A Balanced Act: The Effects of GH–GHR–IGF1 Axis on Mitochondrial Function
 Bowen Hu
Bowen Hu Hongmei Li
Hongmei Li Xiquan Zhang
Xiquan Zhang- 1State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources, South China Agricultural University, Guangzhou, China
- 2Guangdong Provincial Key Lab of AgroAnimal Genomics and Molecular Breeding, Key Lab of Chicken Genetics, Breeding and Reproduction, Ministry of Agriculture, Guangzhou, China
Mitochondrial function is multifaceted in response to cellular energy homeostasis and metabolism, with the generation of adenosine triphosphate (ATP) through the oxidative phosphorylation (OXPHOS) being one of their main functions. Selective elimination of mitochondria by mitophagy, in conjunction with mitochondrial biogenesis, regulates mitochondrial function that is required to meet metabolic demand or stress response. Growth hormone (GH) binds to the GH receptor (GHR) and induces the JAK2/STAT5 pathway to activate the synthesis of insulin-like growth factor 1 (IGF1). The GH–GHR–IGF1 axis has been recognized to play significant roles in somatic growth, including cell proliferation, differentiation, division, and survival. In this review, we describe recent discoveries providing evidence for the contribution of the GH–GHR–IGF1 axis on mitochondrial biogenesis, mitophagy (or autophagy), and mitochondrial function under multiple physiological conditions. This may further improve our understanding of the effects of the GH–GHR–IGF1 axis on mitochondrial function, which may be controlled by the delicate balance between mitochondrial biogenesis and mitophagy. Specifically, we also highlight the challenges that remain in this field.
Introduction
Growth hormone (GH), also known as somatotropin, is an amino acid peptide that, together with prolactin (PRL) and human placental lactogen, belongs to the somatotropin family (Strobl and Thomas, 1994). GH is produced by the pituitary gland, which is under the positive control of the hypothalamic peptide GH-releasing hormone (GHRH) and the negative feedback of somatostatin. GH mediates its functions directly through its receptor (GHR) or indirectly via insulin-like growth factor 1 (IGF1) (Yang et al., 2008). Meanwhile, a complex feedback system can regulate GH secretion, including IGF-1, leptin, and ghrelin along with the central nervous system.
GH receptor, a member of the class I cytokine receptor family, is an amino acid dimeric receptor with an extracellular domain (ECD), a single-pass transmembrane domain (IMD), and a cytoplasmic intracellular domain (ICD) (Dehkhoda et al., 2018). GHR is widely expressed in GH target cells, which can combine with GH to activate diverse signal cascades, including mitogenic signaling through Janus kinase (JAKs) signal transducers and activators of transcription (STATs) (Brooks et al., 2008), mitogen-activated protein kinase (MAPK) (Vanderkuur et al., 1997), phosphoinositide-3-kinase (PI3K)/Protein kinase B (PKB or AKT)/mammalian target of rapamycin (mTOR) pathways (Hayashi and Proud, 2007) and phospholipase C (PLC)/Protein kinase C (PKC) (Bergan et al., 2015). Accordingly, normal function of GHR directly influences the physiological effects of GH. The deficiency in GHR will further disrupt the normal development of the organism and lead to dwarfism phenotype in a wide array of species (Lin et al., 2018).
Insulin-like growth factor 1, the main mediator of GH actions, is an amino acid insulinoid peptide with an amino acid sequence similar to that of proinsulin (positions 1–29 are homologous to insulin B chain and positions 42–62 to insulin A chain). GH combines with GHR to regulate IGF1 production via the JAK2/STAT5 pathway through endocrine and paracrine/autocrine mechanisms (Sjogren et al., 1999; Junnila et al., 2013). IGF1 mainly occurred in the liver and also in several tissues including the brain, testes, skeletal muscle, bone, and cartilage. Meanwhile, the roles of GH and IGF1 are influenced by GH-binding proteins (GHBPs) and IGF-binding proteins (IGFBPs), respectively (Baumann et al., 1988; Duan and Xu, 2005). Analogous to GH, IGF1 acts through its receptor (IGF1R), a tyrosine kinase receptor that can activate multiple pathways including the PI3K/AKT, MAPK, and PLC pathways (Hakuno and Takahashi, 2018). However, GH and IGF-1 have very different roles on glucose and lipid metabolism. GH primarily blocks the action of insulin, promotes lipolysis, and prevents fat production, whereas IGF1 has the opposite effects (Moller and Jorgensen, 2009).
Overall, the GH–GHR–IGF1 axis, part of the somatotropic–hypothalamic–pituitary axis, has been commonly recognized in response to somatic growth, including cell proliferation, differentiation, division, and survival (Figure 1). On the other hand, the GH–GHR–IGF1 axis also plays essential roles in mitochondrial function with an unexpected complexity and versatility regulation mechanisms. In this review, we describe recent discoveries providing evidence for the contribution of the GH–GHR–IGF1 axis on mitochondrial biogenesis, mitophagy (or autophagy), and mitochondrial function under multiple physiological conditions. Based on this integrative view, we also emphasize the remaining challenges in this field. Besides, there is a long list of studies utilizing different cell lines or mice with varying membership of the GH–GHR–IGF1 axis, showing the effects of the GH–GHR–IGF1 axis on aging and cellular senescence. Although important, they were not described primarily in this review.
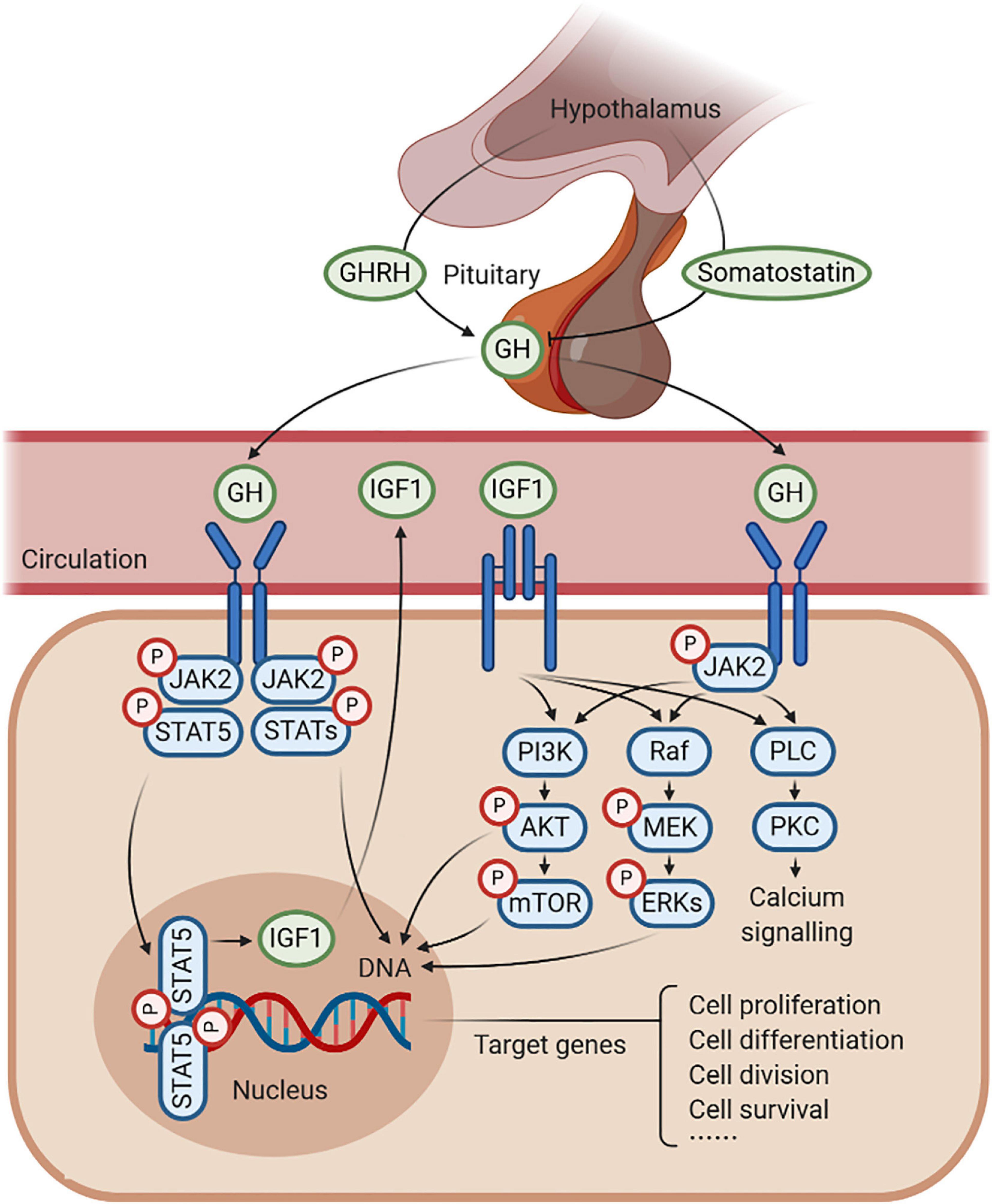
Figure 1. Schematic representation of growth hormone (GH)–GH receptor (GHR)–insulin-like growth factor 1 (IGF1) axis. GH is produced by the pituitary gland, which is under the positive control of the GH-releasing hormone (GHRH) and the negative feedback of somatostatin. GH combines with GHR to regulate the production of IGF1, in response to somatic growth, including cell proliferation, cell differentiation, cell division, and cell survival. In this process, several signaling pathways are activated, such as JAK2/STAT5, MAPK, and PI3K/AKT signaling under an anabolic state (such as during periods of feeding), as well as PLC/PKC signaling under a catabolic state (such as during periods of fasting).
Overview of the Mitochondrial Biogenesis
Mitochondria are double membrane-bound organelles, especially with the generation of adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS) being one of their main functions. The OXPHOS proteins comprise five complexes (I–V) embedded in the inner mitochondrial membrane that are uniquely controlled by mitochondrial DNA (mtDNA) and the nuclear DNA (nDNA). In mammals, the mtDNA encodes only 37 genes, of which 13 are subunits of the OXPHOS (complexes I, III, IV, and V), two are rRNA genes, and 22 are tRNA genes required for mitochondrial protein synthesis.
