 Huan Li1†Rong Tang2†Yang Lou1†Zelin Cui2Wenjing Chen2Qing Hong2
Huan Li1†Rong Tang2†Yang Lou1†Zelin Cui2Wenjing Chen2Qing Hong2 Zhaohuan Zhang1
Zhaohuan Zhang1 Pradeep K. Malakar1Yingjie Pan1,3,4
Pradeep K. Malakar1Yingjie Pan1,3,4 Yong Zhao1,3,4*
Yong Zhao1,3,4*- 1College of Food Science and Technology, Shanghai Ocean University, Shanghai, China
- 2Shanghai General Hospital, Shanghai, China
- 3Laboratory of Quality and Safety Risk Assessment for Aquatic Products on Storage and Preservation (Shanghai), Ministry of Agriculture, Shanghai, China
- 4Shanghai Engineering Research Center of Aquatic-Product Processing and Preservation, Shanghai, China
Vibrio parahaemolyticus is one of the most important pathogen for seafood-borne gastroenteritis in Shanghai and the rest of the world. A total of 42 V. parahaemolyticus strains were isolated from 1900 fecal specimens collected from patients in Shanghai hospital presenting from January 2014 to December 2015. All isolates were evaluated for potential virulence factors [tdh, trh, and type three secretion system (T3SS) genes], typed using multilocus sequence typing (MLST) and screened for antimicrobial resistance phenotype and genotype. And for the first time, the relationship between virulence, genetic diversity and antimicrobial resistance of these isolates were identified. The results showed that 37 isolates carried the tdh gene (88.1%) and only seven isolates were positive for the trh gene. The T3SS1 and T3SS2 genes were detected in all strains and only trh-positive isolates are also containing the T3SS2β genes. MLST analysis of the 42 Shanghai isolates identified 20 sequence types (STs) with 16 novel STs and that these clinical V. parahaemolyticus strains showed high degrees of genetic diversity. All isolates expressed high levels of resistance against Ampicillin (100.0%), Streptomycin (100.0%), Cephazolin (92.9%), Kanamycin (92.8%) and Amikacin (90.5%), and eight out of 38 resistance genes (SHV, tet(B), strA, qnrA, gryA, qnrB, sulI, sulII) were detected in at least two isolates. This study confirms that antimicrobial resistance of clinical V. parahaemolyticus isolates is greater than those of environmental isolates. Furthermore, no clear correlation between antimicrobial resistance and virulence or genetic diversity was found in this study. These results add to epidemiological data of clinical V. parahaemolyticus isolates in Shanghai and highlight the need for additional mechanistic studies, especially antimicrobial resistance, to reduce the burden of disease caused by this pathogen in China.
Introduction
Illnesses caused by foodborne pathogens are an increasingly critical public health concern (Velusamy et al., 2010). Vibrio parahaemolyticus is recognized as a major foodborne pathogen for causing gastroenteritis worldwide, especially in coastal countries and regions (McLaughlin et al., 2005; Su and Liu, 2007; Letchumanan et al., 2014; Hubbard et al., 2016). Clinical symptom of V. parahaemolyticus infections include diarrhea, abdominal cramps, vomiting and fever, which also progresses to septicemia can sometimes lead to death in patients (Scallan et al., 2011; Lopatek et al., 2015). In 1950, this pathogen was first discovered in Japan, which resulted 272 illnesses with 20 deaths (Fujino et al., 1953). From 1997 to 2000, 84 food poisoning outbreaks caused by V. parahaemolyticus were recorded in Spain (Fournier and Ogata, 2005). From 2000 to 2008, it was reported around 35,000 V. parahaemolyticus infections annually in US (Haendiges et al., 2015). In China, during 2003 to 2008, this microorganism has caused 9041 illnesses and 3948 hospitalizations (Wu et al., 2014). It is essential to gather the epidemiological data of V. parahaemolyticus to reduce the burden of disease caused by this pathogen in China.
Virulence of V. parahaemolyticus is primarily attributed to the production of a thermostable direct haemolysin (TDH), TDH-related haemolysin (TRH) and two type III secretion systems, T3SS (Makino et al., 2003; Ritchie et al., 2012; Zhang et al., 2013). TDH and TRH are encoded by the tdh gene and trh gene, respectively (Letchumanan et al., 2014). Although the specific actions of these genes in human infection remain unknown, the relevance between pathogenicity of V. parahaemolyticus and the presence of tdh and trh is well recognized (Broberg et al., 2011; Ceccarelli et al., 2013). Contamination of foods with tdh- and/or trh-positive V. parahaemolyticus strains is considered a public health risk (Pazhani et al., 2014). The two T3SS systems in V. parahaemolyticus are known as T3SS1 and T3SS2 (Wang et al., 2015). T3SS1 is encoded by the first pathogenicity island on chromosome I and is involved in cytotoxicity (Paranjpye et al., 2012). T3SS2 is located on chromosome II and is also encoded by a pathogenicity island. As a newly identified type of secretion system, T3SS2 appears to be associated with enterotoxicity and cytotoxicity, in experiments conducted in vitro and in intestinal cell lines (Ritchie et al., 2012). Currently, it is possible to detect the presence of tdh, trh and T3SS genes in V. parahaemolyticus isolates by PCR-based methods (West et al., 2013).
Vibrio parahaemolyticus strains exhibit high genetic diversity due to high rates of recombination and mutation, which caused potential infection risk for human health (Ludeke et al., 2015). A number of molecular typing methods have been used to determine the molecular epidemiology of V. parahaemolyticus, and these methods include multilocus sequence typing (MLST), serotyping, and pulsed-field gel electrophoresis (PFGE) (Banerjee et al., 2014; Xu et al., 2015). MLST has proven to be powerful tool for investigating the prevalence and diversity of V. parahaemolyticus strains in recent years (Wu et al., 2016). MLST is based on the sequencing of seven housekeeping genes and can be analyzed directly via the internet (Gonzalez-Escalona et al., 2008). MLST is commonly used for identifying the relationship between isolates in public database and has proven to be an important method for investigation of the evolution and epidemiology of V. parahaemolyticus (Banerjee et al., 2014).
Antimicrobials are used in the treatment of infectious diseases and improper or enhanced application of antimicrobials leads to development of antimicrobial resistant (AMR) bacteria (Yano et al., 2014). The Economic Forum for Global Risks indicates that the problem of AMR is projected to be one of the greatest threats to human health in the future (Koser et al., 2014; Blair et al., 2015). The critical factors for the emergence of AMR are antimicrobial resistance genes (ARGs) which can be transferred by the horizontal gene transfer (Thomas and Nielsen, 2005). ARGs are emerging contaminants posing a potential worldwide human health risk (Allen et al., 2010; Lou et al., 2016). It is vital to monitor the AMR and ARGs of V. parahaemolyticus strains, which can be used for disease management and reducing the burden of disease caused by this pathogen.
Shanghai is one of the largest prosperous cities in China with high annual consumption of seafood and many cases of V. parahaemolyticus infections (Zhang and Orth, 2013; Qi et al., 2016), which have become a potential threat for human health. The researches for V. parahaemolyticus strains isolated from aquatic products are widely reported (Wang et al., 2011; Guo et al., 2013; He et al., 2015, 2016; Lou et al., 2016; Hu and Chen, 2016; Yu et al., 2016; Zhang et al., 2017), while studies on clinical isolates has been poorly documented (Zhang and Orth, 2013; Qi et al., 2016).
The main objectives of this study are to monitor the virulence, genetic diversity and antimicrobial susceptibility of clinical V. parahaemolyticus isolates from Shanghai. We hope to provide reliable information, for assessing the genetic traits and the antimicrobial resistance risk of V. parahaemolyticus strains, and for better management of foodborne infections in Shanghai.
Materials and Methods
Specimen Collection and Bacteria Isolation
A total of 1900 fecal specimens were collected by Shanghai hospital from patients who presented with acute diarrhea to gastroenteritis outpatient clinics during the period from January 2014 through to December 2015. These fecal samples were placed in sterile sealed plastic bags and stored at 4°C prior to further analysis. Confirmation of V. parahaemolyticus samples were performed using standard culture methods (ISO, 2007).
