 Zdravka Ivanova1
Zdravka Ivanova1 Gaurav Sablok2
Gaurav Sablok2 Evelina Daskalova1
Evelina Daskalova1 Gergana Zahmanova1
Gergana Zahmanova1 Elena Apostolova1Galina Yahubyan1
Elena Apostolova1Galina Yahubyan1 Vesselin Baev1*
Vesselin Baev1*- 1Department of Plant Physiology and Molecular Biology, University of Plovdiv, Plovdiv, Bulgaria
- 2Plant Functional Biology and Climate Change Cluster, University of Technology at Sydney, Sydney, NSW, Australia
Haberlea rhodopensis is a paleolithic tertiary relict species, best known as a resurrection plant with remarkable tolerance to desiccation. When exposed to severe drought stress, H. rhodopensis shows an ability to maintain the structural integrity of its photosynthetic apparatus, which re-activates easily upon rehydration. We present here the results from the assembly and annotation of the chloroplast (cp) genome of H. rhodopensis, which was further subjected to comparative analysis with the cp genomes of closely related species. H. rhodopensis showed a cp genome size of 153,099 bp, harboring a pair of inverted repeats (IR) of 25,415 bp separated by small and large copy regions (SSC and LSC) of 17,826 and 84,443 bp. The genome structure, gene order, GC content and codon usage are similar to those of the typical angiosperm cp genomes. The genome hosts 137 genes representing 70.66% of the plastome, which includes 86 protein-coding genes, 36 tRNAs, and 4 rRNAs. A comparative plastome analysis with other closely related Lamiales members revealed conserved gene order in the IR and LSC/SSC regions. A phylogenetic analysis based on protein-coding genes from 33 species defines this species as belonging to the Gesneriaceae family. From an evolutionary point of view, a site-specific selection analysis detected positively selected sites in 17 genes, most of which are involved in photosynthesis (e.g., rbcL, ndhF, accD, atpE, etc.). The observed codon substitutions may be interpreted as being a consequence of molecular adaptation to drought stress, which ensures an evolutionary advantage to H. rhodopensis.
Introduction
Chloroplasts are uniparentally inherited organelles in plant cells; they play an important role in many plant cell functions, including photosynthesis, carbon fixation, and stress response. In angiosperms, the chloroplast genome has a conserved quadripartite structure composed of two copies of inverted repeat (IR), one large single copy (LSC), and one small single copy (SSC) (Palmer, 1985). In contrast, extensive loss of the IR copies has been observed in gymnosperms (Wu et al., 2011). Although chloroplast shows evolutionary conservation across the tree of life, an accelerated rate of evolution has been widely observed in particular genes. For example, rbcL, which encodes the large subunit of ribulose-1,5-bisphosphate carboxylase/oxy-genase (RUBISCO) has been shown to play a fundamental role in light-dark state transitions (Morton and Clegg, 1993). It is worth mentioning that in addition to rbcL, chloroplast-encoded low molecular mass subunits of Photosystem II (PSII), including psbI, psbJ, psbL, psbM, and psbTc (Umate et al., 2008) and psbA mRNA translation (Kim and Mullet, 1994), are also under the influence of light transitions. Taking into account these interconnections, it can be assumed that chloroplast represents a major organelle that can be very important when studying the role of desiccation stress.
Genomic organization of plastome in photosynthetic plants comprises up to 88 protein coding genes and, in most eudicots, about 35 structural RNA genes, totaling 100–120 unique genes (Wicke et al., 2011). After the acquisition of chloroplasts, many genes relocated from the ancestral organellar genomes to the nucleus. As remodeled nuclear copies of organelle genes usurped the functions of those located in the organelle, biochemical pathways were transferred entirely from the chloroplasts to the cytosol and the plastid genomes were reduced in size. The relentless influx of organelle DNA into the nucleus has resulted in a decreased organelle autonomy and increased nuclear complexity (Timmis et al., 2004). In turn, the nucleus, depends on signals coming from the chloroplasts that transfer information to the nucleus via “retrograde signaling.” This allows modification of the nuclear gene expression according to the status of the chloroplast (Nott et al., 2006). Besides having a vital role in cellular communication, retrograde signaling plays an important role in the adaptive responses of plants to stress (Sun and Guo, 2016).
Haberlea rhodopensis Friv. belonging of the Gesneriaceae family is a homoiochlorophyllous plant that retains chlorophyll in a readily recoverable form throughout desiccation (Georgieva et al., 2005) and is a tertiary relic species, endemic to the Balkan peninsula. In addition to its homoiochlorophyllous nature, H. rhodopensis has the capability of resurrection (survival of extreme vegetative dehydration), a trait that is of significant importance in global climate change. Desiccation tolerance is one of the most widely described traits studied in this paleoendemic species, and previous studies have shown that light absorption and oxygen intake evolution play a key role in the adaptation of this species to desiccation stress (Heber et al., 2007; Heber, 2012). Drought resistance and rapid recovery of H. rhodopensis after rehydration were attributed to specific characteristics of the chloroplast of this species, including unchanged chlorophyll content, maintenance of chlorophyll–protein complexes, reversible modifications in PSII electron transport, and enhanced dissipation of non-radiative energy (Georgieva et al., 2007; Mihailova et al., 2011). From an evolutionary point of view, the origin of European Gesneriaceae genera has been dated back to the early Oligocene, while the Haberlea lineage emerged in the late Oligocene as suggested by population genetic (ISSRs) analyses of the chloroplast encoded atpB-rbcL. trnH-psbA, and trnL-F genes (Petrova et al., 2015). Despite its importance as a resurrection plant, there is a lack of studies using the chloroplast genome of Haberlea lineage to understand its molecular evolution and resolve the phylogenetic position of H. rhodopensis with respect to Lamiales.
In the present paper, we reconstruct the whole chloroplast genome by using next-generation sequencing and applying a combination of de novo and reference-guided assembly. This will help to delineate the phylogenetic position of this species and to understand the role of natural selection in the adaptation of H. rhodopensis to drought stress. In this study, we report on a 153,099 bp plastome of H. rhodopensis, analyze the genomic features and structure of its genome, and conduct comparative genomic studies to inform an improved understanding of the organelle genome evolution of this resurrection plant.
Materials and Methods
DNA Extraction and Sequencing
Haberlea rhodopensis samples were collected from Rhodopi mountain, Bulgaria (location 42°1′N 24°52′E). Chloroplast DNA was isolated from leaf tissue of 16 individual plants. For an optimal yield of intact chloroplasts, 40/80% Percoll gradient (Chloroplast Isolation Kit – Sigma-Aldrich) was used. Chloroplast DNA was extracted using DNeasy Plant Mini Kit (QIAGEN). Two biological replicates were performed. Library preparation and sequencing were performed at BGI-Shenzhen, China. For each replicate, the isolated DNA was used to generate 100-bp paired-end (PE) libraries with insert size of 170 bp, in accordance with the Illumina Hiseq2000 standard protocol. In our case, the 100 bp paired-end reads are overlapping approximately with 30 bp, thus producing longer (joined) fragments of about 170 bp, corresponding to the insert size. This method of joined pair-end reads was used as it increases the accuracy of the assembly.
Genome Assembly
Prior to de novo genome assembly, raw reads were mapped to the NCBI Viridiplantae chloroplast genomes using BWA to filter the non-chloroplastic reads (Li and Durbin, 2010) in order to avoid contamination of mtDNA and nuclear DNA. Quality assessment of extracted chloroplast reads was performed by FASTX-Toolkit1. High-quality paired-end reads were merged by FLASH (Magoc and Salzberg, 2011). Length distribution and quality of the merged reads are presented in Supplementary Data Sheet S1. Subsequently merged reads were assembled using Abyss version 1.9.0 with k-mer = 55 in single-end mode. Assembly N50, L50 and related statistics were evaluated using QUAST (Gurevich et al., 2013) and assemblies with the most consistent results in terms of assembly evaluation parameters (N50 and L50) retained. QUAST results are shown in Supplementary Data Sheet S1. Assembled contigs were scaffolded using SSPACE 3.0 (Boetzer et al., 2011). Re-ordering of draft genome scaffolds based on comparison to the reference genome of Boea hygrometrica was performed using Abacas 1.3.1 (Assefa et al., 2009) and MUMmer (Kurtz et al., 2004).
Due to the limitations in the assembly process, a single fragment with twice the coverage is expected in the case of duplicated copies. After the identification, assembly, and scaffolding of such a fragment (a contig constituting an IR region), Abacas 1.3.1 was used to align, order and orientate all currently assembled contigs based on the reference plastome of Boea hygrometrica. The resulting output was a pseudomolecule of the draft chloroplast genome, where the IR region contig was represented twice. The key step in this standard approach is that a reference from a closely related species is used (e.g., B. hygrometrica), which ensures that the IR regions are accurately identified and ordered. Subsequently, gaps were filled by GapFiller (Nadalin et al., 2012) with three rounds of iterations. Finished chloroplast assembly was re-aligned to B. hygrometrica to confirm the quadripartite structure representing – LSC, SSC, and two IRs.
Genome Annotation of the Haberlea rhodopensis Chloroplast Genome and Comparative Plastomics
The assembled chloroplast sequence was annotated using Dual Organellar Genome Annotator (DOGMA) (Wyman et al., 2004) with manual start and stop codon validation by using the Sequin tool from NCBI2. The nomenclature of cp genes was used according to Chloroplast Genome Database3 and previously published cp genomes. Transfer RNA (tRNA) genes were identified with DOGMA and the tRNAscan-SE program ver. 1.21 (Schattner et al., 2005). Circular representation of the H. rhodopensis chloroplast genome was generated with OGDRAW tool (Lohse et al., 2013). The complete cp genome of H. rhodopensis was compared with the cp genomes of B. hygrometrica, Lavandula angustifolia, Rosmarinus officinalis, Sesamum indicum, Salvia miltiorrhiza, and Olea europaea using mVista in Shuffle-LAGAN mode and Blast Ring Image Generator (BRIG). H. rhodopensis was set as a reference. The comparison of IR/LSC and IR/SSC regions was conducted using the GenBank genome files for Boea hygrometrica (NC_016468.1), Olea europaea (NC_013707.2) and Sesamum indicum (NC_016433.2) with coordinates for the gene features. IR/SSC border coordinates were obtained from previous studies (Mariotti et al., 2010; Zhang et al., 2012, 2013). For the identification of perfect and compound simple sequence repeats (SSRs), MISA4 was used with a minimum repetitive stretch of 10 nucleotides as mono-, a consecutive stretch of four repeats units to be classified as di- and tri-, and a stretch of three repeat units for each tetra-, penta-, and hexa nucleotide stretches as SSRs. For the identification of the dispersed repeats including the forward and palindromic repeats, REPUTER (Kurtz et al., 2001) was used with parameters (repfind -f -p -l 30 -h 3 -best 10000) and the identified repeat regions are checked with the corresponding genomic coordinates and were annotated.
