PKA inhibition is a central step in D,L-methadone-induced ER Ca2+ release and subsequent apoptosis in acute lymphoblastic leukemia
 Hamza Kamran†
Hamza Kamran†  Jung Kwon Lee
Jung Kwon Lee Ki-Young Lee
Ki-Young Lee- Department of Cell Biology and Anatomy, Arnie Charbonneau Cancer and Alberta Children’s Hospital Research Institutes, University of Calgary, Calgary, AB, Canada
Acute lymphoblastic leukemia (ALL) is a hematologic cancer that mostly affects children. It accounts for over a quarter of ALL pediatric cancers, causing most of the cancer death among children. Previously, we demonstrated that D,L-methadone causes ALL cell apoptosis via μ-opioid receptor 1 (OPRM1)-triggered ER Ca2+ release and decrease in Ca2+ efflux, elevating [Ca2+]i. However, the precise mechanism by which D,L-methadone induces ER Ca2+ release remains to be defined. Here, we show that in ALL cells, D,L-methadone-induced ER Ca2+ release is blocked by inhibition of Gαi, but not Gβϒ, indicating that the process is dependent on Gαi. Activation of adenylyl cyclase (AC) with forskolin or treatment with 8-CPT-cAMP blocks D,L-methadone-induced ER Ca2+ release, indicating that the latter results from Gαi-dependent downregulation of AC and cAMP. The 14–22 amide (myr) PKA inhibitor alone elicits ER Ca2+ release, and subsequent treatment with D,L-methadone does not cause additional ER Ca2+ release, indicating that PKA inhibition is a key step in D,L-methadone-induced ER Ca2+ release and can bypass the D,L-methadone-OPRM1-AC-cAMP step. This is consistent with the decrease in PKA-dependent (i) inhibitory PLCβ3 Ser1105 phosphorylation that leads to PLCβ3 activation and ER Ca2+ release, and (ii) BAD Ser118 phosphorylation, which together ultimately result in caspase activation and apoptosis. Thus, our findings indicate that D,L-methadone-induced ER Ca2+ release and subsequent apoptosis in ALL cells is mediated by Gαi-dependent downregulation of the AC-cAMP-PKA-PLCβ3/BAD pathway. The fact that 14–22 amide (myr) alone effectively kills ALL cells suggests that PKA may be targeted for ALL therapy.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common malignancy in children that is characterized by rapid expansion of immature lymphocytes in the blood and bone marrow (Capria et al., 2020). The development of ALL has been linked to exposure to environmental hazards such as benzene, ionizing radiation, or previous exposures to chemotherapy and radiotherapy, and mutations in one or more of the genes that control hematopoiesis (Midic et al., 2020). Chemotherapeutic drugs have predominantly been used to target leukemic cells through mechanisms that include double-stranded DNA breaks, inhibition of protein and nucleic acid synthesis, and disruption of mitotic cell division (Terwilliger and Abdul-Hay, 2017). However, leukemic cells often develop resistance to chemotherapy drugs, resulting in poor patient prognosis (Zhang et al., 2019). Thus, the search for improved therapy for ALL is imperative.
D,L-methadone is a synthetic opioid that targets neuronal cells through G-protein-coupled receptors, GPCRs. It has conventionally been used as a pain killer and as a treatment for patients suffering from severe addictions to more potent opioids such as heroine and fentanyl (Ali et al., 2017). Like neuronal cells, leukocytes express opioid receptors (Celik et al., 2016) that when stimulated modulate proliferation, chemotaxis, cytokine production and cytotoxicity (Bidlack, 2000; Sharp, 2006; Ninkovic and Roy, 2013). Stimulation of opioid GPCRs causes alteration in receptor conformation, which allows GDP to GTP exchange on G protein α subunit (Gα), one of the heterotrimeric G protein complexes (Gαβγ), resulting in uncoupling of Gα from Gβγ (Al-Hasani and Bruchas, 2011). Both Gβγ dimer and GTP-bound Gα are involved in pain modulation (Celik et al., 2016) and apoptosis (Friesen et al., 2013), respectively, by activating their effectors: Ca2+ or cyclic adenosine monophosphate (cAMP). Dissociated Gβγ stimulates phospholipase C beta (PLCβ) that hydrolyzes phosphatidylinositol-4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol. IP3 triggers endoplasmic reticulum (ER) Ca2+ release via IP3 receptor (IP3R) Ca2+ channel, which in leukocytes leads to secretion of opioid peptides such as Met-enkephalin (Labuz et al., 2009), β-endorphin (Labuz et al., 2010), and dynorphin (Liou et al., 2011) which mediate the pain relief response (Celik et al., 2016). Conversely, GTP-bound Gαi blocks adenylyl cyclase (AC) activity, resulting in reduced [cAMP]i, activation of caspases, and induction of apoptosis (Friesen et al., 2013).
