Cardiomyocyte Proliferation and Maturation: Two Sides of the Same Coin for Heart Regeneration
 Ming-Tao Zhao
Ming-Tao Zhao Shiqiao Ye
Shiqiao Ye Juan Su
Juan Su Vidu Garg
Vidu Garg- 1Center for Cardiovascular Research, The Abigail Wexner Research Institute, Nationwide Children’s Hospital, Columbus, OH, United States
- 2The Heart Center, Nationwide Children’s Hospital, Columbus, OH, United States
- 3Department of Pediatrics, The Ohio State University College of Medicine, Columbus, OH, United States
- 4Department of Molecular Genetics, The Ohio State University, Columbus, OH, United States
In the past few decades, cardiac regeneration has been the central target for restoring the injured heart. In mammals, cardiomyocytes are terminally differentiated and rarely divide during adulthood. Embryonic and fetal cardiomyocytes undergo robust proliferation to form mature heart chambers in order to accommodate the increased workload of a systemic circulation. In contrast, postnatal cardiomyocytes stop dividing and initiate hypertrophic growth by increasing the size of the cardiomyocyte when exposed to increased workload. Extracellular and intracellular signaling pathways control embryonic cardiomyocyte proliferation and postnatal cardiac hypertrophy. Harnessing these pathways could be the future focus for stimulating endogenous cardiac regeneration in response to various pathological stressors. Meanwhile, patient-specific cardiomyocytes derived from autologous induced pluripotent stem cells (iPSCs) could become the major exogenous sources for replenishing the damaged myocardium. Human iPSC-derived cardiomyocytes (iPSC-CMs) are relatively immature and have the potential to increase the population of cells that advance to physiological hypertrophy in the presence of extracellular stimuli. In this review, we discuss how cardiac proliferation and maturation are regulated during embryonic development and postnatal growth, and explore how patient iPSC-CMs could serve as the future seed cells for cardiac cell replacement therapy.
Introduction
Heart failure has been a leading cause of morbidity and mortality in the world, affecting more than 40 million people. About 2% of adults will have heart failure and the percentage rises significantly for people over the age of 65 (Metra and Teerlink, 2017). The most common cause of heart failure is myocardial infarction (MI, also known as heart attack) which is frequently a consequence of coronary artery disease. During myocardial infarction, the blood flow decreases or stops to supply to a part of the heart, which leads to severe damage to cardiac muscle. Unlike skeletal muscle cells, adult cardiomyocytes are not capable of robust proliferation to regenerate the damaged myocardium. Instead, the damaged heart undergoes extensive remodeling to replenish the dead cardiomyocytes with fibrotic scars and pathological hypertrophy, which significantly impairs the normal cardiac function and eventually results in the development of chronic heart failure with either preserved ejection fraction (HFpEF) or reduced ejection fraction (HFrEF) (Swynghedauw, 1999; Borlaug and Paulus, 2011). Currently there is no effective curative regimen for heart failure, though the main treatment strategy focuses on improving the symptoms and preventing the progression of the disease. Regenerating the damaged cardiac muscle is the key point to restore the normal cardiac function in heart failure.
In the past few decades, numerous biomedical and bioengineering endeavors have been sought for heart regeneration (Laflamme and Murry, 2011). Therapeutic intervention strategies can be summarized into two categories: (1) stimulation of the proliferation of endogenous cardiomyocytes; and (2) transplantation of exogenous stem cells that can regenerate the damaged heart. Though amphibians and fish can efficiently regenerate the heart after injury (Poss et al., 2002), human adult heart has very limited ability for regeneration. The mammalian heart has transient regeneration potential shortly after birth (Porrello et al., 2011), and the turnover of human cardiomyocytes becomes less than 1% per year in adulthood (Bergmann et al., 2015). Therefore, the first regeneration strategy seems to be relatively elusive until major obstacles to resume the cell cycle entry of adult cardiomyocytes are overcome. In contrast, exogenous stem cell transplantation tends to be more promising with the recent discovery of human pluripotent stem cells (PSCs) including induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs) (Thomson et al., 1998; Takahashi et al., 2007). While the existence of adult cardiac stem cells has been controversial (van Berlo et al., 2014; Chien et al., 2019), the beneficial effects of adult stem cell therapy could be attributed to an acute inflammatory-based wound-healing response that rejuvenates the infarcted area of the heart (Vagnozzi et al., 2020). Recent studies using human PSC-derived cardiomyocytes and cardiac patches have shown promising therapeutic treatment of myocardial infarction in large animal models, shedding light on the future cardiac regeneration therapies (Chong et al., 2014; Liu et al., 2018).
In this review article, we discuss the regulatory mechanisms that govern the proliferation of embryonic cardiomyocytes and the physiological maturation (hypertrophy) of postnatal cardiomyocytes. We explore the mechanistic insights of cardiomyocyte proliferation and maturation from the perspectives of cardiac differentiation of human PSCs. At the end, we envision the future use of human iPSC-derived cardiomyocytes for treating cardiovascular disease.
Developmental Programs for Embryonic and Fetal Cardiomyocyte Proliferation
The human heart is the first functional organ to form during embryonic development. A fetal heartbeat can be detected as early as 6 weeks after gestation. Shortly after gastrulation and mesoderm commitment, early cardiac mesoderm which is marked by the transcription factor, MESP1, first appears at the posterior side of the embryo along with the primitive streak (Bondue et al., 2008). Cardiac mesoderm further migrates toward the anterior position of the embryo to form the reservoirs for the two cardiac progenitor cell populations: the first heart field (FHF) and the second heart field (SHF). The FHF is located at the cardiac crescent whereas the SHF is posterior to the crescent. The FHF progenitors generate the primitive heart tube and give rise to the left ventricle and parts of atria; the SHF progenitors migrate toward the primitive and looping heart tube and contribute to the right ventricle (RV), parts of the atria, outflow tract (OFT) which will form the aorta and pulmonary artery (Brade et al., 2013; Spater et al., 2014). The expression of the transcription factor, ISL1, distinguishes the FHF (ISL–) and SHF (ISL+) progenitors (Cai et al., 2003; Bu et al., 2009; Paige et al., 2015).
The de novo cardiomyocytes are derived either from the differentiation of early cardiac progenitors or from the proliferation of preexisting cardiomyocytes. Robust cardiomyocyte proliferation is necessary for proper formation of cardiac structures such as myocardial trabeculation and ventricular wall maturation. Multiple signaling pathways have been involved in the control of cardiomyocyte proliferation during this stage. The prominent pathways driving embryonic cardiac proliferation include NOTCH, Neuregulin (NRG), Hippo/Yap, IGF, and Wnt (Figure 1 and Table 1). The intensive crosstalk among these signaling pathways is embedded in the intercellular communication between the developing epicardium, myocardium and endocardium, and thus is highly coordinated to ensure the appropriate heart chamber growth in a spatiotemporal manner.
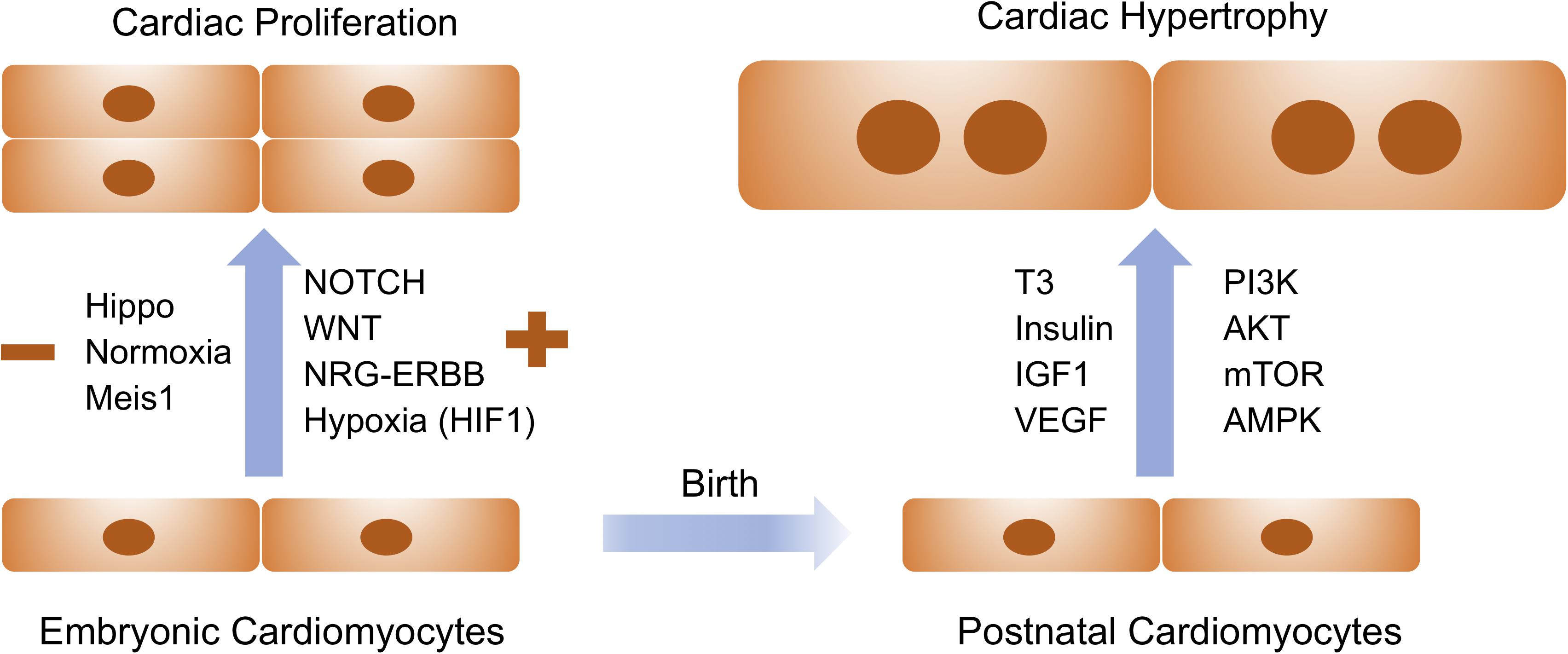
Figure 1. Extracellular signaling and intracellular pathways drive embryonic cardiac proliferation and postnatal cardiac hypertrophy. Signaling molecules listed on the left are positive and negative regulators for embryonic cardiac proliferation whereas those on the right are essential for physiological hypertrophy in postnatal cardiomyocytes. These pathways and molecules could be prospectively manipulated to stimulate cardiomyocyte proliferation and growth for therapeutic interventions of cardiovascular disease.