Molecular Evolution Analysis
For the identification of codon usage patterns, all coding sequences (CDSs) shorter than 300 bp were removed. Filtered CDSs were subsequently used for the estimation of codon usage using CodonW5 with translational table = 11. In addition to the overall codon usage, we further tabulated additional codon usage measures such as Nc (effective number of codons), GC3s (frequency of the GC at the third synonymous position) (Hu et al., 2015). All the calculations of the GC at the first, second, and third position as defined by GC, GC1, GC2, and GC3, respectively, was done using in-house PERL scripts. Estimation of the standard effective number of codon (Nc) was tabulated using the equation N(c) = 2 + s + 29/[s(2) + (1 – s)(2)], where s denotes GC3s (Wright, 1990). Ka/Ks value for each gene was calculated using the KaKs_calculator (Wang et al., 2010) with the following settings: genetic code table 11 (bacterial and plant plastid code); method of calculation: YN. In the results, the indication for Ka/Ks “NA” which appears when Ks = 0 (in cases with no substitutions in the alignment, or 100% match) was replaced in all cases with 0. Taking into account that KaKs Calculator estimates selection using model averaging, we also evaluated the role of the site-specific selection in 34 genes present across the 33 phylogenetically related species (Supplementary Table S1). For the identification of the site-specific selection, trimmed codon alignments were analyzed using Selecton (Stern et al., 2007), taking into account two models: M8 (model of positive selection) and M8a (null model) and likelihood scores estimated by models were evaluated using log-likelihood ratio test (LRT) with degree of freedom = 1. H. rhodopensis was used as a reference sequence to estimate the site-specific selection models in Selecton. We considered that genes with a p-value for the LRT below 5% (i.e., p-value < 0.05) to exhibit signatures of sites under positive selection.
Phylogenetic Profiling
For understanding evolution of the coding regions, codon alignment of the coding regions of H. rhodopensis and 32 other chloroplast genomes (Supplementary Table S1) for genes atpA. atpB. atpE. atpF. atpH. atpI, ndhC. ndhD. ndhE. ndhF. ndhH. ndhI. ndhJ, petA, petB, petD, petG, psaA, psaB, psaC, psaI, psbA, psbB, psbC, psbD, psbE, psbH, psbK, psbL, psbM, psbN, rbcL, rpoA, rpoB, rpoC2 was done using MACSE, which allows the identification of frameshift events (Ranwez et al., 2011). Following codons alignments, alignments were trimmed for the ambiguity and the concatenated alignment was analyzed for phylogenetic reconstruction using the IQTree by Maximum Likelihood method (Nguyen et al., 2015) with 1000 bootstrap replicates. Model selection for the phylogenetic assessment was done based on the Akaike Information Content (AIC) and corrected AIC. Bootstrap values were calculated using the in-built UFBoot within IQTree, and rapid bootstrap calculation and avoid less biased support values estimation (Minh et al., 2013).
RNA Editing
Prediction of the RNA editing events was done using the BLASTX prediction mode of PREPACT26 and all the 17 chloroplast reference genomes were selected to predict the RNA editing events. For the prediction of the RNA Editing events, we have kept only those sites which have 100% prediction probability and are represented across all reference genomes included in PREPACT 2.0. Black font indicates a pre-edited state and red font indicates an editing event to reconstitute a conserved codon in a given reference, respectively.
Data Deposition
The complete chloroplast genome of Haberlea rhodopensis has been submitted to GenBank with accession number KX657870. The raw Illumina reads were submitted to NCBI SRA archive in FASTQ format with accession numbers SRR4428742, SRR4428743, PRJNA348808.
Results and Discussion
Haberlea rhodopensis Chloroplast Genome Organization and Gene Content
The complete cp genome sequence of H. rhodopensis (submitted to GenBank under Acc. No KX657870) is 153,099 bp in length and has the common quadripartite structure found in most land plants. The genome includes a LSC of 84,443 bp (covering 55.2%) and a SSC of 17,826 bp (covering 11.6%), separated by two IRs of 25,415 bp (covering 33.20 %) (Figure 1). Overall, the H. rhodopensis cp genome CG content was 37.8%, with a higher GC content in the IRs regions (43.3%) than in LSC and SSC (35.7 and 31.7%, respectively). Most of the cp genome sequence encodes proteins, tRNAs and rRNAs (51.56, 1.78, and 5.90%, respectively). The remaining regions are non-CDSs, including introns, intergenic spacers and pseudogenes. There are in total 137 genes in the genome, including 86 protein-coding genes, 36 tRNAs genes, 4 rRNAs, 4 pseudogenes, and 21 duplicated genes (Table 1). Of the observed gene space in H. rhodopensis, 80 protein-coding genes, 29 tRNA, and 4 rRNAs were found to be unique while six protein-coding (rpl2, rpl23, ycf2, ycf15, ndhB, rps7), seven tRNAs (trnI-CAU,trnL-CAA, trnV-GAC,trnI-GAU, trnA-UGC,trnR-ACG, trnN-GUU), and four rRNA genes (rrn4.5, rrn5, rrn16, rrn23) were found be duplicated in IRa and IRb (Table 1). Moreover, we identified four pseudogenes (ycf1, ycf68, orf42, orf56). Among the genes that underwent pseudogenization, only ycf1 showed incomplete duplication (in the IRa and SSC junction region), the others – ycf68, orf42, and orf56 showed complete duplication in IRa/IRb suggesting a loss of function due to the accumulation of premature stop codons or truncations. The cp genome has 13 intron-containing genes, of which eight protein-coding genes (rps16, atpF, rpoC1, ycf3, clpP, rpl2, ndhB. ndhA) and five tRNA genes (trnK-UUU, trnL-UAA, trnV-UAC, trnI-GAU. trnA-UGC). Two of the intron-containing genes have two introns (clpP, ycf3) and the other 15 have only one intron (Table 2). In addition, four cases of overlapping genes were observed (psbD-psbC, ndhK-ndhC, atpE-atpB, ycf1-ndhF).
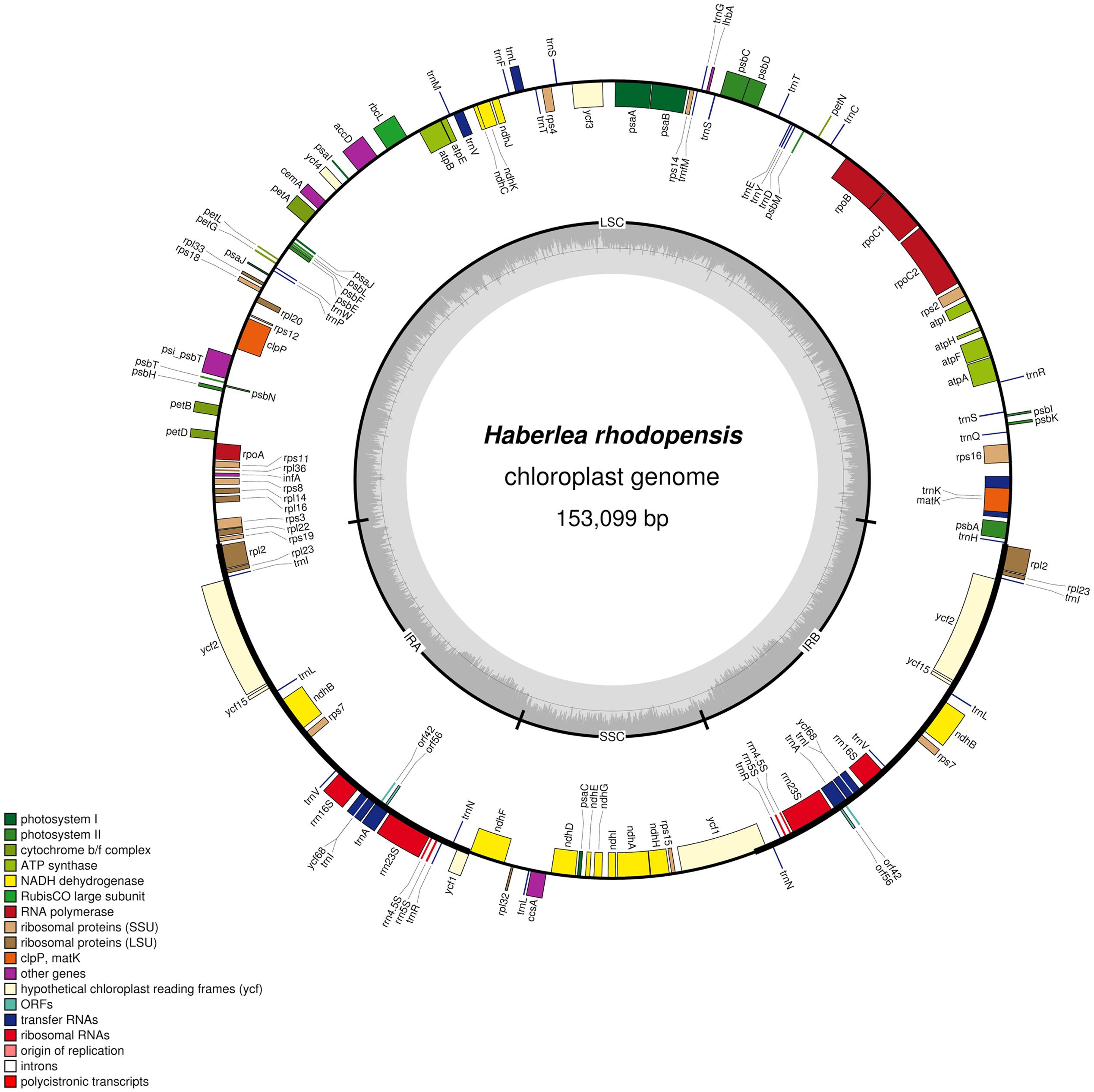
FIGURE 1. Circular map of the chloroplast genome of H. rhodopensis: Genes drawn within the circle are transcribed clockwise, while genes drawn outside are transcribed counterclockwise. Genes belonging to different functional groups are color coded. Dark bold lines show Inverted repeats (IRa, IRb). The dashed area in the inner circle indicates CG content in the chloroplast genome. The map is drawn by OGDRAW.
