HyCas9-12aGEP: an efficient genome editing platform for Corynebacterium glutamicum
 Feng Zhang
Feng Zhang Jin-Yu Wang
Jin-Yu Wang - The Key Laboratory of Industrial Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi, China
Corynebacterium glutamicum plays a crucial role as a significant industrial producer of metabolites. Despite the successful development of CRISPR-Cas9 and CRISPR-Cas12a-assisted genome editing technologies in C. glutamicum, their editing resolution and efficiency are hampered by the diverse on-target activities of guide RNAs (gRNAs). To address this problem, a hybrid CRISPR-Cas9-Cas12a genome editing platform (HyCas9-12aGEP) was developed in C. glutamicum in this study to co-express sgRNA (corresponding to SpCas9 guide RNA), crRNA (corresponding to FnCas12a guide RNA), or hfgRNA (formed by the fusion of sgRNA and crRNA). HyCas9-12aGEP improves the efficiency of mapping active gRNAs and outperforms both CRISPR-Cas9 and CRISPR-Cas12a in genome editing resolution and efficiency. In the experiment involving the deletion of the cg0697-0740 gene segment, an unexpected phenotype was observed, and HyCas9-12aGEP efficiently identified the responsible genotype from more than 40 genes. Here, HyCas9-12aGEP greatly improve our capability in terms of genome reprogramming in C. glutamicum.
Introduction
The enzyme associated with clustered regularly interspaced short palindromic repeats (CRISPR), such as Cas9 or Cas12a, is an RNA-guided endonuclease that utilizes RNA–DNA base-pairing to identify and target foreign DNA within bacteria (Jinek et al., 2012; Zetsche et al., 2015). Guide RNA complexes with Cas9 or Cas12a are potent genome-engineering agents in both eukaryotes and prokaryotes, extensively employed in CRISPR-based methodologies (Cong et al., 2013; Jiang et al., 2013; Yan et al., 2017; Zetsche et al., 2017). For example, genetic engineering has been developed for C. glutamicum (Jiang et al., 2017; Liu et al., 2017; Peng et al., 2017; Wang et al., 2021; Liu et al., 2022). C. glutamicum, strategically engineered for industrial amino acid synthesis, serves as a versatile microorganism capable of producing a diverse range of compounds, including sunscreens, anti-aging sugars, biofuels, and polymers designed for regenerative medicine applications (Becker et al., 2018; Wolf et al., 2021). Despite being regarded as a promising tool for genome engineering in C. glutamicum, CRISPR-Cas9 or CRISPR-Cas12a encounters challenges primarily due to the varied on-target activities of guide RNAs (gRNAs), posing potential obstacles to its successful development.
Cas9 and Cas12a recognize distinct protospacer adjacent motif (PAM) sequences, for instance, SpCas9 and FnCas12a specifically recognize 5′-NGG-3′ and 5′-TTN-3′, respectively (Jinek et al., 2012; Zetsche et al., 2015). A major challenge in CRISPR/Cas9-and Cas12a-mediated genome engineering is that not all guide RNAs (gRNAs) efficiently cleave the DNA (Doench et al., 2014; Jiang et al., 2017; Kim et al., 2018; Creutzburg et al., 2020; Zhang et al., 2020; Corsi et al., 2022). Therefore, the selection and design of active gRNA are critical for CRISPR-Cas9-mediated gene editing. However, the efficacy of guide RNA (gRNA) is impacted by various factors, encompassing gRNA structure (Abdel-Mawgoud and Stephanopoulos, 2020; Creutzburg et al., 2020; Magnusson et al., 2021; Riesenberg et al., 2022), conformational transitions (Chen et al., 2017; Wang et al., 2019; Kim et al., 2020; Talas et al., 2021), R-loop formation (Gong et al., 2018), Cas9 and Cas12a variants (Chen et al., 2017; Guo et al., 2018; Wang et al., 2019; Kim et al., 2020), PAM sequences (Doench et al., 2014), supercoiling (Ivanov et al., 2020), DNA covalent modification (Tao et al., 2017; Vlot et al., 2018; Liu Y et al., 2020; Dong et al., 2021), interactions at non-targeted sites (Sternberg et al., 2014; Moreb et al., 2020), target copy number (Ivanov et al., 2020), and target accessibility (Chen et al., 2016; Horlbeck et al., 2016; Yarrington et al., 2018). Despite extensive screening of large guide RNA (gRNA) libraries and the development of algorithms to predict sequence-dependent gRNA activity (Park et al., 2021; Talas et al., 2021; Xiang et al., 2021), these algorithms exhibit limitations in accurately predicting other datasets, training datasets, or variations across different species (Moreb and Lynch, 2021). Thus, beyond the utilization of current gRNA design tools, the central challenge in gene editing resides in the experimental strategies to promptly identify active gRNAs and enhance their efficacy.
Off-target effects pose a critical challenge in CRISPR-Cas9-based gene editing and disease therapy. These effects are influenced by various factors, encompassing the quality of guide gRNA, the selection of the Cas9 protein, concentrations of both gRNA and Cas protein, cell type, and the choice of target site (Pattanayak et al., 2013; Wienert and Cromer, 2022; Guo et al., 2023). Among these factors, gRNA concentration and the level of Cas9 protein expression are the primary contributors (Hsu et al., 2013; Pattanayak et al., 2013). The effectiveness of the CRISPR-Cas9 system depends on sufficient Cas9 protein expression, as Cas9 serves as the actual nuclease responsible for gene editing. Insufficient Cas9 protein expression can impair its ability to accurately identify and cleave the target DNA, reducing editing efficiency (Pattanayak et al., 2013). Conversely, an excessive Cas9 protein concentration may lead to cleavage at off-target sites with partial sequence similarity, thereby increasing the risk of off-target effects (Hsu et al., 2013). Thus, in CRISPR-Cas9 experiments, it is essential to maintain a balanced Cas9 protein expression level to ensure efficient editing while minimizing the potential for off-target effects.
In this study, we present HyCas9-12aGEP, a system that involves the co-expression of Streptococcus pyogenes (Sp)-Cas9 and Francisella novicida (Fn)-Cas12a nucleases by integrating the Spcas9 and Fncas12a genes into C. glutamicum. This system utilizes 'hybrid fused guide’ (hfg)RNAs, generated by fusing SpCas9 and FnCas12a guide RNAs and expressed from a single promoter. By reducing the expression level of SpCas9, HyCas9-12aGEP mitigates the off-target effects associated with SpCas9. In comparison to conventional CRISPR-Cas9 or Cas12a systems, HyCas9-12aGEP, coupled with hfgRNA, markedly augments guide RNA (gRNA) activity, streamlines the identification of active gRNAs, and enhances the efficiency of gene editing. Additionally, SpCas9 and FnCas12a recognize different PAM sequences, specifically 5′-NGG-3′ and 5′-TTN-3′, respectively. Therefore, the active gRNAs (corresponding to an active PAM) that HyCas9-12aGEP can use are the sum of SpCas9 and FnCas12a, which improves the resolution of gene editing, such as precise substitution of the 149th Glycine with Lysine in the γ-glutamyl kinase encoded by proB. Interestingly, our experiments unveiled a notable phenotype in colonies with a 40.96 kb DNA segment (cg0697-cg0740) deleted from the C. glutamicum genome, exhibiting increased “moisture.” Using HyCas9-12aGEP, genotypes that influence the phenotype were rapidly mapped from a pool of over 40 genes. In summary, HyCas9-12aGEP emerges as a potent tool for genetically modifying C. glutamicum, accelerating research in gene function, and optimizing the production of target products.
Materials and methods
Strains and culture conditions
Strains used in this study are listed in Supplementary Table S1. E. coli JM109, utilized for plasmid cloning, was aerobically cultivated at 37°C in Luria–Bertani (LB) broth. The medium was supplemented accordingly with kanamycin (Kan, 50 μg/mL) or chloramphenicol (Cm, 20 μg/mL). C. glutamicum were cultured at 30°C in LBG medium (LB medium supplemented with 5 g/L glucose). The Epo medium, consisting of LBG supplemented with 3% glycine, 0.1% Tween 80, and 0.4% isoniazid, was utilized for cultivating electroporation-competent cells. LBHIS medium, containing 2.5 g/L yeast extract, 5 g/L tryptone, 5 g/L NaCl, 18.5 g/L Brain Heart Infusion powder, and 91 g/L sorbitol, were employed to obtain transformants of C. glutamicum, following previously described procedures (Xu J et al., 2014). The CM medium (10 g/L yeast extract, 10 g/L beef extract, 10 g/L tryptone, 5 g/L glucose, and 5 g/L NaCl) is utilized for colony phenotype observation on agar plates. Kan (25 μg/mL) was added to LBHIS medium as required.
Plasmid construction
Plasmids utilized in this study are listed in Supplementary Table S2 and Supplementary Data S1. Plasmids were constructed via recombination or T4 DNA ligase. Recombination was conducted using the ClonExpress II and ClonExpress MultiS One Step Cloning Kit (Vazyme, Nanjing, China). Restriction endonucleases and T4 DNA ligase were purchased from TaKaRa (Dalian, China). DNA polymerase and reagents were purchased from Vazyme (Nanjing, China). Gene synthesis and DNA sequencing were provided by GENEWIZ Inc. (Suzhou, China). Primers synthesized by Exsyn-bio (Wuxi, China) and details for constructing plasmids are described in Supplementary Data S1.
Design of gRNAs
The general sgRNAs were designed via online software (http://www.rgenome.net/cas-designer/) (Park J and Kim, 2015). The relevant sgRNA sequences are listed in Supplementary Data S2.
Genome editing in C. glutamicum
The traditional pK18mobsacB–based gene deletion and insertion were performed as previously described (Xu J et al., 2014). HyCas9-12aGEP has been successfully applied in genome editing, and the detail progress are listed in the Supplementary methods (Supplementary Material). In this study, the size of homologous arm (HA) carried by plasmid pZF2 was −1 kb.
The preparation of competent C. glutamicum was carried out following the previously described method with appropriate modifications (Liu et al., 2022). The strains were cultured in 50-mL shake flasks with 10 mL of LBG media for 10–13 h, and then 3 mL was transferred to 100 mL of Epo media in 500-mL shake flasks for 30°C-cultivation. When the △OD600 of the culture reached to 0.4-0.5, the culture were ice-bathed for 15–20 min and were then harvested through 5-min centrifugation at 4°C and 4,000 rpm. The cells were subsequently resuspended in 300–500 μL of 10.0% (v/v) glycerol after washing 3 times using 4°C pre-chilled 10% glycerol. The plasmid was mixed with competent cells and subsequently introduced into an electroporation cuvette. Electroporation was carried out utilizing an GenePulser Xcell™ (Bio-Rad Laboratories, Shanghai, China) with parameter settings of 1800 V, 5 ms, and 1 mm. Subsequently, 800 μL of LBHIS media was immediately added, followed by rapidly 6-min incubation of the suspension at 46°C. The cells were 2-h cultured at 30°C, and then spread on LBHIS plates containing antibiotics for 30°C-incubation until the apperance of colonies.