Previously, it was shown that D,L-methadone induces ALL cell apoptosis through stimulation of an opioid receptor, downregulation of cAMP and upregulation of caspases (Friesen et al., 2008; Friesen et al., 2013): opioid receptor-Gαi-AC-↓[cAMP]i-caspase pathway. Since there are at least four different types of opioid receptors and the precise mechanism through which D,L-methadone induces ALL cell apoptosis was not known, we previously investigated these issues and demonstrated that D,L-methadone-induced ALL apoptosis occurred via the μ-opioid receptor 1 (OPRM1), triggering ER Ca2+ release and decreased Ca2+ efflux, elevating [Ca2+]i that upregulated the calpain-1-Bid-cytochrome C-caspase-3/12 apoptotic pathway (Lee et al., 2021): OPRM1-↑[Ca2+]i-calpain-1-Bid-cyt C-caspase-3/12 pathway. Since cAMP activates protein kinase A (PKA), which phosphorylates (i) PLCβ3 at Ser1105, inhibiting PLCβ3 activation (Yue et al., 1998), and (ii) BAD at Ser118, disrupting BCL-2-BAD interaction, which stimulates the BCL-2 anti-apoptotic activity (Lizcano et al., 2000; Virdee et al., 2000), it is possible that D,L-methadone-induced downregulation of PKA activity due to ↓[cAMP]i may cause reduced phosphorylation of (i) PLCβ3 at Ser1105, stimulating PLCβ3 activity and IP3R-mediated ER Ca2+ release, and (ii) BAD at Ser118, promoting the formation of the BCL-2-BAD complex, which neutralizes the Bcl-2 anti-apoptotic activity, allowing BAK and BAX to form pores in the outer mitochondrial membrane, inducing cyt C release, caspase activation, and apoptosis (Gavathiotis et al., 2008; Westphal et al., 2011). If this holds true, PKA may serve as a link between the two D,L-methadone-induced OPRM1-mediated apoptotic pathways: (i) Gαi-AC-↓[cAMP]i-caspase pathway (Friesen et al., 2008; Friesen et al., 2013) and (ii) ↑[Ca2+]i-calpain-1-Bid-cyt C-caspase-3/12 pathway (Lee et al., 2021).
In this study, we examined the possibility that PKA is a link between the two D,L-methadone-induced apoptotic pathways in leukemic cells. Using SEM and POETIC2 ALL cells, we demonstrate that D,L-methadone-induced ER Ca2+ release occurs through Gαi-dependent downregulation of AC and cAMP. This coincides with a rapid and transient downregulation of PKA-mediated inhibitory phosphorylation of PLCβ3, resulting in PLCβ3 stimulation and subsequent ER Ca2+ release as well as downregulation of PKA-mediated BAD Ser118 phosphorylation, which ultimately results in caspase activation and apoptosis. Our finding that PKA inhibition alone elicited ER Ca2+ release while PKA inhibition together with D,L-methadone treatment does not enhance ER Ca2+ release indicates that PKA, indeed, connects the two proposed D,L-methadone-induced apoptotic pathways in leukemic cells.
Materials and methods
Materials
RPMI 1640 (11875093), Opti-MEM reduced serum (31985062), MEMα (12561056), penicillin-streptomycin (15140122), phosphorylated Ser118 BAD (PA5-12550) and Mag-Fluo-4 a.m. (M14206) were from ThermoFisher Scientific (Burlington, ON, Canada). FBS (89510-186) was from VWR. The preservative-free methadone hydrochloride (NDC 17478-380-20) was from Market Drugs Medical Ltd. (Edmonton, AB). The 2,5-Di- tert-butylhydroquinone (TBHQ, 112976), 14–22 amide (myr) (476485), pertussis toxin (PTx; P7208), and Ponceau S stain (p-3504) were from Sigma-Aldrich (Oakville, ON, Canada). The 8-CPT-cAMP (ab120424) and HRP-conjugated goat antirabbit IgG (ab288151) secondary antibody were from Abcam (Toronto, ON, Canada). Forskolin (66575-29-9) was from Alomone Labs (Jerusalem, Israel). Gallein (sc-202631) and antibodies for total PLCβ3 (D-7), total BAD (C-7), and GAPDH (0411) were from Santa Cruz Biotechnology (Dallas, TX, United States of America). The antibody against phosphorylated Ser1105 PLCβ3 (2484) and HRP-conjugated goat antimouse IgG (7076) secondary antibody were from Cell Signalling (Whitby, ON, Canada).
Cell culture
SEM and POETIC2 cell lines originated from a female 5-year-old patient diagnosed with B-cell ALL (Lee et al., 2019) and a 14-year-old patient with pre-B ALL (Lee et al., 2021), respectively, were gifts from Dr. Aru Narendran, University of Calgary. RPMI1640 and Opti-MEM reduced serum containing 10% fetal bovine serum, and 100 μg/mL penicillin/streptomycin were used to culture the SEM and POETIC2 cells, respectively, at 37°C and CO2 level of 5%. SEM cells stably infected with a retrovirus carrying an empty pRS vector or pRS-shHAP1 were generated as described previously (Lee et al., 2019). SK-N-MC neuroblastoma cells, obtained from an Askin’s tumor in a 14-year-old female patient, were cultured in MEMα containing 10% fetal bovine serum, and 100 μg/mL penicillin/streptomycin at 37°C and CO2 level of 5%.
Measurement of endoplasmic reticulum (ER) Ca2+ release
SEM, POETIC2 and SK-N-MC cells (∼0.5 × 106) grown on 0.2 mg/mL poly-L-ornithine-coated 12 mm glass coverslips were loaded with 2 μM Mag-Fluo-4 a.m. in RPMI media for 45 min. Coverslips were then transferred to a 3.5 cm glass bottom plate containing 1 mL of Ca2+-free Krebs-Ringer-Henseleit (KRH) buffer (25 mM HEPES, pH 7.4, 125 mM NaCl, 5 mM KCl, 6 mM glucose, 1.2 mM MgCl2, and 2 μM EGTA). Ca2+ transients were traced using the DMi8-Film microscope at a magnification of ×20 and the LASX imaging software (Leica Microsystems). The HyD laser for confocal imaging (Leica Microsystems) was used at λEx = 495nm and λEm = 530nm. After obtaining stable baseline ER Ca2+ levels, cells were pre-treated (or not pre-treated) with 0.1 μg/mL pertussis toxin (PTx), 2 μM galleon, 2 μM 14–22 amide (myr), 1 μM 8-CPT-cAMP, or 4 μM forskolin for the indicated period of time, then treated with 2 μg/mL of D,L-methadone or 10 μM TBHQ. The average Ca2+ tracings were measured per sec in 10–12 individual cells following the treatment. The resulting ER Ca2+ release was assessed through changes in ER fluorescence: the signal-to-baseline ratio (SBR), which is simply the F/F0 ratio, where F = fluorescence value after stimulation and F0 = basal or initial fluorescence).