Mitochondrial biogenesis is a self-renewal process by which new mitochondria are produced from the ones already existing. The sophisticated process of mitochondrial biogenesis requires coordination between mtDNA and nDNA, including mtDNA transcription and translation, translation of nDNA-encoded transcripts, membrane recruitment, protein import, and assembly of the OXPHOS complexes (Attardi and Schatz, 1988). So far, it has been widely accepted that the members of the peroxisome proliferative-activated receptor gamma coactivator 1 (PGC1) family of coactivators act as key players in the regulation of energy metabolism and mitochondrial biogenesis. PPARG coactivator 1 alpha (PGC1α) was first described in 1999 (Wu et al., 1999), and PPARG coactivator 1 beta (PGC1β) along with PPARG related-coactivator 1 (PRC) were discovered subsequently (Andersson and Scarpulla, 2001; Lin et al., 2002). These coactivators generally orchestrate with DNA-bound transcription factors, such as nuclear respiratory factors (NRF1, NRF2), estrogen-related receptors (ERRα, ERRβ, and ERRγ), and myocyte enhancer factor 2 (MEF2) to drive the expression of target genes. Furthermore, their regulations on transcription or post-translation level are responsive to meet the multiple metabolic demands induced by physiological signals, senescence, and diseases (Cui et al., 2006; Bellance et al., 2009; D’Errico et al., 2011; Patel et al., 2012; Tsunemi and La Spada, 2012). For instance, PGC1-related coactivators can activate the expression of NRF1, NRF2, and transcription factor A (TFAM, the final effectors of mtDNA transcription and replication) to regulate the expression of respiratory chain and the biogenesis of mitochondria, a process that has been well-documented in previous reviews (Gleyzer et al., 2005; Scarpulla, 2008; Scarpulla et al., 2012; Villena, 2015; Popov, 2020). Therefore, we commonly regard the expression of PGC1α, NRF1, TFAM, and mitochondrial-related genes, as well as mtDNA copy number as the markers for mitochondrial biogenesis. Notably, a recent study has underlined that the relationship between TFAM expression and mitochondrial biogenesis is more complex than is generally appreciated and may be ambiguous in most mammalian cells. As TFAM does not always exhibit parallel with the mtDNA copy number, TFAM should be used judiciously as a marker of mitochondrial biogenesis (Kozhukhar and Alexeyev, 2019).
Overview of the Mitophagy
Autophagy, meaning “self-eating” in Greek, is a process by which portions of cytoplasm, such as the organelles and protein aggregates, are sequestered and subsequently delivered to lysosomes for degradation (Klionsky et al., 2003; Nakatogawa et al., 2009). Three major types of autophagic pathways, macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), have been recognized in eukaryotic cells (Mizushima and Komatsu, 2011). Macroautophagy (hereafter referred to as autophagy) sequesters cytosolic cargo, mainly organelles, by a double membrane vesicle called autophagosome, which is formed through conjugation of specific proteins among themselves in an intricate process. Subsequently, autophagosome is fused with endolysosome to constitute autolysosome, in which the cytosolic cargo is degraded. Although autophagy was first considered to perform in a bulk manner, it is now clear that autophagy is mainly a selective process that originally encounters considerable resistance (Mizumura et al., 2014).
Early in the 20th century, the concept of mitochondrial degradation was proposed by Margaret and Warren Lewis (Lewis and Lewis, 1915). Later, mitochondria sequestered in lysosomes were first observed in rat tissues by electron microscopy studies (Ashford and Porter, 1962; Novikoff and Essner, 1962). Until the beginning of the 21st century, the idea of mitochondrial autophagy was initially termed as “mitophagy” to define the selective elimination of depolarized mitochondria in autophagosome (Scott and Klionsky, 1998; Elmore et al., 2001; Lemasters, 2005). Up to now, mitophagy is considered as a selective form of autophagy to deliver dysfunctional or superfluous mitochondria to the lysosome for degradation, which has exhibited an essential contribution to cell homeostasis under different stimuli or cellular contexts, including cellular differentiation, oxidative stress response, and aging, as well as various disease conditions (Um and Yun, 2017; Palikaras et al., 2018). In yeast, Ohsumi’s and Klionsky’s laboratories first identified that ATG32 acts as the mitophagy-specific receptor, which directly interacts with the selective autophagy adaptor ATG8 via an ATG8-interacting motif (AIM) and ATG11 to form the nascent autophagosome (Kanki et al., 2009; Okamoto et al., 2009). Recently, ATG43 is identified as another mitophagy-specific receptor, which is localized on the mitochondrial outer membrane and binds to ATG8 (Fukuda and Kanki, 2021).
In mammals, the regulation of mitophagy appears to be more complex, both ubiquitin-mediated and receptor-mediated pathways have been described in response to mitophagy. To our knowledge, the most studied and understood mitophagy pathway is mediated by PTEN-induced kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin (Narendra et al., 2008; Vives-Bauza et al., 2010), both of which have been linked to forms of Parkinson’s disease (Kitada et al., 1998; Valente et al., 2004). The complicated mechanisms of canonical and non-canonical PINK1/parkin-mediated mitophagy have been well summarized in previous reviews (Eiyama and Okamoto, 2015; Lazarou et al., 2015; Nguyen et al., 2016; Clark et al., 2020; Malpartida et al., 2020). Moreover, two main types of receptor-mediated mitophagy pathway have been classified as follows in brief: ubiquitin-independent mitophagy receptors, including BCL2-interacting protein 3 (BNIP3) (Quinsay et al., 2010), BCL2-interacting protein 3 like (NIX/BNIP3L) (Sandoval et al., 2008), FUN14 domain-containing 1 (FUNDC1) (Liu et al., 2012), BCL2-like 13 (BCL2L13) (Otsu et al., 2015), autophagy and beclin 1 regulator 1 (AMBRA1) (Strappazzon et al., 2015), FKBP prolyl isomerase 8 (FKBP8) (Bhujabal et al., 2017), prohibitin 2 (PHB2) (Wei et al., 2017), and NLR family member X1 (NLRX1) (Zhang et al., 2019); lipid-mediated mitophagy receptors, including ceramide (Sentelle et al., 2012) and cardiolipin (Li et al., 2015). It has been identified that these mitophagy receptors can directly interact with the autophagy mediators LC3/GABARAP via a conserved LC3-interacting region (LIR) motif or ULK1 protein to form the nascent autophagosome. These intricate processes of receptor-mediated mitophagy pathways also have been well summarized in previous reviews (Liu et al., 2014; Ploumi et al., 2017; Villa et al., 2018; Montava-Garriga and Ganley, 2020). However, whether each type of mitophagy receptor functions in a distinct pathway, or there is cooperation between them under various mitochondrial stresses are still not completely understood.
A Balanced Act of Mitochondrial Biogenesis and Mitophagy
Mitochondrial function is multifaceted in response to cellular energy homeostasis and metabolism, including calcium homeostasis, amino acid metabolism, pyridine synthesis, cellular replication, apoptosis, reactive oxygen species (ROS) production, and senescence (Spinelli and Haigis, 2018). In order to perform these many functions, mitochondria are structured in a dynamic network where, for instance, mitochondria biogenesis, elimination, fission, and fusion are harmoniously orchestrated (Ploumi et al., 2017). Accordingly, maintenance of a healthy mitochondrial network, defined as mitochondrial homeostasis, is critical for normal mitochondrial function during development and even throughout life. Like ancient Chinese philosophy “Ying” and “Yang,” both generation of newly synthesized mitochondria, by mitochondrial biogenesis, and elimination of detrimental and/or superfluous mitochondria, by mitophagy, are predominantly required for maintaining mitochondrial homeostasis. Recent findings have hinted that any abnormality in these two opposing processes can influence the quantity and quality of mitochondria, which will further affect mitochondrial function and the ability of cells to adjust their mitochondrial networks in response to physiological adaptations and stress conditions (Palikaras et al., 2015; Singh et al., 2018; Wang et al., 2019; Yau et al., 2019; Zhou et al., 2019; Chen et al., 2020). At the same time, impaired mitochondrial function and homeostasis are now widely accepted to be associated with multiple aspects of the aging process and age-onset diseases (Lopez-Otin et al., 2013; Mattson and Arumugam, 2018; Akbari et al., 2019).
Therefore, maintaining the healthy function of mitochondria by biogenesis and mitophagy is conducive to cellular life activity. The balance between mitochondrial biogenesis and mitophagy requires delicate regulation to maintain a sustainable mitochondria population in healthy cells (Pickles et al., 2018). Several signaling pathways have been implicated in both mitochondrial biogenesis and mitophagy, and they may play important roles in coordinating these processes (Figure 2). For instance, cyclic-AMP (cAMP) is one of the upstream signals that regulate both mitochondrial biogenesis and mitophagy. cAMP level can regulate the protein kinase A (PKA)-dependent activation of the cAMP response element-binding protein (CREB), which in turn upregulates the expression of PGC1α and inhibits LC3-II (Cherra et al., 2010; Chowanadisai et al., 2010). Also, mammalian target of rapamycin (mTOR) signaling promotes mitochondrial biogenesis by activation of PGC1α and ERRα, and inhibits mitophagy by phosphorylation of ULK1 or inhibition of PINK1 expression and Parkin translocation (Cunningham et al., 2007; Bartolome et al., 2017). Under energy stress condition, AMP-activated protein kinase (AMPK) promotes mitochondrial biogenesis through phosphorylation of SIRT1 to activate PGC1α, and promotes mitophagy through inhibition of mTOR and activation of ULK1 (Herzig and Shaw, 2018). Also, mitogen-activated protein kinase (MAPK) signaling is associated with mitochondrial homeostasis, MAPK1/3 potently inhibit mitochondrial biogenesis (Zhu et al., 2012); however, p38 MAPK exerts a positive regulation on PGC1α, and MAPK1 along with MAPK14 promote both starvation- and hypoxia-induced mitophagy in HeLa cells (Akimoto et al., 2005; Hirota et al., 2015).