Briefly, 25 g of each fecal specimen was homogenized for 2 min in a stomacher 400 with 225 mL of alkaline peptone water (APW; Beijing Land Bridge Technology Company Ltd., Beijing, China) containing 3% NaCl, and incubated at 37°C for 16-18 h. After incubation, a loop from the top 1 cm was streaked onto thiosulfate-citrate-bile salts-sucrose (TCBS; Beijing Land Bridge Technology Company Ltd., Beijing, China) agar plates and incubated at 37°C for 18–24 h. Presumptive individual bacterial colony (green or blue green colony, 2–3 mm in diameter) were grown in 10 ml tryptic soy broth (TSB; Beijing Land Bridge Technology Company Ltd., Beijing, China) supplemented with 3.0% NaCl and incubated at 37°C for 18–24 h. After cultivation, the bacterial liquid and 50% glycerol in the proportion of 1:1 were placed in a glycerol tube and stored at –80°C for further analysis.
DNA Extraction
DNA extraction of all presumptive V. parahaemolyticus isolates was performed using the TIANamp Bacteria DNA Kit (Tiangen Biotech Beijing Co., Ltd, Beijing, China), in accordance to the manufacturer’s recommended protocols and then stored it at –20°C prior to PCR analysis.
Identification of Vibrio parahaemolyticus
The presumptive V. parahaemolyticus isolates were tested for the presence of the species specific gene tlh by using polymerase chain reaction (PCR). Detection of tlh gene was carried out using the primer tlh-F (5- AAA GCG GAT TAT GCA GAA GCA CTG -3) and tlh-R (5- GCT ACT TTC TAG CAT TTT CTC TGC -3) as specified in (Food and Drug Administration [FDA], 2004). The reaction mixture for this PCR assay was performed in 25 μL, containing 1 μL of DNA template, 12.5 μL of PCR Mix (Sangon Biotech, Shanghai, China), 9.5 μL of dd H2O and 1 μL of each primer. The thermal-cycling program is as follows: initial denaturation at 94°C for 3 min, 25 cycles of 94°C for 1 min, 60°C for 1 min and 72°C for 2 min, and a final extension at 72°C for 3 min. Finally, PCR products were analyzed by agarose gel electrophoresis.
We also chose the API 20E system (BioMerieux, Inc., Durham, NC, United States) and DBI-08 (Beijing Land Bridge Technology Company Ltd., Beijing, China) to analyze and identify the V. parahaemolyticus isolates, according to the procedure described by the manufacturer and using V. parahaemolyticus ATCC 33847 as the reference strain (Croci et al., 2007; Li et al., 2016; Xie et al., 2016).
Detection of Virulence-Associated Genes
Detection of the V. parahaemolyticus virulence genes tdh (West et al., 2013) and trh (Nilsson and Turner, 2016) were also performed by PCR. We designed a primer for detection of the ureR gene to study the variation of the trh gene as outlined in Nilsson and Turner, 2016. The ureR gene encodes for the transcriptional activator of the urease gene cluster located immediately upstream from trh and is widely reported to be genetically linked to trh (Nilsson and Turner, 2016). V. parahaemolyticus virulence associated genes of type III secretion system-1 (T3SS1) genes (VP1670 [vscP], VP1686 [putative], VP1689 [vscK] and VP1694 [vscF]), T3SS2α genes (VP1362 [vopB2], VP1339 [vscC2], VP1335 [vscS2] and VP1327 [vopT]) and the T3SS2β genes (vscC2, vopB2, vopC, vscS2) were tested by conventional PCR (Jones et al., 2012). In our study, the oligonucleotide primers were synthesized by Sangon Biotech (Sangon Biotech, Shanghai, China). Particularly worth mentioning is that the V. parahaemolyticus ATCC17802 (trh+) and ATCC33847(tdh+) were used as the reference strains, and distilled water was used as the negative control.
Multilocus Sequence Typing
Seven housekeeping genes, dnaE, gyrB, recA, dtdS, pntA, pyrC, and tnaA (Supplementary Table S1), were used for V. parahaemolyticus characterization under the MLST scheme, PCR fragments were sequenced by Sangon Biotech (Sangon Biotech, Shanghai, China) and alignments of these sequences were determined using DNAMAN. The sequences were analyzed online1 to assign allele numbers and define sequence types (STs). New sequences for alleles and new ST profiles were submitted to the V. parahaemolyticus MLST database. Based on the relatedness of the STs, all of the isolates were subdivided into clonal complexes (CCs) or groups by eBURST program. Nucleotide sequence analyses were evaluated by MEGA5.1 program. In this study, the primary founder of a CC, a single locus variants (SLVs), double locus variants (DLVs), and singletons were defined as described previously (Han et al., 2015).
Antimicrobial Susceptibility Testing
The antibiotic susceptibilities of the 42 isolates were assessed using the disk diffusion method on Mueller Hinton agar (MHA) (OXOID Limited, China) according to the guidelines of the Clinical and Laboratory Standards Institute (CLSI, 2015; Lou et al., 2016). Briefly, Muller–Hinton agar and a panel of 18 antibiotics disks were selected for resistance tests. The 18 common antimicrobials belonging to 6 classes used in this study were: β-lactam (ampicillin: AMP, amoxicillin-clavulanic: AMC, piperacillin: PRL, cefotaxime: CTX, ceftazidime: CAZ, cefoxitin: FOX, cephazolin: KZ, imipenem: IPM, meropenem: MEM), aminoglycoside (amikacin: AK, gentamicin: CN, kanamycin: K, streptomycin: S), tetracycline (tetracycline: TET), quinolone (ciprofloxacin: CIP, levofloxacin: LEV), sulfonamides (trimethoprim-sulfamethoxazole: SXT), chloramphenicol (chloramphenicol: C). The results were expressed as sensitive (S), intermediate (I), or resistant (R) according to the methods of the CLSI (CLSI, 2015). Escherichia coli ATCC 25922 was used as the quality control organism for the antimicrobial susceptibility testing.
Evaluation of Antibiotic Resistance-Encoding Genes
The 38 antibiotic resistance genes (Supplementary Table S2) of six classes of antibiotics were identified by PCR, as previously described (Lou et al., 2016). All obtained PCR products were purified and sequenced by Sangon Biotech (Sangon Biotech, Shanghai, China). The acquired sequences were aligned and analyzed with the BLAST program2.
Results
Prevalence of V. parahaemolyticus
A total of 42 presumptive V. parahaemolyticus strains were isolated from 1900 fecal specimens (2.2%) collected from patients presenting in Shanghai hospital during January, 2014 to December, 2015. The PCR results showed all isolates were positive for the presence of the tlh gene (Table 1), which indicated that these 42 isolates were V. parahaemolyticus strains. And the results of API 20E and DBI-08 provided further evidence to the veracity of PCR outcomes, with all 42 isolates being identified with 99% confidence as V. parahaemolyticus. The demographic characteristics of patients about 42 V. parahaemolyticus isolates are also presented in Table 1. The 42 individuals enrolled in this research included 20 females and 22 males. The male patients’ ages ranged from 13 to 57, while for the female patients, the age distribution was more uniform in women whose median ages above 30 years-old.