Western blot analysis
Lysates of cells (1 × 106) treated with 2 μg/mL D,L-methadone or 4 µM forskolin followed by 2 μg/mL D,L-methadone at the indicated times were resolved by 10% SDS-PAGE and transferred to nitrocellulose membranes (Pall Laboratory, ON), which were then immunostained for pSer1105-PLCβ3 (2484, Cell Signalling), total PLCβ3 (D-7, Santa Cruz Biotechnology), pSer118-BAD(PA5-12550, ThermoFisher Scientific), and total BAD (C-7, Santa Cruz Biotechnology). Membranes were incubated with the indicated antibody (diluted to 1:1,000) overnight at 4°C. After washing three times in tris-buffered saline containing 0.1% triton X-100 (TBS-T), membranes were incubated with an HRP-conjugated secondary antibody [1:10,000; goat antirabbit IgG (Abcam, ab288151) or goat antimouse IgG (Cell signaling, 7076)] in TBS-T for 1 h. Immunoreactive bands were detected by enhanced chemiluminescence and visualized using the Bio-Rad ChemiDoc Imager at the optimal exposure set up. Ratios of pSer1105-PLCβ3 vs. total-PLCβ3 was determined using the NIH ImageJ 1.61 software.
Apoptotic analysis
SEM cells (#; 1 × 104) stably infected with retrovirus carrying either pRS empty vector (#+pRS) or pRS-shHAP1 (#+pRS-shHAP1) were seeded in 96-well plates coated with 0.02% poly-L-ornithine. Cells pre-treated with 2 μg/mL D,L-methadone or 2 μM 14–22 amide (myr), for 8 h were stained with 0.5 μg/mL annexin V-FITC (Invitrogen) and visualized at λex = 485 nm and λem = ×530 nm and ×10 magnification using an IX71 Olympus inverted microscope (Tokyo, Japan). The percentage of apoptotic cells was calculated based on a total of 101–298 cells counted per treatment. Analysis was performed using ImageJ 1.4.1 (NIH, United States). For flow cytometry, cells (0.5 × 106) pre-treated with 2 μM 14–22 amide (myr) then treated with 2 μg/mL D,L-methadone for 12 h were harvested, washed twice with 1× PBS, stained with Annexin V-FITC (2 μL) and propidium iodide (2 μL), and analysed using an Attune NxT flow cytometer (ThermoFisher Scientific, United States).
Statistical analysis
The student’s unpaired, two-tailed t-test was performed at p < 0.05. For experiments that exceeded more than two groups or treatments, one-way Analysis of Variance (ANOVA) with Tukey Honestly Significantly Different (HSD) post hoc tests were conducted to uncover the statistical differences between groups or treatments.
Results
D,L-methadone-induced ER Ca2+ release in leukemic cells is dependent on Gαi but not Gβγ
Since activation of opioid receptors has been shown to cause Gβγ-mediated ER Ca2+ release via PLC in neuroblastoma (Yoon et al., 1999) and leukocytes (Celik et al., 2016; Machelska and Celik, 2018), we tested the potential involvement of Gβγ in D,L-methadone-induced ER Ca2+ release in leukemic cells. To do so, SEM (Lee et al., 2019) and POETIC2 (Lee et al., 2021) leukemic cells loaded with an ER Ca2+ probe (Rossi and Taylor, 2020), Mag-Fluo-4 a.m., were pre-treated with gallein, a potent Gβγ inhibitor (Sanz et al., 2017), then treated with D,L-methadone, and analysed for ER Ca2+ release. Similar analysis was performed in SK-N-MC neuroblastoma cells, which served as positive control. While gallein inhibited D,L-methadone-induced ER Ca2+ release in SK-N-MC neuroblastoma cells (Figure 1A), it did not do so in SEM (Figure 1B) and POETIC2 (Supplementary Figure S1B) leukemic cells, indicating that D,L-methadone-induced ER Ca2+ release in leukemic cells is independent of Gβγ. Previous studies have shown that stimulation of opioid receptors by D,L-methadone activates Gαi, which blocks adenylyl cyclase (AC) activity that in turn reduces [cAMP]i (Friesen et al., 2013; Kang et al., 2017). However, it is not known whether this pathway is involved in D,L-methadone-induced ER Ca2+ release. Therefore, we initially tested the involvement of Gαi in D,L-methadone-induced ER Ca2+ release. Treatment with PTx, a potent Gαi inhibitor (Wu and Wong, 2005), completely abolished D,L-methadone-induced ER Ca2+ release (Figure 1C; Supplementary Figure S1C), indicating that this process involves Gαi.
