates and topologies of the MCMC tree of the RdRp gene, we bound two independent data of the MCMC chains5. The MCMC chain length was 2 × 108 steps and 4 × 108 with sampling every 5,000 steps for the MCMC tree of the RdRp gene. Statistical analyses were performed with the Welch's t-test in Excel 2013.
Results and Discussion
Distribution of GII.2 Genotype during the 2004–2015 Seasons
Four genotypes of the GII.2 strains, including GII.P2-GII.2 (13 strains), GII.Pe-GII.2 (one strain), GII.P12-GII.2 (one strain), and GII.P16-GII.2 (36 strains), were determined by the Norovirus Typing Tool (Figure 1). Of them, GII.P16-GII.2 strains were the most prevalent genotype after 2009. The single GII.P12-GII.2 and GII.Pe-GII.2 strains were detected in 2004 and 2014 respectively. The GII.P2-GII.2 strains were detected throughout the investigation periods.
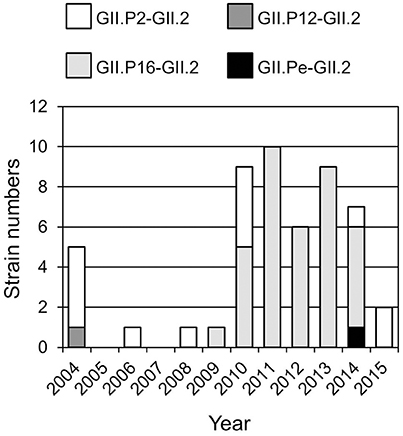
Figure 1. Distribution of GII.2 genotype during the investigation periods (2004–2015 seasons).
Phylogenetic Analysis and Evolutionary Rates of VP1 and RdRp Virus Genes
Based on the VP1 gene sequences, we constructed a time-scale evolutionary tree (Figure 2A). The phylogeny of the VP1 gene showed that GII.2 strains could be divided according to the type of RdRp gene. GII.P16-GII.2 could be subdivided into three clusters of strains in 2009–2010, 2010–2012, and 2012–2014. In addition, the phylogenetic divergence of the GII.P16-GII.2 strains might be wider than that of the GII.P2-GII.2 strains. The tree shows that the most recent common ancestor (MRCA) of the present GII.2 strains appeared in 1956 (mean ± 95% highest posterior densities [HPD]: 1945–1966). Subsequently, GII.P2-GII.2 virus strain emerged in 2000 (mean ± 95% HPD: 1998–2001). Moreover, the GII.P16-GII.2 strains detected in 2010–2012 diverged from a common ancestor of the GII.P2-GII.2 strains at 2002 (mean ± 95% HPD: 2001–2004). The GII.P16-GII.2 strains detected in 2009–2010 and 2012–2014 diverged at 2005 (mean ± 95% HPD: 2004–2007). The evolutionary rate of these VP1 genes was 2.987 × 10−3 substitutions/site/year (mean ± 95% HPD: 2.496–3.486 × 10−3 substitutions/site/year).
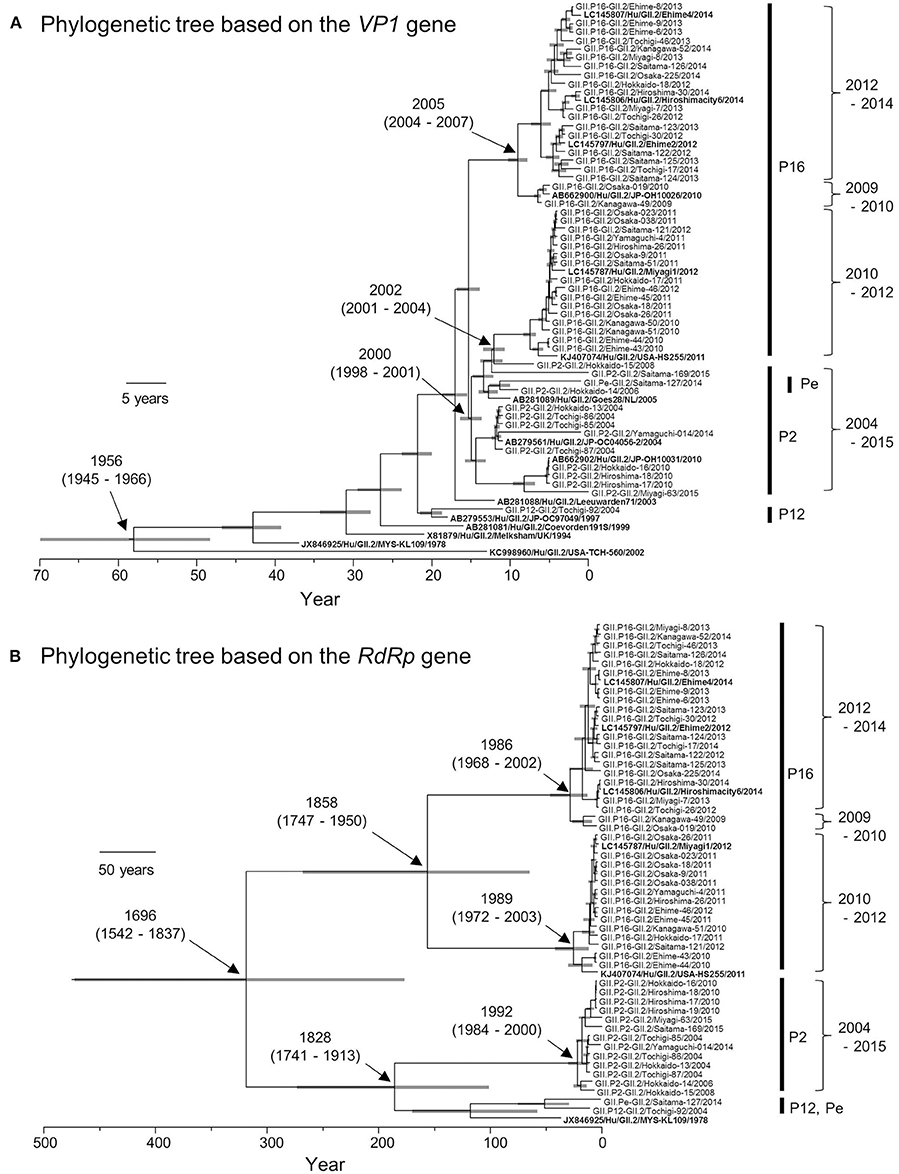