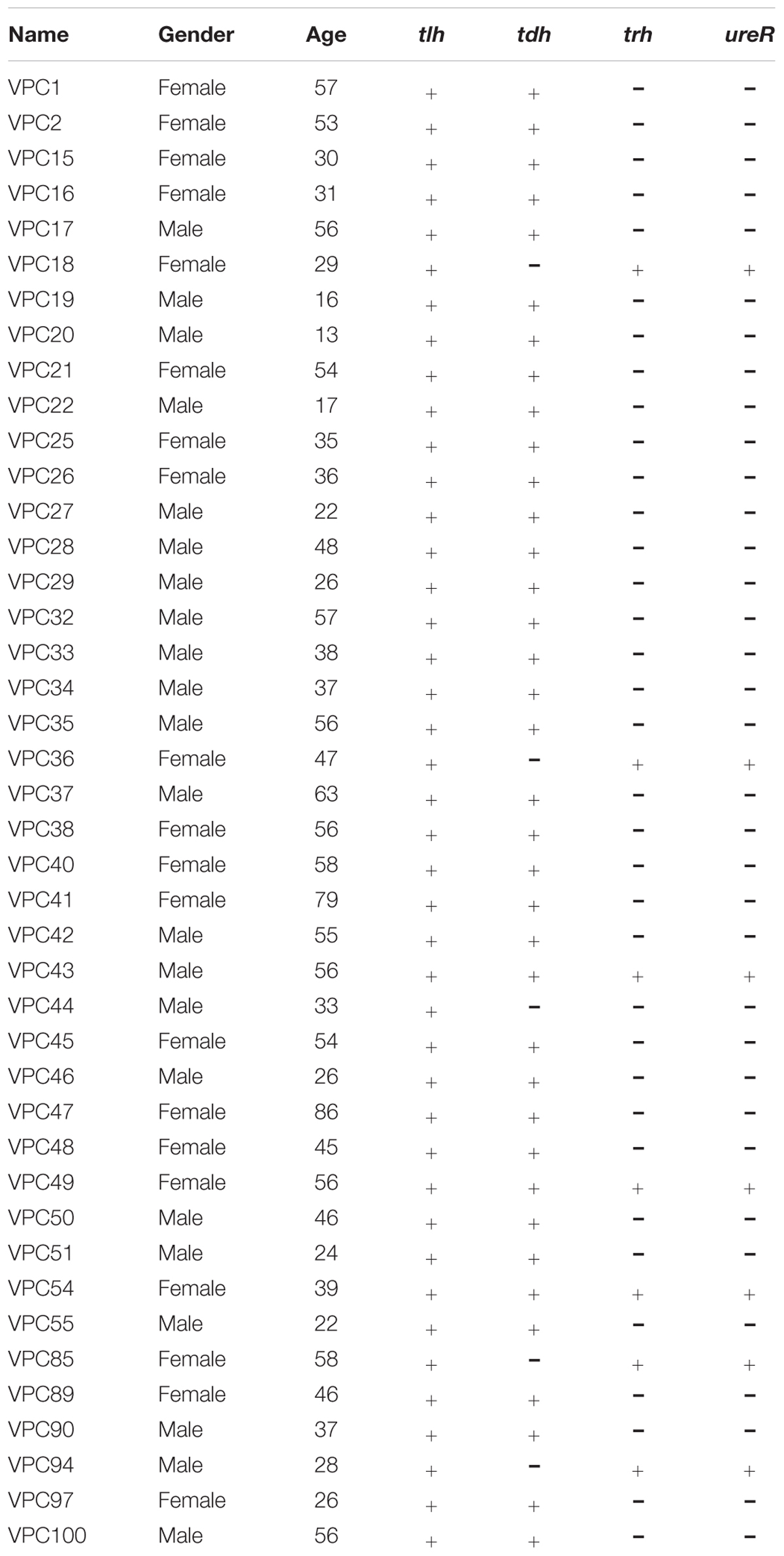
TABLE 1. Information of 42 Vibrio parahaemolyticus clinical isolates.
Distribution of Virulence-Associated Genes
From our study, the hemolysin gene tdh was detected in most of the isolates (88.1%, 37/42), whereas the trh gene was present in only 7 strains (16.7%, 7/42). We further determine the distribution of the trh gene by the ureR gene. The ureR gene and the variable trh gene were observed in the same seven V. parahaemolyticus isolates. Of these, 3 of 42 (7.1%) clinical isolates were positive for both tdh and trh. Only one strains (2.4%, 1/42) from the diarrheal patients contained neither the tdh nor the trh gene.
The two types of T3SS complexes are features in the virulence mechanism of V. parahaemolyticus and the distribution of T3SS genes is presented in Table 2. T3SS1 genes were identified in all of the V. parahaemolyticus isolates and these samples contained all four T3SS1 genes (100%). All thirty-four of the tdh+/trh- and one of the tdh-/trh- clinical isolates contained all four genes of the T3SS2α genes. However, the isolates of tdh+/trh+ and tdh-/trh+ the detected percentage of all T3SS2α genes was only 33.3% and 50.0%, respectively. Additionally, four of the tdh-/trh+ isolates were amplified all four genes of the T3SS2β genes, followed by the isolates of tdh+/trh+ the detected percentage of all T3SS2β genes was 66.7%. Only one remaining tdh+/trh- isolate (VPC89) amplified all four T3SS2β genes but vscC2. As expected, one of the tdh-/trh- clinical isolate was evaluated in negative of all four T3SS2β genes. Overall, the T3SS2α-associated genes were most prevalent in tdh+ isolates (93.5%, 35/37), and the T3SS2β genes were detected prevalent in the trh+ clinical isolates (85.7%, 6/7).
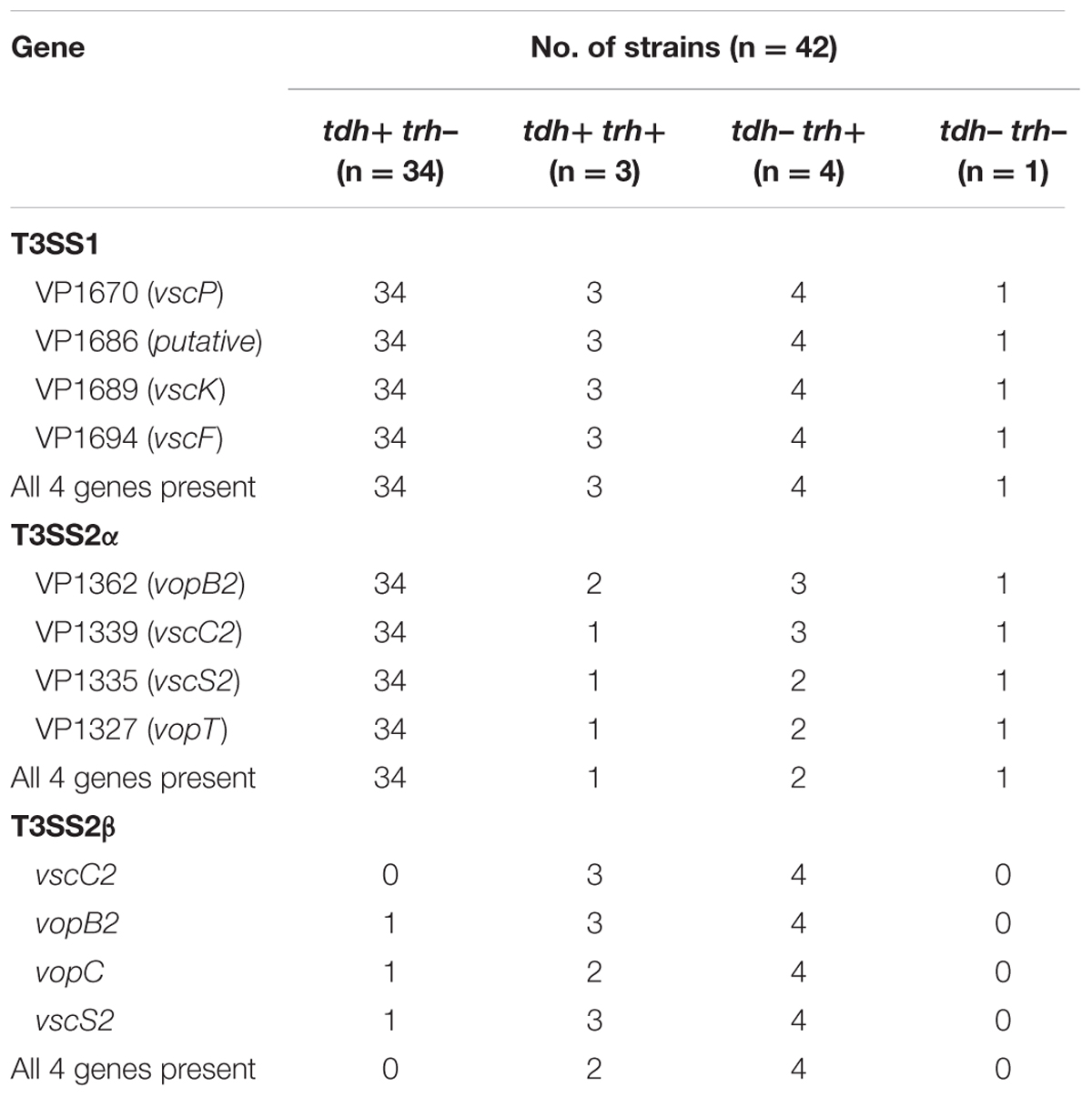
TABLE 2. Distribution of T3SS genes among 42 clinical V. parahaemolyticus isolates.
Multilocus Sequence Typing Analysis
The genetic characteristic of the V. parahaemolyticus isolates was analyzed by MLST. MLST classified the 42 V. parahaemolyticus isolates into 20 different STs (Figure 1), of which 16 ST were novel (ST1457, ST1458, ST1459, ST1460, ST1461, ST1462, ST1463, ST1464, ST1465, ST1466, ST1467, ST1468, ST1469, ST1470, ST1471, and ST1472). The go eBURST algorithm used in our study categorized 20 different STs into 10 singletons, one clone complexes (CC655) and Two groups (Figure 2). Among these, CC655 was the most prevalent clone complexes, including 22 isolates with ST655(50%), ST1464(36.4%), ST3(9.1%) and ST1468(4.5%). In addition, ST655 was the most frequent sequence type, which including 11 isolates (VPC16, VPC19 VPC20, VPC21, VPC22, VPC25, VPC27, VPC29, VPC33, VPC34, VPC97).