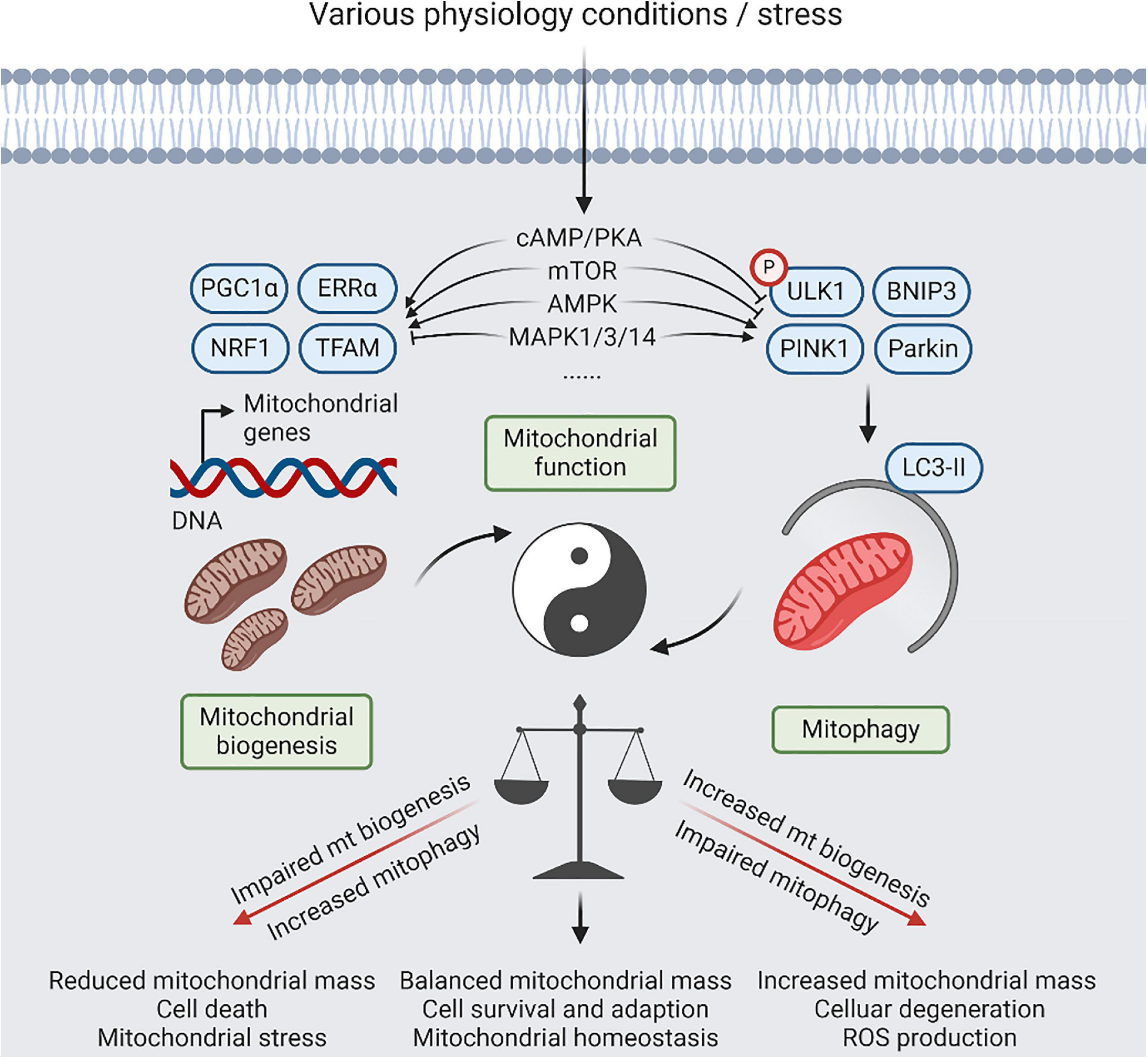
Figure 2. A balanced act of mitophagy and mitochondrial biogenesis. Coordination between mitochondrial (mt) biogenesis and mitophagy results in generation of new synthesized mitochondria, and elimination of detrimental and/or superfluous mitochondria, which is controlled by several signaling pathways, including cyclic-AMP (cAMP)/protein kinase A (PKA), AMP-activated protein kinase (AMPK), mitogen-activated protein kinase (MAPK), and mammalian target of rapamycin (mTOR) signaling, in response to physiological adaptations and stress conditions. Relative to normal condition, increased mitophagy or impaired mt biogenesis will lead to reduced mitochondria mass, contributing to cell death that be dependent on mitochondrial function. Reversely, impaired mitophagy or increased mt biogenesis will lead to imbalanced responses, resulting in increased mitochondria mass, increased reactive oxygen species (ROS) production, and cellular degeneration. However, restoration of mitochondrial homeostasis with increased mitochondrial damage will require simultaneous upregulation of mitophagy and mitochondrial biogenesis.
On the other hand, in addition to its well-known functions, PGC1α, a master regulator of mitochondrial biogenesis, positively regulates mitophagy by directly inducing the expression of transcription factor EB (TFEB) that mediates lysosomal biogenesis (Settembre et al., 2011). NRF2 is also required to couple mitochondrial biogenesis with mitophagy by inducing BNIP3 expression, which is essential for facilitating cancer progression (Riis et al., 2020). Meanwhile, Parkin is involved in the regulation of mitochondrial biogenesis via PARIS, a transcription factor that negatively regulates PGC1a and its target NRF1 or directly enhances TFAM-mediated mitochondrial transcription in proliferating cells (Kuroda et al., 2006; Shin et al., 2011). Furthermore, it was found that the general control of amino acid synthesis 5-like 1 (GCN5L1) negatively regulates both mitochondrial biogenesis and degradation pathways through acting on both PGC1a and TFEB in mouse embryonic fibroblasts (Scott et al., 2014).
So far, many upstream signals have been implicated in both mitochondrial biogenesis and mitophagy as described above. However, there is still a lack of systematic research on whether there are interactions among them. Notably, the versatility of the key regulators of mitochondrial biogenesis (PGC1α, NRF2) and mitophagy (Parkin) may further underline that these two processes are balanced and constrained with each other in order to control mitochondrial function under multiple physiological conditions.
The Effects of Growth Hormone on Mitochondrial Biogenesis
The relationship between GH and mitochondrial biogenesis was initially explored in the early 1970s. The first question is whether the GH performs a direct interaction with mitochondrial membranes (Maddaiah et al., 1970). After administration of radio-labeled bovine GH to rats, radioactive signals were detected in the mitochondria of liver and kidney (Groves et al., 1972). Later, this question was further explored by Mutvei et al. in hypophysectomized rats treated with T3 and/or human GH by the continuous infusion of hormone for 6 days. Intriguingly, they found that T3 exerts a direct effect on mitochondrial biogenesis. However, the high-affinity binding sites for GH are not present in the liver mitochondria; only a few negligible amounts of radio-labeled bovine GH are transported to the mitochondria compared with other subcellular compartments (Mutvei et al., 1989). This compelling evidence demonstrates that mitochondria are not a direct target for GH and/or its receptor.
Growth hormone is considered to be a conducive hormone that enhances mitochondrial biogenesis. Human GH treatment of hypophysectomized rats increases incorporation of leucine in vivo and in vitro, indicating that GH has a significant effect on liver mitochondrial protein synthesis (Maddaiah et al., 1973). Administration of either human or bovine GH restores the cytochrome level and increases the cytochrome oxidase activity in hypophysectomized rats (Maddaiah et al., 1976). Also, administration of bovine GH to hypophysectomized rats partially restores the respiration rate and ATPase activity of liver mitochondria, and increases the heart mitochondrial protein synthesis measured by the incorporation capacity of radioactive leucine (Katkocin et al., 1979; Maddaiah and Clejan, 1986). Furthermore, GH replacement therapy restores the age-associated impairments in the skeletal muscle mitochondrial biogenesis, which is mainly manifested by the increased PGC1α, NRF1, cytochrome c expression, and citrate synthase enzymatic activity (Brioche et al., 2014). Consistently, PGC1α protein level is diminished in bovine GH transgenic (bGH Tg) mice, which overexpresses GH and are short-lived (Al-Regaiey et al., 2005). These investigations are consistent with the results in the early 1970s showing the beneficial effects of GH on mitochondrial biogenesis.
On the other hand, some different views concerning the effects of GH on mitochondrial biogenesis have also been discovered. The expression of PGC1α and OXPHOS activities are increased in the long-living Ames dwarf mice (lack of GH, prolactin, and thyroid-stimulating hormone), demonstrating a negative effect of GH on mitochondrial biogenesis (Westbrook et al., 2009; Brown-Borg et al., 2012). Moreover, Mutvei et al. (1989) suggested that GH is not a major regulator of mammalian mitochondrial biogenesis. They considered that previous results concerning the effect of GH on mitochondrial biogenesis were based on the incorrect interpretations of the data, as increased respiration does not necessarily reflect increased mitochondrial biogenesis. Similarly, it was recently reported that the expression of mitochondria-specific markers (PGC1α, AMPKα, SIRT1, and cytochrome b, etc.) along with the protein levels of electron transport chain (complexes I, II, III, IV, and V) in osteocytes are not significant differences between bGH Tg and control mice (Liu et al., 2019). Taken together, these different results mentioned above indicate that GH may account for complex functions on mitochondrial biogenesis during different physiological conditions in vivo and in vitro.
The Effects of Growth Hormone on Mitophagy
Mitophagy is a well-studied type of cargo-specific autophagy to selectively eliminate mitochondria. However, no direct relationship between GH and mitophagy has been reported. Therefore, in this section, we only summarize the connection between GH and autophagy, which may throw the rope for the future researches.
Early study initially suggested that disruptions of the insulin or GH/IGF1 axis with low insulin and IGF1 levels may enhance autophagy to prevent the age-related mitochondrial degradation and extend the lifespan (Bergamini et al., 2003). Wang and Miller (2012) demonstrated that fibroblasts in Snell mice (which secrete very low amounts of GH, prolactin, and thyroid-stimulating hormone) are more susceptible to autophagy induced by amino acid withdrawal or by oxidative stress than control cells. They also found evidence of reduced mTOR function in dwarf cells under autophagy conditions, which is consistent with the evidence that increased autophagy requires lower mTOR activity (Wang and Miller, 2012). Furthermore, somatostatin analog treatment might induce apoptosis, increase autophagy, and decrease cell proliferation in GH-secreting adenomas (Dagistanli et al., 2018). A recent study also showed that reduced GH signaling in the liver of Snell mice upregulates the CMA (Endicott et al., 2020). These findings indicate a negative regulatory effect of GH on autophagy.
In contrast with these results, lack of the GH secretagogue ghrelin causes lethal hypoglycemia in mice under fasted and fat-depleted state; however, the wild-type mice under the same conditions exhibit a massive increase in plasma GH and hepatic autophagy, suggesting the positive connection between the plasma GH and hepatic autophagy (Zhang et al., 2015). Besides, GH acts through its receptor GHR in the liver to activate autophagy, preserve triglycerides, enhance gluconeogenesis, and prevent hypoglycemia in calorie-restricted mice (Fang et al., 2019). Accordingly, similar to the effects of GH on mitochondrial biogenesis, the findings above indicate the different roles of GH on autophagy during diverse physiological conditions in vivo.
The Effects of Growth Hormone Receptor on Mitochondrial Biogenesis
In recent decades, genetically engineered mouse strains (GHR gene disrupted or knockout mouse) have become vital tools for exploring the various activities of GH and GHR in vivo (List et al., 2020). Similarly, the genetic background of the sex-linked dwarf (SLD) chickens, which are caused by GHR gene mutations, also allows this strain to become a model system to understand the roles of GHR in vivo (Luo et al., 2016).
Early in the 21st century, GHR is considered to exhibit a negative effect on the process of mitochondrial biogenesis in vivo. Al-Regaiey et al. (2005) published a paper in which they described that PGC1α protein level was increased in the liver of male GHRKO mice. The gene expression and protein level of mitochondrial biogenesis markers, including PGC1α, AMPK, SIRT3, eNOS, and MFN2, are increased in GHRKO mice (Gesing et al., 2011a, b). A recent study also showed that the PGC1α protein levels in liver are significantly increased in both male and female GHRKO pigs compared with sex-matched controls (Riedel et al., 2020). However, the decrease in NRF1 and TFAM expression in the skeletal muscles and TFAM expression in kidneys of GHRKO mice was also demonstrated (Gesing et al., 2011b). The authors suggested that the decrease in TFAM may reflect potentially unaltered mtDNA content in GHRKO mice (Gesing et al., 2013). Notably, the increased expression of key regulators of mitochondrial biogenesis in GHRKO mice is not improved further by calorie restriction or visceral fat removal (Gesing et al., 2011b).