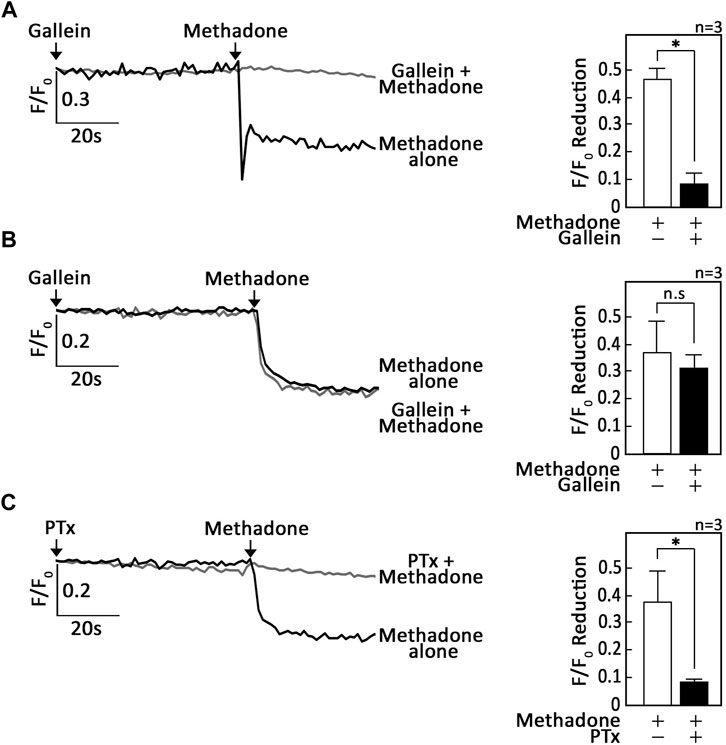
Figure 1. D,L-methadone-induced ER Ca2+ release in leukemic cells is blocked by inhibition of Gαi, but not Gβγ. SK-N-MC neuroblastoma (A) and SEM leukemic (B,C) cells loaded with Mag-Fluo-4 a.m. were subjected to Ca2+ tracings by single-cell Ca2+ imaging. After obtaining stable baseline ER Ca2+ levels, cells were pre-treated (or not pre-treated) with gallein (A,B) or PTx (C) for 60 s, then treated with D,L-methadone for 60 s to analyze ER Ca2+ release. The left panels show the average Ca2+ tracing measured per sec in 10 individual cells after gallein (A,B) or PTx (C) treatment. Data are from one of three independent experiments (n = 3) showing similar results. The charts on the right show the difference in ER Ca2+ release following treatment with D,L-methadone pretreated (or not pretreated) with gallein (A,B) or PTx (C). F/F0 value of 30 s after D,L-methadone addition (left panel) was used to determine F/F0 reduction. Values are means ± SEM of the three independent experiments. *p < 0.025. N.S., not significant.
D,L-methadone-induced ER Ca2+ release in leukemic cells results from downregulation of AC, cAMP and PKA
We then tested whether the Gαi-dependent D,L-methadone-induced ER Ca2+ release involves AC and cAMP. To do so, SEM and POETIC2 leukemic cells loaded with Mag-Fluo-4 a.m. were pre-treated with forskolin, an AC activator, or 8-CPT-cAMP, an exogenous cAMP analogue. Cells were then treated with D,L-methadone and analysed for ER Ca2+ release. As shown in Figures 2A, B (SEM cells) as well as Supplementary Figures S2A, S2B (POETIC2 cells), forskolin and 8-CPT-cAMP, respectively, inhibited D,L-methadone-induced ER Ca2+ release, indicating AC and cAMP involvement in this process.
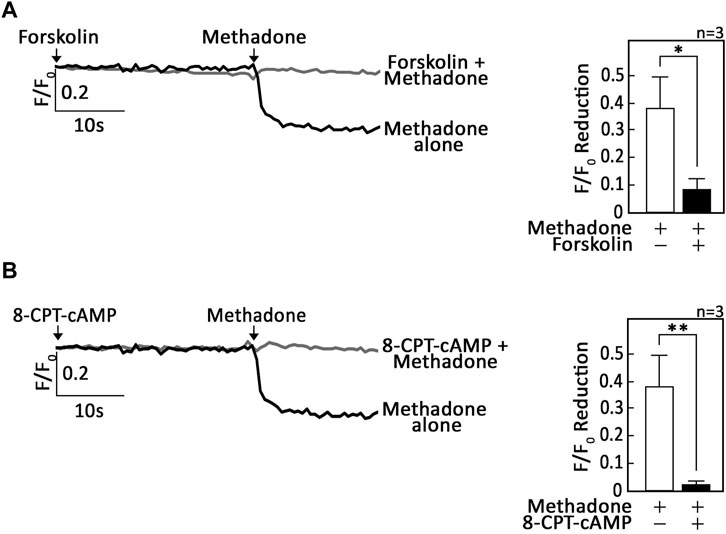
Figure 2. Stimulation of AC with forskolin or treatment with exogenous 8-CPT-cAMP inhibits D,L-methadone-induced ER Ca2+ release. SEM leukemic cells loaded with Mag-Fluo-4 a.m. were subjected to Ca2+ tracings by single-cell Ca2+ imaging. After obtaining stable baseline ER Ca2+ levels, cells were pre-treated (or not pre-treated) with forskolin (A) or 8-CPT-cAMP (B) for 30 s then treated with D,L-methadone for 30 s, to analyze ER Ca2+ release. The left panels show the average Ca2+ tracing measured per sec in 10 individual cells after forskolin (A) or 8-CPT-cAMP (B) treatment. Data are from one of three independent experiments (n = 3) showing similar results. The charts on the right show the difference in ER Ca2+ release following treatment with D,L-methadone pretreated (or not pretreated) with forskolin (A) or 8-CPT-cAMP (B). F/F0 value of 15 s after D,L-methadone addition (left panel) was used to determine F/F0 reduction. Values are means ± SEM of the three independent experiments. *p < 0.025. **p < 0.05.
Since cAMP signaling regulates [Ca2+]i through inhibitory phosphorylation of PLCβ3 by PKA (Liu and Simon, 1996; Ali et al., 1998; Yue et al., 1998), it is possible that OPRM1 stimulation by D,L-methadone also involves PKA inhibition. If so, we expect that treatment with 14–22 amide (myr), a myristylated cell permeable PKA-specific inhibitor (Dalton et al., 2005), alone will evoke ER Ca2+ release. In addition, pre-treatment with 14–22 amide (myr) will not cause further increase in D,L-methadone-induced ER Ca2+ release. Indeed, as shown in Figure 3; Supplementary Figure S3 14-22 amide (myr) alone triggered ER Ca2+ release and subsequent treatment with D,L-methadone did not cause additional ER Ca2+ release. These findings indicate that Gαi-AC-cAMP-dependent D,L-methadone-induced ER Ca2+ release involves PKA inhibition, which can circumvent the upstream D,L-methadone-OPRM1-AC-cAMP step. Treatment with TBHQ, an ER Ca2+ pump inhibitor, caused further ER Ca2+ release, indicating that these cells were viable during analysis (Figure 3; Supplementary Figure S3).
