Mice Overexpressing Both Non-Mutated Human SOD1 and Mutated SOD1G93A Genes: A Competent Experimental Model for Studying Iron Metabolism in Amyotrophic Lateral Sclerosis
 Anna Gajowiak1
Anna Gajowiak1  Agnieszka Styś1
Agnieszka Styś1  Rafał R. Starzyński1*
Rafał R. Starzyński1*  Aleksandra Bednarz2
Aleksandra Bednarz2  Małgorzata Lenartowicz2
Małgorzata Lenartowicz2  Robert Staroń1
Robert Staroń1  Paweł Lipiński1*
Paweł Lipiński1*- 1Department of Molecular Biology, Institute of Genetics and Animal Breeding, Polish Academy of Sciences, Magdalenka, Poland
- 2Department of Genetics and Evolution, Institute of Zoology, Jagiellonian University, Kraków, Poland
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by degeneration and loss of motor neurons in the spinal cord, brainstem and motor cortex. Up to 10% of ALS cases are inherited (familial, fALS) and associated with mutations, frequently in the superoxide dismutase 1 (SOD1) gene. Rodent transgenic models of ALS are often used to elucidate a complex pathogenesis of this disease. Of importance, both ALS patients and animals carrying mutated human SOD1 gene show symptoms of oxidative stress and iron metabolism misregulation. The aim of our study was to characterize changes in iron metabolism in one of the most commonly used models of ALS – transgenic mice overexpressing human mutated SOD1G93A gene. We analyzed the expression of iron-related genes in asymptomatic, 2-month-old and symptomatic, 4-month-old SOD1G93A mice. In parallel, respective age-matched mice overexpressing human non-mutated SOD1 transgene and control mice were analyzed. We demonstrate that the overexpression of both SOD1 and SOD1G93A genes account for a substantial increase in SOD1 protein levels and activity in selected tissues and that not all the changes in iron metabolism genes expression are specific for the overexpression of the mutated form of SOD1.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most widespread motor neuron disease. It is characterized by a progressive and selective degeneration of neurons in motor cortex and lower motor neurons projecting from the brainstem and spinal cord. In consequence, the disease leads to gradual skeletal muscle weakness, atrophy and paralysis and it is usually fatal within 2–3 years from the onset. The most common cause of death among ALS patients is respiratory failure caused by the degeneration of the nerves and muscles that control breathing (Pratt et al., 2012). Although ALS is mostly a sporadic disease (sALS) of generally unknown etiology, approximately 10% of ALS cases, designed as familial ALS (fALS), are due to genetic factors and have an autosomal dominant pattern of transmission (Rothstein, 2009). Mutations found within the SOD1 gene encoding an antioxidant enzyme, (SOD1, CuZn-SOD) were the first established genetic cause of ALS (Rosen et al., 1993) and are nowadays estimated to account for up to 20 and 3% of fALS and sALS cases, respectively. In addition to SOD1 gene mutations, defects in some other genes have also been involved in fALS (Pratt et al., 2012). Importantly, how the various ALS-linked gene products determine similar course of the disease is incompletely understood. The two forms of ALS (fALS and sALS) share similar clinical and pathological features, suggesting common molecular pathogenic mechanisms. Oxidative stress (a condition arising from an imbalance between the production of reactive oxygen species and the efficiency of antioxidant defense systems) has been proposed to be implicated in ALS pathogenesis mostly by interfering with other pathogenic mechanisms, such as motor neuron excitotoxicity, mitochondrial and cytoskeletal dysfunction, protein aggregation, neuroinflammation, and deficit in neutrophic growth factors (Barber et al., 2006). The evidence of the involvement of oxidative stress in ALS pathology originates from studies on ALS patients (Carri et al., 2003) as well as from experiments on rodent models of this disease, which are in vast majority transgenic mice that overexpress mutated human SOD1 gene (Turner and Talbot, 2008). Although in general these mouse models recapitulate human disease, some critical differences exist between the animal and human pathology. An important one concerns an increased enzymatic activity of the SOD1 in some transgenic mice widely used in ALS research (for example SOD1G37R and SOD1G93A), a phenomenon, which does not occur in humans. Of importance, SOD1 is a particular antioxidant enzyme as it converts the superoxide anion radical (O2⋅-) to another reactive oxygen species (ROS) – hydrogen peroxide (H2O2) (McCord and Fridovich, 1969). It seems therefore that the final effect of SOD1 overexpression depends on the balance between the beneficial effects of the O2⋅- scavenging and the destructive effects of H2O2 production. In this context, it is not surprising that the toxicity induced by oxidative stress has been reported in both, SOD1 null mice (Reaume et al., 1996) and in mice overexpressing human wild-type SOD1 gene (Lee et al., 2001).
The commonly accepted paradigm of iron dichotomy in biological systems states that iron is essential for the function of many enzymes and thus it is absolutely fundamental for most biological life forms, but, on the other hand, it is toxic in excess (Pantopoulos et al., 2012). Toxicity of iron is usually explained by its ability to induce oxidative stress through the catalysis of the Fenton reaction that leads to the formation of the hydroxyl radical (⋅OH), a highly destructive oxidant. Both aforementioned ROS interact with iron in the Fenton reaction: O2⋅- as a rate-limiting reducing factor for the pre-existing pool of free iron active in the generation of ⋅OH, and H2O2, as a factor that directly reacts with ferrous iron to yield this radical. Apart from the participation in the Fenton reaction, both H2O2 and O2⋅- have been shown to alter the expression of iron-related genes by various molecular mechanisms (reviewed by Orino et al., 2001).
Misregulation of iron homeostasis in the central nervous system, which results in the pathological iron accumulation and in increased formation of ROS is a frequent phenomenon, largely documented in neurodegenerative diseases (Oshiro et al., 2011; Hadzhieva et al., 2014). The elevated amounts of iron deposits have been also reported in the brain and spinal cord of ALS patients (Kasarskis et al., 1995; Kwan et al., 2012; Ignjatović, 2013) and mice overexpressing mutated human SOD1 gene (Jeong et al., 2009; Lee et al., 2015). Presumably, the most convincing argument supporting pathological involvement of iron accumulation and iron-mediated oxidative stress in the progression of ALS derives from studies showing beneficial effects of iron chelation therapy in transgenic mice overexpressing human mutated SOD1 gene (Jeong et al., 2009; Kupershmidt et al., 2009; Wang et al., 2011; Lee et al., 2015). Based on the analysis of the complex interplay between oxidative stress and disturbed iron homeostasis in these models, Hadzhieva et al., 2014 proposed that in ALS, oxidative stress strongly affects cellular iron balance leading to iron overload in motor neurons, and thus creates a vicious circle to exacerbate oxidative injury. However, it remains unclear, up to what extent SOD1 mouse models of ALS reproduce the mechanisms of oxidative stress induction in human pathology. In particular, an intriguing question remains open: how (if at all) the increased SOD1 activity observed in the CNS of transgenic SOD1G93A and SOD1G37R mice affects their iron metabolism. Of importance, to our knowledge, only these two mouse models were used so far in in vivo studies of iron metabolism dysregulation in ALS. Strictly speaking, it is still unclear, which iron metabolism dysregulation mechanisms can be regarded as a disease-specific (and thus can be used to draw conclusions in humans), and which should be regarded as a secondary, model-associated effects.
Here, to clear up this issue, in addition to mice overexpressing human mutated SOD1G93A gene we used two types of control age-matched mice – wild-type mice and mice overexpressing human wild-type SOD1 gene. In those mice we compared the expression pattern of iron-related genes in their brain stems (Medulla oblongata), spinal cords, skeletal muscles (represented by gastrocnemius and broadest dorsal – Latissimus dorsi muscles) and livers. We have also determined iron content and distribution in their gastrocnemius muscles.
We demonstrate that the overexpression of both SOD1 and SOD1G93A genes accounts for a substantial increase in SOD1 protein levels and activity in selected tissues and that not all the changes in iron metabolism genes expression are specific for the overexpression of the mutated form of SOD1. Furthermore, not all the changes are specific to the disease-affected human tissues. Importantly, among various analyzed genes, only Hmox1, encoding HO1, an important oxidative stress responder was found to be induced solely in mice overexpressing human mutated SOD1 gene and only in tissues known to be affected by ALS. Similarly, iron accumulation in the gastrocnemius muscle was also exclusively restricted to mice carrying SOD1G93A mutation in the symptomatic stage of disease.
Materials and Methods
Mice
Following mice provided by The Jackson Laboratory (Bar Harbor, ME, USA) were used in the study: males [strain B6SJL-Tg(SOD1-G93A)1Gur/J] hemizygous for the SOD1G93A transgene, with transgenic expression of a G93A mutant form of human SOD1 (harboring a single amino acid substitution of glycine to alanine at codon 93) (Gurney et al., 1994), transgenic males [strain B6SJL-Tg(SOD1)2Gur/J] carrying the normal allele of the human SOD1 gene (called SOD1), and males (strain B6129PF2/J) used as controls for genetically engineered mice from the above mentioned strains. SOD1G93A mice exhibit a phenotype similar to ALS in humans. All analyses were performed in age-matched 2- and 4-month-old mice, designed as asymptomatic and symptomatic (showing apparent symptoms of paralysis in the case of mice overexpressing human mutated gene), respectively. The age of mice from the two control groups (wild-type and mice overexpressing human wild-type SOD1) was the same as for the ALS model mice. Mice arrived at the age of 4 weeks and were housed at 24–25°C, relative humidity 50–60% with a light–dark cycle of 12 h. Mice received a standard diet (Labo-feed, Kcynia, Poland) and water ad libitum. Animals were euthanized by intraperitoneal injection of Vetbutal (Biovet, Puławy, Poland). All procedures were conducted according to the guidelines of the Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the Protection of Animals Used for Scientific Purposes.
Measurement of SOD1 Activity
Superoxide dismutase 1 activity in tissue total extracts was measured by gel electrophoresis using the Nitroblue Tetrazolium (NBT)/riboflavin method as described previously (Beauchamp and Fridovich, 1971). Briefly, 15 mg samples of the tissue total extracts were resolved by electrophoresis on 12% polyacrylamide gels under non-denaturing and non-reducing conditions. After electrophoresis, the activity of SOD was visualized by immersion of the gels in staining buffer [50 mM potassium phosphate (pH 7.8), 0.1 mM EDTA, 28 mM TEMED, 3 mM riboflavin, 0.25 mM NBT] for 30 min in the dark at RT. Gels were then exposed to light until the SOD activity bands became visible as bright bands on a dark blue background. The reaction was stopped by rinsing the gels with water. Activities of CuZn-SOD, and Mn-SOD were distinguished by a selective inhibition of the former activity by incubation of gels in a buffer containing 3 mM KCN prior to the staining, as described by Salin and Bridges (1981).
Hematoxylin/Eosin and Prussian Blue Staining of the gastrocnemius Muscle
The gastrocnemius muscle was excised from both hind limbs, dissected from fat and connective tissue, immediately fixed in Bouin’s solution for 24 h, and stored in 70% ethanol before further preparation. After dehydration, muscle was embedded in paraffin and cut into 7 μm sections with a microtome (Reichert-Jung, Germany) on the horizontal plane, transversely to the long axis of the muscle. The sections were placed on a slide and stained with haematoxylin and eosin. The morphology of muscle structure was studied by standard light microscopy (Olympus, type CH2) under × 2 and × 40 objectives.
