Dysregulated expression of death, stress and mitochondrion related genes in the sciatic nerve of presymptomatic SOD1G93A mouse model of Amyotrophic Lateral Sclerosis
 Chrystian J. Alves
Chrystian J. Alves  Jessica R. Maximino
Jessica R. Maximino Gerson Chadi
Gerson Chadi- Department of Neurology, Neuroregeneration Center, University of São Paulo School of Medicine, São Paulo, Brazil
Schwann cells are the main source of paracrine support to motor neurons. Oxidative stress and mitochondrial dysfunction have been correlated to motor neuron death in Amyotrophic Lateral Sclerosis (ALS). Despite the involvement of Schwann cells in early neuromuscular disruption in ALS, detailed molecular events of a dying-back triggering are unknown. Sciatic nerves of presymptomatic (60-day-old) SOD1G93A mice were submitted to a high-density oligonucleotide microarray analysis. DAVID demonstrated the deregulated genes related to death, stress and mitochondrion, which allowed the identification of Cell cycle, ErbB signaling, Tryptophan metabolism and Rig-I-like receptor signaling as the most representative KEGG pathways. The protein-protein interaction networks based upon deregulated genes have identified the top hubs (TRAF2, H2AFX, E2F1, FOXO3, MSH2, NGFR, TGFBR1) and bottlenecks (TRAF2, E2F1, CDKN1B, TWIST1, FOXO3). Schwann cells were enriched from the sciatic nerve of presymptomatic mice using flow cytometry cell sorting. qPCR showed the up regulated (Ngfr, Cdnkn1b, E2f1, Traf2 and Erbb3, H2afx, Cdkn1a, Hspa1, Prdx, Mapk10) and down-regulated (Foxo3, Mtor) genes in the enriched Schwann cells. In conclusion, molecular analyses in the presymptomatic sciatic nerve demonstrated the involvement of death, oxidative stress, and mitochondrial pathways in the Schwann cell non-autonomous mechanisms in the early stages of ALS.
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disease characterized by a selective death of motor neurons of the spinal cord, brainstem and cerebral cortex, leading to progressive paralysis. The patient's death is often due to a respiratory failure, usually within 3–5 years from diagnosis (Kiernan et al., 2011).
ALS pathogenesis is poorly understood and the proposed mechanisms related to neurodegeneration seem to be multifactorial and include mitochondrial dysfunction, oxidative stress, protein aggregation and axonal transport impairment (Boillée et al., 2006a; de Vos et al., 2007). Additionally, mechanisms associated with motor neuron degeneration involve non-neuronal cells (Boillée et al., 2006a,b; Yamanaka et al., 2008; Wang et al., 2011), as seen by toxicity in neuron-glial co-culture experiments and also by the activation of molecular pathways related to paracrine stress signaling (Boillée et al., 2006a,b; Ferraiuolo et al., 2007; Kiernan et al., 2011).
Evidence obtained from studies on early axon and neuromuscular junction events in ALS patients and a mouse model have indicated the presence of peripheral motor neuron dysfunction before the activation of death pathways and the onset of clinical symptoms (Fischer et al., 2004; Gould et al., 2006; Moloney et al., 2014). Indeed, the peripheral events seem to proceed toward soma in a retrograde dying-back manner (Coleman and Perry, 2002; Fischer et al., 2004; Saxena and Caroni, 2007; Rocha et al., 2013). Oxidative stress and compromised mitochondria represent one set of proposed mechanisms underlying peripheral ALS dysfunction (Barber et al., 2006; Cozzolino and Carrì, 2012; Cozzolino et al., 2013).
The Schwann cell is the major functional cell type supporting axonal integrity (Mirsky and Jessen, 1999). Recent evidence suggests that Schwann cells may contribute to ALS distal axonopathy (Fischer et al., 2004; de Winter et al., 2006; Gorlewicz et al., 2009; Keller et al., 2009; Lobsiger et al., 2009; Chen et al., 2010; Verheijen et al., 2014). For instance, the up-regulation of inducible nitric oxide synthase and semaphorin 3A in the Schwann cells close to terminal fibers of the sciatic nerve has been associated with dying-back mechanisms in presymptomatic ALS mice (de Winter et al., 2006; Keller et al., 2009; Chen et al., 2010; Malaspina et al., 2010; Venkova et al., 2014). Furthermore, accumulation of axonal ribosomes in Schwann cells bearing mutant hSOD1 in a presymptomatic phase of ALS mouse model further suggests an early involvment of this glial cell type in the pathogenesis of the disease (Verheijen et al., 2014).
Microarray analyses have been useful in identifying new molecular cues potentially involved in ALS pathogenesis both in postmortem human tissue and also in several clinical stages of experimental animal models of ALS (Olsen et al., 2001; Hensley et al., 2002; Yoshihara et al., 2002; Dangond et al., 2004; Perrin et al., 2005; Ferraiuolo et al., 2007, 2009; Fukada et al., 2007; Vargas et al., 2008; Kudo et al., 2010; Boutahar et al., 2011; Cooper-Knock et al., 2012; de Oliveira et al., 2013, 2014; Maximino et al., 2014). However, there is a lack of information on gene expression in peripheral motor nerves in ALS despite the importance of recently described dying-back events in this disorder. Furthermore, an evaluation of dysregulated genes in specific, enriched cell populations obtained by cell sorting might extend these molecular analyses at cellular level.
By means of a high-density oligonucleotide microarray analysis linked to specific tools capable of identifying distinct cellular components and biological processes, the aim of this work was to determine whether the expression of genes involved in the regulation of death, stress and mitochondrial function was dysregulated in the sciatic nerve of the SOD1G93A mouse model during the presymptomatic stage of ALS. This work has also evaluated the modulation of selected molecules in enriched sciatic nerve-derived Schwann cells, thus detailing the role of these glial cells in the early phase of this disease.
Materials and Methods
Animal and Tissue Sample
Transgenic SOD1G93A mice (The Jackson Laboratory, Bar Harbor, ME, USA) were crossbred and the colony was maintained in a specific pathogen-free environment within the animal facility of the University of São Paulo Medical School (São Paulo, Brazil) as described previously (Gurney, 1994; Scorisa et al., 2010; Alves et al., 2011). Animals were kept under controlled temperature and humidity conditions with a standardized light–dark cycle (lights on at 7:00 a.m. and off at 7:00 p.m.) and free access to food pellets and tap water. Mice were genotyped by PCR amplification of tail extracted DNA which identified the presence of the human SOD1 mutated gene (mSOD1) (Gurney, 1994; Scorisa et al., 2010; Alves et al., 2011). The Transgenic SOD1G93A mice express high number of mutant human SOD1 copies (Gurney, 1994; Verheijen et al., 2014). The study was conducted under protocols approved by the Ethical Animal Care and Use Committee at the University of São Paulo and in accordance with the Guide for the Care and Use of Laboratory Animals adopted by the National Institutes of Health.
Sixty-day-old presymptomatic male SOD1G93A mice and their age-paired wild-type controls (~20–25 g body weight) were used in the experiments. No motor neuron death was seen in any animal at this age (Alves et al., 2011), therefore the animals were chosen for the present presymptomatic analyses. Animals were killed by decapitation and sciatic nerves were removed, frozen and stored at −80°C for further use. Four mice were used in each group for the microarray experiments. The quantitative polymerase chain reaction (qPCR) analyses of sciatic nerves were performed using samples from six different mice from each transgene and wild-type groups.
Immunofluorescence Labelings and Histological Sections of Sciatic Nerve
Four animals per genotype were used for immunofluorescence labelings, according to previous publications (Guzen et al., 2009; Batista et al., 2014). Mice were anesthetized with sodium pentobarbital and euthanized by a transcardiac perfusion with 7 ml isotonic saline at room temperature followed by 35 ml fixation fluid (4°C) over a period of 6 min. The fixative consisted of 4% paraformaldehyde (w/v) in 0.1 M phosphate buffer (pH 6.9). The sciatic nerves were removed, kept in fixative at 4°C for 90 min and rinsed for 24 h in 10% sucrose dissolved in PBS. Sciatic nerves were then frozen in dry ice-cooled (−40°C) isopentane (Sigma) and stored at a −80°C freezer until use. Longitudinal sections (5 μm thick) were obtained using a cryostat (Leica, CM3000, Germany). The sections were initially washed for 2 × 10 min in PBS and then were incubated overnight in PBS containing 0.5% Triton X-100 (Sigma) and 1% bovine albumin serum (BSA, Sigma) with a polyclonal rabbit antibody against microtubule associated protein 2 (MAP2; diluted 1:200; Sigma), growth associated protein 43 (GAP-43; diluted 1:200; Sigma), S100 (diluted 1:200; Abcam) and p75NGF neurotrophin receptor (p75; diluted 1:200; Abcam). After the incubation of the primary antibodies, sections were washed for 2 × 10 min in PBS and incubated for 1 h in the dark at 37°C with a dilution of Alexa Fluor® 488 or 594-conjugated secondary antibodies specific for rabbit (1:200, all from Invitrogen, USA). Preparations were mounted on microscope slides and counterstained with nuclear 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Vector, USA). Digital images were obtained by means of an Olympus BX-51 microscope (Olympus, USA).
In addition, four animals per genotype were processed to histological staining using methylene blue. Briefly, sciatic nerves were fixed in 2.5% glutaraldehyde, pH 7.4 (24 h). After extensive wash, tissues were embedded in araldite and transverse semi-thin sections (0.5 μm thickness) were obtained. The sections were stained with methylene blue and photomicrographed using an Olympus BX-51 microscope (Olympus).
RNA Isolation and Microarray Experiments
The procedures for RNA isolation and microarray experiments with sciatic nerve were described in our previous publication (Maximino et al., 2014). Briefly, RNAs from samples (25 ng) and reference (100 ng) were reverse transcribed by the Low-input RNA Linear Amplification kit and then transcribed to Cy3-labeled (samples) or Cy5-labeled (reference) RNAs according to the manufacturer's instructions (Agilent Technologies, USA) and to our previously described protocols (de Oliveira et al., 2013).
Microarray Analysis
Raw image data were converted to numerical data using the Agilent Feature Extraction Software, version 11.0.1.1, as described in our previous study (Maximino et al., 2014; Alves et al., 2015). Raw signal intensities were normalized using the GeneSpring GX v12.6 software package (Agilent Technologies, USA). After normalization, the probes were tested for differential expression. GeneSpring GX was also used in the statistical analyses of gene expressions between genotypes (SOD1G93A × wild-type), according to previous publications (Smyth, 2004; Fu et al., 2014; Yang et al., 2014; Ryan et al., 2015; Wang et al., 2015). Genes with p < 0.05 were considered differentially expressed. The raw data from hybridizations are available on the Gene Expression Omnibus Database, and the GEO accession number is GSE69450.
Bioinformatic Analysis
The dysregulated genes were submitted for the following analyses to provide information regarding their involvement with specific cellular/molecular pathways related to ALS:
Functional Enrichment Analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7b functional tool (https://david.ncifcrf.gov/) (Huang Da et al., 2008) was used to identify genes related to death, stress and mitochondrial function through the Gene Ontology (GO) annotation database. The DAVID analysis focused on the category: Biological Process and Cellular Component. Stringency (EASE score set to 0.05) parameters were selected to improve confidence in the terms designated as enriched. The Biological Process and Cellular Component terms related to death, stress and mitochondrial function were further organized using the BioVenn tool (http://www.cmbi.ru.nl/cdd/biovenn/) (Hulsen et al., 2008) which identifies common and exclusively expressed genes between lists.
In order to identify over-represented pathways, the GO terms list containing genes related to death, stress and mitochondrion was submitted to the DAVID tool using the Kyoto Encyclopedia of Genes and Genomes (KEGG).
Protein Interaction Network Analysis
The Cytoscape plugin GeneMANIA (Warde-Farley et al., 2010) was used to predict protein interactions from the list of differentially expressed genes in the sciatic nerve of presymptomatic SOD1G93A mice related to death, stress and mitochondrion. The network was generated using only information derived from the pathway and physical interactions categories in GeneMANIA. The connectivity of the nodes contained in the network was achieved by means of the node centrality parameters “degree” and “betweenness,” using the Cytoscape plug-in CentiScaPe (Scardoni et al., 2009). Node degree is a local structure measure in networks that determines the number of edges in each node. Conversely, betweenness centrality is a global structure measure in networks that determines the number of shortest paths passing through a specific node while connecting, directly or indirectly, pairs of nodes (Scardoni et al., 2009). A scatter plot was constructed by inputting node degree and betweenness values for each node in GraphPad Prism 5. The combination of such attributes in the scatter plot allowed the visualization of hubs (nodes with high node degree) and bottlenecks (nodes with high node betweenness). Subnetworks were built using list of genes related to death, stress and mitochondrion separately; scatter plots were also constructed by inputting node degree and betweenness values for each node in GraphPad Prism 5.
Flow Cytometry Sorting of Isolated Schwann Cells and Fibroblast
Schwann cells and fibroblasts were isolated by means of flow cytometry sorting from sciatic nerve explants of presymptomatic SOD1G93A mice and their age-paired wild-type controls as described in our previous publication (Maximino et al., 2014). The sciatic nerve-derived cell suspension was submitted to a double immunolabeling to identify Schwann cells and fibroblasts by means of a fluorescein isothiocyanate (FITC)-conjugated mouse p75NGF Receptor antibody (Abcam, USA) and a fluorescein phychoerythrin (PE-Cy5)-conjugated monoclonal antibody against Thy-1 (Abcam, USA), respectively. The p75NGF Receptor labeling was employed in the cell sorting experiments because it is a well-characterized surface marker for Schwann cells (Niapour et al., 2010). Cells were then analyzed for type and specificity as well as separated on a FACSAria III Cell Sorter (BD Biosciences, USA). A maximum of 106 cells were ressuspended in 500 μl of buffer. Flow cytometry dot plot Schwann cell and fibroblast profiles are shown in Figures 1A–D. Of note, the flow cytometry sorting Schwann cells of ALS mice did not show morphological differences (cell size and cytoplasmic granules) compared to control mice (Figures 1C,D). Also, the Schwann cells of ALS mice expressed high levels of mutant human SOD1, while no signal was seen in the Schwann cells of wild-type mice, as evidenced by PCR (Figure 1E).
