Immunotherapeutic implications on targeting the cytokines produced in rhinovirus-induced immunoreactions
 Le Sang
Le Sang Xia Gong1
Xia Gong1  Linling Zhang
Linling Zhang- 1Department of Medicine, Shaoxing University, Shaoxing City, Zhejiang Province, China
- 2Department of Respiratory Medicine, Shaoxing People’s Hospital, Shaoxing City, Zhejiang Province, China
Rhinovirus is a widespread virus associated with several respiratory diseases, especially asthma exacerbation. Currently, there are no accurate therapies for rhinovirus. Encouragingly, it is found that during rhinovirus-induced immunoreactions the levels of certain cytokines in patients' serum will alter. These cytokines may have pivotal pro-inflammatory or anti-inflammatory effects via their specific mechanisms. Thus far, studies have shown that inhibitions of cytokines such as IL-1, IL-4, IL-5, IL-6, IL-13, IL-18, IL-25, and IL-33 may attenuate rhinovirus-induced immunoreactions, thereby relieving rhinovirus infection. Furthermore, such therapeutics for rhinovirus infection can be applied to viruses of other species, with certain practicability.
Introduction
Rhinovirus (RV), an ubiquitous and widespread respiratory virus, might be crucial in the occurrence and exacerbation of various chronic respiratory diseases, including bronchiectasis, chronic obstructive pulmonary disease, especially asthma exacerbation. Besides, RV is demonstrated as pivotal in wheezing of adults and children. For instance, children who suffer from RV-induced wheezing at an early age are more prone to develop persistent asthma. Also, once asthma is established, RV infections can constantly be a risk factor for acute wheezing illnesses (1, 2). RV is roughly divided into three subtypes (RV-A, B, and C), which have specific receptors, respectively. Generally speaking, it is identified that RV infection activates the Th2 inflammatory response while inhibiting the Th1 inflammatory response (3, 4). Such regularity may indicate that the inhibition of Th2 inflammation can play a role in curing RV infection.
At the same time, it is notable that there are relatively specific changes in pro-inflammatory cytokines and anti-inflammatory cytokines released by immunocytes during RV infection. Therefore, figuring out the relationship between cytokines and RV infection might be helpful for the identification of pathogens. In addition, studies showed that inflammatory response in the host might be attenuated after blocking several pro-inflammatory cytokines (5). Overall, viral infection is considered a self-limited disease. It is reasonable to presume that viral infection can be ameliorated after inhibitions of pro-inflammatory cytokines produced in RV-induced immunoreactions.
This article focuses on the alterations of cytokines during RV-induced immunoreactions and the outcome achieved after inhibitions of these expressed cytokines, trying to search for novel therapies for RV infection.
RV-related respiratory diseases
RV, a non-enveloped ssRNA virus, belongs to the family Picornaviridae, genus Enterovirus, and is considered one of the most common independent pathogenic factors of various respiratory diseases.
RV causes respiratory diseases worldwide, and is prevalent all year round, as evidenced by epidemiological investigations (6). The detection rates in certain countries are shown as 33.2%, 50.4%, and 26%, respectively (7–9). In a study, the pathogens in 592 children with community-acquired pneumonia (CAP) confirmed by radiograph were detected. As a result RV was found as an independent agent in 99 cases (29.0%) and as one of the concurrent agents in 73 cases with two or more viruses infected (40.7%) (10). A small sample study showed that approximately 90% of children who had wheezing triggered by RV when they were 3 years old would have asthma when 6 years old (11). It was found that approximately 20% to 40% of bronchiolitis or acute wheezing in infants were caused by RV which as a pathogen was second only to respiratory syncytial virus (RSV), and approximately 60% of acute exacerbation of chronic obstructive pulmonary disease (AECOPD) was also induced by RV (12–15).
Given RV's threat to human health, it is necessary to elucidate the mechanisms of RV infection and find new therapeutics.
Mechanisms in RV-induced cytokines production
The viral life cycle of RV
Picornaviruses are small, nonenveloped, icosahedral RNA viruses with positive-strand polarity. Even though partial picornavirus infections remain asymptomatic, numerous picornaviruses are crucial human and animal pathogens and may lead to diseases which will affect various organs and systems, such as respiratory and gastrointestinal tracts. Thus, the conclusion for the viral life cycle of RV may help understand the bioactivity of RV more deeply (16, 17).
The process of RV infecting airway epithelium generally contains specific procedures. RV enters the cytoplasm through receptor-mediated endocytosis, and virus RNA (vRNA) is dissociative after viral uncoating. Then the vRNA is transcribed in vesicles with the action of RNA polymerase (RNA-pol). Subsequently, complementary RNA (cRNA) is produced and new vRNA is formed. Ultimately, the new vRNA is translated to form the viral core structure, assembled with the pre-generated capsid, and finally budding and release are completed (Figure 1) (16, 17). Simultaneously, numerous cytokines are released during the process.
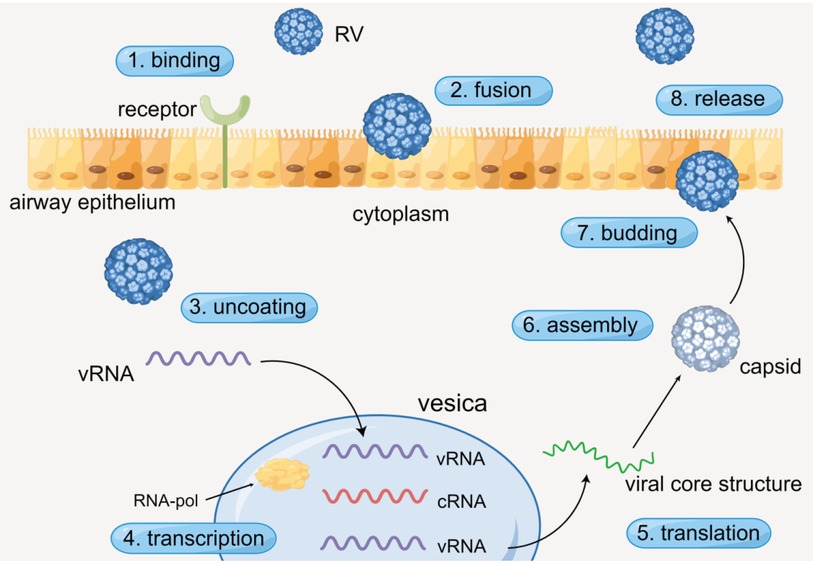
Figure 1. The viral life cycle of RV. By Figdraw.
The innate immune mechanism mediated by the toll-like receptors
When hosts have contact with RV, various cytokines will be produced via the innate immune mechanism mediated by the toll-like receptors. In human airway epithelial cells (HAECs), each of the three types of RV has the distinct receptor: RV-A is taken up by low-density lipoprotein (LDL) receptors, RV-B and much of RV-A are received by intercellular adhesion molecule-1 (ICAM-1), and RV-C is combined by cadherin-related family member 3 (CDHR3) (18, 19). RV enters epithelial cells by receptor-mediated endocytosis, and the ssRNA of RV is recognized by the toll-like receptor (TLR) 7 and 8 in the endosome, which then activates the myeloid differentiation primary response 88 (MYD88) and TIR domain-containing adapter inducing interferon β (TRIF) (1, 19, 20). Besides, RV in the cytoplasm is identified by melanoma differentiation-associated gene 5 (MDA-5) and retinoic acid-inducible gene 1 (RIG-1), activating the mitochondrial anti-viral signaling protein (MAVS) signaling pathway (20–22). MYD88, TRIFF, and MAVS ultimately activate interferon regulatory transfer factor (IRF) 3, 7, and NF-kB, leading to up-regulate the gene expression of cytokines such as IL-1 (IL-1α and IL-1β), IL-6, IL-12, IL-18, and type 1 interferon (INF I) (23–27). The cytokines above subsequently recruit and activate a series of immunocytes such as NK cells, dendritic cells (DC), and T/B cells, causing certain lesions such as airway remodeling (Figure 2) (28–30).