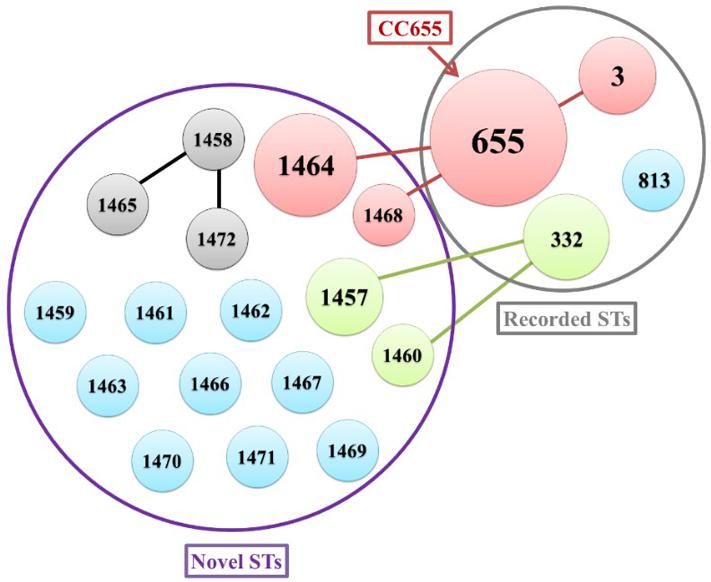
FIGURE 1. Multilocus sequence typing (MLST) of 42 V. parahaemolyticus clinical isolates.
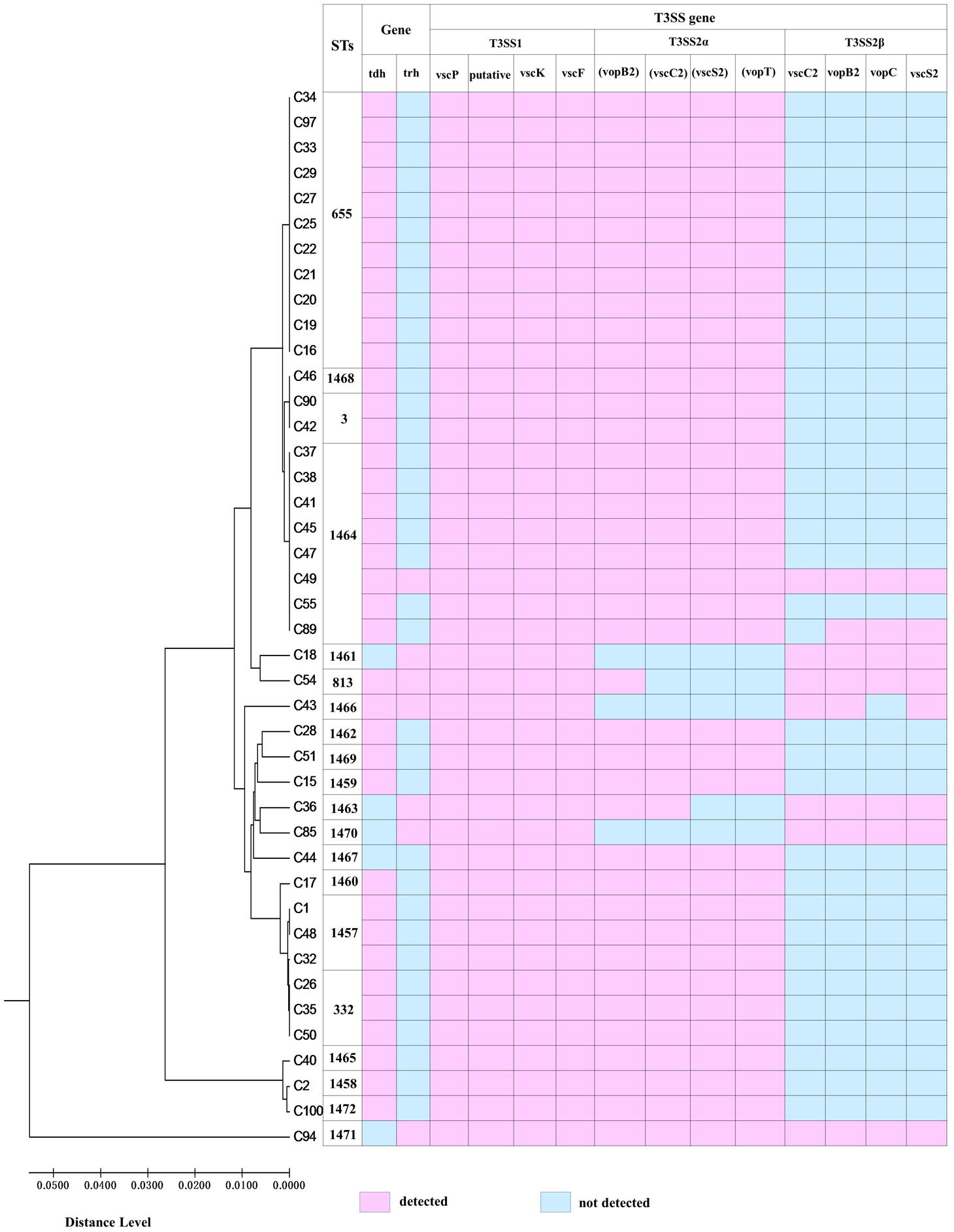
FIGURE 2. The relationship between genetic diversity and virulence-associated genes characterization of isolates. ST, sequence type.
Antimicrobial Resistance Profile
As shown in the Table 3, the isolates of V. parahaemolyticus were detected for different levels of antibiotic resistance. One concern, all V. parahaemolyticus strains were resistant to ampicillin (100%) and streptomycin (100%), followed by Cephazolin (92.9%), Kanamycin (92.9%) and Amikacin (90.5%). Isolates were also commonly resistant to Gentamicin (71.4%), Piperacillin (54.8%), and Cefoxitin (50%). In addition, all the clinical isolates showed susceptibility to six antibiotics, including Chloramphenicol, Cefotaxime, Imipenem, Ceftazidime, Trimethoprim-sulfamethoxazole and Meropenem. And only one V. parahaemolyticus strains (VPC2) were not found in multidrug resistance (MDR, defined as resistance to 3 or more different antimicrobials). So the multidrug resistance rate reaches to 97.6% of all 42 clinical V. parahaemolyticus isolates. Of these, there were 92.9% multidrug-resistant isolates showing resistance to more than five antibiotics. Strikingly, we found that four isolates showed resistance to ten antibiotics.