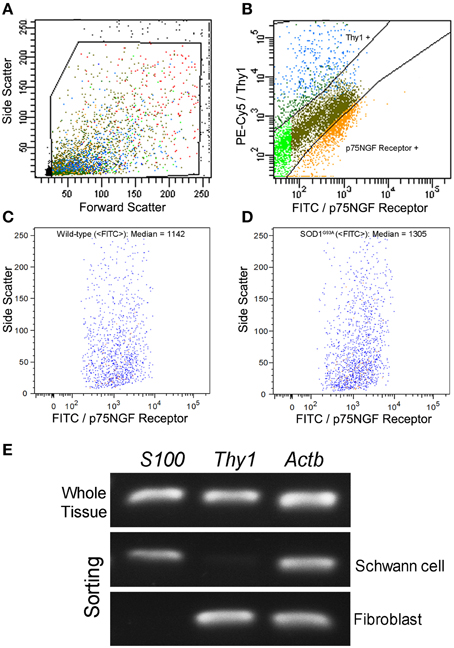
Figure 1. Flow cytometry cell sorting (A–D) and PCR analysis of Schwann cell and fibroblast markers (E) from sciatic nerve of 60-day-old presymptomatic SOD1G93A and the wild-type controls. FITC-conjugated p75NGF Receptor and PE-Cy5-conjugated Thy1 antibodies were employed in the two-color immunolabeling of Schwann cells and fibroblasts, respectively (A,B). Dot plots indicate the total number of events in the sciatic nerve cell suspension, and the dots inside the box represent the excluded doublet and dead profiles, which have been eliminated by morphological criteria, according to previous descriptions (Herzenberg et al., 2006) (A). After morphological criteria, dot plots of Schwann cell and fibroblast profiles (B) were obtained using respective fluorescence filters and the blots inside the boxes represent the specific profiles after discounting the unspecific labeling. Positive p75NGF Receptor and Thy1 cell profiles are shown in corresponding boxes after FITC vs. PE-Cy5 fluorescence intensity plotting (B). Specific profiles-based on morphological criteria were further analyzed in relation to fluorescence criteria and the specific p75NGF Receptor positive Schwann cells profiles were identified in wild-type (median of FITC = 1142; C), and in SOD1G93A mice (median of FITC = 1305; D). Representative bands of PCRs for specific gene markers of Schwann cells (S100), fibroblasts (Thy1), and actin b (Actb) were searched in Schwann cells and fibroblasts enriched samples obtained by flow cytometry sorting of SOD1G93A mice. Mouse whole sciatic nerve sample was used as a positive control (E).
Total RNA from enriched Schwann cells was extracted using Trizol (Life Technologies, USA) according to the manufacturer's protocol. The quantity (NanoDrop 1000 Spectrophotometer) and quality (Agilent 2100 bioanalyzer, RNA 6000 Pico LabChip) of RNAs were analyzed as described in our previous publication (Maximino et al., 2014). Also, the Schwann cell samples were submitted to PCR analyses in order to assess contamination from other cell types.
Schwann Cell Enrichment and hSOD1G93A Verification by PCR
Schwann cells and fibroblast from sciatic nerve of 60-day-old presymptomatic SOD1G93A and their wild-type controls were obtained by fluorescence activated cell sorting and submitted to PCR for sample purity verification. Total RNA from enriched Schwann cells and fibroblast was extracted using Trizol and synthesized in cDNA as described above. Primers to evaluate the presence of Schwann cells (S100) and fibroblast (Thy1) used in the PCR reactions are shown in Table 1, as well as the primer for the internal control (Actb). The reactions were performed to 20 μl final volume, using GoTaq Flexi DNA Polymerase (Promega), according to the manufacturer, and 500 nM of each primer. The protocol for PCRs consisted in 95°C during 5 min, followed by 35 cycles of 95°C during 30 s, 60°C during 30 s, 72°C during 45 s, ending with 72°C in 7 min. Whole sciatic nerve sample was used as a positive control. PCR products were submitted to electrophoresis in 2% agarosis gel containing ethidium bromide for 60 min at 100 V, and then visualized under UV exposure.

Table 1. Information of primers used to evaluate Schwann cell enrichment, demonstration of hSOD1G93A in sciatic nerve and Schwann cell samples by PCR and to SYBR qPCR experiments in the sciatic nerve and Schwann cells isolated by means of flow cytometry sorting of 60-day-old pre-symptomatic SOD1G93A mice.
Sciatic nerve and Schwann cells of 60-day-old presymptomatic SOD1G93A and their wild-type controls were submitted to PCR for verification the presence of human SOD1G93A (hSOD1G93A). The procedures were the same as described above. Primers to evaluate the presence of hSOD1G93A and Actb (internal control) used in the PCR reactions are shown in Table 1.
Quantitative PCR
A subset of genes was chosen for verification of expression patterns based on their possible involvement in ALS/neurodegeneration-related death, stress and mitochondrion or based on their higher level of connectivity (betweenness/degree). qPCR was performed on samples from mouse sciatic nerves and also enriched Schwann cells as previously described (Maximino et al., 2014). Briefly, cDNA was synthesized from 100 ng of total RNA using the Maxima First Strand cDNA Synthesis Kit (Thermo Scientific, USA), according to manufacturer's instructions. qPCR reactions were carried out in duplicate with 10 ng cDNA, using the DyNAmo ColorFlash SYBR Green qPCR kit (Thermo Scientific, USA) and 400 nM of each primer in a final reaction volume of 20 μl. Reactions were run with the Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems). Sequence information regarding the SYBR primers can be found in Table 1. qPCR of Foxo3 was also performed on enriched fibroblasts obtained from the mouse sciatic nerves by means of flow cytometry cell sorting (Figure S1).
Thermocycling conditions for SYBR reactions included an initial denaturation at 95°C for 10 min. Templates were amplified for 40 cycles at 95°C for 15 s, and for additional 40 cycles for at 60°C for 30 s. A dissociation curve was then generated to ensure amplification of a single product, and the verification of no primer dimer formation. A standard curve was generated for each primer pair in order to determine the efficiency of the PCR reaction over a range of template concentrations from 0.032 to 20 ng/μl, using cDNA synthesized from reference mouse RNA. The efficiency for each set of primers was 100 ± 5%. Gene expression was normalized to the expression of Actb and determined using the ΔΔCt mathematical model (Livak and Schmittgen, 2001). Actb was chosen as a housekeeping gene to normalize the qPCR values because the microarray analysis showed no alteration in expression of this gene across samples.
Statistical Analysis
The statistical method employed for the microarray analysis is described in details in the microarray analysis section. Furthermore, a two-tailed unpaired t-test was used to evaluate the level of significance of gene expression independently between the two genotypes (SOD1G93A × wild-type) in the qPCR analyses. Analyses were performed using the GraphPad Prism 5 (San Diego, CA). Data were presented as Means ± Standard Error of Mean (SEM) and significance level was set to p < 0.05.
Results
Qualitative Analyses of Histological Sections and Enriched Schwann Cells of the Sciatic Nerves
No qualitative changes were found regarding morphology of sciatic nerves of presymptomatic ALS mice compared to control at histopathological examination (Figures 2A–J). PCR analysis of sciatic nerve and Schwann cells enriched by flow cytometry showed the presence of hSOD1G93A in the SOD1G93A mice, but not in the wild-type controls (Figures 2K,L). Also, the flow cytometry sorting Schwann cells of ALS mice did not show morphological differences (cell size and cytoplasmic granules) compared to control mice (Figures 1C,D).
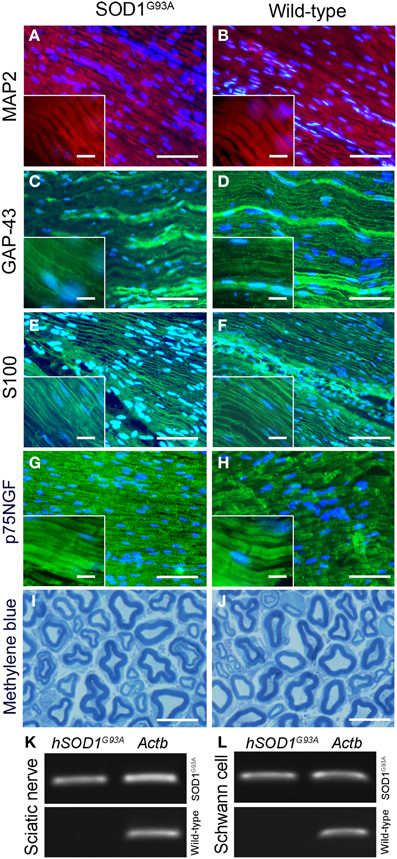
Figure 2. Histopathological analysis (A–J) and molecular evaluation of hSOD1G93A signal (B) in the sciatic nerve and Schwann cell samples of 60-day-old presymptomatic SOD1G93A and aged paired wild-type mice. Immunofluorescence staining of MAP2 (A,B; red), GAP-43 (C,D; green), S100 (E,F; green) and p75NGF (G,H; green) in the sciatic nerve of 60-day-old presymptomatic SOD1G93A mice (B,D,F,H) and their wild-type controls (A,C,E,G). MAP2 and GAP-43 are markers of neuronal fibers; S100 and p75NGF are markers of Schwann cells. Cell nuclei were stained with DAPI (blue). The insert boxes in the bottom left of images show a higher magnification of the cell profiles. Methylene blue staining of Schwann cell myelin sheets of sciatic nerve of 60-day-old presymptomatic SOD1G93A mice (J) and their wild-type controls (I) are also seen. Scale bars: 10 μm. Of note, the same staining pattern was observed for both genotypes (SOD1G93A and wild-type controls) for all cell markers and for the histological sections. Representative bands of PCR for specific gene markers of human SOD1G93A (hSOD1G93A) and actin b (Actb) in sciatic nerve (K) and Schwann cells enriched samples (L) obtained by flow cytometry sorting of SOD1G93A and wild-type control mice.
Verification of Microarray Results by Quantitative PCR
The results of qPCR verification of 10 representative genes in the sciatic nerve of presymptomatic SOD1G93A mice are shown in Table 2. The up and down-regulations of the verified genes in the sciatic nerve of 60-day-old SOD1G93A mice by means of qPCR were coincident and supported the microarray findings of correspondent animal ages (Table S1).
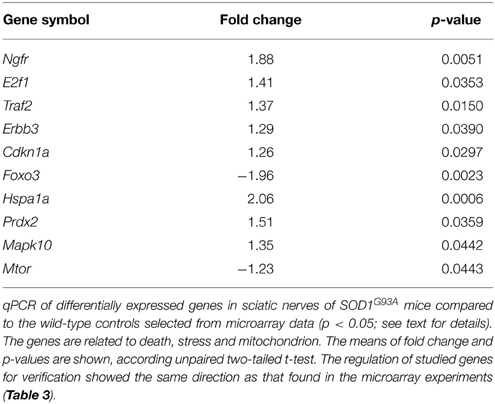
Table 2. qPCR data of selected genes for verification in the sciatic nerve of 60-day-old pre-symptomatic SOD1G93A mice.
Bioinformatics Analysis
Functional Enrichment Analysis
The DAVID analysis of differentially expressed genes in the sciatic nerve of 60-day-old SOD1G93A mice revealed 19 GO terms of Biological Process related to cell death and apoptosis (genes related to Death in Table 3). These GO terms of genes related to Death showed 112 dysregulated genes (46 down and 66 up-regulated genes). Furthermore, DAVID also identified two GO terms of Biological Process related to Stress (Table 3). These GO terms related to Stress showed 66 dysregulated genes (31 down and 35 up-regulated genes). Finally, DAVID identified two GO terms of Cellular Components related to mitochondrial function (Table 3). These GO terms of Mitochondrion showed 143 dysregulated genes (91 down and 52 up-regulated genes). Table 3 shows the down regulated and up regulated genes of Death, Stress, Mitochondrion categories with fold change equal or higher than 1.5. The deregulated genes of these categories with fold change smaller than that are shown in the Table S2 of the Supplementary Material. We have not expected to find gene regulation with high degree of fold change in this stage of presymptomatic events, a period in which dramatic occurrences related to inflammation, neurodegeneration and necrosis are not taking place. The above-mentioned cut-off in the Table 3 is just to facilitate the demonstration of the relatively higher deregulated genes. All analyses of this study were performed without a cut-off.