Re-sequencing analysis
Re-sequencing was conducted to identify off-target occurrences in the edited strains. Referring to the previously described method (Peng et al., 2017). Total DNA from C. glutamicum was extracted following the manufacturer’s protocol provided by Vazyme, Nanjing, China. The assessment of DNA quality involved utilizing the Qubit Fluorometer (Thermo Fisher Scientific, San Jose, CA, United States) for measuring overall mass and the Fragment Analyzer for evaluating DNA integrity. The genomic sequencing was executed utilizing the Illumina HiSeq/Nova 2 × 150bp system (Illumina, San Diego, CA, United States) at the GENEWIZ Inc. (Suzhou, China).
Plasmid curing
For plasmid curing, transformants were grown in antibiotic-free LBG medium at 37°C overnight (−8 h) and subsequently plated on antibiotic-free CM plates. The next day, the culture is diluted and coated into kanamycin-resistant CM plates and cultured at 30°C. Plasmid curing was judged by the colony’s sensitivity to antibiotics. The grown single colonies were transferred to one CM kanamycin-resistant plate and the corresponding CM plate, respectively. Single colonies that cannot be grown in response to resistant plates are plasmid eliminated successfully for the next round of gene editing or other tests.
RT-PCR for mRNA quantification
RT-qPCR assay was performed as described previously (Wang et al., 2020). To determine the transcriptional intensity of Spcas9 or Fncas12a in the seven Spcas9 or Fncas12a-expressing strains, i.e. 9-12, E9-12, L9-12, 9-E12, 9-L12, Cas9-2, and Cas12-2, quantitative reverse transcription PCR (qRT-PCR) was performed using total RNA samples. Total RNA was isolated from the cells with the RNAprep Pure Cell/Bacteria Kit (Tiangen, China). 0.5 μg of the total bacterial RNA was subjected to reverse transcription using the HiScript II Q RT SuperMix Kit (Vazyme). RT-qPCR analysis using the ChamQ Universal SYBR qPCR master mix kit (Vazyme) in a total reaction volume of 20 μL in a CFX96™ Real-Time System (Bio-Rad, Hercules, CA, United States). The 16S rRNA encoding gene was used as an internal control as described previously (Pan et al., 2022). PCR primers used in RT-PCR are listed in Supplementary Data S1.
Analytical methods
Cell growth was calculated by measuring the optical density at 600 nm (OD600) using a spectrophotometer (Shanghai, China). Cell morphology was examined using field emission scanning electron microscopy (FESEM). C. glutamicum cells were harvested by centrifugation, washed thrice with physiological saline (pH 7.0), and subsequently deposited onto a small silicon platelet. After air-drying at room temperature, the cells underwent in-situ fixation with a 2.5% glutaraldehyde solution in a 0.15 M sodium phosphate buffer (pH 7.4) for 10 min. The samples were gold-coated and examined under field emission scanning electron microscopy (FESEM) using a Hitachi SU8220 instrument with an accelerating voltage of 3 kV.
Statistical analysis
All experiments were conducted with three independent replicates. Statistical analysis of the data was carried out using t-tests in SPSS v.25. A significance threshold of p < 0.05 was applied, and the level of significance is denoted as ***p < 0.01.
Results
Exploration of gene editing system to broaden the target range
The success of CRISPR gene editing applications relies significantly on the presence of active gRNAs. SpRY-Cas9 (Walton RT et al., 2020) is an unconstrained near-PAMless genome, and SpCas9-HF1 (Chen et al., 2017) is high-fidelity SpCas9 variant. Thus, the mutant SpRY-HF1 obtained by combining SpRY-Cas9 and SpCas9-HF1 was inferred to increase the density of active gRNAs and to improve editing resolution. Thus, the open reading frame (ORF) of SpCas9 in pFSC (Peng et al., 2017) was replaced with the codon-optimized ORF of SpRY-HF1 (NCBI accession numbers: OP345224) to obtain the plasmid pXMJ19-SpRY-HF1. Subsequently, a plasmid, pFST-porB-HD (Peng et al., 2017), carrying active gRNA targeting porB and two homologous arms of −1 kb each, was electroporated into C. glutamicum ATCC13032 harboring the plasmid pXMJ19-SpRY-HF1. Although SpRY-HF1 exhibited high expression (Supplementary Figure S1A), it indicated a exceeding low counter-selection efficiency (Supplementary Figure S1B), which may be attributed to the excessive number of mutation sites that tend to reduce nuclease activity (Okafor et al., 2019) or increase off-target titration in the genome (Moreb and Lynch, 2021). This results in the extension of the search times for targeted loci, and in turn, leads to a reduction in counter-selection efficiency. Hence, this indicates that the gene editing application of the SpCas9 variant SpRY-HF1 in C. glutamicum still requires further optimization.
Development and optimization of a hybrid CRISPR-Cas9-Cas12a gene editing platform
SpCas9 and FnCas12a recognize different PAM sequences of 5′-NGG-3′ and 5′-TTN-3′ respectively (Jinek et al., 2012; Zetsche et al., 2015). Therefore, incorporating both SpCas9 and FnCas12a into a unified system facilitates the broadening of the targeting range. Considering this, 2 C. glutamicum-E. coli shuttle plasmids were constructed based on the plasmids pFSC (Peng et al., 2017) and pJYS3 (Jiang et al., 2017): pFSC-Cas12a and pJYS3-Cas9, which carried the PlacM-Fncas12a-rrnBT1T2-Spcas9-Ptac cassette and were transformed into C. glutamicum ATCC 13032 (i.e., 13032). However, this yielded no transformants (Supplementary Figure S1B). It has been reported that constitutive expression of plasmid-borne dCas9 from S. pyogenes in C. glutamicum has proven to be unattainable (Cleto et al., 2016). Thus, it was speculated that this may be due to the oversized plasmid vector and the simultaneous expression of the two Cas nucleases, resulting in a superposition of toxicity. To address these challenges, the Ptac-SpCas9-rrnBT1T2 and PlacM-Fncas12a-rrnBT1T2 cassettes were integrated into the 13032 genome at the putA and ldh gene loci, resulting in strains Ptac-9 and L12, respectively (Supplementary Figure S2A). The strains with two Cas-coding gene copies were then constructed to obtain the following: 9–12 with one copy each of Ptac-Spcas9-rrnBT1T2 and PlacM-cas12a-rrnBT1T2, Cas9-2 with two copies of Ptac-Spccas9-rrnBT1T2, and Cas12a-2 with two copies of Fncas12a (Supplementary Figure S2A; Supplementary Table S3). In this growth experiment, it was observed that by integrating a single copy of the Ptac-Spcas9-rrnBT1T2 and PlacM-Fncas12a-rrnBT1T2 into the genome, the growth of strain 13032 was hardly affected, whereas, growth was significantly inhibited when two copies of the Ptac-Spcas9-rrnBT1T2 and PlacM-Fncas12a-rrnBT1T2 were integrated (Supplementary Figure S2B). Hence, nuclease-induced toxicity can be entirely removed in the 13032 when genomic integration uses a single copy of the Ptac-Spcas9-rrnBT1T2 and PlacM-Fncas12a-rrnBT1T2.
Next, the genome-editing performance of the hybrid CRISPR-Cas9-Cas12a system was tested. A temperature-sensitive plasmid, pZF2, was initially designed for expressing the gRNA and harboring homologous arms (HA) (Figure 1A). It has been reported that crRNA-crtYf (Jiang et al., 2017) (crRNA1) and sgRNA-porB (Peng et al., 2017) (sgRNA1) facilitate highly effective gene editing. Thus, crRNA1 and sgRNA1 are assembled into plasmids pZF2 harboring the corresponding homologous arms, obtaining plasmids pZF2-sgRNA1-△porB and pZF2-crRNA1-crtYf*, which are used to knock out the gene porB (0.5 kb) and introduce point mutations at the crtYf site, respectively. The two plasmids were then transformed to 9–12 strain, and the colony PCR results showed that the knockout of porB and the point mutation efficiency of crtYf reached 100% (Figures 1B,C; Supplementary Figure S3), which was comparable to the optimal editing efficiency previously reported (Jiang et al., 2017; Peng et al., 2017). The strains that were successfully edited were subsequently cultured overnight at 37°C, resulting in plasmid curing efficiencies of approximately 100% (Supplementary Figure S4).
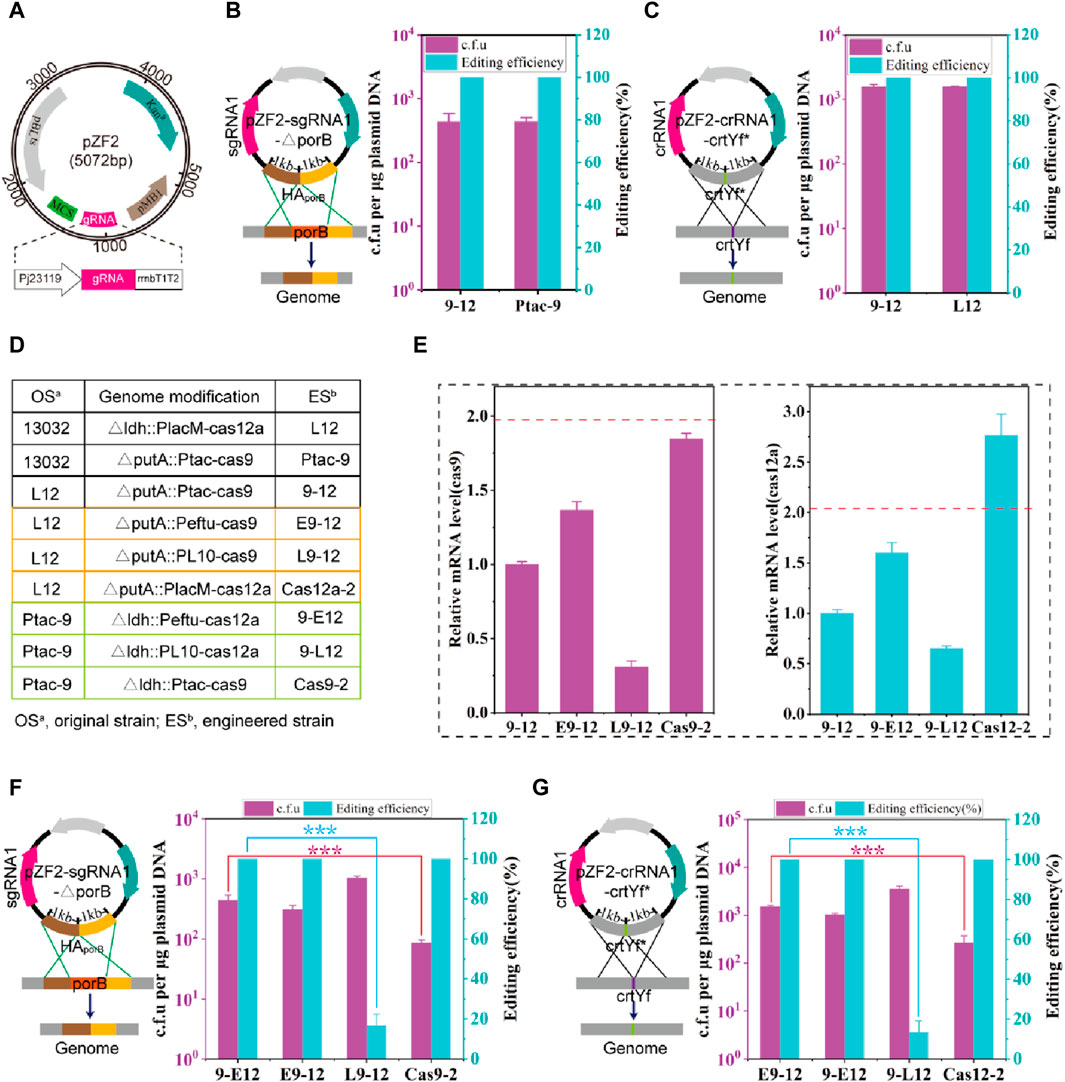
FIGURE 1. Construction and optimization of HyCas9-12aGEP. (A) Schematic diagram of the temperature-sensitive gRNA expression plasmid pZF2. pBL1ts: a temperature sensitive replication derived from the pBL1 replicon of C. glutamicum; Knr: kanamycin resistance gene encoded by the aminoglycoside phosphotransferase gene; pMB1: a replication origin of E. coli; Pj23119: a synthetic constitutive expression promoter. MCS: multiple cloning site, which can be used to assemble homologous repair templates; gRNA: guid RNA; rrnBT1T2: rrnBT1T2 terminator; In this study, sgRNA and crRNA specifically refer to the guide RNAs for SpCas9 and FnCas12a, respectively. (B) Deletion of a 0.5 kb DNA fragment (porB) using the plasmid pZF2-sgRNA1-ΔporB in strains 9-12 and Ptac-9. Plasmid pZF2-sgRNA1-ΔporB expresses sgRNA1 targeting the porB locus and carries a 1 kb homologous arm. (C) Introduction of 20 nucleotide changes using the plasmid pZF2-crRNA1-crtYf* in strains 9-12 and L12. Plasmid pZF2-crRNA1-crtYf* expresses crRNA1 targeting the crtYf locus and carries a 1 kb homologous arm. In the genomes of Ptac-9 and L12 strains, a single copy of the Spcas9 and Fncas12a genes is respectively present. However, the 9–12 strain simultaneously carries a copy of both the Spcas9 and Fncas12a genes. (D) Control of the expression of Spcas9 or Fncas12a genes integrated into the genome through different strength promoters and copy numbers. PlacM: synthetic medium-strength promoter; Ptac: synthetic medium-strength promoter; Peftu: strong promoter; PL10: weak promoter (Yim et al., 2013). (E) The relative transcription level of Spcas9 and Fncas12a gene. The transcription level of Spcas9 in E9-12, L9-12 and Cas9-2 was compared against that of Spcas9 in 9–12; The transcription level of Fncas12a in 9-E12, 9-L12 and Cas12-2 was compared against that of cas12a in 9–12. (F) Impact of Spcas9 gene expression at different strengths on the efficiency of porB deletion. (G) Impact of Fncas12a gene expression at different strengths on the efficiency of introducing 20 nucleotide substitutions in crtYf. Data are presented as mean values+/−SD (n = 3 independent experiments). ***p < 0.01, Student’s two-tailed t-test. Source data underlying Supplementary Figure S3 are provided as a Source Data file.