However, several recent findings are different from the previous results. Our previous study has revealed that the gene expression of mitochondrial biogenesis markers (PGC1α, NRF1, and TFAM) and mtDNA-encoded OXPHOS genes are all downregulated in the skeletal muscle of SLD chickens and GHR knockdown cells (Hu et al., 2019). At the same time, we observed that the enzymatic activities of OXPHOS complexes (complexes I, II, III, and IV) are reduced in the skeletal muscle of SLD chickens and GHR knockdown cells, indicating that GHR exhibits a positive effect on mitochondrial biogenesis. Furthermore, the expression of mitochondria-specific markers (PGC1α, AMPKα, SIRT1, and cytochrome b, etc.) and the protein levels of OXPHOS complexes (complexes I, II, III, IV, and V) in osteocytes are not significantly different between GHRKO and control mice (Liu et al., 2019). This investigation suggested that GHR has no effect on mitochondrial biogenesis, at least in vitro. On the other hand, the gene expression of mitochondrial biogenesis markers (PGC1α, AMPK, SIRT1, NRF2, and MFN2) and mitochondrial activity marker (COXIV) in liver-specific GHRKO (LiGHRKO) and wild-type mice are significantly different between the males and females, suggesting that sexual dimorphism may also play an essential role in regulating the mitochondrial biogenesis (Zawada et al., 2015).
As the middle of the GH–GHR–IGF1 axis, GHR plays a pivotal role in its functions. Taking into account these important observations, it seems that the effects of GH and GHR on mitochondria biogenesis are similar, as they both exhibit a multifaceted feature that are summarized in Table 1. There may be many explanations accounting for this difference. One explanation might be that the roles of GH and GHR on mitochondrial biogenesis may be different among cell-, organ-, and species-specific factors. The other explanation might be that mitochondrial biogenesis per se is not always assayed. The induction or repression of some mitochondrial markers is not always representative of the expansion of the mitochondrial network.
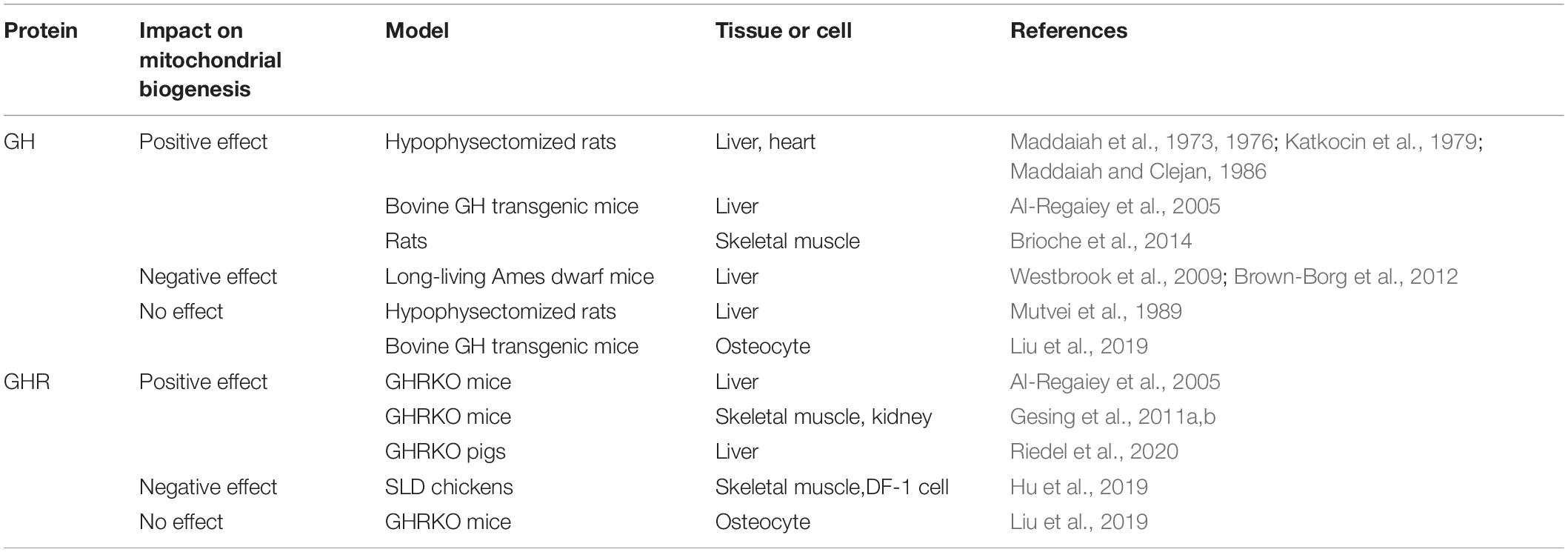
Table 1. The multiple effects of growth hormone (GH) and growth hormone receptor (GHR) on mitochondrial biogenesis.
The Effects of Growth Hormone Receptor on Mitophagy
In recent decades, similar to GH, no direct relationship has been reported between GHR and mitophagy. Therefore, in this part, we merely summarized the connection between GHR and autophagy from sporadic studies. It was found that GHR expression and its protein level are reduced in the skeletal muscle of ATG7 knockout mice, implying a synchronous relationship between GHR and autophagy (Zecchini et al., 2019). Furthermore, the level of the autophagy marker LC3-II is increased in GHRKO osteocytes (Liu et al., 2019). A recent study also showed that LC3-II flux is increased in the liver of GHRKO mice, but unaltered in LiGHRKO mice (Endicott et al., 2020). Up to now, the above two studies suggest a negative regulatory effect of GHR on autophagy. These new findings only reveal a fraction of the relationship between GHR and autophagy. Meanwhile, it is currently observed that these multifunctional effects of GHR on autophagy may be similar to GH. In the future, more research will be needed to deepen our understanding of the relationship between GHR and autophagy, even mitophagy.
The Effects Of Insulin-Like Growth Factor 1 on Mitochondrial Biogenesis
Cells generate new mitochondria when stimulated by extracellular factors to grow and divide. Numerous studies have aimed at assessing the effects of IGF1 on mitochondrial biogenesis. Neuregulin and IGF1 can act synergistically to increase mitochondrial biogenesis and mtDNA replication in primary Schwann cells, which requires both the ERK and PI3K signaling pathways (Echave et al., 2009). This process is mediated by the transcription factor ERRα and is independent of AKT/mTOR signaling pathways. IGF1 also enhances the level of mitochondrial protein involved in signal transduction, protein import and folding, mtDNA transcription, and bioenergetics in Huntington’s disease (HD) striatal cells (Ribeiro et al., 2014). Similarly, the aging rats untreated with IGF1 exhibit a significant mitochondrial dysfunction, including reduced activity of ATPase and complex IV (Garcia-Fernandez et al., 2008; Puche et al., 2008). Furthermore, cMYC regulates the expression of PGC1β in breast cancer cells in response to Her2/IGF1 activation (Chang et al., 2011). There is also evidence showing that IGF1 promotes mitochondrial biogenesis through the induction of PGC1β and PRC, not PGC1α, in vitro (Lyons et al., 2017). Likewise, loss of IGF1 signal reduces the expression of mitochondrial biogenesis markers (PGC1α, TFAM) in the steroidogenic cells of prepubertal testis (Radovic et al., 2019). Overall, these compelling evidences demonstrate that IGF1 acts as a protector in the process of regulating mitochondrial biogenesis.
The Effects of Insulin-Like Growth Factor 1 on Mitophagy
Up to now, numerous studies have reported the relationship between IGF1 and autophagy. Here, we only briefly elucidate as follows. IGF1 inhibits starvation-induced cardiac autophagy via mTOR signaling in vitro, and negatively regulates cardiac autophagy and AMPK activity in vivo (Troncoso et al., 2012). Conversely, high protein levels of IGF1 and its receptors, accompanied by a reduction in AKT/mTOR signaling pathways resulting from resistance exercise training, are associated with increased autophagy activity in aged skeletal muscles (Luo et al., 2013). Also, IGF1 expression is significantly reduced in ATG7 knockout mice, indicating that IGF1 plays a beneficial role in regulating autophagy (Zecchini et al., 2019).
Nevertheless, the role of IGF1 on mitophagy is rarely reported. IGF1 can induce the expression and accumulation of BNIP3 in mitochondria through a PI3K-dependent manner, indicating that IGF1 promotes mitophagy in vitro (Lyons et al., 2017). In mouse and cell models of amyotrophic lateral sclerosis (ALS), IGF1 also strongly protects mitochondria from apoptosis and upregulates mitophagy, as evidenced by a decrease in the p62 level and an increase in the LC3-II level (Wen et al., 2019). Recently, a study further revealed that IGF1-induced BNIP3 expression requires NRF2 to act through downstream transcriptional factors HIF-1α and NRF1 (Riis et al., 2020). These novel findings above strongly demonstrate that IGF1 promotes the process of mitophagy both in vivo and in vitro. However, whether there is an interaction between the mitochondrial biogenesis and mitophagy regulated by IGF1 is still unknown, and further research is needed.
The Effects of GH–GHR–IGF1 Axis on Mitochondrial Function
Many methods have been utilized to measure normal mitochondrial function or dysfunction in different systems. Generally, mitochondrial respiration control, including oxygen consumption rate (OCR) and respiratory control ratio (RCR), is utilized to measure the mitochondrial function in diverse cell populations (Brand and Nicholls, 2011). Mitochondrial membrane potential (ΔΨm) is also used as an indicator for mitochondrial function. Loss of ΔΨm normally indicates mitochondrial dysfunction and is accompanied by increased mitochondrial swelling (Javadov et al., 2006, 2009). Furthermore, reduced ΔΨm will lead to uncoupling of OXPHOS and increases ROS production accompanied by elevated malondialdehyde (MDA) levels and reduced ATP levels (Lebiedzinska et al., 2010; Bai et al., 2017). Notably, numerous studies have revealed a positive correlation between ΔΨm and ROS production in various physiological and pathological scenarios. Mitochondrial proton leakage, mainly due to the decrease in ΔΨm, is considered to counteract mitochondrial ROS production to protect cells from oxidative stress (Turrens, 2003; Mailloux and Harper, 2011).
Until now, a long list of studies utilizing numerous cell types has shown the effects of the GH–GHR–IGF1 axis on mitochondrial function in vivo and in vitro. Early in the 1970s, most of researches had revealed the positive effects of GH on mitochondrial respiration rate and its related enzyme activities in hypophysectomized rats (Maddaiah et al., 1976; Katkocin et al., 1979; Maddaiah and Clejan, 1986). Short-term GH therapy for 3 months increases the activity of succinate dehydrogenase, which represents the mitochondrial function in human quadriceps muscle (Gonzalez et al., 2018). A recent study also demonstrated that the OCR and ATP production is significantly increased in primary osteocytes of bGH Tg mice (Liu et al., 2019). These results suggested that GH has a positive effect on mitochondrial function in vivo. Furthermore, increased OXPHOS activities and oxygen consumption, along with reduced ROS production were found in the long-living Ames dwarf mice (Westbrook et al., 2009). Also, decreased oxygen consumption was observed in bGH Tg mice (Brown-Borg et al., 2012). These findings indicated that GH has a negative effect on mitochondrial function in vivo. In addition, administration of GH by the continuous infusion of hormone for 6 days in hypophysectomized rat liver has no effect on mitochondrial respiration and enzyme activities (Mutvei et al., 1989).