Figure 2. Phylogenetic trees of VP1 (A) and RdRp (B) genes of the genotype GII.2 constructed by the Bayesian MCMC method. We analyzed VP1 gene of 50 strains, and RdRp gene of 49 strains, excluding 100%—matched homologous strains. Reference strains in these trees were indicated in bold letters. Gray bar shows 95% HPD. The scale bar represents actual time (year).
We also constructed a time-scale evolutionary tree of the RdRp gene (Figure 2B). The tree shows that the MRCA of RdRp of the present GII.2 strains was in the year 1696 (mean ± 95% HPD: 1542–1837). The common ancestor of the GII.P16-GII.2 strains diverged in 1858 (mean ± 95% HPD: 1747–1950) and formed two clusters. Moreover, the GII.P16-GII.2 strains detected in 2010–2012 diverged at 1989 (mean ± 95% HPD: 1972–2003), whereas the GII.P16-GII.2 strains detected in 2009–2010 and 2012–2014 diverged at 1986 (mean ± 95% HPD: 1968–2002). The common ancestor of the GII.P2-GII.2, GII.P12-GII.2, and GII.Pe-GII.2 diverged in 1828 (mean ± 95% HPD: 1741–1913). The RdRp gene of GII.P2-GII.2 diverged in 1992 (mean ± 95% HPD: 1984–2000). The evolutionary rate of these RdRp genes was 1.314 × 10−3 substitutions/site/year (mean ± 95% HPD: 0.698–1.95 × 10−3 substitutions/site/year).
Next, we compared the evolutionary rates of the GII.P16-GII.2 and GII.P2-GII.2 strains. To gain statistical significance, we collected the nucleotide sequences of the GII.P2-GII.2 strains (25 strains) from GenBank, but we could not collect a sufficient number of the GII.P12-GII.2 and GII.Pe-GII.2 sequences from the GenBank to reach statistical significance. The evolutionary rate of GII.P16-GII.2 (1.838 × 10−3 substitutions/site/year; mean ± 95% HPD: 1.237–2.456 × 10−3 substitutions/site/year) was greater than that of GII.P2-GII.2 (1.712 × 10−3 substitutions/site/year; mean ± 95% HPD: 0.957–2.41 × 10−3 substitutions/site/year) (p = 7.891 × 10−135).
A previous report suggested that the evolution of VP1 may be influenced by the activities of RdRp (Bull et al., 2010). Collectively, our bioinformatics data also showed that the evolution of the GII.2 VP1 gene was accelerated by a recombination of ORF1, including the RdRp gene. However, additional in vitro studies regarding the mutation rates of RdRp of the GII.P2 and GII.P16 may be needed to clarify the hypothesis of the relationships between and VP1 and RdRp genes in this study. Furthermore, GII.2 variant strains were detected in the present season (2016/17 season), and thus, further genetic studies may be needed to prove this hypothesis.