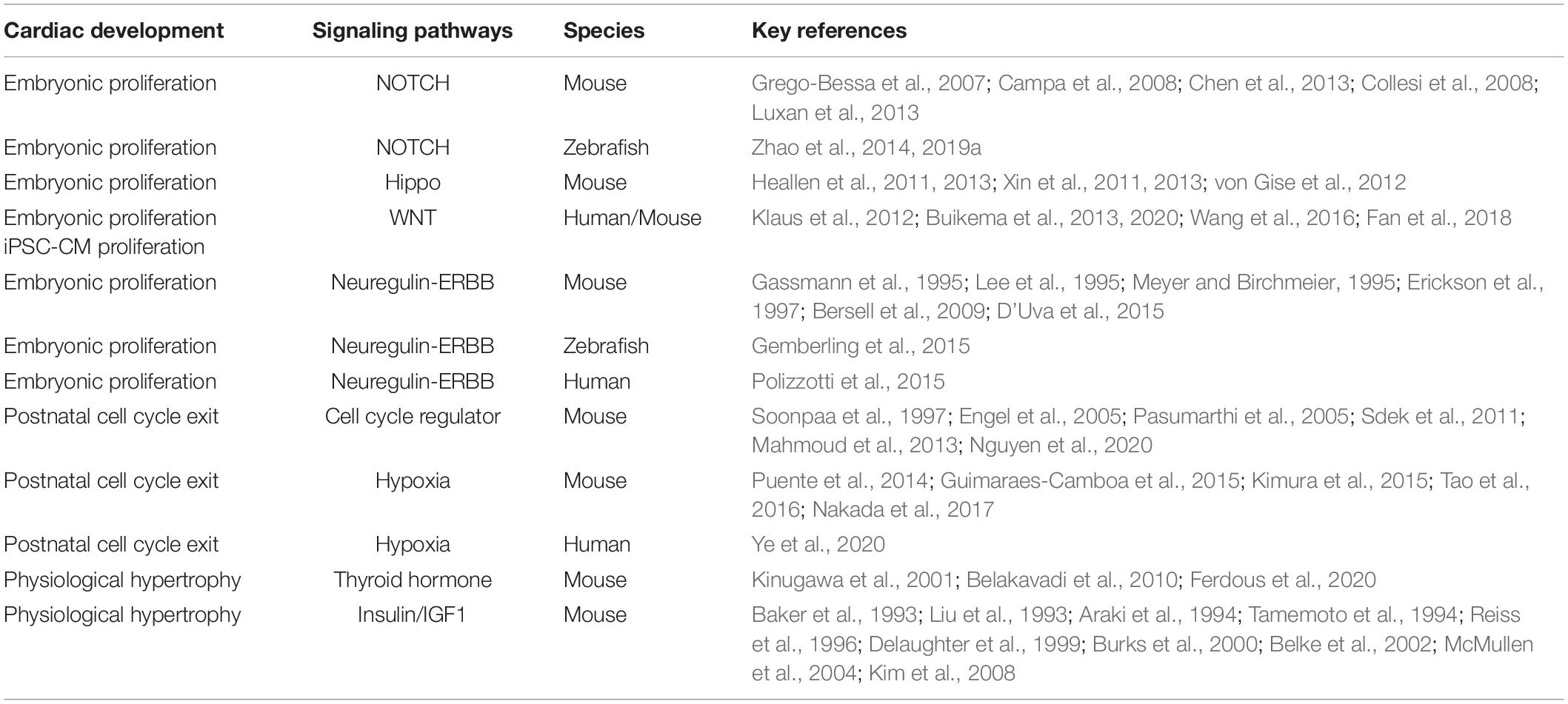
Table 1. Signaling pathways associated with cardiac proliferation and hypertrophic growth. Key references and species of these studies are listed.
NOTCH Signaling Pathway
In mammals, the NOTCH pathway includes 4 type I transmembrane receptors (NOTCH1-4) and 5 type I transmembrane ligands (JAG1, JAG2, DLL1, DLL3, and DLL4). The NOTCH receptors are heterodimeric latent transcriptional regulators and contain EGF-like repeats and Lin12/Notch (LNR) motif in the extracellular domain, and RBPJ associated molecule (RAM) motif, 7 ankyrin motifs, 2 nuclear localization signals, and a transactivation domain (TAD) in the intracellular domain. Upon ligand binding from the neighboring cell, NOTCH receptors are cleaved by the γ-secretase complex, resulting in the release of NOTCH intracellular domain (NICD). The NICD is then translocated to the nucleus, where it binds to RBPJ (CBF in humans) and MAML to form a trimeric complex that converts the RBPJ from a repressor to an activator and initiates the transcriptional activation of downstream target genes such as Hey1 and Hey2 (Siebel and Lendahl, 2017).
During embryonic heart chamber development, endocardial NOTCH signaling is essential for the trabeculation and compaction of ventricular myocardium (Grego-Bessa et al., 2007; MacGrogan et al., 2018). NICD/RBPJ activation stimulates BMP10 activity in the underlying myocardium which triggers the proliferation of cardiomyocytes through the inhibition of the cell cycle inhibitor p57 (Grego-Bessa et al., 2007). Notch1 deletion in mouse embryos leads to reduced cardiomyocyte differentiation and impaired ventricular trabeculation, whereas exogenous Bmp10 expression can rescue the phenotype in Notch1 knockout (KO) embryos. In addition, endocardial deletion of the NOTCH1 negative regulator, FKBP12, leads to ventricular hypertrabeculation and non-compaction, in contrast to the hypotrabeculation in Notch1 KO hearts (Chen et al., 2013). NOTCH signaling also controls the degradation of extracellular matrix (ECM) during the formation of endocardial projections through an antagonistic interaction with Neuregulin1 (NRG1) (Del Monte-Nieto et al., 2018).
Ectopic Notch activation promotes the cell cycle reentry and progression in isolated neonatal cardiomyocytes through transcriptional regulation of the cell cycle regulator cyclin D1 (Campa et al., 2008). In contrast, inhibition of Notch signaling blocks the proliferation of immature cardiomyocyte and induces apoptosis in mammals (Collesi et al., 2008). In zebrafish, Notch receptors are elevated in the endocardium and epicardium in response to ventricular amputation. Notch signaling is required for cardiomyocyte proliferation during zebrafish heart regeneration as suppression of Notch signaling impairs cardiac regeneration and induces scar formation at the amputation site (Zhao et al., 2014). Recent studies have demonstrated that endocardial Notch signaling stimulates cardiomyocyte proliferation in the regenerating zebrafish heart through upregulation of the Wnt signaling antagonists, Wif1 and Notum1b (Zhao et al., 2019a). In humans, mutations in NOTCH signaling components lead to a spectrum of congenital heart defects (CHDs). Pathogenic NOTCH1 mutations are linked to severe forms of CHDs such as hypoplastic left heart syndrome (HLHS) and tetralogy of Fallot (Garg et al., 2005; McBride et al., 2008; Durbin et al., 2017; Page et al., 2019). Germline mutations in MIB1, a NOTCH pathway regulator, cause reduced Notch1 activity and lead to left ventricle non-compaction (LVNC) with abnormal ventricular myocardium development and trabecular maturation (Luxan et al., 2013).
Hippo and Wnt Pathways
Hippo signaling is evolutionarily conserved in Drosophila and vertebrates for controlling tissue growth and organ size. The core components of Hippo pathway in mammals consist of a kinase cascade, MST1/2 (homology of Hippo in mammals) and LATS1/2, as well as the downstream effectors and transcriptional coactivators YAP and TAZ (Ma et al., 2019). The pathway activity modulates the dynamic shuttle of YAP/TAZ between the nucleus and cytoplasm. When the Hippo signaling is inactivated (OFF), the dephosphorylated YAP/TAZ are located in the nucleus and compete with VGLL4 for binding to TEADs. YAP/TAZ/TEAD complex then recruits other transcriptional factors to induce gene transcription. When the Hippo pathway is activated (ON), MST1/2 kinase phosphorylates LATS1/2 which further phosphorylates YAP/TAZ. The phosphorylation leads to the binding of YAP/TAZ to 14-3-3 and their cytoplasmic sequestration as well as degradation by poly-ubiquitination (Pan, 2010; Yu et al., 2015).
In mammals, Hippo pathway is an important regulator to restrain cardiomyocyte proliferation and control heart size (Wang et al., 2018). Genetic deletion of the Hippo pathway component Salv in cardiac lineage gives rise to enlarged heart at birth with increased proliferation of cardiomyocytes (Heallen et al., 2011). Similarly, embryonic knockout of the kinase Mst1/2 or Lats1/2 leads to embryonic lethality with massive cardiac overgrowth. Fetal deletion of the downstream effector Yap1 results in lethal myocardial hypoplasia and reduced cardiomyocyte proliferation. In contrast, YAP1 activation stimulates cardiomyocyte proliferation both in culture and in the intact heart (von Gise et al., 2012). Nevertheless, cardiomyocyte size is normal in these Hippo knockout hearts, suggesting that Hippo signaling pathway suppresses cardiac proliferation but does not affect hypertrophic growth of cardiomyocytes (Heallen et al., 2020). In the postnatal heart, Hippo pathway activation prevents cardiac regeneration in response to injury as Hippo-deficient adult cardiomyocytes reenter the cell cycle and undergo cytokinesis in unstressed conditions (Heallen et al., 2013). In addition, suppression of Hippo pathway enhances adult cardiac regeneration and functional recovery after myocardial infarction and cardiac apex resection. For example, deletion of the Hippo pathway component Salv promotes a reparative genetic program with increased scar border vascularity, reduced fibrosis and enhanced ejection fraction after myocardial infarction. Cardiac specific deletion of the Hippo pathway effector Yap impairs neonatal heart regeneration whereas forced expression of Yap stimulates cardiac regeneration in adult heart (Xin et al., 2013). Gene therapy with short hairpin RNAs targeting Salv improves heart function when administered following myocardial infarction and ischemic heart failure (Leach et al., 2017), suggesting that Hippo pathway could serve as a promising therapeutic target for treating heart failure.