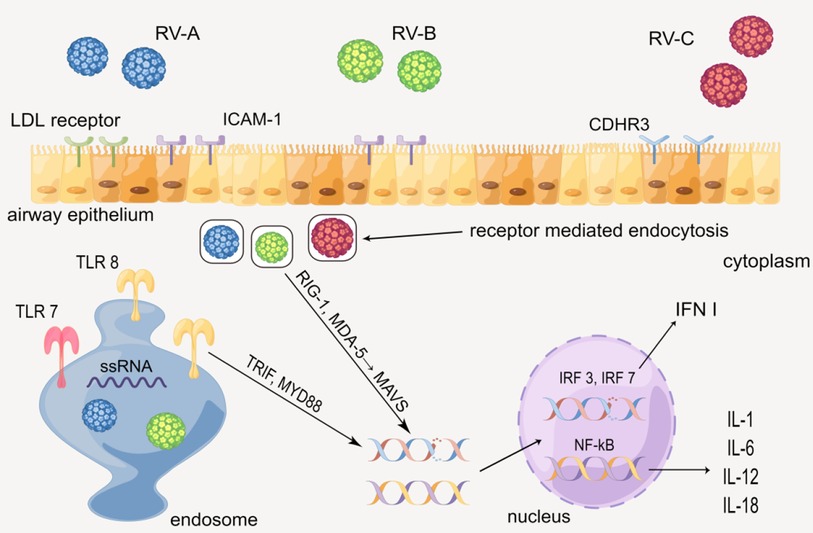
Figure 2. Production of cytokines through the innate immune mechanism mediated by the toll-like receptor. By Figdraw.
The acquired immune mechanism
On the other hand, acquired immunity is also indispensable in the secretion of cytokines. Epithelial cells immediately produce several alarmins such as IL-25, IL-33, and TSLP when RV invades HAECs, then recruiting innate lymphoid cell precursors (ILCP) and leading it to differentiate into ILC2 by up-regulating GATA3, retinoic acid receptor-related orphan receptor a (RORa), and T-cell factor 1 (TCF-1), etc. (31, 32). Subsequently, ILC2 produces Th2 cytokines such as IL-4, IL-5, IL-9, and IL-13. In addition, DC is also necessary for acquired immunity. As a professional antigen-presenting cell, DC presents antigen to naive T cells via the MHC class II molecular pathway. With the action of IL-4, naive T cells differentiate into helper T cells (Th) such as Th2, Th9, and Th17, then various cytokines such as IL-9, IL-17, and IFN are produced. In this process, certain immunocytes such as mast cells will also secrete IL-5, IL-13, and granulocyte-macrophage colony-stimulating factor (GM-CSF) (33). Eventually, sequential cytokines lead to the infiltration of a series of immunocytes and inflammatory response, which might impair lung function ultimately (Figure 3) (26, 29, 34).
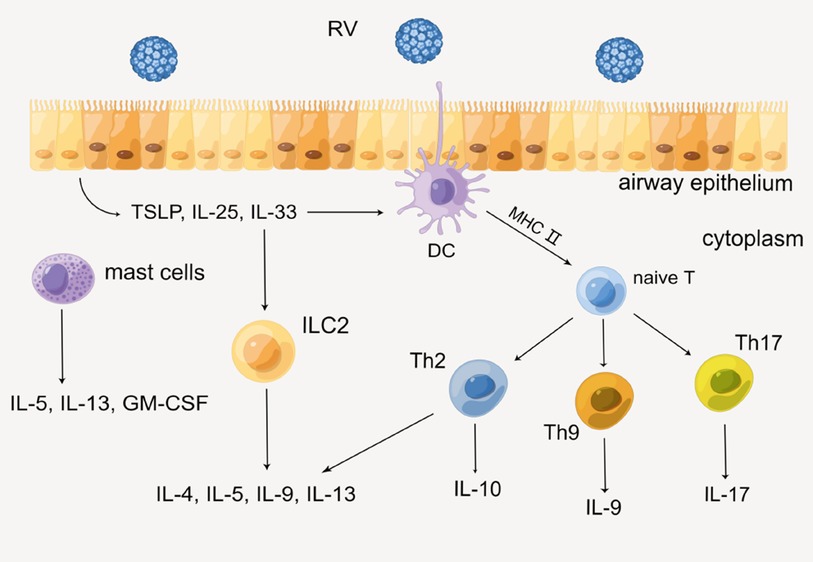
Figure 3. Production of cytokines through the acquired immune mechanism. By Figdraw.
Taken together, the alterations of cytokines during RV infection might be traceable. Therefore, searching the relationship between alterations of cytokines and viral infection might help identify the pathogens. For instance, RV and influenza virus (IFV) are common viruses causing similar diseases and symptoms. The level of IL-5 in patients' serum during RV infection was found to be higher than that of patients infected with IFV. Detecting the level of IL-5 might be utilized to distinguish between RV and IFV infection (35). In addition, a study showed that the level of RV-induced IL-6 generally peaked at 48 h postinfection. As compared with RSV infection, IL-8 increased more rapidly after RV-C infection. Besides, the levels of RV-induced IL-6 and IL-8 were lower than those induced by RSV. Another recent study also showed that the type of immunoreactions depends on RV serotypes. For example, as compared with RV-16, RV-1B produced higher IFN-β, IFN-λ1/3, CXCL10, IL-6, IL-8, and IL-18 levels (36). Accordingly, partially identifying pathogens by comparing the cytokines' alterations might be feasible, which can direct empirical clinical work to an extent (37).
Cytokines produced in RV-induced immunoreactions
Inhibitions of pro-inflammatory cytokines
Therapies for RV infection through anti-interleukin may be practicable. For example, IL-1 is a pivotal inflammatory cytokine leading to neutrophil aggregation, mucus metaplasia, and inflammatory reactions. In the study of Samuel et al, it was found that IL-1α and IL-1β were increased in supernatant and associated with cell death following RV infection (38). It was found that blocking the IL-1 signaling pathway significantly reduced the RV-induced cytokines, consistent with the experimental results of Persson et al. (26). In their research, they found that the main neutrophil chemotactic protein, CXCL1/KC, was less induced at asthma exacerbation in IL-1β knockout mouse models. At the same time, deficiency in IL-1β showed the tendency towards reduced induction of TSLP and IL-33 gene levels. These phenomena may be associated with the amelioration of RV-induced exacerbation of asthma (39–41). Besides, studies showed that dual-specificity phosphatases (DUSP) derived from primary bronchial epithelial cells inhibited IL-1β-mediated inflammatory response, showing the feasibility of therapies for RV infection targeting IL-1. The idea was also confirmed in patients with IFV infection (5, 42, 43). Nowadays, there have been several IL-1β-neutralising biologicals such as anakinra and canakinumab, and their effects in therapies for RV infection still need further clarification.
IL-4 and IL-13 are generally produced in the inflammatory reactions with ILC2 as the core (39). Previous research demonstrated that IL-4 produced by non-T cells was crucial for the aggregation of Th2 cells (33). Another study showed that IL-4 and IL-13 produced remarkable dose-dependent inhibition of RV-induced IFN-β mRNA expression and IFN-β protein release at 8 h after infection, which may partially explain why IL-4/IL-13 promotes the RV infection (44). Meanwhile, a study found that the tight junctions of lung epithelium were destroyed, and immunoreactions against RV were weakened by long-term exposure to IL-13. What's worse, lung function can be damaged (45). Fortunately, targeting IL-4 and IL-13 in therapies for RV infection is highly likely achievable. For instance, blocking IL-13 might alleviate RV-induced diseases in children (46). And the hospitalization rate of asthmatics was reduced after administrating the IL-4 antagonist (dupilumab) and IL-13 antagonist (lebrikizumab or tralokinumab). It is reasonable to assume that L-4 and IL-13 blockade decreases the Th2 response so that they relieve RV-related diseases, which may be verified in the research of David et al. They found that anti-IL-4, anti-IL-13, and anti-IL-4/13 reduced airway inflammation. Besides, it might be more significant to administrate with anti-IL-4/13 concurrently. The mice treated with combined anti-IL-4/13 were more manifestly protected from arteriolar hypertrophy and fibrosis, whereas mice treated with anti-IL-4 less suffered from fibrosis and still had evidence of arteriolar hypertrophy (47). Yet, in the meanwhile, it was shown that IL-4 and 13 antagonists may increase the risk of adverse events as compared with patients utilizing IL-5 and IgE antagonists, needing further study (48).