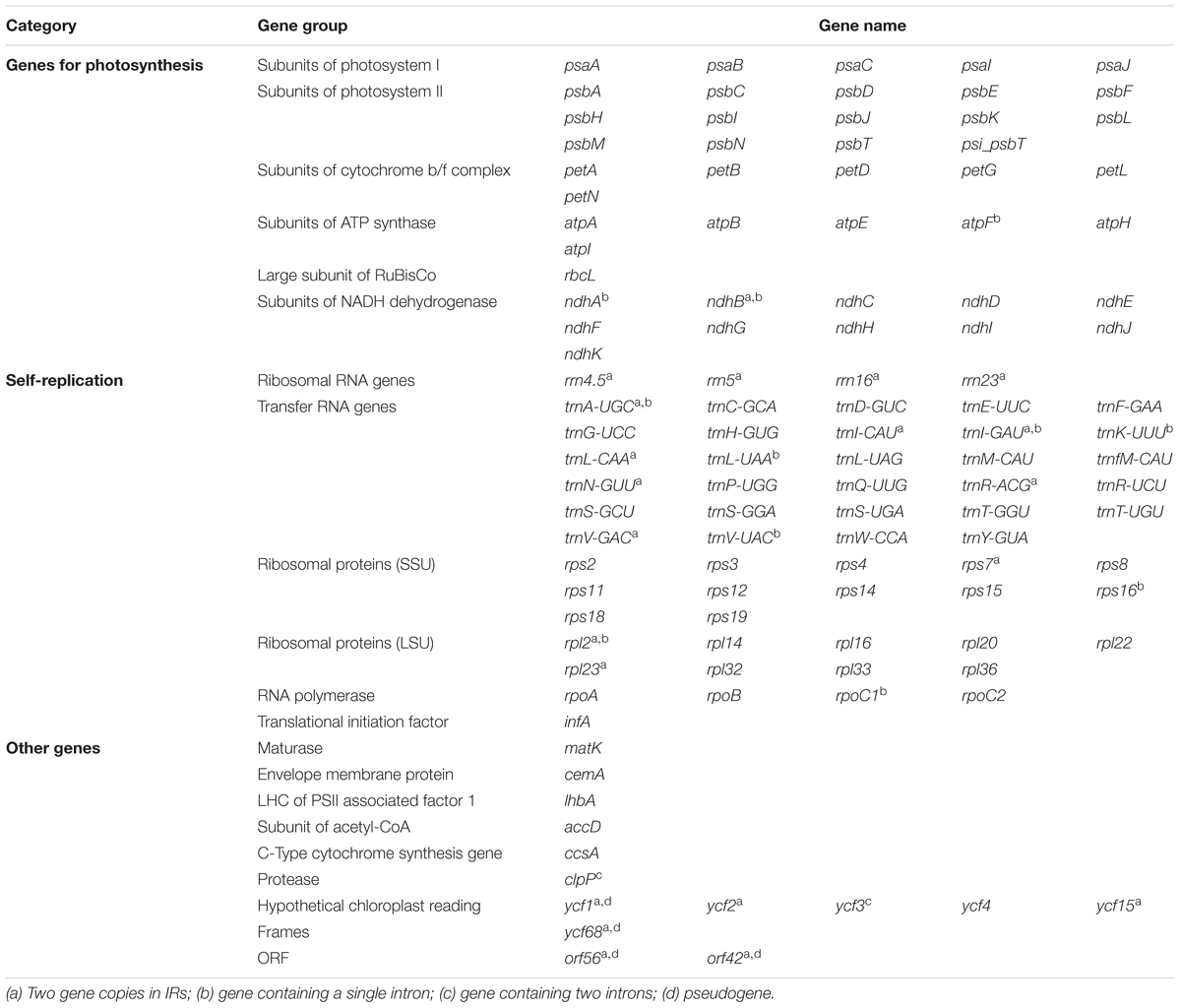
TABLE 1. List of genes encoded by Haberlea rhodopensis chloroplast genome.
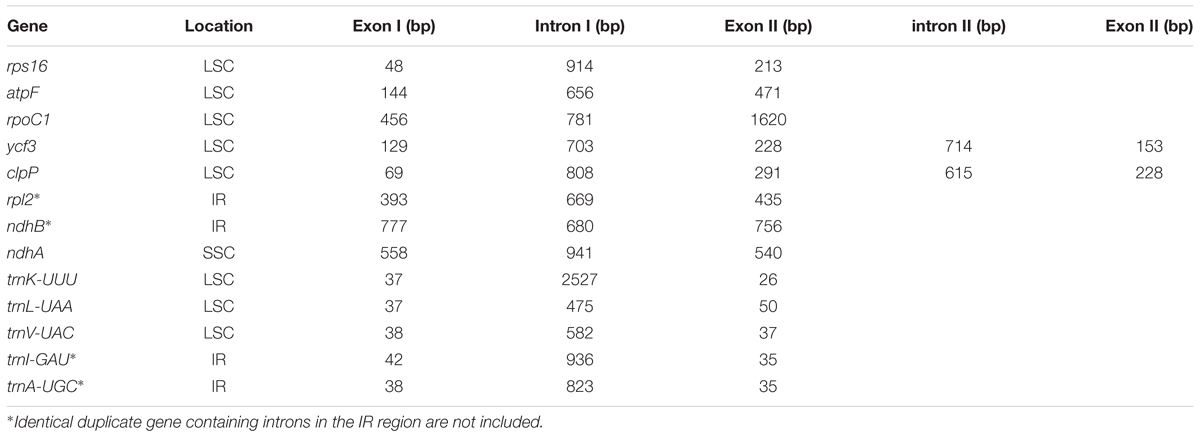
TABLE 2. Location and length of intron-containing genes in Haberlea rhodopensis chloroplast genome.
Plant cells often contain multiple copies of chloroplast genomes (Krebs et al., 1987) which can be regarded as genetic heterogeneity within a population (Yang et al., 2010). We mapped all Illumina reads to the assembled genome to identify the possible polymorphic sites. However, no polymorphisms were recovered, which was also observed in a cp genome from the family Gesneriaceae (B. hygrometrica, Zhang et al., 2012).
Comparative Analysis of the Haberlea rhodopensis Chloroplast Genome
The comparative analysis between the cp genomes of H. rhodopensis and B. hygrometrica is shown in Table 3. As expected, conserved synteny and gene order conservation was observed, which might be due to the family level conservation as both species belong to the family Gesneriaceae. Furthermore, a multiple sequence alignment (MSA) based on mVISTA (Poliakov et al., 2014) and BRIG (Alikhan et al., 2011) (Supplementary Figure S1) was performed among six closely related cp genomes for investigating levels of sequence divergence between them (Figure 2). Pairwise chloroplast genomic alignment between H. rhodopensis and other genomes also revealed a high degree of synteny suggesting the evolutionary conservation of these genomes at the genome-scale level. The complete aligned sequences revealed that Lamiales cp genomes possess high sequence similarity which suggests the genomes are rather conservative, although some divergent regions were found as well. As seen in other flowering plants (Nie et al., 2012; Dong et al., 2013; Liu et al., 2013), coding regions were more conserved than their non-coding counterparts. The most dissimilar coding regions among aligned cp genomes were ycf1, ndhF, accD, ccsA, rbcL, ycf2, and rps19. The ycf1, rbcL, and accD coding regions have also been observed as divergent in plastomes of other angiosperms (Nie et al., 2012; Dong et al., 2013, 2015; Liu et al., 2013; Luo et al., 2014) that makes such genes reliable markers for phylogenetic analysis (Nazareno et al., 2015). In contrast to S. indicum and S. miltiorrhiza, where rps19 is duplicated, B. hygrometrica is the only species in Gesneriaceae that has been reported to contain pseudogene rps19 (Zhang et al., 2012), but is was observed as protein coding gene in H. rhodopensis. The IR regions are less divergent than SSC and LSC regions. The non-coding regions showing higher sequence divergence were petA-psbJ, psbE-petL, ycf4-cemA, atpH-atpI, ndhC-trnV. Due to this fact, some of these chloroplast non-coding regions have been used in phylogenetic studies (Shaw et al., 2007; Wu et al., 2010; Nie et al., 2012). Another interesting observation from the comparative point of view (with the closest member of the same family) is that the genes psbI and rps12 are found in B. hygrometrica as pseudogenes, whereas in H. rhodopensis we found them to be normal protein-coding genes.

TABLE 3. Comparison of genome contents of H. rhodopensis and B. hygrometrica.
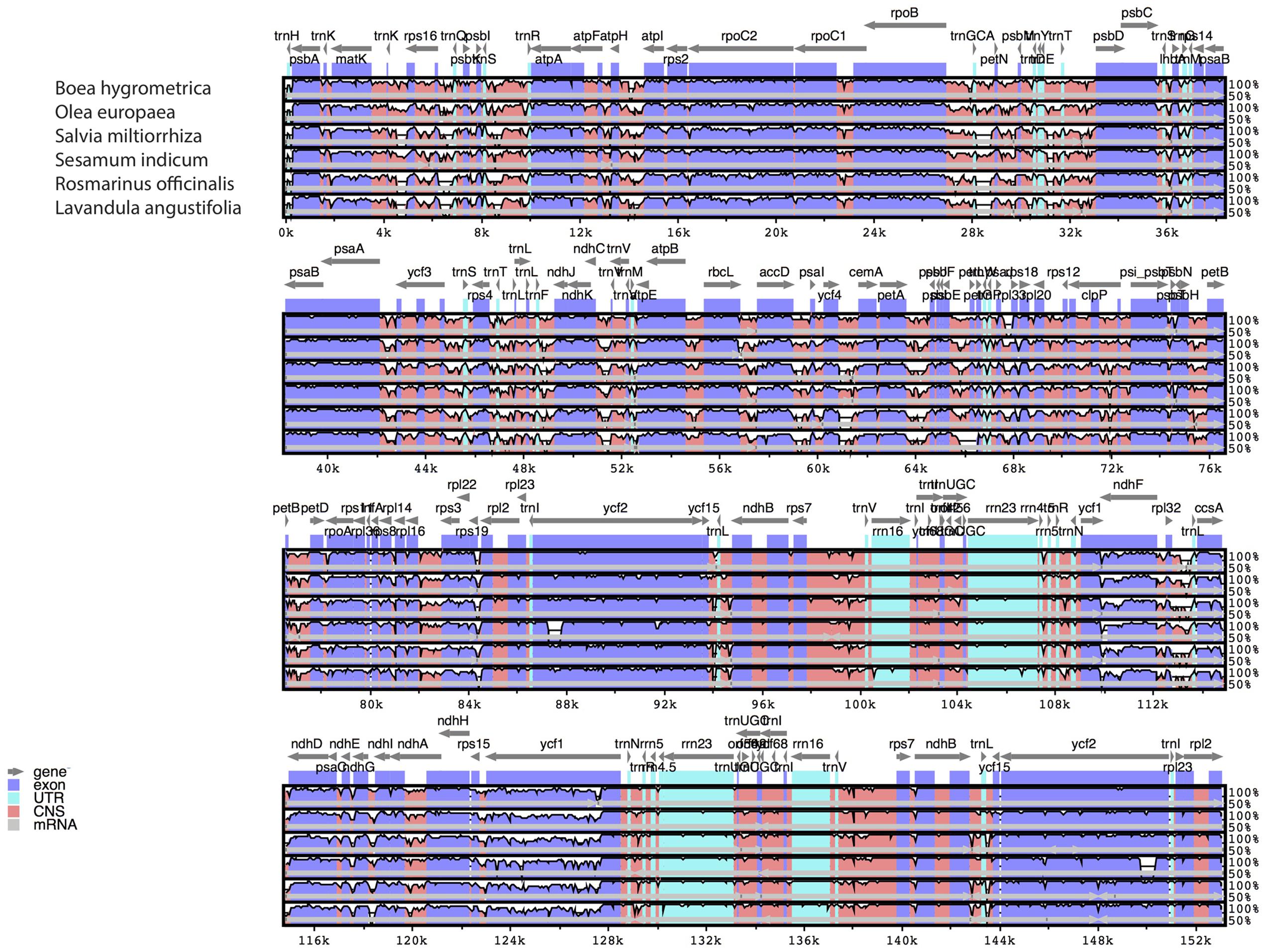
FIGURE 2. Sequence identity plots (mVISTA) among six species of Lamiales, using H. rhodopensis as reference genome. Arrows indicate the annotated genes and their transcriptional direction. Genome regions are color coded as exon, untranslated region (UTR), conserved non-coding sequences (CNS), and mRNA.
The intron-containing genes in H. rhodopensis cp genome are summarized in Table 2. Unlike some very close Lamiales members (S. indicum and S. miltiorrhiza), several introns are lacking in trnG-UCC, rps12, petB, petD, rpl16 in the plastome of H. rhodopensis, similar to B. hygrometrica. Similar structures are reported also in these genes in Tanaecium tetragonolobum (Nazareno et al., 2015).