Non-heme iron staining of the gastrocnemius muscle samples was analyzed using Accustain Iron Deposition Kit (Sigma–Aldrich). After mounting on glass slides, muscle sections were deparaffinized, incubated with working solution containing Perls’ Prussian blue for 30 min, counterstained with pararoseaniline solution for 2 min and analyzed under standard light microscopy (Olympus, type CH2).
RNA Preparation and Real-Time Quantitative RT-PCR
Total RNA was extracted using the TRIzol reagent (Invitrogen). 0.5 μg (spinal cord), 1 μg (gastrocnemius) or 2 μg (liver, broadest dorsal muscle) of total RNA was reverse-transcribed using random hexamers and Transcription First Strand cDNA Synthesis Kit (Roche). Real-time quantitative PCR was performed using the Roche Light Cycler 96 system and the FastStart Essential DNA Green Master Kit (Roche) together with the following primers: TfR1, 5′-TCG CTT ATA TTG GGC AGA CC-3′ (forward) and 5′-CCA TGT TTT GAC CAA TGC TG-3′ (reverse); HO1, 5′-TCT TGC CTG GCT CTC TTC TC-3′ (forward) and 5′- GTC GTG GTC AGT CAA CAT GG -3′ (reverse); 18 S ribosomal RNA (18S), 5′-CTG AGA AAC GGC TAC CAC ATC-3′ (forward) and 5′-CGC TCC CAA GAT CCA ACT AC-3′ (reverse). Data were analyzed with the Light Cycler 3.5 software. mRNA expression was standardized to 18S ribosomal RNA levels mRNA levels, as indicated in legends.
Preparation of Tissues Protein Extracts and Western Blot Analysis
Following mouse tissues were collected: M. oblongata, spinal cord, the gastrocnemius muscle and the L. dorsi (broadest dorsal) muscle, liver and kidney. Total extracts, cytosolic fractions and crude membrane extracts were prepared as described previously (Canonne-Hergaux et al., 1999; Starzyński et al., 2009). Protein concentration was determined spectrophotometrically using the Bio-Rad protein assay. SOD1 was detected in total protein extracts obtained from tissues using rabbit polyclonal anti-superoxide dismutase 1 antibody (Abcam; ab16831). H- (H-Ft) and L-ferritin (L-Ft) subunit expression was analyzed using cytosolic extracts and rabbit antisera raised against, respectively, the recombinant mouse ferritin H and L subunits (kindly provided by Dr. Paolo Santambrogio, San Raffaele Scientific Institute, Milano, Italy). L-Ft was also analyzed using purified rabbit mouse liver ferritin antiserum (kindly provided by Dr. J. Brock, Glasgow University, Glasgow, UK) (Bouton et al., 2002). Fpn, Cp and HO1 levels in crude membrane extracts were detected using, respectively, a rabbit polyclonal antibody raised against mouse Fpn (MTP11-A, Alpha Diagnostics), a rabbit polyclonal antibody raised against human Cp (DAKO) and a rabbit polyclonal antibody raised against rat HO1 (ADI-OSA-150-F, Enzo Life Sciences). The loading controls – β-actin and tubulin in protein extracts were detected using goat polyclonal and mouse monoclonal antibodies, respectively (both from Santa Cruz Biotechnology). Peroxidase-conjugated anti-rabbit, anti-chicken, anti-mouse, or anti-goat secondary antibodies (Santa Cruz Biotechnology) were used. Immunoreactive bands were detected using the Western Bright ECL Western blotting detection kit (Advansta). Tissue extracts from Hmox1-/- or Irp1-/- mice or recombinant L- and H-Ft proteins (kindly provided by Dr. Paolo Santambrogio) were used as controls. Western blot analyses were performed using extracts prepared separately for each mouse (six experimental groups: two age groups and three genotypes for each age; three mice in each group; total 18 extracts per tissue).
Statistical Analysis
All experiments were performed in biological triplicates, and error bars indicate standard deviation. Statistica 10 software was used for all of the statistical analyses. The assays were performed by two way ANOVA and means comparisons were adjusted by Tukey (P < 0.05 and P < 0.01).
Results
Increased SOD1 Expression and Activity in Tissues of Transgenic SOD1 and SOD1G93A Mice
Before investigating iron metabolism genes in mice from 3 experimental groups, first we measured SOD1 activity and expression in tissues affected by ALS (neuronal tissues and skeletal muscles) and in the two tissues important for systemic iron metabolism (liver and kidney). In all analyzed tissues: skeletal muscles (Figure 1A), neuronal tissues (Figure 1B), liver (Figure 1C, left-hand), and kidney (Figure 1C, right-hand) from both transgenic SOD1 and SOD1G93A mice, the superoxide-scavenging activity of SOD1 was markedly higher compared to age-matched 2- and 4-month-old wild-type animals. We found that this increase was due to significantly (P < 0.01) elevated SOD1 protein level, detected by western immunoblotting (Supplementary Table). We found that in spinal cord, M. oblongata (Figure 1B), liver and kidney (Figure 1C) of the transgenic SOD1 mice SOD1 protein was expressed at a higher level than in the corresponding tissues of the ALS mice although these differences did not reach statistical significance. Accordingly, the SOD1 superoxide-scavenging activity was higher in those tissues of mice overexpressing human wt SOD1 gene.
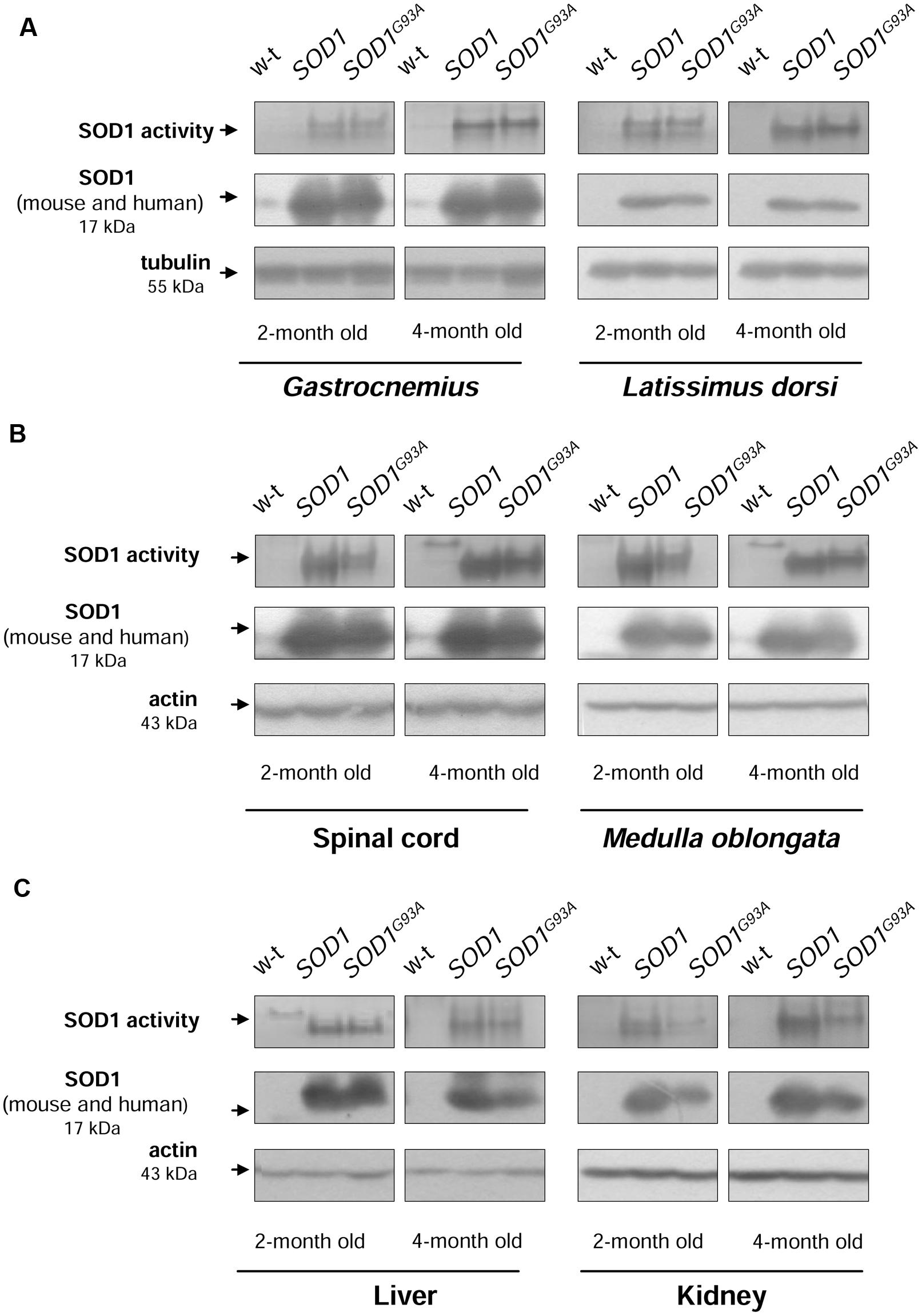
FIGURE 1. CuZn-SOD1 (SOD1) activity and expression in tissues of experimental mice. In-gel activity staining for SOD1 activity in total protein extracts obtained from skeletal muscles (A), neuronal tissues (B), liver, and kidney (C) (upper). Results of Western blot analysis of SOD1 in the same protein extracts are shown in (middle). Blots were also re-probed with monoclonal anti-tubulin or anti-β-actin antibodies from mouse as a loading control (lower). Data are representative for three independent Western blot analyses performed using extracts from 3 mice of each experimental group. The intensity of the SOD1 bands (relative to the intensity of tubulin or actin bands) was quantified with a Molecular Imager using Quantity One software (Bio-Rad) and is shown in arbitrary units to present protein level (Supplementary Table). Results are expressed as mean ± SD for three mice in each experimental group. Significant differences (P < 0.01) in SOD1 protein levels were found between transgenic (SOD1 and SOD1G93A) and wild-type mice in a given tissue and age group.