IL-5 is mainly secreted by Th2 immunocytes, and related to severe asthma. There is a higher level of IL-5 in asthmatics’ serum after RV infection (23, 49). Currently, several monoclonal antibodies against IL-5 show positive effects. For instance, mepolizumab and reslizumab show the ability to inhibit the binding of IL-5 and IL-5R, while benralizumab directly inhibits IL-5Rα (50). The medicine above reduces the eosinophils in patients’ peripheral blood, which might relieve RV-induced asthma. In November 2021, mepolizumab was licensed by China's Drug Administration (NMPA) for utilization to treat adult eosinophilic granulomatous polyangiitis. Similarly, it is believable that treatments for RV infection via anti-interleukin might also have feasibility. In addition, a study showed that after asthmatics utilized IL-5 antagonists, there were more IFN-α secreted by plasmacytoid dendritic cells (pDC) in patients' serum, inspiring that anti-IL-5 can be effective for curing RV-related diseases such as asthma (51).
Neutrophils-mediated epithelial airway damage and airway hyper-responsiveness (AHR), induced by RV, might be triggered by IL-6 and IL-8 (37). IL-6 is produced by several immunocytes such as macrophages and DC. And it has two types of receptors (IL-6R): membrane-bound IL-6R and soluble IL-6R, cooperating with TGF-β to boost the differentiation of Th17 cells (52–54). Regarding IL-6 antagonists, Tocilizumab has been routinely utilized to treat rheumatic immune diseases, showing great effectiveness. Besides, other available IL-6 inhibitors such as sirukumab, olokizumab, and siltuximab are limited to utilization due to their high cost, invasive delivery techniques, and high immunogenicity. Therefore, novel IL-6 inhibitors are required. Generally, there are three types of IL-6 inhibitors, namely IL-6 production inhibitors, IL-6 expression inhibitors, and IL-6 signaling pathway inhibitors. Encouragingly, several novel IL-6 antagonists have been found. For example, phenyl ring might reduce the level of LPS-induced IL-6, steroids from marine organisms might inhibit the IL-6 mRNA expression, epoxyresibufogenin-3-formates might have the pharmacological effect to antagonize IL-6R, and thiophene derivatives might be IL-6-induced STAT3 inhibitors (54). However, even though new IL-6 antagonists emerge endlessly, their clinical application in RV-related diseases lacks enough research.
IL-11, belongs to the IL-6 family, with pro-inflammatory and anti-inflammatory effects concurrently (55, 56). During RV infection, IL-11 is also released (23, 24). IL-11 and α-IL-11R form a compound with glycoprotein 130, activating the downstream. signal pathway, and finally exerting biological effects (57, 58). It is believed that IL-11 is redundant in adults, and may lead to idiopathic pulmonary fibrosis and asthma (59–62). In addition, treatments for RV infection targeting IL-11 show possibilities due to the combination between IL-11 and its receptor is likely to be blocked (61, 63). For instance, a study showed that recombinant mouse IL-11Rα Fc chimeric protein, an IL-11 antagonist, inhibited Th17 cell-mediated neuroinflammatory response (64). However, there is insufficient research on the relationship between IL-11 and RV infection.
IL-15, containing a series of cytokines, is produced by certain antigen-presenting cells such as DC, monocytes, and macrophages during RV infection. The productions of natural kill cells, CD8+ T cells, and IFN-γ are stimulated after IL-15 binds to its receptor, thus attenuating viral infection (65, 66). A study showed that IL-15 isoform suppressed the effect of IL-15 in several inflammatory diseases (67). Yet, the compound of IL-15 and its receptor (sIL-15Rα) had no positive impact on the lung function of asthmatic mice (68).
Previous research showed that after RV infection, the level of IL-17 secreted by various immunocytes including mast cells and macrophages would increase, leading to neutrophil recruitment and AHR, even damaging lung function (69, 70). Currently, the monoclonal antibodies against IL-17 such as secukinumab, lxekizumab, and brodalumab are utilized to cure diseases such as psoriasis, among which secukinumab has been approved by the FDA. Additionally, neutralizing IL-13 and IL-17 simultaneously may positively affect asthmatics from eosinophilia, mucus hyperplasia, and airway hyperreactivity and abolish the neutrophilic inflammation, indicating the potential of blocking IL-13 and IL-17 for therapies for RV infection (47).
IL-18, as an IFN-γ inducible factor related to RV, is produced by inflammasome, and might enhance epithelial cell differentiation (25, 71). The Th2 inflammatory and allergic reactions might be promoted after IL-18 combines with its receptor (IL-18R) (72, 73). And it was found that IL-18 increased in severe asthmatic sputum and serum (74). In addition, the IL-18 axis also exists in severe asthma as proved by machine learning. Also, an endogenous antagonist against IL-18, IL-18 binding protein (IL-18BP), might neutralize IL-18 and IL-18R to inhibit the biological effects (75). Accordingly, blocking IL-18 to ameliorate RV infection can be practicable.
Thus far, research shows that HAECs from asthmatics release IL-25 during RV infection, enhancing Th2 inflammation and closely related to severe asthma (76, 77). Regarding IL-25 antagonists, LNR125 was confirmed. to relieve viral infection by up-regulating type I&III interferon genes and down-regulating type II inflammatory genes such as CCL26, IL1RL1, and IL-25 receptor, verifying that the therapies for RV infection in the basis of anti-IL-25 might be feasible (78).
Several studies show that IL-33 might have the capability of increasing the production of Th2 cytokines such as IL-4, IL-5, and IL-13, and promote RV-induced inflammation (79). For example, IL-33 boosts the release of IL-5 and IL-13 and the virus-specific Th2 inflammation via the IL-33/ST2 (IL-33R) signaling axis in allergic asthmatics (80, 81). Also, IL-33 was detected in the supernatant of HAECs from RV-infected patients, while the phenomenon was entirely repressed after blocking IL-33, indicating that anti-IL-33 might be utilized in treatments for RV infection (39). The results are consistent with the previous conclusion concerning anti-IL-33 (itepekimab) (82). Besides, 25 (OH)-Vitamin D3 [25 (OH)-VitD3] has also been proven to have the effect of antagonizing ST2 (83, 84). In addition, in asthmatic children with RV infection, soluble ST2 which may neutralize IL-33 is significantly reduced by low serum levels of 25 (OH)-VitD3. The low level of 25 (OH)-VitD3 might reduce the production of interferon β (IFN-β) (85). Thus, 25 (OH)-VitD3 can be applied to therapies for RV infection in the future.
Currently, studies show that RV infection might activate peripheral transforming growth factor β (TGF-β), which might subsequently recruit neutrophils and monocytes, and induce Th17 cells and regulatory T cells (Tregs) through downstream. forkhead box P3+ (FOXP3) and retinoic acid-related orphan receptor γ (RORγ), ultimately causing airway remodeling and inflammation. After RV infection, the combination of TGF-β and its receptor on the cell surfaces increases, promoting viral replication and the severity of asthma (52). Besides, it was found that the inflammation was alleviated after the TGF-β pathway was antagonized (86). Therefore, it might be feasible to block TGF-β for therapies for RV infection.
The IFN-β/programmed death ligand-1 (PD-L1) axis also affects asthmatic children with RV infection. A short cohort study showed that in the control group, the IFN released by PBMC was related to the production of PD-L1. Yet, the study also showed that higher IFN-β levels were associated with the lower level of PD-L1 and better lung function (87). To summarize, PD-L1 might be a pro-inflammatory cytokine in RV-related asthmatics. Currently, PD-L1 has been widely utilized in anti-tumor immunotherapies, but whether it can be utilized in therapies for RV infection requires further research (Table 1).
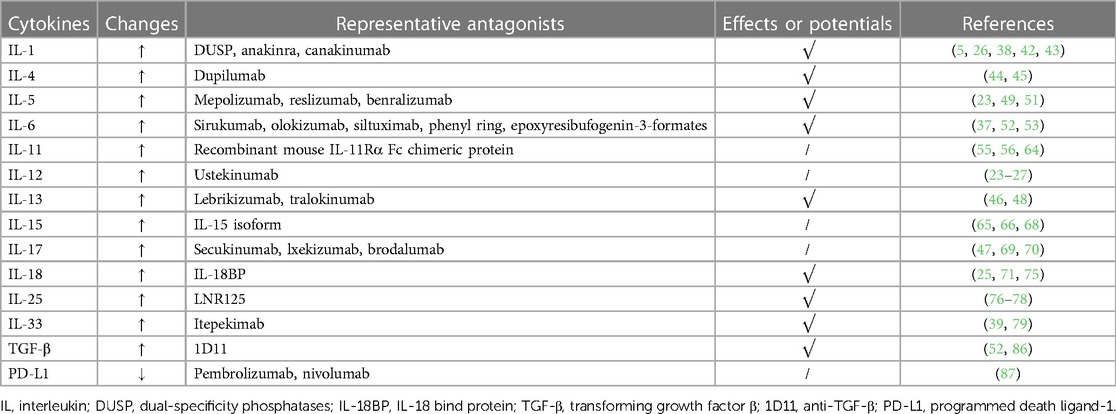
Table 1. Pro-inflammatory cytokines.