The IRs regions are one of the most conserved regions in the cp genomes among all species. The IR boundary contraction and expansion are regarded as evolutionary events and are showed to be the main reason for size variation in cp genomes. These junctions are regarded as an index of chloroplast genome evolution (Zhang et al., 2013). The IR/LSC and IR/SSC regions of H. rhodopensis cp genome were compared to the corresponding regions of the closely related cp genomes of B. hygrometrica, O. europaea, S. indicum (Figure 3). As observed in other chloroplast genome studies (Nazareno et al., 2015), the IR expansion/contraction in Lamiales has led to changes in the structure of the chloroplast genome, contributing to the formation of pseudogenes. The IRa/SSC border extended into ycf1 resulting in à pseudogene in the four compared cp genomes. The length of ycf1 pseudogene was 775 bp in H. rhodopensis, 1,115 bp in O. europaea, 813 bp in B. hygrometrica and 1,012 bp in S. indicum. Moreover, there is overlapping of ycf1 pseudogene and ndhF in H. rhodopensis. B. hygrometrica, O. europaea, and S. indicum. The IRb/SSC region was located in the CDS of ycf1 gene in all compared cp genomes. The trnH genes of compared species were located in LSC region, 0–44 bp apart from the IRb/LSC border. Interestingly, the rps19 gene was situated in LSC region of H. rhodopensis and O. europaea genomes, separated from the IRa/LSC region by 27 and 108 bp, respectively, whereas it was extended into IRa in B. hygrometrica and S. miltiorrhiza and S. indicum.
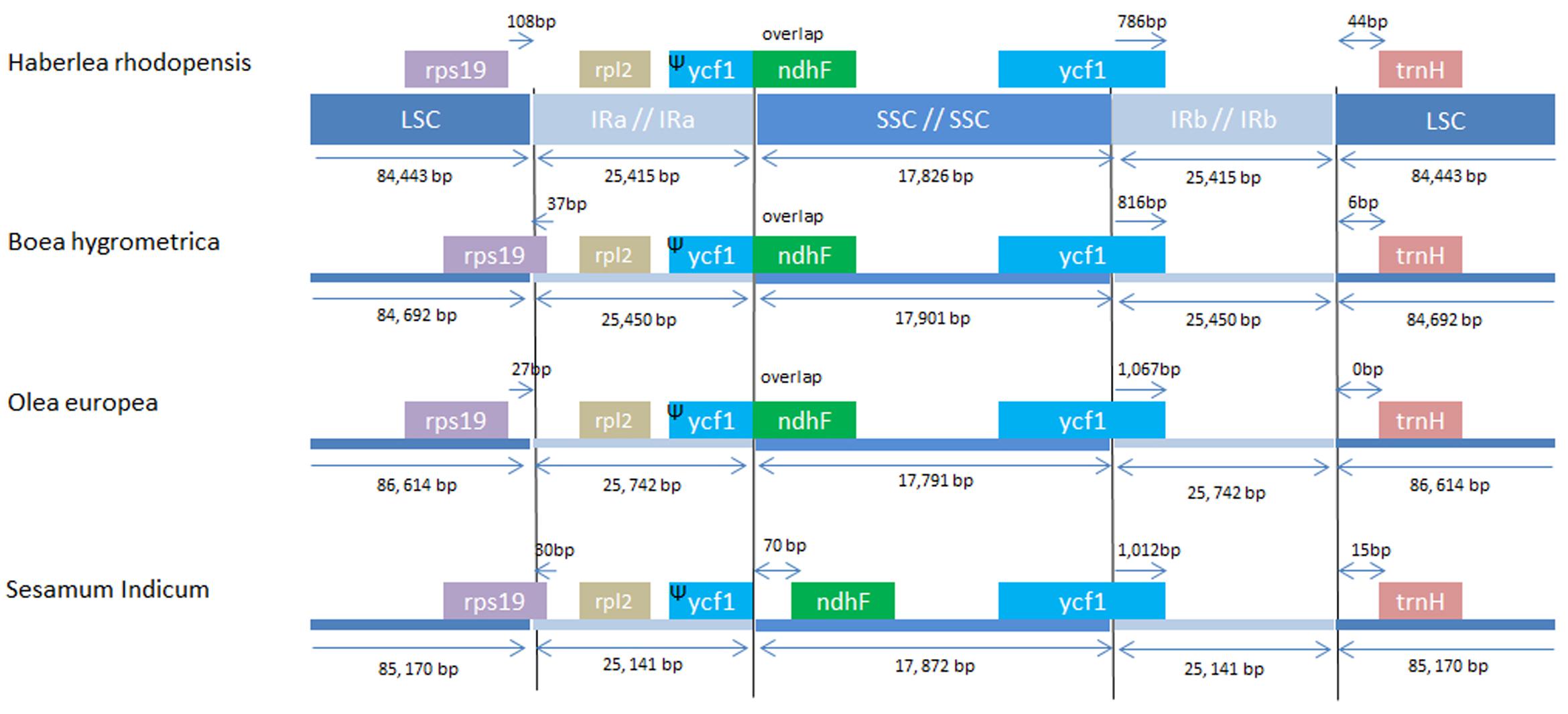
FIGURE 3. Comparison of chloroplast borders of LSC, SSC, and IRs among the species from Lamiales.
Repeat Analysis
Organelle genomes, especially chloroplast, have long been used as a source to understand the phylogenetic relationship of species, mainly due to its uniparental inheritance mode and also due to its rapidly evolving gene content (Sablok et al., 2013). Taking into account the role of the organelle SSRs as important phylogenetic markers, we analyzed the distribution of SSRs with following length thresholds of minimum repetitive units: 10 repeats for mono-, 4 repeats for di- and tri-, and 3 repeats for tetra-, penta-, and hexa-nucleotide repeat patterns. We observed a total of 71 SSRs patterns with 10 SSRs present in compound patterns (Table 4). Among the identified repeat patterns, dinucleotide repeat patterns (AG/CT and AT/TA) formed the most abundant repeat patterns. Our finding agrees with the observation that cp SSRs are generally composed of short polyadenine (polyA) or polythymine (polyT) repeats and rarely contain tandem guanine (G) or cytosine (C) repeats (Kuang et al., 2011). In total, 29 SSRs were found to be present in the genic regions in H. rhodopensis. Among them, seven genes (atpF, rpoC2, rpoA, rpl2, ycf2, ndhB, rrn23) were found to harbor at least two SSRs. It is interesting to see that the number of identified SSRs is low compared to the previously characterized SSRs in organelle genomes (e.g., B. hygrometrica, O. europaea, and S. indicum, Mariotti et al., 2010; Yi and Kim, 2012; Zhang et al., 2012). Additionally, we did not find a large abundance of tri- to tetra-nucleotide repeats, which is in contrast to previous reports (Qian et al., 2013). We did not observe any penta- or hexa-nucleotide repeat patterns in H. rhodopensis. This is similar to the chloroplast genome of Utricularia reniformis belonging to Lamiales, which lacks penta- or hexa-nucleotide repeats (Silva et al., 2016). The identified SSRs, together with the provided primer pairs, could be used for determination of the phylogeography and population structure pattern of H. rhodopensis (Supplementary Table S3).
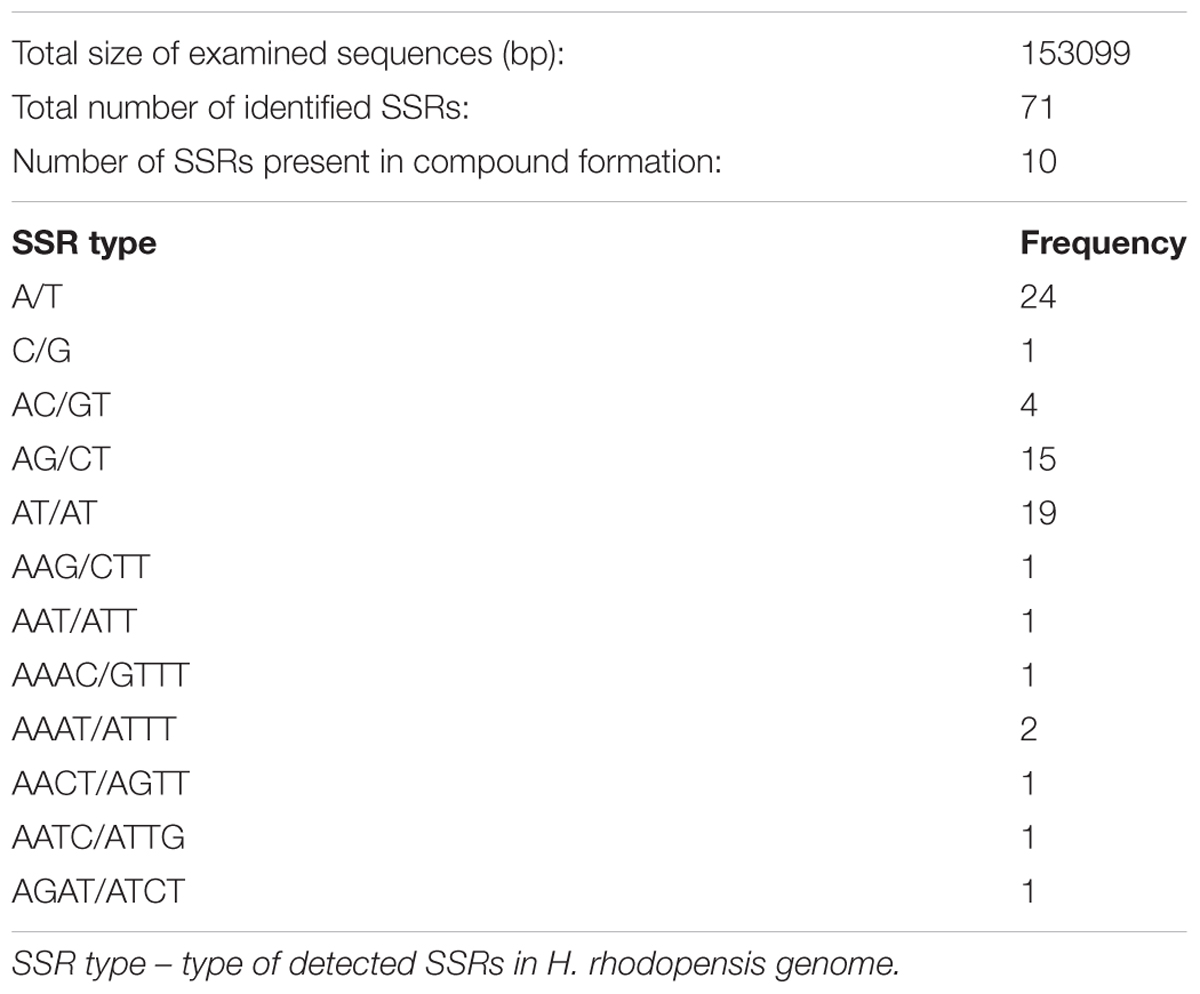
TABLE 4. Cumulative SSR frequency and corresponding primer pairs in Haberlea rhodopensis (SSR search parameters: 1–10; 2–4; 3–4; 4–3; 5–3; 6–3 where 1, 2, 3, 4, 5, and 6 indicate the mono- di-, tri-, tetra-, penta-, and hexa-nucleotide repeats).