Conclusions
Here we report the molecular evolution of the VP1 and RdRp genes in HuNoV GII.2. Our main findings and hypothesis are as follows. (1) Four genotypes of GII.2 (GII.P2-GII.2, GII.P16-GII.2, GII.P12-GII.2, and GII.Pe-GII.2) were detected in Japan in 2004–2015. (2) A common ancestor of the current GII.2 virus strains circulated around 1956. (3) VP1 gene evolution seems to depend on the RdRp gene. The VP1 gene in a prevalent HuNoV genotype GII.2 might evolve uniquely by transfer of the RdRp gene.
Ethics Statement
This study protocol was approved by the National Institute of Infectious Diseases Ethics Committee (No. 532).
Author Contributions
FM designed and performed the research, analyzed the data and wrote the manuscript. KN, YD, and KH performed the research and analyzed the data. FM, SY, YU, MS, MI, NS (Sakon), NS (Shigemoto), RO, and AO contributed samples and analyzed the data; and HK and KK designed and supervised the research, analyzed the data, and wrote the manuscript. All authors contributed, read, and approved the manuscript.
Funding
This work was partly supported by a commissioned project for Research on Emerging and Re-emerging Infectious Diseases from Japan Agency for Medical Research and Development, AMED.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Gary C. Howard (Gladstone Institutes) for a critical review of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00705/full#supplementary-material
Footnotes
1. ^http://www.cdc.gov/norovirus/reporting/calicinet/data.html
2. ^http://www.nih.go.jp/niid/ja/10/2096-weeklygraph/1647-04gastro.html
3. ^http://www.nih.go.jp/niid/ja/iasr-noro.html
References
Bull, R. A., Eden, J. S., Rawlinson, W. D., and White, P. A. (2010). Rapid evolution of pandemic noroviruses of the GII.4 lineage. PLoS Pathog. 6:e1000831. doi: 10.1371/journal.ppat.1000831
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Green, K. Y. (2013). “Caliciviridae: the noroviruses,” in Fields Virology, 6th Edn, eds D. M. Knipe and P. M. Howley (Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins Health), 582–608.
Kobayashi, M., Matsushima, Y., Motoya, T., Sakon, N., Shigemoto, N., Okamoto-Nakagawa, R., et al. (2016). Molecular evolution of the capsid gene in human norovirus genogroup II. Sci. Rep. 6:29400. doi: 10.1038/srep29400
Kobayashi, M., Yoshizumi, S., Kogawa, S., Takahashi, T., Ueki, Y., Shinohara, M., et al. (2015). Molecular evolution of the capsid gene in norovirus genogroup I. Sci. Rep. 5:13806. doi: 10.1038/srep13806
Kroneman, A., Vega, E., Vennema, H., Vinjé, J., White, P. A., Hansman, G., et al. (2013). Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 158, 2059–2068. doi: 10.1007/s00705-013-1708-5
Kroneman, A., Vennema, H., Deforche, K. v d Avoort, H., Peñaranda, S., Oberste, M. S., et al. (2011). An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 51, 121–125. doi: 10.1016/j.jcv.2011.03.006
Matsushima, Y., Ishikawa, M., Shimizu, T., Komane, A., Kasuo, S., Shinohara, M., et al. (2015). Genetic analyses of GII.17 norovirus strains in diarrheal disease outbreaks from December 2014 to March 2015 in Japan reveal a novel polymerase sequence and amino acid substitutions in the capsid region. Euro. Surveill. 20:21173. doi: 10.2807/1560-7917.ES2015.20.26.21173
Keywords: norovirus, capsid, RNA-dependent RNA polymerase, molecular epidemiology, phylogeny, molecular evolution
Citation: Mizukoshi F, Nagasawa K, Doan YH, Haga K, Yoshizumi S, Ueki Y, Shinohara M, Ishikawa M, Sakon N, Shigemoto N, Okamoto-Nakagawa R, Ochi A, Murakami K, Ryo A, Suzuki Y, Katayama K and Kimura H (2017) Molecular Evolution of the RNA-Dependent RNA Polymerase and Capsid Genes of Human Norovirus Genotype GII.2 in Japan during 2004–2015. Front. Microbiol. 8:705. doi: 10.3389/fmicb.2017.00705
Received: 22 February 2017; Accepted: 05 April 2017;
Published: 25 April 2017.
Edited by:
Hirofumi Akari, Kyoto University, JapanReviewed by:
Artur Rzezutka, Veterinary Research Institute, PolandChang-jun Bao, Jiangsu Province Center for Disease Control and Prevention, China
Copyright © 2017 Mizukoshi, Nagasawa, Doan, Haga, Yoshizumi, Ueki, Shinohara, Ishikawa, Sakon, Shigemoto, Okamoto-Nakagawa, Ochi, Murakami, Ryo, Suzuki, Katayama and Kimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuhiko Katayama, katayama@lisci.kitasato-u.ac.jp
Hirokazu Kimura, kimhiro@nih.go.jp