The regulatory roles of Hippo pathway in controlling cardiomyocyte proliferation and regeneration are executed through crosstalk with other signaling pathways (Wang et al., 2018). Active Wnt signaling is essential for silencing Hippo pathway. Cardiac-specific knockout of Salv in mice results in upregulation of Wnt target genes including Sox2 and Snai2 through the interaction between Yap and β-catenin (Heallen et al., 2011). When Hippo pathway is off, YAP enters the nucleus and forms a complex with TEA domain (TEAD) transcription factors and β-catenin/T-cell factor/lymphoid enhancer factor (Tcf/Lef), which binds to the promoters and regulates the transcription of genes associated with cardiac proliferation. In addition, the YAP/β-catenin complex also interacts with the insulin-like growth factor (IGF) signaling pathway. Elevated IGF signaling stimulates phosphoinositide-3-kinase (PI3K) which phosphorylates RACα serine/threonine-protein kinase (AKT). The phosphorylated AKT inhibits the phosphorylation of glycogen synthase kinase 3β (GSK3β) that is a member of the β-catenin destruction complex (Xin et al., 2011). Consequently, β-catenin is stabilized to drive the transcription of genes targeted by the YAP/β-catenin complex and promote cardiomyocyte proliferation and myocardial growth.
Neuregulin-ERBB Signaling Pathway
Neuregulins (NRGs) are encoded by 6 individual genes NRG1, NRG2, NRG3, NRG4, NRG5, and NRG6, and are parts of the epidermal growth factor (EGF) family proteins (Mei and Nave, 2014). They can bind to the ligands of ERBB family of tyrosine kinase receptors (ERBB2, ERBB3, and ERBB4) through an extracellular EGF-like domain. The ERBB receptors consist of an extracellular ligand-binding domain, a transmembrane domain and a cytoplasmic tyrosine kinase domain. NRG1 binds to ERBB3 and ERBB4 (but not ERBB2) and causes the dimerization and activation of ERBB receptors, which subsequently phosphorylate their intracellular domains and provide docking sites for adaptor proteins such as Grb2/Shc for Erk activation and p85 for PI3K activation (Odiete et al., 2012).
NRG/ERBB signaling is essential for multiple aspects of early cardiac development. Disruption of Nrg1, Erbb2, or Erbb4 leads to embryonic lethality by E10 in utero with lack of ventricular trabeculation and a mature ventricular wall (Gassmann et al., 1995; Lee et al., 1995; Meyer and Birchmeier, 1995). During embryonic heart development, Nrg1 is expressed in the endocardium whereas Erbb2 and Erbb4 are expressed in the ventricular myocytes. Erbb3 is expressed in the mesenchymal cells of the endocardial cushions that are essential for cardiac valve formation (Erickson et al., 1997). NRG1 promotes in vitro cell division of mononucleated ventricular cardiomyocytes through the receptor ERBB4 (Bersell et al., 2009). In vivo deletion of Erbb4 suppresses cardiomyocyte proliferation whereas as overexpression of Erbb4 enhances cardiac growth. Intriguingly, NRG1 injection in adult mice stimulates cardiomyocyte proliferation and promotes cardiac functional recovery after myocardial infarction. Recent studies reveal that NRG1 and its receptor ERBB2 can promote the regeneration of cardiac muscle cells in zebrafish, mice and infant heart tissue (Yutzey, 2015). NRG1 is induced in perivascular cells in response to adult heart injury in zebrafish. Myocardial overexpression of Nrg1 enhances cardiomyocyte proliferation whereas Erbb2 suppression disrupts cardiac regeneration in the injured heart. In unstressed zebrafish heart, NRG1 reactivation results in cardiomyocyte dedifferentiation, overt muscle hyperplasia and cardiomegaly (enlarged heart) through overgrowth of wall myocardium (Gemberling et al., 2015). In the mammalian heart, NRG1 co-receptor ERBB2 is essential for cardiomyocyte proliferation at both embryonic and neonatal stages. Reduced Erbb2 expression causes diminished cardiac proliferation in the presence of NRG1. Constitutive overexpression of Erbb2 results in an enlarged heart characterized by extensive cardiomyocyte hypertrophy, dedifferentiation and proliferation, whereas transient induction of ERBB2 promotes cardiomyocyte dedifferentiation and regeneration after myocardial infarction (D’Uva et al., 2015). Therefore, ERBB2 is required and sufficient to trigger postnatal cardiac proliferation and regeneration. Concomitantly, administration of recombinant NRG1 could improve myocardial function in response to injury and stimulate cardiomyocyte proliferation in cultured pediatric myocardium (<6 months old), suggesting a critical therapeutic window for regeneration therapy using NRG1 (Polizzotti et al., 2015).
Regulation of Cardiomyocyte Maturation (Hypertrophy) After Birth
During mammalian embryonic and fetal development, heart growth is primarily driven by the cell division of cardiomyocytes. Shortly after birth, the increase of myocardial volume is transitioning to cardiac hypertrophy (an increase in the size instead of an increase in the number of cardiomyocytes) by which cardiomyocytes exit the cell cycle and become enlarged in both length and width to accommodate an increased cardiac workload (Nakamura and Sadoshima, 2018). In mice, cardiac regeneration capacity is retained 1 day after birth as the postnatal day 0 (P0) or P1 heart is capable of rejuvenating massive cardiac growth from the proliferation of preexisting cardiomyocytes in response to injury (Porrello et al., 2011). However, this regeneration ability diminishes by 7 days of age. In pigs, the neonatal heart is capable of regeneration after myocardial infarction in the first 2 days after birth, but the regeneration potential is lost soon after (Ye et al., 2018; Zhu et al., 2018). Interestingly, a thyroid hormone surge appears to activate the IGF/Akt pathway and initiate a transient but intense proliferative burst of binuclear cardiomyocytes (∼40% increase in cardiomyocyte number) in P15 mouse heart (Naqvi et al., 2014). Nevertheless, this phenomenon is contradicted by other studies where no evidence of an increase in cardiomyocyte number is observed in preadolescent mice (Alkass et al., 2015; Soonpaa et al., 2015).
The regulatory mechanisms governing the postnatal cardiac hypertrophy have been emerging in the past two decades. Extracellular signaling induced by growth hormones (thyroid hormone, IGF1 and VEGF) and mechanical stimuli (blood flow) are transduced to converge on a number of intracellular pathways which regulate the downstream gene expression, protein translation and metabolism that are essential for physiological hypertrophy in postnatal heart (Maillet et al., 2013). In the following section, we discuss the extracellular and intracellular signaling transduction mechanisms by which postnatal cardiomyocytes exit the cell cycle and transit to hypertrophic growth.
Regulation of Cell Cycle Withdrawal in Postnatal Cardiomyocytes
Isotope tracing experiments show that the total number of cardiomyocytes is set within the first week after birth, despite of two waves of non-replicative DNA synthesis mainly contributing to cardiomyocyte multinucleation and nuclear polyploidization in the second and third postnatal weeks (Alkass et al., 2015). In humans, the turnover of cardiomyocytes is highest in early childhood but gradually decreases to less than 1% per year in adulthood (Bergmann et al., 2015). The mechanisms underlying the cell cycle arrest in postnatal cardiomyocytes have been of particularly interest to seek for potential therapeutic interventions for reactivation of cardiac proliferation.
Cell cycle regulators such as cyclins and cyclin-dependent kinases (CDKs) are playing important roles in the arrest of dividing cardiomyocytes (Ahuja et al., 2007). There is a critical cell cycle checkpoint called restriction (R) point in late G1 phase when cells must decide whether to advance through M phase or exit into the quiescent G0 stage. To advance cell cycle progression, cyclin D (cyclin D1, D2, or D3) must be accumulated and translocated to the nucleus, where it forms a complex with CDK4/6. The cyclin D-CDK4/6 complex then phosphorylates the Rb family (Rb, p107, and p130), leading to the release of E2F transcription factors which promote the synthesis of cyclin E (predominately expressed in the G1-S transition stage). Cyclin E interacts with CDK2 to form a cyclin E/CDK2 complex which drives additional Rb phosphorylation to push the cell cycle toward S phase and onward. E2F transcription factors are also essential for transcriptional regulation of genes that are involved in the exit to quiescent G0 phase.
The cell cycle positive regulators such as cyclin D1, CDK4/6, Rb, and E2F transcription factors are highly expressed in fetal cardiomyocytes but are significantly downregulated in neonatal and adult cardiomyocytes, coincident with the cell cycle arrest in postnatal cardiomyocytes (Kang et al., 1997; Flink et al., 1998). Constitutive overexpression of cell cycle regulators promotes DNA synthesis and increases multinucleation in cardiomyocytes, but completion of cytokinesis to generate new cardiomyocytes in adult hearts is relatively difficult to achieve. Transient overexpression of cyclin D1 or cyclin D2 stimulates DNA synthesis in adult hearts and results in infarct regression in response to cardiac injury (Soonpaa et al., 1997; Pasumarthi et al., 2005). Cardiac-specific deletion of p38 MAP kinase triggers the mitosis of neonatal cardiomyocytes whereas activation of p38 blocks fetal cardiomyocyte proliferation (Engel et al., 2005). Double knockouts of Rb and p130 in the mouse heart result in the proliferation of adult cardiomyocytes by blocking their interactions with the heterochromatin protein 1 (HP1) (Sdek et al., 2011). A homeodomain transcription factor, Meis1, is recently identified as a critical regulator for cell cycle arrest in postnatal cardiomyocytes. Genetic deletion of Meis1 extends the proliferation window of postnatal cardiomyocytes while stimulates the re-entry of mitosis in adult cardiomyocytes without detrimental influence in cardiac function. In contrast, overexpression of Meis1 blocks neonatal cardiomyocyte proliferation and heart regeneration under stressed condition, possibly through the transcriptional activation of CDK inhibitors p15, p16, and p21 (Mahmoud et al., 2013). In addition, Meis1 interacts with its co-factor Hoxb13 to cooperatively regulate cardiomyocyte proliferation and maturation. Cardiomyocyte-specific deletion of Hoxb13 defers the postnatal window of cardiac regeneration and reactivates cardiomyocyte cell cycle entry in adult heart, whereas double knockouts of Meis1 and Hoxb13 lead to widespread cardiomyocyte mitosis and improved left ventricle systolic function after myocardial infarction (Nguyen et al., 2020). Recently, the combinatorial expression of four cell cycle regulators CDK1, CDK4, cyclin B1, and cyclin D1 was shown to be sufficient to induce cell division in post-mitotic cardiomyocytes across multiple species including mouse, rat and human (Mohamed et al., 2018). In acute or subacute myocardial infarction, 15–20% adult cardiomyocytes expressing these four factors can go through stable cytokinesis and improve cardiac function, implying a promising therapeutic application for cardiac regeneration.