In addition to the SSRs, we further explored the role of the long repeats as identified by REPUTER (Kurtz et al., 2001). We identified a total of 40 repeats – 18 forward and 22 palindromic (inverted) repeats (Table 5). These repeats were found to be at least 30 bp per unit and the biggest was 59 bp. Around 40% of these repeats fell exclusively into intergenic regions, whereas 60% are situated in genes or at their border. In contrast to some closely related species such as S. indicum with 15 repeats, B. hygrometrica with eight repeats, and O. europaea with three repeats (Nazareno et al., 2015), H. rhodopensis contains a higher number of repeat elements. Most of these repeats exhibit lengths between 30 and 44 bp, while the ycf2 CDS possesses the highest number of repeats (11) and ycf1 has the longest repeats at 59 bp. The localization of forward repeats can be a consequence of the plastomic rearrangements and can provide important clues toward understanding the spatial-temporal organization in Geraniaceae (Huang et al., 2013).
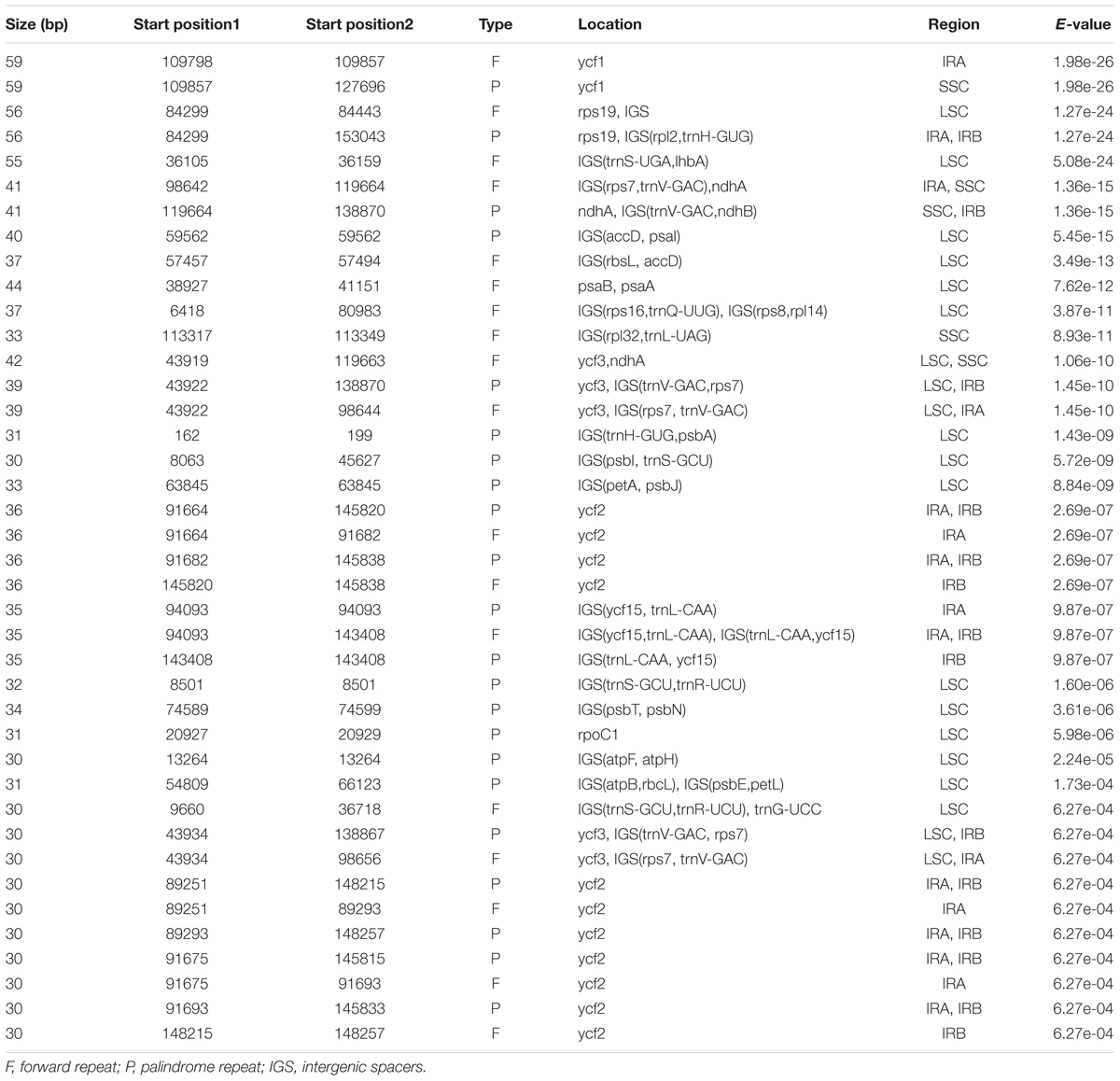
TABLE 5. Distribution and localization of repeat sequences in cpDNA of Haberlea rhodopensis.
Codon Usage and Selection Events in Protein-Coding Genes
Codon usage plays an important part in shaping the plastome evolution. Among the several features that shape codon usage, mutational bias has an essential role in shaping this evolutionary phenomenon (Liu and Xue, 2005). Several features have been shown to affect the codon usage at the mutational and translational levels, however, mutational pressure is the dominant force acting at the level of chloroplast genomes. The strand asymmetry, causing strand-specific bias in organelle genomes, is among the other factors that could contribute to shaping the codon usage bias (Jia and Higgs, 2008). For the estimation of the codon usage, a total of 58 genes were selected based on 300 bp length threshold from H. rhodopensis and B. hygrometrica. Codon usage measures such as Nc, frequency of A, T, G, and C at the third synonymous sites, aromaticity and gravy were estimated (Supplementary Table S2). For the estimation of the mutational bias, we evaluated mutational pressure in H. rhodopensis using Nc plots (Supplementary Figure S2). We observed that most of the genes, with a size threshold of 300 bp, fall below the expected line of Nc thus suggesting that mutational bias is a dominant factor shaping the codon usage patterns in H. rhodopensis (Supplementary Table S2). Furthermore, we evaluated the spearman rank correlation between the Nc and GC3s (R = 0.544; p > 0.0001), which further indicates the role of the mutational pressure in H. rhodopensis. We further evaluated the role of mutational bias in phylogenetically close B. hygrometrica and also observed a strong positive correlation between Nc and GC3s (R = 0.4542; p > 0.0001), in agreement with an important role of mutational bias in members of the Gesneriaceae family.
The synonymous and non-synonymous nucleotide substitution patterns are very important markers in gene evolution studies. In most genes except for the very rapidly evolving ones, non-synonymous nucleotide substitutions have occurred less frequently than synonymous substitutions due to the action of purifying selection. Accordingly, the ratio of Ka/Ks < 1 (especially less than 0.5) indicates purifying selection; Ka/Ks > 1 indicates probable positive selection whereas Ka/Ks values close to 1 indicate neutral evolution, or relaxed selection (Kimura, 1983). We calculated Ka/Ks ratios of H. rhodopensis chloroplast genome versus three closely related Lamiales species: B. hygrometrica, S. indicum, S. miltiorrhiza and the more distantly related Coffea arabica (Supplementary Table S4). Our analysis showed that the Ka/Ks ratios are not region-specific (the IR, SSC, and LSC regions showed comparable values) but are mostly gene-specific. The average Ka/Ks ratio for 72 protein genes analyzed in the five genomes was 0.2139. The most conserved genes with average Ka/Ks values between 0 and 0.01, indicating very strong purifying selection pressure are atpH, psaA, psaB, psaC; psbA, psbC, psbD, psbE, psbF, psbH, psbL, psbM, psbN, psbT, petG, petL, and infA. Model averaging method in KaKs calculator showed average Ka/Ks > 1 for rps12 (encoding the large subunit ribosomal protein 23) and rpl23 (encoding the ribosomal protein S12). The increased Ka/Ks ratios indicative for positive selection could reflect selection pressures specific for H. rhodopensis cp genome. Alternatively, they could be a sign of increased variability of the particular proteins within a broader group of species. In the first case, there could be a relationship between these selection indexes and the resurrection phenotype. To discern which is the case, we performed a cross analysis calculating Ka/Ks ratios between all other possible sequence pairs of sequences compared (C. arabica and S. indicum; C. arabica and S. miltiorrhiza; C. arabica and B. hygrometrica; S. indicum and S. miltiorrhiza; S. indicum and B. hygrometrica; S. miltiorrhiza and B. hygrometrica) for the genes rps12 and rpl23.
The Ka/Ks cross test showed that the positive selection events for these genes were specific for H. rhodopensis. rpl23 is one of the candidates for adaptive evolution. The rpl23 average Ka/Ks value between H. rhodopensis and the other four species was 2.154, whereas the average Ka/Ks value for the rest of the analyzed pairs was 0.298 (Supplementary Table S4). rps12 is another gene with indications for positive selection. This gene showed a very high average Ka/Ks ratio – 2.15 for the pairs of H. rhodopensis vs. the other four species, in comparison to 0.633 for the pairs not including H. rhodopensis.
Both rpl23 and rps12 have been proven to be essential for the chloroplast ribosome (Fleischmann et al., 2011; Tiller and Bock, 2014). The bacterial ortholog of rpl23 in E. coli is located next to the tunnel exit of the large subunit, where it participates in the formation of a cradle-like surface embracing the polypeptide exit region. The ribosome structure and topology are highly conserved between bacteria and chloroplasts. As it was revealed in a three-dimensional cryo-EM map of the spinach 70S chloro-ribosome, the chloroplast rpl23 protein is located at the same position on the chloro-ribosome as its bacterial ortholog and probably performs a similar function (Sharma et al., 2007). The positive selection pressure on rpl23 sequence in H. rhodopensis genome could reflect rapid adaptive changes in the machinery during the biosis–anabiosis transitions of this resurrection species. Interestingly, we did not observe signs of positive selection for this gene in the closely related genome of Boea hygrometrica (another resurrection species) so this is most probably a recent adaptation unique for Haberlea rhodopensis.
A similar rationale could be applied to the rps12 gene, which codes for an important protein of the small ribosome subunit. Studies in Chlamydomonas reveal that Rps12 protein plays an important role in the decoding center of chloroplast ribosomes and its absence leads to a general block of chloroplast translation. Repression of the rps12 gene leads to the arrest of cell growth and induces a response that involves expression changes in nuclear-encoded genes for plastid biogenesis, protein turnover, and stress. This response also leads to the overaccumulation of several plastid transcripts. It is an indication of the existence of complex negative regulatory feedback loops in the chloroplast gene circuitry (Ramundo et al., 2013). The chloroplast ribosomes and translational apparatus are important participants in the retrograde signaling and play a key role in the regulation of important signaling cascades related to plant growth and stress responses (Fleischmann et al., 2011). In this respect, the positive selection signatures on the sequences of the two ribosomal protein genes in Haberlea genome could be regarded and further analyzed in a broader perspective.
Values of Ka/Ks in the range of 0.5 to 1.0 (indicating relaxed selection) were observed for the genes rpl2, rpl32, atpE, psaI, psbI, matK and clpP (Figure 4). The Ka/Ks values of the rest of the genes were between 0.02 and 0.49, which means most of the genes in the cp genome of H. rhodopensis are under purifying selection.