The expression level of Cas proteins is a critical factor influencing editing efficiency (Hsu et al., 2013; Pattanayak et al., 2013). Thus, various strategies, including the replacement of Cas gene promoters and increasing copy numbers (Figure 1D), were employed to optimize the expression levels of Cas genes. A series of C. glutamicum carrying the HyCas9-12aGEP were developed, utilizing either a strong Peftu promoter or a weak PL10 (Yim et al., 2013) promoter to regulate the expression of Spcas9 and Fncas12a (Figure 1D). As expected, the transcription levels of Spcas9 and Fncas12a were consistent with the strength of their respective promoters (Figure 1E). Notably, compared to 9-12, the transcription levels of Fncas12a and Spcas9 in Cas12a-2 and Cas9-2 were increased by 176% and 84%, respectively (Figure 1E), suggesting that the location of genes in the genome affects the expression of genes (Akhtar et al., 2013; Bryant et al., 2014; Goormans et al., 2020). Transformation with 1 μg of pZF2-sgRNA1-△porB into L9-12, produced more than 103 c.f.u, among which −16.6% were correctly edited (Figure 1F). However, compared to 9-E12, Cas9-2 exhibited similar editing efficiency but a significantly reduced number of transformants (Figure 1F). Similar results were observed when the plasmid pZF2-crRNA1-crtYf* was transformed into strains containing cas12a genes controlled by promoters of varying strengths (Figure 1G). These results indicate that insufficient expression levels of SpCas9 and FnCas12a result in a high escape rate that is inadequate for eliminating wild-type cells, while excessively high expression levels of SpCas9 and FnCas12a lead to reduced transformation efficiency. Therefore, the 9–12 strain, which balances transformation efficiency and editing efficiency, was selected for subsequent experiments.
HyCas9-12aGEP improves genome editing resolution
Due to the heterogeneity of gRNA activity, there are instances where no active gRNA is available for specific gene loci, leading to a decrease in the resolution of gene editing. For example, CRISPR-FnCas12a (Jiang et al., 2017) and SpCas9 (Zhang et al., 2020) have been reported to lack suitable gRNAs for specific gene loci in proB and zwf, respectively, resulting in failures to precisely introduce mutations at the target gene sites in C. glutamicum. Thus, this generally lowers the resolution of CRISPR-Cas-based gene editing compared to the theoretical value. However, by leveraging both SpCas9 and FnCas12a nucleases, each recognizing different PAM sequences, HyCas9-12aGEP increases gRNA availability, thereby enhancing gene editing resolution.
The γ-glutamyl kinase encoded by proB is the rate-limiting enzyme in proline synthesis and is subject to feedback inhibition by proline (Perez-Arellano et al., 2006; Wendisch, 2014). Using CRISPR-Cas12a-assisted genome editing, the mutant strain 13032ProBG149K was generated to alleviate the feedback inhibition of proline, involving the introduction of adjacent synonymous mutations (Jiang et al., 2017). However, it has been reported that synonymous mutations do not alter the encoded protein but can influence gene expression (Kudla et al., 2009). Thus, to prevent the introduction of synonymous mutations, theoretically, only 1 PAM is available for FnCas12a to introduce site-directed mutations at the 149th amino acid residue of ProB, while SpCas9 has 3 available PAMs (Figure 2A). Here, crRNA2, sgRNA2 and sgRNA3 are inactivated by mutation of the 149th amino acid residue glycine (GGT) of CgProB in the homologous arm to lysine (AAG). Thus, the plasmids pZF2-crRNA2-ProBG149K, pZF2-sgRNA2-ProBG149K, and pZF2-sgRNA3-ProBG149K are created by assembling crRNA2, sgRNA2, sgRNA3 and homology arms into the plasmid pZF2 (Figure 2B). The FnCas12a-based edited plasmid pZF2-crRNA2-ProBG149K was electroporated into the 9–12 strain, and no transformants containing the ProBG149K mutation were detected (Figure 2C), potentially attributed to insufficient crRNA2 activity (Figure 2C). However, SpCas9-based editing plasmids pZF2-sgRNA2-ProBG149K and pZF2-sgRNA3-ProBG149K were transformed into the 9–12 strain, produced −1.1×103 c.f.u, among which −93.3% and 96.6% were correctly edited, respectively (Figures 2C; Supplementary Figure S5). The mutated transformants were subsequently further verified by sequencing (Figure 2D). Thus, these results suggest that the HyCas9-12aGEP enables more precise mutations than the CRISPR-Cas12a system, thereby improving gene editing resolution. Furthermore, these findings emphasize the importance of active gRNAs in successful gene editing.
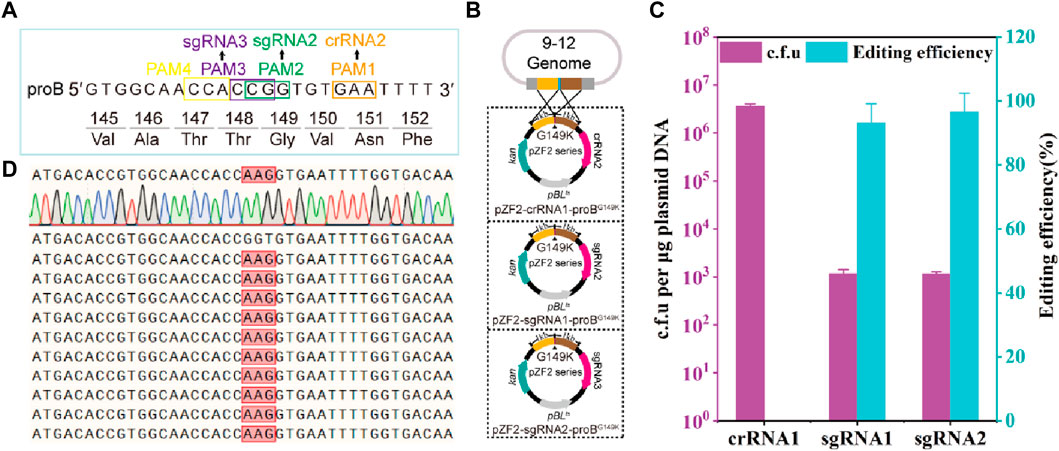
FIGURE 2. HyCas9-12aGEP improves genome editing resolution. (A) DNA sequence near the 149th amino acid residue of the proB gene, encoding γ-glutamyl kinase. PAM1 represents the recognition sites for Cas12a/crRNA2; PAM2 and PAM3 are the recognition sites for Cas9/sgRNA2 and Cas9/sgRNA3, respectively. (B) Schematic representation of precise mutagenesis at the 149th amino acid residue of the proB gene, mediated by crRNA2, sgRNA2, and sgRNA3. A single plasmid carrying a 1 kb homologous arm and the gRNA was transformed into the 9–12 strain. Cas9 and Cas12a/gRNA induce target site cleavage, and the homologous arm with inactivated PAM or seed sequence mutations serves as a template for homologous recombination repair, leading to the precise introduction of an amino acid replacement at the 149th position in ProB. (C) Gene editing assessment of the proB (G149K) mutation mediated by crRNA2, sgRNA2, and sgRNA3 in the 9–12 strain. (D) Ten representative proB recombinants identified by PCR were further sequenced, which revealed the substitution of GGT by AAG for all samples, as expected. Data are presented as mean values+/−SD (n = 3 independent experiments). Source data for Supplementary Figure S5 are provided as a Source Data file.
HyCas9-12aGEP improves the mapping efficiency of active gRNAs
The efficiency of CRISPR-based gene editing is highly dependent on gRNA activity (Figure 2C). Therefore, the selection and design of efficient gRNAs is essential to improve gene editing efficiency. Although tools have been developed to predict gRNA activity in certain hosts, these programs still have limitations in accurately predicting gRNA activity in diverse hosts because different cells and host types are also factors affecting gRNA activity (Moreb and Lynch, 2021). Literature mining is an effective strategy, but the targets of gRNA used in the literature are limited, which is difficult to meet the requirements of different experimental content. Therefore, efficiently mapping active gRNA remains a bottleneck in CRISPR-Cas gene editing experiments.