Utilizations of anti-inflammatory cytokines
Several anti-inflammatory cytokines are pivotal in immunoreactions against RV. It is quite necessary to figure out their alterations and effects due to their potentials for curing RV infection.
The level of GM-CSF is affected by diversified factors. For instance, GM-CSF is produced by epithelial cells, fibroblasts, and mast cells stimulated by RV (88–90). And the production can be inhibited by certain cytokines such as IFN-γ, IL-1β, IL-4, and IL-10 (91–93). GM-CSF activates the downstream, Janus kinase (JAK) 2, STAT5, and the phosphatidylinositol-3-kinase (PI3K) pathway after binding to its receptor, then leading to the recruitment and activation of several inflammatory cells such as monocyte macrophages and DC (94–96). A study showed that inhaled sargramostim (yeast-derived recombinant human GM-CSF) might significantly improve oxygenation in hospitalized COVID-19 patients with hypoxic respiratory failure, and subcutaneous injection of sargramostim might shorten mechanical ventilation of patients with severe sepsis (97, 98). By analogy, GM-CSF probably has the potential for treatments for RV infection.
IFN can also be produced by pDC in RV-induced immunoreactions (99). It appears that IFN-α and IFN-β couple to the same receptor, which is different from the receptor for IFN-λ. Type I IFN (IFN-α and IFN-β) receptors (IFNAR), are coupled to the JAK1 and the Tyrosine kinase 2 (TYK2). After Type I IFN binds to IFNAR, JAK1 and TYK2 are activated, which then phosphorate signal translator and activator of translation (STAT) 1 and 2. The compound of STAT homodimers and interferon-stimulated gene factor (ISGF) 3 forms, eventually inducing ISG transcription in the nucleus (100–102). The process above has the effects against viral infection. Type I IFN also recruits immunocytes such as NK cells and DC, exerting immunifaction (103, 104). In addition, Type I IFN signaling induces the production of the 2′-5′-oligoadenylate synthetase (OAS) family, which includes OAS1, OAS2, OAS3 and OAS-like (OASL) proteins. It was identified that OASL is manifestly induced upon viral infection through the involvement of the RNA sensor, to promote the anti-viral type I IFN response (105). In several studies, deficiency of various IFN has been verified utilizing cultured human bronchial epithelial cells (HBECs) from asthmatics after RV infection in vitro, which leads to the deficiency of IFN-β and IFN-λ in primary bronchial epithelial cells (PBECs) and deficiency of IFNs (γ, α, β and λ) and IL-15 in bronchoalveolar lavage (BAL) cells (23). This result is accorded with that of Lin et al, showing the feasibility of utilizing IFN for alleviating RV infection (106).
IL-10 was identified over two decades ago and the most studied suppressive molecule of the immune system so far. IL-10, found to be secreted by Th2 cells, plays a critical role in anti-inflammatory and autoimmune pathologies by limiting immunoreactions to pathogens (107). Thus far, several studies show that IL-10 is likely to be produced by various cells (108, 109). As an essential anti-inflammatory cytokine family released in RV-induced immunifaction, it includes IL-9, IL-20, IL-22, IL-24, IL-26, IL-28A, IL-28B, and IL-29 (110, 111). The IL-10/IL-10R axis exerts effects through the downstream. mechanism similar to that of Type I IFN (112–114). The IL-10/IL-10R axis triggers a series of signaling cascades mediated by the JAK signal transducer and activator of the transcription (STAT) pathway, especially by STAT3. signaling through the IL-10/IL-10R axis regulates several steps of the immune response, from decreasing cytokine gene expression to inhibiting the antigen-presenting ability of monocytes via down-regulating the MHC class II molecules (115). Besides, IL-10 has been found to have the capability of preventing apoptosis by enhancing the PI3K/Protein Kinase B (AKT) cascade and promoting the expression of anti-apoptotic factors as Bcl-2 and Bcl-xl, while weakening that of caspase-3 (109). In the meantime, IL-10 attenuates the toxicity of pathogens, and reduces the cytokines released by macrophages and DC, thus producing anti-inflammatory effects (116–118). Generally, IL-10 is considered crucial in anti-inflammation. Its clinical application in treating RV infection is worthy of further study.
It was found that under the action of several pro-inflammatory cytokines, HAECs will secrete several IL-1 receptor antagonists (IL-1RA) such as secreted IL-1RA (sIL-1RA) and intracellular IL-1Ra (icIL-1Ra). Among them, Levine et al. proposed that icIL-1Ra type I release from HAEC may modulate IL-1 bioactivity in the airway microenvironment, weakening inflammation subsequently (119). The phenomenon indicates whether there are more endogenous antagonists of pro-inflammatory cytokines in the host, requiring more exploration (Table 2).
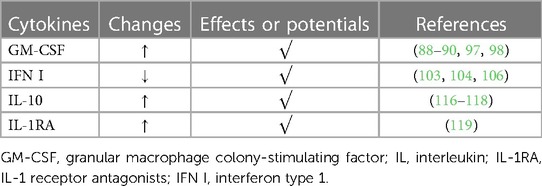
Table 2. Anti-inflammatory cytokines.
Conclusion
The threat from respiratory viruses is nonnegligible due to their capacity to cause various diseases. RV, as a common respiratory virus, is closely related to chronic respiratory diseases, especially asthma exacerbation. Currently, several experiments show that during RV-induced immunoreactions the levels of several cytokines in the hosts' serum will alter, indicating whether it is achievable to identify pathogens according to such alterations. In general, viral infection is known as a self-limited disease, which may drive us to assume viral infection can be ameliorated after the virus-induced immunoreactions are inhibited. Fortunately, researchers find that inflammatory response is inhibited after blocking several pro-inflammatory cytokines such as IL-1, IL-4, IL-5, IL-6, IL-25, and IL-33, showing the feasibility of therapies for RV infection targeting RV-induced cytokines. Several endogenous cytokines such as IL-10, Type I IFN, and IL-1RA might inhibit RV-induced inflammatory response, with practicability in treatments for RV infection.
Future perspective
RV is crucial in respiratory diseases, and also positively associated with asthma severity. However, therapeutics targeting RV are deficient, and it is difficult to identify the similar respiratory pathogens. Searching for novel and accurate methods of pathogenic diagnosis is necessary, which may guide empirical therapies in clinical work. In numerous experiments, researchers have found that the levels of cytokines in patients' serum after RV infection will change. What's more, these cytokines may be antagonized or directly utilized to attenuate RV infection. It is worthy of summarizing such cytokines' changes and searching for therapies for RV infection.
Currently, research shows that several antagonists of pro-inflammatory cytokines have manifest potentials for anti-inflammation. For example, mepolizumab, the IL-5 antagonist, has been widely utilized to treat eosinophilic granuloma. In the future, it might be applied to cure RV-related diseases. Besides, additional statistics concerning RV-induced cytokine alterations are required to further clarify the relationship between cytokines and RV infection.
In general, the pro-inflammatory cytokines mentioned in the article boost inflammation during RV infection. Yet, the opposite conclusion was reached after administrating IL-1β inhibitors such as NLRP3 KO in immature mice, that RV-induced type II immunoreactions were aggravated (120). It indicated that whether several cytokines such as IL-1 were indispensable in establishing immunity in immature individuals, is worthy of more research.
Even though RV is prevalent widely, its virulence is relatively weak. While RV induces relatively specific alterations of cytokines, different viruses may induce similar immune reactions to an extent. Therefore, it is reasonable to propose that therapies for RV infection targeting cytokines can also be applied to other more threatening viruses. For instance, tocilizumab has been approved to treat COVID-19 by the FDA and NMPA, similarly verifying the practicability of such therapies for RV infection.