Pathological Changes and Iron Distribution in the gastrocnemius Muscle from Symptomatic ALS Mice
Skeletal muscle atrophy observed in SOD1 models of ALS is usually considered as a consequence of motor neuron loss, although there is some evidence that it is a direct target of SOD1-mediated pathology (Dobrowolny et al., 2008). We analyzed myopathology of the gastrocnemius muscle upon staining with eosin/hematoxylin. We did not find any abnormalities in this muscle in 2-month-old mice – neither in the wild-type strain, nor in the transgenic ones – SOD1 and SOD1G93A (data not shown). Similarly, no abnormalities were detected in 4-month-old wild-type and transgenic SOD1 animals. In contrast, the gastrocnemius muscle from symptomatic transgenic SOD1G93A mice showed changes in the muscle architecture characteristic for ALS (Figure 2F). In particular, fibers had similar size (Figures 2A,B) in the muscle of 4-month-old control mice (wild-type and SOD1). In contrast, on the muscle section of the 4-month-old SOD1G93A mice we observed grouped angular atrophic muscle fibers (Figure 2F) and among small and degenerative muscle fibers we identified hypertrophic ones (Figures 2C–E). Few characteristic pyknotic nuclear clumbs were observed mainly in the group of degenerated fibers (Figure 2F). Microscopic analysis of gastrocnemius muscle sections from 4-month old wild-type and transgenic SOD1 mice stained for non-heme iron with Perls’ Prussian blue showed no evidence of non-heme iron accumulation (Figures 3A,B). In contarst, non-heme iron accumulation was detected in the gastrocnemius muscles derived from 4-month-old SOD1G93A symptomatic mutants (Figures 3C–E). Deposits of iron had a form of irregular cytoplasmic inclusions, localized in some (not all) muscle fibers close the cell nuclei.
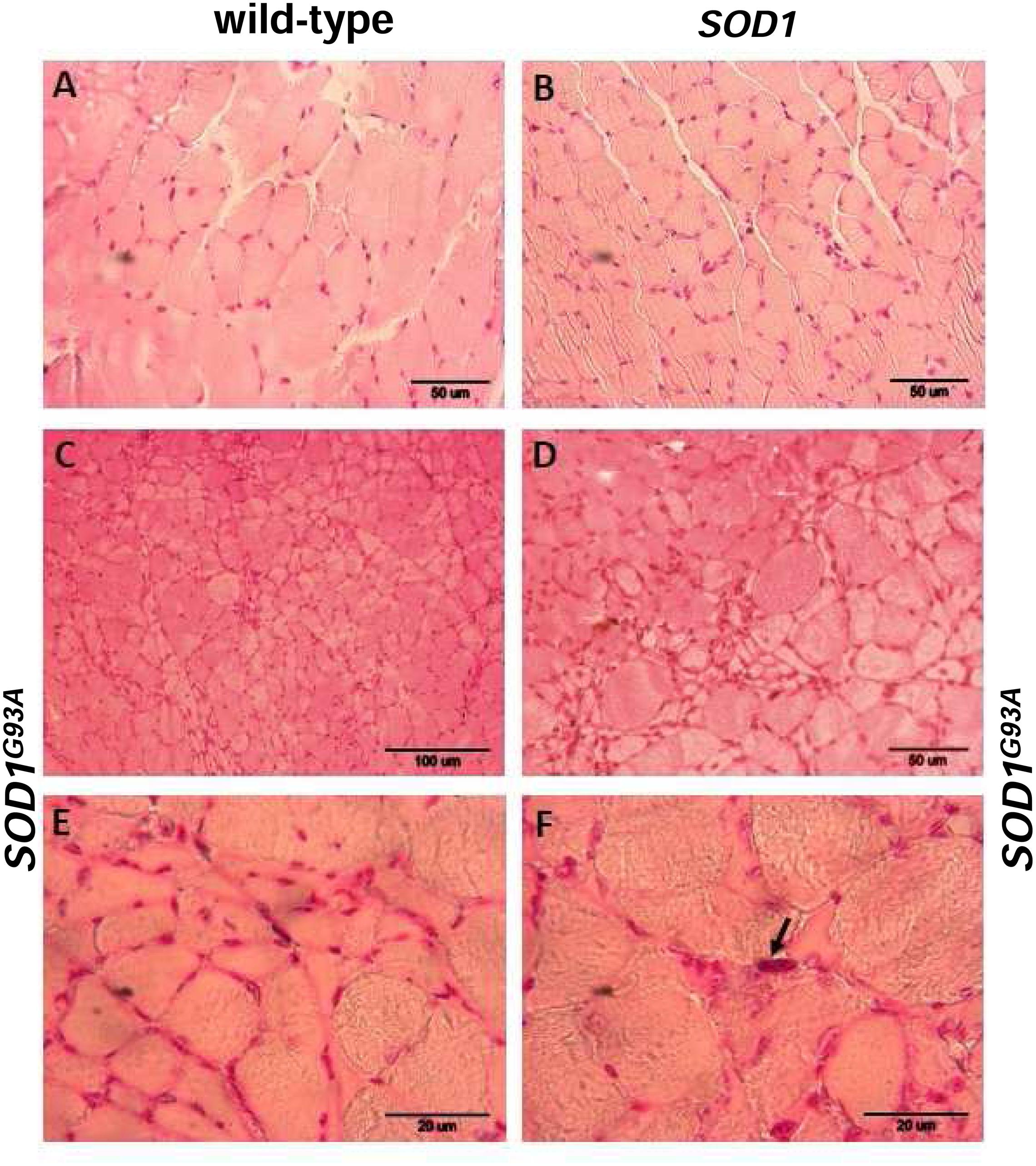
FIGURE 2. Pathological changes in the gastrocnemius muscle of symptomatic ALS mice. Haematoxylin and eosin-stained cross-sections of the gastrocnemius muscle from 4-month-old wild-type (A) and SOD1 transgenic (B) mice show similar regular size of muscle fibers and normal architecture of the muscle; bar = 50 μm. Sections from aged-matched symptomatic SOD1G93A mice show pathological changes in muscle structure characteristic for ALS (C–F). Larger regions of the muscle sections at lower magnification (bar = 100 μm) show characteristic grouped atrophy in the muscle of SOD1G93A mice (C). Muscle tissue from symptomatic SOD1G93A mice shows the presence of mixed fiber types of different size. Group of small and atrophic muscle fibers and presence of hypertrophied fibers in the gastrocnemius muscle from 4-month-old symptomatic mouse (D); bar = 50 μm. Group of the small and angular fibers at a higher magnification (bar = 20 μm) (E). Pyknotic nuclear clumb (arrow) present in the muscle of symptomatic SOD1G93A mice (F); bar = 20 μm.
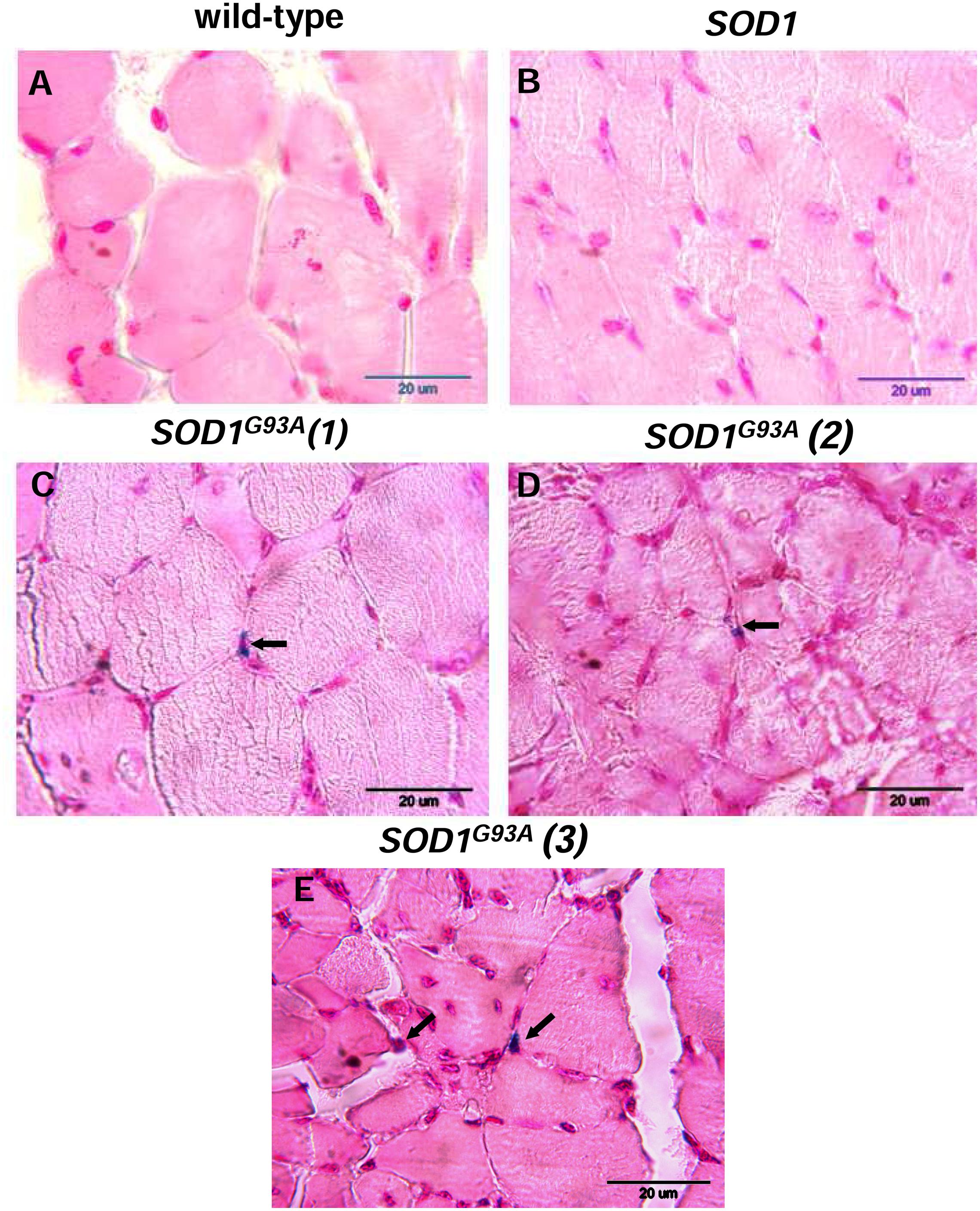
FIGURE 3. Histological examination of iron loading in the gastrocnemius muscle of 4-month-old experimental mice. Non-heme iron deposits were detected by staining with Perls Prussian Blue in the gastrocnemius muscle of wild-type (A) and SOD1 mice (B). Blue staining (indicated by arrows) was present exclusively in the muscle section of symptomatic SOD1G93A mice. Staining of muscle sections from three SOD1G93A mice are shown (C,D,E).
IRP1 Protein Levels are Unaffected in SOD1 and SOD1G93A Mice
Iron regulatory protein 1 (IRP1) is a cytoplasmic bifunctional protein showing either aconitase or trans-regulatory activity involved in the mechanisms that control iron metabolism in mammalian cells. IRP1 lacking its iron-sulfur cluster acts as a trans-regulatory element that modulates the expression of iron-related proteins such as ferritin subunits, TfR1 and Fpn at a post-transcriptional level by binding to specific iron regulatory elements (IREs) on their mRNAs (Wilkinson and Pantopoulos, 2014). We have previously shown that in SOD1 knock-out mice the expression of IRP1 is strongly reduced (Starzyński et al., 2005). On the other hand, the effect of increased SOD1 activity on IRP1 protein level has been investigated in cells overexpressing wild-type SOD1 gene as well as in transgenic SOD1G93A and SOD1G37R ALS cellular (Danzeisen et al., 2006) and mouse (Massignan et al., 2007; Jeong et al., 2009) models, but discrepant data on IRP1 regulation have been reported. As intracellular protein IRP1 level is important for modifications of its IRE-binding affinity (Lipiński et al., 2000; Starzyński et al., 2006) we compared its expression at the protein level in cytosolic extracts obtained from tissues of wild-type, SOD1 and SOD1G93A mice. Our Western blot analysis shows permanently high stability of IRP1 protein in all analyzed tissue tissues obtained from both asymptomatic and symptomatic SOD1G93A mice, in age-matched mice overexpressing wild-type SOD1 gene and in wild-type controls (Figure 4). Our results militate against the possibility that in our model of ALS changes in IRP1 expression may account for its IRE-binding activity and in consequence for the regulation of its target mRNAs.