In addition to the intrinsic control, cell cycle exit in cardiomyocytes is also tightly regulated by the extrinsic signals. Recent studies suggest that oxygen-rich environment is primarily responsible for the cell cycle arrest in postnatal cardiomyocytes (Puente et al., 2014). When newborns are exposed to normoxia (20–21% O2), reactive oxygen species (ROS), oxidative DNA damage and DNA damage response (DDR) markers significant increase in the heart. Intriguingly, reversal of normoxemia to hypoxemia conditions can extend the cardiac regenerative window and delay cell cycle withdrawal in neonatal cardiomyocytes. Genetic lineage tracing has identified rare hypoxic proliferative cardiomyocytes with smaller size, mononucleation and low oxidative DNA damage in neonatal hearts (Kimura et al., 2015). HIF1α is essential for the proliferation of hypoxic fetal cardiomyocytes during mid-gestation as loss-of-function of Hif1α leads to activation of ATF4 and p53 which inhibit cardiomyocyte proliferation (Guimaraes-Camboa et al., 2015). A transcription factor named Pitx2 potentially interacts with the Hippo pathway effector YAP to suppress the ROS levels. Genetic deletion of Pitx2 results in compromised regeneration capacity in response to apex resection in neonatal heart (Tao et al., 2016), suggesting a crosstalk interaction between Hippo and HIF1α pathways in the regulation of cardiomyocyte regeneration. Furthermore, mice exposed to hypoxemia conditions display the reactivation of cardiomyocyte mitosis in the heart through inhibition of oxidative metabolism, decreased ROS production and oxidative DNA damage. One-week exposure to hypoxemia could induce robust myocardial regeneration from preexisting cardiomyocytes with decreased myocardial fibrosis and improved systolic function in response to myocardial infarction (Nakada et al., 2017). Similar beneficial effects of hypoxia on cardiomyocyte cell cycle activation are also observed in human myocardial tissue and iPSC-derived cardiomyocytes (iPSC-CMs), highlighting that hypoxia could serve as a potential therapeutic path forward toward human myocardial regeneration (Sadek and Olson, 2020; Ye et al., 2020).
Physiological Hypertrophy of Cardiomyocytes in Postnatal Heart
During postnatal cardiac hypertrophy, cardiomyocytes become multinucleated, expand their contractile apparatus such as sarcomeres, and increase excitation-contraction coupling efficiency in order to maintain heart function upon an increased workload (Maillet et al., 2013). Physiological hypertrophy results in increased contractile function without interstitial fibrosis or cell death, which is distinct from pathological hypertrophy that leads to cardiomyopathy and heart failure (Nakamura and Sadoshima, 2018). Physiological hypertrophy is initiated by a number of extracellular stimuli which are transduced through multiple intracellular signaling pathways in order to modulate gene expression, protein translation and metabolic remodeling. These physiological signaling pathways include growth hormone, VEGF, thyroid hormone, insulin and IGF1. Note that pathological hypertrophy is regulated by numerous different extracellular signaling pathways and readers can refer to excellent reviews elsewhere (van Berlo et al., 2013; Nakamura and Sadoshima, 2018).
The active form of thyroid hormone T3 (3,5,3′-triiodothyronine) is essential for postnatal cardiac hypertrophy in mammals. Circulating T3 concentration in the blood surges (2,000-fold increase) in the second week after birth. T3 binds to the thyroid hormone receptors TRα and TRβ which are nuclear receptors. T3 binding results in the translocation of TR receptors into the nuclei, where they serve as transcriptional activators for initiating gene transcription (Brent, 2012; Mullur et al., 2014). In rodents, TR-responsive genes include upregulation of α-myosin heavy chain α-MHC (adult muscle isoform, MYH6) and sarcoplasmic reticulum Ca2+ ATPase (SERCA), and downregulation of β-myosin heavy chain β-MHC (fetal muscle isoform, MYH7) and phospholamban (PLN) in postnatal cardiomyocytes (Kinugawa et al., 2001; Belakavadi et al., 2010). T3-dependent gene expression in heart involves the crosstalk with retinoic acid receptors and is coordinated with histone modifications and non-coding RNAs to facilitate the removal of corepressors and recruitment of RNA polymerase II and coactivators (Mullur et al., 2014; Forini et al., 2019). Recent investigations uncover the forkhead box protein O1 (FoxO1) controls the enzyme Dio2 which is responsible for intracellular thyroid hormone metabolism, suggesting that FoxO1-Dio2 axis regulates T3-induced hypertrophic growth of neonatal cardiomyocytes (Ferdous et al., 2020).
Insulin and IGF1 are essential factors for initiating the physiological hypertrophy of postnatal cardiomyocytes. Insulin binds to the insulin receptor (IR) and activates the recruitment and phosphorylation of adaptor proteins such as insulin receptor substrate 1 (IRS1) and IRS2. IGF1 is structurally similar to insulin and binds to IR and IGF1 receptor (IGF1R). IR and IGF1R are tyrosine kinase receptors that activate PI3K-AKT-mTOR signaling pathway through the docking protein IRS1/IRS2, and RAS-RAF-MEK-MAPK signaling axis through the adaptor protein growth factor receptor-bound protein 2 (GRB2), respectively (Saltiel and Kahn, 2001; Taniguchi et al., 2006). Mice null for Irs1/2 or Igf1 display general growth retardation, and disruption of Igf1r results in perinatal lethality and a more severe growth deficiency (45% normal size) (Baker et al., 1993; Liu et al., 1993; Araki et al., 1994; Tamemoto et al., 1994; Burks et al., 2000). Cardiac-specific deletion of insulin receptor leads to reduced cardiomyocyte and heart size by 20–30% and persistent expression of the fetal β-myosin heavy chain isoform in mice (Belke et al., 2002). Overexpression of Igf1 in the heart promotes cardiomyocyte proliferation and cardiac hypertrophy with enlargement of individual cardiac myofibers (Reiss et al., 1996; Delaughter et al., 1999). However, long-term local Igf1 overexpression results in pathological hypertrophy with decreased systolic performance and increased fibrosis. Overexpression of Igf1r in the heart stimulates hypertrophic growth with an increase in cardiomyocyte size and enhanced systolic function, but without evidence of pathological hypertrophy (McMullen et al., 2004). Mice with cardiac-specific double knockouts of Igf1r and Ir appear normal in heart development but show resistance to exercise-induced hypertrophy, implying the essential role of IGF1 signaling in myocardial hypertrophic growth (Kim et al., 2008).
During postnatal cardiac growth, the vascular capillary network in the heart is proportionally expanded to supply sufficient nutrients and oxygen. Increased capillary activity leads to massive release of nitric oxide (NO) which results in the degradation of regulator of G-protein signaling 4 (RGS4), thus activating G protein-coupled receptor (GPCR) mediated downstream signaling pathways such as PI3K and mTOR to promote cardiac hypertrophy (Jaba et al., 2013; Oka et al., 2014). Reciprocally, myocardial hypertrophy also induces angiogenesis in the myocardium through activation of the intracellular signaling HIF1α and transcription factor GATA4. GATA4 is abundantly expressed in cardiomyocytes during early embryonic development and regulates the expression of cardiac-specific genes such as structural and contractile genes essential for hypertrophic growth. Cardiac-specific deletion of Gata4 causes defects in cardiac hypertrophic growth and reduced density of myocardial capillary (Oka et al., 2006; Heineke et al., 2007). In contrast, cardiomyocyte-specific overexpression of Gata4 increases capillary density in the myocardium. Furthermore, HIF1α is induced in the cell that is not receiving enough oxygen (hypoxia), and HIF1α trans-activates VEGF expression to stimulate angiogenesis (Forsythe et al., 1996; Semenza, 2014). VEGF is a critical factor in maintaining the myocardial capillary density during physiological cardiac hypertrophy through regulating angiogenesis (the growth of blood vessels from preexisting vasculature). Inhibition of VEGF signaling leads to suppression of capillary density and angiogenesis, which eventually promotes the transition from physiological hypertrophy to pathological heart failure during pressure overload (Shiojima et al., 2005; Izumiya et al., 2006).
Harness Human iPSC-Derived Cardiomyocytes for Heart Regeneration
In the past few decades, cardiac stem cell therapy has been spearheaded as a potential treatment for myocardial infarction and heart failure. As there are no existing endogenous cardiac stem cells which can produce large number of de novo cardiomyocytes, patient-derived iPSCs possess great promise in cardiac regeneration (Murry and MacLellan, 2020). Human iPSC-derived cardiomyocytes (iPSC-CMs) are autologous and can be augmented for bulk production in vitro and generate billions of beating myocytes for repairing the damaged heart. Though precision generation of subtype-specific cardiomyocytes have been successfully achieved (Zhao et al., 2020), a major hurdle for the clinical application is the relative immature state of iPSC-CMs. Numerous strategies including in vivo transplantation and engineered 3D heart tissue with electromechanical coupling have been applied to improve the structural and functional maturation of iPSC-CMs (Karbassi et al., 2020). These approaches are designed to stimulate the maturation of iPSC-CMs by mimicking embryonic cardiomyocyte proliferation and postnatal hypertrophic growth during normal heart development. Here, we dissect how these cardiomyocyte maturation methodologies reflect the two perspectives of cardiac development: proliferation and maturation.
