Die hard: necroptosis and its impact on age-dependent neuroinflammatory diseases
 Kaitlan Smith
Kaitlan Smith Meagan Colie
Meagan Colie Trinity Moore
Trinity Moore Jonathan C. Schisler
Jonathan C. Schisler- Department of Pharmacology, McAllister Heart Institute, The University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
The pro-inflammatory form of cellular death, necroptosis, is critical to age-related pathologies. Necroptosis primarily functions as an antipathogenic and antitumor biological mechanism by triggering inflammatory pathways within rogue cell bodies, resulting in cell death. Several neurodegenerative conditions have hallmarks of necroptosis, suggesting a potential role for this cell death pathway in the pathogenesis of neuroinflammation and neuronal cell death, likely through the release of pro-inflammatory cytokines that perpetuate inflammatory signaling and neurodegeneration. The receptor-interacting protein kinases 1 and 3 (RIPK1/3) signaling cascade is critical to necroptosis regulation; however, the complete mechanism behind necroptotic activation, regulation, and resolution remains incomplete. In cases where necroptosis is disadvantageous, such as neurodegenerative diseases, we lack effective pharmacological suppressors of necroptosis that could mitigate disease progression. Targeting regulatory proteins within the necroptotic signaling pathway has shown promise; however, the need for specific inhibitors limits therapeutic opportunities. This review focuses on necroptosis and its role in neuroinflammation and neurodegeneration in age-dependent disorders. We comprehensively detail the known necroptotic signaling pathways and potential signaling partners and discuss the ongoing therapeutic efforts in targeting and preventing active necroptotic signaling and their relevance to neuroprotection.
Introduction
Cellular death within multicellular organisms is an active and vital process for regulating organismal homeostasis. Cell death, historically, is categorized by the structural appearance of the plasma membrane: apoptosis (type I), autophagy (type II), and necrosis (type III)(Schweichel and Merker, 1973; Galluzzi et al., 2007). However, several discoveries made in the mid-2000s have challenged this definition. One such discovery was a type of cell death known as necroptosis.
Necroptosis is a “programmed” form of nonapoptotic cell death distinguished by necrotic phenotypes such as cellular swelling and membrane rupture (Hanson, 2016; Dhuriya and Sharma, 2018; Shan et al., 2018). In contrast to caspase-dependent apoptosis, necroptosis is a caspase-independent form of cell death tightly regulated by two effector serine/threonine kinases from the receptor-interacting protein kinase (RIPK) family and the executioner mixed linage kinase domain-like (MLKL) pseudokinase (Vercammen et al., 1998; Sun et al., 1999; Holler et al., 2000; Degterev et al., 2005; Cho et al., 2009; Zhang et al., 2009; Yuan et al., 2019). Necroptosis is a pro-inflammatory form of cell death that is analogous to an injury response similar to the cell death observed in necrosis (Kaczmarek et al., 2013; Dhuriya and Sharma, 2018; Orozco et al., 2019).
Increased necroptosis is associated with the onset and progression of several age-dependent neuroinflammatory diseases (Ofengeim et al., 2015; Caccamo et al., 2017; Yuan et al., 2019; Oñate et al., 2020). However, it remains unclear what activates necroptotic signaling and how necroptosis is directly associated with clinical phenotypes. Importantly, it is crucial to understand the factors surrounding necroptosis in neuroinflammatory diseases as the cell types most affected, neurons, are post-mitotic cells. Given that the hallmarks of necroptosis occur in age-dependent neuroinflammatory diseases, therapies targeting this pathway are potentially valuable in preventing cell loss and localized inflammation.
This article will provide a comprehensive overview of necroptosis, including the historical beginnings and the most recent associated signaling and regulatory mechanisms involved in neuroinflammation. Additionally, we will review the therapeutic compounds used to inhibit necroptosis and their associated outcomes. Lastly, to further promote investigations into this topic, we will discuss the various methods employed to induce necroptosis within in vitro systems to provide a better understanding of how to study necroptotic cell death across laboratory settings.
Necroptosis
The functions and purposes of necroptosis
How cellular death events are defined and how those events are subsequently categorized has been a regular point of contention between scientists (Kroemer et al., 2005; Galluzzi et al., 2007; Galluzzi et al., 2012; Galluzzi et al., 2018). Cellular death, predominately defined by the modality of the plasma membrane, has three classifications: type I, type II, and type III (Schweichel and Merker, 1973; Kroemer et al., 2005; Green and Llambi, 2015). Cells undergoing type I cell death exhibit phenotypes such as cell shrinkage, membrane blebbing, and chromatin condensation (an early definition of apoptosis). Type II cell death involves large intracellular vesicles that result in membrane engulfment and cytoplasm degradation (commonly referred to as autophagy). The last type of cell death, type III, was traditionally thought to occur during times of cellular injury and is characterized by cellular swelling resulting in the rupture of the plasma membrane leading to the loss of organellar structure and chromatin condensation (necrosis) (Schweichel and Merker, 1973; Kroemer et al., 2005; Kroemer et al., 2009; Green and Llambi, 2015).
However, because these categorizations rely on the structural appearance of the plasma membrane and do not account for the biochemical by-products produced by these cell death events, it is no surprise that how we categorize cell death has been routinely challenged (Kroemer et al., 2005; Kroemer et al., 2009; Galluzzi et al., 2007; Galluzzi et al., 2012; Galluzzi et al., 2018). Several discoveries within the last decade have so pushed the boundaries of these cellular death characterizations that the only way cell death can be reliably categorized is in two ways: regulated (programmed) or unregulated (nonprogrammed) cell death (RCD or URCD, respectively) (Galluzzi et al., 2012; Galluzzi et al., 2018).
One such discovery that pushed the boundaries of the original categorizations is the recently discovered form of cell death known as necroptosis. Necroptosis (referred to as TNF-induced necrosis or regulated necrosis in early literature) is a tightly regulated form of RCD that is highly pro-inflammatory (Vercammen et al., 1997; Vercammen et al., 1998; Galluzzi et al., 2012; Galluzzi et al., 2018; Moriwaki and Chan, 2013; Hanson, 2016; Dhuriya and Sharma, 2018). Like apoptosis, necroptosis is a “programmed” event involving signaling cascades but differs by the resulting localized immune response. However, while it is a form of RCD, the cell death event manifests with a necrotic morphotype, making it aptly named necroptosis. Analogous to necrosis, necroptosis is often associated with cell injury and has the connotation of being disadvantageous. However, there is mounting evidence that the induction of necroptosis can also be a vital and beneficial process (Kaczmarek et al., 2013; Dhuriya and Sharma, 2018). For example, upon failing responses to stressful stimuli, necroptosis has been shown to mediate adaptive functions and work against pathogen-mediated infections by serving as an RCD subroute (Dhuriya and Sharma, 2018). Additionally, during development, necroptosis can engage in developmental safeguard programs to aid in the elimination of potentially defective organisms before maturation (Kaczmarek et al., 2013), consistent with a multifaceted role for necroptosis within the human body. As such, we should continue to investigate this complex form of cell death to understand its role in health and disease better.
Canonical necroptosis
Inflammatory insults often initiate necroptotic signaling at the extracellular membrane or intracellular perturbations to the microenvironment (Galluzzi et al., 2018). Regarding the latter, it remains unclear exactly how and which perturbations to the intracellular microenvironment activate necroptotic signaling. In contrast, extracellular signaling occurs via inflammatory mediators and specific death receptors that initiate necroptotic signaling (Galluzzi et al., 2018). These receptors include, but are not limited to, FAS, Tumor Necrosis Factor 1 (TNFR1), pathogen recognition receptors (PRRs) such as toll-like receptors 3 and 4 (TLR3 and TLR4, respectively), the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor 4/5, and Z-DNA binding protein (ZBP1), the later of which is involved in viral induction of necroptosis and will not be discussed in this review (Vercammen et al., 1998; Degterev et al., 2008; Upton et al., 2010; Upton et al., 2012; Kaczmarek et al., 2013; Kaiser et al., 2013; Green et al., 2014; Nahacka et al., 2018).
While significant efforts have parsed out the intracellular necroptotic signaling activators, the field has hesitated to move on from the canonical understanding of the necroptotic signaling cascade. As it stands, the molecular mechanisms and post-translational modifications that follow TNFR1 activation via binding from an inflammatory cytokine, such as tumor necrosis factor-alpha (TNFα), is the most widely investigated necroptotic signaling cascade (Vercammen et al., 1998; Wallach et al., 1999; Vanlangenakker et al., 2011; Hanson, 2016; Shan et al., 2018). Activation of TNFR1 results in the recruitment of RIPK1 to the plasma membrane, where it forms a complex (known as complex 1) with the TNFR1-associated death domain protein (TRADD), TNFR-associated factor 2 (TRAF2), cIAP1/2, and the linear ubiquitin chain assembly complex (LUBAC) (Vanlangenakker et al., 2011; Mifflin et al., 2020; Ye et al., 2023).
Following the formation of complex 1, the cell will meet one of three fates: pro-survival NF-κB signaling, caspase-dependent apoptosis, or caspase-independent pro-inflammatory necroptosis. The determining factor behind cellular survival or death lies with the post-translational modifications of RIPK1. Recent literature summarizes well the impacts and outcomes of RIPK1 post-translational modifications (Mifflin et al., 2020; Ye et al., 2023) that are outside the scope of this review. However, it is important to mention that under conditions where RIPK1 is deubiquitinated, such as in the absence of LUBAC, cIAP1/2, or other caspases, the kinase domain of RIPK1 can be phosphorylated and subsequently activated (Mifflin et al., 2020; Ye et al., 2023).
Through interactions via the RIPK homeotype interaction motif (RHIM), RIPK1 binds and phosphorylates RIPK3 for activation as well (Moriwaki and Chan, 2013; Morgan and Kim, 2022; Riebeling et al., 2022). These autophosphorylation events allow for RIPK1 and RIPK3 to form a complex with the RIPK3 substrate, mixed linage kinase domain-like pseudokinase (MLKL) (Moriwaki and Chan, 2013; Dondelinger et al., 2014). Together, RIPK1, RIPK3, and MLKL form the necrosis-inducing signaling complex known as the “necrosome” (complex IIb) (Ye et al., 2023). Necrosome formation is a crucial step in necroptosis as it is a precursor to MLKL oligomerization. During necrosome formation, active RIPK3 phosphorylates the pseudokinase domain of MLKL, allowing MLKL to become oligomeric. MLKL oligomers are then recruited to the plasma membrane through interactions between the positively charged 4-helical bundle domain on the N-terminal of MLKL and negatively charged phosphatidylinositol phosphates (PIPs) on the plasma membrane (Murphy et al., 2013; Dondelinger et al., 2014; Jacobsen et al., 2016; Petrie et al., 2017). Following MLKL recruitment to the cell membrane, several physiological events take place. These include releasing reactive oxygen species (ROS) from the mitochondria, ion influx, plasma membrane permeabilization, cellular swelling, and eventually rupture, which releases pro-inflammatory cellular contents. These physiological events all result in an inflammatory cell death similar to necrosis, making up the hallmarks of necroptotic cell death (Vercammen et al., 1998; Morgan and Liu, 2013; Dhuriya and Sharma, 2018). Several studies in recent years have identified additional events that trigger necroptosis, and we highlight these non-canonical activation triggers of necroptotic activation in the sections below.
Necroptosis and its impact on neuroinflammation
Necroptosis directly triggers inflammation due to an increase in active transcription of inflammatory cytokines and chemokines, production of reactive oxygen species (ROS), and ultimately a release of these factors as well as the release of damage-associated molecular patterns (DAMPs) upon cell lysis (Figure 1) (Kaczmarek et al., 2013; Orozco et al., 2019). Unlike apoptosis, necroptosis does not involve ordered disassembly of the dying cell. Instead, necroptosis induces rapid disintegration of the affected cell, resulting in a massive release of these highly inflammatory markers. Once in the extracellular space, they activate resident immune cells and bind to and activate pro-inflammatory receptors on neighboring cells, creating a vicious cycle of inflammatory propagation (Figure 1).
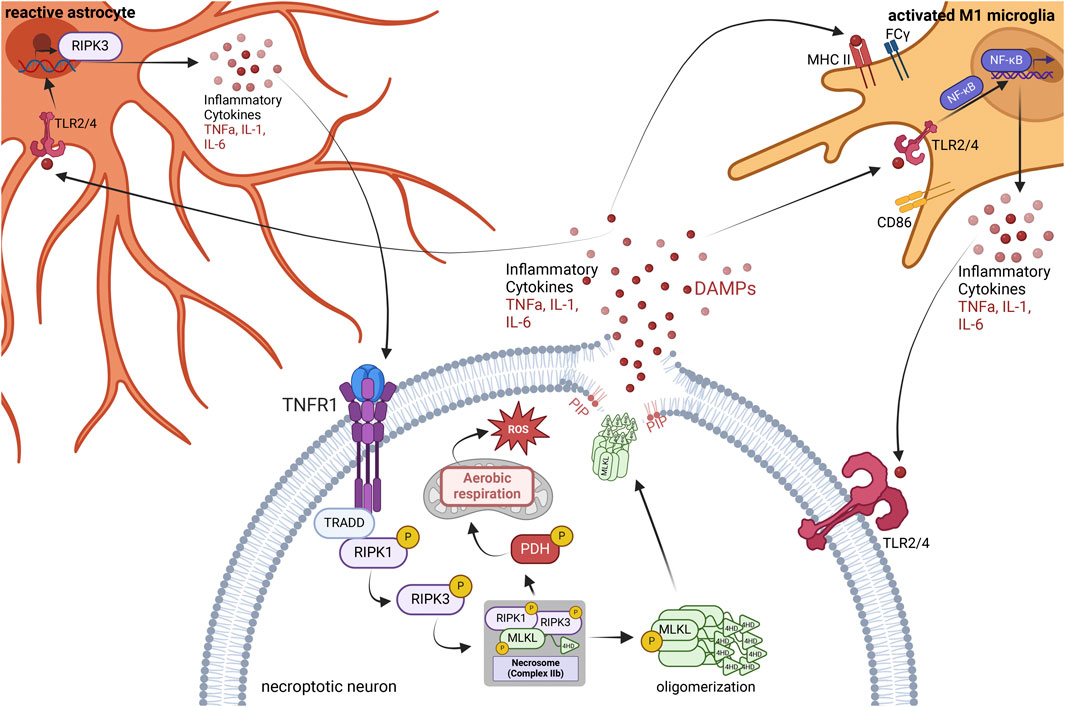
FIGURE 1. Neuroinflammatory consequences of necroptosis. The necroptosis signaling cascade initiates through the binding of TNF-α to the TNFR1. Formation of the necrosome stimulated direct phosphorylation of pyruvate dehydrogenase (PDH), causing mitochondrial aerobic respiration, thereby producing reactive oxygen species (ROS) as a by-product. Oligomerized phospho-MLKL traffics to the phospholipid bilayer, where it attaches to phosphatidylinositol phosphates (PIPs), causing the breakdown of the phospholipid bilayer and creating a pore in the cell membrane, releasing inflammatory cytokines and damage-associated molecular patterns (DAMPs). DAMPs activate the TLR2 and TLR4 receptors of nearby microglia, astrocytes, and other neurons. In microglia, this leads to a pro-inflammatory M1 phenotype where microglia increase expression of MHC II, CD86, and FCγ receptors. Activation of the TLR2/4 receptors in microglia also stimulates NFkB nuclear translocation and increases the transcription and release of pro-inflammatory cytokines. Astrocytes are activated by the binding of DAMPs and inflammatory cytokines to TLR2/4 receptors and other receptors, leading to an increase in RIPK3 transcription and an increase in inflammatory cytokine release. In turn, the plethora of inflammation surrounding the necroptotic neuron spreads to neighboring neurons, activating their pro-inflammatory receptors and necroptosis. Figure created using BioRender.com.
During necroptosis, activated RIPK3 and MLKL trigger the production of ROS via phosphorylation of pyruvate dehydrogenase (PDH), increasing mitochondrial aerobic respiration and its by-product, ROS, leading to oxidative stress (Yang Z. et al., 2018). RIPK3 induces MLKL oligomerization via phosphorylation, causing oligomerized phospho-MLKL to traffic to the cell membrane and bind to phosphatidylinositol phosphates (PIPs) within the phospholipid bilayer, forming pores in the cell membrane (Dondelinger et al., 2014). The cell then swells and lyses, releasing DAMPs, ROS, and inflammatory cytokines into the extracellular space. Activated RIPK1 and RIPK3 also independently induce inflammation in both neurons and glia. Activated RIPK3 induces transcription of genes encoding inflammatory cytokines and chemokines, causing an increase in the translation of these mediators, which continues even once cell membrane integrity has been compromised (Orozco et al., 2019). Factors released due to necroptosis activate nearby astrocytes, increasing RIPK3 transcription and signaling, causing an increase in pro-inflammatory cytokine production and release (Chang et al., 2023). Activation of RIPK1 in both microglia and astrocytes can also induce the production of pro-inflammatory cytokines via the activation of Nuclear Factor-kB (NF-κB) in the absence of active caspases (Huang et al., 2021; Zelic et al., 2021).
From an organismal protection perspective, the release of DAMPs is a warning signal to innate immune cells of an impending threat. However, the release of DAMPs activates the innate immune response and leads to the propagation of inflammation. DAMPs associate with and activate receptors on the surface of antigen-presenting cells (APCs) and induce the maturation of APCs and subsequent production of pro-inflammatory cytokines (Oppenheim and Yang, 2005). Further, mature APCs can travel to lymphoid tissue and trigger an adaptive immune response via antigen presentation to T-cells (Oppenheim and Yang, 2005).