Next, we attempted to establish a method for evaluating gRNA activity through experimental assays. It is widely recognized that non-homologous end joining (NHEJ) and homologous recombination (HR) are the primary repair pathways for DNA double-strand breaks. However, it should be noted that the NHEJ pathway is impaired in C. glutamicum (Resende et al., 2011) (https://www.kegg.jp/kegg/pathway.html). When gRNA guides SpCas9 or FnCas12a to cleavage DNA, in the absence of a homologous repair template, the organism undergoes cell death since the double-stranded DNA break cannot be repaired. Therefore, gRNA activity can be assessed through the transformation efficiency of gRNA expression plasmids lacking homologous repair templates. In other words, higher gRNA activity leads to more efficient SpCas9/gRNA DNA cleavage, resulting in fewer corresponding transformants, and vice versa (Figure 3A). Since FnCas12a can process its own crRNA, transcribed hfgRNA can be processed into independent crRNA and sgRNA, which can orthogonally guide SpCas9 and FnCas12a to target two genomic loci (Figure 3B). This allows a single hfgRNA to assess the targeting activity of two designed gRNAs, doubling the efficiency of identifying active gRNAs. As a proof of concept, nine crRNAs and sgRNAs targeting the zwf, pck, gnd, crtR, alaT, phage CGP3, gdh, crtEb-crtR and esrR loci, were transcribed individually or co-transcribed with their respective nine hfgRNAs. Unexpectedly, we did not observe a significant difference in the counter-selection efficiency of hfgRNA in Ptac-9 and L12 when compared to independently transcribed sgRNA and crRNA (Figures 3C,D). These results suggested that hfgRNA design does not affect the activity of sgRNA or crRNA, thereby confirming that HyCas9-based approaches can indeed improve the efficiency of identifying active gRNAs. Notably, the counter-selection efficiency achieved with hfgRNA in strain 9–12 exhibited a significant enhancement compared to Ptac-9 and L12 (Figure 3E). Similarly, based on the gRNA online design tool (http://www.rgenome.net/cas-designer/), we designed 15 crRNAs and 15 sgRNAs and assembled them into corresponding hfgRNAs (Supplementary Data S1, S2), which can also significantly improve the counter-selection efficiency in 9–12 (Figure 3E). These results suggested that hfgRNA based on HyCas9-12aGEP can also improve the counter-selection efficiency.
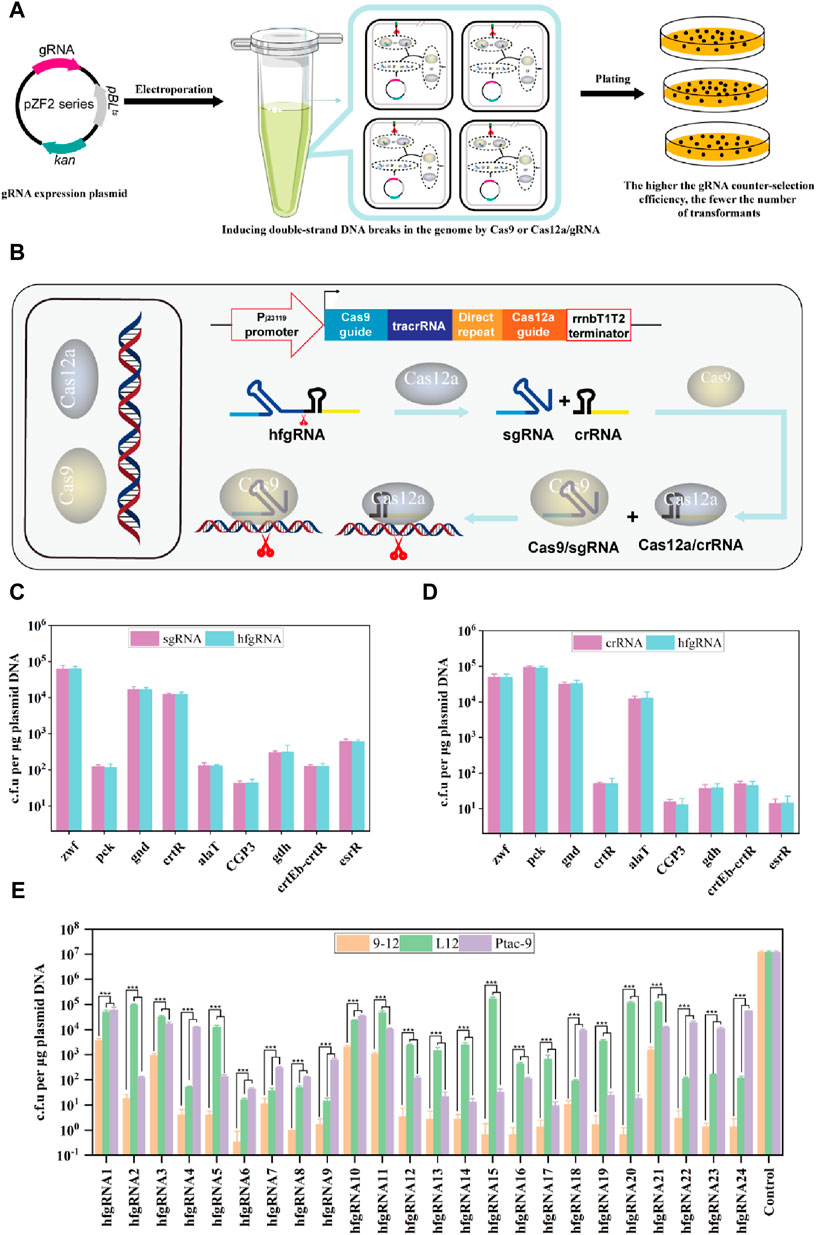
FIGURE 3. Determination of gRNA activity through experimental methods. (A) Schematic representation of gRNA activity assessment based on experiments. I: Plasmids carrying gRNAs were transformed into Ptac-9 (CRISPR-Cas9), L12 (CRISPR-Cas12a), and 9-12 (HyCas9-12aGEP); II: Expressed gRNAs guide Cas9 or Cas12a for genome cleavage; III: The more thorough the cleavage of genomic DNA by Cas9/gRNA or Cas12a/gRNA, the fewer the number of transformants. (B) Schematic representation of dual-target DNA cleavage mediated by hfgRNA in 9–12. Transformed hfgRNA is processed by Cas12a to yield crRNA and sgRNA, guiding Cas12a and Cas9 to simultaneously target two genomic loci. (C, D) Impact of hfgRNA design on the activity of sgRNAs and crRNAs targeting the zwf, pck, gnd, crtR, alaT, CGP3, gdh, crtEb-crtR and esrR loci. Plasmids expressing sgRNA, crRNA, and their respective fusion hfgRNA were separately transformed into Ptac-9 and L12 to determine the number of transformants. (E) Determination of the transformation efficiency of 24 hfgRNAs in Ptac-9, L12, and 9-12. The 24 corresponding target sites for hfgRNAs are provided in Supplementary Data S2. hfgRNA1-9 are used in panels (C) and (D). Data are presented as mean values+/−SD (n = 3 independent experiments). ***p < 0.01, Student’s two-tailed t-test.
HyCas9-12aGEP combined with hfgRNA improves the genome editing efficiency
Based on HyCas9-12aGEP, hfgRNA effectively improves the counter-selection efficiency (Figure 3E). To test hfgRNA gene editing performance, plasmids expressing hfgRNA and harboring two −1 kb HAs were transformed into 9–12, Ptac-9, and L12 strains (Figure 4A). First, hfgRNA4 was used to introduce a stop codon into the crtR gene to inactivate CrtR (CrtRG47Z). Although the site-directed mutation efficiency of hfgRNA4 in Ptac-9 and L12 is as high as 86.9% and 88.4%, respectively, the editing efficiency reached 100% in 9–12 (Figure 4B). Similarly, when hfgRNA5 was used to delete the 0.5 kb alaT gene in 9–12, an editing efficiency of 85.5% was obtained, which was significantly higher than the 50.7% and 1.44% obtained in Ptac-9 and L12 (Figure 4C). Notably, when hfgRNA6 was employed in Ptac-9 and L12 to delete a 219 kb DNA fragment (intact phage CGP3), editing efficiencies of 36.2% and 40.6% were achieved, respectively, slightly exceeding the 34.8% efficiency previously reported (Liu et al., 2022) (Figure 4D). However, when the CGP3 (219 kb) was deleted in strains 9–12, an editing efficiency of 66.6% was achieved, surpassing the levels attainable in Ptac-9 and L12 (Figure 4D).
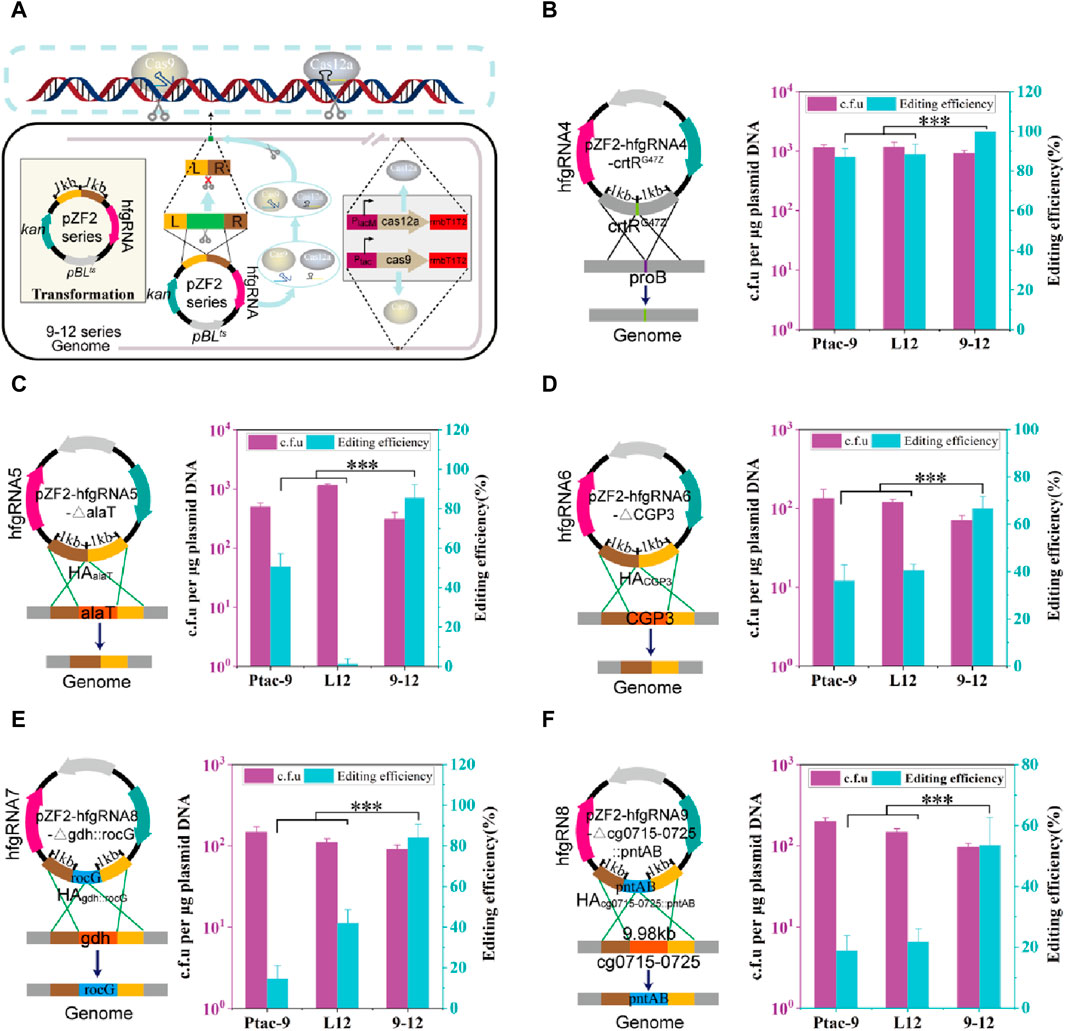
FIGURE 4. Genome editing mediated by hfgRNA in CRISPR-Cas9, CRISPR-Cas12a, and HyCas9-12aGEP. (A) Schematic representation of gene editing mediated by hfgRNA in the HyCas9-12aGEP system. The transcribed hfgRNA is processed by FnCas12a to generate crRNA and sgRNA, which respectively guide FnCas12a and SpCas9 to simultaneously target and cleave two distinct loci in the genome. Homologous recombination is employed for the introduction of the desired mutations. (B–F) Genome editing using hfgRNA in the 9–12 strain harboring HyCas9-12aGEP. hfgRNA4-8 are designed to target crtR, alaT, CGP3, gdh, and crtR/crtEb, for the introduction of mutations including crtRG47Z, ∆alaT (0.5 kb), ∆CGP3 (219 kb), ∆gdhrocG, and ∆cg0715-0725pntAB. CrtRG47Z involves the substitution of the 47th glycine residue in CrtR with a stop codon to deactivate the crtR gene. ∆alaT (0.5 kb): deletion of a 0.5 kb segment of the alaT gene. ∆CGP3 (219 kb): the complete removal of the CGP3 bacteriophage, spanning 219 kb ∆gdhrocG: deletion of the endogenous glutamate dehydrogenase gene gdh (1.34 kb) and concomitant insertion of the expression cassette Ptac-rocG-rrnBT1T2 (1.58 kb), encoding the glutamate dehydrogenase rocG derived from Bacillus subtilis. ∆cg0715-0725pntAB: deletion of the gene cluster cg0715-0725 while inserting the expression cassette PH36-pntAB-rrnBT1T2 (3.5 kb) from E. coli. Data are presented as mean values+/−SD (n = 3 independent experiments). ***p < 0.01, Student’s two-tailed t-test. Source data underlying Supplementary Figure S6 are provided as a Source Data file.