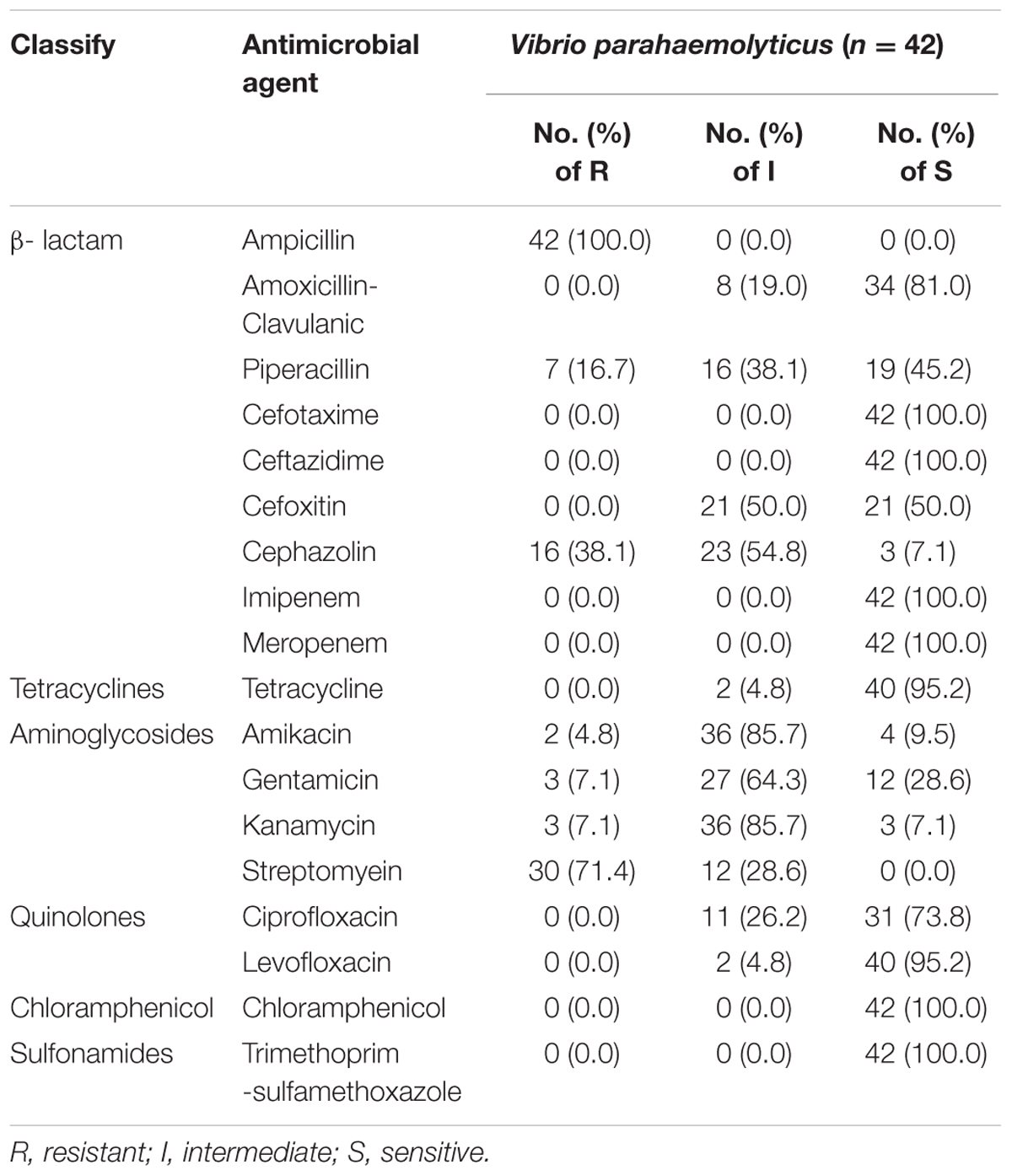
TABLE 3. Antimicrobial resistance profiles of 42 clinical V. parahaemolyticus isolates.
Antimicrobial Resistance Genotypes of V. parahaemolyticus
The 38 antibiotic resistance genes of 6 classes of antibiotics searched in 42 pathogenic V. parahaemolyticus isolates are shown in Supplementary Table S3. Eight out of 38 resistance genes (SHV, tet(B), strA, qnrA, gryA, qnrB, sulI, sulII) were detected in at least one isolates. Notably, all of the clinical isolates carried two or more ARGs evaluated. Among them, tet(B) was the most prevalent gene, with the detection frequencies of 100%, followed by strA, sulI, SHV, qnrA, qnrB gryA, and sulII the detected percentage of them was 92.9, 90.5, 28.6, 28.6, 26.2,19.0, and 4.8%, respectively.
Correlation among Virulence genes, STs, Resistance Phenotype, and Genotype
A minimum spanning tree (MST) of the sequence types (STs) that was constructed based on subtyping information, including sequence type and virulence-associated genes, is shown in Figure 2. As shown in Figure 2, We can see that the most prevalent clonal complexes were CC655, all of them were positive for virulence-related tdh (100%), T3SS1(100%) genes and T3SS2α (100%) genes and the majority of them were negative for the trh (95.5%) and T3SS2β (95.5%) genes. Notably, two distinct lineages of the T3SS2 have been described with a correlation between the presence of tdh with T3SS2α and trh with T3SS2β. From this we can observe that the virulence-related gene of trh and T3SS2β are co-occurrences and disappearance simultaneously. Specifically, the tdh (100%), T3SS1(100%) genes and T3SS2α (100%) genes were detected in all ST655 isolates.
We conclude that correlation between virulence-related genes, AMR phenotypes and genotypes was absent. Likewise, as shown in Figure 3, genetic diversity is not apparently associated with the AMR phenotypes and genotypes.
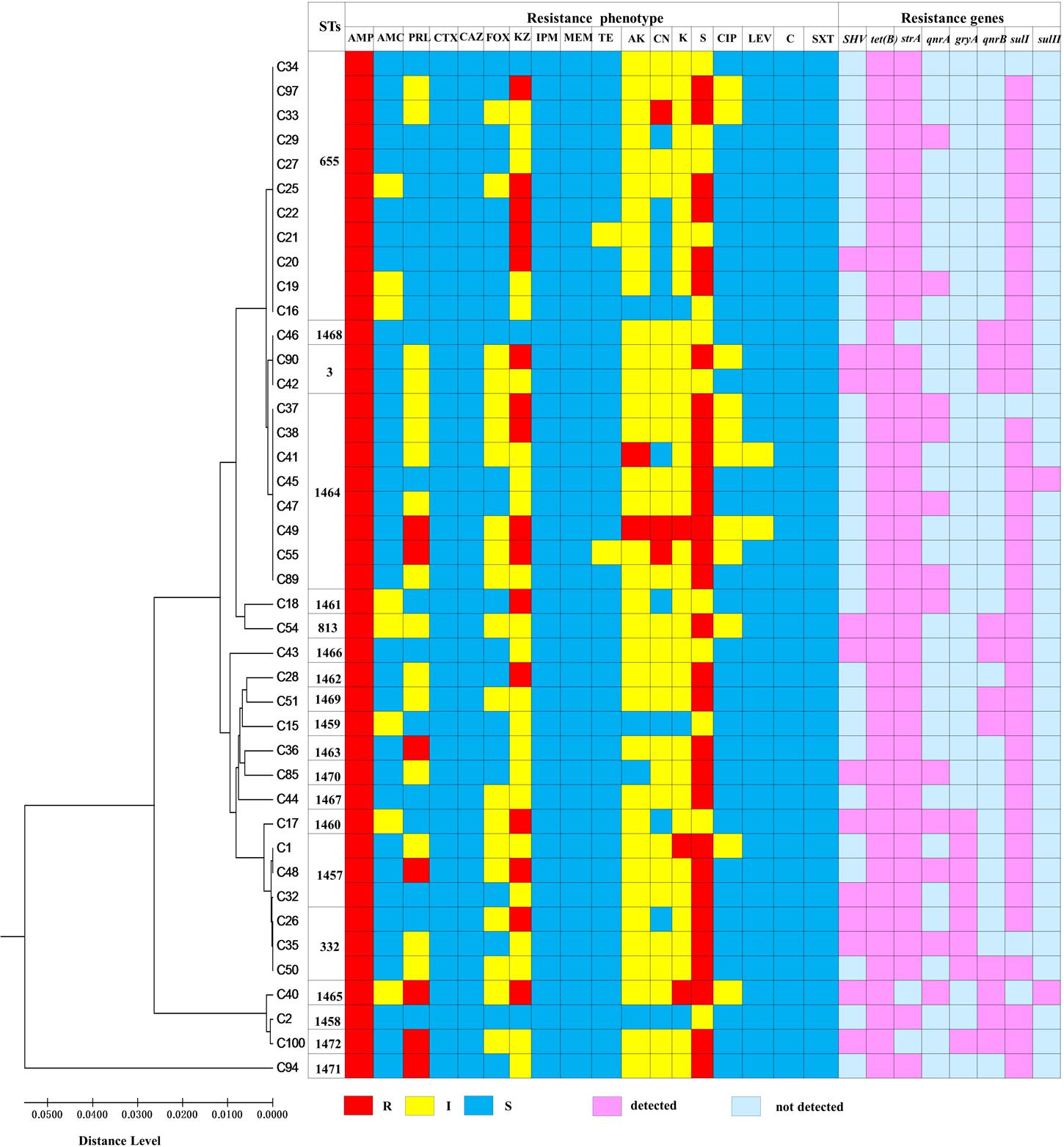
FIGURE 3. The relationship of genetic diversity, resistance phenotypic and genotypic characterization of 42 clinical V. parahaemolyticus isolates. ST, sequence type; R, resistant; I, intermediate; S, sensitive.
Discussion
In this study, we analyzed 1900 fecal specimens collected from Shanghai hospital from January, 2014 to December, 2015 and isolated 42 V. parahaemolyticus strains, with an isolation rate of 2.2%. Compared to other studies in China, this rate is significantly less than the 6.0% reported for southern coastal China in 2007–2012 (Li et al., 2014) and the 8.1% observed in southeastern China in 2009–2013 (Chen et al., 2016). Compared to other developing countries, our result is also less than the clinical V. parahaemolyticus isolation rate of 5.1% detected in Northwestern Mexico from 2004 to 2010 (Velazquez-Roman et al., 2012). That indicates the current status of V. parahaemolyticus infection in Shanghai is better than other regions.