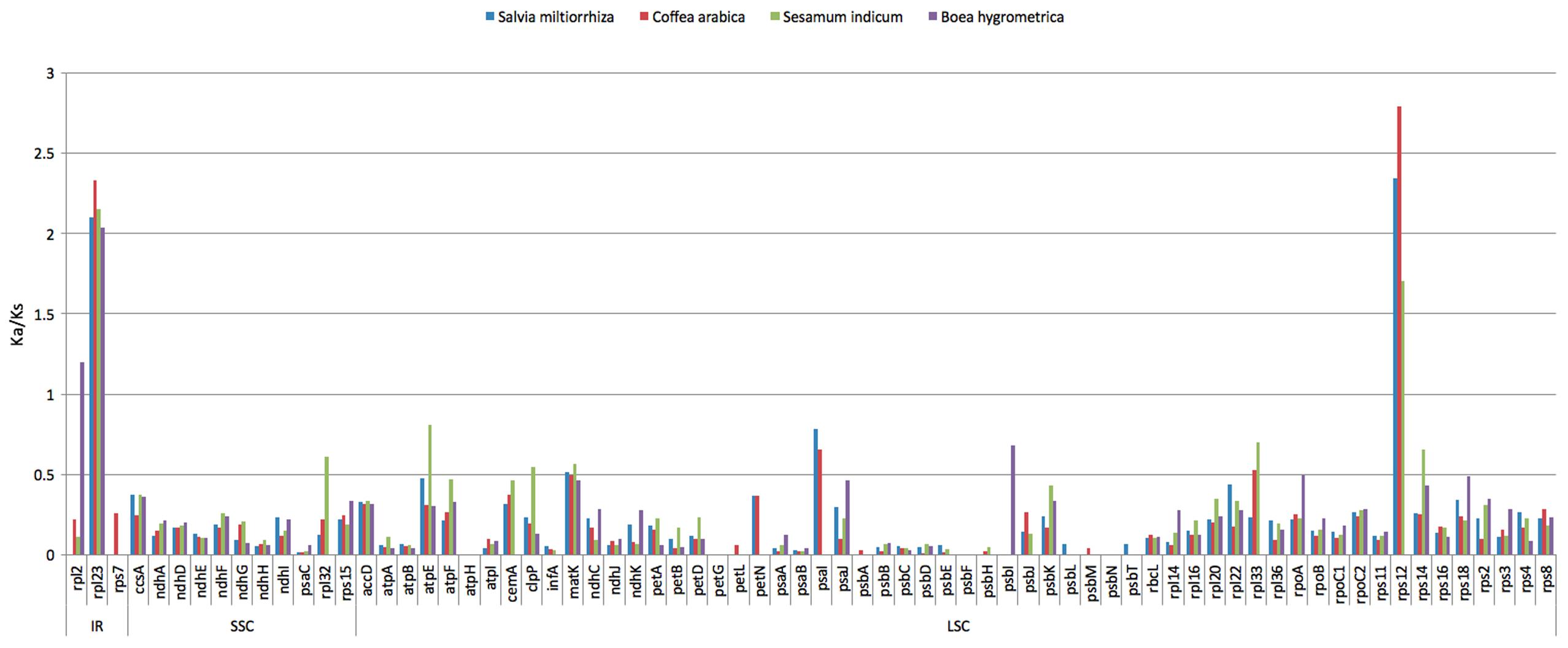
FIGURE 4. The Ka/Ks ratio of 72 protein-coding genes of four cp genomes for comparison with H. rhodopensis.
H. rhodopensis is interesting with its homoiochlorophyllous resurrection phenotype, and with the rapid recovery of its photosynthetic levels upon rehydration. The possible relaxed selection pressure on psaI and psbI, which encode integral thylakoid membrane proteins, may reflect recent adaptations of the photosynthetic apparatus of this species to repeated desiccation and rehydration. The relaxed selection for the ATP synthase subunit atpE, along with the presence of positively selected sites in its sequence (see below) could also be related to adaptations to desiccation stress.
In addition to the Ka/Ks analysis, we also identified the site-specific selection events using the Selecton (Stern et al., 2007). Selecton analysis revealed a total of 17 genes displaying site-specific selection (Table 6). Interestingly, rbcL was found to harbor 13 sites under positive selection. This gene has been previously used to establish the diverse phylogenetic relationships in the resurrections plants in Selaginellaceae (Korall and Kenrick, 2002). rbcL plays an important role as a modulator of photosynthetic electron transport and is essential for photosynthesis (Allahverdiyeva et al., 2005). Previous estimates of the desiccation in resurrection plants indicate a physiological basis of the slow recovery of the assimilatory action of the photosynthetic carbon (Schwab et al., 1989). Identification of the positive sites in this study could lead to the establishment of the photosynthetic mutants that could lead to the physiological understanding of the desiccation in resurrection plants. In addition, to rbcL, we also observed site-specific selection in atpE, which is a co-transcriptionally coupled gene with atpB (Chotewutmontri and Barkan, 2016) and plays a critical role in the activation and de-activation of the CF0CF1, which is a H+ translocating ATPase. A key feature of the homoiochlorophyllous resurrection plants is their ability to maintain the integrity of the photosynthetic apparatus and its re-activation following the rehydration. High rates of evolvability in atpE might indicate the role of selection in fine-tuning the demands of the rapid activation of the ATP synthase and to increase the co-transcription of the atpE with atpB to meet the metabolic demand for ATP and NADPH in resurrection plants (Rott et al., 2011). Furthermore, we also detected site-specific selection events in accD, which have been shown to affect the plant fitness by altering the acetyl-CoA carboxylase production (Madoka et al., 2002). In addition to these genes, we observed site-specific selection in ndhF, which is critical to the onset and delay in senescence. The observed site-specific selection events in the ndhF gene might affect the translational output of ndhF gene and thus may play a role in the stress adaptation of H. rhodopensis by delaying drought-induced senescence. It is worth mentioning that previously ndhF mutants (DeltandhF) with the plastid ndhF gene knocked-out in transgenic tobacco showed 30-day-delayed senescence (Zapata et al., 2005).
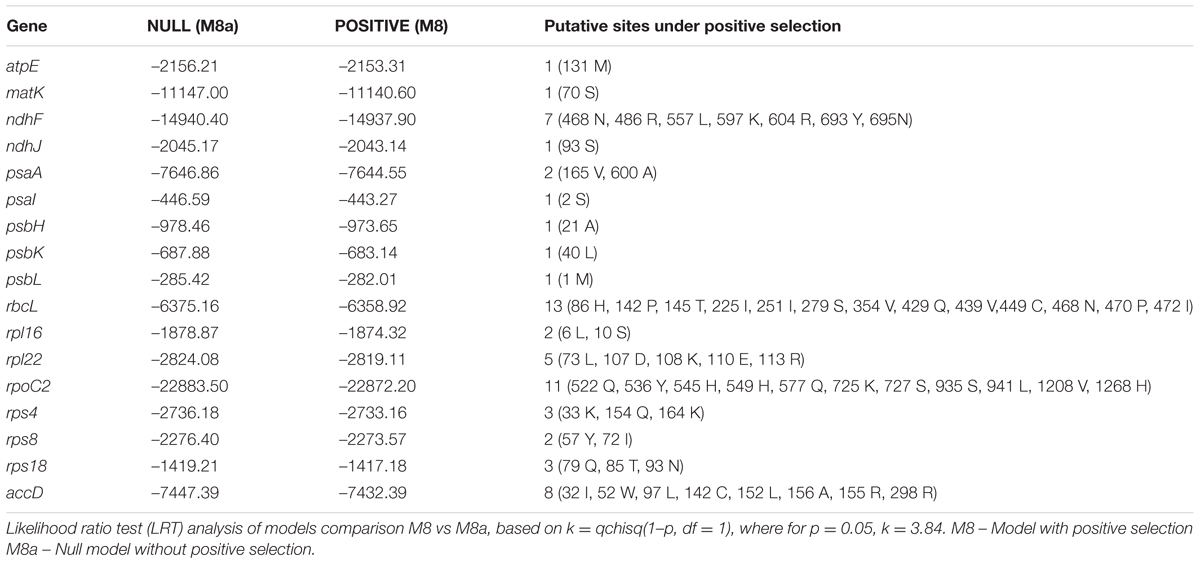
TABLE 6. Positive selection sites identified with selection with df = 1.
Phylogenetic Analysis
Organelle genome sequencing plays a key role in deciphering the evolutionary phylogenomics and cladistics of plants species. For the phylogenetic reconstruction, we performed a concatenate codon-based sequence alignment of the atpA, atpB, atpE, atpF, atpH, atpI, ndhC, ndhD, ndhE, ndhF, ndhH, ndhI, ndhJ, petA, petB, petD, petG, psaA, psaB, psaC, psaI, psbA, psbB, psbC, psbD, psbE, psbH, psbK, psbL, psbM, psbN, rbcL, rpoA, rpoB, and rpoC2 (Full list of species used in the phylogenetic analysis is provided in Supplementary Table S1). The basis of selection of these genomes for the comparative analysis is due to the fact that these genomes share the same evolutionary clade thus making them phylogenetically close. Additionally, chloroplasts from Lamiales were chosen as a closest relative outgroup to the sequenced chloroplast genome in this study, i.e., H. rhodopensis. For phylogenetic inferences, Arabidopsis thaliana has also been included as a distant outgroup. Model estimation revealed GTR+G4 on the basis of the Akaike Information Criterion (–lnL = 165400.957), Corrected Akaike Information Criterion (–lnL = 335191.641) and Bayesian Information Criterion (–lnL = 352321.906). For the maximum-likelihood analysis, 1000 bootstrap replicates were evaluated and the consensus tree from IQTree (Nguyen et al., 2015) was re-rooted using Arabidopsis thaliana as an outgroup, revealing the monophyletic origin of the H. rhodopensis which clusters with B. hygrometrica (Figure 5) (Supplementary Data Sheet S2). IQtree consensus tree supported the monophyletic origin of H. rhodopensis, which can be further used to understand the phylogenetic clade and evolution of Gesneriaceae species.