Next, we tested the editing efficiency of hfgRNA in insertion. To optimize chemical and biofuel production, strategies in cofactor engineering have been devised, including adjustments in cofactor supply and modifications to reactants’ cofactor preference, ensuring redox balance (Wang et al., 2017). Thus, hfgRNA7 was used to delete the NADPH-dependent glutamate dehydrogenase gene gdh (1.34 kb) deletion in C. glutamicum, while insert the NADH-dependent glutamate dehydrogenase gene rocG (1.58 kb) from Bacillus subtilis 168. The editing efficiency of hfgRNA5 in Ptac-9 and L12 was 14.5% and 42.0%, respectively, but it was as high as 84.0% in 9–12 (Figure 4E). Finally, we also test whether hfgRNA8 can achieve deletion of large fragments (9.98 kb) while inserting the E. coli-derived pntAB (3.5 kb) operon. The membrane-bound transhydrogenase encoded by pntAB is advantageous for enhancing the supply of NADPH, thereby promoting the synthesis of high-value metabolic products (Kleine et al., 2017). Notably, hfgRNA6 achieved a deletion-insertion efficiency of 53.6% in 9–12 strain, significantly higher than Ptac-9 and L12, which yielded efficiencies of 18.8% and 21.7%, respectively (Figure 4F). In conclusion, the above results suggested that hfgRNA significantly improves the efficiency of genome editing based on HyCas9-12aGEP.
Application of the HyCas9-12aGEP for efficient phenotype-genotype mapping
Large fragment genome editing can be used to study the function of genes and noncoding regions. By deleting large DNA fragments, researchers can gain profound insights into how these regions impact an organism’s growth, development, and physiological processes. The complete crtREBIYe/fEb gene cluster (cg0717-0725) is an important factor in the yellow color of C. glutamicum, and deletion of the cg0717-0725 may cause the color of the organism to change (Figure 5A). Here, hfgRNA8 was used to performed large fragment gene deletion testing of cg0715-0725 (9.98 kb), cg0715-0736 (20.02 kb) and cg0697-cg0740 (40.96 kb) (Figure 5B). For deletion of cg0715-0725, transformants of two colony morphologies, white and yellow, were obtained (Supplementary Figure S7A). The white single clones were picked for PCR verification, and the results showed that they were all successfully edited strains (Supplementary Figure S7B). Hence, the success of editing can be determined by the color of individual colonies (Supplementary Figures S7B, S8A). As expected, hfgRNA8 significantly improved editing efficiency in 9–12 compared to Ptac-9 and L12 (Figure 5C; Supplementary Table S3). Remarkably, the deletion of 40.9 kb failed to yield detectable successfully edited strains in L12 and Ptac-9, whereas 9–12 maintained a remarkable editing efficiency of 38.1%. Unexpectedly, the deletion of the 40.9 kb fragment altered the colony morphology, rendering it more “moisture” (Figure 5D). However, no significant changes in the morphology of the bacteria were found by electron microscopy (Figure 5E). Therefore, we inferred that this phenotype may be caused by significant metabolic changes.
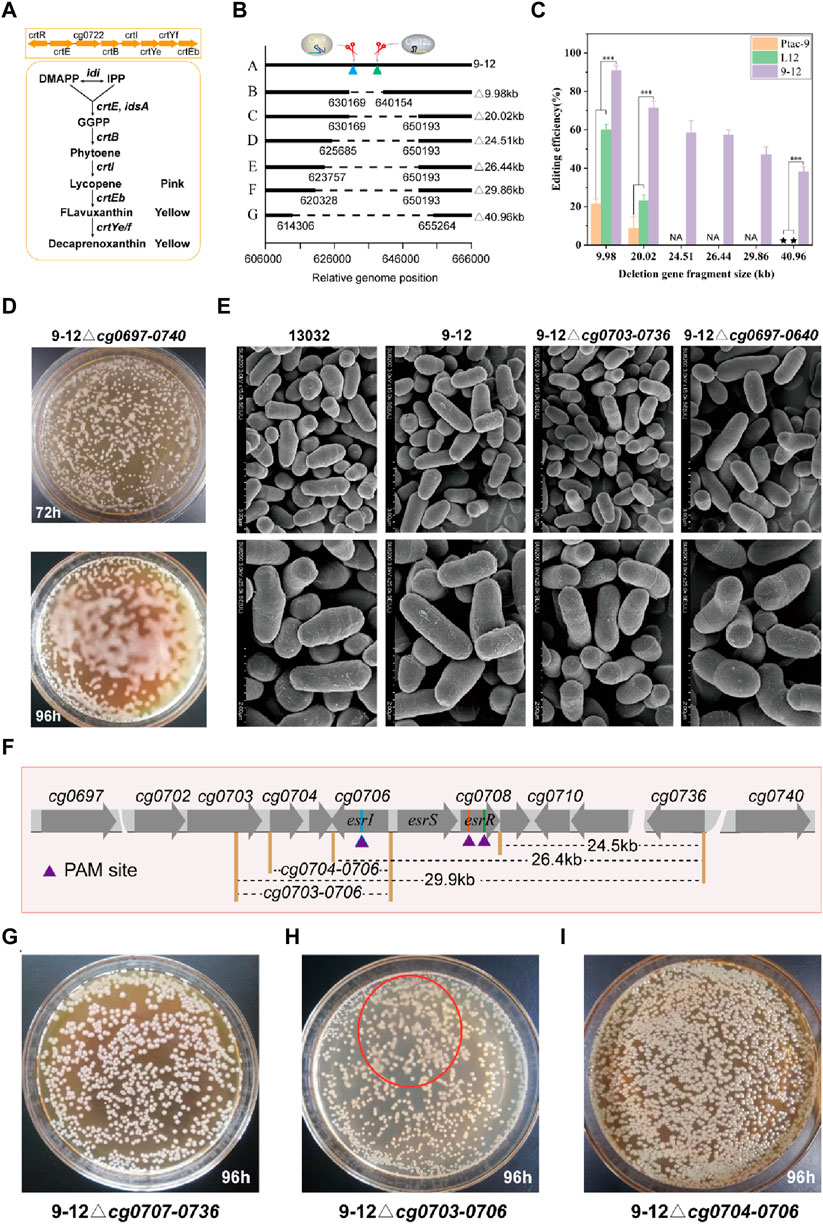
FIGURE 5. Application of HyCas9-12aGEP for phenotype-genotype mapping via large DNA segment deletion. (A) The reaction catalyzed by the crtEBIYe/fEb gene cluster for terpenoid biosynthesis. DMAPP: dimethylallyl pyrophosphate; IPP: isopentenyl pyrophosphate; GGPP: geranylgeranyl diphosphate; (B) Relative genomic positions of cg0725-0725 (9.98 kb), cg0715-0736 (20.02 kb), cg0710-0736 (24.51 kb), cg0707-0736 (26.44 kb), cg0703-0736 (29.86 kb), and cg0697-0740 (40.96 kb) in C. glutamicum ATCC13032. (C) Deletion efficiency of cg0725-0725 (9.98 kb), cg0715-36 (20.02 kb), cg0710-0736 (24.51 kb), cg0707-0736 (26.44 kb), cg0703-0736 (29.86 kb), and cg0697-0740 (40.96 kb) guided by hfgRNA8. (D) Phenotypic changes induced by the deletion of cg0697-0740 (40.96 kb) in C. glutamicum. Deletion of cg0697-0740 (40.96 kb) leads to increased ‘moisture’ of C. glutamicum on CM agar plates. (E) Scanning electron microscopy results of five different genotypes of (C) glutamicum, including 13032, 9-12, 9-12△cg0703-0736, and 9-12△cg0697-0740. (F) Phenotype-genotype mapping strategy. Rapid phenotype-genotype mapping is achieved through a strategy of halving the number of genes each time to pinpoint the genotypes responsible for the observed phenotypic changes. (G–I) Colony phenotypes of 9–12△cg0707-0736, 9-12△cg0704-0706, and 9-12△cg0703-0706 on CM agar plates after 96 h, respectively. The red ellipse indicates the corresponding ‘moisture’ colony phenotype observed in 9–12△cg0703-0706. Data are presented as mean values+/−SD (n = 3 independent experiments). ***p < 0.01, Student’s two-tailed t-test. Source data underlying Supplementary Figure S7 and Supplementary Table S3 are provided as a Source Data file.
Next, we performed gradual gene fragment deletions to identify phenotype-influencing genes (Figure 5F). Since the expected phenotype didn't emerge with the 20 kb gene fragment knockout, the gene affecting the phenotype likely resides in the 20–40 kb region. Deleting the 29.86 kb DNA fragment (47.2% editing efficiency) revealed the phenotype (Supplementary Figure S8B), pinpointing the gene affecting it in the 20–30 kb range. Notably, the three-component system EsrISR (encoded by cg0706-0708) regulates a cell envelope stress response in C. glutamicum (Kleine et al., 2017). However, sequential deletions of cg0710-0736 (24.51 kb) and cg0707-0736 (26.44 kb) fragments did not induce the same phenotype (Figure 5G), which suggested that the EsrISR three-component system has an extremely limited contribution to this phenotype, and deletion experiments of cg0706-0708 using hfgRNA9 also support this finding (Supplementary Figures S8C, D). Thus, only cg0703-0706 remained unevaluated. Finally, crRNA11 was employed to target esrI (cg0706) and facilitate the deletion of cg0703-0706, resulting in the strain 9–12△cg0703-0706 (Supplementary Figure S8D), which exhibited a milder version of the phenotype compared to the 29.86 kb and 40.96 kb deletions (Figure 5H). Moreover, 9-12△cg0704-0706 strain did not yield the expected phenotype (Figure 5I; Supplementary Figure S8D). Thus, it can be inferred that the deletion of cg0703 is responsible for this phenotype, but the deletion of cg0704-0736 also greatly contributed to it. The precise regulatory mechanism can be further elucidated through subsequent transcriptomic and metabolomic analyses. In conclusion, based on HyCas9-12aGEP, hfgRNA enables efficient phenotype-genotype mapping.