The resident innate immune cells of the central nervous system are microglia and astrocytes (Ransohoff and Brown, 2012; Miller, 2018; Linnerbauer et al., 2020; Singh, 2022). Microglia become activated upon binding to DAMPs and take on a pro-inflammatory M1-like phenotype, leading to an upregulation in the expression of MHC II, CD86, and FCγ receptors (Figure 1) (Cherry et al., 2014). This response aims to promote the targeting and killing of rogue cells or invading organisms and aid in the phagocytosis of cellular debris. Once this is complete, the microglia typically transition into an anti-inflammatory M2-like phenotype to promote repair of the damaged area. However, with chronic exposure to DAMPs and activation, microglia will remain in the pro-inflammatory M1-like phenotype and do more harm than good (Cherry et al., 2014). Interestingly, DAMPs and other factors released as a by-product of necroptosis activate astrocytes, increasing astrocytic RIPK3 transcription and signaling, further driving inflammatory transcription and neurotoxicity (Figure 1) (Chang et al., 2023). Therefore, necroptosis activates the pro-inflammatory states of both microglia and astrocytes in the brain.
Concomitantly, DAMPs trigger the death of nearby cells by activating pattern recognition receptors such as Toll-like receptors (TLRs) (Kaczmarek et al., 2013). TLRs are present in microglia, astrocytes, and neurons alike. When activated via the binding of a ligand such as a DAMP, TLR2, and TLR4 specifically induce NFkB translocation to the nucleus (Zhang and Ghosh, 2001) and promote the transcription and translation of pro-inflammatory cytokines such as IL-6 and TNF⍺ (Lin et al., 2022). TNF-alpha is a potent activator of cell death in the absence of active caspases, and an activator of necroptosis specifically (Zhu et al., 2018). The release of these inflammatory cytokines from nearby necroptotic cells drives the propagation of inflammation (Figure 1).
Unsurprisingly, necroptosis is present in numerous diseases and disorders, such as ischemic stroke, neurodegenerative disorders, myocardial infarction, pulmonary diseases, liver injuries, and inflammatory bowel disease (Khoury et al., 2020). Importantly, in diseases involving the loss of non-mitotic, irreplaceable cells such as neurons or cardiomyocytes, it is crucial to understand the factors surrounding the activation and inhibition of necroptosis.
Necroptosis and its role in age-dependent neuroinflammatory diseases
Necroptosis appears to contribute to several age-dependent neuroinflammatory diseases, such as Alzheimer’s disease, Parkinson’s disease, ataxic disorders, and more. These diseases manifest with age or in adulthood, accompanied by neuroinflammation and neurodegeneration. However, necroptosis has also been observed independent of disease in aged mammals. Studies suggest this is due to sterile inflammation associated with aging and cellular senescence (Royce et al., 2019), highlighting the essential need for studies of necroptosis in disease and in the process of “normal” aging. One such study found that an endogenous inhibitor of RIPK1, TAK1, is reported to decrease with age and thus promote RIPK1 activation and subsequent inflammation (Xu et al., 2018). Similarly, expression of RIPK3, RIPK1, and phospho-MLKL are elevated with age in neurons of the cortex and hippocampus of otherwise healthy aged mice, further supporting the idea that necroptosis can occur due to aging in the absence of disease (Thadathil et al., 2021). Below, we focus on the involvement of necroptosis in age-dependent neuroinflammatory diseases.
Spinocerebellar Ataxia
Ataxia is an umbrella term that describes various neurological conditions that result in the lack of muscle control and coordination of voluntary movements. Spinocerebellar Ataxia (SCA), in particular, is a type of ataxia caused by autosomal mutations that result in adult-onset cerebellar atrophy and brainstem degeneration (Edamakanti et al., 2023). SCAs are chronic and progressive, and those diagnosed currently have no treatment options available (Edamakanti et al., 2023). SCA patients display a range of clinical phenotypes including, but not limited to, loss of coordination and balance, ataxic gait, hand tremors, dysarthria, hypogonadism, slurred speech, accelerated aging, and multisystemic neurodegeneration (Genis et al., 2018; De Michele et al., 2019; Mitroshina et al., 2022). While there are over 40 recognized types of SCAs, to date, 1:10,000 individuals are diagnosed with SCA(Mitroshina et al., 2022).
While the role necroptosis and the subsequent neuroinflammation have on SCA disease progression and severity still stands to be explored, an ever-growing number of studies suggest both play an active role in the manifestation of SCA onset and clinical phenotypes. It has been demonstrated through a handful of studies that targeting inflammatory pathways in SCA patients appears to have a therapeutic effect. Indeed, Park et al. found that reducing Bergmann glia inflammation via the inhibiting the c-Jun N-terminal kinase (JNK) resulted in both pathological and behavioral improvements in SCA1 knock-in mice (Edamakanti et al., 2023), consistent with the finding that a low abundance of Bergmann glia correlated with SCA1 diagnoses, suggesting that Bergmann glia loss can contribute to Purkinje cell degeneration (Shiwaku et al., 2013).
Similarly, efforts made to inhibit microglia activation using small molecule inhibitors or growth factors such as TFG- β1 resulted in suppressed cytokine activity in SCA3 patients and CA rat models, suggesting that microglia have an active role in promoting cerebellar vulnerability in ataxias (Ferro et al., 2019; Cao et al., 2020; Chiu et al., 2020). While the role necroptosis plays within this group of diseases has been less explored, Saeidikhoo et al. demonstrated that implantation of Sertoli cells resulted in a reduction of RIPK3-positive population, resulting in a neuroprotective effect which enhanced motor function and behavioral characteristics in ataxic rat models by counteracting necroptosis in cerebellar ataxia (Saeidikhoo et al., 2020). Moreover, studies using the Atxn2-CAG100-KIN transgenic mouse model of ataxia demonstrated that increased expression of RIPK1 and RIPK3 results in active necroptotic signaling in SCA2 and SCA28(Canet-Pons et al., 2021; Mitroshina et al., 2022). Increased RIPK1 and RIPK3 expression may also be relevant to Spinocerebellar Ataxia Autosomal Recessive 16 (SCAR16) and a dominant form of spinocerebellar ataxia, SCA48 (Genis et al., 2018; Schuster et al., 2018; De Michele et al., 2019). Both SCAR16 and SCA48 map to coding mutations in STUB1, the gene that encodes for the carboxyl terminus of heat-shock 70 interacting protein (CHIP)(Genis et al., 2018; Schuster et al., 2018; Shi et al., 2018; Madrigal et al., 2019). RIPK1 and RIPK3, critical kinases involved in the induction and regulation of necroptosis, are known substrates of CHIP’s ubiquitin ligase activity (Seo et al., 2016). Several studies suggest that the accelerated aging and cerebellar degeneration observed in SCAR16 and SCA48 patients result from an inability to adapt to metabolic stressors, potentially promoting an increased sensitivity to necroptosis (Seo et al., 2016; Shi et al., 2018; Madrigal et al., 2019) As such, the activation of necroptotic signaling and the subsequent neuroinflammation that follows may have a significant role in the SCA disease progression and manifestation and warrants further investigation.
Alzheimer’s disease
Alzheimer’s disease (AD) is an age-related form of dementia affecting nearly 6 million individuals in the United States today. In AD, abnormal amounts of amyloid beta protein accumulate and form plaques in the extracellular space around neurons. The abundance of amyloid beta oligomers in post-mortem human AD brain tissue coincided with increased RIPK1 activation and phospho-MLKL expression (Salvadores et al., 2022). In-vitro models showed that conditioned media from microglia exposed to amyloid-beta oligomers triggered neuronal necroptosis through TNF-alpha signaling. Pharmacological inhibition of RIPK3 using GSK872 and RIPK1 using Nec-1s ameliorated the neurodegenerative effects of amyloid beta oligomers. Thus, these findings support the contribution of microglial activation in the activation of necroptosis in AD (Salvadores et al., 2022).
Often accompanying plaque formation are increases in hyperphosphorylated tau, a microtubule-stabilizing protein. Hyperphosphorylated tau aggregates into neurofibrillary tangles or NFTs within the neurons and disrupts neuronal infrastructure and cellular signaling, causing inflammation and eventual neuronal death. The severity of AD is categorized into Braak stages by the location of NFTs within the brain, with higher Braak stages indicating more severe NFT infiltration (Scheltens et al., 2021). Evidence of increased necroptosis markers, RIPK3 and MLKL, were found in human AD brains and necrosome formation (Caccamo et al., 2017) and positively correlated with the level of patient Braak stage in human samples, cognitive deficits, and a higher degree of neuron loss in AD-model mice (Caccamo et al., 2017).
The pathophysiology of neuronal death in AD remains unclear; however, recent studies suggest an essential role for necroptosis. For example, in cultured mouse hippocampal neurons, hyperphosphorylated tau induced necroptosis and subsequent inflammation by stimulating elevated expression of pro-inflammatory cytokines via NF-κB signaling (Dong et al., 2022). Importantly, activation of NF-κB by phosphorylated Tau seems to be dependent on RIPK1/RIPK3/MLKL activity, and tau pathology improved with RIPK3 inhibition in the Tau P301S AD model mouse (Dong et al., 2022). The long noncoding RNA MEG3, found to induce necroptosis in cultured neurons, is upregulated in AD patient brain samples (Balusu et al., 2023). Experimental downregulation of MEG3 and either pharmacologic or genetic inhibition of necroptosis rescued neuronal loss in ex-vivo human neurons.
Studies suggest that levels of pro-inflammatory markers associated with necroptosis and cell death, IL-1 and TNF-alpha, are also elevated in AD patients (Zheng et al., 2016), including one study where elevated levels of TNF associated with increased necroptosis in post-mortem-AD-brains (Jayaraman et al., 2021). Additionally, there is a significant focus on the involvement of RIPK1 as a driver of necroptosis and inflammation in AD. RIPK1 expression and activation are elevated in the AD brain and correspond with amyloid beta and NFT levels, mitochondrial dysfunction, and neuronal loss (Li et al., 2022), as well as in the dysfunction of microglia (Ofengeim et al., 2017). Interestingly, microglia in post-mortem AD brains had increased expression of RIPK1(Salvadores et al., 2022). Typically, microglia are responsible for phagocytosis of abnormal cells or protein debris in the brain. However, in AD, microglia do not phagocytose amyloid beta protein, allowing the pathological plaques to build up in the brain and cause inflammation. Significantly, inhibiting RIPK1 in microglia promoted amyloid beta protein degradation (Ofengeim et al., 2017).
Another co-pathology of AD is granulovacuolar degeneration (GVD), represented by the formation of neuronal cytoplasmic vacuolar lesions. Within GVD lesions of neurons from post-mortem human AD brain samples, components of active necrosome formation were detected and colocalized with markers of GVD (Koper et al., 2020). The number of neurons positive for markers of GVD correlated with the level of NFT pathology and was inversely proportional to the density of neurons in layer III of the frontal cortex. This data suggests a role of necroptosis in GVD formation as a result of AD pathology. Limbic-predominant age-related TDP-43 encephalopathy neuropathological changes (LATE-NC) is another AD-associated pathology observed in up to 70% of symptomatic AD cases (Koper et al., 2022). In TDP-43 positive neurons from AD cases, an increase in phospho-MLKL and GVD were observed compared to TDP-43 negative AD neurons. Concomitantly, an increase in neuronal loss in the CA1 region of the hippocampus was observed in TDP-43 positive AD cases, suggesting that LATE-NC in AD contributes to GVD-mediated necroptosis and neurodegeneration (Koper et al., 2022).
Parkinson’s disease
Parkinson’s disease (PD) is an age-related form of neurodegeneration involving the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), an area of the brain that releases the neurotransmitter dopamine to the neurons in the striatum, which project to the basal ganglia (Ni and Ernst, 2022). Dopamine signaling to the basal ganglia is necessary for signaling to the thalamus and motor cortex, both movement control centers of the brain. Degeneration of these dopaminergic neurons impairs dopamine release and norepinephrine release, causing muscle tremors, stiffness, impaired balance and coordination, irregular blood pressure, and fatigue in affected individuals. Another hallmark of PD is the buildup of α-synuclein protofibrils that aggregate and lead to dysregulation of neuronal homeostasis and death (Stefanis, 2012). Notably, the cause of neuronal death in PD remains largely unknown. Recent studies point to necroptosis as a potential driver of dopaminergic neuron loss in PD. Similar to AD, markers of necroptosis RIPK1, RIPK3, and MLKL were elevated in the SNpc of post-mortem PD patients, suggesting necroptosis may have been active in these neurons before death (Oñate et al., 2020). In addition to brain tissue, essential necroptotic-related genes were identified in PD patients’ peripheral blood lymphocytes, again suggesting the presence of necroptosis in PD (Lei et al., 2023). Indeed, several studies have shown that RIPK1, RIPK3, and MLKL induce neuroinflammation, thereby mediating necroptotic degeneration of dopaminergic neurons in models of PD (Lin et al., 2020; Kim et al., 2023). One study found that pre-formed α-synuclein fibrils activate astrocytes via RIPK1/RIPK3-dependent activation of NF-κB, leading to increased transcription of pro-inflammatory factors and decreased phagocytic function and neurotoxicity (Chou et al., 2021). The presence of α-synuclein aggregates also induces neuroinflammation in PD via activation of the NLRP3 inflammasome and TLR2 (Araújo et al., 2022). Microglia become activated by the abundance of α-synuclein, prompting the release of pro-inflammatory IL-1β, which activates NLRP3 (Codolo et al., 2013). Aggregated α-synuclein also activates TLR2, a pro-inflammatory receptor that leads to NFkB nuclear translocation, initiating increased expression and release of pro-inflammatory cytokines (Gustot et al., 2015). Though NLRP3 inflammasome activation is commonly associated with pyroptosis, several studies demonstrated how RIPK3 activates NLRP3, and inflammasome formation frequently occurs alongside necroptosis activation, though the two are not dependent on one another (Kang et al., 2014; Lawlor et al., 2015; Guo et al., 2019). However, this association suggests an overlap between inflammation in PD and necroptosis. Pharmacological inhibition of RIPK1 in PD protected dopaminergic neurons against injury and mitochondrial toxicity-related necroptosis (Wu et al., 2015; Iannielli et al., 2018).
Huntington’s disease
Huntington’s disease (HD), one of the most common genetic age-related dementias today (MacDonald, 1993), is caused by abnormal repeats of the DNA sequence “CAG” that encodes the amino acid glutamine, resulting in the huntingtin protein carrying a long track of polyglutamine residues. Abundantly expressed in neurons throughout the CNS, mutant huntingtin aggregates and disrupts axonal transport and signaling (Sharp et al., 1995). As time passes, HD progresses in severity and leads to increased neuroinflammation, motor function disturbances, cognitive decline, and psychosis (Saba et al., 2022). Although it is unclear if necroptosis plays a causal role in the etiology of HD neuronal death, a limited number of reports detailed the effect of necroptosis inhibitors in HD models. Inhibition of RIPK1 in cultured striatal neurons and an HD transgenic mouse protected against necroptotic cell death due to huntingtin and alleviated motor function deficits (Zhu et al., 2011). Another study showed that stem cell implantation to the striatum of HD model rats prevented necroptosis and assisted in maintaining striatal volume (Bayat et al., 2021). Though few, these studies shed light on necroptosis as a potential driver of neurodegeneration in HD.
Frontotemporal dementia
Frontotemporal dementia (FTD) is another common age-related form of dementia caused by a buildup of aberrant transactive response DNA-binding protein 43 (TDP-43) and tau tangles in the frontal and temporal lobes of the brain. This results in progressive neurodegeneration of both lobes and leads to behavioral abnormalities, movement disorders, and, in some cases, aphasia. The presence of abundant TDP-43 and tau tangles in the frontal cortex leads to prolonged microglial and astrocyte activation, likely accompanied by an increase in neuroinflammation (McCauley and Baloh, 2019). Interestingly, FTD is associated with a partial loss of function of both TANK-binding kinase 1 (TBK1), a serine/threonine protein kinase, and Transforming growth factor-β activated kinase 1 (TAK1), a member of the MAPK family. Both kinases target RIPK1, an activator of necroptosis for inhibition (Xu et al., 2018). However, dysregulation of both TBK1 and TAK1 in FTD suggests that unchecked RIPK1 causes activation of necroptosis and neuronal death. Indeed, another study revealed that markers of necroptosis were elevated in spinal oligodendrocytes of FTD model mice, suggesting that degeneration of those neurons is caused by necroptosis (Wang et al., 2018).