Author contributions
LS: Writing – original draft. XG: Writing – review & editing. YH: Writing – review & editing. LZ: Writing – review & editing. JS: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
RV, rhinovirus; IL, interleukin; CAP, community-acquired pneumonia; RSV, respiratory syncytial virus; AECOPD, acute exacerbation of chronic obstructive pulmonary disease; vRNA, virus RNA; RNA-pol, RNA polymerase; cRNA, complementary RNA; HAECs, human airway epithelial cells; HBECs, human bronchial epithelial cells; PBECs, primary bronchial epithelial cells; BAL, bronchoalveolar lavage; LDL, low-density lipoprotein; ICAM-1, intercellular adhesion molecule-1; CDHR3, cadherin-related family member 3; TLR, toll-like receptor; MYD88, myeloid differentiation primary response 88; TRIF, TIR domain-containing adapter inducing interferon β; MDA-5, melanoma differentiation-associated gene 5; RIG-1, retinoic acid-inducible gene 1; MAVS, mitochondrial anti-viral signaling protein; IRF, interferon regulatory transfer factor; IFN I, interferon type 1; DC, dendritic cells; ILCP, lymphoid cell precursors; RORa, retinoic acid receptor-related orphan receptor a; TCF-1, T-cell factor 1; Th, helper T cells; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFV, influenza virus; DUSP, dual-specificity phosphatases; NMPA, China's Drug Administration; pDC, plasmacytoid dendritic cells; AHR, airway hyper-responsiveness; IL-18BP, IL-18 bind protein; 25 (OH)-VitD3, 25 (OH)-Vitamin D3; IFN-β, interferon β; TGF-β, transforming growth factor β; Tregs, regulatory T cells; FOXP3, forkhead box P3+; RORγ, retinoic acid-related orphan receptor γ; PD-L1, programmed death ligand-1; IFNAR, IFN-α/β receivers; JAK, janus kinase; PI3K, phosphatidylinositol-3-kinase; AKT, protein kinase B; TYK2, tyrosine kinase 2; STAT, signal translator and activator of translation; ISGF, interferon-stimulated gene factor; OAS, oligoadenylate synthetase; OASL, OAS-like; IL-1RA, IL-1 receptor antagonists; sIL-1RA, secreted IL-1RA; icIL-1Ra, intracellular IL-1Ra; 1D11, anti-TGF-β.
References
1. Jacobs S, Lamson D, St George K, Walsh T. Human rhinoviruses. Clin Microbiol Rev. (2013) 26(1):135–62. doi: 10.1128/CMR.00077-12
2. Singh M, Lee SH, Porter P, Xu C, Ohno A, Atmar RL, et al. Human rhinovirus proteinase 2A induces TH1 and TH2 immunity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2010) 125(6):1369–78. doi: 10.1016/j.jaci.2010.02.035
3. Fedele G, Schiavoni I, Nenna R, Pierangeli A, Frassanito A, Leone P, et al. Analysis of the immune response in infants hospitalized with viral bronchiolitis shows different Th1/Th2 profiles associated with respiratory syncytial virus and human rhinovirus. Pediatr Allergy Immunol. (2018) 29(5):555–7. doi: 10.1111/pai.12919
4. Yuan X, Li Y, Shen Y, Yang J, Jin Y. Clinical and Th1/Th2 immune response features of hospitalized children with human rhinovirus infection. J Med Virol. (2020) 92(1):26–33. doi: 10.1002/jmv.25587
5. Manley GCA, Stokes CA, Marsh EK, Sabroe I, Parker LC. DUSP10 negatively regulates the inflammatory response to rhinovirus through interleukin-1β signaling. J Virol. (2019) 93(2):01659–18. doi: 10.1128/JVI.01659-18
6. Jartti T, Lee WM, Pappas T, Evans M, Lemanske RF Jr, Gern JE. Serial viral infections in infants with recurrent respiratory illnesses. Eur Respir J. (2008) 32(2):314–20. doi: 10.1183/09031936.00161907
7. Hartiala M, Lahti E, Forsstrom V, Vuorinen T, Ruuskanen O, Peltola V. Characteristics of hospitalized rhinovirus-associated community-acquired pneumonia in children, Finland, 2003–2014. Front Med (Lausanne). (2019) 6:235. doi: 10.3389/fmed.2019.00235
8. Ko F, Chan P, Chan R, Chan K, Ip A, Kwok A, et al. Rhinovirus and Bacteria synergistically induce IL-17C release from human airway epithelial cells to promote neutrophil recruitment. Respir Res. (2019) 20(1):210. doi: 10.1186/s12931-019-1181-0
9. Linam WM, Marrero EM, Honeycutt MD, Wisdom CM, Gaspar A, Vijayan V. Focusing on families and visitors reduces healthcare associated respiratory viral infections in a neonatal intensive care unit. Pediatr Qual Saf. (2019) 4(6):e242. doi: 10.1097/pq9.0000000000000242
10. Esposito S, Daleno C, Tagliabue C, Scala A, Tenconi R, Borzani I, et al. Impact of rhinoviruses on pediatric community-acquired pneumonia. Eur J Clin Microbiol Infect Dis. (2012) 31(7):1637–45. doi: 10.1007/s10096-011-1487-4
11. Jackson DJ, Gangnon RE, Evans MD, Roberg KA, Anderson EL, Pappas TE, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. (2008) 178(7):667–72. doi: 10.1164/rccm.200802-309OC
12. George SN, Garcha DS, Mackay AJ, Patel AR, Singh R, Sapsford RJ, et al. Human rhinovirus infection during naturally occurring COPD exacerbations. Eur Respir J. (2014) 44(1):87–96. doi: 10.1183/09031936.00223113
13. Jartti T, Lehtinen P, Vuorinen T, Ruuskanen O. Bronchiolitis: age and previous wheezing episodes are linked to viral etiology and atopic characteristics. Pediatr Infect Dis J. (2009) 28(4):311–7. doi: 10.1097/INF.0b013e31818ee0c1
14. Marguet C, Lubrano M, Gueudin M, Le Roux P, Deschildre A, Forget C, et al. In very young infants severity of acute bronchiolitis depends on carried viruses. PLoS One. (2009) 4(2):25. doi: 10.1371/journal.pone.0004596
15. Midulla F, Scagnolari C, Bonci E, Pierangeli A, Antonelli G, De Angelis D, et al. Respiratory syncytial virus, human bocavirus and rhinovirus bronchiolitis in infants. Arch Dis Child. (2010) 95(1):35–41. doi: 10.1136/adc.2008.153361
16. Chase AJ, Semler BL. Viral subversion of host functions for picornavirus translation and RNA replication. Future Virol. (2012) 7(2):179–91. doi: 10.2217/fvl.12.2
17. Zell R. Picornaviridae—the ever-growing virus family. Arch Virol. (2017) 163(2):299–317. doi: 10.1007/s00705-017-3614-8
18. Basnet S, Palmenberg AC, Gern JE. Rhinoviruses and their receptors. Chest. (2019) 155(5):1018–25. doi: 10.1016/j.chest.2018.12.012
19. Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, et al. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc Natl Acad Sci U S A. (2015) 112(17):5485–90. doi: 10.1073/pnas.1421178112
20. Triantafilou K, Vakakis E, Richer EA, Evans GL, Villiers JP, Triantafilou M. Human rhinovirus recognition in non-immune cells is mediated by toll-like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune response. Virulence. (2011) 2(1):22–9. doi: 10.4161/viru.2.1.13807
21. Sykes A, Edwards R, Macintyre J, del Rosario A, Bakhsoliani E, Trujillo Torralbo MB, et al. Rhinovirus 16-induced IFN-α and IFN-β are deficient in bronchoalveolar lavage cells in asthmatic patients. J Allergy Clin Immunol. (2012) 129(6):1506–14. doi: 10.1016/j.jaci.2012.03.044
22. Wang Q, Nagarkar DR, Bowman ER, Schneider D, Gosangi B, Lei J, et al. Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J Immunol. (2009) 183(11):6989–97. doi: 10.4049/jimmunol.0901386
23. Hansel TT, Tunstall T, Trujillo Torralbo MB, Shamji B, Del Rosario A, Dhariwal J, et al. A comprehensive evaluation of nasal and bronchial cytokines and chemokines following experimental rhinovirus infection in allergic asthma: increased interferons (IFN-γ and IFN-λ) and type 2 inflammation (IL-5 and IL-13). EBioMedicine. (2017) 19(–):128–38. doi: 10.1016/j.ebiom.2017.03.033