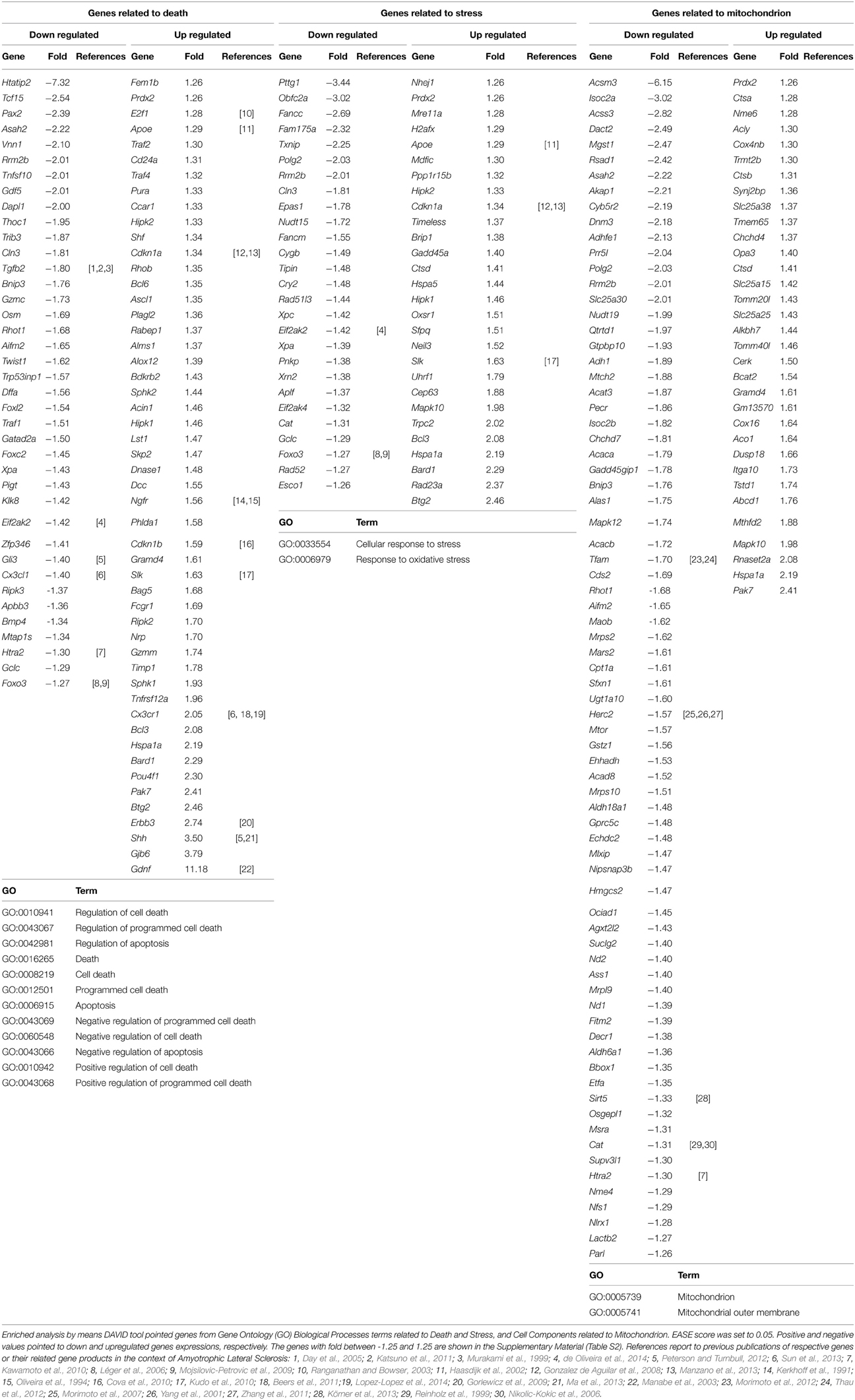
Table 3. Differentially expressed genes related to death, stress and mitochondrion in the sciatic nerve of 60-day-old SOD1G93A mice.
A VEEN diagram of those dysregulated genes related to Death and Stress and Mitochondrion showed 17 genes belonging to both Death and Stress groups of genes, 9 genes belonging to both Death and Mitochondrion groups of genes, 5 genes belonging to both Stress and Mitochondrion groups of genes, and finally 3 genes belonging to the three, Death, Stress, and Mitochondrion groups of genes (Figure 3).
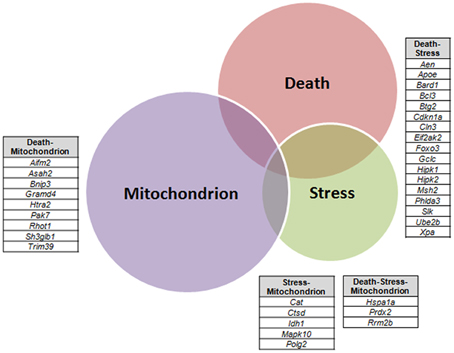
Figure 3. Venn diagram of differentially expressed genes related to Death, Stress and Mitochondrion categories in sciatic nerve (60-day-old) of SOD1G93A animals compared to wild-type controls by means of microarray experiments. The lists of genes of the enriched categories were obtained by means of DAVID tool based on Biological Process and Cellular Component Ontology (EASE score set to 0.05), which identified 112 differentially expressed genes in Death, 66 genes in Stress, and 143 genes in Mitochondrion categories. Venn diagram demonstrates genes which are common to Death and Stress (17 genes), Death and Mitochondrion (nine genes), and also Stress and Mitochondrion (nine genes). Three genes (Hspa1a, Prdx2, Rrm2b) were present in the three categories (Death, Stress, and Mitochondrion).
The DAVID analysis of dysregulated genes related to Death, Stress and Mitochondrion shown in Table 3 are identified with 16 KEGG pathways (Table 4). Furthermore, those identified KEGG pathways that have been previously described in the literature as being related to ALS include RIG-I-like receptor signaling (2 down and 3 up-regulated genes; Figure 4), tryptophan metabolism (5 down-regulated genes; Figure 4), ErbB signaling (1 down and 5 up-regulated genes; Figure 4) and cell cycle (2 down and 5 up-regulated genes; Figure 4). Of note, Cancer, Small cell lung cancer, the Adipocytokine signaling pathway and Chronic myeloid leukemia pathways are likely not related to ALS and were not included in Table 4.
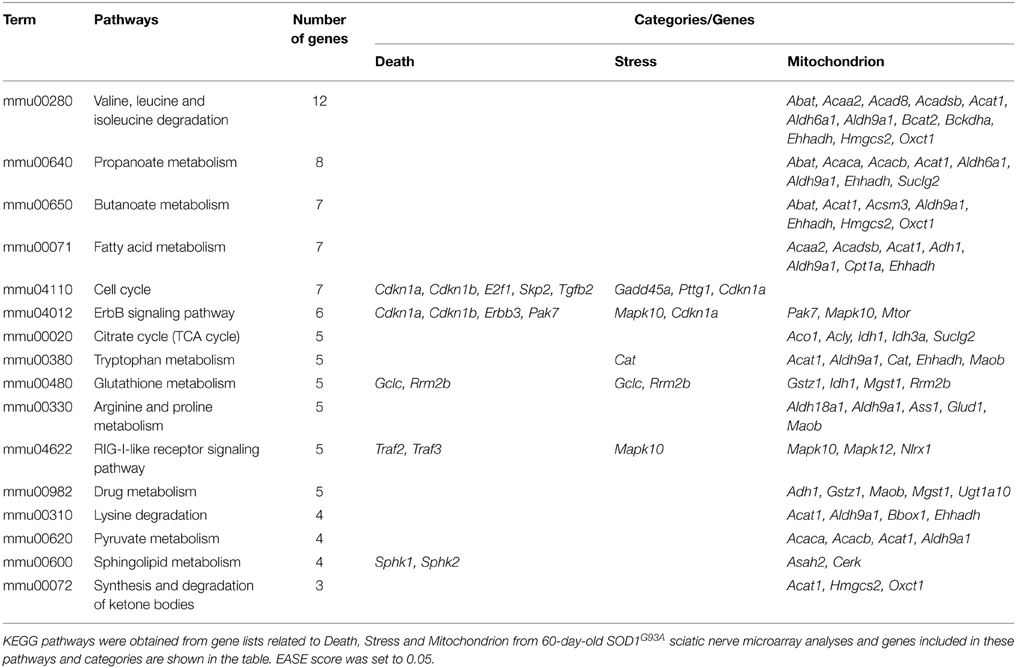
Table 4. KEGG pathways obtained from gene lists related to Death, Stress and Mitochondrion from 60-day-old SOD1G93A sciatic nerve microarray analyses.
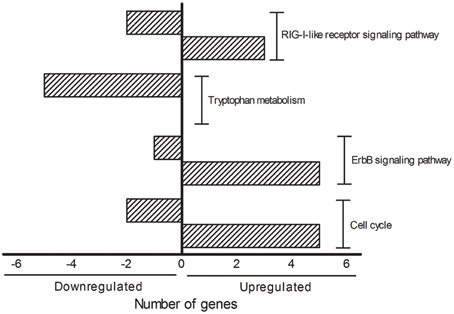
Figure 4. KEGG pathways showing the number of transcripts up (right side bars with positive values) and down-regulated (left side bars with negative values). The figure is representative of the KEGG pathways (EASE score was set to 0.05) obtained from the GO terms list containing genes related to death, stress and mitochondrion that have been already mentioned in the ALS literature. The categories were composed by seven genes (Cell cycle), six genes (ErbB signaling pathway), five genes (Tryptophan metabolism and RIG-I-like receptor signaling pathway).
Protein Interaction Network Analysis from Dysregulated Genes
Protein interaction network analysis using dysregulated genes related to Death, Stress and Mitochondrion showed the hubs (Figure 5A) TRAF2 (16 connectors), H2AFX (8 connectors) and E2F1, FOXO3, MSH2, NGFR, TGFBR1 (7 connectors). Furthermore, the network analysis showed five bottlenecks (TRAF2, E2F1, CDKN1B, TWIST1, and FOXO3). The scatter plot of the values of the hubs node degree vs. the values of node betweenness is shown in Figure 5B. Of note, TRAF2, E2F1, CDKN1B, FOXO3, and H2AFX occupied the highest positions in the scatter plot (Figure 5B). Networks and scatter plots produced from the analyses of the specific genes of Death, Stress and Mitochondrion categories were shown in Figures S2–S5 of Supplementary Material.
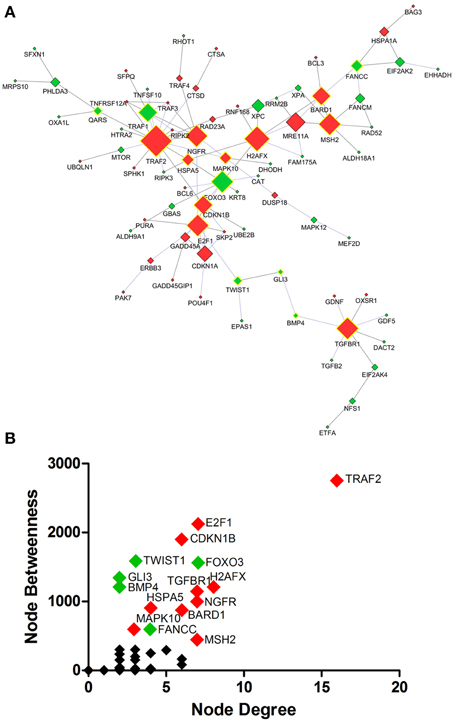
Figure 5. Protein interaction network developed from differentially expressed genes related to Death, Stress and Mitochondrion of sciatic nerves from SOD1G93A mice (A). Scatter plot of the correlation between highest values of node degree (hubs) and node betweenness (bottlenecks) is shown in (B), as described in the text. Up and down-regulated genes are represented respectively as red and green diamonds. Nodes with the highest values for node degree (number of connections) and node betweenness (number of shortest paths) are represented with a yellow border. Of note from this analysis, the genes E2f1, Foxo3, Gli3, Ngfr, Cdkn1a or their related products were already described in the context of ALS.
Schwann Cell and qPCR Experiments
The results of selected genes for verification in the mouse sciatic nerves by qPCR are shown in the Table 2. qPCR analyses of gene expression from the enriched Schwann cells isolated from sciatic nerves of presymptomatic 60-day-old SOD1G93A mice identified a number of up-regulated genes including Ngfr (6.85-fold), Cdkn1b (1.51-fold), E2f1 (1.89-fold), Traf2 (1.31-fold) and Erbb3 (1.38-fold) related to Death (Figure 6A). The gene H2afx (1.42-fold) of the Stress group was up-regulated. The genes Cdkn1a (1.95-fold) and Foxo3 (-1.67-fold) from the Death and Stress groups were up- and down regulated, respectively. The genes Hspa1a (4.48-fold) and Prdx2 (1.48-fold) of the Death, Stress and Mitochondrion groups were up-regulated. Mapk10 (1.42-fold) of the Stress and Mitochondrion groups was up-regulated and Mtor (−1.86-fold), related to mitochondrion, was down regulated (Figure 6B). Interestingly, the expression of Foxo3 was not altered in the enriched fibroblasts of the mouse sciatic nerve (Figure S1).
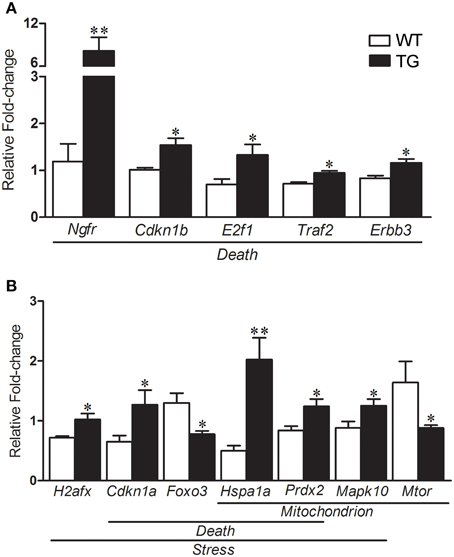
Figure 6. Relative fold change values of selected dysregulated genes related to death, stress and mitochondrion by qPCR in enriched sciatic nerve-derived Schwann cell samples of 60-day-old SOD1G93A (TG) mice compared to the age matched wild-type (WT) mice. Schwann cells were enriched by means of flow cytometry cell sorting technique from the sciatic nerves of the mice. Significant increases in expression are seen in genes related to Death (Ngfr, Cdkn1b, E2f1, Traf2, and Erbb3; A). Up-regulated H2afx is related to Stress category (B). Down-regulated Cdkn1a and Foxo3 are related to Death and Stress categories respectively (B). Up-regulated Hspa1 and Prdx belonged to Death, Stress and Mitochondrion categories (B). Up-regulated Mapk10 (Stress and Mitochondrion categories) and down-regulated Mtor (Mitochondrion) were also shown down-regulated (B). A pool of the non-ALS group was used as reference samples with a reference value of 1, see text for details. Means ± SEM; n = 6 for each group; * and **p-values indicates the < 0.05 and < 0.01 levels of significance, respectively, according to unpaired two-tailed t-test.