These discrepancy results can also be observed on GHR functions. GHR ablation is detrimental to osteocyte and fibroblast mitochondrial function (Liu et al., 2019). Consistently, mitochondrial function is impeded in the skeletal muscle of SLD chickens and GHR knockdown cells, indicating that GHR promotes mitochondrial function in vivo and in vitro (Hu et al., 2019). However, these data are in conflict with a previous report that demonstrated an enhanced mitochondrial function in the liver, muscle, heart, kidneys, and brain of aged GHRKO mice (Brown-Borg et al., 2009). Also, the abundance of three tricarboxylic acid cycle (TCA) cycle enzymes (isocitrate dehydrogenase, fumarase, and malate dehydrogenase) is significantly increased in the GHRKO pigs’ liver proteome, suggesting that GHR inhibits mitochondrial function in vivo (Riedel et al., 2020). Due to the lack of GH and GHR on the regulation of mitophagy, we proposed that these discrepant results of GH and GHR on mitochondrial function may be explained by the coordinated regulation mechanism between mitochondrial biogenesis and mitophagy to meet different physiological conditions in vivo and in vitro.
In the last 10 years, most evidence has revealed that IGF1 is regarded as a protector for mitochondrial function under several diseases and stress conditions. IGF1 not only protects cardiomyocytes from hypoxia/reoxygenation injury by stabilizing ΔΨm and reducing ROS damage but also alleviates mitochondria dysfunction in cardiomyocytes from high-fat diet mice (Pi et al., 2007; Zhang et al., 2012). In neuronal cells, IGF1 protects against prion diseases caused by mitochondrial dysfunction and increased ROS production via inhibition of Bax translocation (Park et al., 2012). Consistently, IGF1 increases ΔΨm in HD striatal cells in a PI3K/AKT-dependent manner (Ribeiro et al., 2014). Activation of IGF1 signaling pathways also ameliorates O2 consumption and ΔΨm in HD lymphoblasts (Naia et al., 2015). Furthermore, regulation of astrocytic mitochondrial function and redox status by IGF1 is essential to maintain astrocytic function and coordinate hippocampal-dependent spatial learning (Logan et al., 2018). Knockdown of the IGF1 leads to a reduction in ΔΨm and alterations in mitochondrial morphology in ALS mice (Wen et al., 2019). In addition, IGF1 activates AMPK to augment mitochondrial function (OCR and ATP production) in sensory neurons in type-1 diabetes (Aghanoori et al., 2019). Of note is that a recent study revealing that induced liver IGF1 knockout can impair hippocampal mitochondrial OXPHOS coupling efficiency and reduce cortex ATP levels (Pharaoh et al., 2020). However, IGF1 has no significant impact on muscle mitochondrial function, indicating that the deficiency of IGF1 in male mice has different roles on tissue mitochondrial function between the center and periphery circulation (Pharaoh et al., 2020). Taken together, these findings strongly demonstrate that IGF1 promotes mitochondrial function to restore various diseases and stress conditions, such as improving metabolism and exerting mitochondrial protection, hepatoprotective as well as neuroprotective effects.
Accordingly, Sadaba et al. (2016) suggested that one of the newest targets to recover mitochondrial dysfunction could be the administration of low doses of IGF1. This is supported by compelling evidences that IGF1 replacement therapy is able to restore mitochondrial dysfunction observed in untreated cirrhotic rats and in IGF1 partial deficiency mice (Perez et al., 2008; Olleros et al., 2017).
Future Perspectives
To sum up, it is generally believed that IGF1 enhances mitochondrial function by promoting both mitochondrial biogenesis and mitophagy under several conditions of metabolic or mitochondrial stress. Accordingly, regulation of IGF1 secretion may have a therapeutic potential in the protection of mitochondrial function for treating many mitochondria-related diseases. In Figure 3, we delineate the hypothetical mechanism of the GH–GHR–IGF1 axis, which may be mostly mediated by IGF1, on the regulation of mitochondrial biogenesis, mitophagy, and mitochondrial function based on the results of studies in the recent decades.
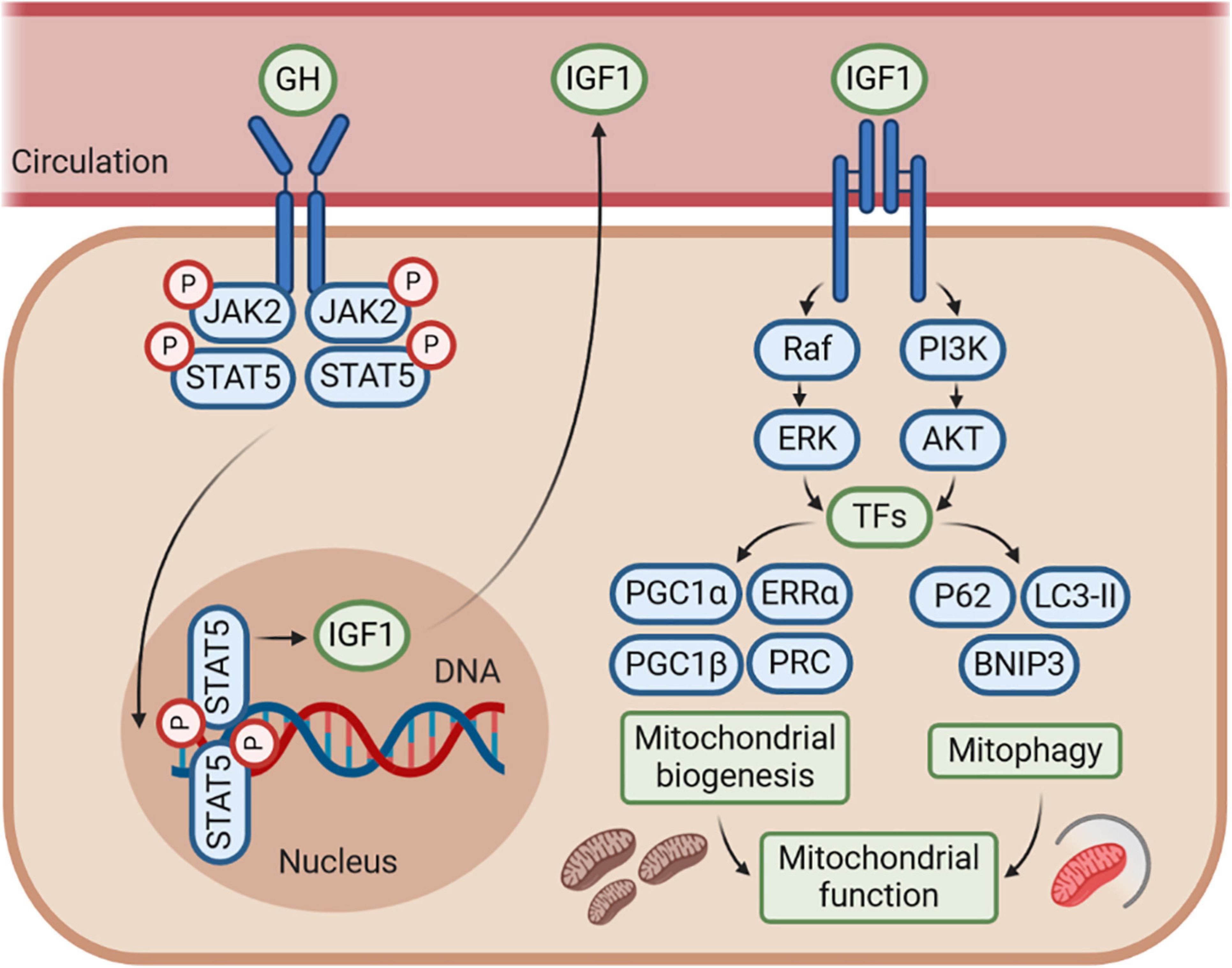
Figure 3. The GH–GHR–IGF1 axis may control mitochondrial function by regulating mitochondrial biogenesis and mitophagy. The effects of GH–GHR–IGF1 axis on mitochondrial function may be mostly mediated by IGF1, which stimulates several signaling pathways, including phosphoinositide-3-kinase (PI3K)/AKT and Ras/Raf/MAPK signaling, to further activate numerous transcription factors (TFs). This cascade leads to transcriptional activity of genes involved in both mitochondrial biogenesis and mitophagy. Accordingly, IGF1 is commonly considered to enhance mitochondrial function by promoting mitochondrial biogenesis and mitophagy under several conditions of metabolic or mitochondrial stress. However, whether there is an interaction between the mitochondrial biogenesis and mitophagy regulated by IGF1 is still unknown, and further research is needed.
On the other hand, the effects of GH and GHR on mitochondrial function are multifaceted, which may be induced by the differences between cell-, organ-, and species-dependent features or various physiological conditions in vivo and in vitro. The findings above may lead us to think deeply that GH may exert its multiple effects on mitochondrial function under the direct control by its receptor GHR. However, whether GHR functions as a control valve is still currently lacking in research. Moreover, the molecular basis of GH and GHR on mitophagy is not well understood. In the future, more research is needed to improve our understanding of the effects of GH and GHR on mitochondrial function. This intricate biological process may be explained by the balance between mitochondrial biogenesis and mitophagy under different physiological conditions.