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s Disease, is a rare age-related neurodegenerative disease involving the progressive idiopathic loss of motor neurons in the brain, leading to a lack of signaling to the muscles in the body, resulting in movement dysfunction, muscle atrophy, and an eventual loss of all voluntary muscle control. Like PD, the means of cell death in motor neurons in ALS has yet to be clearly defined. However, in recent years, necroptosis has gained some momentum as a potential causative agent for motor neuron death in ALS, however, critical drivers of necroptosis have not been identified in all ALS studies (Dermentzaki et al., 2019; Wang et al., 2020; Dominguez et al., 2021). Re et al. reported that astrocytes from ALS-affected individuals induce death of co-cultured motor neurons in a RIPK1 and MLKL-dependent manner (Re et al., 2014). Another group showed that RIPK1 and RIPK3 play a significant role in axonal degeneration of motor neurons in a mouse model of ALS and human post-mortem brain samples from ALS patients (Ito et al., 2016). In a mouse model of ALS carrying mutations in superoxide dismutase 1 (SOD1), found in familial ALS, elevated levels of RIPK1 and RIPK3 were observed, as well as axonal degeneration similar to that of Optineurin (OPTN) null mouse models (Saccon et al., 2013; Ito et al., 2016). OPTN acts as a suppressor of RIPK1-dependent signaling that regulates the degradative ubiquitination of RIPK1. Mutant OPTN, seen in ALS, suppresses RIPK1 ubiquitin-mediated degradation and is associated with increased pro-inflammatory cytokines, axonal degeneration, and necroptosis (Ito et al., 2016; Toth and Atkin, 2018). However, RIPK1 inhibition arrested TNF-induced necroptosis in OPTN knockout oligodendrocytes, further supporting the role of RIPK1 in necroptotic degeneration of neurons in ALS (Ito et al., 2016; Politi and Przedborski, 2016; Greco et al., 2018).
Similar to FTD, mutations in TBK1 are common in ALS and promote RIPK1-dependent necroptosis in the central nervous system via a loss of suppressive phosphorylation of RIPK1(Xu et al., 2018). In addition to OPTN and TBK1 mutations, loss-of-function mutations such as p.Arg261His in the gene NEK1, which encodes a serine/threonine kinase, are involved in approximately 3% of ALS cases (Kenna et al., 2016). Mutant NEK1 promoted degradation of critical modulators of RIPK1, thus rendering endothelial cells of the cerebral vasculature susceptible to RIPK1-dependent necroptosis (Wang et al., 2021).
Several studies have shown that inflammatory cytokines and neuroinflammation are abundant in ALS and contribute to ALS pathogenesis (McGeer and McGeer, 2002; Zürcher et al., 2015; Chen X. et al., 2018; Huang et al., 2020; Tortelli et al., 2020). Similar to AD, researchers in ALS mouse models identified a subset of pro-inflammatory microglia regulated by RIPK1, and inhibiting RIPK1 activity downstream of the TNF receptor decreased the abundance of these pro-inflammatory microglia (Mifflin et al., 2021). Further, genetic and pharmacological inhibition of necroptosis in ALS models reduced oligodendrocyte death, axonal degeneration, and neuroinflammation (Ito et al., 2016; Royce et al., 2019). Using a pharmacological inhibitor of necroptosis in mouse models of ALS also improved vertical rearing activity, delayed the onset of motor dysfunction, and extended lifespan (Ito et al., 2016). This evidence suggests a prominent role for RIPK1, RIPK3, and necroptosis in the pathogenesis and progression of ALS and related neuroinflammation.
Multiple Sclerosis (MS) is an autoimmune disease that involves the loss of myelinating oligodendrocytes, causing progressive degeneration of the protective myelin sheath of neurons in the CNS, resulting in neuronal damage and axonal degeneration. Though MS, while considered an age-dependent neurodegenerative disease, does not manifest as late in life as AD, FTD, or PD, tending to manifest between twenty and 40 years of age. MS is a neuroinflammatory disease, given the elevated presence of TNF-alpha in active lesions, blood serum, and CSF of MS patients; moreover, TNF-alpha correlates with MS severity and progression (Sharief and Hentges, 1991). Given TNF-alpha’s role as a primary activator of necroptosis, several studies have investigated the role of necroptosis in the loss of oligodendrocytes and neuroinflammatory signaling in MS. Indeed, protein expression of RIPK1, RIPK3, and oligomerized phospho-MLKL were observed in post-mortem brain tissue lysates of MS patients, as well as at specific lesion sites in the brain (Ofengeim et al., 2015; Picon et al., 2021; Zelic et al., 2021). The abundance of necrosome formation correlated with the rapidity of MS disease progression in these samples (Picon et al., 2021). One study suggested that the means for oligodendrocyte degeneration was related to microglia and astrocyte activation following necroptotic stimulation (Zelic et al., 2021). This group showed that oligodendrocytes were reduced in a RIPK1-dependent manner when co-cultured with necroptosis-activated microglia and astrocytes. Indeed, RIPK1 inhibition in a mouse model of MS arrested disease progression and suppressed pro-inflammatory RIPK1-dependent signaling in microglia and astrocytes, as well as prevented necroptosis in oligodendrocytes (Ofengeim et al., 2015; Zelic et al., 2021).
Similarly, RIPK3 knockout mice showed decreased susceptibility to necroptosis-induced demyelination and reduced pro-inflammatory microglia and astrocytes (Ofengeim et al., 2015). Inhibition of RIPK1 and RIPK3 also protected oligodendrocytes from TNF-alpha-induced necroptosis in vitro (Ofengeim et al., 2015). This evidence supports the involvement of necroptotic activators in the pathology and progression of MS and places inhibitors of necroptosis as potential therapeutics for the management of MS.
The mediators, RIPK1, RIPK3, and MLKL, and canonical and non-canonical activation routes of necroptosis contribute to cell death and neuroinflammation in many age-related neurodegenerative diseases. Given the evidence of the presence of necroptosis in these highly prevalent diseases, routes of necroptotic inhibition are becoming highly sought after to both inhibit the further loss of cells and prevent further inflammation. Indeed, several studies have utilized various methods of necroptotic inhibition in disease with overall positive results.
Expanding the canonical necroptotic pathway
While the foundation of the necroptotic signaling pathway may still be in its relative infancy, new strides have revealed new routes of necroptotic activation and associated signaling partners that expand the necroptotic signaling pathway. The following section will discuss the additional secondary signaling partners linked to active necroptotic signaling (Figure 2) by acting as either a direct or indirect activator due to regulatory dysfunction. We hope our review informs the field on the expanded necroptotic signaling pathway while specific and selective therapies to treat age-dependent neuroinflammatory diseases continue to evolve.
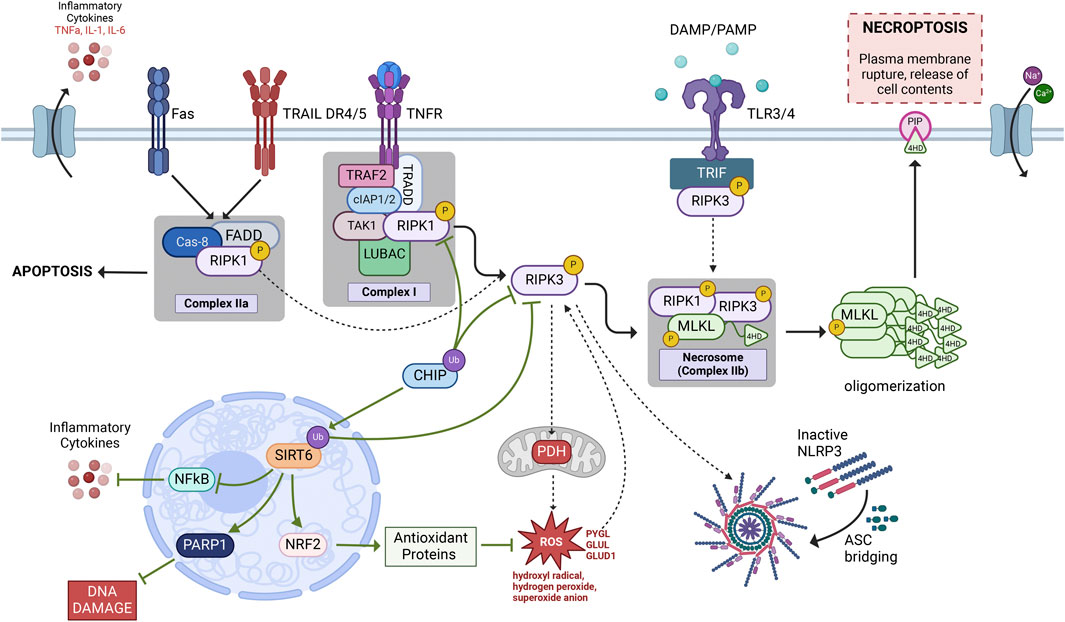
FIGURE 2. Necroptosis signaling pathway. The most widely investigated necroptotic signaling cascade is initiated by an inflammatory cytokine, such as TNF-α, binding to and activating its receptor, TNFR1. Following TNFR1 activation, the TNFR1-associated Death Domain (TRADD), which forms a complex with RIPK1, cIAP1/2, TRAF2, TAK1, and LUBAC known as Complex 1. Post-translational modifications of RIPK1 determine which singlaing pathway is activated thereafter. In conditions where RIPK1 is phosphorylated it can bind and phosphorylate RIPK3 via their shared RHIM domains. Phosphorylated RIPK1/3 then bind and phosphorylate MLKL to form a necrosis-inducing complex known as the necrosome (Complex IIb). Within the necrosome, RIPK1/3 phosphorylation of MLKL allows for MLKL to form oligomers that are then recruited to the plasma membrane through interactions between the positively charged 4-helical bundle domain on the N-terminal of MLKL and negatively charged phosphatidylinositol phosphates (PIPs) resulting in cellular rupture and leakage of cell contents, a terminal step in necroptotic cell death. The solid black arrows represent our rendention of the current conconical necroptotic signaling casade while the dashed arrows represent the additional signaling cascades explained throughout this review that may lead to necroptotic activation. Green arrows denote neuroprotetive signlaing partners that may aid in necroptotic regulation as discussed in this review. Figure created using Biorender.com.
Reactive oxygen species
Reactive oxygen species (ROS) have long been associated with, and are considered a hallmark of, necroptosis (Yang Z. et al., 2018). However, the findings of a recent study concluded that mitochondrial ROS are not involved in the necroptotic cell death events of HT-29, SVEC, and 3T3 cells (Zhang et al., 2017). This surprising observation led researchers to wonder what the actual role of ROS was in necroptosis and what the implications were on necroptotic signaling. They found that mitochondria are essential for the induction of TNF-induced necroptosis in most cell types (Zhang et al., 2017). More specifically, RIPK1 was the primary target of ROS, and the oxidation of three crucial cysteine residues resulted in RIPK1 autophosphorylation promoting the recruitment of RIPK3, leading to functional necrosome formation (Zhang et al., 2017). While mitochondrial ROS is commonly associated with aerobic respiration and known to participate in necroptosis (Turrens, 2003; Yang Z. et al., 2018), the work done by the Zhang et al. group found that TNF-induced ROS production is RIPK3-dependent (Zhang et al., 2017). The resulting model proposed that following TNF-induced necroptosis, the metabolic enzymes glycogen phosphorylase (PYGL), glutamate-ammonia ligase (GLUL), and glutamate dehydrogenase 1 (GLUD1) are targeted by RIPK3 via a positive feedback loop allowing for the production and regulation of ROS (Zhang et al., 2009; Yang Z. et al., 2018; Morgan and Kim, 2022).
Additionally, pyruvate dehydrogenase complex (PDC or PDH), an enzyme that controls aerobic respiration by linking glycolysis to the Krebs cycle, can be activated by RIPK3 (Zhang et al., 2009; Zhang et al., 2017; Yang Z. et al., 2018). The RIPK3-MLKL-containing necrosome translocates to the mitochondria, where it phosphorylates the E3 subunit of PDH, subsequently activating it. Once activated, this complex increases aerobic respiration and, subsequently, mitochondrial ROS, leading to an intracellular increase in oxidative stress and necroptosis (Yang J. et al., 2018). The role of ROS in metabolism and respiration is well understood; however, we still have a novice understanding of its impact and importance on necroptosis. Because of ROS’s critical role in promoting neuroinflammation, the mechanism behind RIPK3-dependent ROS needs further investigation.
Receptors
Fas, also known as APO-1 or CD95, is a receptor that belongs to the TNF family of receptors activated by its distinct ligand (FasL). Holler et al. were the first group to discover Fas-mediated RIPK1-dependent necroptosis (Holler et al., 2000). However, due to the discovery of TNF-induced necroptosis, investigations into the impact and outcomes of Fas-induced necroptosis appeared to be cast aside. Because of this, as Morgan et al. point out, much of the information about Fas-induced necroptosis has primarily been inferred by comparing its well-mapped-out role in apoptosis with the information we have on the TNF counterpart mechanism (Morgan and Kim, 2022).
Indeed, the mechanism behind Fas-induced necroptosis appears to behave similarly to TNF-induced necroptosis. Following FasL binding, Fas promotes the formation of the apoptotic death complex, complex IIa, comprised of FADD, caspase 8, and RIPK1 (Gupta et al., 2004; Ofengeim and Yuan, 2013; Liu et al., 2021). FADD and RIPK1 are recruited to FAS through their death domains, while caspase-8 is chaperoned directly into the receptor signaling complex (Morgan and Liu, 2013). While secondary complexes also occur in response to FasL, it remains unknown whether these are essential for Fas-induced necroptosis (Morgan and Kim, 2022). Active apoptotic signaling initiates following complex IIa formation (Holler et al., 2000). However, in conditions where cIAP is present, and caspase-8 is absent, Fas/FasL can promote the necrosome formation in a RIPK3-dependent manner (Geserick et al., 2009; Ofengeim and Yuan, 2013; Dhuriya and Sharma, 2018; Liu et al., 2021). Of interest, unlike the TNF-mediated necroptotic pathway, FADD is required for the Fas-mediated necroptosis as FADD-deficient cell types resist both necroptotic and apoptotic cell death events (Yeh et al., 1998; Holler et al., 2000; Lawrence and Chow, 2005). Morgan et al. argue that FADD supports RIPK1 recruitment to form complex IIa (Morgan and Kim, 2022). Little else is known about Fas-induced necroptosis and the physiological relevance of this mechanism; however, the findings made by Krzyzowska et al. that Fas/FasL can lead to excessive neuroinflammation following HSV-1 infection (Krzyzowska et al., 2021) suggests that Fas may have a much more significant role in neuroinflammation and necroptosis and should continue to be studied.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor 4/5 (DR4/5) is a cell surface receptor that belongs to the TNF receptor superfamily 10A (TNFRSF10A) that processes an intracellular death domain. Similar to the scant literature linking the Fas receptor and necroptosis, how TRAIL, specifically DR4/5, induces necroptosis is predominantly inferred or predicted. Indeed, TRAIL-induced necroptosis may behave very similarly to Fas-induced necroptosis (Morgan and Kim, 2022). Similar to TNF-induced necroptosis and Fas-induced necroptosis, TRAIL initiates necroptosis following the inhibition of cIAP and/or caspase-8 or under conditions where TAK1 is deficient (Dondelinger et al., 2013). TRAIL-induced necroptotic signaling requires FADD as the mechanisms behind the complex recruitment to the receptor are FADD-dependent (Kuang et al., 2000; Sprick et al., 2000). In alignment with Fas-induced necroptosis, because of TRAIL’s dependency on FADD, RIPK1 plays an important and essential role in the induction and activation of necroptotic signaling (Jouan-Lanhouet et al., 2012). Much like TNF-induced necroptosis, TRAIL-induced necroptosis can be regulated by the linear ubiquitination of RIPK1 (Lafont et al., 2017). While there are secondary downstream complexes from TRAIL, it is unknown whether these complexes are vital to the necroptotic signaling cascade (Morgan and Kim, 2022). While the mechanism behind TRAIL-induced necroptosis still leaves much to the imagination, the recent findings that TRAIL promotes neuroinflammation and degeneration in alcohol use disorders make this receptor an attractive target that should be further investigated (Qin et al., 2021).
Toll-like receptors 3 and 4 (TLR 3/4) belong to the family of PRRs and specialize in the recognition of pathogen-associated molecule patterns (PAMPs) and DAMPs (Kumar, 2019; Sameer and Nissar, 2021). Behind TNF-induced necroptosis, TLR-induced necroptosis via immune cell activation in response to bacterial lipopolysaccharide (LPS) is the next-best studied pathway. While TLR3/4 excels at initiating pro-inflammatory immune response through NF-κB activation, they have also been shown to induce necroptosis in developmental diseases (Liu et al., 2021) and cancer (Cen et al., 2018). The ability of these two receptors to induce necroptotic signaling stems from an identical signaling mechanism. Following the activation of the cytosolic adaptor Toll/IL-1 receptor domain-containing adaptor protein inducing interferon-β (TICAM-1, also referred to as TRIF) by LPS, TLR3/4 recruits RIPK3 through TRIF’s RHIM domain, similar to that of RIPK1 (Kaiser et al., 2013). When bound to TLR3/4, the TRIF-RIPK3 complex is formed, directly activating MLKL and necroptosis after that (Kaiser et al., 2013; Morgan and Kim, 2022). Of interest, while RIPK3 is essential for LPS-mediated necroptosis, RIPK1 does not appear to be required, making this a RIPK1-independent form of necroptosis (He et al., 2011; Lim et al., 2019). As seen in the TNF pathway, caspase-8 cleaves TRIF and can inhibit TLR-induced necroptosis (Rebsamen et al., 2008) or cIAP (McComb et al., 2012). Given the role TLR3/4 has in pro-inflammatory signaling and mainly due to its ability to induce necroptosis independent of RIPK1, making it an intrinsic mechanism, TLR-induced necroptosis needs more consideration in age-related neuroinflammatory diseases.