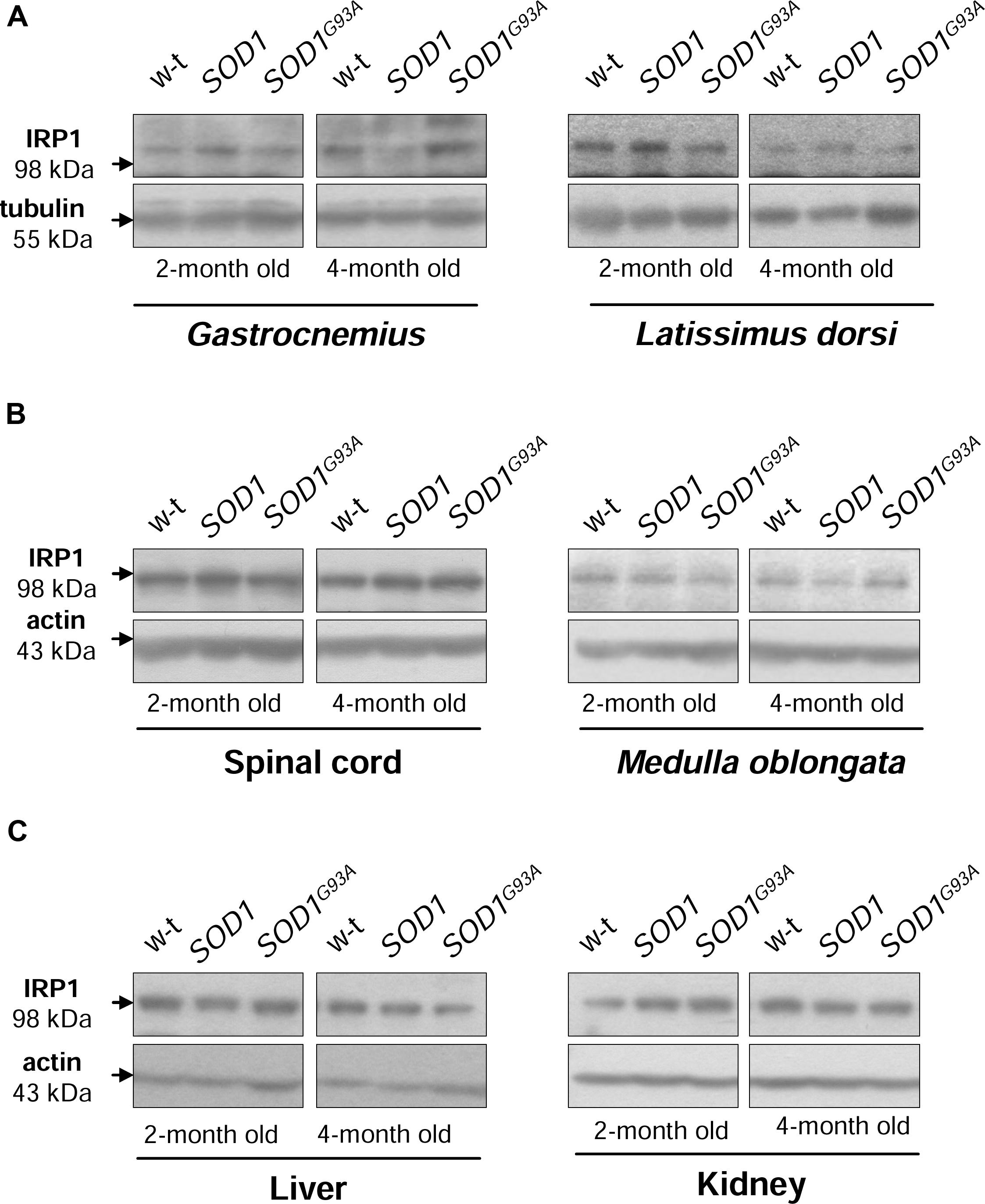
FIGURE 4. Wild-type, SOD1 and SOD1G93A mice show similar IRP1 protein levels in all examined tissues. IRP1 levels were assessed in total skeletal muscle (A), neuronal tissues (B), hepatic and renal (C) extracts by Western blotting as described under “Material and Methods”. Blots were also re-probed with monoclonal anti-tubulin or anti-β-actin antibodies from mouse as a loading control. Data are representative for three independent Western blot analyses performed using extracts from 3 mice of each experimental group. The intensity of the IRP1 bands (relative to the intensity of tubulin or actin bands) was quantified with a Molecular Imager using Quantity One software (Bio-Rad) and is shown in arbitrary units to present protein level (Supplementary Table).
Increased H-Ft Protein Levels in SOD1 and SOD1G93A Mice
Ferritin level within the cell is a hallmark of intracellular iron accumulation (Harrison and Arosio, 1996). Among two types of subunits forming a hollow shell of the protein, H-ferritin possessing ferroxidase activity has been also shown to be regulated by oxidants and to prevent pro-oxidant labile iron from exacerbating oxidative stress (Orino et al., 2001). Bearing this in mind and referring to the concept of the involvement of oxidative stress in ALS pathology, we aimed to check whether changes in ferritin expression in tissues are specific for SOD1G93A mouse model of ALS. We determined both H- and L-ferritin expression at the protein level (Figures 5A–C). We noticed a substantial (from ∼3- to 25-fold depending on tissue, Supplementary Table) upregulation of H-ferritin protein in all analyzed tissues in both SOD1 and SOD1G93A mice. This increase has been already observed in 2-month-old mice, strongly suggesting that it is rather caused by the enhanced SOD1 activity and is not directly linked strictly to the ALS pathology. In all tissues L-ferritin protein levels remained largely unaffected (Figures 5A–C).
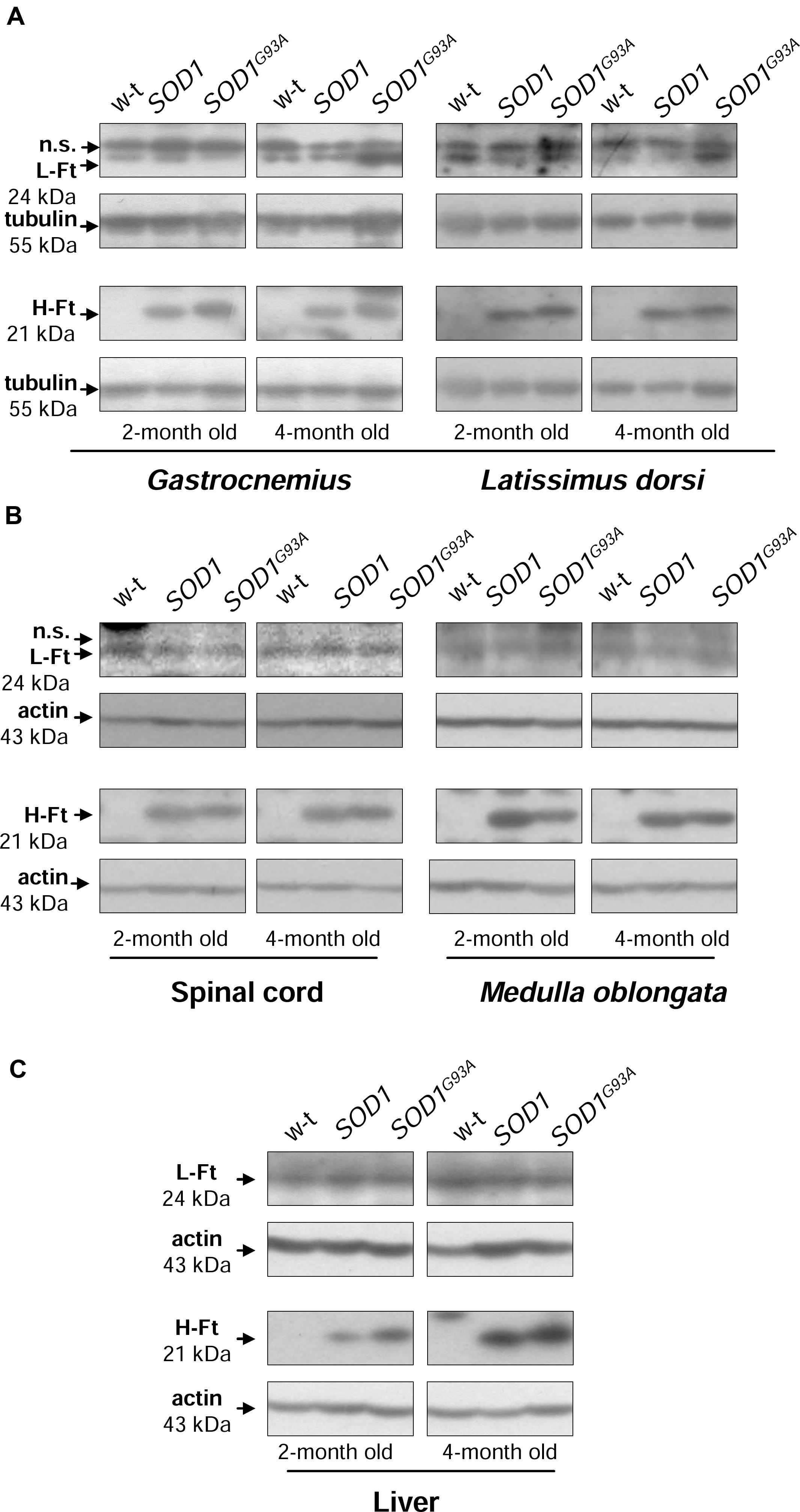
FIGURE 5. Analysis of ferritin protein levels in tissues of experimental mice. Western blot analysis of L-Ft and H-Ft protein levels in skeletal muscle (A), neuronal tissues (B), and liver (C) cytosolic extracts. Tubulin and actin were used as a loading controls, as indicated. n.s. – non-specific band detected with purified rabbit mouse liver ferritin antiserum. Data are representative for three independent Western blot analyses performed using extracts from 3 mice of each experimental group. The intensity of the H- and L-Ft bands (relative to the intensity of tubulin or actin bands) was quantified with a molecular Imager using Quantity One software (Bio-Rad) and is shown in arbitrary units to present proteins level (Supplementary Table). Statistically significant differences (P < 0.01) in H-Ft protein levels were found between transgenic (SOD1 and SOD1G93A) and wild-type mice in a given tissue and age group.