24. Jazaeri S, Goldsmith AM, Jarman CR, Lee J, Hershenson MB, Lewis TC. Nasal interferon responses to community rhinovirus infections are similar in controls and children with asthma. Ann Allergy Asthma Immunol. (2021) 126(6):690–5. doi: 10.1016/j.anai.2021.01.023
25. Piper SC, Ferguson J, Kay L, Parker LC, Sabroe I, Sleeman MA, et al. The role of interleukin-1 and interleukin-18 in pro-inflammatory and anti-viral responses to rhinovirus in primary bronchial epithelial cells. PLoS One. (2013) 8(5):e63365. doi: 10.1371/journal.pone.0063365
26. Shariff S, Shelfoon C, Holden NS, Traves SL, Wiehler S, Kooi C, et al. Human rhinovirus infection of epithelial cells modulates airway smooth muscle migration. Am J Respir Cell Mol Biol. (2017) 56(6):796–803. doi: 10.1165/rcmb.2016-0252OC
27. Stokes CA, Ismail S, Dick EP, Bennett JA, Johnston SL, Edwards MR, et al. Role of interleukin-1 and MyD88-dependent signaling in rhinovirus infection. J Virol. (2011) 85(15):7912–21. doi: 10.1128/JVI.02649-10
28. Bosco A, Wiehler S, Proud D. Interferon regulatory factor 7 regulates airway epithelial cell responses to human rhinovirus infection. BMC Genomics. (2016) 17(76):016–2405. doi: 10.1186/s12864-016-2405-z
29. Leigh R, Oyelusi W, Wiehler S, Koetzler R, Zaheer RS, Newton R, et al. Human rhinovirus infection enhances airway epithelial cell production of growth factors involved in airway remodeling. J Allergy Clin Immunol. (2008) 121(5):1238–45. doi: 10.1016/j.jaci.2008.01.067
30. Yang Z, Mitländer H, Vuorinen T, Finotto S. Mechanism of rhinovirus immunity and asthma. Front Immunol. (2021) 12:731846. doi: 10.3389/fimmu.2021.731846
31. Gold MJ, Antignano F, Halim TY, Hirota JA, Blanchet MR, Zaph C, et al. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J Allergy Clin Immunol. (2014) 133(4):1142–8. doi: 10.1016/j.jaci.2014.02.033
32. Li Y, Chen S, Chi Y, Yang Y, Chen X, Wang H, et al. Kinetics of the accumulation of group 2 innate lymphoid cells in IL-33-induced and IL-25-induced murine models of asthma: a potential role for the chemokine CXCL16. Cell Mol Immunol. (2019) 16(1):75–86. doi: 10.1038/s41423-018-0182-0
33. Voehringer D, Reese TA, Huang X, Shinkai K, Locksley RM. Type 2 immunity is controlled by IL-4/IL-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J Exp Med. (2006) 203(6):1435–46. doi: 10.1084/jem.20052448
34. Akdis CA, Arkwright PD, Brüggen MC, Busse W, Gadina M, Guttman Yassky E, et al. Type 2 immunity in the skin and lungs. Allergy. (2020) 75(7):1582–605. doi: 10.1111/all.14318
35. To KKW, Lu L, Fong CHY, Wu AKL, Mok KY, Yip CCY, et al. Rhinovirus respiratory tract infection in hospitalized adult patients is associated with TH2 response irrespective of asthma. J Infect. (2018) 76(5):465–74. doi: 10.1016/j.jinf.2018.02.005
36. Kim JH, Jang JY, Jang YJ. Human rhinovirus serotypes induces different immune responses. Virol J. (2021) 18(1):232. doi: 10.1186/s12985-021-01701-1
37. Vandini S, Calamelli E, Faldella G, Lanari M. Immune and inflammatory response in bronchiolitis due to respiratory syncytial virus and rhinovirus infections in infants. Paediatr Respir Rev. (2017) 24:60–4. doi: 10.1016/j.prrv.2016.11.006
38. Montgomery ST, Frey DL, Mall MA, Stick SM, Kicic A. Rhinovirus infection is associated with airway epithelial cell necrosis and inflammation via interleukin-1 in young children with cystic fibrosis. Front Immunol. (2020) 11:596. doi: 10.3389/fimmu.2020.00596
39. Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo Torralbo MB, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. (2014) 190(12):1373–82. doi: 10.1164/rccm.201406-1039OC
40. Mahmutovic Persson I, Menzel M, Ramu S, Cerps S, Akbarshahi H, Uller L. IL-1β mediates lung neutrophilia and IL-33 expression in a mouse model of viral-induced asthma exacerbation. Respir Res. (2018) 19(1):018–0725. doi: 10.1186/s12931-018-0725-z
41. Uller L, Leino M, Bedke N, Sammut D, Green B, Lau L, et al. Double-stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon- in bronchial epithelial cells from donors with asthma. Thorax. (2010) 65(7):626–32. doi: 10.1136/thx.2009.125930
42. Bawazeer AO, Rosli S, Harpur CM, Docherty CA, Mansell A, Tate MD. Interleukin-1β exacerbates disease and is a potential therapeutic target to reduce pulmonary inflammation during severe influenza A virus infection. Immunol Cell Biol. (2021) 99(7):737–48. doi: 10.1111/imcb.12459
43. Tate MD, Ong JDH, Dowling JK, McAuley JL, Robertson AB, Latz E, et al. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci Rep. (2016) 6:27912. doi: 10.1038/srep27912
44. Contoli M, Ito K, Padovani A, Poletti D, Marku B, Edwards MR, et al. Th2 cytokines impair innate immune responses to rhinovirus in respiratory epithelial cells. Allergy. (2015) 70(8):910–20. doi: 10.1111/all.12627
45. Huang Z, Liu J, Ong H, Yuan T, Zhou X, Wang J, et al. Interleukin-13 alters tight junction proteins expression thereby compromising barrier function and dampens rhinovirus induced immune responses in nasal epithelium. Front Cell Dev Biol. (2020) 8:572749. doi: 10.3389/fcell.2020.572749
46. Caballero MT, Hijano DR, Acosta PL, Mateu CG, Marcone DN, Linder JE, et al. Interleukin-13 associates with life-threatening rhinovirus infections in infants and young children. Pediatr Pulmonol. (2018) 53(6):787–95. doi: 10.1002/ppul.23998
47. Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, et al. TH2 And TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. (2015) 7(301):301ra129. doi: 10.1126/scitranslmed.aab3142
48. Gallagher A, Edwards M, Nair P, Drew S, Vyas A, Sharma R, et al. Anti-interleukin-13 and anti-interleukin-4 agents versus placebo, anti-interleukin-5 or anti-immunoglobulin-E agents, for people with asthma. Cochrane Database Syst Rev. (2021) 10(10):CD012929. doi: 10.1002/14651858.CD012929.pub2
49. Bhalla A, Mukherjee M, Nair P. Airway eosinophilopoietic and autoimmune mechanisms of eosinophilia in severe asthma. Immunol Allergy Clin North Am. (2018) 38(4):639–54. doi: 10.1016/j.iac.2018.06.003
50. Hillas G, Fouka E, Papaioannou AI. Antibodies targeting the interleukin-5 signaling pathway used as add-on therapy for patients with severe eosinophilic asthma: a review of the mechanism of action, efficacy, and safety of the subcutaneously administered agents, mepolizumab and benralizumab. Expert Rev Respir Med. (2020) 14(4):353–65. doi: 10.1080/17476348.2020.1718495
51. Dill McFarland KA, Schwartz JT, Zhao H, Shao B, Fulkerson PC, Altman MC, et al. Eosinophil-mediated suppression and anti-IL-5 enhancement of plasmacytoid dendritic cell interferon responses in asthma. J Allergy Clin Immunol. (2022) 150(3):666–75. doi: 10.1016/j.jaci.2022.03.025
52. Bielor C, Sopel N, Maier A, Blau A, Sharma H, Vuorinen T, et al. Role of TGF-β in anti-rhinovirus immune responses in asthmatic patients. J Allergy Clin Immunol. (2017) 140(1):283–6. doi: 10.1016/j.jaci.2016.10.049
53. Kaiser L, Fritz RS, Straus SE, Gubareva L, Hayden FG. Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J Med Virol. (2001) 64(3):262–8. doi: 10.1002/jmv.1045
54. Kaur S, Bansal Y, Kumar R, Bansal G. A panoramic review of IL-6: structure, pathophysiological roles and inhibitors. Bioorg Med Chem. (2020) 28(5):115327. doi: 10.1016/j.bmc.2020.115327