NOD-like Receptor Protein 3 (NLRP3) belongs to the family of intracellular PRRs. NLRP3 elicits inflammatory responses within cells, independent of cell death, through its ability to form a complex known as the inflammasome (Conos et al., 2017). Canonically, the apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC) also helps form the inflammasome by bridging NLRP3 proteins to form the complex (Compan et al., 2015). However, following a study in 2014, MLKL can trigger NLRP3 activation, leading to the formation of the inflammasome (Kang et al., 2014), suggesting NLRP3 may play a role in necroptotic signaling. Along with MLKL’s direct activation of NLRP3, low levels of intracellular potassium have been linked to NLRP3 action, thought to be explained by the potassium efflux that occurs when MLKL oligomers puncture the plasma membrane, allowing for disruption in ion gradients (Conos et al., 2017) further linking NLRP3 and necroptosis. In addition to MLKL’s involvement in forming the NLRP3 inflammasome, in 2015, Lawlor et al. found a link between RIPK3 and NLRP3 independent of MLKL (Lawlor et al., 2015), further supported by recent findings that suggested that RIPK3 actively participates in NLRP3 inflammasome activation and that inhibiting RIPK3 resulted in inhibited NLRP3 inflammasome activity (Chen J. X. et al., 2018; Xue et al., 2022). While our understanding of the role that NLRP3 plays in necroptosis is still developing, NLRP3’s ability to mediate the cell’s intrinsic inflammatory response makes it a popular protein to study concerning necroptosis and neuroinflammation.
Transcription factors
Nuclear factor-kB (NF-κB) is a transcription factor that regulates many genes involved in the cellular immune response and plays a crucial role in inflammation and various forms of PCD, including necroptosis (Park and Hong, 2016; Verzella et al., 2020). As stated earlier, following complex 1 formation, the cell is shepherded into pro-survival NF-κB signaling, caspase-dependent apoptosis, or caspase-independent pro-inflammatory necroptosis (Ye et al., 2023). As Mifflin et al. and Xe et al. described, RIPK1 ubiquitination chiefly determines the fate of complex 1 signaling (Mifflin et al., 2020; Ye et al., 2023). After RIPK1 polyubiquitination, complex 1 recruits TAK and IKK complexes, resulting in direct RIPK1 phosphorylation (Delanghe et al., 2020). This direct phosphorylation inhibits RIPK1 kinase activity, activating NF-κB and promoting inflammatory factor production and cell survival (Delanghe et al., 2020). While the role that RIPK1 plays in NF-κB activation following canonical RIPK1 ubiquitination is well understood, the interactions between RIPK1 and NF-κB in the non-canonical necroptotic signaling cascade are less defined (Verzella et al., 2020). Morgan et al. also point out that the role of RIPK3 in NF-κB activation is even less defined and contradictory and surmised that noncanonical NF-κB activity may require RIPK3 in specific cell types following a 2015 study that demonstrated impaired nuclear translocation of the RelB-p50 heterodimer of NF-κB in RIPK3 knockout cells. Thus, noncanonical NF-κB activity requires RIPK3 in specific cell types (Moriwaki et al., 2014; Morgan and Kim, 2022). While NF-κB and its role in necroptosis remains vague, the NF-κB-dependent regulation of pro-inflammatory genes, such as chemokines and cytokines, and known interactions with RIPK1 make NF-κB an exciting factor to study in the context of necroptotic signaling and neuroinflammation.
Regulatory proteins
Carboxyl Terminus of HSC70-Interacting Protein (CHIP) is a critical regulator of protein quality control (PQC)(Ballinger et al., 1999; Rosser et al., 2007; Min et al., 2008). While expressed throughout the body, CHIP is most abundant in tissues containing terminally differentiated cells, such as Purkinje cells, an inhibitory class of neurons located in the cerebellum (Shi et al., 2014; Schuster et al., 2018; Madrigal et al., 2019). Because of this, CHIP has been thought to be neuroprotective in multiple models of proteinopathies due to its central role in maintaining PQC during aging (Min et al., 2008). CHIP is particularly well positioned to regulate PQC due to its ability to function as both a co-chaperone protein and ubiquitin ligase enzyme. When acting as a co-chaperone, CHIP interacts with heat shock proteins (HSP) to stabilize substrates and promote refolding (Ballinger et al., 1999). Alternatively, when functioning as a ubiquitin ligase, CHIP targets and ubiquitinates terminally defective proteins through the utilization of the ubiquitin-proteasome system (Ballinger et al., 1999; Rosser et al., 2007; Seo et al., 2016). Because of CHIP’s ability to be dual functioning, it is not surprising that CHIP has been linked to several age-related protein accumulation disorders (Min et al., 2008; Schisler et al., 2013; Ronnebaum et al., 2014; Shi et al., 2014; Palubinsky et al., 2015; Madrigal et al., 2019) Both RIPK1 and RIPK3, which are critical kinases involved in the induction and regulation of necroptosis, are known CHIP substrates (Seo et al., 2016). Seo et al. demonstrated that the protein stability of RIPK3 is regulated by CHIP-mediated ubiquitylation and degraded in a lysosomal-dependent manner. Similarly, RIPK1 expression was also negatively regulated by CHIP-mediated ubiquitylation and subsequent proteasome degradation. These landmark findings were the first to suggest that CHIP was a critical regulator of RIPK1 and RIPK3 expression and validated CHIP’s role in regulating necrosome formation and necroptosis (Seo et al., 2016). Recent studies have discovered that the molecular chaperone, heat shock protein 90 kDA alpha family class A member 1 (best known as HSP90), which commonly interacts with CHIP through its tetratricopeptide repeat domain, plays a critical role in MLKL oligomerization and translocation further suggesting that CHIP may play a much more significant role in necroptotic regulation (Jacobsen et al., 2016; Marunouchi et al., 2021).
Sirtuins (Sirt 1–7) are a family of NAD+ -dependent histone deacetylases that play a role in protection against age-related diseases such as cancer (Fu et al., 2020) and neurodegeneration (Sebastián et al., 2012). One of these sirtuins, Sirtuin 6 (SIRT6), a histone deacetylase and mono-ADP-ribosyltransferase located in the nucleus of cells, has therapeutic effects on cellular processes such as inflammation, aging, metabolism, stress-resilience, and cell death (Fu et al., 2020). SIRT6 has a modulatory role in both apoptosis and necroptosis. Regarding necroptosis, SIRT6 suppresses the necroptotic signaling pathway, inhibiting cell death and preventing the inflammatory immune response (Fu et al., 2020). Overexpression of SIRT6 blocks the activation of RIPK3 and subsequent MLKL activation, while a knockdown of SIRT6 results in cell senescence and apoptosis (Chen J. et al., 2018). SIRT6 also has many anti-inflammatory, cell-protective properties that could potentially reduce inflammation caused by necroptosis. As a mono-ADP-ribosyltransferase, it can target and activate the transcription of DNA damage repair genes and antioxidant genes. SIRT6 activates Nuclear factor erythroid 2-related factor 2 (Nrf2) via mono-ADP-ribosylation of a subunit of the BAF chromatin remodeling complex, which drives the expression of antioxidant proteins (Rezazadeh et al., 2019). SIRT6 also activates poly-ADP-ribose polymerase-1 (PARP-1) by mono-ADP-ribosylation of lysine residue 521, thus promoting DNA damage repair (Mao et al., 2011). Using its deacetylase properties, Sirt6 can also inhibit the transcription of the pro-inflammatory Nuclear Factor kappa B (NF-κB). To do this, SIRT6 deacetylates lysine 9 of histone H3 at the promoter of the NF-κB target gene, which suppresses the release of inflammatory cytokines, as well as modulating macrophage inflammatory phenotypes (Kawahara et al., 2009; Lee et al., 2017; Koo et al., 2019). Though SIRT6 poses a vital role in maintaining cellular health in the face of age-related necroptosis, studies have shown that SIRT6 does indeed decrease with age and in neurodegenerative diseases such as Alzheimer’s disease (Liang et al., 2008; Kaluski et al., 2017; Garcia-Venzor and Toiber, 2021).
For the field to grow and design more effective therapies, we must expand our view of the necroptotic signaling pathway and consider what other factors may consistently influence this high-proinflammatory pathway. As such, each signaling partner spelled out above has a known role with the systems that activate and regulate necroptosis. Further identifying key necroptotic players at the transcriptional, metabolic, intra-, and extracellular levels will open new avenues to target necroptotic signaling to aid neuroprotection therapeutically.
Necroptotic inhibitors
The role of necroptosis in influencing and promoting age-dependent neuroinflammation makes it an attractive signaling cascade for therapeutic targeting. However, despite numerous efforts and investigations, a specific and efficacious therapeutic remains to be discovered. Below, we comprehensively discuss the therapeutic compounds used to target the RIPK1-RIPK3-MLKL signaling cascade and their impact. These compounds range from small molecule inhibitors directly binding to their target protein to compounds that impact expression levels. We hope our collation of studies promotes continued investigation of compounds to treat age-dependent neuroinflammatory diseases.
RIPK1 inhibitors
RIPK1 inhibitors are the most widely investigated and available class of inhibitors within the necroptotic signaling cascade and have proved to be neuroprotective in age-dependent neuroinflammatory diseases such as AD, MS, and ALS (Ofengeim et al., 2015; Rubinsztein, 2017; Mifflin et al., 2021). Ideally, RIPK1 inhibitors designed to treat these diseases must be able to penetrate the blood-brain barrier to target specific cell types in the brain. There have been several RIPK1 inhibitors tested in clinical trials with differing degrees of success. Denali Therapeutics has been a leader in this market. Their first compound, DNL104, was identified as a specific, brain-penetrant inhibitor of RIPK1 phosphorylation and underwent Phase 1 clinical trials for the treatment of AD and ALS (Grievink et al., 2020). Post-dose liver toxicity effects found in the healthy volunteers, not associated with RIPK1 inhibition, halted the trial but provided strong evidence for RIPK1 inhibition in the CNS (Grievink et al., 2020). Following this, Denali Therapeutics then went on to initiate Phase 1 clinical trials for the treatment of AD and ALS with SAR443060 (also known as DNL747), another CNS-penetrant RIPK1 inhibitor. Although DNL747 did appear to inhibit peripheral phosphorylated RIPK1 levels, and was well-tolerated, the drug showed potential dose-limiting toxicities in preclinical long-term toxicology studies (Vissers et al., 2022). As of January 2024, Denali’s partner company, Sanofi, is currently in Phase 2 clinical trials with another CNS-penetrant RIPK1 inhibitor, SAR443820 (also known as DNL788), that is being tested in ALS and MS patients (Clinical trial IDs NCT05237284 and NCT05630547, respectively). There have been additional RIPK1 inhibitors that have gone through clinical trials, such as GSK2982772, which is a selective inhibitor of RIPK1 activity and was tested for the treatment of chronic inflammatory diseases such as ulcerative colitis (UC) and rheumatoid arthritis (RA) (Weisel et al., 2021a; Weisel et al., 2021b). Although the drug did not appear to have any significant safety or tolerability issues, in both the UC and RA trials, it did not appear to inhibit RIPK1 activity levels compared to the placebo, indicating that there may be issues with clinical efficacy when using GSK2982772 as a monotherapy ((Weisel et al., 2021a; Weisel et al., 2021b).
Additionally, SAR443122 (also known as DNL758 and eclitasertib) is a RIPK1 inhibitor that is currently in Phase 2 clinical trials for the treatment of peripheral inflammatory diseases such as, cutaneous lupus erythematosus (CLE), and ulcerative colitis (UC) (Clinical trial IDs: NCT04781816, NCT05588843) and was studied as a treatment for COVID-19 (Clot et al., 2021). As of January 2024, this drug is still undergoing clinical trials. While there has been significant headway in discovering and testing brain-penetrant RIPK1 inhibitors for the treatment of age-dependent neuroinflammatory diseases, it remains to be seen if a RIPK1 inhibitor can be both safe and efficacious enough to aid in the treatment of these diseases. As such, below, we outline the various RIPK1 inhibitors that have shown promise in laboratory settings, as these untested compounds could have clinical efficacy.
Necrostatins are a group of chemical compounds named for their ability to block necrotic cell death (Vandenabeele et al., 2013). Necrostatin-1 (Nec-1) was initially identified as a compound that blocks necrotic cell death in human and murine cells (Degterev et al., 2005). Nec-1 has since been identified more specifically as an inhibitor of RIPK1 activity, making it a therapeutic target for many necroptosis diseases (Degterev et al., 2008).Nec-1 has a very short half-life (about 1–2 h in rats) and has off-target effects, making it challenging for clinical applications (Muller et al., 2005; Geng et al., 2017). However, a stable analog of Nec-1, Nec-1s, does not produce the same off-target effects, making it more specific for RIPK1 activity (Takahashi et al., 2012). The discovery of Nec-1 and Nec-1s has proven crucial in targeting RIPK1 and has provided new insight into therapeutics that can help mediate the pathway to necroptosis. It has been demonstrated in numerous studies that both Nec-1 and Nec-1s can alleviate various pathophysiological changes associated with age-dependent neuroinflammatory diseases and can protect various neuronal cell types against necroptotic stimuli (Zhu et al., 2011; Wu et al., 2015; Ito et al., 2016; Yang et al., 2017; Iannielli et al., 2018). In addition to Nec-1, the recently identified Nec-34 has also shown promise as a RIPK1 inhibitor. While Nec-1s works as an allosteric inhibitor of RIPK1, Nec-34 stabilizes RIPK1 in its inactive conformation by binding to specific pockets within the RIPK1 kinase domain. Interestingly, Meng et al. noted that Nec-34 and Nec-1s could be used as a combination therapy to inhibit RIPK1 in vitro and in vivo, suggesting a better avenue of RIPK1 inhibition that can be utilized for the treatment of RIPK1-mediated disorders (Meng et al., 2021).
Geldanamycin (GA) is an antibiotic that works by selectively binding to a binding pocket on HSP90 chaperone protein, which prevents the binding to its client protein, one of which is RIPK1, leading to a reduction in RIPK1 protein level expression (Xie et al., 2011). Chen et al. demonstrated that primary cortical neurons, when treated with GA following ischemic injury via oxygen deprivation, showed resistance to cell death stimuli and that GA appeared to aid in neuroprotection by decreasing HSP90 protein levels, leading to the instability of RIPK1 promoting protection against neuronal injury (Chen et al., 2012). However, while GA has shown promise in its ability to be neuroprotective, RIPK1 is not the only client protein of HSP90. As such, GA can not be classified as a specific inhibitor and would need further investigation to determine its off-target effects.
Oxazolone is a compound historically used to induce colitis as a model for studying inflammatory bowel disease (IBD)(Meroni et al., 2018). However, following the screening of a small library of new molecules for potential inhibitors of necroptosis, Oliveria et al. found that Oxazolone (Oxa 12) could inhibit zVAD-fmk-induced necroptosis in mouse microglial cells (Oliveira et al., 2018). Additionally, Oxa12 can prevent the terminal steps of necroptosis by binding to a known inhibitory region of RIPK1, inhibiting necrosome formation, MLKL phosphorylation, and subsequent necrosome assembly (Oliveira et al., 2018). Oxa12 reduced the expression of specific cytokines and transcription factors involved in eliciting inflammatory responses such as TNF⍺, IL-1β, and NF-κB in glial cells, indicating the potential of Oxa12 to be therapeutic in age-dependent neuroinflammatory diseases (Oliveira et al., 2018).
RIPK3 inhibitors
GSK’872/840 GSK’872 was identified as a small molecule RIPK3 inhibitor through screening of a small molecule library and found to reduce RIPK3-dependent necrosis independent of the upstream activator, RIPK1(Kaiser et al., 2013). GSK’872 acts by binding to the kinase domain of RIPK3 to prevent RIPK3 kinase activity and inhibit the RHIM-dependent signaling that is necessary for MLKL binding to RIPK3 and necrosome formation (Mandal et al., 2014; Riebeling et al., 2022). GSK’840 also inhibits RIPK3 through a similar mechanism (Mandal et al., 2014). Though GSK’872 and GSK’840 inhibit RIPK3 and necroptosis, once bound to RIPK3, they have been shown to elicit a conformational change that promotes the recruitment of RIPK1 via the RHIM domain leading to the activation of casape 8 resulting in apoptosis (Mandal et al., 2014). However, the FDA-approved cancer treatment drug Ibrutinib was recently identified as a RIPK3 inhibitor that acts similarly to GSK’872 (Huang et al., 2023). Ibrutinib inhibits the phosphorylation and autophosphorylation of RIPK3 independent of RIPK1 (Huang et al., 2023). Unlike GSK’872 or GSK’840, Ibrutinib inhibits necroptosis without enforcing the apoptosis-inducing RIPK3 conformational change, making it a more ideal inhibitor of RIPK3 and TNF⍺-induced necroptotic cell death. This finding points to the potential of these compounds to be therapeutic against necroptotic stimuli and highlights the need for further investigation.
Zharp-99 and Compound 38 are two recently identified RIPK3 inhibitors that have proven more potent than GSK ‘872, discovered by researchers supported by Accro Bioscience Inc. Zharp-99 was identified as a small molecule inhibitor of RIPK3 activity showing potent cellular efficacy for blocking necroptosis induced by TNF⍺ signaling and TLR signaling from pathogen infection (Xia et al., 2020). Zharp-99 selectively inhibits RIPK3 while having no effect on RIPK1 activity and is a more potent inhibitor than GSK ‘872 (Xia et al., 2020). From the same group, compound 38 was identified as a potent inhibitor of TNF⍺ induced necroptosis through blocking RIPK3 kinase activity (Wu et al., 2021). Compound 38 proved more potent than GSK ‘872 and appeared stable with favorable safety testing in vitro, making it an attractive therapeutic for future studies (Wu et al., 2021).