The thermostable direct haemolysin (TDH), the TDH-related haemolysin (TRH) and the two type III secretion systems (T3SS1 and T3SS2) are recognized as major virulence factors in V. parahaemolyticus (Ceccarelli et al., 2013; Letchumanan et al., 2014; Raghunath, 2015). In our study, the hemolysin gene tdh was detected in 88.1% V. parahaemolyticus isolates, whereas the trh gene was present in only 7 strains. And we found that one of the clinical isolates is co-negative for tdh and trh gene. This finding is consistent with prior studies that not all clinical strains harbor these genes (Okuda et al., 1997). All V. parahaemolyticus strains in this study contains the T3SS1 component genes, which is also consistent with a previous study (Jones et al., 2012). The T3SS2 contains two gene clusters, T3SS2α and T3SS2β (Ritchie et al., 2012; Wang et al., 2015), which is closely related to tdh-positive and trh-positive V. parahaemolyticus, respectively (Jones et al., 2012). However, in this study, T3SS2α genes didn’t appear with tdh+ genes simultaneously, which indicates that there is high genetic heterogeneity in V. parahaemolyticus T3SSs.
The genetic diversity of V. parahaemolyticus was also investigated using MLST. Compared to other molecular methods, such as the identification of known virulence genes, phylogenetic analysis of housekeeping genes, microarray, and PFGE, MLST give a better understanding of the genetic relationships among V. parahaemolyticus isolates (Perez-Losada et al., 2013; Hazen et al., 2015; Ludeke et al., 2015). In this study, 42 V. parahaemolyticus isolates were classified into 20 sequence types (STs) with 16 novel STs. The high proportion of novel STs indicated a high genetic diversity of V. parahaemolyticus strains, and shows that the information content in the MLST database on this strain is still evolving. Thus, more MLST surveillances should be performed in China and the rest of world, to contributed to better understanding the genetic diversity of V. parahaemolyticus. Furthermore, ST655 was the most frequent hypotype in our study, which was clustered into the major clonal complex, CC655. Previous studies have been reported that ST3 belonged to the most prevalent clonal complex CC3, which is widely distributed and plays an important role in V. parahaemolyticus infections in multiple countries (Fuenzalida et al., 2006; Martinez-Urtaza et al., 2013; Turner et al., 2013; Haendiges et al., 2015; Han et al., 2016). There is only one locus difference between ST3 and ST655, which indicates that these STs are closely related. We recommend further research on the pandemic clonal complexes CC655 containing ST655 for management of V. parahaemolyticus infections.
Antibiotic treatment is necessary for controlling V. parahaemolyticus infections, but overuse of antibiotics has led to the generation and distribution of antimicrobial-resistant bacteria, which is becoming a major concern for human health (Ji et al., 2011; Shaw et al., 2014; Blair et al., 2015). This study also investigated the antimicrobial resistance phenotype and genotype of the 42 clinical V. parahaemolyticus strains. All isolates showed a high level of resistance against Ampicillin (100.0%), Streptomycin (100.0%), Cephazolin (92.9%), Kanamycin (92.8%), and Amikacin (90.5%), and eight out of 38 resistance genes (SHV, tet(B), strA, qnrA, gryA, qnrB, sulI, sulII) were detected in at least two isolates. According to this study and some previous researches (Chen et al., 2016; Xie et al., 2017), the antimicrobial resistance of clinical V. parahaemolyticus was significantly higher than that of environmental strains which were isolated from water (Shaw et al., 2014), aquatic products (Letchumanan et al., 2015; Lou et al., 2016; Yu et al., 2016; Xie et al., 2017) or ready-to-eat foods (Xie et al., 2016). As the human gastrointestinal tract is a conducive environment for promoting horizontal ARGs transfer (Hu et al., 2013; Theethakaew et al., 2013), we speculate that the complex gastrointestinal environment may accelerate the acquisition of antimicrobial resistance in V. parahaemolytiucs. The microevolution mechanisms for the different rates of acquisition of antibiotic resistance between clinical and environmental pathogens should be studied further.
Conclusion
This study is the first comprehensive research describing the virulence, genetic diversity, antibiotic resistance phenotype, and genotype of V. parahaemolyticus from diarrhea patients in Shanghai. The study reveals that tdh, trh and T3SS genes are of equal importance as virulence associated factors. MLST analysis showed that the novel loci and STs points to high genetic diversity of V. parahaemolyticus strains isolated in Shanghai. ST655 was the most prevalent STs and this ST could have evolved from the global pandemic ST3. The antimicrobial resistance profiles indicated that the multidrug-resistant isolates were also widespread and measures to contain or slowdown the emergence of drug-resistant strains should be a top priority in China. These results add to the epidemiological data of clinical V. parahaemolyticus isolates in Shanghai and highlight the need for more AMR type research for managing the burden of disease caused by this pathogen in China.
Author Contributions
HL, RT, and YL contributed equally to carrying out the experiments and writing the draft manuscript. YZ (Corresponding Author), YP, PM, and ZZ provided support for experimental design and editing of final manuscript. ZC, WC, QH assisted in completing the experiments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (31271870), the project of Science and Technology Commission of Shanghai Municipality (14320502100), Key Project of Shanghai Agriculture Prosperity through Science and Technology [2014(3-5), 2015(4-8), and 2016 (1-1)], the “Dawn” Program of Shanghai Education Commission (15SG48).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01043/full#supplementary-material
Footnotes
References
Allen, H. K., Donato, J., Wang, H. H., Cloud-Hansen, K. A., Davies, J., Handelsman, J., et al. (2010). Call of the wild: antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 8, 251–259. doi: 10.1038/nrmicro2312
Banerjee, S. K., Kearney, A. K., Nadon, C. A., Peterson, C. L., Tyler, K., Bakouche, L., et al. (2014). Phenotypic and genotypic characterization of Canadian clinical isolates of Vibrio parahaemolyticus collected from 2000 to 2009. J. Clin. Microbiol. 52, 1081–1088. doi: 10.1128/JCM.03047-13
Blair, J. M. A., Webber, M. A., Baylay, A. J., Ogbolu, D. O., and Piddock, L. J. V. (2015). Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51. doi: 10.1038/nrmicro3380
Broberg, C. A., Calder, T. J., and Orth, K. (2011). Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect. 13, 992–1001. doi: 10.1016/j.micinf.2011.06.013
Ceccarelli, D., Hasan, N. A., Huq, A., and Colwell, R. R. (2013). Distribution and dynamics of epidemic and pandemic Vibrio parahaemolyticus virulence factors. Front. Cell. Infect. Microbiol. 3:97. doi: 10.3389/fcimb.2013.00097
Chen, Y., Chen, X., Yu, F., Wu, M., Wang, R., Zheng, S., et al. (2016). Serology, virulence, antimicrobial susceptibility and molecular characteristics of clinical Vibrio parahaemolyticus strains circulating in southeastern China from 2009 to 2013. Clin. Microbiol. Infect. 258, e9–e16. doi: 10.1016/j.cmi.2015.11.003
CLSI (2015). Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Fifth Informational Supplement, M100-S25. Wayne, PA: Clinical and Laboratory Standards Institute.
Croci, L., Suffredini, E., Cozzi, L., Toti, L., Ottaviani, D., Pruzzo, C., et al. (2007). Comparison of different biochemical and molecular methods for the identification of Vibrio parahaemolyticus. J. Appl. Microbiol. 102, 229–237.
Food and Drug Administration [FDA] (2004). Bacteriological Analytical Manual, 8th Edn. Arlington, VA: Food and Drug Administration, AOAC International.
Fournier, P. E., and Ogata, H. (2005). Pandemic Vibrio parahaemolyticus O3: K6. Europe. Emerg. Infect. Dis. 11, 1317–1318. doi: 10.3201/eid1108.050322
Fuenzalida, L., Hernandez, C., Toro, J., Rioseco, M. L., Romero, J., Espejo, R. T., et al. (2006). Vibrio parahaemolyticus in shellfish and clinical samples during two large epidemics of diarrhoea in southern Chile. Environ. Microbiol. 8, 675–683.
Fujino, T., Okuno, Y., Nakada, D., Aoyama, A., Mukai, T., and Ueho, T. (1953). On the bacteriological examination of shirasu food poisoning. Med. J. Osaka Univ. 4, 299–304.