Author Contributions
BH wrote the manuscript. HL and XZ put forward a lot of invaluable opinions for revision and finalized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the China Agriculture Research System (CARS-41-G03) and National Natural Science Foundation of China (31401046).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aghanoori, M. R., Smith, D. R., Shariati-Ievari, S., Ajisebutu, A., Nguyen, A., Desmond, F., et al. (2019). Insulin-like growth factor-1 activates AMPK to augment mitochondrial function and correct neuronal metabolism in sensory neurons in type 1 diabetes. Mol. Metab. 20, 149–165. doi: 10.1016/j.molmet.2018.11.008
Akbari, M., Kirkwood, T., and Bohr, V. A. (2019). Mitochondria in the signaling pathways that control longevity and health span. Ageing Res. Rev. 54:100940. doi: 10.1016/j.arr.2019.100940
Akimoto, T., Pohnert, S. C., Li, P., Zhang, M., Gumbs, C., Rosenberg, P. B., et al. (2005). Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 280, 19587–19593. doi: 10.1074/jbc.M408862200
Al-Regaiey, K. A., Masternak, M. M., Bonkowski, M., Sun, L., and Bartke, A. (2005). Long-lived growth hormone receptor knockout mice: interaction of reduced insulin-like growth factor i/insulin signaling and caloric restriction. Endocrinology 146, 851–860. doi: 10.1210/en.2004-1120
Andersson, U., and Scarpulla, R. C. (2001). Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell Biol. 21, 3738–3749. doi: 10.1128/MCB.21.11.3738-3749.2001
Ashford, T. P., and Porter, K. R. (1962). Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 12, 198–202. doi: 10.1083/jcb.12.1.198
Attardi, G., and Schatz, G. (1988). Biogenesis of Mitochondria. Annu. Rev. Cell Biol. 4, 289–333. doi: 10.1146/annurev.cb.04.110188.001445
Bai, F., Guo, F., Jiang, T., Wei, H., Zhou, H., Yin, H., et al. (2017). Arachidonyl-2-Chloroethylamide Alleviates Cerebral Ischemia Injury Through Glycogen Synthase Kinase-3beta-Mediated Mitochondrial Biogenesis and Functional Improvement. Mol. Neurobiol. 54, 1240–1253. doi: 10.1007/s12035-016-9731-7
Bartolome, A., Garcia-Aguilar, A., Asahara, S. I., Kido, Y., Guillen, C., Pajvani, U. B., et al. (2017). MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell Biol. 37:17. doi: 10.1128/MCB.00441-17
Baumann, G., Amburn, K., and Shaw, M. A. (1988). The circulating growth hormone (GH)-binding protein complex: a major constituent of plasma GH in man. Endocrinology 122, 976–984. doi: 10.1210/endo-122-3-976
Bellance, N., Benard, G., Furt, F., Begueret, H., Smolkova, K., Passerieux, E., et al. (2009). Bioenergetics of lung tumors: alteration of mitochondrial biogenesis and respiratory capacity. Int. J. Biochem. Cell Biol. 41, 2566–2577. doi: 10.1016/j.biocel.2009.08.012
Bergamini, E., Cavallini, G., Donati, A., and Gori, Z. (2003). The anti-ageing effects of caloric restriction may involve stimulation of macroautophagy and lysosomal degradation, and can be intensified pharmacologically. Biomed. Pharmacother. 57, 203–208. doi: 10.1016/s0753-3322(03)00048-9
Bergan, H. E., Kittilson, J. D., and Sheridan, M. A. (2015). Nutritional state modulates growth hormone-stimulated lipolysis. Gen. Comp. Endocrinol. 21, 1–9. doi: 10.1016/j.ygcen.2015.04.017
Bhujabal, Z., Birgisdottir, A. B., Sjottem, E., Brenne, H. B., Overvatn, A., Habisov, S., et al. (2017). FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 18, 947–961. doi: 10.15252/embr.201643147
Brand, M. D., and Nicholls, D. G. (2011). Assessing mitochondrial dysfunction in cells. Biochem. J. 435, 297–312. doi: 10.1042/BJ20110162
Brioche, T., Kireev, R. A., Cuesta, S., Gratas-Delamarche, A., Tresguerres, J. A., Gomez-Cabrera, M. C., et al. (2014). Growth hormone replacement therapy prevents sarcopenia by a dual mechanism: improvement of protein balance and of antioxidant defenses. J. Gerontol. Biol. Sci. Med. Sci. 69, 1186–1198. doi: 10.1093/gerona/glt187
Brooks, A. J., Wooh, J. W., Tunny, K. A., and Waters, M. J. (2008). Growth hormone receptor; mechanism of action. Int. J. Biochem. Cell Biol. 40, 1984–1989. doi: 10.1016/j.biocel.2007.07.008
Brown-Borg, H. M., Johnson, W. T., and Rakoczy, S. G. (2012). Expression of oxidative phosphorylation components in mitochondria of long-living Ames dwarf mice. Age 34, 43–57. doi: 10.1007/s11357-011-9212-x
Brown-Borg, H. M., Rakoczy, S. G., Sharma, S., and Bartke, A. (2009). Long-living growth hormone receptor knockout mice: potential mechanisms of altered stress resistance. Exp. Gerontol. 44, 10–19. doi: 10.1016/j.exger.2008.07.002
Chang, C. Y., Kazmin, D., Jasper, J. S., Kunder, R., Zuercher, W. J., and McDonnell, D. P. (2011). The metabolic regulator ERRalpha, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell 20, 500–510. doi: 10.1016/j.ccr.2011.08.023
Chen, K. G., Kang, R. R., Sun, Q., Liu, C., Ma, Z., Liu, K., et al. (2020). Resveratrol ameliorates disorders of mitochondrial biogenesis and mitophagy in rats continuously exposed to benzo(a)pyrene from embryonic development through adolescence. Toxicology 442:152532. doi: 10.1016/j.tox.2020.152532
Cherra, S. R., Kulich, S. M., Uechi, G., Balasubramani, M., Mountzouris, J., Day, B. W., et al. (2010). Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol. 190, 533–539. doi: 10.1083/jcb.201002108
Chowanadisai, W., Bauerly, K. A., Tchaparian, E., Wong, A., Cortopassi, G. A., and Rucker, R. B. (2010). Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1alpha expression. J. Biol. Chem. 285, 142–152. doi: 10.1074/jbc.M109.030130
Clark, E. H., Vazquez, D. L. T. A., Hoshikawa, T., and Briston, T. (2020). Targeting mitophagy in Parkinson’s disease. J. Biol. Chem. 296:100209. doi: 10.1074/jbc.REV120.014294
Cui, L., Jeong, H., Borovecki, F., Parkhurst, C. N., Tanese, N., and Krainc, D. (2006). Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69. doi: 10.1016/j.cell.2006.09.015
Cunningham, J. T., Rodgers, J. T., Arlow, D. H., Vazquez, F., Mootha, V. K., and Puigserver, P. (2007). mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450, 736–740. doi: 10.1038/nature06322
Dagistanli, F. K., Ozkaya, H. M., Kucukyoruk, B., Biceroglu, H., Metin, D., Gazioglu, N., et al. (2018). Preoperative Somatostatin Analogue Treatment Might Trigger Apoptosis and Autophagy in Tumor Tissues of Patients with Acromegaly: A Pilot Study. Exp. Clin. Endocrinol. Diabet. 126, 168–175. doi: 10.1055/s-0042-107243
Dehkhoda, F., Lee, C., Medina, J., and Brooks, A. J. (2018). The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects. Front. Endocrinol. 9:35. doi: 10.3389/fendo.2018.00035
D’Errico, I., Salvatore, L., Murzilli, S., Lo, S. G., Latorre, D., Martelli, N., et al. (2011). Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1alpha) is a metabolic regulator of intestinal epithelial cell fate. Proc. Natl. Acad. Sci. U S A. 108, 6603–6608. doi: 10.1073/pnas.1016354108
Duan, C., and Xu, Q. (2005). Roles of insulin-like growth factor (IGF) binding proteins in regulating IGF actions. Gen. Comp. Endocrinol. 142, 44–52. doi: 10.1016/j.ygcen.2004.12.022
Echave, P., Machado-da-Silva, G., Arkell, R. S., Duchen, M. R., Jacobson, J., Mitter, R., et al. (2009). Extracellular growth factors and mitogens cooperate to drive mitochondrial biogenesis. J. Cell Sci. 122(Pt 24), 4516–4525. doi: 10.1242/jcs.049734
Eiyama, A., and Okamoto, K. (2015). PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 33, 95–101. doi: 10.1016/j.ceb.2015.01.002
Elmore, S. P., Qian, T., Grissom, S. F., and Lemasters, J. J. (2001). The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 15, 2286–2287. doi: 10.1096/fj.01-0206fje
Endicott, S. J., Boynton, D. J., Beckmann, L. J., and Miller, R. A. (2020). Long-lived mice with reduced growth hormone signaling have a constitutive upregulation of hepatic chaperone-mediated autophagy. Autophagy 2020:1725378. doi: 10.1080/15548627.2020.1725378
Fang, F., Shi, X., Brown, M. S., Goldstein, J. L., and Liang, G. (2019). Growth hormone acts on liver to stimulate autophagy, support glucose production, and preserve blood glucose in chronically starved mice. Proc. Natl. Acad. Sci. U S A. 116, 7449–7454. doi: 10.1073/pnas.1901867116
Fukuda, T., and Kanki, T. (2021). Atg43, a novel autophagy-related protein, serves as a mitophagy receptor to bridge mitochondria with phagophores in fission yeast. Autophagy 2021:1874662. doi: 10.1080/15548627.2021.1874662
Garcia-Fernandez, M., Delgado, G., Puche, J. E., Gonzalez-Baron, S., and Castilla, C. I. (2008). Low doses of insulin-like growth factor I improve insulin resistance, lipid metabolism, and oxidative damage in aging rats. Endocrinology 149, 2433–2442. doi: 10.1210/en.2007-1190
Gesing, A., Bartke, A., Wang, F., Karbownik-Lewinska, M., and Masternak, M. M. (2011a). Key regulators of mitochondrial biogenesis are increased in kidneys of growth hormone receptor knockout (GHRKO) mice. Cell Biochem. Funct. 29, 459–467. doi: 10.1002/cbf.1773
Gesing, A., Masternak, M. M., Lewinski, A., Karbownik-Lewinska, M., Kopchick, J. J., and Bartke, A. (2013). Decreased levels of proapoptotic factors and increased key regulators of mitochondrial biogenesis constitute new potential beneficial features of long-lived growth hormone receptor gene-disrupted mice. J. Gerontol. Biol. Sci. Med. Sci. 68, 639–651. doi: 10.1093/gerona/gls231
Gesing, A., Masternak, M. M., Wang, F., Joseph, A. M., Leeuwenburgh, C., Westbrook, R., et al. (2011b). Expression of key regulators of mitochondrial biogenesis in growth hormone receptor knockout (GHRKO) mice is enhanced but is not further improved by other potential life-extending interventions. J. Gerontol. Biol. Sci. Med. Sci. 66, 1062–1076. doi: 10.1093/gerona/glr080
Gleyzer, N., Vercauteren, K., and Scarpulla, R. C. (2005). Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell Biol. 25, 1354–1366. doi: 10.1128/MCB.25.4.1354-1366.2005
Gonzalez, S., Sathyapalan, T., Javed, Z., and Atkin, S. L. (2018). Effects of Growth Hormone Replacement on Peripheral Muscle and Exercise Capacity in Severe Growth Hormone Deficiency. Front. Endocrinol. 9:56. doi: 10.3389/fendo.2018.00056
Groves, W. E., Houts, G. E., and Bayse, G. S. (1972). Subcellular distribution of 125 I-labeled bovine growth hormone in rat liver and kidney. Biochim. Biophys. Acta 264, 472–480. doi: 10.1016/0304-4165(72)90010-4
Hakuno, F., and Takahashi, S. I. (2018). IGF1 receptor signaling pathways. J. Mol. Endocrinol. 61, T69–T86. doi: 10.1530/JME-17-0311
Hayashi, A. A., and Proud, C. G. (2007). The rapid activation of protein synthesis by growth hormone requires signaling through mTOR. Am. J. Physiol. Endocrinol. Metab. 292, E1647–E1655. doi: 10.1152/ajpendo.00674.2006
Herzig, S., and Shaw, R. J. (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135. doi: 10.1038/nrm.2017.95
Hirota, Y., Yamashita, S., Kurihara, Y., Jin, X., Aihara, M., Saigusa, T., et al. (2015). Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 11, 332–343. doi: 10.1080/15548627.2015.1023047
Hu, B., Hu, S., Yang, M., Liao, Z., Zhang, D., Luo, Q., et al. (2019). Growth Hormone Receptor Gene is Essential for Chicken Mitochondrial Function In Vivo and In Vitro. Int. J. Mol. Sci. 20:20071608. doi: 10.3390/ijms20071608
Javadov, S., Baetz, D., Rajapurohitam, V., Zeidan, A., Kirshenbaum, L. A., and Karmazyn, M. (2006). Antihypertrophic effect of Na+/H+ exchanger isoform 1 inhibition is mediated by reduced mitogen-activated protein kinase activation secondary to improved mitochondrial integrity and decreased generation of mitochondrial-derived reactive oxygen species. J. Pharmacol. Exp. Ther. 317, 1036–1043. doi: 10.1124/jpet.105.100107
Javadov, S., Karmazyn, M., and Escobales, N. (2009). Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J. Pharmacol. Exp. Ther. 330, 670–678. doi: 10.1124/jpet.109.153213
Junnila, R. K., List, E. O., Berryman, D. E., Murrey, J. W., and Kopchick, J. J. (2013). The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 9, 366–376. doi: 10.1038/nrendo.2013.67
Kanki, T., Wang, K., Cao, Y., Baba, M., and Klionsky, D. J. (2009). Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell 17, 98–109. doi: 10.1016/j.devcel.2009.06.014
Katkocin, D. M., Gupta, K. M., Collipp, P. J., and Maddaiah, V. T. (1979). Effects of growth hormone on respiration and ATPase activity of rat liver and heart mitochondria. Biochem. Med. 22, 134–144. doi: 10.1016/0006-2944(79)90046-2
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Klionsky, D. J., Cregg, J. M., Dunn, W. J., Emr, S. D., Sakai, Y., Sandoval, I. V., et al. (2003). A unified nomenclature for yeast autophagy-related genes. Dev. Cell 5, 539–545. doi: 10.1016/s1534-5807(03)00296-x
Kozhukhar, N., and Alexeyev, M. F. (2019). Limited predictive value of TFAM in mitochondrial biogenesis. Mitochondrion 49, 156–165. doi: 10.1016/j.mito.2019.08.001
Kuroda, Y., Mitsui, T., Kunishige, M., Shono, M., Akaike, M., Azuma, H., et al. (2006). Parkin enhances mitochondrial biogenesis in proliferating cells. Hum. Mol. Genet. 15, 883–895. doi: 10.1093/hmg/ddl006
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lebiedzinska, M., Karkucinska-Wieckowska, A., Giorgi, C., Karczmarewicz, E., Pronicka, E., Pinton, P., et al. (2010). Oxidative stress-dependent p66Shc phosphorylation in skin fibroblasts of children with mitochondrial disorders. Biochim. Biophys. Acta 1797, 952–960. doi: 10.1016/j.bbabio.2010.03.005
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5. doi: 10.1089/rej.2005.8.3
Lewis, M. R., and Lewis, W. H. (1915). Mitochondria (and other cytoplasmic structures) in tissue cultures. Am. J. Anatomy 17, 339–401.