Current cardiac differentiation protocols mostly rely on manipulation of distinct signaling pathways to achieve robust cardiomyocyte generation (Protze et al., 2019; Zhao et al., 2020). In the early stage of directed differentiation, PSCs are induced to cardiac mesoderm and then cardiac progenitors by sequential modulation of WNT signaling pathway (Zhao et al., 2017). Cardiac progenitors are then directed to generate committed cardiomyocytes which resemble embryonic cardiomyocytes. There is a burst of cardiomyocyte proliferation to form ventricular chambers during embryonic heart development, which is modulated by the combinatorial interactions of signaling molecules such as NOTCH1, BMP10, NRG1, WNT, and VEGF (Foglia and Poss, 2016). An inhibitor CHIR99021 blocks GSK3-mediated degradation of β-catenin and leads to the nuclear translocation of β-catenin and activation of Wnt signaling target genes. Addition of CHIR99021 results in robust proliferation of mouse and human PSC-derived cardiomyocytes (Figure 2), consistent with the essential roles of the Wnt/β-catenin pathway for ventricular cardiomyocyte proliferation during embryonic development (Klaus et al., 2012; Buikema et al., 2013, 2020). In addition, Wnt signaling-induced cardiomyocyte proliferation is also observed in human neonatal cardiomyocytes and mouse adult cardiomyocytes (Wang et al., 2016; Fan et al., 2018). As NOTCH and Hippo pathways control cardiomyocyte proliferation through regulating Wnt/β-catenin pathway, manipulation of these pathway activities could boost the proliferation of early human iPSC-CMs to generate clinical-level magnitude for repairing the damaged myocardium.
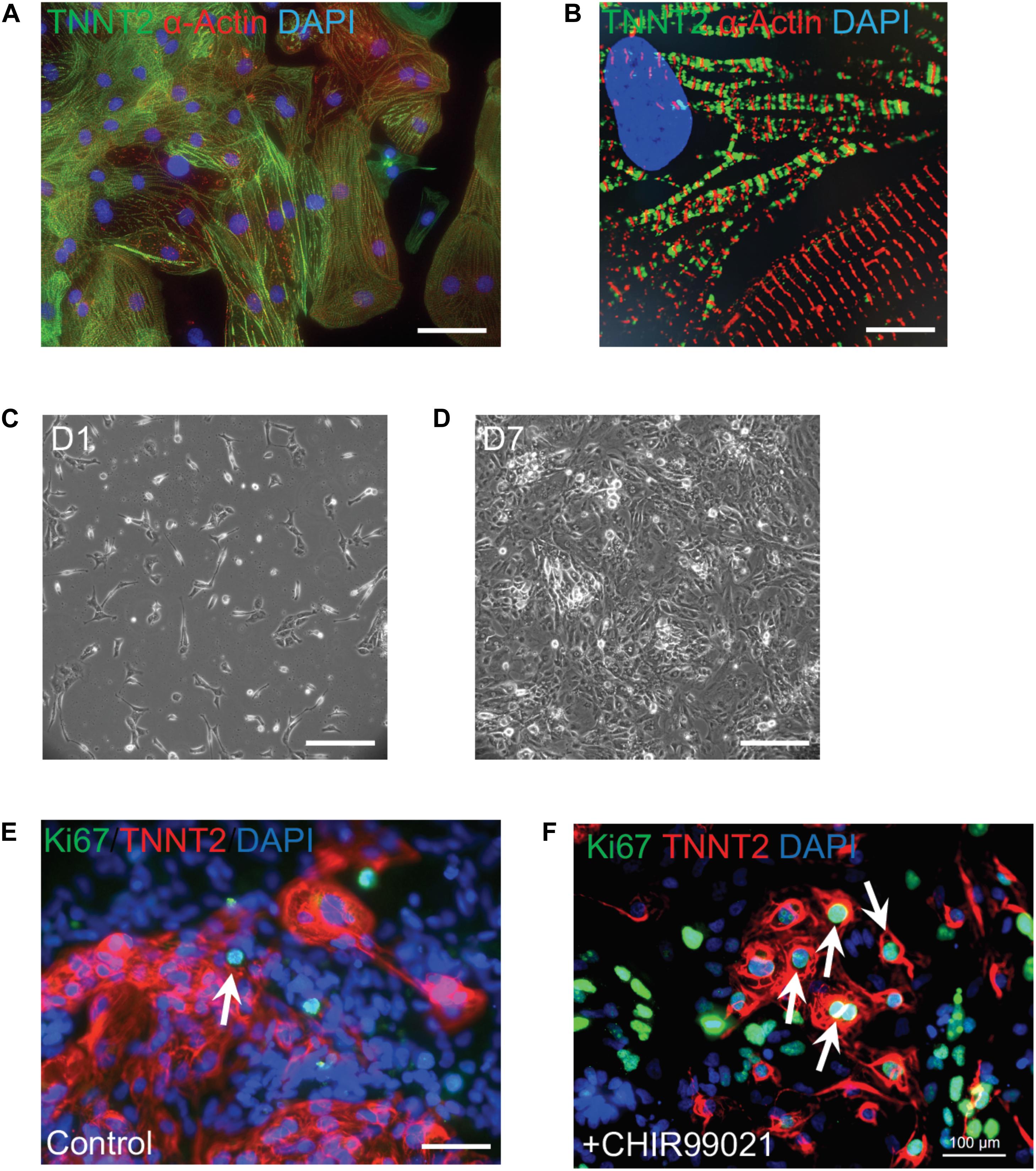
Figure 2. Robust proliferation of human iPSC-CMs is promoted by WNT signaling activation. (A) Typical morphology of human iPSC-CMs stained with antibodies against cardiac troponin T (TNNT2, green) and α-actinin (red). Nuclei were counterstained with DAPI (blue). (B) Zoom-in sarcomere structure of human iPSC-CMs with intercalated TNNT2 (green) and α-actinin (red). (C,D) Robust proliferation of human iPSC-CMs from Day 1 (D1,C) to D7 (D) in the presence of 2 μM of a WNT activator (CHIR99021). (E,F) Dramatic increase of dividing CMs is propelled by CHIR99021. Cells were stained with Ki67 (green) and TNNT2 (red). Nuclei were counterstained with DAPI (blue). Double positive cells (indicated by white arrows) are dividing cardiomyocytes. The percentage of Ki67+ TNNT2+ cells is increased in CHIR99021-treated iPSC-CMs (F) compared to the controls (E). Scale bars: 10 μm (B); 100 μm (A,E,F); 200 μm (C,D).
In vivo, cardiomyocytes undergo structural and functional maturation accompanied with embryonic proliferation and postnatal hypertrophy. However, under current 2D differentiation protocols, human iPSC-CMs are relatively immature and resemble embryonic/fetal cardiomyocytes due to the absence of physical and environmental cues essential for the induction of physiological hypertrophy (Karbassi et al., 2020). Human iPSC-CMs are small (5–10 μm in diameter) and misaligned, compared to the rod-like shape (150 μm in length) and well-aligned adult cardiomyocytes (Snir et al., 2003; Yang et al., 2014). In addition, human iPSC-CMs generally do not have well-formed myofibrils and T-tubules, polyploidy, polarized intercalated discs or abundant mitochondria (Table 2). At the electrophysiological level, human iPSC-CMs beat spontaneously due to high levels of hyperpolarization-activated cyclic nucleotide-gate channel 4 (HCN4) present in the plasma membrane, whereas adult ventricular cardiomyocytes are usually quiescent unless receiving electric signaling from the depolarization of an adjacent cell. At the metabolic level, iPSC-CMs primarily rely on the energy release from glycolysis while in adult cardiomyocytes most (85%) of the energy supplies are from β-oxidation of fatty acids. At the gene expression level, human iPSC-CMs initially express the fetal isoform of myosin heavy chain (α-MHC, MYH6) and can gradually switch to the adult isoform (β-MHC, MYH7) dominant after long period of culture.
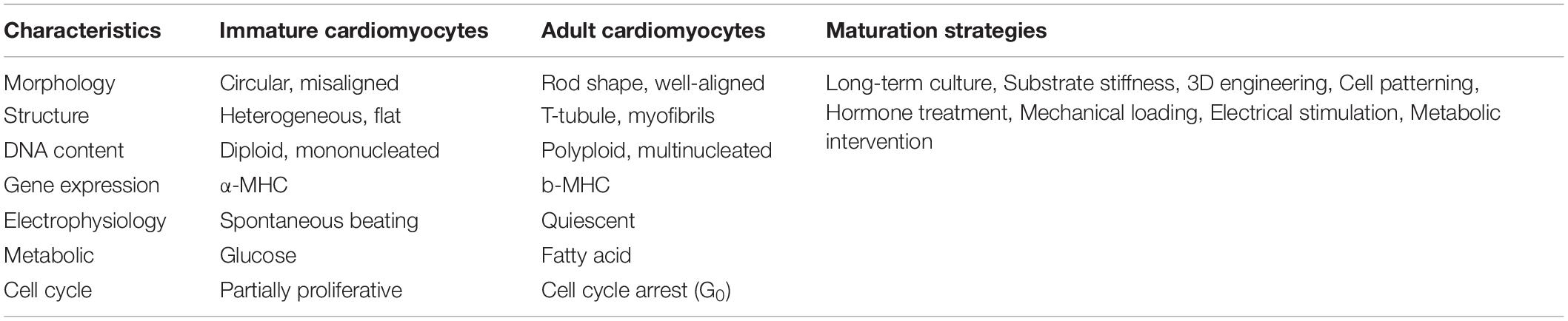
Table 2. Characteristic differences between human iPSC-CMs and adult cardiomyocytes, and multiple strategies to advance the maturation of iPSC-CMs.