Dabrafenib is a compound initially identified as a B-RAF inhibitor approved to treat metastatic melanoma. However, previous studies have demonstrated that Dabrafenib can prevent necroptotic signaling due to its ability to act as a RIPK3 inhibitor (Menzies et al., 2012; Cruz et al., 2018). Dabrafenib inhibits RIPK3 through competitive binding with ATP (Li et al., 2014), as it has already undergone clinical trials and received FDA approval for its use in other diseases, it makes Dabrafenib an attractive therapeutic for inhibiting RIPK3-mediated necroptotic signaling. However, further studies need to determine the brain penetrance of Dabrafenib, and its non-specific/off-target effects may limit its use.
MLKL inhibitors
Necrosulfonamide (NSA) is a compound that inhibits necroptosis by blocking the phosphorylation of MLKL, the executioner protein involved in necroptotic signaling. NSA can only inhibit necroptosis via an MLKL-dependent pathway and does not affect its upstream signaling partners, RIPK1 or RIPK3 (Sun et al., 2012). Because MLKL activation is the last step before necrosome formation and MLKL oligomerization, the terminal structure that punctures the cell membrane resulting in the distinctive necroptotic phenotype, MLKL is likely the most specific target for inhibiting necroptosis (Motawi et al., 2020). Treatment with NSA inhibits the activation of MLKL with no effect on RIPK1 or RIPK3 levels, preventing the formation of the necrosome and membrane translocation of the MLKL oligomer, thereby inhibiting necroptosis. However, NSA may not be specific to MLKL, as studies have found that it can also inhibit the pore-forming protein gasdermin D (GSDMD) involved in pyroptosis (Rathkey et al., 2018). NSA was neuroprotective in ischemic brain injury (Zhou et al., 2017) and spinal cord injury (Jiao et al., 2019) models, but its lack of specificity towards the necroptotic pathway requires further testing for use as a therapeutic in the context of neuronal injuries or necroptotic disease models.
Xanthine 35 and 36 are another class of MLKL inhibitors with increased specificity to MLKL. Xanthine 35 (TC13172 35) is a compound that was found to bind to a cysteine on the 4-helical bundle domain on the N-terminal of MLKL, inhibiting its oligomerization and membrane translocation, a similar mechanism of action to NSA (Yan et al., 2017). Xanthine 36 blocks MLKL oligomerization by stabilizing the packing structures within MLKL, as destabilizing these structures drives increased MLKL activity (Rübbelke et al., 2020). Xanthine 35 and 36 target MLKL without affecting RIPK1 and RIPK3, but their potential off-target toxicity may limit their efficacy. Further studies must test their clinical relevance, especially in age-dependent neuroinflammatory diseases (Gardner et al., 2023).
Combination inhibitors
B-RAF inhibitors are another group of FDA-approved drugs investigated for inhibiting necroptosis. Ponatinib is a B-RAF inhibitor identified as a RIPK1 and RIPK3 inhibitor in a drug repurposing screen for potential necroptotic inhibitors (Fauster et al., 2015). Like Dabrafenib, Ponatinib is another approved anti-cancer treatment. Still, due to its non-specificity and potential side effects when not used for cancer treatment, further studies need to be done to test its efficacy as a necroptotic inhibitor.
Sorafenib is another B-RAF inhibitor like the ones mentioned above, but unlike dabrafenib, this drug has been identified as a combination inhibitor of RIPK1 and RIPK3 activity (Feldmann et al., 2017). Sorafenib is an FDA-approved drug for the treatment of acute leukemia. It also inhibits Smac mimetic/TNF⍺-induced necroptosis in acute leukemia cells by inhibiting the kinase activity of RIPK1 and RIPK3, preventing necrosome formation (Feldmann et al., 2017). Beyond leukemia, Sorafenib conferred protection against an acute inflammatory response to RIPK1/3-mediated necroptosis (Martens et al., 2017).
Additionally, Quizartinib, another FDA-approved anti-cancer drug, was identified as an inhibitor of RIPK1, RIPK3, and MLKL phosphorylation (Huang et al., 2023). While the specific target and mechanism of action for quizartinib is still unknown, it is a promising inhibitor of TNF⍺-induced necroptosis. Like other B-RAF inhibitors previously mentioned, this class of inhibitors is an attractive therapeutic option for targeting the necroptotic pathway due to numerous FDA approvals. However, further in vivo studies will need to test the drug’s efficacy in neuroinflammatory-driven diseases.
Celastrol, a root extract from the Chinese herb Tripterygium wilfordii, has been shown to mediate several inflammatory diseases such as rheumatoid arthritis, osteoarthritis (Gao et al., 2020; Jing et al., 2021), and autoimmune hepatitis (Shi et al., 2022). It has also been shown to be neuroprotective in Parkinson’s disease (Zhang et al., 2021), acute ischemic stroke-induced brain injury (Cao et al., 2023), and spinal cord injury models (Dai et al., 2019). Celastrol may act through inhibition of the NF-κB signaling pathway by reducing NF-κB protein levels, as demonstrated in cells treated with high concentrations of the extract (Liang et al., 2023). Celastrol has also demonstrated its ability to inhibit the phosphorylation of RIPK1, RIPK3, and MLKL, thereby preventing necrosome formation and inhibiting necroptosis (Liang et al., 2023). Though reports on necroptosis-driven studies are few, the anti-inflammatory and anti-oxidative effects of the drug are well known. Given its recent use as a treatment for many inflammatory and neuronal diseases, Celastrol could be a potent new therapeutic for age-dependent neuroinflammatory diseases.
The therapeutics listed above have demonstrated some capacity to inhibit necroptosis in vitro or in vivo, some of which have already undergone clinical trials and received FDA approval. Concomitantly, most of these therapeutics also act as potent anti-inflammatory drugs, an essential component needed for mediating age-dependent neuroinflammatory diseases. It is important to note that small-molecule screenings of both FDA-approved and not yet approved compound libraries continue to identify more necroptotic inhibitors (Kaiser et al., 2013; Fauster et al., 2015; Huang et al., 2023), holding promise that new drugs may emerge for age-dependent neuroinflammatory diseases.
In Vitro models of induced necroptosis
One of the most significant rate-limiting steps preventing us from discovering more efficacious therapeutics that can inhibit necroptotic signaling and aid in neuroprotection is our limited ability to induce physiologically relevant necroptotic signaling using in vitro models. To further promote compound development and testing and to aid in the continued exploration of the potential signaling partners we mapped out above, we discuss below the various methodologies employed to study necroptosis in vitro. By discussing these methodologies, we hope to make the various methods of testing necroptotic signaling more transparent and inspire the creation of better models to investigate necroptosis so that the studies of age-related neuroinflammatory diseases may continue.
Small molecule cocktails
The most popular method to induce necroptosis in vitro models involves using a medley of small molecules. While various iterations of these small molecule cocktails have been used to investigate necroptotic signaling, there are two standard consistencies among them, which are the presence of an inflammatory cytokine and a caspase inhibitor (Vercammen et al., 1998; Cho et al., 2009; Laukens et al., 2011; Seo et al., 2016; Galluzzi et al., 2018; Chen et al., 2020). Using these small molecule cocktails to induce necroptosis is an ideal method to inhibit other programmed cell death pathways, specifically the apoptotic pathway, while allowing unabated necroptotic signaling. However, the true strength behind this model lies in the idea that when used in combination with small molecule therapeutics, cell death can be prevented, allowing for this model to also act as a screening platform to investigate the therapeutic potential of necroptotic inhibitors (Seo et al., 2016). In the early 2000s, the now gold standard small molecule cocktail, commonly referred to as TCZ, compromised of the inflammatory cytokine TNF⍺, the pan-caspase inhibitor z-VAD-FMK, and the protein synthesis inhibitor cycloheximide was introduced (Cho et al., 2009; Seo et al., 2016). This model has since been reimagined using the IAP antagonist, SMAC/DIABLO mimetic, in place of cycloheximide, referred to as TSZ, as it promotes TNF-induced, RIPK1-dependent, and caspase-independent necroptosis in apoptosis-resistant cell types (Laukens et al., 2011). However, there is not a one-size-fits-all approach with either the TCZ or TSZ models, and there tend to be variations in methods depending on cell type (Cho et al., 2009; Laukens et al., 2011; Seo et al., 2016; Chen et al., 2020). As such, we have outlined the differences in (Table 1) to provide a better understanding of how to use the TCZ/TSZ model best to study necroptotic signaling-associated therapeutic candidates further.
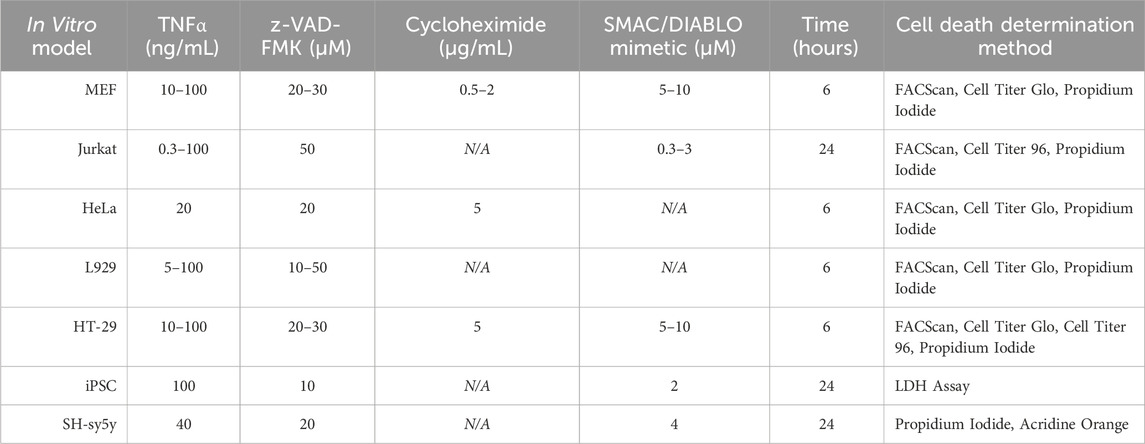
TABLE 1. Concentrations of small molecule inhibitors used to induce necroptosis in various in vitro models.
Oxygen-glucose deprivation
Oxygen-glucose deprivation (OGD) is an in vitro model for ischemic stroke. It acts as a potent activator of cell inflammation and is effective on any cell or tissue culture system. Cells or tissue can be plated and maintained in normal conditions, and then, to induce OGD, the culture media is wholly replaced with glucose-free media and moved to a low-oxygen incubator (<1% O2) for a predetermined time. Cells can then be harvested and analyzed, or glucose-containing media and oxygen are re-introduced, simulating reperfusion. Reperfusion after transient OGD mimics the restoration of blood flow to ischemic areas, which also increases inflammation and cell death as reperfusion disrupts the membrane permeability of affected cells and leads to cell death (Shi et al., 2016). OGD in cells results in the upregulation of both RIPK1 and RIPK3, as well as an increase in the release of TNF-alpha and IL-18 (Vieira et al., 2014; Yang J. et al., 2018; Guo et al., 2022), indicating that necroptosis is occurring in cells as a result of OGD.
Shikonin
Shikonin is a compound derived from the root of the shikonin plant and has historically been used in Traditional Chinese Medicine to treat inflammation, infection, and cancer (Wang et al., 2019). Recently, Shikonin and its naturally occurring analogs appear to evade cancer drug resistance through necroptosis induction (Xuan and Hu, 2009). All of the Shikonin analogs contain a naphthoquinone core structure, which researchers believe causes this compound class to induce necroptosis (Xuan and Hu, 2009). Though the mechanism of how Shikonin induces necroptosis is not entirely understood, at least in non-small cell lung cancer cells, the drug induces necroptosis in a RIPK1-dependent matter (Kim et al., 2017). RIPK1 protein expression levels increased in non-small cell lung cancer cells treated with Shikonin, an effect blocked by Nec-1 (Kim et al., 2017). Shikonin also induced necroptosis in glioma cells and increased ROS production and RIPK1/3 expression levels (Lu et al., 2017). Specific RIPK1 and RIPK3 inhibitors (Nec-1 and GSK’872, respectively) were sufficient to block Shikonin-induced necroptotic induction and ROS overproduction in glioma cells, indicating that Shikonin acts through a RIPK1/RIPK3-dependent necroptotic pathway (Lu et al., 2017). The complete mechanism of how Shikonin induces necroptosis is still not completely understood. However, these recent studies establish a solid foundation for how Shikonin can induce necroptosis in vitro. As such, researchers should reconsider Shikonin as a method to induce necroptosis as it may prove to be more physiologically relevant with less off-target effects than the methods listed above.
Conclusion
Necroptosis and its role in the progression and severity of age-dependent neuroinflammatory diseases is an ever-growing topic of discussion that demands more research, from the basic to the translational level. Targeting necroptotic signaling partners has potential therapeutic benefits across neuroinflammatory diseases. However, the signaling pathways activating necroptosis and any associated crosstalk need further exploration. Future studies should seek to provide the field with a clearer picture of the necroptotic machinery involved in age-dependent neuroinflammatory disease and how that machinery impacts disease progression and severity. We hope these findings will lead to the discovery of newer and more specific therapeutic targets that will provide hope and a better quality of life for those diagnosed with an age-dependent neuroinflammatory disease.
Author contributions
KS: Conceptualization, Writing–original draft, Conceptualization, Writing–original draft, MC: Writing–original draft, Writing–original draft, TM: Writing–original draft, Writing–original draft, JS: Conceptualization, Funding acquisition, Writing–review and editing, Conceptualization, Funding acquisition, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The National Institutes of Health supported this work by grants R01AG066710 and R01AG061188 to JS.