Li, X. X., Tsoi, B., Li, Y. F., Kurihara, H., and He, R. R. (2015). Cardiolipin and its different properties in mitophagy and apoptosis. J. Histochem. Cytochem. 63, 301–311. doi: 10.1369/0022155415574818
Lin, J., Puigserver, P., Donovan, J., Tarr, P., and Spiegelman, B. M. (2002). Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem. 277, 1645–1648. doi: 10.1074/jbc.C100631200
Lin, S., Li, C., Li, C., and Zhang, X. (2018). Growth Hormone Receptor Mutations Related to Individual Dwarfism. Int. J. Mol. Sci. 19:19051433. doi: 10.3390/ijms19051433
List, E. O., Duran-Ortiz, S., and Kopchick, J. J. (2020). Effects of tissue-specific GH receptor knockouts in mice. Mol. Cell Endocrinol. 515:110919. doi: 10.1016/j.mce.2020.110919
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Liu, L., Sakakibara, K., Chen, Q., and Okamoto, K. (2014). Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 24, 787–795. doi: 10.1038/cr.2014.75
Liu, Z., Solesio, M. E., Schaffler, M. B., Frikha-Benayed, D., Rosen, C. J., Werner, H., et al. (2019). Mitochondrial Function Is Compromised in Cortical Bone Osteocytes of Long-Lived Growth Hormone Receptor Null Mice. J. Bone Miner Res. 34, 106–122. doi: 10.1002/jbmr.3573
Logan, S., Pharaoh, G. A., Marlin, M. C., Masser, D. R., Matsuzaki, S., Wronowski, B., et al. (2018). Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-beta uptake in astrocytes. Mol. Metab. 9, 141–155. doi: 10.1016/j.molmet.2018.01.013
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Luo, L., Lu, A. M., Wang, Y., Hong, A., Chen, Y., Hu, J., et al. (2013). Chronic resistance training activates autophagy and reduces apoptosis of muscle cells by modulating IGF-1 and its receptors, Akt/mTOR and Akt/FOXO3a signaling in aged rats. Exp. Gerontol. 48, 427–436. doi: 10.1016/j.exger.2013.02.009
Luo, W., Lin, S., Li, G., Nie, Q., and Zhang, X. (2016). Integrative Analyses of miRNA-mRNA Interactions Reveal let-7b, miR-128 and MAPK Pathway Involvement in Muscle Mass Loss in Sex-Linked Dwarf Chickens. Int. J. Mol. Sci. 17:276. doi: 10.3390/ijms17030276
Lyons, A., Coleman, M., Riis, S., Favre, C., O’Flanagan, C. H., Zhdanov, A. V., et al. (2017). Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J. Biol. Chem. 292, 16983–16998. doi: 10.1074/jbc.M117.792838
Maddaiah, V. T., and Clejan, S. (1986). Growth hormone and liver mitochondria: time course of effects on respiration and fatty acid composition in hypophysectomized rats. Endocrinology 119, 250–252. doi: 10.1210/endo-119-1-250
Maddaiah, V. T., Rezvani, I., Chen, S. Y., and Collipp, P. J. (1970). Distribution of 3 H-acetyl human growth hormone in subcellular fractions of rat liver. Biochem. Med. 4, 492–499. doi: 10.1016/0006-2944(70)90079-7
Maddaiah, V. T., Sharma, R. K., Balachandar, V., Rezvani, I., Collipp, P. J., and Chen, S. Y. (1973). Effect of growth hormone on mitochondrial protein synthesis. J. Biol. Chem. 248, 4263–4268.
Maddaiah, V. T., Weston, C. L., Chen, S. Y., and Collipp, P. J. (1976). Growth hormone and liver mitochondria. Effects on cytochromes and some enzymes. Arch. Biochem. Biophys. 173, 225–230. doi: 10.1016/0003-9861(76)90253-8
Mailloux, R. J., and Harper, M. E. (2011). Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 51, 1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022
Malpartida, A. B., Williamson, M., Narendra, D. P., Wade-Martins, R., and Ryan, B. J. (2020). Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2020:7. doi: 10.1016/j.tibs.2020.11.007
Mattson, M. P., and Arumugam, T. V. (2018). Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 27, 1176–1199. doi: 10.1016/j.cmet.2018.05.011
Mizumura, K., Choi, A. M., and Ryter, S. W. (2014). Emerging role of selective autophagy in human diseases. Front. Pharmacol. 5:244. doi: 10.3389/fphar.2014.00244
Mizushima, N., and Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026
Moller, N., and Jorgensen, J. O. (2009). Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr. Rev. 30, 152–177. doi: 10.1210/er.2008-0027
Montava-Garriga, L., and Ganley, I. G. (2020). Outstanding Questions in Mitophagy: What We Do and Do Not Know. J. Mol. Biol. 432, 206–230. doi: 10.1016/j.jmb.2019.06.032
Mutvei, A., Husman, B., Andersson, G., and Nelson, B. D. (1989). Thyroid hormone and not growth hormone is the principle regulator of mammalian mitochondrial biogenesis. Acta Endocrinol. 121, 223–228. doi: 10.1530/acta.0.1210223
Naia, L., Ferreira, I. L., Cunha-Oliveira, T., Duarte, A. I., Ribeiro, M., Rosenstock, T. R., et al. (2015). Activation of IGF-1 and insulin signaling pathways ameliorate mitochondrial function and energy metabolism in Huntington’s Disease human lymphoblasts. Mol. Neurobiol. 51, 331–348. doi: 10.1007/s12035-014-8735-4
Nakatogawa, H., Suzuki, K., Kamada, Y., and Ohsumi, Y. (2009). Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 10, 458–467. doi: 10.1038/nrm2708
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Nguyen, T. N., Padman, B. S., and Lazarou, M. (2016). Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 26, 733–744. doi: 10.1016/j.tcb.2016.05.008
Novikoff, A. B., and Essner, E. (1962). Cytolysomes and mitochondrial degeneration. J. Cell Biol. 15, 140–146. doi: 10.1083/jcb.15.1.140
Okamoto, K., Kondo-Okamoto, N., and Ohsumi, Y. (2009). Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 17, 87–97. doi: 10.1016/j.devcel.2009.06.013
Olleros, S. M., Sadaba, M. C., Martin-Estal, I., Munoz, U., Sebal, N. C., and Castilla-Cortazar, I. (2017). The single IGF-1 partial deficiency is responsible for mitochondrial dysfunction and is restored by IGF-1 replacement therapy. Growth Horm. IGF Res. 35, 21–32. doi: 10.1016/j.ghir.2017.05.007
Otsu, K., Murakawa, T., and Yamaguchi, O. (2015). BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy 11, 1932–1933. doi: 10.1080/15548627.2015.1084459
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. doi: 10.1038/s41556-018-0176-2
Park, Y. G., Jeong, J. K., Moon, M. H., Lee, J. H., Lee, Y. J., Seol, J. W., et al. (2012). Insulin-like growth factor-1 protects against prion peptide-induced cell death in neuronal cells via inhibition of Bax translocation. Int. J. Mol. Med. 30, 1069–1074. doi: 10.3892/ijmm.2012.1087
Patel, V. P., Defranco, D. B., and Chu, C. T. (2012). Altered transcription factor trafficking in oxidatively-stressed neuronal cells. Biochim. Biophys. Acta 1822, 1773–1782. doi: 10.1016/j.bbadis.2012.08.002
Perez, R., Garcia-Fernandez, M., Diaz-Sanchez, M., Puche, J. E., Delgado, G., Conchillo, M., et al. (2008). Mitochondrial protection by low doses of insulin-like growth factor- I in experimental cirrhosis. World J. Gastroenterol. 14, 2731–2739. doi: 10.3748/wjg.14.2731
Pharaoh, G., Owen, D., Yeganeh, A., Premkumar, P., Farley, J., Bhaskaran, S., et al. (2020). Disparate Central and Peripheral Effects of Circulating IGF-1 Deficiency on Tissue Mitochondrial Function. Mol. Neurobiol. 57, 1317–1331. doi: 10.1007/s12035-019-01821-4
Pi, Y., Goldenthal, M. J., and Marin-Garcia, J. (2007). Mitochondrial involvement in IGF-1 induced protection of cardiomyocytes against hypoxia/reoxygenation injury. Mol. Cell Biochem. 301, 181–189. doi: 10.1007/s11010-007-9410-0
Pickles, S., Vigie, P., and Youle, R. J. (2018). Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 28, R170–R185. doi: 10.1016/j.cub.2018.01.004
Ploumi, C., Daskalaki, I., and Tavernarakis, N. (2017). Mitochondrial biogenesis and clearance: a balancing act. FEBS J. 284, 183–195. doi: 10.1111/febs.13820
Popov, L. D. (2020). Mitochondrial biogenesis: An update. J. Cell Mol. Med. 24, 4892–4899. doi: 10.1111/jcmm.15194
Puche, J. E., Garcia-Fernandez, M., Muntane, J., Rioja, J., Gonzalez-Baron, S., and Castilla, C. I. (2008). Low doses of insulin-like growth factor-I induce mitochondrial protection in aging rats. Endocrinology 149, 2620–2627. doi: 10.1210/en.2007-1563
Quinsay, M. N., Thomas, R. L., Lee, Y., and Gustafsson, A. B. (2010). Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 6, 855–862. doi: 10.4161/auto.6.7.13005
Radovic, S. M., Starovlah, I. M., Capo, I., Miljkovic, D., Nef, S., Kostic, T. S., et al. (2019). Insulin/IGF1 signaling regulates the mitochondrial biogenesis markers in steroidogenic cells of prepubertal testis, but not ovary. Biol. Reprod. 100, 253–267. doi: 10.1093/biolre/ioy177
Ribeiro, M., Rosenstock, T. R., Oliveira, A. M., Oliveira, C. R., and Rego, A. C. (2014). Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 74, 129–144. doi: 10.1016/j.freeradbiomed.2014.06.023