Despite the immaturity of iPSC-CMs, a number of approaches have been successfully used to advance iPSC-CMs toward a mature stage. Current state-of-the-art strategies include long-term culture, in vivo transplantation, 3D engineering heart tissue (EHT) coupled with electrical stimulation and mechanical stress, metabolic intervention and hormone treatment (Table 2). Of note, 3D EHT method appears to be most efficient as it can reconstruct the biophysical stimuli and intercellular crosstalk which are essential for the physiological hypertrophy of postnatal cardiomyocytes (Feric and Radisic, 2016; Scuderi and Butcher, 2017). Human cardiac tissues made from early-stage iPSC-CMs and supporting fibroblasts can advance to an adult myocardial-like state after a few weeks of 3D training under physical conditioning and electrical stimulation (Ronaldson-Bouchard et al., 2018). They show adult-like gene expression profiles, well-organized sarcomere structures, presence of transverse tubules, a positive force-frequency relationship and high-density mitochondria. Using a Biowire chip seeded with cell-hydrogel mixture, Zhao et al. (2019b) have constructed a platform which can generate atrial- and ventricular-specific cardiac tissue by combining the directed cell differentiation and electric field conditioning, which is very close to the simulation of distinct electric signaling in adult heart chambers.
Conclusion and Perspectives
Extracellular signaling pathways governing the proliferation of embryonic and fetal mammalian cardiomyocytes have been discovered in the past few decades. Of these pathways, neuregulin and Hippo pathways have shown fascinating pre-clinical outcomes in stimulating endogenous cardiomyocyte proliferation in response to myocardial infarction. In addition, hypoxia-induced HIF1α pathway could be the next phase of therapeutic targets for myocardial regeneration, as HIF1α not only stimulates cell cycle reentry of postnatal cardiomyocyte but also promotes capillary angiogenesis to advance hypertrophic growth of cardiomyocytes. Concomitantly, human iPSC-CMs are promising exogenous sources for cardiac stem cell therapy due to the robust production and controlled progression toward adult-like phenotypes. Combined with CRISPR/Cas9-mediated genome editing (Doudna, 2020), patient iPSC-CMs are more defined and prone to genetic engineering, and exert advantages of beneficial clinical effects compared to adult stem cell therapy. Recent 3D heart printing and cardiac organoid technologies using iPSC-CMs have placed another hope for cardiac regeneration (Lee et al., 2019; Mills et al., 2019). Future thrives on innovative therapeutic strategies will be resorted to the knowledge that we are gleaning on the mechanistic control of cardiac differentiation, growth and regeneration.
Author Contributions
M-TZ: conception and design, figure preparation, manuscript writing, and final approval of the manuscript. SY and JS: figure preparation and final approval of the manuscript. VG: manuscript writing and final approval of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the American Heart Association (AHA) Career Development Award 18CDA34110293 (M-TZ), Additional Ventures Innovation Fund (M-TZ and VG), and National Institute of Health (NIH) grants R01 HL121797, R01 HL144009, and R01HL132801 (VG). M-TZ was also supported by startup funds from the Abigail Wexner Research Institute at Nationwide Children’s Hospital.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahuja, P., Sdek, P., and Maclellan, W. R. (2007). Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 87, 521–544. doi: 10.1152/physrev.00032.2006
Alkass, K., Panula, J., Westman, M., Wu, T. D., Guerquin-Kern, J. L., and Bergmann, O. (2015). No evidence for cardiomyocyte number expansion in preadolescent mice. Cell 163, 1026–1036. doi: 10.1016/j.cell.2015.10.035
Araki, E., Lipes, M. A., Patti, M. E., Bruning, J. C., Haag, B. III, Johnson, R. S., et al. (1994). Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 372, 186–190. doi: 10.1038/372186a0
Baker, J., Liu, J. P., Robertson, E. J., and Efstratiadis, A. (1993). Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75, 73–82. doi: 10.1016/s0092-8674(05)80085-6
Belakavadi, M., Saunders, J., Weisleder, N., Raghava, P. S., and Fondell, J. D. (2010). Repression of cardiac phospholamban gene expression is mediated by thyroid hormone receptor-{alpha}1 and involves targeted covalent histone modifications. Endocrinology 151, 2946–2956. doi: 10.1210/en.2009-1241
Belke, D. D., Betuing, S., Tuttle, M. J., Graveleau, C., Young, M. E., Pham, M., et al. (2002). Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Invest. 109, 629–639. doi: 10.1172/jci0213946
Bergmann, O., Zdunek, S., Felker, A., Salehpour, M., Alkass, K., Bernard, S., et al. (2015). Dynamics of cell generation and turnover in the human heart. Cell 161, 1566–1575. doi: 10.1016/j.cell.2015.05.026
Bersell, K., Arab, S., Haring, B., and Kuhn, B. (2009). Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138, 257–270. doi: 10.1016/j.cell.2009.04.060
Bondue, A., Lapouge, G., Paulissen, C., Semeraro, C., Iacovino, M., Kyba, M., et al. (2008). Mesp1 acts as a master regulator of multipotent cardiovascular progenitor specification. Cell Stem Cell 3, 69–84. doi: 10.1016/j.stem.2008.06.009
Borlaug, B. A., and Paulus, W. J. (2011). Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur. Heart J. 32, 670–679. doi: 10.1093/eurheartj/ehq426
Brade, T., Pane, L. S., Moretti, A., Chien, K. R., and Laugwitz, K. L. (2013). Embryonic heart progenitors and cardiogenesis. Cold Spring Harb. Perspect. Med. 3:a013847. doi: 10.1101/cshperspect.a013847
Bu, L., Jiang, X., Martin-Puig, S., Caron, L., Zhu, S., Shao, Y., et al. (2009). Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 460, 113–117. doi: 10.1038/nature08191
Buikema, J. W., Lee, S., Goodyer, W. R., Maas, R. G., Chirikian, O., Li, G., et al. (2020). Wnt activation and reduced cell-cell contact synergistically induce massive expansion of functional human iPSC-derived cardiomyocytes. Cell Stem Cell 27, 50–63.e5.
Buikema, J. W., Mady, A. S., Mittal, N. V., Atmanli, A., Caron, L., Doevendans, P. A., et al. (2013). Wnt/beta-catenin signaling directs the regional expansion of first and second heart field-derived ventricular cardiomyocytes. Development 140, 4165–4176. doi: 10.1242/dev.099325
Burks, D. J., Font De Mora, J., Schubert, M., Withers, D. J., Myers, M. G., Towery, H. H., et al. (2000). IRS-2 pathways integrate female reproduction and energy homeostasis. Nature 407, 377–382. doi: 10.1038/35030105
Cai, C. L., Liang, X., Shi, Y., Chu, P. H., Pfaff, S. L., Chen, J., et al. (2003). Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 5, 877–889. doi: 10.1016/s1534-5807(03)00363-0
Campa, V. M., Gutierrez-Lanza, R., Cerignoli, F., Diaz-Trelles, R., Nelson, B., Tsuji, T., et al. (2008). Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J. Cell Biol. 183, 129–141. doi: 10.1083/jcb.200806104
Chen, H., Zhang, W., Sun, X., Yoshimoto, M., Chen, Z., Zhu, W., et al. (2013). Fkbp1a controls ventricular myocardium trabeculation and compaction by regulating endocardial Notch1 activity. Development 140, 1946–1957. doi: 10.1242/dev.089920
Chien, K. R., Frisen, J., Fritsche-Danielson, R., Melton, D. A., Murry, C. E., and Weissman, I. L. (2019). Regenerating the field of cardiovascular cell therapy. Nat. Biotechnol. 37, 232–237. doi: 10.1038/s41587-019-0042-1
Chong, J. J., Yang, X., Don, C. W., Minami, E., Liu, Y. W., Weyers, J. J., et al. (2014). Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 510, 273–277.
Collesi, C., Zentilin, L., Sinagra, G., and Giacca, M. (2008). Notch1 signaling stimulates proliferation of immature cardiomyocytes. J. Cell Biol. 183, 117–128. doi: 10.1083/jcb.200806091
Del Monte-Nieto, G., Ramialison, M., Adam, A. A. S., Wu, B., Aharonov, A., D’Uva, G., et al. (2018). Control of cardiac jelly dynamics by NOTCH1 and NRG1 defines the building plan for trabeculation. Nature 557, 439–445. doi: 10.1038/s41586-018-0110-6
Delaughter, M. C., Taffet, G. E., Fiorotto, M. L., Entman, M. L., and Schwartz, R. J. (1999). Local insulin-like growth factor I expression induces physiologic, then pathologic, cardiac hypertrophy in transgenic mice. FASEB J. 13, 1923–1929. doi: 10.1096/fasebj.13.14.1923
Doudna, J. A. (2020). The promise and challenge of therapeutic genome editing. Nature 578, 229–236. doi: 10.1038/s41586-020-1978-5
Durbin, M. D., Cadar, A. G., Williams, C. H., Guo, Y., Bichell, D. P., Su, Y. R., et al. (2017). Hypoplastic left heart syndrome sequencing reveals a novel NOTCH1 mutation in a family with single ventricle defects. Pediatr. Cardiol. 38, 1232–1240. doi: 10.1007/s00246-017-1650-5
D’Uva, G., Aharonov, A., Lauriola, M., Kain, D., Yahalom-Ronen, Y., Carvalho, S., et al. (2015). ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 17, 627–638. doi: 10.1038/ncb3149
Engel, F. B., Schebesta, M., Duong, M. T., Lu, G., Ren, S., Madwed, J. B., et al. (2005). p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 19, 1175–1187. doi: 10.1101/gad.1306705
Erickson, S. L., O’shea, K. S., Ghaboosi, N., Loverro, L., Frantz, G., Bauer, M., et al. (1997). ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2-and heregulin-deficient mice. Development 124, 4999–5011.
Fan, Y., Ho, B. X., Pang, J. K. S., Pek, N. M. Q., Hor, J. H., Ng, S. Y., et al. (2018). Wnt/beta-catenin-mediated signaling re-activates proliferation of matured cardiomyocytes. Stem Cell Res. Ther. 9:338.
Ferdous, A., Wang, Z. V., Luo, Y., Li, D. L., Luo, X., Schiattarella, G. G., et al. (2020). FoxO1-Dio2 signaling axis governs cardiomyocyte thyroid hormone metabolism and hypertrophic growth. Nat. Commun. 11:2551.