Acknowledgments
We thank members of the JS Laboratory and the administration teams at the McAllister Heart Institute and the Department of Pharmacology. Figures were created using BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Araújo, B., Caridade-Silva, R., Soares-Guedes, C., Martins-Macedo, J., Gomes, E. D., Monteiro, S., et al. (2022). Neuroinflammation and Parkinsons disease-from neurodegeneration to therapeutic opportunities. Cells 11 (18), 2908. doi:10.3390/cells11182908
Ballinger, C. A., Connell, P., Wu, Y., Hu, Z., Thompson, L. J., Yin, L. Y., et al. (1999). Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19 (6), 4535–4545. doi:10.1128/MCB.19.6.4535
Balusu, S., Horré, K., Thrupp, N., Craessaerts, K., Snellinx, A., Serneels, L., et al. (2023). MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer’s disease. Science 381 (6663), 1176–1182. doi:10.1126/science.abp9556
Bayat, A.-H., Saeidikhoo, S., Ebrahimi, V., Mesgar, S., Joneidi, M., Soltani, R., et al. (2021). Bilateral striatal transplantation of human olfactory stem cells ameliorates motor function, prevents necroptosis-induced cell death and improves striatal volume in the rat model of Huntington’s disease. J. Chem. Neuroanat. 112, 101903. doi:10.1016/j.jchemneu.2020.101903
Caccamo, A., Branca, C., Piras, I. S., Ferreira, E., Huentelman, M. J., Liang, W. S., et al. (2017). Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 20 (9), 1236–1246. doi:10.1038/nn.4608
Canet-Pons, J., Sen, N. E., Arsović, A., Almaguer-Mederos, L. E., Halbach, M. V., Key, J., et al. (2021). Atxn2-CAG100-KnockIn mouse spinal cord shows progressive TDP43 pathology associated with cholesterol biosynthesis suppression. Neurobiol. Dis. 152, 105289. doi:10.1016/j.nbd.2021.105289
Cao, B.-B., Zhang, X. X., Liu, Z., Qiu, Y. H., and Peng, Y. P. (2020). TGF-β1 provides neuroprotection via inhibition of microglial activation in 3-acetylpyridine-induced cerebellar ataxia model rats. Front. Neurosci. 14, 187. doi:10.3389/fnins.2020.00187
Cao, F., Wang, Y., Song, Y., Xu, F., Xie, Q., Jiang, M., et al. (2023). Celastrol treatment ameliorated acute ischemic stroke-induced brain injury by microglial injury inhibition and Nrf2/HO-1 pathway activations. BioMed Res. Int. 2023, 1076522. doi:10.1155/2023/1076522
Cen, X., Liu, S., and Cheng, K. (2018). The role of toll-like receptor in inflammation and tumor immunity. Front. Pharmacol. 9, 878. doi:10.3389/fphar.2018.00878
Chang, N. P., DaPrano, E. M., Evans, W. R., Nissenbaum, M., McCourt, M., Alzate, D., et al. (2023) “Neuronal DAMPs exacerbate neurodegeneration via astrocytic RIPK3 signaling.,” BioRxiv. doi:10.1101/2023.07.21.550097
Chen, J., Xie, J. J., Jin, M. Y., Gu, Y. T., Wu, C. C., Guo, W. J., et al. (2018). Sirt6 overexpression suppresses senescence and apoptosis of nucleus pulposus cells by inducing autophagy in a model of intervertebral disc degeneration. Cell death Dis. 9 (2), 56. doi:10.1038/s41419-017-0085-5
Chen, J. X., Wang, S., Fu, R., Zhou, M., Zhang, T., Pan, W., et al. (2018). RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. J. Transl. Med. 16 (1), 233. doi:10.1186/s12967-018-1606-4
Chen, M., Huang, M., Chen, W., Jiang, X., Su, W., Wang, X., et al. (2020). Smac mimetic promotes TNF-α to induce apoptosis of gallbladder carcinoma cells. Cell. Signal. 72, 109654. doi:10.1016/j.cellsig.2020.109654
Chen, W.-W., Yu, H., Fan, H. B., Zhang, C. C., Zhang, M., Zhang, C., et al. (2012). RIP1 mediates the protection of geldanamycin on neuronal injury induced by oxygen-glucose deprivation combined with zVAD in primary cortical neurons. J. Neurochem. 120 (1), 70–77. doi:10.1111/j.1471-4159.2011.07526.x
Chen, X., Hu, Y., Cao, Z., Liu, Q., and Cheng, Y. (2018). Cerebrospinal fluid inflammatory cytokine aberrations in Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis: a systematic review and meta-analysis. Front. Immunol. 9, 2122. doi:10.3389/fimmu.2018.02122
Cherry, J. D., Olschowka, J. A., and O’Banion, M. K. (2014). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 11, 98. doi:10.1186/1742-2094-11-98
Chiu, Y.-J., Lin, S. A., Chen, W. L., Lin, T. H., Lin, C. H., Yao, C. F., et al. (2020). Pathomechanism characterization and potential therapeutics identification for SCA3 targeting neuroinflammation. Aging 12 (23), 23619–23646. doi:10.18632/aging.103700
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137 (6), 1112–1123. doi:10.1016/j.cell.2009.05.037
Chou, T.-W., Chang, N. P., Krishnagiri, M., Patel, A. P., Lindman, M., Angel, J. P., et al. (2021). Fibrillar α-synuclein induces neurotoxic astrocyte activation via RIP kinase signaling and NF-κB. Cell death Dis. 12 (8), 756. doi:10.1038/s41419-021-04049-0
Clot, P.-F., Farenc, C., Suratt, B. T., Krahnke, T., Tardat, A., Florian, P., et al. (2024). Immunomodulatory and clinical effects of receptor-interacting protein kinase 1 (RIPK1) inhibitor eclitasertib (SAR443122) in patients with severe COVID-19: a phase 1b, randomized, double-blinded, placebo-controlled study. Respir. Res. 25, 107. doi:10.1186/s12931-024-02670-z
Codolo, G., Plotegher, N., Pozzobon, T., Brucale, M., Tessari, I., Bubacco, L., et al. (2013). Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. Plos One 8 (1), e55375. doi:10.1371/journal.pone.0055375
Compan, V., Martín-Sánchez, F., Baroja-Mazo, A., López-Castejón, G., Gomez, A. I., Verkhratsky, A., et al. (2015). Apoptosis-associated speck-like protein containing a CARD forms specks but does not activate caspase-1 in the absence of NLRP3 during macrophage swelling. J. Immunol. 194 (3), 1261–1273. doi:10.4049/jimmunol.1301676
Conos, S. A., Chen, K. W., De Nardo, D., Hara, H., Whitehead, L., Núñez, G., et al. (2017). Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. U. S. A. 114 (6), E961–E969. doi:10.1073/pnas.1613305114
Cruz, S. A., Qin, Z., Stewart, A. F. R., and Chen, H. H. (2018). Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury. Neural Regen. Res. 13 (2), 252–256. doi:10.4103/1673-5374.226394
Dai, W., Wang, X., Teng, H., Li, C., Wang, B., and Wang, J. (2019). Celastrol inhibits microglial pyroptosis and attenuates inflammatory reaction in acute spinal cord injury rats. Int. Immunopharmacol. 66, 215–223. doi:10.1016/j.intimp.2018.11.029
De Michele, G., Galatolo, D., Salvatore, E., Cocozza, S., Barghigiani, M., et al. (2019). Spinocerebellar ataxia 48 presenting with ataxia associated with cognitive, psychiatric, and extrapyramidal features: a report of two Italian families. Park. Relat. Disord. 65, 91–96. doi:10.1016/j.parkreldis.2019.05.001
Degterev, A., Hitomi, J., Germscheid, M., Ch’en, I. L., Korkina, O., Teng, X., et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4 (5), 313–321. doi:10.1038/nchembio.83
Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., et al. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1 (2), 112–119. doi:10.1038/nchembio711
Delanghe, T., Dondelinger, Y., and Bertrand, M. J. M. (2020). RIPK1 kinase-dependent death: a symphony of phosphorylation events. Trends Cell Biol. 30 (3), 189–200. doi:10.1016/j.tcb.2019.12.009
Dermentzaki, G., Politi, K. A., Lu, L., Mishra, V., Pérez-Torres, E. J., Sosunov, A. A., et al. (2019). Deletion of ripk3 prevents motor neuron death in vitro but not in vivo. eNeuro 6 (1), 0308. doi:10.1523/ENEURO.0308-18.2018
Dhuriya, Y. K., and Sharma, D. (2018). Necroptosis: a regulated inflammatory mode of cell death. J. Neuroinflammation 15 (1), 199. doi:10.1186/s12974-018-1235-0
Dominguez, S., Varfolomeev, E., Brendza, R., Stark, K., Tea, J., Imperio, J., et al. (2021). Genetic inactivation of RIP1 kinase does not ameliorate disease in a mouse model of ALS. Cell Death Differ. 28 (3), 915–931. doi:10.1038/s41418-020-00625-7
Dondelinger, Y., Aguileta, M. A., Goossens, V., Dubuisson, C., Grootjans, S., Dejardin, E., et al. (2013). RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 20 (10), 1381–1392. doi:10.1038/cdd.2013.94
Dondelinger, Y., Declercq, W., Montessuit, S., Roelandt, R., Goncalves, A., Bruggeman, I., et al. (2014). MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7 (4), 971–981. doi:10.1016/j.celrep.2014.04.026
Dong, Y., Yu, H., Li, X., Bian, K., Zheng, Y., Dai, M., et al. (2022). Hyperphosphorylated tau mediates neuronal death by inducing necroptosis and inflammation in Alzheimer’s disease. J. Neuroinflammation 19 (1), 205. doi:10.1186/s12974-022-02567-y
Edamakanti, C. R., Mohan, V., and Opal, P. (2023). Reactive Bergmann glia play a central role in spinocerebellar ataxia inflammation via the JNK pathway. J. Neuroinflammation 20 (1), 126. doi:10.1186/s12974-023-02801-1
Fauster, A., Rebsamen, M., Huber, K. V. M., Bigenzahn, J. W., Stukalov, A., Lardeau, C. H., et al. (2015). A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell death Dis. 6 (5), e1767. doi:10.1038/cddis.2015.130
Feldmann, F., Schenk, B., Martens, S., Vandenabeele, P., and Fulda, S. (2017). Sorafenib inhibits therapeutic induction of necroptosis in acute leukemia cells. Oncotarget 8 (40), 68208–68220. doi:10.18632/oncotarget.19919
Ferro, A., Sheeler, C., Rosa, J. G., and Cvetanovic, M. (2019). Role of microglia in ataxias. J. Mol. Biol. 431 (9), 1792–1804. doi:10.1016/j.jmb.2019.01.016
Fu, W., Li, H., Fu, H., Zhao, S., Shi, W., Sun, M., et al. (2020). The SIRT3 and SIRT6 promote prostate cancer progression by inhibiting necroptosis-mediated innate immune response. J. Immunol. Res. 2020, 8820355. doi:10.1155/2020/8820355
Galluzzi, L., Maiuri, M. C., Vitale, I., Zischka, H., Castedo, M., Zitvogel, L., et al. (2007). Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 14 (7), 1237–1243. doi:10.1038/sj.cdd.4402148
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25 (3), 486–541. doi:10.1038/s41418-017-0012-4
Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., et al. (2012). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19 (1), 107–120. doi:10.1038/cdd.2011.96
Gao, Q., Qin, H., Zhu, L., Li, D., and Hao, X. (2020). Celastrol attenuates collagen-induced arthritis via inhibiting oxidative stress in rats. Int. Immunopharmacol. 84, 106527. doi:10.1016/j.intimp.2020.106527
Garcia-Venzor, A., and Toiber, D. (2021). SIRT6 through the brain evolution, development, and aging. Front. aging Neurosci. 13, 747989. doi:10.3389/fnagi.2021.747989
Gardner, C. R., Davies, K. A., Zhang, Y., Brzozowski, M., Czabotar, P. E., Murphy, J. M., et al. (2023). From (tool)bench to bedside: the potential of necroptosis inhibitors. J. Med. Chem. 66 (4), 2361–2385. doi:10.1021/acs.jmedchem.2c01621
Geng, F., Yin, H., Li, Z., Li, Q., Wang, Z., and Yu, J. (2017). Quantitative analysis of necrostatin-1, a necroptosis inhibitor by LC-MS/MS and the study of its pharmacokinetics and bioavailability. Biomed. Pharmacother. 95, 1479–1485. doi:10.1016/j.biopha.2017.09.063
Genis, D., Ortega-Cubero, S., San Nicolás, H., Corral, J., Gardenyes, J., de Jorge, L., et al. (2018). Heterozygous STUB1 mutation causes familial ataxia with cognitive affective syndrome (SCA48). Neurology 91 (21), e1998. doi:10.1212/WNL.0000000000006550
Geserick, P., Hupe, M., Moulin, M., Wong, W. W. L., Feoktistova, M., Kellert, B., et al. (2009). Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 187 (7), 1037–1054. doi:10.1083/jcb.200904158
Greco, V., Spalloni, A., Corasolla Carregari, V., Pieroni, L., Persichilli, S., Mercuri, N. B., et al. (2018). Proteomics and toxicity analysis of spinal-cord primary cultures upon hydrogen sulfide treatment. Antioxidants (Basel) 7 (7), 87. doi:10.3390/antiox7070087
Green, D. R., Galluzzi, L., and Kroemer, G. (2014). Cell biology. Metabolic control of cell death. Science 345 (6203), 1250256. doi:10.1126/science.1250256
Green, D. R., and Llambi, F. (2015). Cell death signaling. Cold Spring Harb. Perspect. Biol. 7 (12), a006080. doi:10.1101/cshperspect.a006080
Grievink, H. W., Heuberger, J. A. A. C., Huang, F., Chaudhary, R., Birkhoff, W. A. J., Tonn, G. R., et al. (2020). DNL104, a centrally penetrant RIPK1 inhibitor, inhibits RIP1 kinase phosphorylation in a randomized phase I ascending dose study in healthy volunteers. Clin. Pharmacol. Ther. 107 (2), 406–414. doi:10.1002/cpt.1615
Guo, C., Fu, R., Zhou, M., Wang, S., Huang, Y., Hu, H., et al. (2019). Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J. Autoimmun. 103, 102286. doi:10.1016/j.jaut.2019.05.014
Guo, R., Chen, P., Fu, T., Zhang, R., Zhu, Y., Jin, N., et al. (2022). Activation of delta-opioid receptor protects ARPE19 cells against oxygen-glucose deprivation/reoxygenation-induced necroptosis and apoptosis by inhibiting the release of TNF-α. J. Ophthalmol. 2022, 2285663. doi:10.1155/2022/2285663
Gupta, S., Kim, C., Yel, L., and Gollapudi, S. (2004). A role of fas-associated death domain (FADD) in increased apoptosis in aged humans. J. Clin. Immunol. 24 (1), 24–29. doi:10.1023/B:JOCI.0000018059.56924.99
Gustot, A., Gallea, J. I., Sarroukh, R., Celej, M. S., Ruysschaert, J. M., and Raussens, V. (2015). Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem. J. 471 (3), 323–333. doi:10.1042/BJ20150617
Hanson, B. (2016). Necroptosis: a new way of dying? Cancer Biol. Ther. 17 (9), 899–910. doi:10.1080/15384047.2016.1210732
He, S., Liang, Y., Shao, F., and Wang, X. (2011). Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. U. S. A. 108 (50), 20054–20059. doi:10.1073/pnas.1116302108
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1 (6), 489–495. doi:10.1038/82732
Huang, F., Liang, J., Lin, Y., Chen, Y., Hu, F., Feng, J., et al. (2023). Repurposing of Ibrutinib and Quizartinib as potent inhibitors of necroptosis. Commun. Biol. 6 (1), 972. doi:10.1038/s42003-023-05353-5
Huang, F., Zhu, Y., Hsiao-Nakamoto, J., Tang, X., Dugas, J. C., Moscovitch-Lopatin, M., et al. (2020). Longitudinal biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. neurology 7 (7), 1103–1116. doi:10.1002/acn3.51078
Huang, X., Tan, S., Li, Y., Cao, S., Li, X., Pan, H., et al. (2021). Caspase inhibition prolongs inflammation by promoting a signaling complex with activated RIPK1. J. Cell Biol. 220 (6), e202007127. doi:10.1083/jcb.202007127
Iannielli, A., Bido, S., Folladori, L., Segnali, A., Cancellieri, C., Maresca, A., et al. (2018). Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep. 22 (8), 2066–2079. doi:10.1016/j.celrep.2018.01.089
Ito, Y., Ofengeim, D., Najafov, A., Das, S., Saberi, S., Li, Y., et al. (2016). RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353 (6299), 603–608. doi:10.1126/science.aaf6803
Jacobsen, A. V., Lowes, K. N., Tanzer, M. C., Lucet, I. S., Hildebrand, J. M., Petrie, E. J., et al. (2016). HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell death Dis. 7 (1), e2051. doi:10.1038/cddis.2015.386
Jayaraman, A., Htike, T. T., James, R., Picon, C., and Reynolds, R. (2021). TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocampus. Acta neuropathol. Commun. 9 (1), 159. doi:10.1186/s40478-021-01264-w
Jiao, J., Wang, Y., Ren, P., Sun, S., and Wu, M. (2019). Necrosulfonamide ameliorates neurological impairment in spinal cord injury by improving antioxidative capacity. Front. Pharmacol. 10, 1538. doi:10.3389/fphar.2019.01538
Jing, M., Yang, J., Zhang, L., Liu, J., Xu, S., Wang, M., et al. (2021). Celastrol inhibits rheumatoid arthritis through the ROS-NF-κB-NLRP3 inflammasome axis. Int. Immunopharmacol. 98, 107879. doi:10.1016/j.intimp.2021.107879
Jouan-Lanhouet, S., Arshad, M. I., Piquet-Pellorce, C., Martin-Chouly, C., Le Moigne-Muller, G., Van Herreweghe, F., et al. (2012). TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 19 (12), 2003–2014. doi:10.1038/cdd.2012.90
Kaczmarek, A., Vandenabeele, P., and Krysko, D. V. (2013). Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38 (2), 209–223. doi:10.1016/j.immuni.2013.02.003
Kaiser, W. J., Sridharan, H., Huang, C., Mandal, P., Upton, J. W., Gough, P. J., et al. (2013). Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288 (43), 31268–31279. doi:10.1074/jbc.M113.462341