Applicability of HyCas9-12aGEP in other Corynebacterium species
To determine whether HyCas9-12aGEP can be constructed in other species of Corynebacterium except for strain 13032, the HyCas9-12aGEP was introduced into strains C. glutamicum S9114, C. glutamicum ATCC 13869, C. glutamicum LG-3, C. glutamicum N-77, and C. glutamicum I31-5 by sequentially integrating the Cas12a and SpCas9 genes at the ldh and putA locus of these strains genome. Plasmids pZF2-sgRNA1-△porB and pZF2-crRNA1-crtYf* were transformed to a series of strains harboring HyCas9-12aGEP to evaluate the editing efficiency. Although differences in transformation efficiency were observed among different strains harboring HyCas9-12aGEP (Figure 6A), all six tested strains exhibited comparable editing efficiency for the target loci (Figure 6B). Hence, these results suggested that the HyCas9-12aGEP system can be applied to other species of Corynebacterium.
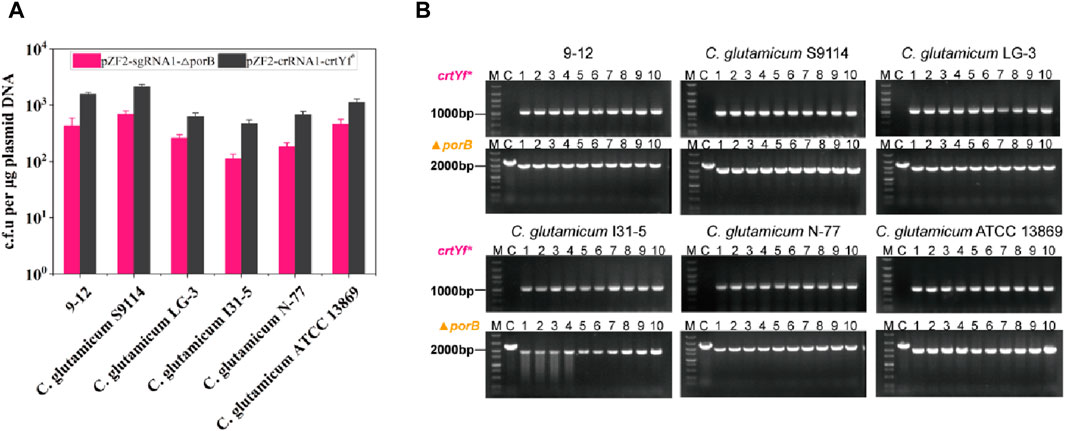
FIGURE 6. Applications of HyCas9-12aGEP in other Corynebacterium species. (A) Growth of Corynebacterium strains carrying the integrated CRISPR-Cas9-Cas12a system transformed with pZF2-sgRNA1-△porB and pZF2-sgRNA1-crtYf*. Experiments were performed in duplicates. (B) Ten transformant colonies of the indicated Corynebacterium strains derived from pZF2-sgRNA1-△porB and pZF2-sgRNA1-crtYf*-based HyCas9-12aGEP recombining were analyzed by colony PCR. A DNA ladder (DL 5000 DNA Marker, Vazyme Biotech Co.,Ltd) was used as a marker.
Off target analysis of HyCas9-12aGEP
To analysis the off-target effect in C. glutamicum after gene editing by HyCas9-12aGEP, genome resequencing was performed to identify all the single nucleotide polymorphism (SNP) and insertions and deletions (Indel). The strains analyzed included 9-12△porB (porB-deleted strain), 9-12△cg0697-0740 (cg0697-0740-deleted strain) and the 9-12△gdh::rocG (gdh-deleted and rocG-inserted strain), with wild-type 13032 serving as the negative control. Furthermore, to investigate whether the SpCas9 and FnCas12a protein induce off-target effects in the absence of gRNA, the SNP and Indel profiles of the 9–12 strain harboring SpCas9 and FnCas12a proteins were also examined. The results indicated that no off-target mutations were identified in the 9–12 strain containing SpCas9 and FnCas12a proteins. That is, in comparison to the wild-type strain, only two Indels (corresponding to ldh and putA deletions) and two intergenic Indels (corresponding to Spcas9 and Fncas12a integrations) were detected, with no SNPs observed in this strain. Meanwhile, No SNP and Indel were identified in the 9-12△cg0697-0740 and 9-12△gdh::rocG strains (Supplementary Table S4). Notely, compared to previous reports where 1 Indel with 1 base deleted was identified during the resequencing of porB-deleted strains (Peng et al., 2017), we did not observe any SNP and Indel in the 9-12△porB strains (Supplementary Table S4). The results suggested that the genomic integration of the SpCas9 gene effectively reduces the off-target effects of SpCas9.
Discussion
HyCas9-12aGEP in C. glutamicum was developed and optimized to improve the resolution of genome editing. Leveraging hfgRNA design, HyCas9-12aGEP outperforms both CRISPR-Cas9 and CRISPR-Cas12a in terms of gene editing efficiency and active gRNA mapping. HyCas9-12aGEP’s robust capability for large-fragment editing enables rapid identification of genotype-phenotype relationships among over 40 genes. Integrating SpCas9 expression into the genome effectively minimizes off-target editing. In summary, HyCas9-12aGEP streamlines gene editing in C. glutamicum, enhancing accessibility and promoting a more sustainable, efficient, and precise biological production process.
SpRY-HF1, an unconstrained near-PAMless and high-fidelity SpCas9 variant, was inferred to have low nuclease activity in C. glutamicum as it is the active gRNA for SpCas9 but not for SpRY-HF1 and its expression level in C. glutamicum is also comparable to SpCas9. SpCas9/gRNA exhibits stable DNA binding with just an 8-9 bp match to the PAM-proximal region (Singh et al., 2018). However, the SpRY-Cas9 variant, which is generally unrestricted by PAM sites, indicates that a large number of SpRY-Cas9/sgRNA complexes will be titrated on the numerous homologous sequences in the genome. In effect, there were significant reductions in effective SpRY-Cas9/gRNA complex concentrations, which prolong the time for it to search for the target site. Consequently, this decreases the editing activity of the gRNA (Moreb and Lynch, 2021). Meanwhile, the conformational state of the HNH nuclease domain directly controls the DNA cleavage activity, in which the DNA cleavage efficiency is proportional to the extent of the activated conformation of the HNH domain (Sternberg et al., 2015). For SpCas9, the HNH-activated conformation is closely linked to the unwinding state of the DNA target, accounting for 78%–100% of the unwinding portion of all DNA targets (Dagdas YS et al., 2017). SpCas9-HF1 also has a reduced ability to unwind target DNA (Okafor et al., 2019), which inevitably inhibits DNA cleavage activity. The titration effect of SpRY-Cas9 and the reduced cleavage efficiency of high-fidelity SpCas9-HF1 may have potentially resulted in significant reductions of SpRY-HF1 nuclease activity in C. glutamicum.
We attempted unsuccessfully to transform plasmids pFSC-Cas12a and PYJS3-Cas9, which co-express SpCas9 and FnCas12a, into C. glutamicum. Plasmids expressing dCas9 in constitutive forms have been challenging to transform in C. glutamicum (Cleto et al., 2016). However, optimizing promoters and ribosomal binding sites (RBS) has significantly improved transformation efficiency by reducing SpCas9 and FnCas12a expression (Cleto et al., 2016; Liu et al., 2017; Li et al., 2020). Furthermore, integrating one copy of the SpCas9 gene into the genome has effectively mitigated the toxicity of SpCas9 proteins with minimal impact on bacterial growth (Wang et al., 2018). The plasmid copy number is governed by the replicon, with pJYS3 and pFSC plasmids’ replicon, pBL1, having a range of 10–30 copies (Pátek and Nešvera, 2013). This elevated SpCas9 or FnCas12a expression resulting from the multi-copy cas gene could potentially lead to cytotoxicity. Therefore, this explains why a single copy integration of the Spcas9 or Fncas12a gene does not result in cytotoxicity, while having two copies of either Spcas9 or Fncas12a genes does. Although high concentrations of SpCas9 proteins can induce cytotoxicity (Cleto et al., 2016) and off-target effects (Hsu et al., 2013; Pattanayak et al., 2013), low-level SpCas9 or FnCas12a expression reduces editing efficiency (Wang et al., 2018) (Figures 1E,F). Therefore, optimizing the expression levels of SpCas9 or FnCas12a proteins is a crucial step in enhancing gene editing efficiency.
The target sites (PAM sites of 5′-NGG-3′ and 5′-TTN-3′) that HyCas9-12aGEP can use are the sum of SpCas9 and FnCas12a, thereby improving gene editing resolution compared to CRISPR-Cas9 or Cas12a. Although this study demonstrates that the SpCas9 system can accurately introduce mutations at amino acid 149 of ProB, FnCas12a cannot. However, we also found that the gRNA activity of SpCas9 is diverse in our experiments (Figure 2C, Figures 3C–E), consistent with previous reports (Doench et al., 2014; Moreb and Lynch, 2021; Corsi et al., 2022). In addition, Cas12a has been reported to be more efficient at cutting covalently modified DNA duplexes than Cas9 (Vlot et al., 2018; Dong et al., 2021). Covalent DNA modifications are prevalent across organisms, giving Cas12a an edge over Cas9 when targeting sites within covalently modified genomes. Based on these analyses, the HyCas9-12aGEP improved the resolution of gene editing compared to the CRISPR-Cas9 and Cas12a gene-editing systems. With the discovery of novel Cas proteins (Pausch et al., 2020; Karvelis et al., 2021; Ozcan et al., 2021; Wu et al., 2021; Tsuchida et al., 2022) and application of modified high-performance variants (Kleinstiver et al., 2019; DeWeirdt et al., 2021) in C. glutamicum, integration into the genome can further improve gene editing resolution and editing efficiency to build higher-dimensional CRISPR systems.
Based on hfgRNA, HyCas9-12aGEP significantly improves gene editing efficiency compared to classical CRISPR-Cas9 and CRISPR-Cas12a gene editing tools. Previously described CRISPR-Cas9 or Cas12a-based multi-targeting strategies co-expressed pairs of Cas9 (Liu et al., 2017; Horlbeck et al., 2018; Zhao et al., 2018; Zhao et al., 2020) or Cas12a gRNA (Campa et al., 2019; Liu et al., 2019), however, these approaches have limited efficiency, especially in synchronous targeting. Meanwhile, some systems employing orthologous Staphylococcus aureus Cas9 and SpCas9 enzymes (Najm et al., 2017; Boettcher et al., 2018) or SpCas9 and Lachnospiraceae bacterium Cas12a (Gonatopoulos-Pournatzis et al., 2020), like HyCas9-12aGEP, have increased editing efficiency, possibly due to reduced recombination through the use of different transactivating CRISPR RNAs. Taken together, HyCas9-12aGEP greatly improve our capability in terms of genome reprogramming in C. glutamicum. Furthermore, we envision that HyCas9-12aGEP is also applicable to other microorganisms.