55. Lee CG, Hartl D, Matsuura H, Dunlop FM, Scotney PD, Fabri LJ, et al. Endogenous IL-11 signaling is essential in Th2- and IL-13-induced inflammation and mucus production. Am J Respir Cell Mol Biol. (2008) 39(6):739–46. doi: 10.1165/rcmb.2008-0053OC
56. Trepicchio WL, Wang L, Bozza M, Dorner AJ. IL-11 regulates macrophage effector function through the inhibition of nuclear factor-kappaB. J Immunol. (1997) 159(11):5661–70. doi: 10.4049/jimmunol.159.11.5661
57. Ernst M, Putoczki TL. Molecular pathways: IL11 as a tumor-promoting cytokine-translational implications for cancers. Clin Cancer Res. (2014) 20(22):5579–88. doi: 10.1158/1078-0432.CCR-13-2492
58. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. (2003) 374(Pt 1):1–20. doi: 10.1042/BJ20030407
59. Minshall E, Chakir J, Laviolette M, Molet S, Zhu Z, Olivenstein R, et al. IL-11 expression is increased in severe asthma: association with epithelial cells and eosinophils. J Allergy Clin Immunol. (2000) 105(2 Pt 1):232–8. doi: 10.1016/s0091-6749(00)90070-8
60. Ng B, Dong J, D' Agostino G, Viswanathan S, Widjaja AA, Lim WW, et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci Transl Med. (2019) 11(511):eaaw1237. doi: 10.1126/scitranslmed.aaw1237
61. Ng B, Dong J, Viswanathan S, Widjaja AA, Paleja BS, Adami E, et al. Fibroblast-specific IL11 signaling drives chronic inflammation in murine fibrotic lung disease. Faseb J. (2020) 34(9):11802–15. doi: 10.1096/fj.202001045RR
62. Nieminen P, Morgan NV, Fenwick AL, Parmanen S, Veistinen L, Mikkola ML, et al. Inactivation of IL11 signaling causes craniosynostosis, delayed tooth eruption, and supernumerary teeth. Am J Hum Genet. (2011) 89(1):67–81. doi: 10.1016/j.ajhg.2011.05.024
63. Metcalfe RD, Aizel K, Zlatic CO, Nguyen PM, Morton CJ, Lio DS, et al. The structure of the extracellular domains of human interleukin 11α receptor reveals mechanisms of cytokine engagement. J Biol Chem. (2020) 295(24):8285–301. doi: 10.1074/jbc.RA119.012351
64. Zhang X, Kiapour N, Kapoor S, Merrill JR, Xia Y, Ban W, et al. IL-11 antagonist suppresses Th17 cell-mediated neuroinflammation and demyelination in a mouse model of relapsing-remitting multiple sclerosis. Clin Immunol. (2018) 197:45–53. doi: 10.1016/j.clim.2018.08.006
65. Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Ralpha recycles and presents IL-15 in trans to neighboring cells. Immunity. (2002) 17(5):537–47. doi: 10.1016/s1074-7613(02)00429-6
66. Jayaraman A, Jackson DJ, Message SD, Pearson RM, Aniscenko J, Caramori G, et al. IL-15 complexes induce NK- and T-cell responses independent of type I IFN signaling during rhinovirus infection. Mucosal Immunol. (2014) 7(5):1151–64. doi: 10.1038/mi.2014.2
67. Zhao L, Hu B, Zhang Y, Song Y, Lin D, Liu Y, et al. An activation-induced IL-15 isoform is a natural antagonist for IL-15 function. Sci Rep. (2016) 6:25822. doi: 10.1038/srep25822
68. Moui A, Klein M, Hassoun D, Dijoux E, Cheminant MA, Magnan A, et al. The IL-15/sIL-15Rα complex modulates immunity without effect on asthma features in mouse. Respir Res. (2020) 21(1):33. doi: 10.1186/s12931-020-1301-x
69. Jamieson KC, Traves SL, Kooi C, Wiehler S, Dumonceaux CJ, Maciejewski BA, et al. Rhinovirus and Bacteria synergistically induce IL-17C release from human airway epithelial cells to promote neutrophil Recruitment. J Immunol. (2019) 202(1):160–70. doi: 10.4049/jimmunol.1800547
70. Liu X, Nguyen TH, Sokulsky L, Li X, Garcia Netto K, Hsu AC, et al. IL-17A is a common and critical driver of impaired lung function and immunopathology induced by influenza virus, rhinovirus and respiratory syncytial virus. Respirology. (2021) 26(11):1049–59. doi: 10.1111/resp.14141
71. Haddad A, Gaudet M, Plesa M, Allakhverdi Z, Mogas AK, Audusseau S, et al. Neutrophils from severe asthmatic patients induce epithelial to mesenchymal transition in healthy bronchial epithelial cells. Respir Res. (2019) 20(1):019–1186. doi: 10.1186/s12931-019-1186-8
72. Yoshimoto T, Mizutani H, Tsutsui H, Noben Trauth N, Yamanaka K, Tanaka M, et al. IL-18 induction of IgE: dependence on CD4+ T cells, IL-4 and STAT6. Nat Immunol. (2000) 1(2):132–7. doi: 10.1038/77811
73. Yoshimoto T, Tsutsui H, Tominaga K, Hoshino K, Okamura H, Akira S, et al. IL-18, although antiallergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Proc Natl Acad Sci U S A. (1999) 96(24):13962–6. doi: 10.1073/pnas.96.24.13962
74. Ghebre MA, Pang PH, Desai D, Hargadon B, Newby C, Woods J, et al. Severe exacerbations in moderate-to-severe asthmatics are associated with increased pro-inflammatory and type 1 mediators in sputum and serum. BMC Pulm Med. (2019) 19(1):019–0906. doi: 10.1186/s12890-019-0906-7
75. Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. (1999) 10(1):127–36. doi: 10.1016/s1074-7613(00)80013-8
76. Beale J, Jayaraman A, Jackson DJ, Macintyre JDR, Edwards MR, Walton RP, et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med. (2014) 6(256):3009124. doi: 10.1126/scitranslmed.3009124
77. Paplińska GM, Grabczak EM, Dąbrowska M, Hermanowicz Salamon J, Proboszcz M, Nejman Gryz P, et al. Sputum interleukin-25 correlates with asthma severity: a preliminary study. Postepy Dermatol Alergol. (2018) 35(5):462–9. doi: 10.5114/ada.2017.71428
78. Williams TC, Loo SL, Nichol KS, Reid AT, Veerati PC, Esneau C, et al. IL-25 blockade augments antiviral immunity during respiratory virus infection. Commun Biol. (2022) 5(1):022–03367. doi: 10.1038/s42003-022-03367-z
79. Gajewski A, Gawrysiak M, Szewczyk R, Gulbas I, Likońska A, Michlewska S, et al. IL-33 augments the effect of rhinovirus HRV16 on inflammatory activity of human lung vascular endothelium-possible implications for rhinoviral asthma exacerbations. Allergy. (2021) 76(7):2282–5. doi: 10.1111/all.14806
80. Gimenes JA, Srivastava V, ReddyVari H, Kotnala S, Mishra R, Farazuddin M, et al. Rhinovirus-induces progression of lung disease in a mouse model of COPD via IL-33/ST2 signaling axis. Clin Sci. (2019) 133(8):983–96. doi: 10.1042/CS20181088
81. Jurak LM, Xi Y, Landgraf M, Carroll ML, Murray L, Upham JW. Interleukin 33 selectively augments rhinovirus-induced type 2 immune responses in asthmatic but not healthy people. Front Immunol. (2018) 9:1895. doi: 10.3389/fimmu.2018.01895
82. Wechsler ME, Ruddy MK, Pavord ID, Israel E, Rabe KF, Ford LB, et al. Efficacy and safety of itepekimab in patients with moderate-to-severe asthma. N Engl J Med. (2021) 385(18):1656–68. doi: 10.1056/NEJMoa2024257
83. Pfeffer PE, Chen YH, Woszczek G, Matthews NC, Chevretton E, Gupta A, et al. Vitamin D enhances production of soluble ST2, inhibiting the action of IL-33. J Allergy Clin Immunol. (2015) 135(3):824–7. doi: 10.1016/j.jaci.2014.09.044
84. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23(5):479–90. doi: 10.1016/j.immuni.2005.09.015
85. Haag P, Sharma H, Rauh M, Zimmermann T, Vuorinen T, Papadopoulos NG, et al. Soluble ST2 regulation by rhinovirus and 25(OH)-vitamin D3 in the blood of asthmatic children. Clin Exp Immunol. (2018) 193(2):207–20. doi: 10.1111/cei.13135