Kaluski, S., Portillo, M., Besnard, A., Stein, D., Einav, M., Zhong, L., et al. (2017). Neuroprotective functions for the histone deacetylase SIRT6. Cell Rep. 18 (13), 3052–3062. doi:10.1016/j.celrep.2017.03.008
Kang, T.-B., Yang, S. H., Toth, B., Kovalenko, A., and Wallach, D. (2014). Activation of the NLRP3 inflammasome by proteins that signal for necroptosis. Methods Enzym. 545, 67–81. doi:10.1016/B978-0-12-801430-1.00003-2
Kawahara, T. L. A., Michishita, E., Adler, A. S., Damian, M., Berber, E., Lin, M., et al. (2009). SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136 (1), 62–74. doi:10.1016/j.cell.2008.10.052
Kenna, K. P., van Doormaal, P. T. C., Dekker, A. M., Ticozzi, N., Kenna, B. J., Diekstra, F. P., et al. (2016). NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 48 (9), 1037–1042. doi:10.1038/ng.3626
Khoury, M. K., Gupta, K., Franco, S. R., and Liu, B. (2020). Necroptosis in the pathophysiology of disease. Am. J. Pathology 190 (2), 272–285. doi:10.1016/j.ajpath.2019.10.012
Kim, D.-Y., Leem, Y. H., Park, J. S., Park, J. E., Park, J. M., Kang, J. L., et al. (2023). RIPK1 regulates microglial activation in lipopolysaccharide-induced neuroinflammation and MPTP-induced Parkinson’s disease mouse models. Cells 12 (3), 417. doi:10.3390/cells12030417
Kim, H.-J., Hwang, K. E., Park, D. S., Oh, S. H., Jun, H. Y., Yoon, K. H., et al. (2017). Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J. Transl. Med. 15 (1), 123. doi:10.1186/s12967-017-1223-7
Koo, J.-H., Jang, H. Y., Lee, Y., Moon, Y. J., Bae, E. J., Yun, S. K., et al. (2019). Myeloid cell-specific sirtuin 6 deficiency delays wound healing in mice by modulating inflammation and macrophage phenotypes. Exp. Mol. Med. 51 (4), 1–10. doi:10.1038/s12276-019-0248-9
Koper, M. J., Tomé, S. O., Gawor, K., Belet, A., Van Schoor, E., Schaeverbeke, J., et al. (2022). LATE-NC aggravates GVD-mediated necroptosis in Alzheimer’s disease. Acta neuropathol. Commun. 10 (1), 128. doi:10.1186/s40478-022-01432-6
Koper, M. J., Van Schoor, E., Ospitalieri, S., Vandenberghe, R., Vandenbulcke, M., von Arnim, C. A. F., et al. (2020). Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathol. 139 (3), 463–484. doi:10.1007/s00401-019-02103-y
Kroemer, G., El-Deiry, W. S., Golstein, P., Peter, M. E., Vaux, D., Vandenabeele, P., et al. (2005). Classification of cell death: recommendations of the nomenclature committee on cell death. Cell Death Differ. 12 (Suppl. 2), 1463–1467. doi:10.1038/sj.cdd.4401724
Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., et al. (2009). Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 16 (1), 3–11. doi:10.1038/cdd.2008.150
Krzyzowska, M., Kowalczyk, A., Skulska, K., Thörn, K., and Eriksson, K. (2021). Fas/FasL contributes to HSV-1 brain infection and neuroinflammation. Front. Immunol. 12, 714821. doi:10.3389/fimmu.2021.714821
Kuang, A. A., Diehl, G. E., Zhang, J., and Winoto, A. (2000). FADD is required for DR4-and DR5-mediated apoptosis: lack of trail-induced apoptosis in FADD-deficient mouse embryonic fibroblasts. J. Biol. Chem. 275 (33), 25065–25068. doi:10.1074/jbc.C000284200
Kumar, V. (2019). Toll-like receptors in the pathogenesis of neuroinflammation. J. Neuroimmunol. 332, 16–30. doi:10.1016/j.jneuroim.2019.03.012
Lafont, E., Kantari-Mimoun, C., Draber, P., De Miguel, D., Hartwig, T., Reichert, M., et al. (2017). The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 36 (9), 1147–1166. doi:10.15252/embj.201695699
Laukens, B., Jennewein, C., Schenk, B., Vanlangenakker, N., Schier, A., Cristofanon, S., et al. (2011). Smac mimetic bypasses apoptosis resistance in FADD- or caspase-8-deficient cells by priming for tumor necrosis factor α-induced necroptosis. Neoplasia 13 (10), 971–979. doi:10.1593/neo.11610
Lawlor, K. E., Khan, N., Mildenhall, A., Gerlic, M., Croker, B. A., D’Cruz, A. A., et al. (2015). RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282. doi:10.1038/ncomms7282
Lawrence, C. P., and Chow, S. C. (2005). FADD deficiency sensitises Jurkat T cells to TNF-alpha-dependent necrosis during activation-induced cell death. FEBS Lett. 579 (28), 6465–6472. doi:10.1016/j.febslet.2005.10.041
Lee, Y., Ka, S. O., Cha, H. N., Chae, Y. N., Kim, M. K., Park, S. Y., et al. (2017). Myeloid sirtuin 6 deficiency causes insulin resistance in high-fat diet-fed mice by eliciting macrophage polarization toward an M1 phenotype. Diabetes 66 (10), 2659–2668. doi:10.2337/db16-1446
Lei, C., Zhongyan, Z., Wenting, S., Jing, Z., Liyun, Q., Hongyi, H., et al. (2023). Identification of necroptosis-related genes in Parkinson’s disease by integrated bioinformatics analysis and experimental validation. Front. Neurosci. 17, 1097293. doi:10.3389/fnins.2023.1097293
Li, J. X., Feng, J. M., Wang, Y., Li, X. H., Chen, X. X., Su, Y., et al. (2014). The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell death Dis. 5 (6), e1278. doi:10.1038/cddis.2014.241
Li, S., Qu, L., Wang, X., and Kong, L. (2022). Novel insights into RIPK1 as a promising target for future Alzheimer’s disease treatment. Pharmacol. Ther. 231, 107979. doi:10.1016/j.pharmthera.2021.107979
Liang, Q.-Q., Shi, Z. J., Yuan, T., Chen, S. Y., Li, Y. P., Zhang, H. R., et al. (2023). Celastrol inhibits necroptosis by attenuating the RIPK1/RIPK3/MLKL pathway and confers protection against acute pancreatitis in mice. Int. Immunopharmacol. 117, 109974. doi:10.1016/j.intimp.2023.109974
Liang, W. S., Dunckley, T., Beach, T. G., Grover, A., Mastroeni, D., Ramsey, K., et al. (2008). Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiol. Genomics 33 (2), 240–256. doi:10.1152/physiolgenomics.00242.2007
Lim, J., Park, H., Heisler, J., Maculins, T., Roose-Girma, M., Xu, M., et al. (2019). Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. eLife 8, e44452. doi:10.7554/eLife.44452
Lin, M.-M., Liu, N., Qin, Z. H., and Wang, Y. (2022). Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol. Sin. 43 (10), 2439–2447. doi:10.1038/s41401-022-00879-6
Lin, Q.-S., Chen, P., Wang, W. X., Lin, C. C., Zhou, Y., Yu, L. H., et al. (2020). RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson disease. Lab. Investig. 100 (3), 503–511. doi:10.1038/s41374-019-0319-5
Linnerbauer, M., Wheeler, M. A., and Quintana, F. J. (2020). Astrocyte crosstalk in CNS inflammation. Neuron 108 (4), 608–622. doi:10.1016/j.neuron.2020.08.012
Liu, X., Xie, X., Ren, Y., Shao, Z., Zhang, N., Li, L., et al. (2021). The role of necroptosis in disease and treatment. MedComm 2 (4), 730–755. doi:10.1002/mco2.108
Lu, B., Gong, X., Wang, Z. Q., Ding, Y., Wang, C., Luo, T. F., et al. (2017). Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta Pharmacol. Sin. 38 (11), 1543–1553. doi:10.1038/aps.2017.112
MacDonald, M. E. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72 (6), 971–983. doi:10.1016/0092-8674(93)90585-e
Madrigal, S. C., McNeil, Z., Sanchez-Hodge, R., Shi, C. H., Patterson, C., Scaglione, K. M., et al. (2019). Changes in protein function underlie the disease spectrum in patients with CHIP mutations. J. Biol. Chem. 294 (50), 19236–19245. doi:10.1074/jbc.RA119.011173
Mandal, P., Berger, S. B., Pillay, S., Moriwaki, K., Huang, C., Guo, H., et al. (2014). RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56 (4), 481–495. doi:10.1016/j.molcel.2014.10.021
Mao, Z., Hine, C., Tian, X., Van Meter, M., Au, M., Vaidya, A., et al. (2011). SIRT6 promotes DNA repair under stress by activating PARP1. Science 332 (6036), 1443–1446. doi:10.1126/science.1202723
Martens, S., Jeong, M., Tonnus, W., Feldmann, F., Hofmans, S., Goossens, V., et al. (2017). Sorafenib tosylate inhibits directly necrosome complex formation and protects in mouse models of inflammation and tissue injury. Cell death Dis. 8 (6), e2904. doi:10.1038/cddis.2017.298
Marunouchi, T., Nishiumi, C., Iinuma, S., Yano, E., and Tanonaka, K. (2021). Effects of Hsp90 inhibitor on the RIP1-RIP3-MLKL pathway during the development of heart failure in mice. Eur. J. Pharmacol. 898, 173987. doi:10.1016/j.ejphar.2021.173987
McCauley, M. E., and Baloh, R. H. (2019). Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 137 (5), 715–730. doi:10.1007/s00401-018-1933-9
McComb, S., Cheung, H. H., Korneluk, R. G., Wang, S., Krishnan, L., and Sad, S. (2012). cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. 19 (11), 1791–1801. doi:10.1038/cdd.2012.59
McGeer, P. L., and McGeer, E. G. (2002). Inflammatory processes in amyotrophic lateral sclerosis. Muscle and Nerve 26 (4), 459–470. doi:10.1002/mus.10191
Meng, H., Wu, G., Zhao, X., Wang, A., Li, D., Tong, Y., et al. (2021). Discovery of a cooperative mode of inhibiting RIPK1 kinase. Cell Discov. 7 (1), 41. doi:10.1038/s41421-021-00278-x
Menzies, A. M., Long, G. V., and Murali, R. (2012). Dabrafenib and its potential for the treatment of metastatic melanoma. Drug Des. Dev. Ther. 6, 391–405. doi:10.2147/DDDT.S38998
Meroni, E., Stakenborg, N., Gomez-Pinilla, P. J., De Hertogh, G., Goverse, G., Matteoli, G., et al. (2018). Functional characterization of oxazolone-induced colitis and survival improvement by vagus nerve stimulation. Plos One 13 (5), e0197487. doi:10.1371/journal.pone.0197487
Mifflin, L., Hu, Z., Dufort, C., Hession, C. C., Walker, A. J., Niu, K., et al. (2021). A RIPK1-regulated inflammatory microglial state in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 118 (13), e2025102118. doi:10.1073/pnas.2025102118
Mifflin, L., Ofengeim, D., and Yuan, J. (2020). Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 19 (8), 553–571. doi:10.1038/s41573-020-0071-y
Miller, S. J. (2018). Astrocyte heterogeneity in the adult central nervous system. Front. Cell. Neurosci. 12, 401. doi:10.3389/fncel.2018.00401
Min, J.-N., Whaley, R. A., Sharpless, N. E., Lockyer, P., Portbury, A. L., and Patterson, C. (2008). CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol. Cell. Biol. 28 (12), 4018–4025. doi:10.1128/MCB.00296-08
Mitroshina, E. V., Saviuk, M., and Vedunova, M. V. (2022). Necroptosis in CNS diseases: focus on astrocytes. Front. aging Neurosci. 14, 1016053. doi:10.3389/fnagi.2022.1016053
Morgan, M. J., and Kim, Y.-S. (2022). Roles of RIPK3 in necroptosis, cell signaling, and disease. Exp. Mol. Med. 54 (10), 1695–1704. doi:10.1038/s12276-022-00868-z
Morgan, M. J., and Liu, Z.-G. (2013). Programmed cell death with a necrotic-like phenotype. Biomol. concepts 4 (3), 259–275. doi:10.1515/bmc-2012-0056
Moriwaki, K., Balaji, S., McQuade, T., Malhotra, N., Kang, J., and Chan, F. K. M. (2014). The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity 41 (4), 567–578. doi:10.1016/j.immuni.2014.09.016
Moriwaki, K., and Chan, F.K.-M. (2013). RIP3: a molecular switch for necrosis and inflammation. Genes and Dev. 27 (15), 1640–1649. doi:10.1101/gad.223321.113
Motawi, T. M. K., Abdel-Nasser, Z. M., and Shahin, N. N. (2020). Ameliorative effect of necrosulfonamide in a rat model of Alzheimer’s disease: targeting mixed lineage kinase domain-like protein-mediated necroptosis. ACS Chem. Neurosci. 11 (20), 3386–3397. doi:10.1021/acschemneuro.0c00516
Muller, A. J., DuHadaway, J. B., Donover, P. S., Sutanto-Ward, E., and Prendergast, G. C. (2005). Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 11 (3), 312–319. doi:10.1038/nm1196
Murphy, J. M., Czabotar, P. E., Hildebrand, J. M., Lucet, I. S., Zhang, J. G., Alvarez-Diaz, S., et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39 (3), 443–453. doi:10.1016/j.immuni.2013.06.018
Nahacka, Z., Svadlenka, J., Peterka, M., Ksandrova, M., Benesova, S., Neuzil, J., et al. (2018). TRAIL induces apoptosis but not necroptosis in colorectal and pancreatic cancer cells preferentially via the TRAIL-R2/DR5 receptor. Mol. cell Res. 1865 (3), 522–531. doi:10.1016/j.bbamcr.2017.12.006
Ni, A., and Ernst, C. (2022). Evidence that substantia nigra pars compacta dopaminergic neurons are selectively vulnerable to oxidative stress because they are highly metabolically active. Front. Cell. Neurosci. 16, 826193. doi:10.3389/fncel.2022.826193
Ofengeim, D., Ito, Y., Najafov, A., Zhang, Y., Shan, B., DeWitt, J. P., et al. (2015). Activation of necroptosis in multiple sclerosis. Cell Rep. 10 (11), 1836–1849. doi:10.1016/j.celrep.2015.02.051
Ofengeim, D., Mazzitelli, S., Ito, Y., DeWitt, J. P., Mifflin, L., Zou, C., et al. (2017). RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 114 (41), E8788–E8797. doi:10.1073/pnas.1714175114
Ofengeim, D., and Yuan, J. (2013). Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 14 (11), 727–736. doi:10.1038/nrm3683
Oliveira, S. R., Dionísio, P. A., Brito, H., Franco, L., Rodrigues, C. A. B., Guedes, R. C., et al. (2018). Phenotypic screening identifies a new oxazolone inhibitor of necroptosis and neuroinflammation. Cell death Discov. 4, 10. doi:10.1038/s41420-018-0067-0
Oñate, M., Catenaccio, A., Salvadores, N., Saquel, C., Martinez, A., Moreno-Gonzalez, I., et al. (2020). The necroptosis machinery mediates axonal degeneration in a model of Parkinson disease. Cell Death Differ. 27 (4), 1169–1185. doi:10.1038/s41418-019-0408-4
Oppenheim, J. J., and Yang, D. (2005). Alarmins: chemotactic activators of immune responses. Curr. Opin. Immunol. 17 (4), 359–365. doi:10.1016/j.coi.2005.06.002
Orozco, S. L., Daniels, B. P., Yatim, N., Messmer, M. N., Quarato, G., Chen-Harris, H., et al. (2019). RIPK3 activation leads to cytokine synthesis that continues after loss of cell membrane integrity. Cell Rep. 28 (9), 2275–2287. doi:10.1016/j.celrep.2019.07.077
Palubinsky, A. M., Stankowski, J. N., Kale, A. C., Codreanu, S. G., Singer, R. J., Liebler, D. C., et al. (2015). CHIP is an essential determinant of neuronal mitochondrial stress signaling. Antioxidants Redox Signal. 23 (6), 535–549. doi:10.1089/ars.2014.6102
Park, M. H., and Hong, J. T. (2016). Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 5 (2), 15. doi:10.3390/cells5020015
Petrie, E. J., Hildebrand, J. M., and Murphy, J. M. (2017). Insane in the membrane: a structural perspective of MLKL function in necroptosis. Immunol. Cell Biol. 95 (2), 152–159. doi:10.1038/icb.2016.125
Picon, C., Jayaraman, A., James, R., Beck, C., Gallego, P., Witte, M. E., et al. (2021). Neuron-specific activation of necroptosis signaling in multiple sclerosis cortical grey matter. Acta Neuropathol. 141 (4), 585–604. doi:10.1007/s00401-021-02274-7
Politi, K., and Przedborski, S. (2016). Axonal degeneration: RIPK1 multitasking in ALS. Curr. Biol. 26 (20), R932–R934. doi:10.1016/j.cub.2016.08.052
Qin, L., Zou, J., Barnett, A., Vetreno, R. P., Crews, F. T., and Coleman, L. G. (2021). TRAIL mediates neuronal death in aud: a link between neuroinflammation and neurodegeneration. Int. J. Mol. Sci. 22 (5), 2547. doi:10.3390/ijms22052547
Ransohoff, R. M., and Brown, M. A. (2012). Innate immunity in the central nervous system. J. Clin. Investigation 122 (4), 1164–1171. doi:10.1172/JCI58644
Rathkey, J. K., Zhao, J., Liu, Z., Chen, Y., Yang, J., Kondolf, H. C., et al. (2018). Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 3 (26), eaat2738. doi:10.1126/sciimmunol.aat2738
Re, D. B., Le Verche, V., Yu, C., Amoroso, M. W., Politi, K. A., Phani, S., et al. (2014). Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81 (5), 1001–1008. doi:10.1016/j.neuron.2014.01.011
Rebsamen, M., Meylan, E., Curran, J., and Tschopp, J. (2008). The antiviral adaptor proteins Cardif and Trif are processed and inactivated by caspases. Cell Death Differ. 15 (11), 1804–1811. doi:10.1038/cdd.2008.119
Rezazadeh, S., Yang, D., Tombline, G., Simon, M., Regan, S. P., Seluanov, A., et al. (2019). SIRT6 promotes transcription of a subset of NRF2 targets by mono-ADP-ribosylating BAF170. Nucleic Acids Res. 47 (15), 7914–7928. doi:10.1093/nar/gkz528
Riebeling, T., Kunzendorf, U., and Krautwald, S. (2022). The role of RHIM in necroptosis. Biochem. Soc. Trans. 50 (4), 1197–1205. doi:10.1042/BST20220535
Ronnebaum, S. M., Patterson, C., and Schisler, J. C. (2014). Emerging evidence of coding mutations in the ubiquitin-proteasome system associated with cerebellar ataxias. Hum. Genome Var. 1, 14018. doi:10.1038/hgv.2014.18