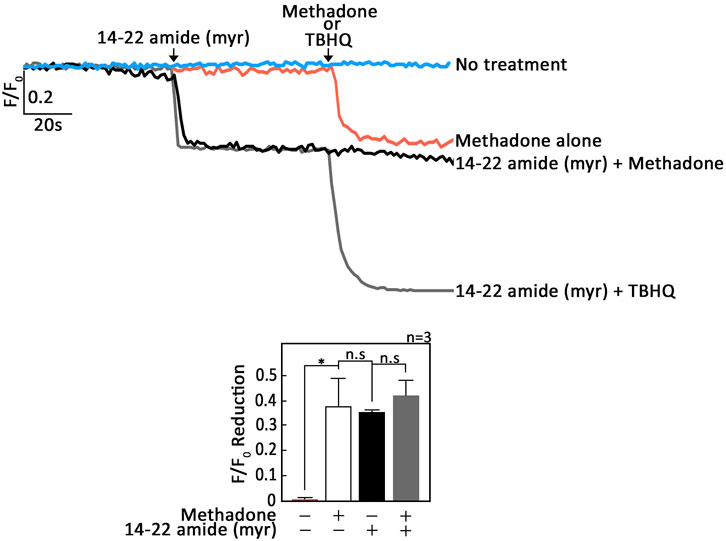
Figure 3. Treatment with 14–22 amide (myr) alone elicits ER Ca2+ release in leukemic cells and subsequent treatment with D,L-methadone does not cause additional ER Ca2+ release. SEM leukemic cells loaded with Mag-Fluo-4 a.m. were subjected to Ca2+ tracing by single-cell Ca2+ imaging. After obtaining stable baseline ER Ca2+ levels, cells were pretreated (or not pretreated) with 14–22 amide (myr) for 60 s then treated with D,L-methadone or TBHQ for 60 s, to analyze ER Ca2+ release. The left panels show the average Ca2+ tracing measured per sec in 10 individual cells after 14–22 amide (myr) treatment. Treatment with TBHQ, an ER Ca2+ pump inhibitor, caused further ER Ca2+ release, indicating that these cells were viable during analysis. Data are from one of three independent experiments (n = 3) showing similar results. The chart on the bottom shows the difference in ER Ca2+ release following treatment with D,L-methadone pretreated (or not pretreated) with 14–22 amide (myr). F/F0 value of 30 s after D,L-methadone and/or 14–22 amide (myr) addition (left panel) was used to determine F/F0 reduction. Values are means ± SEM of the three independent experiments. *p < 0.025. N.S., not significant.
D,L-methadone-induced ER Ca2+ release in leukemic cells is associated with downregulation of PKA-mediated (i) PLCβ3 inhibitory phosphorylation at Ser1105 and (ii) BAD phosphorylation at Ser118
Since stimulation of opioid receptors causes ER Ca2+ release via PLC (Yoon et al., 1999; Machelska and Celik, 2018), we examined whether D,L-methadone downregulates PKA-mediated inhibitory phosphorylation of PLCβ3 at Ser1105, which is necessary for inducing PLC-IP3/IP3R-mediated ER Ca2+ release (Zhong et al., 2008). To do so, lysates of cells treated with D,L-methadone were subjected to SDS-PAGE and immunoblotting for pSer1105-PLCβ3. As shown in Figure 4A; Supplementary Figures S4A, S6A, D,L-methadone drastically reduced the level of pSer1105-PLCβ3 after 15 s of treatment, which coincides with the prompt ER Ca2+ release upon D,L-methadone treatment as shown in Figures 1B, C; Supplementary Figure S1. As expected, pre-treatment with forskolin did not affect D,L-methadone-induced PLCβ3 phosphorylation at Ser1105 (Figure 4B; Supplementary Figures S4A, S6B), which is consistent with the loss of D,L-methadone-induced ER Ca2+ release upon forskolin treament (Figure 2A; Supplementary Figure S2A). Since cAMP activates protein kinase A (PKA), which phosphorylates BAD at Ser118, disrupting BCL-2-BAD interaction, which stimulates the BCL-2 anti-apoptotic activity (Lizcano et al., 2000; Virdee et al., 2000), we sought to examine whether D,L-methadone also downregulates BAD phosphorylation at Ser118. Figure 4A; Supplementary Figures S4, S6A shows that as with pSer1105-PLCβ3, the level of pSer118-BAD was reduced immediately upon D,L-methadone treatment. Pre-treatment with forskolin did not affect D,L-methadone-induced BAD phosphorylation at Ser118 (Figure 4B; Supplementary Figures S4, S6B). To determine the significance of PKA inhibition in PLCβ3 activation, lysates of cells pre-treated with 14–22 amide (myr) then treated with D,L-methadone were subjected to SDS-PAGE and immunoblotting for pSer1105-PLCβ3 and pSer118-BAD. As shown in Figure 4C; Supplementary Figures S5, S6C, treatment with D,L-methadone or 14–22 amide (myr) considerably reduced the phosphorylation of PLCβ3 at Ser1105 and BAD at Ser118, indicating that such phosphorylation occurs via PKA. Our observation that 14–22 amide (myr) alone caused considerable decrease in PLCβ3 phosphorylation at Ser1105 and subsequent treatment with D,L-methadone did not cause further decrease in PLCβ3 phosphorylation at Ser1105 is consistent with our finding that 14–22 amide (myr) alone triggered ER Ca2+ release, and suggests that D,L-methadone-induced OPRM1-mediated ER Ca2+ release occurs via the Gαi-AC-cAMP-PKA-PLC pathway.