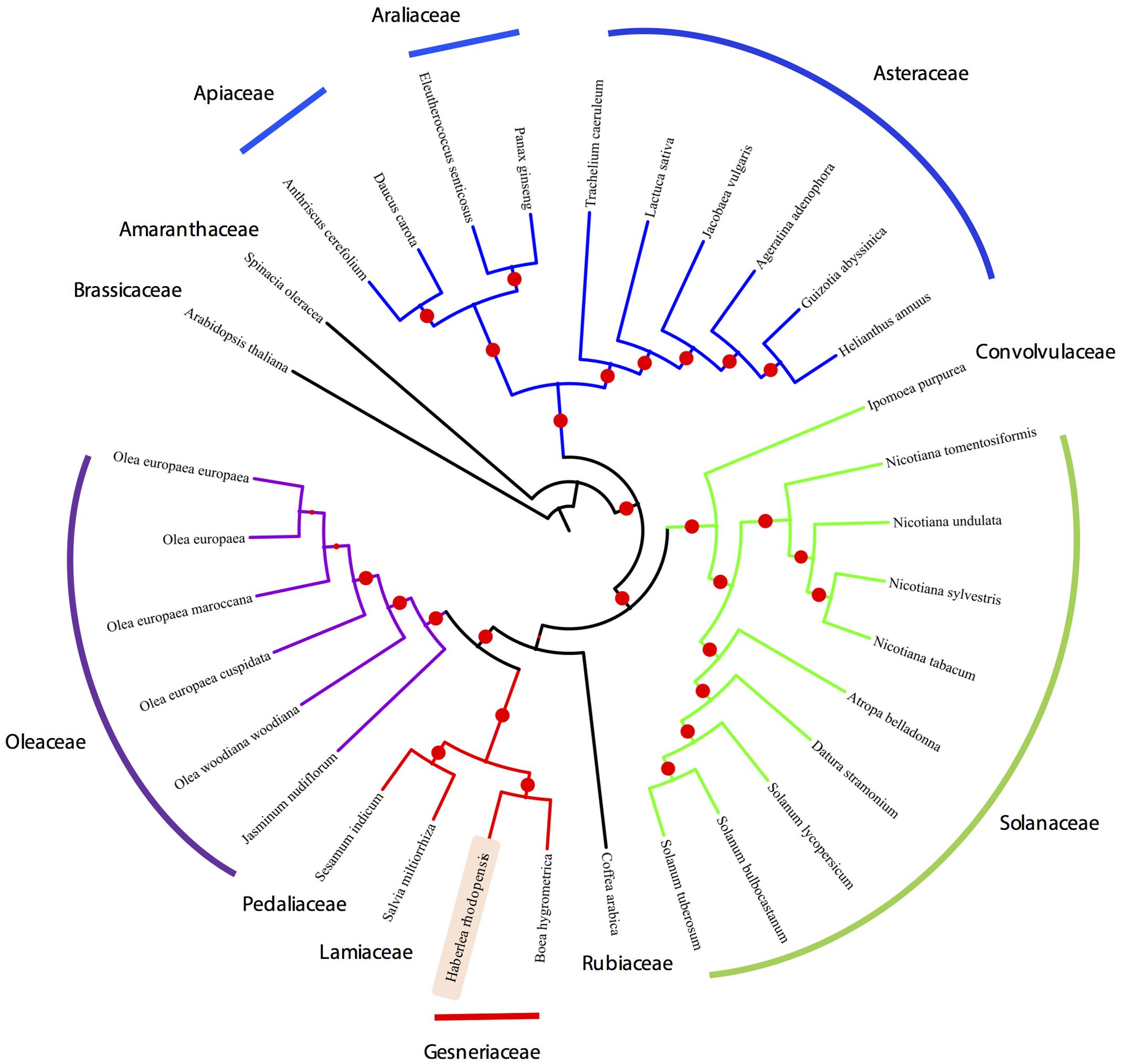
FIGURE 5. Phylogenetic tree based on 35 protein-coding genes from 33 species and H. rhodopensis phylogenetic placement.
RNA Editing Events
RNA editing is a posttranscriptional process, which mainly involves the conversion of cytidine to uridine and forms an important part of the RNA maturation process (Chen et al., 2011; Hein et al., 2016). The RNA editing factors such as CRR28 and RARE1 have been widely shown to affect the cytidine-to-uridine conversion in ndhBeU467PL, ndhDeU878SL, and accDeU794SL (Hein et al., 2016). Using PREPACT2 and parameters described as per (Hein et al., 2016), we identified a total of 17 editing sites with 100% representation across the 17 in-built organelle genomes in PREPACT2 (Supplementary Figure S3). In accordance with the recent studies (Hein et al., 2016), we also observed the most C-U RNA editing events in ndh gene, which are known to act as electro-regulators to adjust the ROS accumulation in cyclic photosynthesis electron transporters (Martín and Sabater, 2010). Although the loss of the editing events ndhBeU467PL and ndhDeU878SL has been observed in previous phylogenomic studies (Hein et al., 2016), these events were now identified in H. rhodopensis. In angiosperms, accDeU794SL has shown to be substantially absent in around 50% of the angiosperms. However, a loss of the accDeU794SL was not observed in the case of H. rhodopensis. Taking into account the conservation of the editing events, it can be presumed that resurrection plants maintain the conservatory editing events. Since H. rhodopensis is a desiccation-tolerant species, the identified RNA editing events could be used in future for understanding the role of the RNA editing in desiccation stress and further to understand the physiological behavior of resurrection plants.
Conclusion
In summary, we present for the first time the complete chloroplast genome of Haberlea rhodopensis, a tertiary relict and Balkan endemic resurrection species from the Gesneriaceae family. The genome sequencing, assembly, annotation, and the comparative analysis, reveal that the cp genomes of H. rhodopensis and B. hygrometrica share a quadruple structure, gene order, GC content, and codon usage features, similar to those of Lamiaceae cp genomes. The phylogenetic analysis supports the monophyletic origin of Haberlea rhodopensis and can be used as a reliable phylogenetic framework to understand the evolution of the Gesneriaceae species. The analysis of H. rhodopensis cp genome uncovers several intriguing features, which can be used as a basis for the understanding the resurrection tolerance of this plant. Specifically, the H. rhodopensis cp genome harbors 137 genes, of which 86 are protein-coding. The results of a site-specific selection analysis point to positively selected sites in several chloroplast genes, such as atpE, rbcL, psbI, psbA, ndhH, and accD. The observed specific cp genomic features of Haberlea rhodopensis may be interpreted as being a consequence of molecular adaptation to drought stress, which awards an evolutionary advantage to this species.
The chloroplast genome reported in this study will make it possible to understand the function of the specific sites under selection, by developing site-directed mutagenesis assays or by point mutations at those sites. Furthermore, protein modeling will be needed in the future to analyze subtle site-specific changes resulting from recent adaptations of the photosynthetic apparatus of this species to repeated desiccation and rehydration.
Author Contributions
ZI, GS, and VB analyzed the data; EA and GZ conducted the experimental part of the work; GY and ED contributed to the analysis; ZI, ED, and VB conceived the project and secured the funding; ZI, GS, and VB wrote the paper with contributions from all co-authors. All co-authors have read and approved the manuscript.
Funding
This work was supported the Ministry of Education and Science, Bulgaria, grant BG051PO001-3.3-05/0001.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00204/full#supplementary-material
FIGURE S1 | Genome comparison of six chloroplast genomes to the H. rhodopensis chloroplast genome. Produced alignments are color coded based on the similarity score. The first outer ring is consisting of protein-coding genes based on (corresponding to) the H. rhodopensis chloroplast genome.
FIGURE S2 | Estimation of the standard effective number of codon (Nc) was tabulated using the equation N(c) = 2 + s + 29/[s(2) + (1 – s)(2)], where s denotes GC3s (Wright, 1990).
FIGURE S3 | RNA editing output from PREPACT2 tool.
TABLE S1 | List of the chloroplast genomes used for phylogenetic analysis.
TABLE S2 | CodonW output for genes ≥ 300 bp.
TABLE S3 | Table showing SSRs with primer pairs.
TABLE S4 | Ka/Ks analysis table.
DATA SHEET S1 | Merged read quality, length distribution and QUAST results.
DATA SHEET S2 | IQtree consensus tree.
Footnotes
- ^http://hannonlab.cshl.edu/fastx_toolkit/
- ^http://www.ncbi.nlm.nih.gov/Sequin
- ^http://chloroplast.cbio.psu.edu
- ^http://pgrc.ipk-gatersleben.de/misa/
- ^http://codonw.sourceforge.net/
- ^http://www.prepact.de/prepact-main.php
References
Alikhan, N.-F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Allahverdiyeva, Y., Mamedov, F., Mäenpää, P., Vass, I., and Aro, E. M. (2005). Modulation of photosynthetic electron transport in the absence of terminal electron acceptors: characterization of the rbcL deletion mutant of tobacco. Biochim. Biophys. Acta 1709, 69–83. doi: 10.1016/j.bbabio.2005.06.004
Assefa, S., Keane, T. M., Otto, T. D., Newbold, C., and Berriman, M. (2009). ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25, 1968–1969. doi: 10.1093/bioinformatics/btp347
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Chen, H., Deng, L., Jiang, Y., Lu, P., and Yu, J. (2011). RNA editing sites exist in protein-coding genes in the chloroplast genome of Cycas taitungensis. J. Integr. Plant Biol. 53, 961–970. doi: 10.1111/j.1744-7909.2011.01082.x
Chotewutmontri, P., and Barkan, A. (2016). Dynamics of chloroplast translation during chloroplast differentiation in maize. PLoS Genet. 12:e1006106. doi: 10.1371/journal.pgen.1006106
Dong, W., Xu, C., Cheng, T., and Zhou, S. (2013). Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in saxifragales. PLoS ONE 8:e77965. doi: 10.1371/journal.pone.0077965
Dong, W., Xu, C., Li, C., Sun, J., Zuo, Y., Shi, S., et al. (2015). ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5:8348. doi: 10.1038/srep08348
Fleischmann, T. T., Scharff, L. B., Alkatib, S., Hasdorf, S., Schottler, M. A., and Bock, R. (2011). Nonessential plastid-encoded ribosomal proteins in tobacco: a developmental role for plastid translation and implications for reductive genome evolution. Plant Cell 23, 3137–3155. doi: 10.1105/tpc.111.088906
Georgieva, K., Maslenkova, L., Peeva, V., Markovska, Y., Stefanov, D., and Tuba, Z. (2005). Comparative study on the changes in photosynthetic activity of the homoiochlorophyllous desiccation-tolerant Haberlea rhodopensis and desiccation-sensitive spinach leaves during desiccation and rehydration. Photosynth. Res. 85, 191–203. doi: 10.1007/s11120-005-2440-0
Georgieva, K., Szigeti, Z., Sarvari, E., Gaspar, L., Maslenkova, L., Peeva, V., et al. (2007). Photosynthetic activity of homoiochlorophyllous desiccation tolerant plant Haberlea rhodopensis during dehydration and rehydration. Planta 225, 955–964. doi: 10.1007/s00425-006-0396-8
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Heber, U. (2012). Conservation and dissipation of light energy in desiccation-tolerant photoautotrophs, two sides of the same coin. Photosynth. Res. 113, 5–13. doi: 10.1007/s11120-012-9738-5
Heber, U., Azarkovich, M., and Shuvalov, V. (2007). Activation of mechanisms of photoprotection by desiccation and by light: poikilohydric photoautotrophs. J. Exp. Bot. 58, 2745–2759. doi: 10.1093/jxb/erm139
Hein, A., Polsakiewicz, M., and Knoop, V. (2016). Frequent chloroplast RNA editing in early-branching flowering plants: pilot studies on angiosperm-wide coexistence of editing sites and their nuclear specificity factors. BMC Evol. Biol. 16:23. doi: 10.1186/s12862-016-0589-0
Hu, S., Sablok, G., Wang, B., Qu, D., Barbaro, E., Viola, R., et al. (2015). Plastome organization and evolution of chloroplast genes in Cardamine species adapted to contrasting habitats. BMC Genomics 16:306. doi: 10.1186/s12864-015-1498-0
Huang, Y. Y., Matzke, A. J. M., and Matzke, M. (2013). Complete sequence and comparative analysis of the chloroplast genome of coconut palm (Cocos nucifera). PLoS ONE 8:e74736. doi: 10.1371/journal.pone.0074736
Jia, W., and Higgs, P. G. (2008). Codon usage in mitochondrial genomes: distinguishing context-dependent mutation from translational selection. Mol. Biol. Evol. 25, 339–351. doi: 10.1093/molbev/msm259
Kim, J., and Mullet, J. E. (1994). Ribosome-binding sites on chloroplast rbcL and psbA mRNAs and light-induced initiation of D1 translation. Plant Mol. Biol. 25, 437–448.