Rosser, M. F. N., Washburn, E., Muchowski, P. J., Patterson, C., and Cyr, D. M. (2007). Chaperone functions of the E3 ubiquitin ligase CHIP. J. Biol. Chem. 282 (31), 22267–22277. doi:10.1074/jbc.M700513200
Royce, G. H., Brown-Borg, H. M., and Deepa, S. S. (2019). The potential role of necroptosis in inflammaging and aging. GeroScience 41 (6), 795–811. doi:10.1007/s11357-019-00131-w
Rübbelke, M., Fiegen, D., Bauer, M., Binder, F., Hamilton, J., King, J., et al. (2020). Locking mixed-lineage kinase domain-like protein in its auto-inhibited state prevents necroptosis. Proc. Natl. Acad. Sci. U. S. A. 117 (52), 33272–33281. doi:10.1073/pnas.2017406117
Rubinsztein, D. C. (2017). RIPK1 promotes inflammation and β-amyloid accumulation in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 114 (41), 10813–10814. doi:10.1073/pnas.1715241114
Saba, J., Couselo, F. L., Bruno, J., Carniglia, L., Durand, D., Lasaga, M., et al. (2022). Neuroinflammation in huntington’s disease: a starring role for astrocyte and microglia. Curr. Neuropharmacol. 20 (6), 1116–1143. doi:10.2174/1570159X19666211201094608
Saccon, R. A., Bunton-Stasyshyn, R. K. A., Fisher, E. M. C., and Fratta, P. (2013). Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain A J. Neurology 136 (Pt 8), 2342–2358. doi:10.1093/brain/awt097
Saeidikhoo, S., Ezi, S., Khatmi, A., Aghajanpour, F., Soltani, R., Abdollahifar, M. A., et al. (2020). Effect of Sertoli cell transplantation on reducing neuroinflammation-induced necroptosis and improving motor coordination in the rat model of cerebellar ataxia induced by 3-acetylpyridine. J. Mol. Neurosci. 70 (7), 1153–1163. doi:10.1007/s12031-020-01522-x
Salvadores, N., Moreno-Gonzalez, I., Gamez, N., Quiroz, G., Vegas-Gomez, L., Escandón, M., et al. (2022). Aβ oligomers trigger necroptosis-mediated neurodegeneration via microglia activation in Alzheimer’s disease. Acta neuropathol. Commun. 10 (1), 31. doi:10.1186/s40478-022-01332-9
Sameer, A. S., and Nissar, S. (2021). Toll-like receptors (TLRs): structure, functions, signaling, and role of their polymorphisms in colorectal cancer susceptibility. BioMed Res. Int. 2021, 1157023. doi:10.1155/2021/1157023
Scheltens, P., De Strooper, B., Kivipelto, M., Holstege, H., Chételat, G., Teunissen, C. E., et al. (2021). Alzheimer’s disease. Lancet 397 (10284), 1577–1590. doi:10.1016/S0140-6736(20)32205-4
Schisler, J. C., Rubel, C. E., Zhang, C., Lockyer, P., Cyr, D. M., and Patterson, C. (2013). CHIP protects against cardiac pressure overload through regulation of AMPK. J. Clin. Investigation 123 (8), 3588–3599. doi:10.1172/JCI69080
Schuster, S., Schelling, Y., Synofzik, M., Höflinger, P., Schöls, L., and Hauser, S. (2018). Establishment of STUB1/CHIP mutant induced pluripotent stem cells (iPSCs) from a patient with Gordon Holmes syndrome/SCAR16. Stem Cell Res. 29, 166–169. doi:10.1016/j.scr.2018.04.001
Schweichel, J. U., and Merker, H. J. (1973). The morphology of various types of cell death in prenatal tissues. Teratology 7 (3), 253–266. doi:10.1002/tera.1420070306
Sebastián, C., Satterstrom, F. K., Haigis, M. C., and Mostoslavsky, R. (2012). From sirtuin biology to human diseases: an update. J. Biol. Chem. 287 (51), 42444–42452. doi:10.1074/jbc.R112.402768
Seo, J., Lee, E. W., Sung, H., Seong, D., Dondelinger, Y., Shin, J., et al. (2016). CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 18 (3), 291–302. doi:10.1038/ncb3314
Shan, B., Pan, H., Najafov, A., and Yuan, J. (2018). Necroptosis in development and diseases. Genes and Dev. 32 (5–6), 327–340. doi:10.1101/gad.312561.118
Sharief, M. K., and Hentges, R. (1991). Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N. Engl. J. Med. 325 (7), 467–472. doi:10.1056/NEJM199108153250704
Sharp, A. H., Loev, S. J., Schilling, G., Li, S. H., Li, X. J., Bao, J., et al. (1995). Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron 14 (5), 1065–1074. doi:10.1016/0896-6273(95)90345-3
Shi, C.-H., Rubel, C., Soss, S. E., Sanchez-Hodge, R., Zhang, S., Madrigal, S. C., et al. (2018). Disrupted structure and aberrant function of CHIP mediates the loss of motor and cognitive function in preclinical models of SCAR16. PLoS Genet. 14 (9), e1007664. doi:10.1371/journal.pgen.1007664
Shi, C.-H., Schisler, J. C., Rubel, C. E., Tan, S., Song, B., McDonough, H., et al. (2014). Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum. Mol. Genet. 23 (4), 1013–1024. doi:10.1093/hmg/ddt497
Shi, F.-L., Ni, S. T., Luo, S. Q., Hu, B., Xu, R., Liu, S. Y., et al. (2022). Dimethyl fumarate ameliorates autoimmune hepatitis in mice by blocking NLRP3 inflammasome activation. Int. Immunopharmacol. 108, 108867. doi:10.1016/j.intimp.2022.108867
Shi, Z., Wu, D., Yao, J. P., Yao, X., Huang, Z., Li, P., et al. (2016). Protection against oxygen-glucose deprivation/reperfusion injury in cortical neurons by combining omega-3 polyunsaturated acid with lyciumbarbarum polysaccharide. Nutrients 8 (1), 41. doi:10.3390/nu8010041
Shiwaku, H., Yagishita, S., Eishi, Y., and Okazawa, H. (2013). Bergmann glia are reduced in spinocerebellar ataxia type 1. Neuroreport 24 (11), 620–625. doi:10.1097/WNR.0b013e32836347b7
Singh, D. (2022). Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J. Neuroinflammation 19 (1), 206. doi:10.1186/s12974-022-02565-0
Sprick, M. R., Weigand, M. A., Rieser, E., Rauch, C. T., Juo, P., Blenis, J., et al. (2000). FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 12 (6), 599–609. doi:10.1016/S1074-7613(00)80211-3
Stefanis, L. (2012). α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2 (2), a009399. doi:10.1101/cshperspect.a009399
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148 (1–2), 213–227. doi:10.1016/j.cell.2011.11.031
Sun, X., Lee, J., Navas, T., Baldwin, D. T., Stewart, T. A., and Dixit, V. M. (1999). RIP3, a novel apoptosis-inducing kinase. J. Biol. Chem. 274 (24), 16871–16875. doi:10.1074/jbc.274.24.16871
Takahashi, N., Duprez, L., Grootjans, S., Cauwels, A., Nerinckx, W., DuHadaway, J. B., et al. (2012). Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell death Dis. 3 (11), e437. doi:10.1038/cddis.2012.176
Thadathil, N., Nicklas, E. H., Mohammed, S., Lewis, T. L., Richardson, A., and Deepa, S. S. (2021). Necroptosis increases with age in the brain and contributes to age-related neuroinflammation. GeroScience 43 (5), 2345–2361. doi:10.1007/s11357-021-00448-5
Tortelli, R., Zecca, C., Piccininni, M., Benmahamed, S., Dell'Abate, M. T., Barulli, M. R., et al. (2020). Plasma inflammatory cytokines are elevated in ALS. Front. neurology 11, 552295. doi:10.3389/fneur.2020.552295
Toth, R. P., and Atkin, J. D. (2018). Dysfunction of optineurin in amyotrophic lateral sclerosis and glaucoma. Front. Immunol. 9, 1017. doi:10.3389/fimmu.2018.01017
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiology 552 (Pt 2), 335–344. doi:10.1113/jphysiol.2003.049478
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2010). Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7 (4), 302–313. doi:10.1016/j.chom.2010.03.006
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2012). DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11 (3), 290–297. doi:10.1016/j.chom.2012.01.016
Vandenabeele, P., Grootjans, S., Callewaert, N., and Takahashi, N. (2013). Necrostatin-1 blocks both RIPK1 and Ido: consequences for the study of cell death in experimental disease models. Cell Death Differ. 20 (2), 185–187. doi:10.1038/cdd.2012.151
Vanlangenakker, N., Bertrand, M. J. M., Bogaert, P., Vandenabeele, P., and Vanden Berghe, T. (2011). TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell death Dis. 2 (11), e230. doi:10.1038/cddis.2011.111
Vercammen, D., Beyaert, R., Denecker, G., Goossens, V., Van Loo, G., Declercq, W., et al. (1998). Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187 (9), 1477–1485. doi:10.1084/jem.187.9.1477
Vercammen, D., Vandenabeele, P., Beyaert, R., Declercq, W., and Fiers, W. (1997). Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine 9 (11), 801–808. doi:10.1006/cyto.1997.0252
Verzella, D., Pescatore, A., Capece, D., Vecchiotti, D., Ursini, M. V., Franzoso, G., et al. (2020). Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell death Dis. 11 (3), 210. doi:10.1038/s41419-020-2399-y
Vieira, M., Fernandes, J., Carreto, L., Anuncibay-Soto, B., Santos, M., Han, J., et al. (2014). Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol. Dis. 68, 26–36. doi:10.1016/j.nbd.2014.04.002
Vissers, M. F. J. M., Heuberger, J. A. A. C., Groeneveld, G. J., Oude Nijhuis, J., De Deyn, P. P., Hadi, S., et al. (2022). Safety, pharmacokinetics and target engagement of novel RIPK1 inhibitor SAR443060 (DNL747) for neurodegenerative disorders: randomized, placebo-controlled, double-blind phase I/Ib studies in healthy subjects and patients. Clin. Transl. Sci. 15 (8), 2010–2023. doi:10.1111/cts.13317
Wallach, D., Varfolomeev, E. E., Malinin, N. L., Goltsev, Y. V., Kovalenko, A. V., and Boldin, M. P. (1999). Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 17, 331–367. doi:10.1146/annurev.immunol.17.1.331
Wang, F., Yao, X., Zhang, Y., and Tang, J. (2019). Synthesis, biological function and evaluation of Shikonin in cancer therapy. Fitoterapia 134, 329–339. doi:10.1016/j.fitote.2019.03.005
Wang, H., Qi, W., Zou, C., Xie, Z., Zhang, M., Naito, M. G., et al. (2021). NEK1-mediated retromer trafficking promotes blood-brain barrier integrity by regulating glucose metabolism and RIPK1 activation. Nat. Commun. 12 (1), 4826. doi:10.1038/s41467-021-25157-7
Wang, J., Ho, W. Y., Lim, K., Feng, J., Tucker-Kellogg, G., Nave, K. A., et al. (2018). Cell-autonomous requirement of TDP-43, an ALS/FTD signature protein, for oligodendrocyte survival and myelination. Proc. Natl. Acad. Sci. U. S. A. 115 (46), E10941–E10950. doi:10.1073/pnas.1809821115
Wang, T., Perera, N. D., Chiam, M. D. F., Cuic, B., Wanniarachchillage, N., Tomas, D., et al. (2020). Necroptosis is dispensable for motor neuron degeneration in a mouse model of ALS. Cell Death Differ. 27 (5), 1728–1739. doi:10.1038/s41418-019-0457-8
Weisel, K., Berger, S., Thorn, K., Taylor, P. C., Peterfy, C., Siddall, H., et al. (2021b). A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res. Ther. 23 (1), 85. doi:10.1186/s13075-021-02468-0
Weisel, K., Scott, N., Berger, S., Wang, S., Brown, K., Powell, M., et al. (2021a). A randomised, placebo-controlled study of RIPK1 inhibitor GSK2982772 in patients with active ulcerative colitis. BMJ open Gastroenterol. 8 (1), e000680. doi:10.1136/bmjgast-2021-000680
Wu, J.-R., Wang, J., Zhou, S. K., Yang, L., Yin, J. l., Cao, J. P., et al. (2015). Necrostatin-1 protection of dopaminergic neurons. Neural Regen. Res. 10 (7), 1120–1124. doi:10.4103/1673-5374.160108
Wu, S., Xu, C., Xia, K., Lin, Y., Tian, S., Ma, H., et al. (2021). Ring closure strategy leads to potent RIPK3 inhibitors. Eur. J. Med. Chem. 217, 113327. doi:10.1016/j.ejmech.2021.113327
Xia, K., Zhu, F., Yang, C., Wu, S., Lin, Y., Ma, H., et al. (2020). Discovery of a potent RIPK3 inhibitor for the amelioration of necroptosis-associated inflammatory injury. Front. cell Dev. Biol. 8, 606119. doi:10.3389/fcell.2020.606119
Xie, Q., Wondergem, R., Shen, Y., Cavey, G., Ke, J., Thompson, R., et al. (2011). Benzoquinone ansamycin 17AAG binds to mitochondrial voltage-dependent anion channel and inhibits cell invasion. Proc. Natl. Acad. Sci. U. S. A. 108 (10), 4105–4110. doi:10.1073/pnas.1015181108
Xu, D., Jin, T., Zhu, H., Chen, H., Ofengeim, D., Zou, C., et al. (2018). TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174 (6), 1477–1491. doi:10.1016/j.cell.2018.07.041
Xuan, Y., and Hu, X. (2009). Naturally-occurring shikonin analogues--a class of necroptotic inducers that circumvent cancer drug resistance. Cancer Lett. 274 (2), 233–242. doi:10.1016/j.canlet.2008.09.029
Xue, S., Cao, Z. X., Wang, J. N., Zhao, Q. X., Han, J., Yang, W. J., et al. (2022). Receptor-interacting protein kinase 3 inhibition relieves mechanical allodynia and suppresses NLRP3 inflammasome and NF-κB in a rat model of spinal cord injury. Front. Mol. Neurosci. 15, 861312. doi:10.3389/fnmol.2022.861312
Yan, B., Liu, L., Huang, S., Ren, Y., Wang, H., Yao, Z., et al. (2017). Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem. Commun. 53 (26), 3637–3640. doi:10.1039/c7cc00667e
Yang, J., Zhao, Y., Zhang, L., Fan, H., Qi, C., Zhang, K., et al. (2018). RIPK3/MLKL-Mediated neuronal necroptosis modulates the M1/M2 polarization of microglia/macrophages in the ischemic cortex. Cereb. Cortex 28 (7), 2622–2635. doi:10.1093/cercor/bhy089
Yang, S.-H., Lee, D. K., Shin, J., Lee, S., Baek, S., Kim, J., et al. (2017). Nec-1 alleviates cognitive impairment with reduction of Aβ and tau abnormalities in APP/PS1 mice. EMBO Mol. Med. 9 (1), 61–77. doi:10.15252/emmm.201606566
Yang, Z., Wang, Y., Zhang, Y., He, X., Zhong, C. Q., Ni, H., et al. (2018). RIP3 targets pyruvate dehydrogenase complex to increase aerobic respiration in TNF-induced necroptosis. Nat. Cell Biol. 20 (2), 186–197. doi:10.1038/s41556-017-0022-y
Ye, K., Chen, Z., and Xu, Y. (2023). The double-edged functions of necroptosis. Cell death Dis. 14 (2), 163. doi:10.1038/s41419-023-05691-6
Yeh, W. C., de la Pompa, J. L., McCurrach, M. E., Shu, H. B., Elia, A. J., Shahinian, A., et al. (1998). FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279 (5358), 1954–1958. doi:10.1126/science.279.5358.1954
Yuan, J., Amin, P., and Ofengeim, D. (2019). Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20 (1), 19–33. doi:10.1038/s41583-018-0093-1
Zelic, M., Pontarelli, F., Woodworth, L., Zhu, C., Mahan, A., Ren, Y., et al. (2021). RIPK1 activation mediates neuroinflammation and disease progression in multiple sclerosis. Cell Rep. 35 (6), 109112. doi:10.1016/j.celrep.2021.109112
Zhang, C., Zhao, M., Wang, B., Su, Z., Guo, B., Qin, L., et al. (2021). The Nrf2-NLRP3-caspase-1 axis mediates the neuroprotective effects of Celastrol in Parkinson’s disease. Redox Biol. 47, 102134. doi:10.1016/j.redox.2021.102134
Zhang, D.-W., Shao, J., Lin, J., Zhang, N., Lu, B. J., Lin, S. C., et al. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325 (5938), 332–336. doi:10.1126/science.1172308
Zhang, G., and Ghosh, S. (2001). Toll-like receptor-mediated NF-kappaB activation: a phylogenetically conserved paradigm in innate immunity. J. Clin. Investigation 107 (1), 13–19. doi:10.1172/JCI11837
Zhang, Y., Su, S. S., Zhao, S., Yang, Z., Zhong, C. Q., Chen, X., et al. (2017). RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 8, 14329. doi:10.1038/ncomms14329
Zheng, C., Zhou, X.-W., and Wang, J.-Z. (2016). The dual roles of cytokines in Alzheimer's disease: update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 5, 7. doi:10.1186/s40035-016-0054-4
Zhou, Y., Zhou, B., Tu, H., Tang, Y., Xu, C., Chen, Y., et al. (2017). The degradation of mixed lineage kinase domain-like protein promotes neuroprotection after ischemic brain injury. Oncotarget 8 (40), 68393–68401. doi:10.18632/oncotarget.19416
Zhu, K., Liang, W., Ma, Z., Xu, D., Cao, S., Lu, X., et al. (2018). Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell death Dis. 9 (5), 500. doi:10.1038/s41419-018-0524-y
Zhu, S., Zhang, Y., Bai, G., and Li, H. (2011). Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington’s disease. Cell death Dis. 2 (1), e115. doi:10.1038/cddis.2010.94
Keywords: necroptosis, neuroinflammation, neurodegeneration, inflammation, cellular death
Citation: Smith K, Colie M, Moore T and Schisler JC (2024) Die hard: necroptosis and its impact on age-dependent neuroinflammatory diseases. Front. Cell. Death 3:1348153. doi: 10.3389/fceld.2024.1348153
Received: 01 December 2023; Accepted: 13 February 2024;
Published: 08 March 2024.
Edited by:
Maria Tanzer, The University of Melbourne, AustraliaReviewed by:
Kezhou Zhu, University of Michigan, United StatesMatija Zelic, Sanofi Genzyme, United States
Copyright © 2024 Smith, Colie, Moore and Schisler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan C. Schisler, schisler@unc.edu
†These authors have contributed equally to this work