Similar Expression Patterns of Iron Importer and Exporter Genes in SOD1, SOD1G93A and Wild-type Mice
The cellular iron status is largely influenced by the balance between iron influx to the cell and its export from the cell to the extracellular environment. Therefore in selected tissues (such as muscles, spinal cord, and liver) of mice from three experimental groups we determined the expression of the main iron importer gene, TfR1. Of importance, TfR1 is predominantly regulated at the level of its transcript stability via the post-transcriptional system IRP/IRE (Wilkinson and Pantopoulos, 2014). As show in Figures 6A–D the TfR1 mRNA levels measured by real-time quantitative RT-PCR show tendency to decrease in all analyzed tissues of SOD1G93A mice compared with wild-type and SOD1 mice. However, the only statistically significant decrease in TfR1 mRNA level was found in the case of L. dorsi muscle of SOD1G93A symptomatic mice (Figure 6B). As regards proteins cooperating in iron export – Fpn and Cp, their levels in all analyzed tissues of SOD1G93A and SOD1 mice assessed by Western blotting are the same as those found in control animals (wild-type and SOD1) in the two analyzed periods (Figures 7A–D).
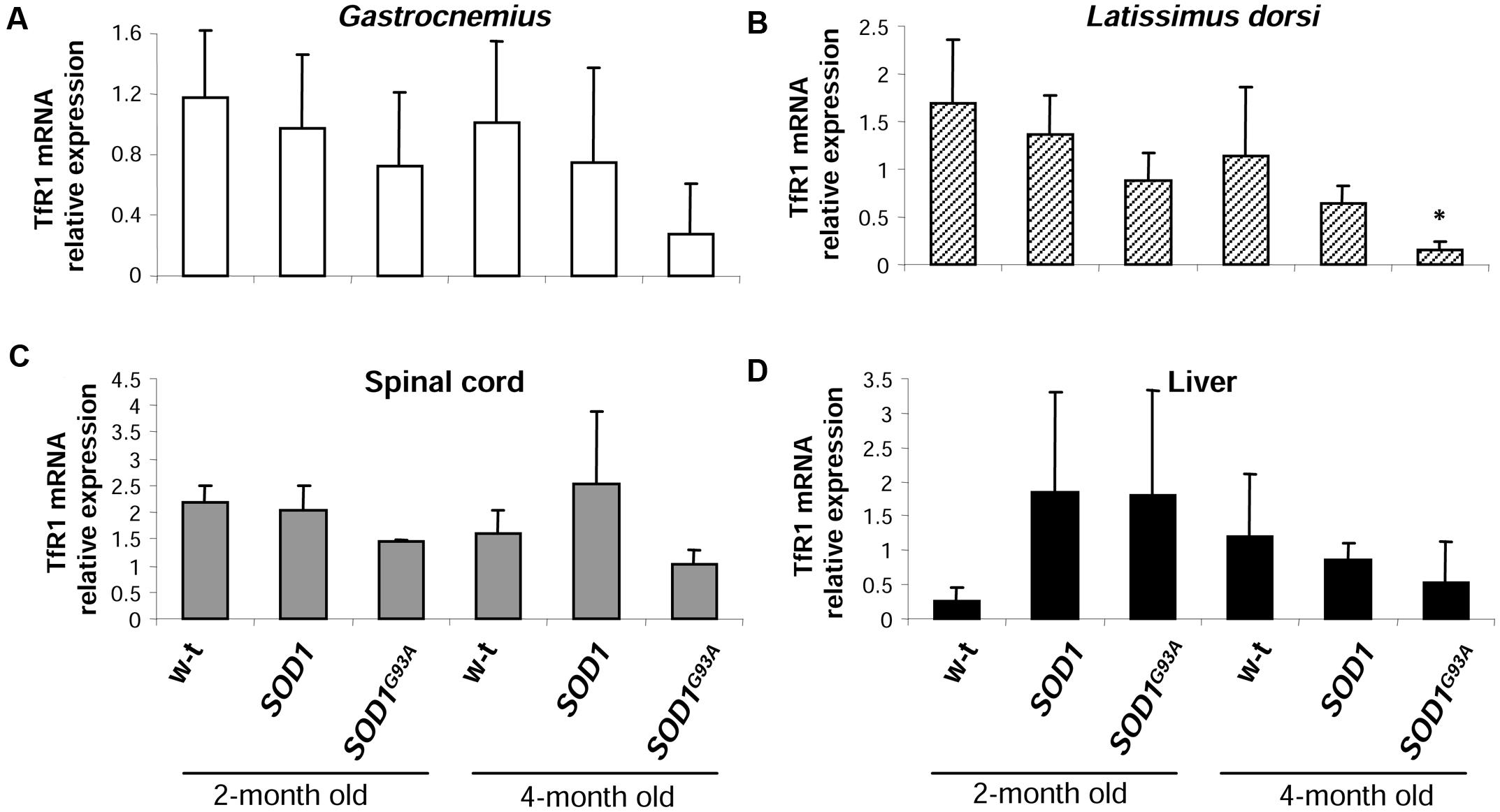
FIGURE 6. Analysis of TfR1 mRNA abundance in tissues of experimental mice. TfR1 mRNA abundance in skeletal muscles (A,B), spinal cord (C), and liver (D) was measured by real-time RT-PCR as described under “Materials and methods.” The histograms display TfR1 mRNA levels after normalization to 18S ribosomal RNA levels. Each column represents the mean ± SD of three biological amplification reactions. RNA from each mouse was extracted separately using TRIZOL reagent, reverse-transcribed and amplified as described in M&M; therefore, 18 cDNA samples (biological triplicates for each condition) were finally used in the analysis of gene expression in each tissue. ∗, significant difference versus wild-type in the group of symptomatic, 4-month-old mice (P < 0.05).
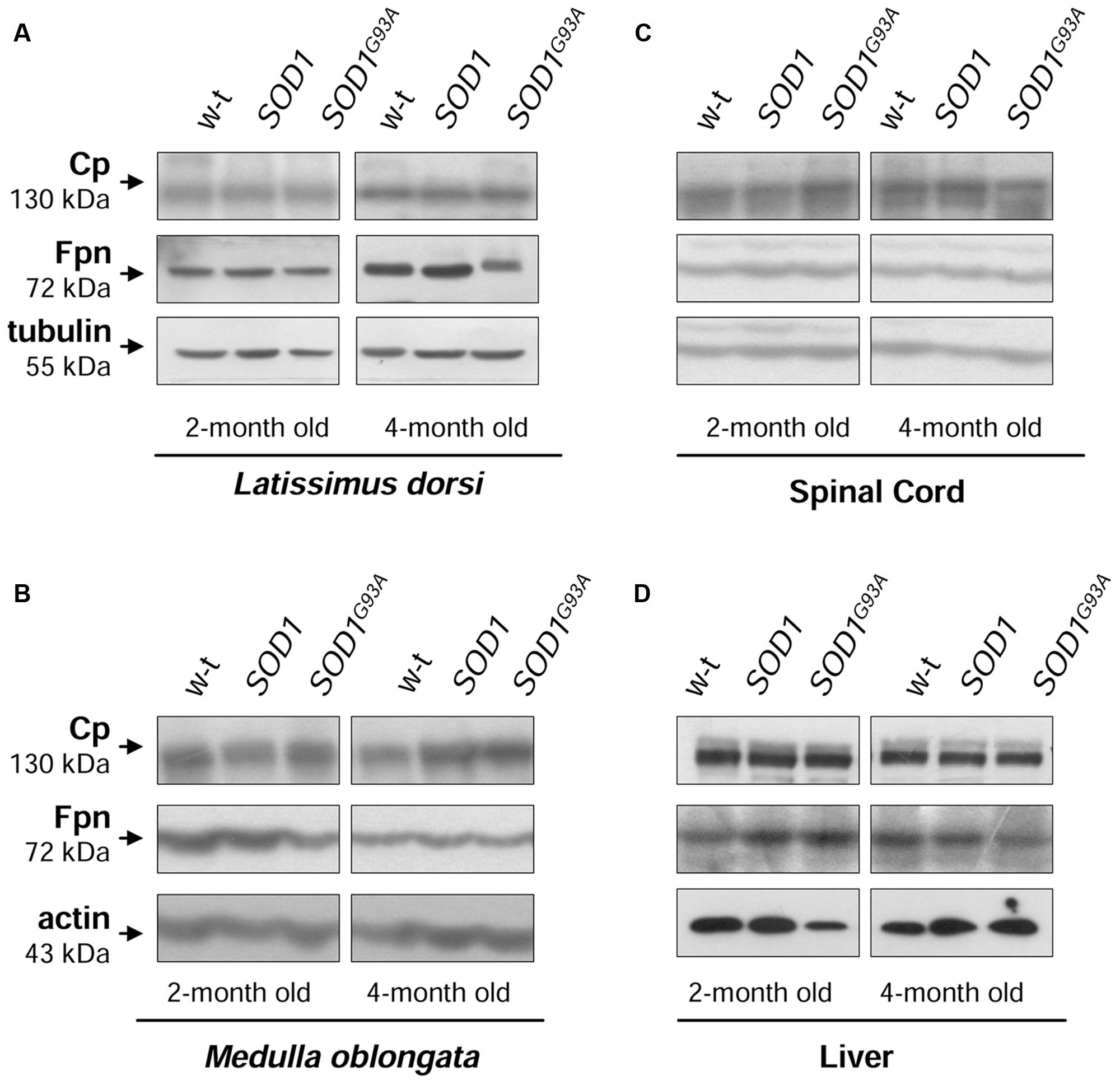
FIGURE 7. Analysis of Cp and Fpn protein levels in tissues of experimental mice. Western blot analysis of Cp and Fpn protein levels in the Latissimus dorsi muscle (A), neuronal tissues (B,C), and liver (D) membrane extracts Tubulin and actin were used as loading controls. Data are representative for three independent Western blot analyses performed using extracts from three mice of each experimental group). The intensity of the IRP1 bands (relative to the intensity of tubulin or actin bands) was quantified with a Molecular Imager using Quantity One software (Bio-Rad) and is shown in arbitrary units to present proteins levels (Supplementary Table).
Heme Oxygenase 1 (HO1) mRNA and Protein Levels are Specifically Increased in Muscles and Spinal Cord of Symptomatic SODG93A Mice
Activation of HO1, a heme-degrading, inducible enzyme responsive to a wide range of cellular stimuli, is considered to convey adaptive responses to various stress conditions including oxidative stress (Grochot-Przeczek et al., 2012). Surprisingly, HO1 expression has been intermittently investigated in ALS pathology (Dwyer et al., 1998). Here we show that Hmox1 gene is specifically induced at both mRNA and protein levels in muscles (Figures 8A,B) and spinal cord (Figure 8C) of symptomatic SODG93A mice. In the gastrocnemius muscle the up-regulation of Hmox1 gene was already observed in asymptomatic mice (Figure 8A). Importantly, in the liver of SOD1G93A mice, tissue not affected by ALS pathology, we did not observe any HO1 up-regulation, neither at the mRNA nor at the protein levels (Figure 8D).