Kimura, M. (1983). The neutral theory of molecular evolution and the world view of the neutralists. Genome 31, 24–31.
Korall, P., and Kenrick, P. (2002). Phylogenetic relationships in Selaginellaceae based on rbcL sequences. Am. J. Bot. 89, 506–517. doi: 10.3732/ajb.89.3.506
Krebs, K. E., Sihag, R., and Bendich, A. J. (1987). Why do chloroplasts and mitochondria contain so many copies of their genome? BioEssays 6, 279–282. doi: 10.1002/bies.950060608
Kuang, D.-Y., Wu, H., Wang, Y.-L., Gao, L.-M., Zhang, S.-Z., and Lu, L. (2011). Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome 54, 663–673. doi: 10.1139/G11-026
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Liu, Q., and Xue, Q. (2005). Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J. Genet. 84, 55–62. doi: 10.1007/BF02715890
Liu, Y., Huo, N., Dong, L., Wang, Y., Zhang, S., Young, H. A., et al. (2013). Complete chloroplast genome sequences of Mongolia medicine Artemisia frigida and phylogenetic relationships with other plants. PLoS ONE 8:e57533. doi: 10.1371/journal.pone.0057533
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, 575–581. doi: 10.1093/nar/gkt289
Luo, J., Hou, B. W., Niu, Z. T., Liu, W., Xue, Q. Y., and Ding, X. Y. (2014). Comparative chloroplast genomes of photosynthetic orchids: insights into evolution of the Orchidaceae and development of molecular markers for phylogenetic applications. PLoS ONE 9:e99016. doi: 10.1371/journal.pone.0099016
Madoka, Y., Tomizawa, K. I., Mizoi, J., Nishida, I., Nagano, Y., and Sasaki, Y. (2002). Chloroplast transformation with modified accD operon increases acetyl-CoA carboxylase and causes extension of leaf longevity and increase in seed yield in tobacco. Plant Cell Physiol. 43, 1518–1525. doi: 10.1093/pcp/pcf172
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mariotti, R., Cultrera, N. G. M., Díez, C. M., Baldoni, L., and Rubini, A. (2010). Identification of new polymorphic regions and differentiation of cultivated olives (Olea europaea L.) through plastome sequence comparison. BMC Plant Biol. 10:211. doi: 10.1186/1471-2229-10-211
Martín, M., and Sabater, B. (2010). Plastid ndh genes in plant evolution. Plant Physiol. Biochem. 48, 636–645. doi: 10.1016/j.plaphy.2010.04.009
Mihailova, G., Petkova, S., Buchel, C., and Georgieva, K. (2011). Desiccation of the resurrection plant Haberlea rhodopensis at high temperature. Photosynth. Res. 108, 5–13. doi: 10.1007/s11120-011-9644-2
Minh, B. Q., Nguyen, M. A. T., and Von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Morton, B. R., and Clegg, M. T. (1993). A chloroplast DNA mutational hotspot and gene conversion in a noncoding region near rbcL in the grass family (Poaceae). Curr. Genet. 24, 357–365. doi: 10.1007/BF00336789
Nadalin, F., Vezzi, F., and Policriti, A. (2012). GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13(Suppl. 1):S8. doi: 10.1186/1471-2105-13-S14-S8
Nazareno, A. G., Carlsen, M., and Lohmann, L. G. (2015). Complete chloroplast genome of Tanaecium tetragonolobum: the first Bignoniaceae plastome. PLoS ONE 10:e129930. doi: 10.1371/journal.pone.0129930
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nie, X., Lv, S., Zhang, Y., Du, X., Wang, L., Biradar, S. S., et al. (2012). Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 7:e36869. doi: 10.1371/journal.pone.0036869
Nott, A., Jung, H.-S., Koussevitzky, S., and Chory, J. (2006). Plastid-to-nucleus retrograde signaling. Annu. Rev. Plant Biol. 57, 739–759. doi: 10.1146/annurev.arplant.57.032905.105310
Palmer, J. D. (1985). Comparative organization of chloroplast genomes. Annu. Rev. Genet. 19, 325–354. doi: 10.1146/annurev.genet.19.1.325
Petrova, G., Moyankova, D., Nishii, K., Forrest, L., Tsiripidis, I., Drouzas, A. D., et al. (2015). The European paleoendemic Haberlea rhodopensis (Gesneriaceae) has an oligocene origin and a pleistocene diversification and occurs in a long-persisting refugial area in Southeastern Europe. Int. J. Plant Sci. 176, 499–514. doi: 10.1086/681990
Poliakov, A., Foong, J., Brudno, M., and Dubchak, I. (2014). GenomeVISTA-an integrated software package for whole-genome alignment and visualization. Bioinformatics 30, 2654–2655. doi: 10.1093/bioinformatics/btu355
Qian, J., Song, J., Gao, H., Zhu, Y., Xu, J., Pang, X., et al. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 8:e57607. doi: 10.1371/journal.pone.0057607
Ramundo, S., Rahire, M., Schaad, O., and Rochaix, J.-D. (2013). Repression of essential chloroplast genes reveals new signaling pathways and regulatory feedback loops in chlamydomonas. Plant Cell 25, 167–186. doi: 10.1105/tpc.112.103051
Ranwez, V., Harispe, S., Delsuc, F., and Douzery, E. J. P. (2011). MACSE: multiple alignment of coding SEquences accounting for frameshifts and stop codons. PLoS ONE 6:e22594. doi: 10.1371/journal.pone.0022594
Rott, M., Martins, N. F., Thiele, W., Lein, W., Bock, R., Kramer, D. M., et al. (2011). ATP synthase repression in tobacco restricts photosynthetic electron transport, CO2 assimilation, and plant growth by overacidification of the thylakoid lumen. Plant Cell 23, 304–321. doi: 10.1105/tpc.110.079111
Sablok, G., Mudunuri, S. B., Patnana, S., Popova, M., Fares, M. A., and La Porta, N. (2013). Chloromitossrdb: open source repository of perfect and imperfect repeats in organelle genomes for evolutionary genomics. DNA Res. 20, 127–133. doi: 10.1093/dnares/dss038
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, 686–689. doi: 10.1093/nar/gki366
Schwab, K. B., Schreiber, U., and Heber, U. (1989). Response of photosynthesis and respiration of resurrection plants to desiccation and rehydration. Planta 177, 217–227. doi: 10.1007/BF00392810
Sharma, M. R., Wilson, D. N., Datta, P. P., Barat, C., Schluenzen, F., Fucini, P., et al. (2007). Cryo-EM study of the spinach chloroplast ribosome reveals the structural and functional roles of plastid-specific ribosomal proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 19315–19320. doi: 10.1073/pnas.0709856104
Shaw, J., Lickey, E. B., Schilling, E. E., and Small, R. L. (2007). Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am. J. Bot. 94, 275–288. doi: 10.3732/ajb.94.3.275
Silva, S. R., Diaz, Y. C. A., Penha, H. A., Pinheiro, D. G., Fernandes, C. C., Miranda, V. F. O., et al. (2016). The chloroplast genome of Utricularia reniformis sheds light on the evolution of the ndh gene complex of terrestrial carnivorous plants from the Lentibulariaceae family. PLoS ONE 11:e0165176. doi: 10.1371/journal.pone.0165176
Stern, A., Doron-Faigenboim, A., Erez, E., Martz, E., Bacharach, E., and Pupko, T. (2007). Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res. 35, 506–511. doi: 10.1093/nar/gkm382
Sun, A.-Z., and Guo, F.-Q. (2016). Chloroplast retrograde regulation of heat stress responses in plants. Front. Plant Sci. 7:398. doi: 10.3389/fpls.2016.00398
Tiller, N., and Bock, R. (2014). The translational apparatus of plastids and its role in plant development. Mol. Plant 7, 1105–1120. doi: 10.1093/mp/ssu022
Timmis, J. N., Ayliffe, M. A., Huang, C. Y., and Martin, W. (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 5, 123–135. doi: 10.1038/nrg1271
Umate, P., Fellerer, C., Schwenkert, S., Zoryan, M., Eichacker, L. A., Sadanandam, A., et al. (2008). Impact of PsbTc on forward and back electron flow, assembly, and phosphorylation patterns of photosystem II in tobacco. Plant Physiol. 148, 1342–1353. doi: 10.1104/pp.108.126060
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., and Yu, J. (2010). KaKs_calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Wicke, S., Schneeweiss, G. M., DePamphilis, C. W., Muller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wright, F. (1990). The “effective number of codons” used in a gene. Gene 87, 23–29. doi: 10.1016/0378-1119(90)90491-9
Wu, C. S., Wang, Y. N., Hsu, C. Y., Lin, C. P., and Chaw, S. M. (2011). Loss of different inverted repeat copies from the chloroplast genomes of pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 3, 1284–1295. doi: 10.1093/gbe/evr095
Wu, F.-H., Chan, M.-T., Liao, D.-C., Hsu, C.-T., Lee, Y.-W., Daniell, H., et al. (2010). Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 10:68. doi: 10.1186/1471-2229-10-68
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yang, M., Zhang, X., Liu, G., Yin, Y., Chen, K., Yun, Q., et al. (2010). The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 5:e12762. doi: 10.1371/journal.pone.0012762
Yi, D. K., and Kim, K. J. (2012). Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PLoS ONE 7:e35872. doi: 10.1371/journal.pone.0035872
Zapata, J. M., Guéra, A., Esteban-Carrasco, A., Martín, M., and Sabater, B. (2005). Chloroplasts regulate leaf senescence: delayed senescence in transgenic ndhF-defective tobacco. Cell Death Differ. 12, 1277–1284. doi: 10.1038/sj.cdd.4401657
Zhang, H., Li, C., Miao, H., and Xiong, S. (2013). Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PLoS ONE 8:e80508. doi: 10.1371/journal.pone.0080508
Keywords: Haberlea rhodopensis, desiccation stress, chloroplast genome, SSR, site-specific selection, rbcL
Citation: Ivanova Z, Sablok G, Daskalova E, Zahmanova G, Apostolova E, Yahubyan G and Baev V (2017) Chloroplast Genome Analysis of Resurrection Tertiary Relict Haberlea rhodopensis Highlights Genes Important for Desiccation Stress Response. Front. Plant Sci. 8:204. doi: 10.3389/fpls.2017.00204
Received: 26 October 2016; Accepted: 03 February 2017;
Published: 20 February 2017.
Edited by:
Miguel Arenas, Institute of Molecular Pathology and Immunology of the University of Porto, PortugalReviewed by:
Antonio González-Martín, Complutense University of Madrid, SpainVitor C. Sousa, Institute of Ecology and Evolution, Switzerland
Yingjuan Su, Sun Yat-sen University, China
Copyright © 2017 Ivanova, Sablok, Daskalova, Zahmanova, Apostolova, Yahubyan and Baev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vesselin Baev, vebaev@plantgene.eu