Feric, N. T., and Radisic, M. (2016). Maturing human pluripotent stem cell-derived cardiomyocytes in human engineered cardiac tissues. Adv. Drug Deliv. Rev. 96, 110–134. doi: 10.1016/j.addr.2015.04.019
Flink, I. L., Oana, S., Maitra, N., Bahl, J. J., and Morkin, E. (1998). Changes in E2F complexes containing retinoblastoma protein family members and increased cyclin-dependent kinase inhibitor activities during terminal differentiation of cardiomyocytes. J. Mol. Cell. Cardiol. 30, 563–578. doi: 10.1006/jmcc.1997.0620
Foglia, M. J., and Poss, K. D. (2016). Building and re-building the heart by cardiomyocyte proliferation. Development 143, 729–740. doi: 10.1242/dev.132910
Forini, F., Nicolini, G., Pitto, L., and Iervasi, G. (2019). Novel insight into the epigenetic and post-transcriptional control of cardiac gene expression by thyroid hormone. Front. Endocrinol. 10:601. doi: 10.3389/fendo.2019.00601
Forsythe, J. A., Jiang, B. H., Iyer, N. V., Agani, F., Leung, S. W., Koos, R. D., et al. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613. doi: 10.1128/mcb.16.9.4604
Garg, V., Muth, A. N., Ransom, J. F., Schluterman, M. K., Barnes, R., King, I. N., et al. (2005). Mutations in NOTCH1 cause aortic valve disease. Nature 437, 270–274. doi: 10.1038/nature03940
Gassmann, M., Casagranda, F., Orioli, D., Simon, H., Lai, C., Klein, R., et al. (1995). Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 378, 390–394. doi: 10.1038/378390a0
Gemberling, M., Karra, R., Dickson, A. L., and Poss, K. D. (2015). Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. eLife 4:e05871.
Grego-Bessa, J., Luna-Zurita, L., Del Monte, G., Bolos, V., Melgar, P., Arandilla, A., et al. (2007). Notch signaling is essential for ventricular chamber development. Dev. Cell 12, 415–429. doi: 10.1016/j.devcel.2006.12.011
Guimaraes-Camboa, N., Stowe, J., Aneas, I., Sakabe, N., Cattaneo, P., Henderson, L., et al. (2015). HIF1alpha represses cell stress pathways to allow proliferation of hypoxic fetal cardiomyocytes. Dev. Cell 33, 507–521. doi: 10.1016/j.devcel.2015.04.021
Heallen, T., Morikawa, Y., Leach, J., Tao, G., Willerson, J. T., Johnson, R. L., et al. (2013). Hippo signaling impedes adult heart regeneration. Development 140, 4683–4690. doi: 10.1242/dev.102798
Heallen, T., Zhang, M., Wang, J., Bonilla-Claudio, M., Klysik, E., Johnson, R. L., et al. (2011). Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461. doi: 10.1126/science.1199010
Heallen, T. R., Kadow, Z. A., Wang, J., and Martin, J. F. (2020). Determinants of cardiac growth and size. Cold Spring Harb. Perspect. Biol. 12:a037150. doi: 10.1101/cshperspect.a037150
Heineke, J., Auger-Messier, M., Xu, J., Oka, T., Sargent, M. A., York, A., et al. (2007). Cardiomyocyte GATA4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J. Clin. Invest. 117, 3198–3210. doi: 10.1172/jci32573
Izumiya, Y., Shiojima, I., Sato, K., Sawyer, D. B., Colucci, W. S., and Walsh, K. (2006). Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension 47, 887–893. doi: 10.1161/01.hyp.0000215207.54689.31
Jaba, I. M., Zhuang, Z. W., Li, N., Jiang, Y., Martin, K. A., Sinusas, A. J., et al. (2013). NO triggers RGS4 degradation to coordinate angiogenesis and cardiomyocyte growth. J. Clin. Invest. 123, 1718–1731. doi: 10.1172/jci65112
Kang, M. J., Kim, J. S., Chae, S. W., Koh, K. N., and Koh, G. Y. (1997). Cyclins and cyclin dependent kinases during cardiac development. Mol. Cells 7, 360–366.
Karbassi, E., Fenix, A., Marchiano, S., Muraoka, N., Nakamura, K., Yang, X., et al. (2020). Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 17, 341–359. doi: 10.1038/s41569-019-0331-x
Kim, J., Wende, A. R., Sena, S., Theobald, H. A., Soto, J., Sloan, C., et al. (2008). Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol. Endocrinol. 22, 2531–2543. doi: 10.1210/me.2008-0265
Kimura, W., Xiao, F., Canseco, D. C., Muralidhar, S., Thet, S., Zhang, H. M., et al. (2015). Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 523, 226–230. doi: 10.1038/nature14582
Kinugawa, K., Yonekura, K., Ribeiro, R. C., Eto, Y., Aoyagi, T., Baxter, J. D., et al. (2001). Regulation of thyroid hormone receptor isoforms in physiological and pathological cardiac hypertrophy. Circ. Res. 89, 591–598. doi: 10.1161/hh1901.096706
Klaus, A., Muller, M., Schulz, H., Saga, Y., Martin, J. F., and Birchmeier, W. (2012). Wnt/beta-catenin and Bmp signals control distinct sets of transcription factors in cardiac progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 109, 10921–10926. doi: 10.1073/pnas.1121236109
Leach, J. P., Heallen, T., Zhang, M., Rahmani, M., Morikawa, Y., Hill, M. C., et al. (2017). Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 550, 260–264. doi: 10.1038/nature24045
Lee, A., Hudson, A. R., Shiwarski, D. J., Tashman, J. W., Hinton, T. J., Yerneni, S., et al. (2019). 3D bioprinting of collagen to rebuild components of the human heart. Science 365, 482–487. doi: 10.1126/science.aav9051
Lee, K. F., Simon, H., Chen, H., Bates, B., Hung, M. C., and Hauser, C. (1995). Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 378, 394–398. doi: 10.1038/378394a0
Liu, J. P., Baker, J., Perkins, A. S., Robertson, E. J., and Efstratiadis, A. (1993). Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75, 59–72. doi: 10.1016/s0092-8674(05)80084-4
Liu, Y. W., Chen, B., Yang, X., Fugate, J. A., Kalucki, F. A., Futakuchi-Tsuchida, A., et al. (2018). Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat. Biotechnol. 36, 597–605. doi: 10.1038/nbt.4162
Luxan, G., Casanova, J. C., Martinez-Poveda, B., Prados, B., D’amato, G., MacGrogan, D., et al. (2013). Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat. Med. 19, 193–201. doi: 10.1038/nm.3046
Ma, S., Meng, Z., Chen, R., and Guan, K. L. (2019). The hippo pathway: biology and pathophysiology. Annu. Rev. Biochem. 88, 577–604. doi: 10.1146/annurev-biochem-013118-111829
MacGrogan, D., Munch, J., and De La Pompa, J. L. (2018). Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat. Rev. Cardiol. 15, 685–704. doi: 10.1038/s41569-018-0100-2
Mahmoud, A. I., Kocabas, F., Muralidhar, S. A., Kimura, W., Koura, A. S., Thet, S., et al. (2013). Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 497, 249–253. doi: 10.1038/nature12054
Maillet, M., Van Berlo, J. H., and Molkentin, J. D. (2013). Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 14, 38–48. doi: 10.1038/nrm3495
McBride, K. L., Riley, M. F., Zender, G. A., Fitzgerald-Butt, S. M., Towbin, J. A., Belmont, J. W., et al. (2008). NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand-induced signaling. Hum. Mol. Genet. 17, 2886–2893. doi: 10.1093/hmg/ddn187
McMullen, J. R., Shioi, T., Huang, W. Y., Zhang, L., Tarnavski, O., Bisping, E., et al. (2004). The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J. Biol. Chem. 279, 4782–4793. doi: 10.1074/jbc.m310405200
Mei, L., and Nave, K. A. (2014). Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 83, 27–49. doi: 10.1016/j.neuron.2014.06.007
Meyer, D., and Birchmeier, C. (1995). Multiple essential functions of neuregulin in development. Nature 378, 386–390. doi: 10.1038/378386a0
Mills, R. J., Parker, B. L., Quaife-Ryan, G. A., Voges, H. K., Needham, E. J., Bornot, A., et al. (2019). Drug screening in human PSC-cardiac organoids identifies pro-proliferative compounds acting via the mevalonate pathway. Cell Stem Cell 24, 895–907.e6.
Mohamed, T. M. A., Ang, Y. S., Radzinsky, E., Zhou, P., Huang, Y., Elfenbein, A., et al. (2018). Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell 173, 104–116.e12.
Mullur, R., Liu, Y. Y., and Brent, G. A. (2014). Thyroid hormone regulation of metabolism. Physiol. Rev. 94, 355–382.
Murry, C. E., and MacLellan, W. R. (2020). Stem cells and the heart-the road ahead. Science 367, 854–855. doi: 10.1126/science.aaz3650
Nakada, Y., Canseco, D. C., Thet, S., Abdisalaam, S., Asaithamby, A., Santos, C. X., et al. (2017). Hypoxia induces heart regeneration in adult mice. Nature 541, 222–227. doi: 10.1038/nature20173
Nakamura, M., and Sadoshima, J. (2018). Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 15, 387–407. doi: 10.1038/s41569-018-0007-y
Naqvi, N., Li, M., Calvert, J. W., Tejada, T., Lambert, J. P., Wu, J., et al. (2014). A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 157, 795–807. doi: 10.1016/j.cell.2014.03.035
Nguyen, N. U. N., Canseco, D. C., Xiao, F., Nakada, Y., Li, S., Lam, N. T., et al. (2020). A calcineurin-Hoxb13 axis regulates growth mode of mammalian cardiomyocytes. Nature 582, 271–276. doi: 10.1038/s41586-020-2228-6
Odiete, O., Hill, M. F., and Sawyer, D. B. (2012). Neuregulin in cardiovascular development and disease. Circ. Res. 111, 1376–1385. doi: 10.1161/circresaha.112.267286
Oka, T., Akazawa, H., Naito, A. T., and Komuro, I. (2014). Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ. Res. 114, 565–571. doi: 10.1161/circresaha.114.300507
Oka, T., Maillet, M., Watt, A. J., Schwartz, R. J., Aronow, B. J., Duncan, S. A., et al. (2006). Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ. Res. 98, 837–845. doi: 10.1161/01.res.0000215985.18538.c4
Page, D. J., Miossec, M. J., Williams, S. G., Monaghan, R. M., Fotiou, E., Cordell, H. J., et al. (2019). whole exome sequencing reveals the major genetic contributors to nonsyndromic Tetralogy of Fallot. Circ. Res. 124, 553–563.