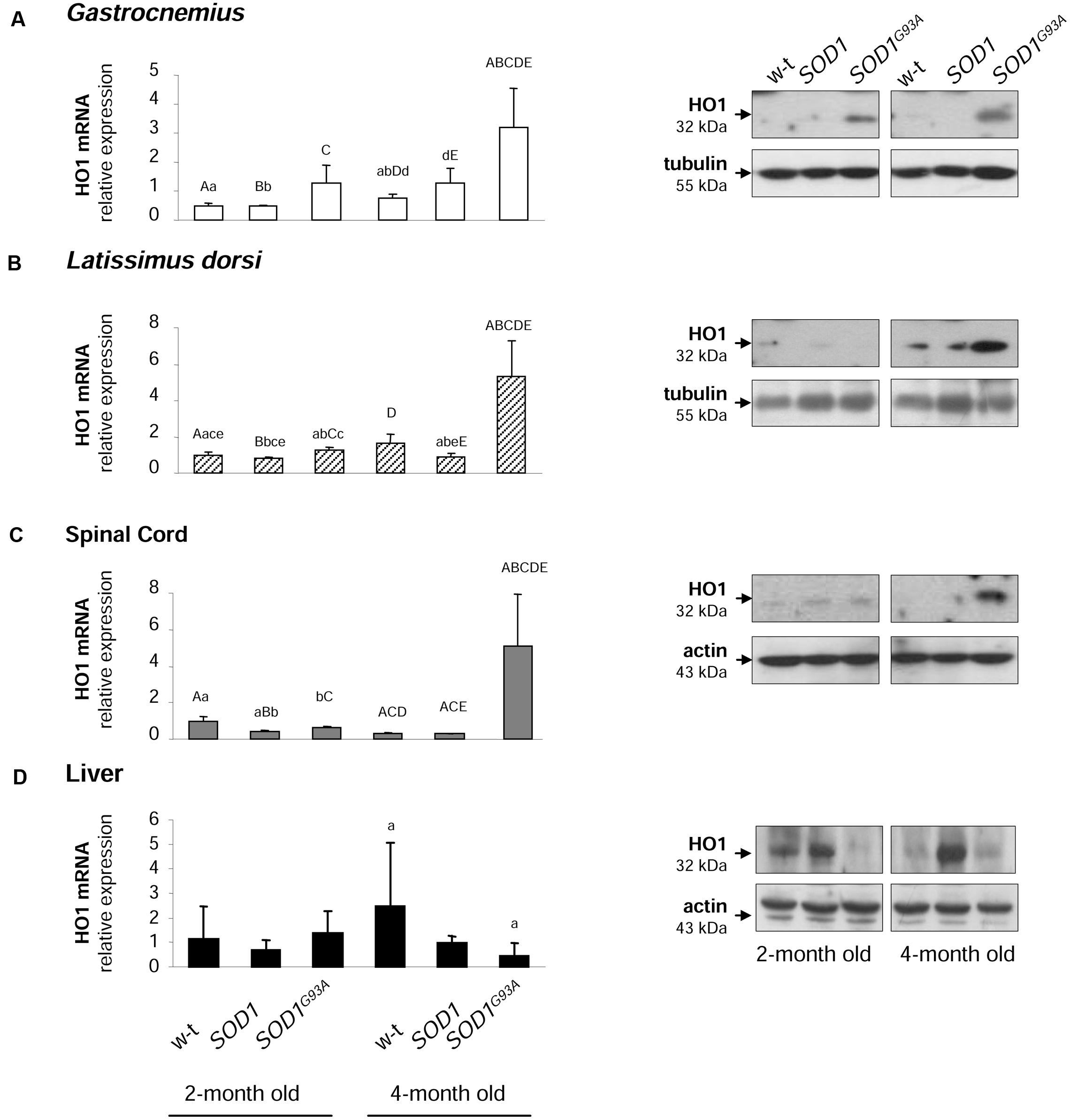
FIGURE 8. Analysis of HO1 mRNA abundance and protein levels in tissues of experimental mice. HO1 transcript abundance was measured in skeletal muscle (A,B), neuronal tissues (C), and liver (D) (left-hand) by real-time RT-PCR as described in M&M. The histograms display HO1 mRNA levels after normalization to 18S ribosomal RNA levels. Each column represents the mean ± SD of three biological amplification reactions. RNA from each mouse was extracted separately using TRIZOL reagent, reverse-transcribed and amplified as described in M&M; therefore, 18 cDNA samples (biological triplicates for each condition) were finally used in the analysis of gene expression in each tissue. The same capital and small letter indicates groups between which statistically significant differences were found at P < 0.01 and P < 0.05, respectively. Western blot analysis of HO1 protein levels was performed in membrane extracts obtained from the above-mentioned tissues (right-hand). Data are representative for three independent Western blot analyses performed using extracts from three mice of each experimental group. The intensity of the HO1 bands (relative to the intensity of tubulin or actin bands) was quantified with a molecular Imager using Quantity One software (Bio-Rad) and is shown in arbitrary units to present protein level. Results are expressed as mean ± SD for three mice in each experimental group.
Discussion
Among antioxidant enzymes the family of ubiquitous SODs deserves special attention because the reaction catalyzed by SODs converts one ROS (O2⋅-) to another (H2O2), and thus these enzymes play a subtle role in the regulation of ROS balance (Zelko et al., 2002). Accordingly, both deficiency and overexpression of Cu,Zn-SOD (SOD1), mostly cytosolic enzyme, induce oxidative stress in a wide range of organisms (Reaume et al., 1996; Lee et al., 2001; Missirlis et al., 2003; Starzyński et al., 2009). On the other hand, it is tempting to propose that enhanced SOD1 activity may lead to the excessive H2O2 formation, and subsequent iron-related exacerbation of oxidative stress through the redox reaction of H2O2 with ferrous iron but also through the H2O2-dependent disturbance of iron homeostasis regulatory mechanisms (Orino et al., 2001).
Identification of mutations in SOD1 gene as causative ones in familial amyotrophic lateral sclerosis (fALS) (Rosen et al., 1993) was immediately followed by the generation of transgenic mice constitutively overexpressing mutated human SOD1 (Gurney et al., 1994). Nowadays these animals are widely used in ALS research (Turner and Talbot, 2008). However, investigation of some pathogenic aspects of this disease in rodent models of mutant SOD1-mediated fALS requires special consideration of increased SOD1 activity, which does not occur in human pathology. It seems that this remark is of particular importance when studying a potential role of iron in the pathogenesis of ALS. Misregulated iron homeostasis that promotes excessive oxidative stress in the motor neurons has been postulated to contribute to the disease pathogenesis (Carri et al., 2003). This proposal is now strengthened by several lines of evidence coming from studies on ALS patients (Kasarskis et al., 1995; Mitchell et al., 2010; Kwan et al., 2012; Ignjatović, 2013), animal ALS models (Jeong et al., 2009; Wang et al., 2011; Lee et al., 2015) and cellular ALS models (Danzeisen et al., 2006; Hadzhieva et al., 2013). Transgenic SOD1G93A (Wang et al., 2011; Lee et al., 2015) and SOD1G37R (Jeong et al., 2009) mice, abundantly expressing active mutant SOD1 and showing fast and slow progression of the disease, respectively, are the most intensely studied in vivo models for investigating the contribution of iron to the pathogenesis of ALS. However, in these studies the results obtained from mice overexpressing mutated SOD1 gene were not compared with those from transgenic mice overexpressing human wild-type SOD1.
In our study, we adopted an experimental approach aimed at comparing changes in iron metabolism in 2-month-old and 4-month-old age-matched mice carrying mutated SOD1G93A and non-mutated human SOD1 genes, as well as in control non-transgenic mice. We show that in all analyzed tissues the expression and SOD1 activity was similar in both SOD1G93A and SOD1 mice and highly raised in both of the transgenic strains in comparison to the wild-type animals. Our results stay in accordance with the prediction of a high copy number of the transgene in SOD1G93A mice (Gurney et al., 1994). Considering the evidence that skeletal muscle weakness and atrophy, followed by paralysis is a final major cause of disability and death in ALS (Pratt et al., 2012; Pansarasa et al., 2014), we first wanted to determine histological hallmarks of myopathology in mice from three experimental groups. Importantly, we found pathological changes in the gastrocnemius muscle morphology only in symptomatic SOD1G93A mice, which indicates that the enhanced activity of SOD1 in those animals is not sufficient for the induction of skeletal muscle pathology, and which confirms progressive character of the disease. Our findings corroborate results of a comprehensive examination of various aspects of muscle pathology from animal SOD1 models of ALS (Pansarasa et al., 2014) and from clinically confirmed ALS patients (Léger et al., 2006). The impairment of iron metabolism that leads to excessive accumulation of this metal in tissues altered in ALS has been mainly demonstrated in relation to neuronal system (Kasarskis et al., 1995; Kwan et al., 2012; Ignjatović, 2013). Here, we provide histological evidence that among mice from 3 experimental groups analyzed at two stages of the disease, deposits of iron were strictly specific for skeletal muscles of symptomatic SOD1G93A animals. Accordingly, progressive increase in muscle iron content has been shown in SOD1G93A rats from the disease onset up to the end-stage of ALS (Halon et al., 2014).
Enhanced ferritin level reflects iron accumulation but may be also considered as an element of the adaptive response to oxidative stress that limits the bioavailability of the free iron, which participates in the generation of ⋅OH. Our analysis of H- and L-ferritin expression clearly shows that only H-Ft is upregulated at the protein level in SOD1G93A mice, not only in tissues primarily affected by ALS (muscles, M. oblongata, and spinal cord) but also in the liver, a tissue, which is not directly related to the ALS pathology. Unexpectedly, similar increase in H ferritin chains expression was also observed in all analyzed tissues of mice that overexpress a wild-type human SOD1 gene. This uniform ferritin upregulation in SOD1G93A and SOD1 mice strongly suggests that it results from oxidative stress induced by increased SOD1 activity. Although increased ferritin expression has been reported in skeletal muscles of SOD1G93A rats (Halon et al., 2010, 2014), in spinal cord of SOD1G93A mice (Massignan et al., 2007) and microglia of SOD1G37R mice (Jeong et al., 2009), the question of the specificity of this upregulation associated with ALS pathology remains uncertain as in all those studies only wild-type animals were used as controls. Ferritin is commonly considered as a hallmark of cellular iron status, and an increase in ferritin mirrors iron accumulation. Our observation of both iron deposits and high ferritin levels in the gastrocnemius muscle of symptomatic SOD1G93A mice seems to confirm this rule. On the other hand, as shown in this study, high level of cytosolic ferritin in the gastrocnemius muscle of SOD1 mice is not associated with detectable iron deposits and, as shown by others, iron accumulation in neurons of SOD1G37R mice is not correlated with any increase in intracellular ferritin content (Jeong et al., 2009). All these data strongly suggest that ferritin may not be a valuable indicator of intracellular iron status in SOD1 ALS models. Regulation of ferritin subunits is mediated by various factors at various regulatory levels such as transcription (Torti and Torti, 2002), translation (Wilkinson and Pantopoulos, 2014) and protein stability (De Domenico et al., 2006). In addition, both ferritin subunits are up-regulated by iron via iron regulatory proteins (IRP1 and IRP2) (Wilkinson and Pantopoulos, 2014). We have previously shown that IRP1 protein level is strongly (by 80%) down-regulated in SOD1 knockout mice (Starzyński et al., 2005). Here, we demonstrate that under the opposite conditions of a marked increase in SOD1 activity – in SOD1G93A and SOD1 mice, IRP1 level is unchanged. Accordingly, analysis of IRP1 in the spinal cord of SOD1G93A mice (Jeong et al., 2009) and in U373 MG glial cells overexpressing SOD1G93A and wild-type SOD1 (Danzeisen et al., 2006) show no changes in IRP1 protein expression (based on the EMSA assay with 2% ME). In contrast to these findings, proteomic analysis of the spinal cord of presymptomatic ALS SOD1G93A mice demonstrated nearly threefold reduction in IRP1 protein level when compared to wild-type mice. However, this decrease has not been validated by direct evaluation of IRP1 level by Western blot analysis (Massignan et al., 2007). Interestingly, the two aforementioned studies (Danzeisen et al., 2006; Jeong et al., 2009) show that increased activity of SOD1 results in the activation of IRP1, i.e., in inducing its ability to bind to the IRE (iron responsive element) sequences contained in mRNAs encoding several iron-related proteins such as ferritin chains, TfR1 and Fpn (Wilkinson and Pantopoulos, 2014). It is tempting to propose that such regulation of IRP1 IRE-binding activity is mediated by H2O2 (Martins et al., 1995; Pantopoulos and Hentze, 1995) produced under conditions of increased SOD1 activity. Nevertheless, according to the principles of the IRP/IRE system, this increased binding of IRP1 to the IREs located in the 5′UTR of H-Ft, L-Ft and Fpn mRNAs, should inhibit their translation (Wilkinson and Pantopoulos, 2014). Therefore, considering that ferritin has been shown to be increased in animals and cells overexpressing SOD1G93A, multiple but yet unknown mechanisms may override the IRP1-mediated Ft inhibition in ALS.