Riedel, E. O., Hinrichs, A., Kemter, E., Dahlhoff, M., Backman, M., Rathkolb, B., et al. (2020). Functional changes of the liver in the absence of growth hormone (GH) action - Proteomic and metabolomic insights from a GH receptor deficient pig model. Mol. Metab. 36:100978. doi: 10.1016/j.molmet.2020.100978
Riis, S., Murray, J. B., and O’Connor, R. (2020). IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3beta-Nrf2-BNIP3 Pathway. Cells 9:9010147. doi: 10.3390/cells9010147
Sadaba, M. C., Martin-Estal, I., Puche, J. E., and Castilla-Cortazar, I. (2016). Insulin-like growth factor 1 (IGF-1) therapy: Mitochondrial dysfunction and diseases. Biochim. Biophys. Acta 1862, 1267–1278. doi: 10.1016/j.bbadis.2016.03.010
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Scarpulla, R. C. (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638. doi: 10.1152/physrev.00025.2007
Scarpulla, R. C., Vega, R. B., and Kelly, D. P. (2012). Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 23, 459–466. doi: 10.1016/j.tem.2012.06.006
Scott, I., Webster, B. R., Chan, C. K., Okonkwo, J. U., Han, K., and Sack, M. N. (2014). GCN5-like protein 1 (GCN5L1) controls mitochondrial content through coordinated regulation of mitochondrial biogenesis and mitophagy. J. Biol. Chem. 289, 2864–2872. doi: 10.1074/jbc.M113.521641
Scott, S. V., and Klionsky, D. J. (1998). Delivery of proteins and organelles to the vacuole from the cytoplasm. Curr. Opin. Cell Biol. 10, 523–529. doi: 10.1016/s0955-0674(98)80068-9
Sentelle, R. D., Senkal, C. E., Jiang, W., Ponnusamy, S., Gencer, S., Selvam, S. P., et al. (2012). Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 8, 831–838. doi: 10.1038/nchembio.1059
Settembre, C., Di Malta, C., Polito, V. A., Garcia, A. M., Vetrini, F., Erdin, S., et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. doi: 10.1126/science.1204592
Shin, J. H., Ko, H. S., Kang, H., Lee, Y., Lee, Y. I., Pletinkova, O., et al. (2011). PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 144, 689–702. doi: 10.1016/j.cell.2011.02.010
Singh, B. K., Sinha, R. A., Tripathi, M., Mendoza, A., Ohba, K., Sy, J., et al. (2018). Thyroid hormone receptor and ERRalpha coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci. Signal 11:5855. doi: 10.1126/scisignal.aam5855
Sjogren, K., Liu, J. L., Blad, K., Skrtic, S., Vidal, O., Wallenius, V., et al. (1999). Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc. Natl. Acad. Sci. U S A. 96, 7088–7092. doi: 10.1073/pnas.96.12.7088
Spinelli, J. B., and Haigis, M. C. (2018). The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 20, 745–754. doi: 10.1038/s41556-018-0124-1
Strappazzon, F., Nazio, F., Corrado, M., Cianfanelli, V., Romagnoli, A., Fimia, G. M., et al. (2015). AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 22, 419–432. doi: 10.1038/cdd.2014.139
Troncoso, R., Vicencio, J. M., Parra, V., Nemchenko, A., Kawashima, Y., Del, C. A., et al. (2012). Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc. Res. 93, 320–329. doi: 10.1093/cvr/cvr321
Tsunemi, T., and La Spada, A. R. (2012). PGC-1alpha at the intersection of bioenergetics regulation and neuron function: from Huntington’s disease to Parkinson’s disease and beyond. Prog. Neurobiol. 97, 142–151. doi: 10.1016/j.pneurobio.2011.10.004
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552(Pt 2), 335–344. doi: 10.1113/jphysiol.2003.049478
Um, J. H., and Yun, J. (2017). Emerging role of mitophagy in human diseases and physiology. BMB Rep. 50, 299–307. doi: 10.5483/bmbrep.2017.50.6.056
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Vanderkuur, J. A., Butch, E. R., Waters, S. B., Pessin, J. E., Guan, K. L., and Carter-Su, C. (1997). Signaling molecules involved in coupling growth hormone receptor to mitogen-activated protein kinase activation. Endocrinology 138, 4301–4307. doi: 10.1210/endo.138.10.5453
Villa, E., Marchetti, S., and Ricci, J. E. (2018). No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 28, 882–895. doi: 10.1016/j.tcb.2018.07.004
Villena, J. A. (2015). New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 282, 647–672. doi: 10.1111/febs.13175
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., de Vries, R. L., Kim, J., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U S A. 107, 378–383. doi: 10.1073/pnas.0911187107
Wang, M., and Miller, R. A. (2012). Augmented autophagy pathways and MTOR modulation in fibroblasts from long-lived mutant mice. Autophagy 8, 1273–1274. doi: 10.4161/auto.20917
Wang, W., Wang, M., Ruan, Y., Tan, J., Wang, H., Yang, T., et al. (2019). Ginkgolic Acids Impair Mitochondrial Function by Decreasing Mitochondrial Biogenesis and Promoting FUNDC1-Dependent Mitophagy. J. Agric. Food Chem. 67, 10097–10106. doi: 10.1021/acs.jafc.9b04178
Wei, Y., Chiang, W. C., Sumpter, R. J., Mishra, P., and Levine, B. (2017). Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 168, 224–238. doi: 10.1016/j.cell.2016.11.042
Wen, D., Cui, C., Duan, W., Wang, W., Wang, Y., Liu, Y., et al. (2019). The role of insulin-like growth factor 1 in ALS cell and mouse models: A mitochondrial protector. Brain Res. Bull. 144, 1–13. doi: 10.1016/j.brainresbull.2018.09.015
Westbrook, R., Bonkowski, M. S., Strader, A. D., and Bartke, A. (2009). Alterations in oxygen consumption, respiratory quotient, and heat production in long-lived GHRKO and Ames dwarf mice, and short-lived bGH transgenic mice. J. Gerontol. Biol. Sci. Med. Sci. 64, 443–451. doi: 10.1093/gerona/gln075
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124. doi: 10.1016/S0092-8674(00)80611-X
Yang, N., Langenheim, J. F., Wang, X., Jiang, J., Chen, W. Y., and Frank, S. J. (2008). Activation of growth hormone receptors by growth hormone and growth hormone antagonist dimers: insights into receptor triggering. Mol. Endocrinol. 22, 978–988. doi: 10.1210/me.2007-0424
Yau, W. W., Singh, B. K., Lesmana, R., Zhou, J., Sinha, R. A., Wong, K. A., et al. (2019). Thyroid hormone (T3) stimulates brown adipose tissue activation via mitochondrial biogenesis and MTOR-mediated mitophagy. Autophagy 15, 131–150. doi: 10.1080/15548627.2018.1511263
Zawada, I., Masternak, M. M., List, E. O., Stout, M. B., Berryman, D. E., Lewinski, A., et al. (2015). Gene expression of key regulators of mitochondrial biogenesis is sex dependent in mice with growth hormone receptor deletion in liver. Aging 7, 195–204. doi: 10.18632/aging.100733
Zecchini, S., Giovarelli, M., Perrotta, C., Morisi, F., Touvier, T., Di Renzo, I., et al. (2019). Autophagy controls neonatal myogenesis by regulating the GH-IGF1 system through a NFE2L2- and DDIT3-mediated mechanism. Autophagy 15, 58–77. doi: 10.1080/15548627.2018.1507439
Zhang, Y., Fang, F., Goldstein, J. L., Brown, M. S., and Zhao, T. J. (2015). Reduced autophagy in livers of fasted, fat-depleted, ghrelin-deficient mice: reversal by growth hormone. Proc. Natl. Acad. Sci. U S A. 112, 1226–1231. doi: 10.1073/pnas.1423643112
Zhang, Y., Yao, Y., Qiu, X., Wang, G., Hu, Z., Chen, S., et al. (2019). Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 20, 433–446. doi: 10.1038/s41590-019-0324-2
Zhang, Y., Yuan, M., Bradley, K. M., Dong, F., Anversa, P., and Ren, J. (2012). Insulin-like growth factor 1 alleviates high-fat diet-induced myocardial contractile dysfunction: role of insulin signaling and mitochondrial function. Hypertension 59, 680–693. doi: 10.1161/HYPERTENSIONAHA.111.181867
Zhou, D., Zhou, M., Wang, Z., Fu, Y., Jia, M., Wang, X., et al. (2019). PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 10:524. doi: 10.1038/s41419-019-1754-3
Keywords: growth hormone, growth hormone receptor, insulin-like growth factor 1, mitochondrial biogenesis, mitophagy, mitochondrial function
Citation: Hu B, Li H and Zhang X (2021) A Balanced Act: The Effects of GH–GHR–IGF1 Axis on Mitochondrial Function. Front. Cell Dev. Biol. 9:630248. doi: 10.3389/fcell.2021.630248
Received: 17 November 2020; Accepted: 16 February 2021;
Published: 18 March 2021.
Edited by:
Martin Van Der Laan, Saarland University, GermanyReviewed by:
Eirini Lionaki, Foundation for Research and Technology Hellas (FORTH), GreeceDu Feng, Guangzhou Medical University, China
Copyright © 2021 Hu, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongmei Li, hongmeili@scau.edu.cn; Xiquan Zhang, xqzhang@scau.edu.cn