86. Pozdzik AA, Giordano L, Li G, Antoine MH, Quellard N, Godet J, et al. Blocking TGF-β signaling pathway preserves mitochondrial proteostasis and reduces early activation of PDGFRβ+ pericytes in aristolochic acid induced acute kidney injury in wistar male rats. PLoS One. (2016) 11(7):0157288. doi: 10.1371/journal.pone.0157288
87. Kölle J, Haag P, Vuorinen T, Alexander K, Rauh M, Zimmermann T, et al. Respiratory infections regulated blood cells IFN-β-PD-L1 pathway in pediatric asthma. Immun Inflamm Dis. (2020) 8(3):310–9. doi: 10.1002/iid3.307
88. Hamilton JA. GM-CSF-Dependent inflammatory pathways. Front Immunol. (2019) 10:2055. doi: 10.3389/fimmu.2019.02055
89. Lang FM, Lee KM, Teijaro JR, Becher B, Hamilton JA. GM-CSF-based treatments in COVID-19: reconciling opposing therapeutic approaches. Nat Rev Immunol. (2020) 20(8):507–14. doi: 10.1038/s41577-020-0357-7
90. Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don't know. Cell Res. (2006) 16(2):126–33. doi: 10.1038/sj.cr.7310017
91. Duhen T, Campbell DJ. IL-1β promotes the differentiation of polyfunctional human CCR6+CXCR3+ Th1/17 cells that are specific for pathogenic and commensal microbes. J Immunol. (2014) 193(1):120–9. doi: 10.4049/jimmunol.1302734
92. Quill H, Gaur A, Phipps RP. Prostaglandin E2-dependent induction of granulocyte-macrophage colony-stimulating factor secretion by cloned murine helper T cells. J Immunol. (1989) 142(3):813–8. doi: 10.4049/jimmunol.142.3.813
93. Shiomi A, Usui T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediators Inflamm. (2015) 568543(10):8. doi: 10.1155/2015/568543
94. Becher B, Tugues S, Greter M. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity. (2016) 45(5):963–73. doi: 10.1016/j.immuni.2016.10.026
95. Bonfield TL, Farver CF, Barna BP, Malur A, Abraham S, Raychaudhuri B, et al. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. Am J Respir Cell Mol Biol. (2003) 29(6):677–82. doi: 10.1165/rcmb.2003-0148OC
96. Zhan Y, Lew AM, Chopin M. The pleiotropic effects of the GM-CSF rheostat on myeloid cell differentiation and function: more than a numbers game. Front Immunol. (2019) 10:2679. doi: 10.3389/fimmu.2019.02679
97. Bosteels C, Damme KV, Leeuw ED, Declercq J, Maes B, Bosteels V, et al. Early treatment with inhaled GM-CSF improves oxygenation and anti-viral immunity in COVID-19 induced lung injury–a randomized clinical trial. Preprint. (2021). doi: 10.21203/rs.3.rs-959220/v1
98. Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J, et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med. (2009) 180(7):640–8. doi: 10.1164/rccm.200903-0363OC
99. Siegal FP, Kadowaki N, Shodell M, Fitzgerald Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. (1999) 284(5421):1835–7. doi: 10.1126/science.284.5421.1835
100. Fu XY, Kessler DS, Veals SA, Levy DE, Darnell JE. ISGF3, The transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc Natl Acad Sci U S A. (1990) 87(21):8555–9. doi: 10.1073/pnas.87.21.8555
101. Heim MH, Kerr IM, Stark GR, Darnell JE. Contribution of STAT SH2 groups to specific interferon signaling by the jak-STAT pathway. Science. (1995) 267(5202):1347–9. doi: 10.1126/science.7871432
102. Levy DE, Kessler DS, Pine R, Darnell JE. Cytoplasmic activation of ISGF3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. (1989) 3(9):1362–71. doi: 10.1101/gad.3.9.1362
103. Hammad H, Lambrecht BN. The basic immunology of asthma. Cell. (2021) 184(9):2521–2. doi: 10.1016/j.cell.2021.04.019
104. Kirchberger S, Majdic O, Stockl J. Modulation of the immune system by human rhinoviruses. Int Arch Allergy Immunol. (2007) 142(1):1–10. doi: 10.1159/000095993
105. Leisching G, Wiid I, Baker B. The association of OASL and type I interferons in the pathogenesis and survival of intracellular replicating bacterial Species. Front Cell Infect Microbiol. (2017) 7:196. doi: 10.3389/fcimb.2017.00196
106. Lin T, Lo C, Tsao K, Chang P, Kuo C, Lo Y, et al. Impaired interferon-α expression in plasmacytoid dendritic cells in asthma. Immun Inflamm Dis. (2021) 9(1):183–95. doi: 10.1002/iid3.376
107. Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. (1989) 170(6):2081–95. doi: 10.1084/jem.170.6.2081
108. Hulsmans M, Sager HB, Roh JD, Valero Muñoz M, Houstis NE, Iwamoto Y, et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. (2018) 215(2):423–40. doi: 10.1084/jem.20171274
109. Lobo SD, Carriche GM, Castro AG, Roque S, Saraiva M. Balancing the immune response in the brain: iL-10 and its regulation. J Neuroinflammation. (2016) 13(1):016–0763. doi: 10.1186/s12974-016-0763-8
110. Ouyang W, O'Garra A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity. (2019) 50(4):871–91. doi: 10.1016/j.immuni.2019.03.020
111. Stöckl J, Vetr H, Majdic O, Zlabinger G, Kuechler E, Knapp W. Human major group rhinoviruses downmodulate the accessory function of monocytes by inducing IL-10. J Clin Invest. (1999) 104(7):957–65. doi: 10.1172/JCI7255
112. Engelhardt KR, Grimbacher B. IL-10 in humans: lessons from the gut, IL-10/IL-10 receptor deficiencies, and IL-10 polymorphisms. Curr Top Microbiol Immunol. (2014) 380:1–18. doi: 10.1007/978-3-662-43492-5_1
113. Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. (1998) 93(3):373–83. doi: 10.1016/s0092-8674(00)81166-6
114. Weber Nordt RM, Riley JK, Greenlund AC, Moore KW, Darnell JE, Schreiber RD. Stat3 recruitment by two distinct ligand-induced, tyrosine-phosphorylated docking sites in the interleukin-10 receptor intracellular domain. J Biol Chem. (1996) 271(44):27954–61. doi: 10.1074/jbc.271.44.27954
115. Waal Malefyt R, Haanen J, Spits H, Roncarolo MG, Velde A, Figdor C, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. (1991) 174(4):915–24. doi: 10.1084/jem.174.4.915
116. Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O' Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. (1991) 147(11):3815–22. doi: 10.4049/jimmunol.147.11.3815
117. Gazzinelli RT, Oswald IP, James SL, Sher A. IL-10 inhibits parasite killing and nitrogen oxide production by IFN-gamma-activated macrophages. J Immunol. (1992) 148(6):1792–6. doi: 10.4049/jimmunol.148.6.1792
118. Macatonia SE, Doherty TM, Knight SC, O' Garra A. Differential effect of IL-10 on dendritic cell-induced T cell proliferation and IFN-gamma production. J Immunol. (1993) 150(9):3755–65. doi: 10.4049/jimmunol.150.9.3755
119. Levine SJ, Wu T, Shelhamer JH. Extracellular release of the type I intracellular IL-1 receptor antagonist from human airway epithelial cells: differential effects of IL-4, IL-13, IFN-gamma, and corticosteroids. J Immunol. (1997) 158(12):5949–57. doi: 10.4049/jimmunol.158.12.5949
Keywords: rhinovirus, rhinovirus-related diseases, immunoreactions, cytokines, therapies for rhinovirus infection
Citation: Sang L, Gong X, Huang Y, Zhang L and Sun J (2024) Immunotherapeutic implications on targeting the cytokines produced in rhinovirus-induced immunoreactions. Front. Allergy 5:1427762. doi: 10.3389/falgy.2024.1427762
Received: 4 May 2024; Accepted: 13 May 2024;
Published: 27 May 2024.
Edited by:
Cemal Cingi, Eskişehir Osmangazi University, TürkiyeReviewed by:
Felicia Manole, Hiperclinica Medlife Bucharest Romania, RomaniaOguzhan Oguz, Nisantasi University, Türkiye
© 2024 Sang, Gong, Huang, Zhang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Sun, 2002sunjian@163.com