Discussion
Triggering Mechanisms of Motor Neuron Death in ALS
Several mechanisms have been proposed as triggers for both autonomous and non-autonomous events related to motor neuron death in ALS. Events of oxidative stress, neuroimmune reactions, protein aggregation, glutamate excitotoxicity, mitochondrial dysfunction and impaired axonal transport in ALS are currently under investigation (Bruijn et al., 2004; Boillée et al., 2006a; de Vos et al., 2007; Jaiswal and Keller, 2009; Redler and Dokholyan, 2012).
The initial triggering and the secondary reactive events associated with motor neurons and their neighboring glial cells are unknown (Tapia, 2014). For instance, early findings related to morpho/physiological changes have been described in presymptomatic phases of the ALS mouse model (Boillée et al., 2006a; Ferraiuolo et al., 2007; Alves et al., 2011; de Oliveira et al., 2013, 2014; Maximino et al., 2014). Of note, fragmentation of the Golgi apparatus (Mourelatos et al., 1996), vacuolization of mitochondria (Bendotti et al., 2001), deficits in axonal transport (Ikenaka et al., 2012), endoplasmic reticulum stress (Tadic et al., 2014), the activation of glial cells (microglia and astrocytes) (Nagai et al., 2007; Graber et al., 2010) and electrophysiological changes (Quinlan, 2011) have all been described within motor neurons and neighboring cells at early postnatal ages. Technological advances have facilitated the discovery of molecular events underlying both autonomous (Wada et al., 2012) and non-autonomous (Ferraiuolo, 2014) mechanisms possibly related to early presymptomatic neuronal toxicity in ALS (Arbour et al., 2015; Saba et al., 2015).
Evidence indicates the existence of very early events, possibly anticipating the motor neuron death, that take place peripherally, i.e., in the motor nerve, motor nerve terminals, neuromuscular junction and muscle (Rocha et al., 2013; de Oliveira et al., 2014; Moloney et al., 2014). In fact, very early electrophysiological events in the motor nerve precede cell body disappearance in the spinal cord as well as the onset of clinical symptoms in ALS (Alves et al., 2011). Absence of neuronal death or morphological alterations in the motor neuron cell bodies has been described in the 60-days-old presymptomatic SOD1 mice (Fischer et al., 2004; Gould et al., 2006; Casas et al., 2013).
Remarkably, early morphological, biochemical and molecular changes in peripheral non-neuronal cells, i.e., Schwann cells and skeletal muscles, are emerging as dying-back mechanisms of motor neuron degeneration in ALS (de Winter et al., 2006; Dupuis and Loeffler, 2009; Dupuis et al., 2009; Keller et al., 2009; Narai et al., 2009; Chen et al., 2010; Dadon-Nachum et al., 2011; Venkova et al., 2014). In fact, the involvement of Schwann cells, which maintain close morphological/physiological relationships with motor axons, has gained more attention in ALS (de Winter et al., 2006; Keller et al., 2009; Chen et al., 2010; Maximino et al., 2014; Venkova et al., 2014). Our work has contributed to a more detailed understanding of the mechanisms involved in the dying-back events associated with motor nerves in ALS by performing a large-scale gene profiling analysis utilizing a microarray analyses in the sciatic nerve of presymptomatic ALS mice.
A deficit in the paracrine trophic interactions between Schwann cells and motor neurons in presymptomatic ALS is a logical choice for further study since Schwann cells offer the main source of trophic stimuli for the maintenance of mature motor neurons (Bhatheja and Field, 2006) and for the regeneration of their fibers after injury (Gupta et al., 2005). In support of this hypothesis, mSOD1 gene expression in distal Schwann cells has been suggested to interfere with the trophic maintenance of motor axon projections (Inoue et al., 2003). Also, reduction of Schwann cell-derived insulin growth factor-1 and cilliary neurotrophic factor in sciatic nerves of both ALS mice and patients have been correlated to disease worsening (Lee et al., 1996; Lobsiger et al., 2009).
In addition to a deficit in paracrine trophic factor maintenance, the presence of toxic factors from Schwann cells affecting motor neurons at early presymptomatic stages in ALS is an additional matter for investigation. Indeed, our DAVID analyses of dysregulated gene expression in sciatic nerves of presymptomatic SOD1G93A mouse model highlighted the GO categories death, stress and mitochondrion, which may be related to the above mentioned possibilities. Of note, the findings of a high number of dysregulated genes related to death, with many also included in the stress and mitochondrial function categories, indicated a complex regulation of these events before motor neuron death, as described for other neurodegenerative disorders (Friedman et al., 1996; Gatzinsky et al., 2003; Pun et al., 2006; Campana, 2007; Lobsiger et al., 2009; Nobbio et al., 2009a,b). Conversely, negative and positive gene regulation related to death and anti-apoptosis mechanisms were also identified in several categories of dysregulated genes, indicating a concomitant regulation of cell toxicity/maintenance before neurodegeneration during the presymptomatic stages in ALS. It should be emphasized that the present description of gene profiling in the sciatic nerve in the presymptomatic ALS mouse model may represent predominantly the regulation of Schwann cells transcripts. In fact, as discussed below, the qPCR verification of selected genes in the mouse sciatic nerve tissue has confirmed the microarray results. qPCR results of selected genes in the enriched Schwann cells were also coincident to microarray. Conversely, the regulation of Foxo3 in the enriched fibroblasts of the sciatic nerve was in the opposite direction.
Mitochondrial/Oxidative Stress Mechanisms Related to Presymptomatic Motor Neuron Impairment
Motor neurons are susceptible to reactive oxygen species and oxidative stress due to their high metabolic rates and decreased ability to buffer calcium (Shaw and Eggett, 2000). Long axons and increased functional activity also confer a high susceptibility to mitochondrial impairments in motor neurons (Barber et al., 2006; Pizzuti and Petrucci, 2011; Cozzolino et al., 2013).
Altered morphology of motor neuron mitochondria has been described in an ALS mouse model (Parone et al., 2013) as well as in humans (Hirano et al., 1984b) at both histological (Hirano et al., 1984a,b) and ultrastructural (Sasaki and Iwata, 1996) levels. Of note, swollen and vacuolated mitochondria were found in motor neurons, glial cells and muscles in ALS (Afifi et al., 1966; Atsumi, 1981; Siklós et al., 1996; Sasaki et al., 2007; Cassina et al., 2008).
Furthermore, oxidative stress has been described in several regions of the brain (Ferrante et al., 1997; Bogdanov et al., 1998, 2000), in the spinal cord (Shaw et al., 1995; Andrus et al., 1998; Liu et al., 1998, 2004; Shibata et al., 2002) and in the skeletal muscle (Mahoney et al., 2006) of both ALS mice and humans. Altered levels of reactive oxygen species within the spinal cord mitochondria of ALS mice (Jung et al., 2002) and patients (Bogdanov et al., 2000) support a possible impairment of the electron transport chain and energy defects in this disorder. Indeed, mitochondrion-induced damage to motor neurons has previously been mentioned as an early ALS signal (Wong et al., 1995; Mattiazzi et al., 2002; Kirkinezos et al., 2005; Cassina et al., 2008; Loizzo et al., 2010). Furthermore, peroxynitrite and superoxide overload in reactive astrocytes and microglia in in vitro models of ALS lead to protein impairment-induced motor neuron damage (Hensley et al., 2006; Dadon-Nachum et al., 2011). These studies all underline the critical involvement of mitochondrial dysfunction in early autonomous and non-autonomous pathogenesis of ALS.
Interestingly, increases in inducible nitric oxide synthase and peroxynitrite in Schwann cells and motor axons of paranodal regions in presymptomatic ALS mice were associated with local mitochondrial reactive oxygen species formation (Chen et al., 2010). Furthermore, Schwann cell-induced trophic support failure and Schwann cell-induced mitochondrial toxicity to motor neurons have both been correlated with high levels of mSOD1 in those peripheral glia of presymptomatic SOD1G93A ALS mice (Gould et al., 2006). Thus, Schwann cell mitochondrion/oxidative stress mechanisms seem to play a key role during the early stages of ALS.
KEGG Pathways Related to Death, Stress and Mitochondrion in ALS
Our DAVID analyses identified genes associated with death, stress and mitochondrial function from the complete list of dysregulated genes of the sciatic nerve from 60-day-old presymptomatic ALS mice before the onset of neurological impairment.
Dysregulated genes of death signaling (Olsen et al., 2001; Hensley et al., 2002; Yoshihara et al., 2002; Dangond et al., 2004; Ferraiuolo et al., 2007; Guipponi et al., 2010) and also those of stress and mitochondrion-related signaling (Dangond et al., 2004; D'arrigo et al., 2010; Guipponi et al., 2010; Bernardini et al., 2013) have been described in ALS models (spinal cord and motor neurons) and in patients (e.g., from post mortem spinal cord and skeletal muscle biopsy) by means of microarray technology. Thus, our work supplements those cited by providing a large-scale profile of genes related to death, stress and mitochondrial function in the peripheral motor nerves of presymptomatic ALS mice.
KEGG pathways from the dysregulated genes associated with death, stress and mitochondrial function of sciatic nerves of presymptomatic ALS mice identified valine, leucine and isoleucine degradation and propionate metabolism as pathways with the highest number of dysregulated genes. Of note, impairments in valine, leucine and isoleucine metabolism (Ilzecka et al., 2003) but not in propionate metabolism, have been described in the context of ALS. Furthermore, fatty acid metabolism, citrate cycle, gluthatione metabolism, arginine and proline metabolism, pyruvate metabolism, sphingolipid metabolism, synthesis and degradation of ketone bodies, and beta-alanine metabolism pathways have been correlated with the onset of ALS (Ariga et al., 1998; Schulz et al., 2000; Cutler et al., 2002; Zhao et al., 2006; Nunn and Ponnusamy, 2009; Panov et al., 2011; Dormann et al., 2012; Zhang and Chook, 2012; de Munck et al., 2013; Yip et al., 2013; Allen et al., 2014). Additionally, dysregulated genes related to RIG-I-like receptor signaling (Day et al., 2005; Katsuno et al., 2011; Zhu et al., 2014), tryptophan metabolism (Sandyk, 2006), ErbB signaling (Gorlewicz et al., 2009), and cell cycle (Manzano et al., 2013) have been reported in ALS (Gorlewicz et al., 2009; Manzano et al., 2013).
Interestingly, the majority of dysregulated genes of the KEGG pathways RIG-I-like receptor signaling, ErbB signaling and cell cycle were found to be up-regulated. Conversely, all genes of tryptophan metabolism were down regulated in the sciatic nerve of presymptomatic ALS mice. The understanding on how such a complex regulation of important singling pathways may represent early triggering or reactive responses in the dying-back mechanisms in ALS is a matter for further analyses. RIG-I-like receptor genes are regulated in ALS, however, the exact mechanisms by which innate immune activation may drive neuronal death in neurodegenerative disorders are far from elucidated. In particular, the role of RIG-I-like receptor gene products in those mechanisms underlying glial cell activation, misfolded proteins/aberrantly localized nucleic acids and mitochondrion signaling-induced autophagy remain unclear (Kawai and Akira, 2008; Tal and Iwasaki, 2009; Heneka et al., 2014; Ying et al., 2015). Furthermore, the involvement of ErbB signaling in ALS was raised with the description of disrupted neuregulin-ErbB4 pathway signaling in presynaptic synapses of less resistant spinal cord motor neurons (Takahashi et al., 2013), but not in the resistant oculomotor neurons in human ALS (Gallart-Palau et al., 2014). Furthermore, ErbB signaling may influence non-autonomous microglial ALS mechanisms (Falls, 2003; Esper et al., 2006; Song et al., 2012) as well as Schwann cell-induced motor axon terminal changes in ALS (Gorlewicz et al., 2009).
The large-scale down-regulation of genes associated with the tryptophan metabolism pathway may be related to serotonin's ability to modulate glutamate motor neuron transmission (Palchaudhuri and Flügge, 2005), and thus excitotoxicity in ALS. Indeed, evidence of serotonin deregulation has been obtained from studies on ALS patients (Sandyk, 2006).
As discussed below, cell cycle gene regulation in the sciatic nerve in presymptomatic phases of ALS might disrupt the normal interactions between reactive Schwann cells and motor axons. In fact, cell cycle impairments have been described in neurodegenerative diseases (Van Leeuwen and Hoozemans, 2015; Wojsiat et al., 2015) and the interaction between mSOD1 and cyclin regulators seems to contribute to autonomous and non-autonomous mechanisms in ALS (Nguyen et al., 2003; Ranganathan and Bowser, 2003; Cova et al., 2010).
Schwann Cell Genes Related to Death, Stress and Mitochondrial Function in the Sciatic Nerve of Presymptomatic ALS Mouse Model
Our study demonstrated the existence of highly interconnected genes from the lists of dysregulated genes involved in death and stress signaling as well as mitochondrial function in the sciatic nerve of presymptomatic ALS mice. The regulation of these genes was further evaluated in the enriched Schwann cells from the ALS mice using qPCR.
Traf2, E2f1, and Cdkn1b from the list of genes of GO biological process related to death were identified as nodes with highest values for node degree (number of connections) and node betweenness (number of shortest paths) in the Protein interaction network analysis.
Mutant SOD1 led to TNF-α pathway activation in the absence of inflammation in ALS (Carter et al., 2009), an event that may also occur in Schwann cells (Au and Yeh, 2007) based on the results of Traf2 up-regulation in ALS Schwann cells described in our work.