An efficient gene editing system should not only have reliable and efficient on-target gene editing efficiency, but also should produce minimal off-target effects. It is reported that high levels of Cas9 protein expression would increase the off-target effects (Hsu et al., 2013). In this study, the expression of SpCas9/FnCas12a protein was reduced by integrating Spcas9/Fncas12a gene into genome, which was beneficial to reduce the probability of off-target. Indeed, our resequencing results confirmed that HyCas9-12aGEP did not induce off-target editing at the previously identified porB off-target sites (Peng et al., 2017). Although the use of hfgRNA in HyCas9-12aGEP for gene editing theoretically raises the probability of off-target effects, our resequencing results for 9-12△porB, 9-12△cg0697-0740 and 9-12△gdh::rocG strains did not reveal any off-target editing. In the further research, it will theoretically contribute to reducing the potential for off-target effects by replacing the high-fidelity Cas9 and Cas12a variants (Chen et al., 2017).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
FZ: Writing–review and editing, Writing–original draft, Validation, Software, Methodology, Investigation, Formal Analysis, Data curation. J-YW: Software, Data curation, Writing–review and editing, Formal Analysis. C-LL: Writing–review and editing, Formal Analysis. W-GZ: Writing–review and editing, Funding acquisition, Formal Analysis.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key Research and Development Program of China (No. 2021YFC2100900).
Acknowledgments
We thank to Prof. Z. H. Bai from National Engineering Laboratory for Cereal Fermentation Technology, Jiangnan University for donating the plasmids pFSC and pFST-porB.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2024.1327172/full#supplementary-material
References
Abdel-Mawgoud, A. M., and Stephanopoulos, G. (2020). Improving CRISPR/Cas9-mediated genome editing efficiency in Yarrowia lipolytica using direct tRNA-sgRNA fusions. Metab. Eng. 62, 106–115. doi:10.1016/j.ymben.2020.07.008
Akhtar, W., de Jong, J., Pindyurin, A., Pagie, L., Meuleman, W., de Ridder, J., et al. (2013). Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 154, 914–927. doi:10.1016/j.cell.2013.07.018
Becker, J., Rohles, C. M., and Wittmann, C. (2018). Metabolically engineered Corynebacterium glutamicum for bio-based production of chemicals, fuels, materials, and healthcare products. Metab. Eng. 50, 122–141. doi:10.1016/j.ymben.2018.07.008
Boettcher, M., Tian, R., Blau, J. A., Markegard, E., Wagner, R. T., Wu, D., et al. (2018). Dual gene activation and knockout screen reveals directional dependencies in genetic networks. Nat. Biotechnol. 36, 170–178. doi:10.1038/nbt.4062
Bryant, J. A., Sellars, L. E., Busby, S. J., and Lee, D. J. (2014). Chromosome position effects on gene expression in Escherichia coli K-12. Nucleic Acids Res. 42, 11383–11392. doi:10.1093/nar/gku828
Campa, C. C., Weisbach, N. R., Santinha, A. J., Incarnato, D., and Platt, R. J. (2019). Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 16, 887–893. doi:10.1038/s41592-019-0508-6
Chen, J. S., Dagdas, Y. S., Kleinstiver, B. P., Welch, M. M., Sousa, A. A., Harrington, L. B., et al. (2017). Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410. doi:10.1038/nature24268
Chen, X., Rinsma, M., Janssen, J. M., Liu, J., Maggio, I., and Gonçalves, M. A. (2016). Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 44, 6482–6492. doi:10.1093/nar/gkw524
Cleto, S., Jensen, J. V. K., Wendisch, V. F., and Lu, T. K. (2016). Corynebacterium glutamicum metabolic engineering with CRISPR interference (CRISPRi). ACS Synth. Biol. 5, 375–385. doi:10.1021/acssynbio.5b00216
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/cas systems. Science 339, 819–823. doi:10.1126/science.1231143
Corsi, G. I., Qu, K., Alkan, F., Pan, X., Luo, Y., and Gorodkin, J. (2022). CRISPR/Cas9 gRNA activity depends on free energy changes and on the target PAM context. Nat. Commun. 13, 3006. doi:10.1038/s41467-022-30515-0
Creutzburg, S. C. A., Wu, W. Y., Mohanraju, P., Swartjes, T., Alkan, F., Gorodkin, J., et al. (2020). Good guide, bad guide: spacer sequence-dependent cleavage efficiency of Cas12a. Nucleic Acids Res. 48, 3228–3243. doi:10.1093/nar/gkz1240
Dagdas Ys, C. J., Sternberg, S. H., Doudna, J. A., and Yildiz, A. (2017). A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci. Adv. 3, eaao0027. doi:10.1126/sciadv.aao0027
DeWeirdt, P. C., Sanson, K. R., Sangree, A. K., Hegde, M., Hanna, R. E., Feeley, M. N., et al. (2021). Optimization of AsCas12a for combinatorial genetic screens in human cells. Nat. Biotechnol. 39, 94–104. doi:10.1038/s41587-020-0600-6
Doench, J. G., Hartenian, E., Graham, D. B., Tothova, Z., Hegde, M., Smith, I., et al. (2014). Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267. doi:10.1038/nbt.3026
Dong, J., Chen, C., Liu, Y., Zhu, J., Li, M., Rao, V. B., et al. (2021). Engineering T4 bacteriophage for in vivo display by type V CRISPR-cas genome editing. ACS Synth. Biol. 10, 2639–2648. doi:10.1021/acssynbio.1c00251
Gonatopoulos-Pournatzis, T., Aregger, M., Brown, K. R., Farhangmehr, S., Braunschweig, U., Ward, H. N., et al. (2020). Genetic interaction mapping and exon-resolution functional genomics with a hybrid Cas9–Cas12a platform. Nat. Biotechnol. 38, 638–648. doi:10.1038/s41587-020-0437-z
Gong, S., Yu, H. H., Johnson, K. A., and Taylor, D. W. (2018). DNA unwinding is the primary determinant of CRISPR-cas9 activity. Cell Rep. 22, 359–371. doi:10.1016/j.celrep.2017.12.041
Goormans, A. R., Snoeck, N., Decadt, H., Vermeulen, K., Peters, G., Coussement, P., et al. (2020). Comprehensive study on Escherichia coli genomic expression: does position really matter? Metab. Eng. 62, 10–19. doi:10.1016/j.ymben.2020.07.007
Guo, C., Ma, X., Gao, F., and Guo, Y. (2023). Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 11, 1143157. doi:10.3389/fbioe.2023.1143157
Guo, J., Wang, T., Guan, C., Liu, B., Luo, C., Xie, Z., et al. (2018). Improved sgRNA design in bacteria via genome-wide activity profiling. Nucleic Acids Res. 46, 7052–7069. doi:10.1093/nar/gky572
Horlbeck, M. A., Witkowsky, L. B., Guglielmi, B., Replogle, J. M., Gilbert, L. A., Villalta, J. E., et al. (2016). Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife 5, e12677. doi:10.7554/elife.12677
Horlbeck, M. A., Xu, A., Wang, M., Bennett, N. K., Park, C. Y., Bogdanoff, D., et al. (2018). Mapping the genetic landscape of human cells. Cell 174, 953–967. doi:10.1016/j.cell.2018.06.010
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832. doi:10.1038/nbt.2647
Ivanov, I. E., Wright, A. V., Cofsky, J. C., Aris, K. D. P., Doudna, J. A., and Bryant, Z. (2020). Cas9 interrogates DNA in discrete steps modulated by mismatches and supercoiling. Proc. Natl. Acad. Sci. U. S. A. 117, 5853–5860. doi:10.1073/pnas.1913445117
Jiang, W., Bikard, D., Cox, D., Zhang, F., and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239. doi:10.1038/nbt.2508
Jiang, Y., Qian, F., Yang, J., Liu, Y., Dong, F., Xu, C., et al. (2017). CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat. Commun. 8, 15179–15189. doi:10.1038/ncomms15179
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi:10.1126/science.1225829
Karvelis, T., Druteika, G., Bigelyte, G., Budre, K., Zedaveinyte, R., Silanskas, A., et al. (2021). Transposon-associated TnpB is a programmable RNA-guided DNA endonuclease. Nature 599, 692–696. doi:10.1038/s41586-021-04058-1
Kim, H. K., Min, S., Song, M., Jung, S., Choi, J. W., Kim, Y., et al. (2018). Deep learning improves prediction of CRISPR–Cpf1 guide RNA activity. Nat. Biotechnol. 36, 239–241. doi:10.1038/nbt.4061
Kim, N., Kim, H. K., Lee, S., Seo, J. H., Choi, J. W., Park, J., et al. (2020). Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 38, 1328–1336. doi:10.1038/s41587-020-0537-9
Kleine, B., Chattopadhyay, A., Polen, T., Pinto, D., Mascher, T., Bott, M., et al. (2017). The three-component system EsrISR regulates a cell envelope stress response in Corynebacterium glutamicum. Mol. Microbiol. 106, 719–741. doi:10.1111/mmi.13839
Kleinstiver, B. P., Sousa, A. A., Walton, R. T., Tak, Y. E., Hsu, J. Y., Clement, K., et al. (2019). Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol. 37, 276–282. doi:10.1038/s41587-018-0011-0
Kudla, G., Murray, A. W., Tollervey, D., and Plotkin, J. B. (2009). Coding-sequence determinants of gene expression in Escherichia coli. Science 324, 255–258. doi:10.1126/science.1170160
Li, M., Chen, J., Wang, Y., Liu, J., Huang, J., Chen, N., et al. (2020). Efficient multiplex gene repression by CRISPR-dCpf1 in Corynebacterium glutamicum. Front. Bioeng. Biotechnol. 8, 357. doi:10.3389/fbioe.2020.00357
Liu, J., Liu, M., Shi, T., Sun, G., Gao, N., Zhao, X., et al. (2022). CRISPR-assisted rational flux-tuning and arrayed CRISPRi screening of an L-proline exporter for L-proline hyperproduction. Nat. Commun. 13, 891–906. doi:10.1038/s41467-022-28501-7
Liu, J., Srinivasan, S., Li, C. Y., Ho, I. L., Rose, J., Shaheen, M., et al. (2019). Pooled library screening with multiplexed Cpf1 library. Nat. Commun. 10, 3144. doi:10.1038/s41467-019-10963-x
Liu, J., Wang, Y., Lu, Y., Zheng, P., Sun, J., and Ma, Y. (2017). Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb. Cell Fact. 16, 205. doi:10.1186/s12934-017-0815-5
Liu Y, D. L., Dong, J., Chen, C., Zhu, J., Rao, V. B., Tao, P., et al. (2020). Covalent modifications of the bacteriophage genome confer a degree of resistance to bacterial CRISPR systems. J. Virol. 94, e01630. doi:10.1128/jvi.01630-20
Magnusson, J. P., Rios, A. R., Wu, L., and Qi, L. S. (2021). Enhanced Cas12a multi-gene regulation using a CRISPR array separator. Elife 10, e66406. doi:10.7554/elife.66406
Moreb, E. A., Hutmacher, M., and Lynch, M. D. (2020). CRISPR-cas "Non-Target" sites inhibit on-target cutting rates. CRISPR J. 3, 550–561. doi:10.1089/crispr.2020.0065
Moreb, E. A., and Lynch, M. D. (2021). Genome dependent Cas9/gRNA search time underlies sequence dependent gRNA activity. Nat. Commun. 12, 5034. doi:10.1038/s41467-021-25339-3
Najm, F. J., Strand, C., Donovan, K. F., Hegde, M., Sanson, K. R., Vaimberg, E. W., et al. (2017). Orthologous CRISPR–Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 36, 179–189. doi:10.1038/nbt.4048
Okafor, I. C., Singh, D., Wang, Y., Jung, M., Wang, H., Mallon, J., et al. (2019). Single molecule analysis of effects of non-canonical guide RNAs and specificity-enhancing mutations on Cas9-induced DNA unwinding. Nucleic Acids Res. 47, 11880–11888. doi:10.1093/nar/gkz1058
Ozcan, A., Krajeski, R., Ioannidi, E., Lee, B., Gardner, A., Makarova, K. S., et al. (2021). Programmable RNA targeting with the single-protein CRISPR effector Cas7-11. Nature 597, 720–725. doi:10.1038/s41586-021-03886-5
Pan, X., Tang, M., You, J., Osire, T., Sun, C., Fu, W., et al. (2022). PsrA is a novel regulator contributes to antibiotic synthesis, bacterial virulence, cell motility and extracellular polysaccharides production in Serratia marcescens. Nucleic Acids Res. 50, 127–148. doi:10.1093/nar/gkab1186
Park, J., Lim, J. M., Jung, I., Heo, S. J., Park, J., Chang, Y., et al. (2021). Recording of elapsed time and temporal information about biological events using Cas9. Cell 184, 1047–1063. doi:10.1016/j.cell.2021.01.014
Park J, B. S., and Kim, J. S. (2015). Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 31, 4014–4016. doi:10.1093/bioinformatics/btv537
Pátek, M., and Nešvera, J. (2013). Corynebacterium glutamicum Biology and Biotechnology. Berlin, Germany: Springer, 51–88.