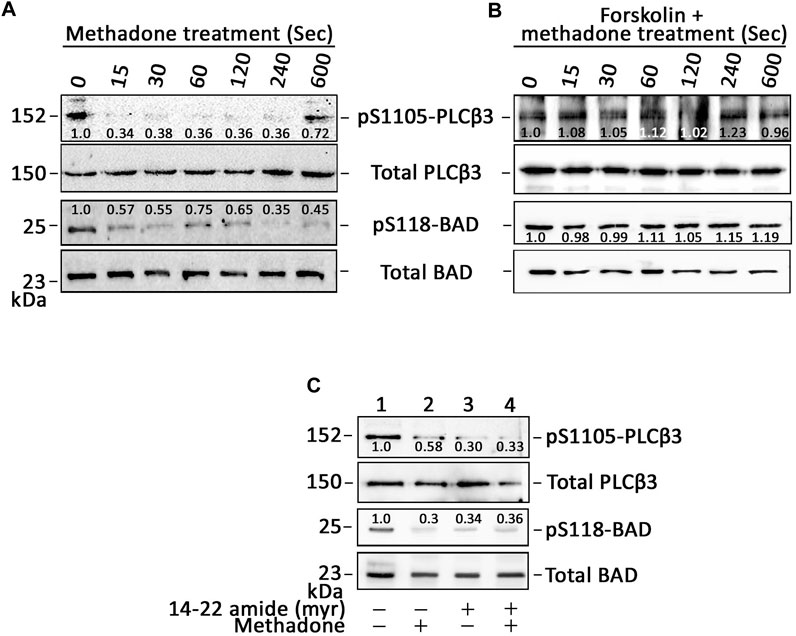
Figure 4. D,L-methadone-induced ER Ca2+ release in leukemic cells is associated with downregulation of PLCβ3 Ser1105 inhibitory phosphorylation and BAD phosphorylation at Ser118. (A–C) Lysates of SEM cells pre-treated (or not pre-treated; A) with forskolin (B) or 14–22 amide (myr) (C) then treated with D,L-methadone were subjected to SDS-PAGE and immunoblotting for pSer1105-PLCβ3 and total PLCβ3, and/or pSer118-BAD and total BAD. In C, lysates of cells treated with D,L-methadone for 120 s were used. The numbers under the pSer1105-PLCβ3 and pSer118-BAD bands represent the relative intensity ratios of pSer1105-PLCβ3 or pSer118-BAD vs. total PLCβ3 or BAD bands, respectively, with values at time 0 normalized to 1. Representative blots are from one of three independent experiments (n = 3) showing similar results.
PKA inhibition alone triggers leukemic cell apoptosis
As our data indicated that PKA inhibition is a central step in D,L-methadone-induced apoptosis that involves PLCβ3 activation and BAD Ser118 phosphorylation, we sought to examine whether treatment with 14–22 amide (myr) alone causes leukemic cell apoptosis. To do so, cells treated with 14–22 amide(myr) for 8 h were stained with FITC-annexin V and subjected to fluorescence microscopy. The percentage of apoptotic cells was calculated. For negative control, we used SEM cells (#) stably depleted of HAP1 by infection with retrovirus carrying pRS-shHAP1 (#+pRS-shHAP1). Lack of HAP1 (Figure 5A), a key component of the functional IP3R/HAP1/Htt ternary complex (Tang et al., 2003; Lee et al., 2019), inhibits ER Ca2+ release upon stimulation with D,L-methadone or treatment with 14–22 amide (myr) (Figure 5B). In contrast, D,L-methadone and 14–22 amide (myr) cause a similar extent of ER Ca2+ release in #+pRS cells (Figure 5B). As shown in Figure 6, #+pRS cells show greater apoptosis compared to #+pRS-shHAP1 cells upon treatment with 14–22 amide (myr) (17.7% vs. 9.8%) or D,L-methadone (11.6% vs. 4.5%). It is interesting that 14–22 amide (myr) causes greater apoptosis compared to D,L-methadone in both #+pRS and #+pRS-shHAP1 cells (17.7% vs. 11.6% and 9.8% vs. 4.5%, respectively). Similar results were also obtained in POETIC2 cells (Supplementary Figure S7). Together, these findings indicate that PKA inhibition by 14–22 amide (myr) effectively kills ALL cells.
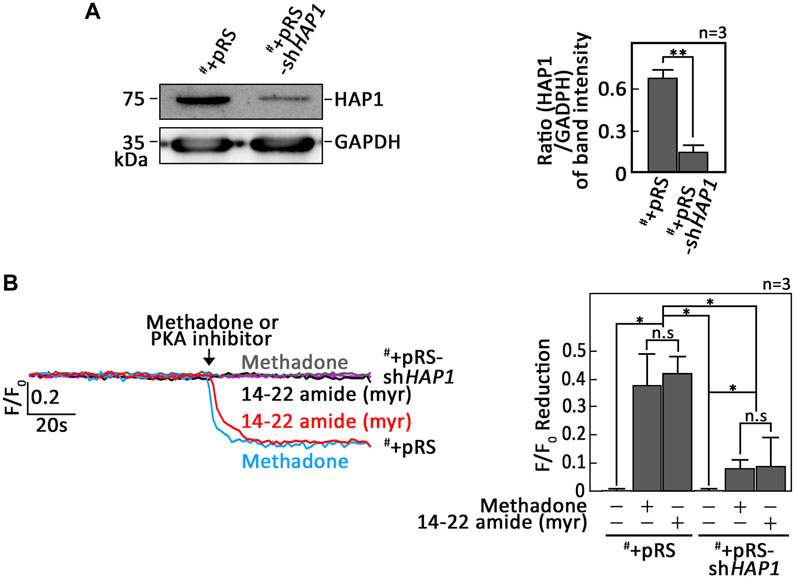
Figure 5. Leukemic cells stably depleted of HAP1 inhibit ER Ca2+ release upon stimulation with D,L-methadone, or treatment with 14–22 amide (myr). (A) Lysates of SEM cells (#) infected with retrovirus carrying pRS empty vector (#+pRS) or pRS-shHAP1 (#+pRS-shHAP1) were resolved by SDS-PAGE and immunoblotted for HAP1. GAPDH blot was used as loading control. The right panel shows the ratios of HAP1 vs. GAPDH levels, which were measured by densitometric analysis of the blots using NIH ImageJ 1.61. GAPDH levels were normalized to 1.0. Standard deviation of the HAP1 vs. GAPDH ratio was calculated from the three sets of experiments. (B) #+pRS and #+pRS-shHAP1 cells loaded with Mag-Fluo-4 a.m. were subjected to Ca2+ tracing by single-cell Ca2+ imaging. After obtaining stable baseline ER Ca2+ levels, cells were (or not treated: purple colored trace) treated with 14–22 amide (myr) or D,L-methadone for 60 s, to analyze ER Ca2+ release. The left panel shows the average Ca2+ tracing per sec from 10 individual cells before and after treatment. Data are from one of three independent experiments (n = 3) showing similar results. The chart on the right shows the difference in ER Ca2+ release in #+pRS and #+pRS-shHAP1 cells following treatment with 14–22 amide (myr) or D,L-methadone. F/F0 value of 30 s after D,L-methadone or 14–22 amide (myr) addition (left panel) was used to determine F/F0 reduction. Values are means ± SEM of the three independent experiments. *p < 0.025. **p < 0.05. N.S., not significant.