TNFα is a potent regulator of Schwann cell division (Chandross et al., 1996) and activation (Bonetti et al., 2000). Moreover, despite a lack of histopathological descriptions of peripheral nerves from ALS subjects and animal models (Fischer et al., 2004; Kano et al., 2012), there are no reports describing local Schwann cell division or death. However, up-regulation of markers of glial activation indicated an early process of Schwann cell reaction in ALS peripheral nerves (Keller et al., 2009; Malaspina et al., 2010). Cell division is a common feature of activated central glia (Pekny and Nilsson, 2005; Dheen et al., 2007). Therefore, it might be possible that the absence of cell division would disrupt normal functions of activated Schwann cells in presymptomatic stages of ALS.
It is likely that the up-regulation Cdkn1b and E2f-1 may lead to intracellular mobilization of cell-cycle proteins and transcriptional regulators in ALS glia (Weinberg, 1995; Ranganathan and Bowser, 2003) and also in ALS activated Schwann cells. These events may interfere with the balance between death/survival signaling pathways in TNFα-activated Schwann cells (Tang et al., 2013), possibly altering the normal functions of activated Schwann cells and leading to a failure of trophic surveillance and/or to toxicity signaling in ALS (Ranganathan and Bowser, 2003; Cova et al., 2010). Therefore, the absence of hyperplasia may impair substantially the paracrine trophic mechanisms of TNFα-activated Schwann cells with motor neurons in ALS.
Other nodes with high values for connections and betweenness described in the Protein interaction network analysis support a non-autonomous mechanism of activated Schwann cells in ALS. The nodes Ngfr, Erbb3, and H2afx of dysregulated genes from the death list further indicated the presence of impaired paracrine trophic functions of Schwann cells in ALS. The up-regulation of Ngfr and Erbb3, which encode the high-affinity neurotrophin receptor TRKA and the neuregulin-associated tyrosine kinase receptor, respectively, could be involved in Schwann cell paracrine functions (Wang et al., 1996; Lyons et al., 2005; Adilakshmi et al., 2011) as well as in early, complex mechanisms of axonal retraction and neuromuscular junction alterations in ALS prior to motor neuron degeneration (Kerkhoff et al., 1991; Gorlewicz et al., 2009). Furthermore, the up-regulation of H2afx in Schwann cells might correlate with the described Schwann cell participation in the pathogenesis of ALS since H2AFX-induced DNA damage in reactive astrocytes has been associated with impaired paracrine glial functions in other neurodegenerative disorder (Simpson et al., 2010).
Foxo3 and Cdkn1a are present in the death and stress lists of genes and were also found to be dysregulated in the enriched Schwann cells from the sciatic nerve of 60-day-old presymptomatic ALS mice. Indeed, the encoded products of these genes have been investigated in ALS skeletal muscles but not in the ALS motor nerve (Léger et al., 2006; Gonzalez de Aguilar et al., 2008; Manzano et al., 2013). Foxo3 showed high levels of betweenness and degree and FOXO3 has been studied as a catabolic target in ALS skeletal muscles (Léger et al., 2006). Conversely, Cdkn1a dysregulation was described in muscles of ALS mice (Gonzalez de Aguilar et al., 2008) and CDKN1A, a cyclin-dependent kinase inhibitor, interferes with satellite cell-induced myoplasticity in ALS skeletal muscles (Manzano et al., 2013). The above findings provide support for the involvement of Foxo3 and Cdkn1a in the dying-back Schwann cell mechanisms in ALS, a matter that should be further investigated.
Hspa1a, Prdx2, and Rrm2b were present in three GO lists, the death, stress, and mitochondrial function categories. Hspa1a and Prdx2 were also found to be dysregulated in the enriched Schwann cells from the sciatic nerves of presymptomatic ALS mice. Those three genes and their encoded products have not been described in the context of ALS. Moreover, up-regulation of Hspa1a and Prdx2 were found in other neurodegenerative disorders and their encoded products have been studied in the context of therapeutic strategies (Muchowski and Wacker, 2005; Ali et al., 2010; Gestwicki and Garza, 2012). Furthermore, RRM2B-related mitochondrial diseases are frequently inherited and associated with neurological symptoms (Horga et al., 2014). Conversely, non-inherited mitochondrial dysfunctions have been suggested as possible mechanisms underlying the pathogenesis of ALS (discussed above). Thus, mitochondria/oxidative stress impairing Schwann cell paracrine regulation in the presymptomatic phases of ALS is a matter for further investigation.
Mtor, identified in the mitochondrion list of dysregulated genes, was down-regulated in the enriched Schwann cells from presymptomatic SOD1 mice, a change which is in line with descriptions of MTOR reduction in spinal cords of ALS rodents (Morimoto et al., 2007). MTOR reduction induced by PI3-K and Akt/PKB signaling (Nagano et al., 2002) worsened ALS pathology in ALS transgenic mice (Zhang et al., 2011). Because MTOR activation leads to neuroprotection in ALS (Saxena et al., 2013), it is a matter of further investigation whether modulation of MTOR signaling in Schwann cells could counteract the degenerative processes associated with this disease.
Finally, Mapk10 of mitochondrion and stress categories was found to be up-regulated in the enriched Schwann cells from sciatic nerves of presymptomatic ALS mice. A MAPK10/JNK3 truncation mutation has previously only been associated with cognitive disorders (Kunde et al., 2013). Furthermore, Mapk10 itself has not been investigated as a factor in the pathogenesis of ALS, despite the general consensus that deregulated MAPK signaling contributes to ALS (Kim and Choi, 2010; Xia et al., 2015). Nevertheless, the involvement of p38MAPK and TNFα receptors in the non-autonomous microglial toxicity in ALS (Veglianese et al., 2006) raised the possibility of a similar mechanism in ALS Schwann cells, an issue that should be explored further.
In conclusion, our large-scale gene profiling reveled the presence of death, stress (with emphasis on oxidative stress), and mitochondrial function pathway signaling taking place in the sciatic nerve of presymptomatic ALS mice. The regulation of highly interconnected genes in ALS Schwann cells indicated the involvement of these pathways in the non-autonomous mechanisms of that peripheral glial cell type in the early stages of the disorder.
Author Contributions
JM and GC designed the study. CA and JM performed the experiments. GC interpreted the results. All authors wrote, read and approved the final manuscript. GC is responsible for the ALS Brazil Project of the University of São Paulo School of Medicine.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank professor Robert Carlone of Brock University for reading the manuscript and for his valuable suggestions and we also thank Hatylas Azevedo for helping us with the Protein interaction network analysis. This work was supported by grants from the São Paulo Research Foundation (FAPESP; #2010/20457-7) and National Council for Scientific and Technological Development (CNPq).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fncel.2015.00332
References
Adilakshmi, T., Ness-Myers, J., Madrid-Aliste, C., Fiser, A., and Tapinos, N. (2011). A nuclear variant of ErbB3 receptor tyrosine kinase regulates ezrin distribution and Schwann cell myelination. J. Neurosci. 31, 5106–5119. doi: 10.1523/JNEUROSCI.5635-10.2011
Afifi, A. K., Aleu, F. P., Goodgold, J., and Mackay, B. (1966). Ultrastructure of atrophic muscle in amyotrophic lateral sclerosis. Neurology 16, 475–481 doi: 10.1212/WNL.16.5.475
Ali, Y. O., Kitay, B. M., and Zhai, R. G. (2010). Dealing with misfolded proteins: examining the neuroprotective role of molecular chaperones in neurodegeneration. Molecules 15, 6859–6887. doi: 10.3390/molecules15106859
Allen, S. P., Rajan, S., Duffy, L., Mortiboys, H., Higginbottom, A., Grierson, A. J., et al. (2014). Superoxide dismutase 1 mutation in a cellular model of amyotrophic lateral sclerosis shifts energy generation from oxidative phosphorylation to glycolysis. Neurobiol. Aging 35, 1499–1509. doi: 10.1016/j.neurobiolaging.2013.11.025
Alves, C. J., Dariolli, R., Jorge, F. M. D. H., Monteiro, M. R., Maximino, J. R., Martins, R. S., et al. (2015). Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front. Cell. Neurosci. 9:289. doi: 10.3389/fncel.2015.00289
Alves, C. J., de Santana, L. P., Dos Santos, A. J., de Oliveira, G. P., Duobles, T., Scorisa, J. M., et al. (2011). Early motor and electrophysiological changes in transgenic mouse model of amyotrophic lateral sclerosis and gender differences on clinical outcome. Brain Res. 1394, 90–104. doi: 10.1016/j.brainres.2011.02.060
Andrus, P. K., Fleck, T. J., Gurney, M. E., and Hall, E. D. (1998). Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 71, 2041–2048. doi: 10.1046/j.1471-4159.1998.71052041.x
Arbour, D., Tremblay, E., Martineau, E., Julien, J. P., and Robitaille, R. (2015). Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J. Neurosci. 35, 688–706. doi: 10.1523/JNEUROSCI.1379-14.2015
Ariga, T., Jarvis, W. D., and Yu, R. K. (1998). Role of sphingolipid-mediated cell death in neurodegenerative diseases. J. Lipid. Res. 39, 1–16
Atsumi, T. (1981). The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 55, 193–198 doi: 10.1007/BF00691318
Au, P. Y., and Yeh, W. C. (2007). Physiological roles and mechanisms of signaling by TRAF2 and TRAF5. Adv. Exp. Med. Biol. 597, 32–47. doi: 10.1007/978-0-387-70630-6_3
Barber, S. C., Mead, R. J., and Shaw, P. J. (2006). Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 1762, 1051–1067. doi: 10.1016/j.bbadis.2006.03.008
Batista, C. M., Bianqui, L. L., Zanon, B. B., Ivo, M. M., Oliveira, G. P., Maximino, J. R., et al. (2014). Behavioral improvement and regulation of molecules related to neuroplasticity in ischemic rat spinal cord treated with PEDF. Neural. Plast. 2014:451639. doi: 10.1155/2014/451639
Beers, D. R., Zhao, W., Liao, B., Kano, O., Wang, J., Huang, A., et al. (2011). Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav. Immun. 25, 1025–1035. doi: 10.1016/j.bbi.2010.12.008
Bendotti, C., Calvaresi, N., Chiveri, L., Prelle, A., Moggio, M., Braga, M., et al. (2001). Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J. Neurol. Sci. 191, 25–33. doi: 10.1016/S0022-510X(01)00627-X
Bernardini, C., Censi, F., Lattanzi, W., Barba, M., Calcagnini, G., Giuliani, A., et al. (2013). Mitochondrial network genes in the skeletal muscle of amyotrophic lateral sclerosis patients. PLoS ONE 8:e57739. doi: 10.1371/journal.pone.0057739
Bhatheja, K., and Field, J. (2006). Schwann cells: origins and role in axonal maintenance and regeneration. Int. J. Biochem. Cell Biol. 38, 1995–1999. doi: 10.1016/j.biocel.2006.05.007
Bogdanov, M. B., Ramos, L. E., Xu, Z., and Beal, M. F. (1998). Elevated “hydroxyl radical” generation in vivo in an animal model of amyotrophic lateral sclerosis. J. Neurochem. 71, 1321–1324. doi: 10.1046/j.1471-4159.1998.71031321.x
Bogdanov, M., Brown, R. H., Matson, W., Smart, R., Hayden, D., O'donnell, H., et al. (2000). Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 29, 652–658. doi: 10.1016/S0891-5849(00)00349-X
Boillée, S., Vande Velde, C., and Cleveland, D. W. (2006a). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. doi: 10.1016/j.neuron.2006.09.018
Boillée, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006b). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Bonetti, B., Valdo, P., Stegagno, C., Tanel, R., Zanusso, G. L., Ramarli, D., et al. (2000). Tumor necrosis factor alpha and human Schwann cells: signalling and phenotype modulation without cell death. J. Neuropathol. Exp. Neurol. 59, 74–84.