Paige, S. L., Plonowska, K., Xu, A., and Wu, S. M. (2015). Molecular regulation of cardiomyocyte differentiation. Circ. Res. 116, 341–353. doi: 10.1161/circresaha.116.302752
Pasumarthi, K. B., Nakajima, H., Nakajima, H. O., Soonpaa, M. H., and Field, L. J. (2005). Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ. Res. 96, 110–118. doi: 10.1161/01.res.0000152326.91223.4f
Polizzotti, B. D., Ganapathy, B., Walsh, S., Choudhury, S., Ammanamanchi, N., Bennett, D. G., et al. (2015). Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci. Transl. Med. 7:281ra245.
Porrello, E. R., Mahmoud, A. I., Simpson, E., Hill, J. A., Richardson, J. A., Olson, E. N., et al. (2011). Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080. doi: 10.1126/science.1200708
Poss, K. D., Wilson, L. G., and Keating, M. T. (2002). Heart regeneration in zebrafish. Science 298, 2188–2190. doi: 10.1126/science.1077857
Protze, S. I., Lee, J. H., and Keller, G. M. (2019). Human pluripotent stem cell-derived cardiovascular cells: from developmental biology to therapeutic applications. Cell Stem Cell 25, 311–327. doi: 10.1016/j.stem.2019.07.010
Puente, B. N., Kimura, W., Muralidhar, S. A., Moon, J., Amatruda, J. F., Phelps, K. L., et al. (2014). The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157, 565–579. doi: 10.1016/j.cell.2014.03.032
Reiss, K., Cheng, W., Ferber, A., Kajstura, J., Li, P., Li, B., et al. (1996). Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 93, 8630–8635. doi: 10.1073/pnas.93.16.8630
Ronaldson-Bouchard, K., Ma, S. P., Yeager, K., Chen, T., Song, L., Sirabella, D., et al. (2018). Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243. doi: 10.1038/s41586-018-0016-3
Sadek, H., and Olson, E. N. (2020). Toward the goal of human heart regeneration. Cell Stem Cell 26, 7–16. doi: 10.1016/j.stem.2019.12.004
Saltiel, A. R., and Kahn, C. R. (2001). Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806. doi: 10.1038/414799a
Scuderi, G. J., and Butcher, J. (2017). Naturally engineered maturation of cardiomyocytes. Front. Cell Dev. Biol. 5:50.
Sdek, P., Zhao, P., Wang, Y., Huang, C. J., Ko, C. Y., Butler, P. C., et al. (2011). Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. J. Cell Biol. 194, 407–423. doi: 10.1083/jcb.201012049
Semenza, G. L. (2014). Hypoxia-inducible factor 1 and cardiovascular disease. Annu. Rev. Physiol. 76, 39–56. doi: 10.1146/annurev-physiol-021113-170322
Shiojima, I., Sato, K., Izumiya, Y., Schiekofer, S., Ito, M., Liao, R., et al. (2005). Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest. 115, 2108–2118. doi: 10.1172/jci24682
Siebel, C., and Lendahl, U. (2017). Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 97, 1235–1294. doi: 10.1152/physrev.00005.2017
Snir, M., Kehat, I., Gepstein, A., Coleman, R., Itskovitz-Eldor, J., Livne, E., et al. (2003). Assessment of the ultrastructural and proliferative properties of human embryonic stem cell-derived cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 285, H2355–H2363.
Soonpaa, M. H., Koh, G. Y., Pajak, L., Jing, S., Wang, H., Franklin, M. T., et al. (1997). Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J. Clin. Invest. 99, 2644–2654. doi: 10.1172/jci119453
Soonpaa, M. H., Zebrowski, D. C., Platt, C., Rosenzweig, A., Engel, F. B., and Field, L. J. (2015). Cardiomyocyte cell-cycle activity during preadolescence. Cell 163, 781–782. doi: 10.1016/j.cell.2015.10.037
Spater, D., Hansson, E. M., Zangi, L., and Chien, K. R. (2014). How to make a cardiomyocyte. Development 141, 4418–4431. doi: 10.1242/dev.091538
Swynghedauw, B. (1999). Molecular mechanisms of myocardial remodeling. Physiol. Rev. 79, 215–262. doi: 10.1152/physrev.1999.79.1.215
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Tamemoto, H., Kadowaki, T., Tobe, K., Yagi, T., Sakura, H., Hayakawa, T., et al. (1994). Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 372, 182–186. doi: 10.1038/372182a0
Taniguchi, C. M., Emanuelli, B., and Kahn, C. R. (2006). Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96. doi: 10.1038/nrm1837
Tao, G., Kahr, P. C., Morikawa, Y., Zhang, M., Rahmani, M., Heallen, T. R., et al. (2016). Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 534, 119–123. doi: 10.1038/nature17959
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. doi: 10.1126/science.282.5391.1145
Vagnozzi, R. J., Maillet, M., Sargent, M. A., Khalil, H., Johansen, A. K. Z., Schwanekamp, J. A., et al. (2020). An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 577, 405–409. doi: 10.1038/s41586-019-1802-2
van Berlo, J. H., Kanisicak, O., Maillet, M., Vagnozzi, R. J., Karch, J., Lin, S. C., et al. (2014). c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509, 337–341. doi: 10.1038/nature13309
van Berlo, J. H., Maillet, M., and Molkentin, J. D. (2013). Signaling effectors underlying pathologic growth and remodeling of the heart. J. Clin. Invest. 123, 37–45. doi: 10.1172/jci62839
von Gise, A., Lin, Z., Schlegelmilch, K., Honor, L. B., Pan, G. M., Buck, J. N., et al. (2012). YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 109, 2394–2399. doi: 10.1073/pnas.1116136109
Wang, J., Liu, S., Heallen, T., and Martin, J. F. (2018). The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 15, 672–684. doi: 10.1038/s41569-018-0063-3
Wang, S., Ye, L., Li, M., Liu, J., Jiang, C., Hong, H., et al. (2016). GSK-3beta Inhibitor CHIR-99021 Promotes Proliferation Through Upregulating beta-Catenin in Neonatal Atrial Human Cardiomyocytes. J Cardiovasc Pharmacol 68, 425–432. doi: 10.1097/fjc.0000000000000429
Xin, M., Kim, Y., Sutherland, L. B., Murakami, M., Qi, X., Mcanally, J., et al. (2013). Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 13839–13844. doi: 10.1073/pnas.1313192110
Xin, M., Kim, Y., Sutherland, L. B., Qi, X., Mcanally, J., Schwartz, R. J., et al. (2011). Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 4:ra70. doi: 10.1126/scisignal.2002278
Yang, X., Pabon, L., and Murry, C. E. (2014). Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 114, 511–523. doi: 10.1161/circresaha.114.300558
Ye, L., D’agostino, G., Loo, S. J., Wang, C. X., Su, L. P., Tan, S. H., et al. (2018). Early regenerative capacity in the porcine heart. Circulation 138, 2798–2808. doi: 10.1161/circulationaha.117.031542
Ye, L., Qiu, L., Feng, B., Jiang, C., Huang, Y., Zhang, H., et al. (2020). Role of blood oxygen saturation during post-natal human cardiomyocyte cell cycle activities. JACC Basic Transl. Sci. 5, 447–460. doi: 10.1016/j.jacbts.2020.02.008
Yu, F. X., Zhao, B., and Guan, K. L. (2015). Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163, 811–828. doi: 10.1016/j.cell.2015.10.044
Yutzey, K. E. (2015). Regenerative biology: neuregulin 1 makes heart muscle. Nature 520, 445–446. doi: 10.1038/520445a
Zhao, L., Ben-Yair, R., Burns, C. E., and Burns, C. G. (2019a). Endocardial notch signaling promotes cardiomyocyte proliferation in the regenerating Zebrafish heart through Wnt Pathway Antagonism. Cell Rep. 26, 546–554.e5.
Zhao, L., Borikova, A. L., Ben-Yair, R., Guner-Ataman, B., Macrae, C. A., Lee, R. T., et al. (2014). Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc. Natl. Acad. Sci. U.S.A. 111, 1403–1408. doi: 10.1073/pnas.1311705111
Zhao, M. T., Chen, H., Liu, Q., Shao, N. Y., Sayed, N., Wo, H. T., et al. (2017). Molecular and functional resemblance of differentiated cells derived from isogenic human iPSCs and SCNT-derived ESCs. Proc. Natl. Acad. Sci. U.S.A. 114, E11111–E11120.
Zhao, M. T., Shao, N. Y., and Garg, V. (2020). Subtype-specific cardiomyocytes for precision medicine: Where are we now? Stem Cells. doi: 10.1002/stem.3178 [Epub ahead of print].
Zhao, Y., Rafatian, N., Feric, N. T., Cox, B. J., Aschar-Sobbi, R., Wang, E. Y., et al. (2019b). A platform for generation of chamber-specific cardiac tissues and disease modeling. Cell 176, 913–927.e18.
Keywords: cardiomyocyte proliferation, cardiomyocyte maturation, cardiomyocyte hypertrophy, induced pluriopotent stem cells, cardiac stem cell therapy, cardiac regeneration
Citation: Zhao M-T, Ye S, Su J and Garg V (2020) Cardiomyocyte Proliferation and Maturation: Two Sides of the Same Coin for Heart Regeneration. Front. Cell Dev. Biol. 8:594226. doi: 10.3389/fcell.2020.594226
Received: 12 August 2020; Accepted: 25 September 2020;
Published: 15 October 2020.
Edited by:
Ge Tao, Medical University of South Carolina, United StatesCopyright © 2020 Zhao, Ye, Su and Garg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming-Tao Zhao, mingtao.zhao@nationwidechildrens.org