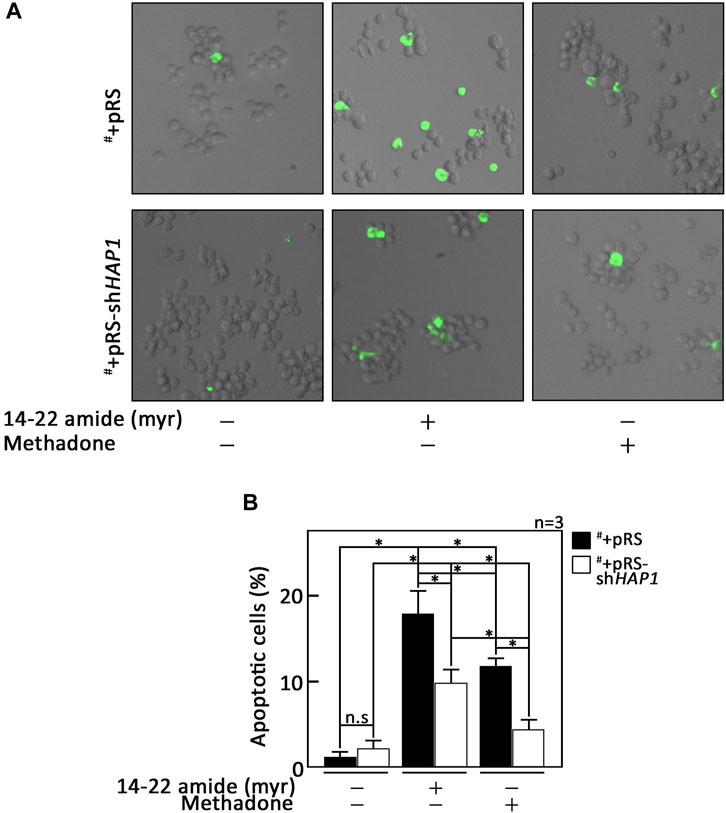
Figure 6. PKA inhibition alone causes leukemic cell apoptosis. (A) #+pRS and #+pRS-shHAP1 cells treated with 14–22 amide (myr) or D,L-methadone for 8 h were stained with FITC-annexin V and subjected to fluorescence microscopy. (B) The percentage of apoptotic cells was calculated based on a total of 101–298 cells counted per treatment. Values are means ± SEM from three independent experiments (n = 3) that showed similar results. *p < 0.025. N.S., not significant.
Discussion
In recent years, the molecular mechanism by which D,L-methadone induces leukemic cell apoptosis has been investigated (Friesen et al., 2013; Lee et al., 2021). Friesen et al. (2013) have shown that D,L-methadone, which stimulates opioid receptors, causes ALL cell apoptosis through Gαi-AC, decrease in [cAMP]i and subsequent activation of caspase-9 and -3 (Figure 7A, in red). However, the identity of the specific opioid receptor(s) targeted by D,L-methadone was not defined. Through unbiased genome-wide RNAi screening and knockdown studies, we previously found that presence or absence of the μ1 opioid receptor subtype, OPRM1, determines the fate of ALL cells following L-asparaginase treatment, i.e., presence leads to apoptosis while absence leads to survival or resistance (Kang et al., 2017). In further studies, we found that OPRM1 is targeted by D,L-methadone to induce leukemic cell apoptosis (Lee et al., 2021). In fact, OPRM1 loss inhibits D,L-methadone-induced ALL cell apoptosis (Lee et al., 2021). As shown in Figures 7A, D,L-methadone activation of OPRM1 causes a lethal rise in [Ca2+]i by stimulating IP3R-induced ER Ca2+ release through PLCβ and decreasing Ca2+ efflux, leading to upregulation of the Ca2+-mediated calpain-1-Bid-cyt C-caspase-3/12 apoptotic pathway (Figure 7A, in black) (Lee et al., 2021). Based on the model that D,L-methadone causes Gβγ-mediated [Ca2+]i increase in leukemic cells (Friesen et al., 2013), and the fact that in leukocytes (Celik et al., 2016; Machelska and Celik, 2018) and neuroblastoma cells (Yoon et al., 1999), opioid causes an increase in [Ca2+]i through Gβγ, it is possible that PLCβ-mediated ER Ca2+ release is triggered through Gβγ-coupled OPRM1 (Figure 7A). Since reduced [cAMP]i, resulting from Gαi-mediated inhibition of AC, causes loss of PKA activation, which (i) stimulates PLCβ3 and subsequent IP3/IP3R-mediated ER Ca2+ release (Yue et al., 1998) and (ii) promotes the formation of the BCL-2-BAD complex, which neutralizes the Bcl-2 anti-apoptotic activity, allowing BAK and BAX to form pores in the outer mitochondrial membrane, inducing cyt C release, caspase activation, and apoptosis (Gavathiotis et al., 2008; Westphal et al., 2011), it is also possible that PKA inhibition, not Gβγ activation, causes this process following stimulation with D,L-methadone (Figure 7A). The current study tested these possibilities.