Gonzalez-Escalona, N., Martinez-Urtaza, J., Romero, J., Espejo, R. T., Jaykus, L. A., Depaola, A., et al. (2008). Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 190, 2831–2840. doi: 10.1128/JB.01808-07
Guo, D., Zhang, Z., Tang, X., Wang, J., Pan, Y., and Zhao, Y. (2013). Antimicrobial resistance and molecular typing of pathogenic Vibrio parahaemolyticus isolated from seafood in Shanghai retail markets. J. Pure Appl. Microbiol. 7, 3085–3090.
Haendiges, J., Timme, R., Allard, M. W., Myers, R. A., Brown, E. W., and Gonzalez-Escalona, N. (2015). Characterization of Vibrio parahaemolyticus clinical strains from Maryland (2012–2013) and comparisons to a locally and globally diverse V. parahaemolyticus strains by whole-genome sequence analysis. Front. Microbiol. 6:125. doi: 10.3389/fmicb.2015.00125
Han, C. X., Tang, H., Ren, C. L., Zhu, X. P., and Han, D. S. (2016). Sero-prevalence and genetic diversity of pandemic V. parahaemolyticus strains occurring at a global scale. Front. Microbiol. 7:567. doi: 10.3389/fmicb.2016.00567
Han, D., Tang, H., Ren, C., Wang, G. Z., Zhou, L., Han, C. X., et al. (2015). Prevalence and genetic diversity of clinical Vibrio parahaemolyticus isolates from China, revealed by multilocus sequence typing scheme. Front. Microbiol. 6:291. doi: 10.3389/fmicb.2015.00291
Hazen, T. H., Lafon, P. C., Garrett, N. M., Lowe, T. M., Silberger, D. J., Rowe, L., et al. (2015). Insights into the environmental reservoir of pathogenic Vibrio parahaemolyticus using comparative genomics. Front. Microbiol. 6:204. doi: 10.3389/fmicb.2015.00204
He, Y., Jin, L., Sun, F., Hu, Q., and Chen, L. (2016). Antibiotic and heavy-metal resistance of Vibrio parahaemolyticus. Environ. Sci. Pollut. Res. Int. 23, 15033–15040. doi: 10.1007/s11356-016-6614-4
He, Y., Wang, H., and Chen, L. (2015). Comparative secretomics reveals novel virulence-associated factors of Vibrio parahaemolyticus. Front. Microbiol. 6:707. doi: 10.3389/fmicb.2015.00707
Hu, Q., and Chen, L. (2016). Virulence and antibiotic and heavy metal resistance of Vibrio parahaemolyticus isolated from crustaceans and shellfish in Shanghai. China. J. Food Prot. 79, 1371–1377. doi: 10.4315/0362-028X.JFP-16-031
Hu, Y., Yang, X., Qin, J., Lu, N., Cheng, G., Wu, N., et al. (2013). Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat. Commun. 4:2151. doi: 10.1038/ncomms3151
Hubbard, T. P., Chao, M. C., Abel, S., Blondela, J. C., Wieschc, P. A. Z., Zhou, X. H., et al. (2016). Genetic analysis of Vibrio parahaemolyticus intestinal colonization. Proc. Natl. Acad. Sci. U.S.A. 113, 6283–6288. doi: 10.1073/pnas.1601718113
ISO (2007). In Microbiology of Food and Animal Feeding Stuffs-horizontal Method for the Detection of Potentially Enteropathogenic Vibrio spp.-Part 1. Detection of Vibrio parahaemolyticus and Vibrio cholerae, 1st Edn. Geneva: International Organization for Standardization.
Ji, H., Chen, Y., Guo, Y., Liu, X. M., Wen, J., Liu, H., et al. (2011). Occurrence and characteristics of Vibrio vulnificus in retail marine shrimp in China. Food Control 22, 1935–1940. doi: 10.1016/j.foodcont.2011.05.006
Jones, J. L., Ludeke, C. H. M., Bowers, J. C., Garrett, N., Fischer, M., Parsons, M. B., et al. (2012). Biochemical, serological, and virulence characterization of clinical and oyster Vibrio parahaemolyticus isolates. J. Clin. Microbiol. 50, 2343–2352 doi: 10.1128/JCM.00196-12
Koser, C. U., Ellington, M. J., and Peacock, S. J. (2014). Whole-genome sequencing to control antimicrobial resistance. Trends Genet. 30, 401–407. doi: 10.1016/j.tig.2014.07.003
Letchumanan, V., Chan, K. G., and Lee, L. H. (2014). Vibrio parahaemolyticus: a review on the pathogenesis, prevalence, and advance molecular identification techniques. Front. Microbiol. 5:705. doi: 10.3389/fmicb.2014.00705
Letchumanan, V., Yin, W. F., Lee, L. H., and Chan, K. G. (2015). Prevalence and antimicrobial susceptibility of Vibrio parahaemolyticus isolated from retail shrimps in Malaysia. Front. Microbiol. 6:33. doi: 10.3389/fmicb.2015.00033
Li, R., Chiou, J., Chan, E. W. C., and Chen, S. (2016). A novel PCR-based approach for accurate identification of Vibrio parahaemolyticus. Front. Microbiol. 7:44 doi: 10.3389/fmicb.2016.00044
Li, Y., Xie, X., Shi, X., Lin, X., Mou, J., Chen, Q., et al. (2014). Vibrio parahaemolyticus, southern coastal region of China, 2007–2012. Emerg. Infect. Dis. 20, 685–688. doi: 10.3201/eid2004.130744
Lopatek, M., Wieczorek, K., and Osek, J. (2015). Prevalence and antimicrobial resistance of Vibrio parahaemolyticus isolated from raw shellfish in Poland. J. Food Prot. 78, 1029–1033. doi: 10.4315/0362-028X.JFP-14-437
Lou, Y., Liu, H., Zhang, Z., Pan, Y., and Zhao, Y. (2016). Mismatch between antimicrobial resistance phenotype and genotype of pathogenic Vibrio parahaemolyticus isolated from seafood. Food Control 59, 207–211. doi: 10.1016/j.foodcont.2015.04.039
Ludeke, C. H. M., Gonzalez-Escalona, N., Fischer, M., and Jones, J. L. (2015). Examination of clinical and environmental Vibrio parahaemolyticus isolates by multi-locus sequence typing (MLST) and multiple-locus variable-number tandem-repeat analysis (MLVA). Front. Microbiol. 6:564. doi: 10.3389/fmicb.2015.00564
Makino, K., Oshima, K., Kurokawa, K., Yokoyama, K., Uda, T., Tagomori, K., et al. (2003). Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet 361, 743–749. doi: 10.1016/S0140-6736(03)12659-1
Martinez-Urtaza, J., Baker-Austin, C., Jones, J. L., Newton, A. E., DePaola, A. (2013). Spread of Pacific Northwest Vibrio parahaemolyticus strain. N. Engl. J. Med. 369, 1573–1574. doi: 10.1056/NEJMc1305535
McLaughlin, J. B., DePaola, A., Bopp, C. A., Martinek, K. A., Napolilli, N. P., Allison C. G., et al. (2005). Outbreak of Vibrio parahaemolyticus gastroenteritis associated with Alaskan oysters. N. Engl. J. Med. 353, 1463–1470. doi: 10.1056/NEJMoa051594
Nilsson, W. B., and Turner, J. W. (2016). The thermostable direct hemolysin-related hemolysin (trh) gene of Vibrio parahaemolyticus: sequence variation and implications for detection and function. J. Microbiol. Methods 126, 1–7. doi: 10.1016/j.mimet.2016.04.007
Okuda, J., Ishibashi, M., Hayakawa, E., Nishino, T., Takeda, Y., Mukhopadhyay, A. K., et al. (1997). Emergence of a unique O3: K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from Southeast Asian travelers arriving in Japan. J. Clin. Microbiol. 35, 3150–3155.