TfR1 is a cell surface protein, providing iron bound to transferrin to the cells via endocytosis (Pantopoulos et al., 2012). Its expression is regulated by the IRP/IRE system in an opposite manner to Ft and Fpn. This means that activated IRP1 stabilizes TfR1 transcript by binding to several IRE motifs located in its 3′UTR (Wilkinson and Pantopoulos, 2014). The expression of TfR1 transcript in all analyzed ALS target tissues, such as skeletal muscles (gastrocnemius and L. dorsi) and spinal cord of SOD1G93A mice shows the tendency to be decreased when compared with SOD1 and WT animals. Results of previous studies showing the TfR1 expression in SOD1 ALS animal and cellular models are not conclusive: cellular studies report the increase of TfR1 at both mRNA and protein levels (Danzeisen et al., 2006; Hadzhieva et al., 2013), but TfR1 protein expression in cervical cord of SOD1G37R mice has been shown unchanged (Jeong et al., 2009). Again, as in the case of Ft, the increase in TfR1 levels were reported in human glioblastoma astrocytoma cells overexpressing mutated and WT SOD1 gene, which denotes no specificity of the regulation for the ALS pathology (Danzeisen et al., 2006).
An alternatively spliced, glycosylphosphatidylinositol (GPI) anchored form of Cp is a copper-dependent ferroxidase that oxidizes ferrous iron exported from the cells by Fpn, and thus enables binding of ferric iron to transferrin. The cooperation of this protein tandem is crucial for protecting the central nervous (CNS) system from toxic iron deposition. Accordingly, aceruloplasminemia in humans results in iron accumulation in the CNS and neurodegeneration (Hellman and Gitlin, 2002). Likewise, reduced Fpn expression was also associated with increased iron content in central nervous system cells under inflammatory conditions (Urrutia et al., 2013). Here, we show that neither the expression of GPI-Cp nor that of Fpn is altered in skeletal muscles and spinal cord of SOD1G93A mice. A recent study showed Fpn decrease in the soleus but not in the tibialis muscle of SOD1G93A rats (Halon et al., 2014). Authors argue that Fpn downregulation in the soleus muscle is due to the increased hepcidin, a peptide that binds Fpn and induces its degradation (Ganz and Nemeth, 2012). However, they do not explain why the systemic effect of hepcidin activity is restricted only to the one type of muscles. As regards Cp, to our knowledge, the expression of membrane anchored form was not studied in ALS, however, increased concentration of its soluble form (sCp, especially relative abundance of more basic sCp isoforms) has been reported in cerebrospinal fluid of ALS patients (Conti et al., 2008). Although serum Cp pattern has been proposed as disease feature, our results do not support the role of Cp in ALS pathogenesis.
The rationale for analyzing the expression of HO1 encoded by Hmox1 gene in both SOD1 and SOD1G93A mice is that this ubiquitous enzyme is induced by a great number of stress stimuli (including oxidative stress) and that is strictly connected with intracellular and systemic iron metabolism (Kovtunovych et al., 2010; Starzyński et al., 2013). HO1 mediates a broad range of beneficial effects by catalyzing the first and rate-limiting step in the heme degradation pathway, resulting in the formation of biologically active molecules such iron, carbon monoxide (CO) and biliverdin (Grochot-Przeczek et al., 2012). In consequence, increased HO1 expression has been considered as a cytoprotective, adaptive cellular response under conditions of oxidative stress and inflammation occurring inter alia in several neurodegenerative diseases (Schipper et al., 2009). Importantly, in this study we have shown that HO1 expression is increased in tissues of the transgenic SOD1G93A mice. Among few genes fundamental for cellular iron homeostasis and analyzed in this study, the upregulation of Hmox1 gene seems to be strictly specific for skeletal muscles and neuronal tissues of symptomatic SOD1G93A mice. The only exception is gastrocnemius muscle, where HO1 protein level is increased also in asymptomatic mice overexpressing mutant SOD1. Of importance, this muscle, in contrast to the broadest dorsal muscle, is made up of mostly fast twitch (type II) muscle fibers, that are preferentially affected by ALS (Atkin et al., 2005). Therefore, it is tempting to speculate, that the oxidative stress precedes the onset of iron metabolism dysregulation in ALS. In contrast to our results, previous studies do not provide any evidence of increased HO1 expression in the spinal cord of SOD1G93A mice (Dwyer et al., 1998). The reason of this discrepancy is not known. Our analysis of Hmox1 gene expression in SOD1G93A mice includes the evaluation of both HO1 mRNA and protein levels in various tissues and unambiguously shows a homogenous pattern of Hmox1 gene induction. It has been postulated that cytoprotective effect of HO1 requires the co-induction of H-ferritin chain, which limits the pro-oxidant effect of the free iron released from the protoporphyrin IX ring of heme following HO1-mediated enzymatic reaction (Gozzelino and Soares, 2014). Coming back to the concept of the iron-mediated toxicity in ALS, it is tempting to propose that in SOD1G93A mice HO1 acts together with ferritin to limit the reactivity of heme-derived intracellular iron as it was originally demonstrated in endothelium under hemolytic conditions (Balla et al., 1992).
Author Contributions
AG supported the design of the study, performed the research, and analyzed data. AS designed and performed the research, analyzed data, and contributed to the writing of the paper. RS contributed to the conception of the work, performed the research and analyzed data. AB performed the research and analyzed data. ML supported the design of the study, performed the research, revised the work for important intellectual content. RS performed the research, analyzed data, and drafted the work. PL designed the study and wrote the paper. All authors read and approved the final manuscript.
Funding
This work was supported by NCN grant no. 2011/01/B/N223/00632.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fnmol.2015.00082
Abbreviations
ALS, amyotrophic lateral sclerosis; Cp, ceruloplasmin; Fpn, ferroportin; H-Ft, H feritin chain; L-Ft, L ferritin chain; HO1, heme oxygenase 1; IRP1, iron regulatory protein 1; SOD, superoxide dismutase; TfR1, transferrin receptor 1.
References
Atkin, J. D., Scott, R. L., West, J. M., Lopes, E., Quah, A. K., and Cheema, S. S. (2005). Properties of slow– and fast-twitch muscle fibres in a mouse model of amyotrophic lateral sclerosis. Neuromuscul. Disord. 2005, 377–388. doi: 10.1016/j.nmd.2005.02.005
Balla, G., Jacob, H. S., Balla, J., Rosenberg, M., Nath, K., Apple, F., et al. (1992). Ferritin: a cytoprotective antioxidant strategem of endothelium. J. Biol. Chem 267, 18148–18153.
Barber, S. C., Mead, R. J., and Shaw, P. J. (2006). Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 1762, 1051–1067. doi: 10.1016/j.bbadis.2006.03.008
Beauchamp, C., and Fridovich, I. (1971). Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 44, 276–287. doi: 10.1016/0003-2697(71)90370-8
Bouton, C., Chauveau, M. J., Lazereg, S., and Drapier, J. C. (2002). Recycling of RNA binding iron regulatory protein 1 into an aconitase after nitric oxide removal depends on mitochondrial ATP. J. Biol. Chem. 277, 31220–31227. doi: 10.1074/jbc.M203276200
Canonne-Hergaux, F., Gruenheid, S., Ponka, P., and Gros, P. (1999). Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood 93, 4406–4417.