Boutahar, N., Wierinckx, A., Camdessanche, J. P., Antoine, J. C., Reynaud, E., Lassabliere, F., et al. (2011). Differential effect of oxidative or excitotoxic stress on the transcriptional profile of amyotrophic lateral sclerosis-linked mutant SOD1 cultured neurons. J. Neurosci. Res. 89, 1439–1450. doi: 10.1002/jnr.22672
Bruijn, L. I., Miller, T. M., and Cleveland, D. W. (2004). Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 27, 723–749 doi: 10.1146/annurev.neuro.27.070203.144244
Campana, W. M. (2007). Schwann cells: activated peripheral glia and their role in neuropathic pain. Brain Behav. Immun. 21, 522–527. doi: 10.1016/j.bbi.2006.12.008
Carter, B. J., Anklesaria, P., Choi, S., and Engelhardt, J. F. (2009). Redox modifier genes and pathways in amyotrophic lateral sclerosis. Antioxid. Redox. Signal. 11, 1569–1586. doi: 10.1089/ars.2008.2414
Casas, C., Herrando-Grabulosa, M., Manzano, R., Mancuso, R., Osta, R., and Navarro, X. (2013). Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav. 3, 145–158. doi: 10.1002/brb3.104
Cassina, P., Cassina, A., Pehar, M., Castellanos, R., Gandelman, M., de León, A., et al. (2008). Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J. Neurosci. 28, 4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008
Chandross, K. J., Spray, D. C., Cohen, R. I., Kumar, N. M., Kremer, M., Dermietzel, R., et al. (1996). TNF alpha inhibits Schwann cell proliferation, connexin46 expression, and gap junctional communication. Mol. Cell Neurosci. 7, 479–500. doi: 10.1006/mcne.1996.0035
Chen, K., Northington, F. J., and Martin, L. J. (2010). Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct. Funct. 214, 219–234. doi: 10.1007/s00429-009-0226-4
Coleman, M. P., and Perry, V. H. (2002). Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 25, 532–537. doi: 10.1016/S0166-2236(02)02255-5
Cooper-Knock, J., Kirby, J., Ferraiuolo, L., Heath, P. R., Rattray, M., and Shaw, P. J. (2012). Gene expression profiling in human neurodegenerative disease. Nat. Rev. Neurol. 8, 518–530. doi: 10.1038/nrneurol.2012.156
Cova, E., Ghiroldi, A., Guareschi, S., Mazzini, G., Gagliardi, S., Davin, A., et al. (2010). G93A SOD1 alters cell cycle in a cellular model of Amyotrophic Lateral Sclerosis. Cell Signal. 22, 1477–1484. doi: 10.1016/j.cellsig.2010.05.016
Cozzolino, M., and Carrì, M. T. (2012). Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66. doi: 10.1016/j.pneurobio.2011.06.003
Cozzolino, M., Ferri, A., Valle, C., and Carrì, M. T. (2013). Mitochondria and ALS: implications from novel genes and pathways. Mol. Cell Neurosci. 55, 44–49. doi: 10.1016/j.mcn.2012.06.001
Cutler, R. G., Pedersen, W. A., Camandola, S., Rothstein, J. D., and Mattson, M. P. (2002). Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 52, 448–457. doi: 10.1002/ana.10312
Dadon-Nachum, M., Melamed, E., and Offen, D. (2011). The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 43, 470–477. doi: 10.1007/s12031-010-9467-1
Dangond, F., Hwang, D., Camelo, S., Pasinelli, P., Frosch, M. P., Stephanopoulos, G., et al. (2004). Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol. Genomics 16, 229–239. doi: 10.1152/physiolgenomics.00087.2001
D'arrigo, A., Colavito, D., Pena-Altamira, E., Fabris, M., Dam, M., Contestabile, A., et al. (2010). Transcriptional profiling in the lumbar spinal cord of a mouse model of amyotrophic lateral sclerosis: a role for wild-type superoxide dismutase 1 in sporadic disease? J. Mol. Neurosci. 41, 404–415. doi: 10.1007/s12031-010-9332-2
Day, W. A., Koishi, K., Nukuda, H., and Mclennan, I. S. (2005). Transforming growth factor-beta 2 causes an acute improvement in the motor performance of transgenic ALS mice. Neurobiol. Dis. 19, 323–330. doi: 10.1016/j.nbd.2005.01.010
de Munck, E., Muñoz-Sáez, E., Miguel, B. G., Solas, M. T., Ojeda, I., Martinez, A., et al. (2013). beta-N-methylamino-l-alanine causes neurological and pathological phenotypes mimicking Amyotrophic Lateral Sclerosis (ALS): the first step towards an experimental model for sporadic ALS. Environ. Toxicol. Pharmacol. 36, 243–255. doi: 10.1016/j.etap.2013.04.007
de Oliveira, G. P., Alves, C. J., and Chadi, G. (2013). Early gene expression changes in spinal cord from SOD1(G93A) Amyotrophic Lateral Sclerosis animal model. Front. Cell. Neurosci. 7:216. doi: 10.3389/fncel.2013.00216
de Oliveira, G. P., Maximino, J. R., Maschietto, M., Zanoteli, E., Puga, R. D., Lima, L., et al. (2014). Early gene expression changes in skeletal muscle from SOD1(G93A) amyotrophic lateral sclerosis animal model. Cell. Mol. Neurobiol. 34, 451–462. doi: 10.1007/s10571-014-0029-x
de Vos, K. J., Chapman, A. L., Tennant, M. E., Manser, C., Tudor, E. L., Lau, K. F., et al. (2007). Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 16, 2720–2728. doi: 10.1093/hmg/ddm226
de Winter, F., Vo, T., Stam, F. J., Wisman, L. A., Bär, P. R., Niclou, S. P., et al. (2006). The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol. Cell. Neurosci. 32, 102–117. doi: 10.1016/j.mcn.2006.03.002
Dheen, S. T., Kaur, C., and Ling, E. A. (2007). Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 14, 1189–1197. doi: 10.2174/092986707780597961
Dormann, D., Madl, T., Valori, C. F., Bentmann, E., Tahirovic, S., Abou-Ajram, C., et al. (2012). Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 31, 4258–4275. doi: 10.1038/emboj.2012.261
Dupuis, L., Gonzalez de Aguilar, J. L., Echaniz-Laguna, A., Eschbach, J., Rene, F., Oudart, H., et al. (2009). Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 4:e5390. doi: 10.1371/journal.pone.0005390
Dupuis, L., and Loeffler, J. P. (2009). Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models. Curr. Opin. Pharmacol. 9, 341–346. doi: 10.1016/j.coph.2009.03.007
Esper, R. M., Pankonin, M. S., and Loeb, J. A. (2006). Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res. Rev. 51, 161–175. doi: 10.1016/j.brainresrev.2005.11.006
Falls, D. L. (2003). Neuregulins and the neuromuscular system: 10 years of answers and questions. J. Neurocytol. 32, 619–647. doi: 10.1023/B:NEUR.0000020614.83883.be
Ferraiuolo, L. (2014). The non-cell-autonomous component of ALS: new in vitro models and future challenges. Biochem. Soc. Trans. 42, 1270–1274. doi: 10.1042/BST20140168
Ferraiuolo, L., de Bono, J. P., Heath, P. R., Holden, H., Kasher, P., Channon, K. M., et al. (2009). Transcriptional response of the neuromuscular system to exercise training and potential implications for ALS. J. Neurochem. 109, 1714–1724. doi: 10.1111/j.1471-4159.2009.06080.x
Ferraiuolo, L., Heath, P. R., Holden, H., Kasher, P., Kirby, J., and Shaw, P. J. (2007). Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci. 27, 9201–9219. doi: 10.1523/JNEUROSCI.1470-07.2007
Ferrante, R. J., Shinobu, L. A., Schulz, J. B., Matthews, R. T., Thomas, C. E., Kowall, N. W., et al. (1997). Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann. Neurol. 42, 326–334. doi: 10.1002/ana.410420309
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Friedman, H. C., Jelsma, T. N., Bray, G. M., and Aguayo, A. J. (1996). A distinct pattern of trophic factor expression in myelin-deficient nerves of Trembler mice: implications for trophic support by Schwann cells. J. Neurosci. 16, 5344–5350
Fu, J., Allen, W., Xia, A., Ma, Z., and Qi, X. (2014). Identification of biomarkers in breast cancer by gene expression profiling using human tissues. Genom Data 2, 299–301. doi: 10.1016/j.gdata.2014.09.004
Fukada, Y., Yasui, K., Kitayama, M., Doi, K., Nakano, T., Watanabe, Y., et al. (2007). Gene expression analysis of the murine model of amyotrophic lateral sclerosis: studies of the Leu126delTT mutation in SOD1. Brain Res. 1160, 1–10. doi: 10.1016/j.brainres.2007.05.044
Gallart-Palau, X., Tarabal, O., Casanovas, A., Sábado, J., Correa, F. J., Hereu, M., et al. (2014). Neuregulin-1 is concentrated in the postsynaptic subsurface cistern of C-bouton inputs to alpha-motoneurons and altered during motoneuron diseases. FASEB J. 28, 3618–3632. doi: 10.1096/fj.13-248583
Gatzinsky, K. P., Holtmann, B., Daraie, B., Berthold, C. H., and Sendtner, M. (2003). Early onset of degenerative changes at nodes of Ranvier in alpha-motor axons of Cntf null (-/-) mutant mice. Glia 42, 340–349. doi: 10.1002/glia.10221
Gestwicki, J. E., and Garza, D. (2012). Protein quality control in neurodegenerative disease. Prog. Mol. Biol. Transl. Sci. 107, 327–353. doi: 10.1016/B978-0-12-385883-2.00003-5
Gonzalez de Aguilar, J. L., Niederhauser-Wiederkehr, C., Halter, B., de Tapia, M., Di Scala, F., Demougin, P., et al. (2008). Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model. Physiol. Genomics 32, 207–218. doi: 10.1152/physiolgenomics.00017.2007
Gorlewicz, A., Wlodarczyk, J., Wilczek, E., Gawlak, M., Cabaj, A., Majczynski, H., et al. (2009). CD44 is expressed in non-myelinating Schwann cells of the adult rat, and may play a role in neurodegeneration-induced glial plasticity at the neuromuscular junction. Neurobiol. Dis. 34, 245–258 doi: 10.1016/j.nbd.2009.01.011
Gould, T. W., Buss, R. R., Vinsant, S., Prevette, D., Sun, W., Knudson, C. M., et al. (2006). Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J. Neurosci. 26, 8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006
Graber, D. J., Hickey, W. F., and Harris, B. T. (2010). Progressive changes in microglia and macrophages in spinal cord and peripheral nerve in the transgenic rat model of amyotrophic lateral sclerosis. J. Neuroinflamm. 7:8. doi: 10.1186/1742-2094-7-8
Guipponi, M., Li, Q. X., Hyde, L., Beissbarth, T., Smyth, G. K., Masters, C. L., et al. (2010). SAGE analysis of genes differentially expressed in presymptomatic TgSOD1G93A transgenic mice identified cellular processes involved in early stage of ALS pathology. J. Mol. Neurosci. 41, 172–182. doi: 10.1007/s12031-009-9317-1
Gupta, R., Gray, M., Chao, T., Bear, D., Modafferi, E., and Mozaffar, T. (2005). Schwann cells upregulate vascular endothelial growth factor secondary to chronic nerve compression injury. Muscle Nerve 31, 452–460. doi: 10.1002/mus.20272
Gurney, M. E. (1994). Transgenic-mouse model of amyotrophic lateral sclerosis. N. Engl. J. Med. 331, 1721–1722. doi: 10.1056/NEJM199412223312516
Guzen, F. P., de Almeida Leme, R. J., de Andrade, M. S., de Luca, B. A., and Chadi, G. (2009). Glial cell line-derived neurotrophic factor added to a sciatic nerve fragment grafted in a spinal cord gap ameliorates motor impairments in rats and increases local axonal growth. Restor. Neurol. Neurosci. 27, 1–16. doi: 10.3233/RNN-2009-0454
Haasdijk, E. D., Vlug, A., Mulder, M. T., and Jaarsma, D. (2002). Increased apolipoprotein E expression correlates with the onset of neuronal degeneration in the spinal cord of G93A-SOD1 mice. Neurosci. Lett. 335, 29–33. doi: 10.1016/S0304-3940(02)01159-X
Heneka, M. T., Kummer, M. P., and Latz, E. (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477. doi: 10.1038/nri3705
Hensley, K., Abdel-Moaty, H., Hunter, J., Mhatre, M., Mou, S., Nguyen, K., et al. (2006). Primary glia expressing the G93A-SOD1 mutation present a neuroinflammatory phenotype and provide a cellular system for studies of glial inflammation. J. Neuroinflamm. 3:2. doi: 10.1186/1742-2094-3-2
Hensley, K., Floyd, R. A., Gordon, B., Mou, S., Pye, Q. N., Stewart, C., et al. (2002). Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. J. Neurochem. 82, 365–374. doi: 10.1046/j.1471-4159.2002.00968.x
Herzenberg, L. A., Tung, J., Moore, W. A., and Parks, D. R. (2006). Interpreting flow cytometry data: a guide for the perplexed. Nat. Immunol. 7, 681–685. doi: 10.1038/ni0706-681
Hirano, A., Donnenfeld, H., Sasaki, S., and Nakano, I. (1984a). Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 43, 461–470. doi: 10.1097/00005072-198409000-00001
Hirano, A., Nakano, I., Kurland, L. T., Mulder, D. W., Holley, P. W., and Saccomanno, G. (1984b). Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 43, 471–480 doi: 10.1097/00005072-198409000-00002
Horga, A., Pitceathly, R. D., Blake, J. C., Woodward, C. E., Zapater, P., Fratter, C., et al. (2014). Peripheral neuropathy predicts nuclear gene defect in patients with mitochondrial ophthalmoplegia. Brain 137, 3200–3212. doi: 10.1093/brain/awu279
Huang Da, W., Sherman, B. T., Stephens, R., Baseler, M. W., Lane, H. C., and Lempicki, R. A. (2008). DAVID gene ID conversion tool. Bioinformation 2, 428–430. doi: 10.6026/97320630002428
Hulsen, T., de Vlieg, J., and Alkema, W. (2008). BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9:488. doi: 10.1186/1471-2164-9-488
Ikenaka, K., Katsuno, M., Kawai, K., Ishigaki, S., Tanaka, F., and Sobue, G. (2012). Disruption of axonal transport in motor neuron diseases. Int. J. Mol. Sci. 13, 1225–1238. doi: 10.3390/ijms13011225