Paranjpye, R., Hamel, O. S., Stojanovski, A., and Liermann, M. (2012). Genetic diversity of clinical and environmental Vibrio parahaemolyticus strains from the Pacific Northwest. Appl. Environ. Microbiol. 78, 8631–8638. doi: 10.1128/AEM.01531-12
Pazhani, G. P., Bhowmik, S. K., Ghosh, S., Guin, S., Dutta, S., Rajendran, K., et al. (2014). Trends in the epidemiology of pandemic and non-pandemic strains of Vibrio parahaemolyticus isolated from diarrheal patients in Kolkata. India. PLoS Negl. Trop. Dis. 8, e2815. doi: 10.1371/journal.pntd.0002815
Perez-Losada, M., Cabezas, P., Castro-Nallar, E., and Crandall, K. A. (2013). Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Infect. Genet. Evol. 16, 38–53. doi: 10.1016/j.meegid.2013.01.009
Qi, X. L., Wang, H. X., Bu, S. R., Xu, X. G., Wu, X. Y., and Lin, D. F. (2016). Incidence rates and clinical symptoms of Salmonella, Vibrio parahaemolyticus, and Shigella infections in China, 1998–2013. J. Infect. Chemother. 10, 127–133. doi: 10.3855/jidc.6835
Raghunath, P. (2015). Roles of thermostable direct hemolysin (TDH) and TDH-related hemolysin (TRH) in Vibrio parahaemolyticus. Front. Microbiol. 5:805. doi: 10.3389/fmicb.2014.00805
Ritchie, J. M., Rui, H., Zhou, X., Iida, T., Kodoma, T., Ito, S., et al. (2012). Inflammation and disintegration of intestinal villi in an experimental model for Vibrio parahaemolyticus-induced diarrhea. PLoS Pathog. 8:e1002593. doi: 10.1371/journal.ppat.1002593
Scallan, E., Hoekstra, R. M., Angulo, F. J., Angulo, F. J., Tauxe, R. V., Widdowson, M. A., et al. (2011). Foodborne illness acquired in the United States-major pathogens. Emerg. Infect. Dis. 17, 7–15. doi: 10.3201/eid1701.P11101
Shaw, K. S., Goldstein, R. E. R., He, X., Jacobs, J. M., Crump, B. C., Sapkota, A. R., et al. (2014). Antimicrobial susceptibility of Vibrio vulnificus and Vibrio parahaemolyticus recovered from recreational and commercial areas of Chesapeake Bay and Maryland Coastal Bays. PLoS ONE 9:e89616. doi: 10.1371/journal.pone.0089616
Su, Y. C., and Liu, C. (2007). Vibrio parahaemolyticus: a concern of seafood safety. Food Microbiol. 24, 549–558. doi: 10.1016/j.fm.2007.01.005
Theethakaew, C., Feil, E. J., Castillo-Ramírez, S., Aanensen, D. M., and Suthienkul, O. (2013). Genetic relationships of Vibrio parahaemolyticus isolates from clinical, human carrier and environmental sources in Thailand determined by multilocus sequence analysis. Appl. Environ. Microbiol. 79, 2358–2370. doi: 10.1128/AEM.03067-12
Thomas, C. M., and Nielsen, K. M. (2005). Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 3, 711–721. doi: 10.1038/nrmicro1234
Turner, J. W., Paranjpye, R. N., Landis, E. D., Biryukov, S. V., Gonzalez-Escalona, N., Nilsson, W. B., et al. (2013). Population structure of clinical and environmental Vibrio parahaemolyticus from the Pacific Northwest coast of the United States. PLoS ONE 8:e55726. doi: 10.1371/journal.pone.0055726
Velazquez-Roman, J., Leon-Sicairos, N., Flores-Villasenor, H., Villafana-Rauda, S., and Canizalez-Roman, A. (2012). Association of pandemic Vibrio parahaemolyticus O3: K6 present in the coastal environment of Northwest Mexico with cases of recurrent diarrhea between 2004 and 2010. Appl. Environ. Microbiol. 78, 1794–1803. doi: 10.1128/AEM.06953-11
Velusamy, V., Arshak, K., Korostynska, O., Oliwa, K., and Adley, C. (2010). An overview of foodborne pathogen detection: in the perspective of biosensors. Biotechnol. Adv. 28, 232–254. doi: 10.1016/j.biotechadv.2009.12.004
Wang, H., Zhang, J., Jiang, T., Bao, Y., and Zhou, X. (2011). Insufficiency of the Kanagawa hemolytic test for detecting pathogenic Vibrio parahaemolyticus in Shanghai. China. Diagn. Microbiol. Infect. Dis. 69, 7–11. doi: 10.1016/j.diagmicrobio.2010.08.016
Wang, R., Zhong, Y., Gu, X., Yuan, J., Saeed, A. F., Wang, S. H., et al. (2015). The pathogenesis, detection, and prevention of Vibrio parahaemolyticus. Front. Microbiol. 6:144. doi: 10.3389/fmicb.2015.00144
West, C. K. G., Klein, S. L., and Lovell, C. R. (2013). High frequency of virulence factor genes tdh, trh, and tlh in Vibrio parahaemolyticus strains isolated from a pristine estuary. Appl. Environ. Microbiol. 79, 2247–2252. doi: 10.1128/AEM.03792-12
Wu, S., Wu, Q., Zhang, J., Chen, M., and Guo, W. (2016). Analysis of multilocus sequence typing and virulence characterization of Listeria monocytogenes isolates from Chinese retail ready-to-eat food. Front. Microbiol. 7:168. doi: 10.3389/fmicb.2016.00168
Wu, Y., Wen, J., Ma, Y., Ma, X. C., and Chen, Y. (2014). Epidemiology of foodborne disease outbreaks caused by Vibrio parahaemolyticus, China, 2003–2008. Food Control 46, 197–202. doi: 10.1016/j.foodcont.2014.05.023
Xie, T., Wu, Q., Zhang, J., Xu, X., and Cheng, J. (2017). Comparison of Vibrio parahaemolyticus isolates from aquatic products and clinical by antibiotic susceptibility, virulence, and molecular characterisation. Food Control 71, 315–321. doi: 10.1016/j.foodcont.2016.06.046
Xie, T., Xu, X., Wu, Q., Zhang, J., and Cheng, J. (2016). Prevalence, molecular characterization, and antibiotic susceptibility of Vibrio parahaemolyticus from ready-to-eat foods in China. Front. Microbiol. 7:549. doi: 10.3389/fmicb.2016.00549
Xu, F., Ilyas, S., Hall, J. A., Jones, S. H., Cooper, V. S., Whistler, C. A., et al. (2015). Genetic characterization of clinical and environmental Vibrio parahaemolyticus from the Northeast USA reveals emerging resident and non-indigenous pathogen lineages. Front. Microbiol. 6:272. doi: 10.3389/fmicb.2015.00272
Yano, Y., Hamano, K., Satomi, M., Tsutsui, I., Ban, M., Aue-umneoy, D., et al. (2014). Prevalence and antimicrobial susceptibility of Vibrio species related to food safety isolated from shrimp cultured at inland ponds in Thailand. Food Control 38, 30–36. doi: 10.1016/j.foodcont.2013.09.019
Yu, Q., Niu, M., Yu, M., Liu, Y., Wang, D., Shi, X., et al. (2016). Prevalence and antimicrobial susceptibility of Vibrio parahaemolyticus isolated from retail shellfish in Shanghai. Food Control 60, 263–268. doi: 10.1016/j.foodcont.2015.08.005
Zhang, H., Sun, S., Shi, W., Cui, L., and Gu, Q. (2013). Serotype, virulence, and genetic traits of foodborne and clinical Vibrio parahaemolyticus isolates in Shanghai. China. Foodborne Pathog. Dis. 10, 796–804. doi: 10.1089/fpd.2012.1378
Zhang, L., and Orth, K. (2013). Virulence determinants for Vibrio parahaemolyticus infection. Curr. Opin. Microbiol. 16, 70–77. doi: 10.1016/j.mib.2013.02.002
Keywords: Vibrio parahaemolyticus, diarrhoea samples, virulence, genetic diversity, antimicrobial susceptibility
Citation: Li H, Tang R, Lou Y, Cui Z, Chen W, Hong Q, Zhang Z, Malakar PK, Pan Y and Zhao Y (2017) A Comprehensive Epidemiological Research for Clinical Vibrio parahaemolyticus in Shanghai. Front. Microbiol. 8:1043. doi: 10.3389/fmicb.2017.01043
Received: 01 March 2017; Accepted: 24 May 2017;
Published: 08 June 2017.
Edited by:
Pendru Raghunath, Texila American University, GuyanaReviewed by:
Anca Ioana Nicolau, Dunarea de Jos University, RomaniaGonçalo Nieto Almeida, Instituto Nacional de Investigação Agrária e Veterinária, Portugal
Copyright © 2017 Li, Tang, Lou, Cui, Chen, Hong, Zhang, Malakar, Pan and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:Yong Zhao, yzhao@shou.edu.cn
†These authors have contributed equally to this work.