Carri, M. T., Ferri, A., Cozzolino, M., Calabrese, L., and Rotilio, G. (2003). Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Brain Res. Bull. 61, 365–374. doi: 10.1016/S0361-9230(03)00179-5
Conti, A., Iannaccone, S., Sferrazza, B., De Monte, L., Cappa, S., Franciotta, D., et al. (2008). Differential expression of ceruloplasmin isoforms in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Proteomics Clin. Appl. 2, 1628–1637. doi: 10.1002/prca.200780081
Danzeisen, R., Achsel, T., Bederke, U., Cozzolino, M., Crosio, C., Ferri, A., et al. (2006). Superoxide dismutase 1 modulates expression of transferrin receptor. J. Biol. Inorg. Chem. 11, 489–498. doi: 10.1007/s00775-006-0099-4
De Domenico, I., Vaughn, M. B., Li, L., Bagley, D., Musci, G., Ward, D. M., et al. (2006). Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 25, 5396–5404. doi: 10.1038/sj.emboj.7601409
Dobrowolny, G., Aucello, M., Rizzuto, E., Beccafico, S., Mammucari, C., Boncompagni, S., et al. (2008). Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 8, 425–436. doi: 10.1016/j.cmet.2008.09.002
Dwyer, B. E., Lu, S. Y., and Nishimura, R. N. (1998). Heme oxygenase in the experimental ALS mouse. Exp. Neurol. 150, 206–212. doi: 10.1006/exnr.1997.6763
Ganz, T., and Nemeth, E. (2012). Hepcidin and iron homeostasis. Biochim. Biophys. Acta 1823, 1434–1443. doi: 10.1016/j.bbamcr.2012.01.014
Gozzelino, R., and Soares, M. P. (2014). Coupling heme and iron metabolism via ferritin H chain. Antioxid. Redox Signal. 20, 1754–1769. doi: 10.1089/ars.2013.5666
Grochot-Przeczek, A., Dulak, J., and Jozkowicz, A. (2012). Haem oxygenase-1: non-canonical roles in physiology and pathology. Clin. Sci. (Lond.) 122, 93–103. doi: 10.1042/CS20110147
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Hadzhieva, M., Kirches, E., and Mawrin, C. (2014). Review: iron metabolism and the role of iron in neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 40, 240–257. doi: 10.1111/nan.12096
Hadzhieva, M., Kirches, E., Wilisch-Neumann, A., Pachow, D., Wallesch, M., Schoenfeld, P., et al. (2013). Dysregulation of iron protein expression in the G93A model of amyotrophic lateral sclerosis. Neuroscience 230, 94–101. doi: 10.1016/j.neuroscience.2012.11.021
Halon, M., Kaczor, J. J., Ziolkowski, W., Flis, D. J., Borkowska, A., Popowska, U., et al. (2014). Changes in skeletal muscle iron metabolism outpace amyotrophic lateral sclerosis onset in transgenic rats bearing the G93A hmSOD1 gene mutation. Free Radic. Res. 48, 1363–1370. doi: 10.3109/10715762.2014.955484
Halon, M., Sielicka-Dudzin, A., Wozniak, M., Ziolkowski, W., Nyka, W., Herbik, M., et al. (2010). Up-regulation of ferritin ubiquitination in skeletal muscle of transgenic rats bearing the G93A hmSOD1 gene mutation. Neuromuscul. Disord. 20, 29–33. doi: 10.1016/j.nmd.2009.08.014
Harrison, P. M., and Arosio, P. (1996). The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1275, 161–203. doi: 10.1016/0005-2728(96)00022-9
Hellman, N. E., and Gitlin, J. D. (2002). Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 22, 439–458. doi: 10.1146/annurev.nutr.22.012502.114457
Ignjatović, A., Stević, Z., Lavrnić, S., Daković, M., and Bačić, G. (2013). Brain iron MRI: a biomarker for amyotrophic lateral sclerosis. J. Magn. Reson. Imaging 38, 1472–1479. doi: 10.1002/jmri.24121
Jeong, S. Y., Rathore, K. I., Schulz, K., Ponka, P., Arosio, P., and David, S. (2009). Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 29, 610–619. doi: 10.1523/JNEUROSCI.5443-08.2009
Kasarskis, E. J., Tandon, L., Lovell, M. A., and Ehmann, W. D. (1995). Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: a preliminary study. J. Neurol. Sci. 130, 203–208. doi: 10.1016/0022-510X(95)00037-3
Kovtunovych, G., Eckhaus, M. A., Ghosh, M. C., Ollivierre-Wilson, H., and Rouault, T. A. (2010). Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood 116, 6054–6062. doi: 10.1182/blood-2010-03-272138
Kupershmidt, L., Weinreb, O., Amit, T., Mandel, S., Carri, M. T., and Youdim, M. B. (2009). Neuroprotective and neuritogenic activities of novel multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 23, 3766–3779. doi: 10.1096/fj.09-130047
Kwan, J. Y., Jeong, S. Y., Van Gelderen, P., Deng, H. X., Quezado, M. M., Danielian, L. E., et al. (2012). Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PLoS ONE 7:e35241. doi: 10.1371/journal.pone.0035241
Lee, J. K., Shin, J. H., Gwag, B. J., and Choi, E. J. (2015). Iron accumulation promotes TACE-mediated TNF-α secretion and neurodegeneration in a mouse model of ALS. Neurobiol. Dis. 80, 63–69. doi: 10.1016/j.nbd.2015.05.009
Lee, M. H., Hyun, D.-H., Jenner, P., and Halliwell, B. (2001). Effect of overexpression of wild-type and mutant Cu/Zn-superoxide dismutases on oxidative damage and antioxidant defences: relevance to Down’s syndrome and familial amyotrophic lateral sclerosis. J. Neurochem. 76, 957–965. doi: 10.1046/j.1471-4159.2001.00107.x
Léger, B., Vergani, L., Sorarù, G., Hespel, P., Derave, W., Gobelet, C., et al. (2006). Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J. 20, 583–585.
Lipiński, P., Drapier, J. C., Oliveira, L., Retmańska, H., Sochanowicz, B., and Kruszewski, M. (2000). Intracellular iron status as a hallmark of mammalian cell susceptibility to oxidative stress: a study of L5178Y mouse lymphoma cell lines differentially sensitive to H(2)O(2). Blood 95, 2960–2966.
Martins, E. A., Robalinho, R. L., and Meneghini, R. (1995). Oxidative stress induces activation of a cytosolic protein responsible for control of iron uptake. Arch. Biochem. Biophys. 316, 128–134. doi: 10.1006/abbi.1995.1019
Massignan, T., Casoni, F., Basso, M., Stefanazzi, P., Biasini, E., Tortarolo, M., et al. (2007). Proteomic analysis of spinal cord of presymptomatic amyotrophic lateral sclerosis G93A SOD1 mouse. Biochem. Biophys. Res. Commun. 353, 719–725. doi: 10.1016/j.bbrc.2006.12.075
McCord, J. M., and Fridovich, I. (1969). Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244, 6049–6055.
Missirlis, F., Hu, J., Kirby, K., Hilliker, A. J., Rouault, T. A., and Phillips, J. (2003). Compartment-specific protection of iron-sulfur proteins by superoxide dismutase. J. Biol. Chem. 278, 47365–47369. doi: 10.1074/jbc.M307700200
Mitchell, R. M., Simmons, Z., Beard, J. L., Stephens, H. E., and Connor, J. R. (2010). Plasma biomarkers associated with ALS and their relationship to iron homeostasis. Muscle Nerve 42, 95–103. doi: 10.1002/mus.21625
Orino, K., Lehman, L., Tsuji, Y., Ayaki, H., Torti, S. V., and Torti, F. M. (2001). Ferritin and the response to oxidative stress. Biochem. J. 357, 241–247. doi: 10.1042/bj3570241
Oshiro, S., Morioka, M. S., and Kikuchi, M. (2011). Dysregulation of iron metabolism in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Adv. Pharmacol. Sci. 2011:378278. doi: 10.1155/2011/378278
Pansarasa, O., Rossi, D., Berardinelli, A., and Cereda, C. (2014). Amyotrophic lateral sclerosis and skeletal muscle: an update. Mol. Neurobiol. 49, 984–990. doi: 10.1007/s12035-013-8578-4
Pantopoulos, K., and Hentze, M. W. (1995). Rapid responses to oxidative stress mediated by iron regulatory protein. EMBO J. 14, 2917–2924.
Pantopoulos, K., Porwal, S. K., Tartakoff, A., and Devireddy, L. (2012). Mechanisms of mammalian iron homeostasis. Biochemistry 51, 5705–5724. doi: 10.1021/bi300752r
Pratt, A. J., Getzoff, E. D., and Perry, J. J. (2012). Amyotrophic lateral sclerosis: update and new developments. Degener. Neurol. Neuromuscul. Dis. 2012, 1–14.
Reaume, A. G., Elliott, J. L., Hoffman, E. K., Kowall, N. W., Ferrante, R. J., Siwek, D. F., et al. (1996). Motor neurons in Cu/Zn superoxide dismutase deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 13, 43–47. doi: 10.1038/ng0596-43
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rothstein, J. D. (2009). Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 65(Suppl. 1), S3–S9. doi: 10.1002/ana.21543
Salin, M. L., and Bridges, S. M. (1981). Absence of the iron-containing superoxide dismutase in mitochondria from mustard (Brassica campestris). Biochem. J. 195, 229–233. doi: 10.1042/bj1950229
Schipper, H. M., Song, W., Zukor, H., Hascalovici, J. R., and Zeligman, D. (2009). Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. J. Neurochem. 110, 469–485. doi: 10.1111/j.1471-4159.2009.06160.x
Starzyński, R. R., Canonne-Hergaux, F., Lenartowicz, M., Krzeptowski, W., Willemetz, A., Styś, A., et al. (2013). Ferroportin expression in haem oxygenase 1-deficient mice. Biochem. J. 449, 69–78. doi: 10.1042/BJ20121139
Starzyński, R. R., Canonne-Hergaux, F., Willemetz, A., Gralak, M. A., Woliński, J., Styś, A., et al. (2009). Haemolytic anaemia and alterations in hepatic iron metabolism in aged mice lacking Cu,Zn-superoxide dismutase. Biochem. J. 420, 383–390. doi: 10.1042/BJ20082137
Starzyński, R. R., Gonçalves, A. S., Muzeau, F., Tyrolczyk, Z., Smuda, E., Drapier, J. C., et al. (2006). STAT5 proteins are involved in down-regulation of iron regulatory protein 1 gene expression by nitric oxide. Biochem. J. 400, 367–375. doi: 10.1042/BJ20060623
Starzyński, R. R., Lipiński, P., Drapier, J. C., Diet, A., Smuda, E., Bartłomiejczyk, T., et al. (2005). Down-regulation of iron regulatory protein 1 activities and expression in superoxide dismutase 1 knock-out mice is not associated with alterations in iron metabolism. J. Biol. Chem. 280, 4207–4212. doi: 10.1074/jbc.M411055200
Torti, F. M., and Torti, S. V. (2002). Regulation of ferritin genes and protein. Blood 99, 3505–3516. doi: 10.1182/blood.V99.10.3505
Turner, B. J., and Talbot, K. (2008). Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 85, 94–134. doi: 10.1016/j.pneurobio.2008.01.001
Urrutia, P., Aguirre, P., Esparza, A., Tapia, V., Mena, N. P., Arredondo, M., et al. (2013). Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 126, 541–549. doi: 10.1111/jnc.12244
Wang, Q., Zhang, X., Chen, S., Zhang, X., Zhang, S., Youdium, M., et al. (2011). Prevention of motor neuron degeneration by novel iron chelators in SOD1(G93A) transgenic mice of amyotrophic lateral sclerosis. Neurodegener. Dis. 8, 310–321. doi: 10.1159/000323469
Wilkinson, N., and Pantopoulos, K. (2014). The IRP/IRE system in vivo: insights from mouse models. Front. Pharmacol. 5:176. doi: 10.3389/fphar.2014.00176
Keywords: ALS, SOD1, G93A, neurons, iron, skeletal muscle, heme oxygenase 1, oxidative stress
Citation: Gajowiak A, Styś A, Starzyński RR, Bednarz A, Lenartowicz M, Staroń R and Lipiński P (2016) Mice Overexpressing Both Non-Mutated Human SOD1 and Mutated SOD1G93A Genes: A Competent Experimental Model for Studying Iron Metabolism in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 8:82. doi: 10.3389/fnmol.2015.00082
Received: 23 September 2015; Accepted: 11 December 2015;
Published: 06 January 2016.
Edited by:
Torben Moos, Aalborg University, DenmarkReviewed by:
Sandra Blaess, University of Bonn, GermanyAndreas Martin Grabrucker, Ulm University, Germany
Copyright © 2016 Gajowiak, Styś, Starzyński, Bednarz, Lenartowicz, Staroń and Lipiński. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paweł Lipiński, p.lipinski@ighz.pl; Rafał R. Starzyński, r.starzynski@ighz.pl