Ilzecka, J., Stelmasiak, Z., Solski, J., Wawrzycki, S., and Szpetnar, M. (2003). Plasma amino acids percentages in amyotrophic lateral sclerosis patients. Neurol. Sci. 24, 293–295. doi: 10.1007/s10072-003-0161-8
Inoue, H., Tsukita, K., Iwasato, T., Suzuki, Y., Tomioka, M., Tateno, M., et al. (2003). The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J. 22, 6665–6674. doi: 10.1093/emboj/cdg634
Jaiswal, M. K., and Keller, B. U. (2009). Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol. Pharmacol. 75, 478–489. doi: 10.1124/mol.108.050831
Jung, C., Higgins, C. M., and Xu, Z. (2002). A quantitative histochemical assay for activities of mitochondrial electron transport chain complexes in mouse spinal cord sections. J. Neurosci. Methods 114, 165–172. doi: 10.1016/S0165-0270(01)00524-6
Kano, O., Beers, D. R., Henkel, J. S., and Appel, S. H. (2012). Peripheral nerve inflammation in ALS mice: cause or consequence. Neurology 78, 833–835. doi: 10.1212/WNL.0b013e318249f776
Katsuno, M., Adachi, H., Banno, H., Suzuki, K., Tanaka, F., and Sobue, G. (2011). Transforming growth factor-beta signaling in motor neuron diseases. Curr. Mol. Med. 11, 48–56. doi: 10.2174/156652411794474356
Kawai, T., and Akira, S. (2008). Toll-like receptor and RIG-I-like receptor signaling. Ann. N.Y. Acad. Sci. 1143, 1–20. doi: 10.1196/annals.1443.020
Kawamoto, Y., Ito, H., Kobayashi, Y., Suzuki, Y., Akiguchi, I., Fujimura, H., et al. (2010). HtrA2/Omi-immunoreactive intraneuronal inclusions in the anterior horn of patients with sporadic and Cu/Zn superoxide dismutase (SOD1) mutant amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 36, 331–344. doi: 10.1111/j.1365-2990.2010.01075.x
Keller, A. F., Gravel, M., and Kriz, J. (2009). Live imaging of amyotrophic lateral sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP in Schwann cells. Glia 57, 1130–1142. doi: 10.1002/glia.20836
Kerkhoff, H., Jennekens, F. G., Troost, D., and Veldman, H. (1991). Nerve growth factor receptor immunostaining in the spinal cord and peripheral nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 81, 649–656. doi: 10.1007/BF00296375
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kim, E. K., and Choi, E. J. (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405. doi: 10.1016/j.bbadis.2009.12.009
Kirkinezos, I. G., Bacman, S. R., Hernandez, D., Oca-Cossio, J., Arias, L. J., Perez-Pinzon, M. A., et al. (2005). Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J. Neurosci. 25, 164–172. doi: 10.1523/JNEUROSCI.3829-04.2005
Körner, S., Böselt, S., Thau, N., Rath, K. J., Dengler, R., and Petri, S. (2013). Differential sirtuin expression patterns in amyotrophic lateral sclerosis (ALS) postmortem tissue: neuroprotective or neurotoxic properties of sirtuins in ALS? Neurodegener. Dis. 11, 141–152. doi: 10.1159/000338048
Kudo, L. C., Parfenova, L., Vi, N., Lau, K., Pomakian, J., Valdmanis, P., et al. (2010). Integrative gene-tissue microarray-based approach for identification of human disease biomarkers: application to amyotrophic lateral sclerosis. Hum. Mol. Genet 19, 3233–3253. doi: 10.1093/hmg/ddq232
Kunde, S. A., Rademacher, N., Tzschach, A., Wiedersberg, E., Ullmann, R., Kalscheuer, V. M., et al. (2013). Characterisation of de novo MAPK10/JNK3 truncation mutations associated with cognitive disorders in two unrelated patients. Hum. Genet 132, 461–471. doi: 10.1007/s00439-012-1260-5
Lee, D. A., Gross, L., Wittrock, D. A., and Windebank, A. J. (1996). Localization and expression of ciliary neurotrophic factor (CNTF) in postmortem sciatic nerve from patients with motor neuron disease and diabetic neuropathy. J. Neuropathol. Exp. Neurol. 55, 915–923 doi: 10.1097/00005072-199608000-00007
Léger, B., Vergani, L., Sorarù, G., Hespel, P., Derave, W., Gobelet, C., et al. (2006). Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J. 20, 583–585. doi: 10.1096/fj.05-5249fje
Liu, J., Lillo, C., Jonsson, P. A., Vande Velde, C., Ward, C. M., Miller, T. M., et al. (2004). Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43, 5–17. doi: 10.1016/j.neuron.2004.06.016
Liu, R., Althaus, J. S., Ellerbrock, B. R., Becker, D. A., and Gurney, M. E. (1998). Enhanced oxygen radical production in a transgenic mouse model of familial amyotrophic lateral sclerosis. Ann. Neurol. 44, 763–770. doi: 10.1002/ana.410440510
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lobsiger, C. S., Boillee, S., Mcalonis-Downes, M., Khan, A. M., Feltri, M. L., Yamanaka, K., et al. (2009). Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc. Natl. Acad. Sci. U.S.A. 106, 4465–4470. doi: 10.1073/pnas.0813339106
Loizzo, S., Pieri, M., Ferri, A., Carri, M. T., Zona, C., Fortuna, A., et al. (2010). Dynamic NAD(P)H post-synaptic autofluorescence signals for the assessment of mitochondrial function in a neurodegenerative disease: monitoring the primary motor cortex of G93A mice, an amyotrophic lateral sclerosis model. Mitochondrion 10, 108–114. doi: 10.1016/j.mito.2009.11.001
Lopez-Lopez, A., Gamez, J., Syriani, E., Morales, M., Salvado, M., Rodriguez, M. J., et al. (2014). CX3CR1 is a modifying gene of survival and progression in amyotrophic lateral sclerosis. PLoS ONE 9:e96528. doi: 10.1371/journal.pone.0096528
Lyons, D. A., Pogoda, H. M., Voas, M. G., Woods, I. G., Diamond, B., Nix, R., et al. (2005). erbb3 and erbb2 are essential for schwann cell migration and myelination in zebrafish. Curr. Biol. 15, 513–524. doi: 10.1016/j.cub.2005.02.030
Ma, X., Turnbull, P., Peterson, R., and Turnbull, J. (2013). Trophic and proliferative effects of Shh on motor neurons in embryonic spinal cord culture from wildtype and G93A SOD1 mice. BMC Neurosci. 14:119. doi: 10.1186/1471-2202-14-119
Mahoney, D. J., Kaczor, J. J., Bourgeois, J., Yasuda, N., and Tarnopolsky, M. A. (2006). Oxidative stress and antioxidant enzyme upregulation in SOD1-G93A mouse skeletal muscle. Muscle Nerve 33, 809–816. doi: 10.1002/mus.20542
Malaspina, A., Ngoh, S. F., Ward, R. E., Hall, J. C., Tai, F. W., Yip, P. K., et al. (2010). Activation transcription factor-3 activation and the development of spinal cord degeneration in a rat model of amyotrophic lateral sclerosis. Neuroscience 169, 812–827. doi: 10.1016/j.neuroscience.2010.04.053
Manabe, Y., Nagano, I., Gazi, M. S., Murakami, T., Shiote, M., Shoji, M., et al. (2003). Glial cell line-derived neurotrophic factor protein prevents motor neuron loss of transgenic model mice for amyotrophic lateral sclerosis. Neurol. Res. 25, 195–200. doi: 10.1179/016164103101201193
Manzano, R., Toivonen, J. M., Calvo, A. C., Olivan, S., Zaragoza, P., Rodellar, C., et al. (2013). Altered in vitro proliferation of mouse SOD1-G93A skeletal muscle satellite cells. Neurodegener. Dis. 11, 153–164. doi: 10.1159/000338061
Mattiazzi, M., D'aurelio, M., Gajewski, C. D., Martushova, K., Kiaei, M., Beal, M. F., et al. (2002). Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 277, 29626–29633. doi: 10.1074/jbc.M203065200
Maximino, J. R., de Oliveira, G. P., Alves, C. J., and Chadi, G. (2014). Deregulated expression of cytoskeleton related genes in the spinal cord and sciatic nerve of presymptomatic SOD1(G93A) Amyotrophic Lateral Sclerosis mouse model. Front. Cell. Neurosci. 8:148. doi: 10.3389/fncel.2014.00148
Mirsky, R., and Jessen, K. R. (1999). The neurobiology of Schwann cells. Brain Pathol. 9, 293–311. doi: 10.1111/j.1750-3639.1999.tb00228.x
Mojsilovic-Petrovic, J., Nedelsky, N., Boccitto, M., Mano, I., Georgiades, S. N., Zhou, W., et al. (2009). FOXO3a is broadly neuroprotective in vitro and in vivo against insults implicated in motor neuron diseases. J. Neurosci. 29, 8236–8247. doi: 10.1523/JNEUROSCI.1805-09.2009
Moloney, E. B., de Winter, F., and Verhaagen, J. (2014). ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 8:252. doi: 10.3389/fnins.2014.00252
Morimoto, N., Miyazaki, K., Kurata, T., Ikeda, Y., Matsuura, T., Kang, D., et al. (2012). Effect of mitochondrial transcription factor a overexpression on motor neurons in amyotrophic lateral sclerosis model mice. J. Neurosci. Res. 90, 1200–1208. doi: 10.1002/jnr.23000
Morimoto, N., Nagai, M., Ohta, Y., Miyazaki, K., Kurata, T., Morimoto, M., et al. (2007). Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1167, 112–117. doi: 10.1016/j.brainres.2007.06.045
Mourelatos, Z., Gonatas, N. K., Stieber, A., Gurney, M. E., and Dal Canto, M. C. (1996). The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc. Natl. Acad. Sci. U.S.A. 93, 5472–5477 doi: 10.1073/pnas.93.11.5472
Muchowski, P. J., and Wacker, J. L. (2005). Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 6, 11–22. doi: 10.1038/nrn1587
Murakami, N., Mclennan, I. S., Nonaka, I., Koishi, K., Baker, C., and Hammond-Tooke, G. (1999). Transforming growth factor-beta2 is elevated in skeletal muscle disorders. Muscle Nerve 22, 889–898.
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nagano, I., Murakami, T., Manabe, Y., and Abe, K. (2002). Early decrease of survival factors and DNA repair enzyme in spinal motor neurons of presymptomatic transgenic mice that express a mutant SOD1 gene. Life Sci. 72, 541–548. doi: 10.1016/S0024-3205(02)02249-X
Narai, H., Manabe, Y., Nagai, M., Nagano, I., Ohta, Y., Murakami, T., et al. (2009). Early detachment of neuromuscular junction proteins in ALS mice with SODG93A mutation. Neurol. Int. 1:e16. doi: 10.4081/ni.2009.e16
Nguyen, M. D., Boudreau, M., Kriz, J., Couillard-Despres, S., Kaplan, D. R., and Julien, J. P. (2003). Cell cycle regulators in the neuronal death pathway of amyotrophic lateral sclerosis caused by mutant superoxide dismutase 1. J. Neurosci. 23, 2131–2140.
Niapour, A., Karamali, F., Karbalaie, K., Kiani, A., Mardani, M., Nasr-Esfahani, M. H., et al. (2010). Novel method to obtain highly enriched cultures of adult rat Schwann cells. Biotechnol. Lett. 32, 781–786. doi: 10.1007/s10529-010-0230-z
Nikolic-Kokic, A., Stevic, Z., Blagojevic, D., Davidovic, B., Jones, D. R., and Spasic, M. B. (2006). Alterations in anti-oxidative defence enzymes in erythrocytes from sporadic amyotrophic lateral sclerosis (SALS) and familial ALS patients. Clin. Chem. Lab. Med. 44, 589–593. doi: 10.1515/CCLM.2006.111
Nobbio, L., Fiorese, F., Vigo, T., Cilli, M., Gherardi, G., Grandis, M., et al. (2009a). Impaired expression of ciliary neurotrophic factor in Charcot-Marie-Tooth type 1A neuropathy. J. Neuropathol. Exp. Neurol. 68, 441–455. doi: 10.1097/NEN.0b013e31819fa6ba
Nobbio, L., Sturla, L., Fiorese, F., Usai, C., Basile, G., Moreschi, I., et al. (2009b). P2X7-mediated increased intracellular calcium causes functional derangement in Schwann cells from rats with CMT1A neuropathy. J. Biol. Chem. 284, 23146–23158. doi: 10.1074/jbc.M109.027128
Nunn, P. B., and Ponnusamy, M. (2009). Beta-N-methylaminoalanine (BMAA): metabolism and metabolic effects in model systems and in neural and other tissues of the rat in vitro. Toxicon 54, 85–94. doi: 10.1016/j.toxicon.2009.03.008
Oliveira, A. S., Isozaki, E., Younger, D., Gabbai, A. A., and Hays, A. P. (1994). Expression of HLA-DR in peripheral nerve of amyotrophic lateral sclerosis. Arq. Neuropsiquiatr. 52, 493–500. doi: 10.1590/S0004-282X1994000400007
Olsen, M. K., Roberds, S. L., Ellerbrock, B. R., Fleck, T. J., Mckinley, D. K., and Gurney, M. E. (2001). Disease mechanisms revealed by transcription profiling in SOD1-G93A transgenic mouse spinal cord. Ann. Neurol. 50, 730–740 doi: 10.1002/ana.1252
Palchaudhuri, M., and Flügge, G. (2005). 5-HT1A receptor expression in pyramidal neurons of cortical and limbic brain regions. Cell Tissue Res. 321, 159–172. doi: 10.1007/s00441-005-1112-x
Panov, A., Kubalik, N., Zinchenko, N., Hemendinger, R., Dikalov, S., and Bonkovsky, H. L. (2011). Respiration and ROS production in brain and spinal cord mitochondria of transgenic rats with mutant G93a Cu/Zn-superoxide dismutase gene. Neurobiol. Dis. 44, 53–62. doi: 10.1016/j.nbd.2011.06.003
Parone, P. A., Da Cruz, S., Han, J. S., Mcalonis-Downes, M., Vetto, A. P., Lee, S. K., et al. (2013). Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J. Neurosci. 33, 4657–4671. doi: 10.1523/JNEUROSCI.1119-12.2013
Pekny, M., and Nilsson, M. (2005). Astrocyte activation and reactive gliosis. Glia 50, 427–434. doi: 10.1002/glia.20207
Perrin, F. E., Boisset, G., Docquier, M., Schaad, O., Descombes, P., and Kato, A. C. (2005). No widespread induction of cell death genes occurs in pure motoneurons in an amyotrophic lateral sclerosis mouse model. Hum. Mol. Genet. 14, 3309–3320. doi: 10.1093/hmg/ddi357
Peterson, R., and Turnbull, J. (2012). Sonic hedgehog is cytoprotective against oxidative challenge in a cellular model of amyotrophic lateral sclerosis. J. Mol. Neurosci. 47, 31–41. doi: 10.1007/s12031-011-9660-x
Pizzuti, A., and Petrucci, S. (2011). Mitochondrial disfunction as a cause of ALS. Arch Ital Biol 149, 113–119.