Pattanayak, V., Lin, S., Guilinger, J. P., Ma, E., Doudna, J. A., and Liu, D. R. (2013). High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 31, 839–843. doi:10.1038/nbt.2673
Pausch, P., Al-Shayeb, B., Bisom-Rapp, E., Tsuchida, C. A., Li, Z., Cress, B. F., et al. (2020). CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science 369, 333–337. doi:10.1126/science.abb1400
Peng, F., Wang, X., Sun, Y., Dong, G., Yang, Y., Liu, X., et al. (2017). Efficient gene editing in Corynebacterium glutamicum using the CRISPR/Cas9 system. Microb. Cell Fact. 16, 201. doi:10.1186/s12934-017-0814-6
Perez-Arellano, I., Rubio, V., and Cervera, J. (2006). Mapping active site residues in glutamate-5-kinase. The substrate glutamate and the feed-back inhibitor proline bind at overlapping sites. FEBS Lett. 580, 6247–6253. doi:10.1016/j.febslet.2006.10.031
Resende, B. C., Rebelato, A., D'Afonseca, V., Santos, A., Stutzman, T., Azevedo, V., et al. (2011). DNA repair in Corynebacterium model. Gene 482, 1–7. doi:10.1016/j.gene.2011.03.008
Riesenberg, S., Helmbrecht, N., Kanis, P., Maricic, T., and Paabo, S. (2022). Improved gRNA secondary structures allow editing of target sites resistant to CRISPR-Cas9 cleavage. Nat. Commun. 13, 489. doi:10.1038/s41467-022-28137-7
Singh, D., Wang, Y., Mallon, J., Yang, O., Fei, J., Poddar, A., et al. (2018). Mechanisms of improved specificity of engineered Cas9s revealed by single-molecule FRET analysis. Nat. Struct. Mol. Biol. 25, 347–354. doi:10.1038/s41594-018-0051-7
Sternberg, S. H., LaFrance, B., Kaplan, M., and Doudna, J. A. (2015). Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113. doi:10.1038/nature15544
Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., and Doudna, J. A. (2014). DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67. doi:10.1038/nature13011
Talas, A., Huszár, K., Kulcsár, P. I., Varga, J. K., Varga, É., Tóth, E., et al. (2021). A method for characterizing Cas9 variants via a one-million target sequence library of self-targeting sgRNAs. Nucleic Acids Res. 49, e31. doi:10.1093/nar/gkaa1220
Tao, P., Wu, X., Tang, W. C., Zhu, J., and Rao, V. (2017). Engineering of bacteriophage T4 genome using CRISPR-cas9. ACS Synth. Biol. 6, 1952–1961. doi:10.1021/acssynbio.7b00179
Tsuchida, C. A., Zhang, S., Doost, M. S., Zhao, Y., Wang, J., O’Brien, E., et al. (2022). Chimeric CRISPR-CasX enzymes and guide RNAs for improved genome editing activity. Mol. Cell 82, 1199–1209. doi:10.1016/j.molcel.2022.02.002
Vlot, M., Houkes, J., Lochs, S. J., Swarts, D. C., Zheng, P., Kunne, T., et al. (2018). Bacteriophage DNA glucosylation impairs target DNA binding by type I and II but not by type V CRISPR-Cas effector complexes. Nucleic Acids Res. 46, 873–885. doi:10.1093/nar/gkx1264
Walton Rt, C. K., Whittaker, M. N., and Kleinstiver, B. P. (2020). Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296. doi:10.1126/science.aba8853
Wang, B., Hu, Q., Zhang, Y., Shi, R., Chai, X., Liu, Z., et al. (2018). A RecET-assisted CRISPR–Cas9 genome editing in Corynebacterium glutamicum. Microb. Cell Fact. 17, 63. doi:10.1186/s12934-018-0910-2
Wang, D., Zhang, C., Wang, B., Li, B., Wang, Q., Liu, D., et al. (2019). Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 10, 4284. doi:10.1038/s41467-019-12281-8
Wang, M., Chen, B., Fang, Y., and Tan, T. (2017). Cofactor engineering for more efficient production of chemicals and biofuels. Biotechnol. Adv. 35, 1032–1039. doi:10.1016/j.biotechadv.2017.09.008
Wang, Y., Cheng, H., Liu, Y., Liu, Y., Wen, X., Zhang, K., et al. (2021). In-situ generation of large numbers of genetic combinations for metabolic reprogramming via CRISPR-guided base editing. Nat. Commun. 12, 678. doi:10.1038/s41467-021-21003-y
Wang, Y. Y., Shi, K., Chen, P., Zhang, F., Xu, J. Z., and Zhang, W. G. (2020). Rational modification of the carbon metabolism of Corynebacterium glutamicum to enhance L-leucine production. J. Ind. Microbiol. Biotechnol. 47, 485–495. doi:10.1007/s10295-020-02282-8
Wendisch, V. F. (2014). Microbial production of amino acids and derived chemicals: synthetic biology approaches to strain development. Curr. Opin. Biotechnol. 30, 51–58. doi:10.1016/j.copbio.2014.05.004
Wienert, B., and Cromer, M. K. (2022). CRISPR nuclease off-target activity and mitigation strategies. Front. Genome Ed. 4, 1050507. doi:10.3389/fgeed.2022.1050507
Wolf, S., Becker, J., Tsuge, Y., Kawaguchi, H., Kondo, A., Marienhagen, J., et al. (2021). Advances in metabolic engineering of Corynebacterium glutamicum to produce high-value active ingredients for food, feed, human health, and well-being. Essays Biochem. 65, 197–212. doi:10.1042/ebc20200134
Wu, Z., Zhang, Y., Yu, H., Pan, D., Wang, Y., Wang, Y., et al. (2021). Programmed genome editing by a miniature CRISPR-Cas12f nuclease. Nat. Chem. Biol. 17, 1132–1138. doi:10.1038/s41589-021-00868-6
Xiang, X., Corsi, G. I., Anthon, C., Qu, K., Pan, X., Liang, X., et al. (2021). Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat. Commun. 12, 3238. doi:10.1038/s41467-021-23576-0
Xu J, X. X., Zhang, J., Guo, Y., Qian, H., and Zhang, W. (2014). A method for gene amplification and simultaneous deletion in Corynebacterium glutamicum genome without any genetic markers. Plasmid 72, 9–17. doi:10.1016/j.plasmid.2014.02.001
Yan, M. Y., Yan, H. Q., Ren, G. X., Zhao, J. P., Guo, X. P., and Sun, Y. C. (2017). CRISPR-Cas12a-Assisted recombineering in bacteria. Appl. Environ. Microbiol. 83, e00947–e00917. doi:10.1128/aem.00947-17
Yarrington, R. M., Verma, S., Schwartz, S., Trautman, J. K., and Carroll, D. (2018). Nucleosomes inhibit target cleavage by CRISPR-Cas9 in vivo. Proc. Natl. Acad. Sci. U. S. A. 115, 9351–9358. doi:10.1073/pnas.1810062115
Yim, S. S., An, S. J., Kang, M., Lee, J., and Jeong, K. J. (2013). Isolation of fully synthetic promoters for high-level gene expression in Corynebacterium glutamicum. Biotechnol. Bioeng. 110, 2959–2969. doi:10.1002/bit.24954
Zetsche, B., Gootenberg, J., Abudayyeh, O., Slaymaker, I., Makarova, K., Essletzbichler, P., et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell 163, 759–771. doi:10.1016/j.cell.2015.09.038
Zetsche, B., Heidenreich, M., Mohanraju, P., Fedorova, I., Kneppers, J., DeGennaro, E. M., et al. (2017). Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 35, 31–34. doi:10.1038/nbt.3737
Zhang, J., Qian, F., Dong, F., Wang, Q., Yang, J., Jiang, Y., et al. (2020). De novo engineering of Corynebacterium glutamicum for l-proline production. ACS Synth. Biol. 9, 1897–1906. doi:10.1021/acssynbio.0c00249
Zhao, D., Badur, M. G., Luebeck, J., Magaña, J. H., Birmingham, A., Sasik, R., et al. (2018). Combinatorial CRISPR-cas9 metabolic screens reveal critical redox control points dependent on the KEAP1-NRF2 regulatory Axis. Mol. Cell 69, 699–708. doi:10.1016/j.molcel.2018.01.017
Keywords: active gRNA, editing resolution, hfgRNA, editing efficiency, Corynebacterium glutamicum
Citation: Zhang F, Wang J-Y, Li C-L and Zhang W-G (2024) HyCas9-12aGEP: an efficient genome editing platform for Corynebacterium glutamicum. Front. Bioeng. Biotechnol. 12:1327172. doi: 10.3389/fbioe.2024.1327172
Received: 24 October 2023; Accepted: 27 February 2024;
Published: 12 March 2024.
Edited by:
Sanjay Vashee, J. Craig Venter Institute, United StatesReviewed by:
Yota Tsuge, Kanazawa University, JapanHaiwei Mou, Wistar Institute, United States
Jia Liu, ShanghaiTech University, China
Copyright © 2024 Zhang, Wang, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Guo Zhang, zhangwg@jiangnan.edu.cn; Feng Zhang, xyzfjn@126.com