Pun, S., Santos, A. F., Saxena, S., Xu, L., and Caroni, P. (2006). Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 9, 408–419. doi: 10.1038/nn1653
Quinlan, K. A. (2011). Links between electrophysiological and molecular pathology of amyotrophic lateral sclerosis. Integr. Comp. Biol. 51, 913–925. doi: 10.1093/icb/icr116
Ranganathan, S., and Bowser, R. (2003). Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am. J. Pathol. 162, 823–835. doi: 10.1016/S0002-9440(10)63879-5
Redler, R. L., and Dokholyan, N. V. (2012). The complex molecular biology of amyotrophic lateral sclerosis (ALS). Prog. Mol. Biol. Transl. Sci. 107, 215–262. doi: 10.1016/B978-0-12-385883-2.00002-3
Reinholz, M. M., Merkle, C. M., and Poduslo, J. F. (1999). Therapeutic benefits of putrescine-modified catalase in a transgenic mouse model of familial amyotrophic lateral sclerosis. Exp. Neurol. 159, 204–216. doi: 10.1006/exnr.1999.7142
Rocha, M. C., Pousinha, P. A., Correia, A. M., Sebastiao, A. M., and Ribeiro, J. A. (2013). Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS ONE 8:e73846. doi: 10.1371/journal.pone.0073846
Ryan, J. C., Wu, Q., and Shoemaker, R. C. (2015). Transcriptomic signatures in whole blood of patients who acquire a chronic inflammatory response syndrome (CIRS) following an exposure to the marine toxin ciguatoxin. BMC Med. Genomics 8:15. doi: 10.1186/s12920-015-0089-x
Saba, L., Viscomi, M. T., Caioli, S., Pignataro, A., Bisicchia, E., Pieri, M., et al. (2015). Altered functionality, morphology, and vesicular glutamate transporter expression of cortical motor neurons from a presymptomatic mouse model of amyotrophic lateral sclerosis. Cereb. Cortex. doi: 10.1093/cercor/bhu317. [Epub ahead of print].
Sandyk, R. (2006). Serotonergic mechanisms in amyotrophic lateral sclerosis. Int. J. Neurosci. 116, 775–826. doi: 10.1080/00207450600754087
Sasaki, S., Horie, Y., and Iwata, M. (2007). Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 114, 633–639. doi: 10.1007/s00401-007-0299-1
Sasaki, S., and Iwata, M. (1996). Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 204, 53–56. doi: 10.1016/0304-3940(96)12314-4
Saxena, S., and Caroni, P. (2007). Mechanisms of axon degeneration: from development to disease. Prog. Neurobiol. 83, 174–191. doi: 10.1016/j.pneurobio.2007.07.007
Saxena, S., Roselli, F., Singh, K., Leptien, K., Julien, J. P., Gros-Louis, F., et al. (2013). Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80, 80–96. doi: 10.1016/j.neuron.2013.07.027
Scardoni, G., Petterlini, M., and Laudanna, C. (2009). Analyzing biological network parameters with CentiScaPe. Bioinformatics 25, 2857–2859. doi: 10.1093/bioinformatics/btp517
Schulz, J. B., Lindenau, J., Seyfried, J., and Dichgans, J. (2000). Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 267, 4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x
Scorisa, J. M., Duobles, T., Oliveira, G. P., Maximino, J. R., and Chadi, G. (2010). The review of the methods to obtain non-neuronal cells to study glial influence on Amyotrophic Lateral Sclerosis pathophysiology at molecular level in vitro. Acta Cir. Bras. 25, 281–289. doi: 10.1590/S0102-86502010000300011
Shaw, P. J., and Eggett, C. J. (2000). Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J. Neurol. 247(Suppl. 1), I17–I27 doi: 10.1007/bf03161151
Shaw, P. J., Ince, P. G., Falkous, G., and Mantle, D. (1995). Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann. Neurol. 38, 691–695. doi: 10.1002/ana.410380424
Shibata, N., Hirano, A., Hedley-Whyte, E. T., Dal Canto, M. C., Nagai, R., Uchida, K., et al. (2002). Selective formation of certain advanced glycation end products in spinal cord astrocytes of humans and mice with superoxide dismutase-1 mutation. Acta Neuropathol. 104, 171–178. doi: 10.1007/s00401-002-0537-5
Siklós, L., Engelhardt, J., Harati, Y., Smith, R. G., Joó, F., and Appel, S. H. (1996). Ultrastructural evidence for altered calcium in motor nerve terminals in amyotropic lateral sclerosis. Ann. Neurol. 39, 203–216. doi: 10.1002/ana.410390210
Simpson, J. E., Ince, P. G., Haynes, L. J., Theaker, R., Gelsthorpe, C., Baxter, L., et al. (2010). Population variation in oxidative stress and astrocyte DNA damage in relation to Alzheimer-type pathology in the ageing brain. Neuropathol. Appl. Neurobiol. 36, 25–40. doi: 10.1111/j.1365-2990.2009.01030.x
Smyth, G. K. (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3. doi: 10.2202/1544-6115.1027
Song, F., Chiang, P., Wang, J., Ravits, J., and Loeb, J. A. (2012). Aberrant neuregulin 1 signaling in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 71, 104–115. doi: 10.1097/NEN.0b013e3182423c43
Sun, H., Bénardais, K., Stanslowsky, N., Thau-Habermann, N., Hensel, N., Huang, D., et al. (2013). Therapeutic potential of mesenchymal stromal cells and MSC conditioned medium in Amyotrophic Lateral Sclerosis (ALS)–in vitro evidence from primary motor neuron cultures, NSC-34 cells, astrocytes and microglia. PLoS ONE 8:e72926. doi: 10.1371/journal.pone.0072926
Tadic, V., Prell, T., Lautenschlaeger, J., and Grosskreutz, J. (2014). The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 8:147. doi: 10.3389/fncel.2014.00147
Takahashi, Y., Fukuda, Y., Yoshimura, J., Toyoda, A., Kurppa, K., Moritoyo, H., et al. (2013). ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am. J. Hum. Genet. 93, 900–905. doi: 10.1016/j.ajhg.2013.09.008
Tal, M. C., and Iwasaki, A. (2009). Autophagic control of RLR signaling. Autophagy 5, 749–750. doi: 10.4161/auto.5.5.8789
Tang, X., Wang, Y., Zhou, S., Qian, T., and Gu, X. (2013). Signaling pathways regulating dose-dependent dual effects of TNF-alpha on primary cultured Schwann cells. Mol. Cell Biochem. 378, 237–246. doi: 10.1007/s11010-013-1614-x
Tapia, R. (2014). Cellular and molecular mechanisms of motor neuron death in amyotrophic lateral sclerosis: a perspective. Front. Cell. Neurosci. 8:241. doi: 10.3389/fncel.2014.00241
Thau, N., Knippenberg, S., Körner, S., Rath, K. J., Dengler, R., and Petri, S. (2012). Decreased mRNA expression of PGC-1alpha and PGC-1alpha-regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J. Neuropathol. Exp. Neurol. 71, 1064–1074. doi: 10.1097/NEN.0b013e318275df4b
Van Leeuwen, L. A., and Hoozemans, J. J. (2015). Physiological and pathophysiological functions of cell cycle proteins in post-mitotic neurons: implications for Alzheimer's disease. Acta Neuropathol. 129, 511–525. doi: 10.1007/s00401-015-1382-7
Vargas, M. R., Pehar, M., Díaz-Amarilla, P. J., Beckman, J. S., and Barbeito, L. (2008). Transcriptional profile of primary astrocytes expressing ALS-linked mutant SOD1. J. Neurosci. Res. 86, 3515–3525. doi: 10.1002/jnr.21797
Veglianese, P., Lo Coco, D., Bao Cutrona, M., Magnoni, R., Pennacchini, D., Pozzi, B., et al. (2006). Activation of the p38MAPK cascade is associated with upregulation of TNF alpha receptors in the spinal motor neurons of mouse models of familial ALS. Mol. Cell. Neurosci. 31, 218–231. doi: 10.1016/j.mcn.2005.09.009
Venkova, K., Christov, A., Kamaluddin, Z., Kobalka, P., Siddiqui, S., and Hensley, K. (2014). Semaphorin 3A signaling through neuropilin-1 is an early trigger for distal axonopathy in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 73, 702–713. doi: 10.1097/NEN.0000000000000086
Verheijen, M. H., Peviani, M., Hendricusdottir, R., Bell, E. M., Lammens, M., Smit, A. B., et al. (2014). Increased axonal ribosome numbers is an early event in the pathogenesis of amyotrophic lateral sclerosis. PLoS ONE 9:e87255. doi: 10.1371/journal.pone.0087255
Wada, T., Goparaju, S. K., Tooi, N., Inoue, H., Takahashi, R., Nakatsuji, N., et al. (2012). Amyotrophic lateral sclerosis model derived from human embryonic stem cells overexpressing mutant superoxide dismutase 1. Stem Cells Transl. Med. 1, 396–402. doi: 10.5966/sctm.2011-0061
Wang, J., Yu, H., Ye, L., Jin, L., Yu, M., and Lv, Y. (2015). Integrated regulatory mechanisms of miRNAs and targeted genes involved in colorectal cancer. Int. J. Clin. Exp. Pathol. 8, 517–529.
Wang, R., Yang, B., and Zhang, D. (2011). Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia 59, 946–958. doi: 10.1002/glia.21167
Wang, Z. H., Walter, G. F., and Gerhard, L. (1996). The expression of nerve growth factor receptor on Schwann cells and the effect of these cells on the regeneration of axons in traumatically injured human spinal cord. Acta Neuropathol. 91, 180–184 doi: 10.1007/s004010050411
Warde-Farley, D., Donaldson, S. L., Comes, O., Zuberi, K., Badrawi, R., Chao, P., et al. (2010). The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, W214–W220. doi: 10.1093/nar/gkq537
Weinberg, R. A. (1995). The retinoblastoma protein and cell cycle control. Cell 81, 323–330. doi: 10.1016/0092-8674(95)90385-2
Wojsiat, J., Prandelli, C., Laskowska-Kaszub, K., Martin-Requero, A., and Wojda, U. (2015). Oxidative stress and aberrant cell cycle in Alzheimer's disease Lymphocytes: diagnostic prospects. J. Alzheimers Dis. 46, 329–350. doi: 10.3233/JAD-141977
Wong, P. C., Pardo, C. A., Borchelt, D. R., Lee, M. K., Copeland, N. G., Jenkins, N. A., et al. (1995). An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105–1116 doi: 10.1016/0896-6273(95)90259-7
Xia, Q., Hu, Q., Wang, H., Yang, H., Gao, F., Ren, H., et al. (2015). Induction of COX-2-PGE2 synthesis by activation of the MAPK/ERK pathway contributes to neuronal death triggered by TDP-43-depleted microglia. Cell Death Dis. 6:e1702. doi: 10.1038/cddis.2015.69
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253. doi: 10.1038/nn2047
Yang, D., Parrish, R. S., and Brock, G. N. (2014). Empirical evaluation of consistency and accuracy of methods to detect differentially expressed genes based on microarray data. Comput. Biol. Med. 46, 1–10. doi: 10.1016/j.compbiomed.2013.12.002
Yang, Y., Hentati, A., Deng, H. X., Dabbagh, O., Sasaki, T., Hirano, M., et al. (2001). The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 29, 160–165. doi: 10.1038/ng1001-160
Ying, H., Turturro, S., Nguyen, T., Shen, X., Zelkha, R., Johnson, E. C., et al. (2015). Induction of autophagy in rats upon overexpression of wild-type and mutant optineurin gene. BMC Cell. Biol. 16, 14. doi: 10.1186/s12860-015-0060-x
Yip, P. K., Pizzasegola, C., Gladman, S., Biggio, M. L., Marino, M., Jayasinghe, M., et al. (2013). The omega-3 fatty acid eicosapentaenoic acid accelerates disease progression in a model of amyotrophic lateral sclerosis. PLoS ONE 8:e61626. doi: 10.1371/journal.pone.0061626
Yoshihara, T., Ishigaki, S., Yamamoto, M., Liang, Y., Niwa, J., Takeuchi, H., et al. (2002). Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 80, 158–167. doi: 10.1046/j.0022-3042.2001.00683.x
Zhang, X., Li, L., Chen, S., Yang, D., Wang, Y., Wang, Z., et al. (2011). Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 7, 412–425 doi: 10.4161/auto.7.4.14541
Zhang, Z. C., and Chook, Y. M. (2012). Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the Fused in Sarcoma protein (FUS). Proc. Natl. Acad. Sci. U.S.A. 109, 12017–12021. doi: 10.1073/pnas.1207247109
Zhao, Z., Lange, D. J., Voustianiouk, A., Macgrogan, D., Ho, L., Suh, J., et al. (2006). A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 7:29. doi: 10.1186/1471-2202-7-29
Keywords: ALS, SOD1G93A, pre-symptomatic, sciatic nerve, microarray, flow cytometry sorting, Schwann cells, network analysis
Citation: Alves CJ, Maximino JR and Chadi G (2015) Dysregulated expression of death, stress and mitochondrion related genes in the sciatic nerve of presymptomatic SOD1G93A mouse model of Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 9:332. doi: 10.3389/fncel.2015.00332
Received: 05 June 2015; Accepted: 10 August 2015;
Published: 01 September 2015.
Edited by:
Manoj Kumar Jaiswal, Center for Neuroscience and Regenerative Medicine, USAReviewed by:
Robert Weissert, University of Regensburg, GermanyLei Liu, University of Florida, USA
Hakan Muyderman, Flinders University, Australia
Copyright © 2015 Alves, Maximino and Chadi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gerson Chadi, Department of Neurology, Neuroregeneration Center, University of São Paulo School of Medicine, Av. Dr. Arnaldo, 455, 2nd Floor, Room 2119, 01246-903 São Paulo, Brazil, gerchadi@usp.br