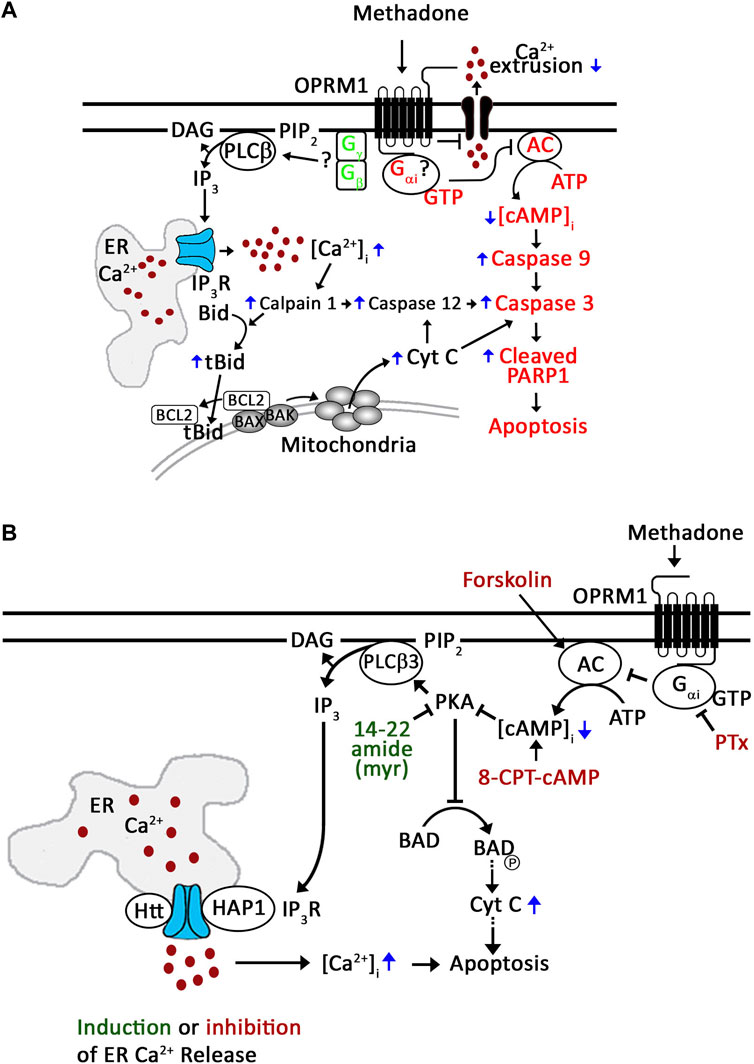
Figure 7. Proposed mechanism for D,L-methadone-induced OPRM1-mediated apoptosis in leukemic cells. (A) Friesen et al. (2013) has shown that D,L-methadone induces leukemic cell apoptosis through the Gαi-AC-↓[cAMP]i-caspase pathway (in red). Our previous studies showed that D,L-methadone specifically stimulates the opioid receptor mu1 subtype, OPRM1, in leukemic cells (Lee et al., 2021); D,L-methadone activation of OPRM1 causes increased [Ca2+]i by enhancing IP3R-mediated ER Ca2+ release and decreasing Ca2+ efflux, upregulating the Ca2+-mediated calpain-1-Bid-cyt C-caspase-3/12 apoptotic pathway (in black) (Lee et al., 2021). Together, these findings point to two D,L-methadone-induced OPRM-mediated apoptotic pathways: (i) Gαi-AC-↓[cAMP]i-caspase pathway (Friesen et al., 2013) and (ii) ↑[Ca2+]i-calpain-1-Bid-cyt C-caspase-3/12 pathway (Lee et al., 2021). Since activation of opioid receptors causes Gβγ-mediated ER Ca2+ release via PLC in neuroblastoma cells (Yoon et al., 1999) and leukocytes (Celik et al., 2016; Machelska and Celik, 2018), in the latter pathway, we proposed the involvement of Gβγ (in green) in D,L-methadone-induced ER Ca2+ release in leukemic cells (Lee et al., 2021). (B) In the current study, we demonstrate that D,L-methadone-induced ER Ca2+ release in ALL cells results from Gαi-dependent downregulation of AC and cAMP. PKA inhibition elicits ER Ca2+ release and overrides D,L-methadone-induced ER Ca2+ release, indicating that PKA inhibition is a central step in D,L-methadone-induced ER Ca2+ release. This is consistent with decrease in PKA-dependent (i) inhibitory phosphorylation of PLCβ3 at Ser1105 that leads to PLCβ3 activation and ER Ca2+ release, and (ii) BAD Ser118 phosphorylation, which ultimately result in caspase activation and apoptosis. Thus, D,L-methadone-induced ER Ca2+ release and subsequent apoptosis in ALL cells is mediated by Gαi-dependent downregulation of the AC-cAMP-PKA-PLCβ3/BAD pathway. Small molecular activators/inhibitors shown in red inhibit ER Ca2+ release; 14–22 amide (myr) shown in green induces ER Ca2+ release.
Upon testing the possibility that Gβγ-coupled OPRM1 triggers PLCβ3-mediated ER Ca2+ release, we found that PTx, but not gallein, inhibited D,L-methadone-induced ER Ca2+ release, indicating that this process is dependent on Gαi but not Gβγ. This was corroborated by our data showing that stimulation of AC with forskolin (Insel and Ostrom, 2003), and treatment with exogenous 8-CPT-cAMP inhibited D,L-methadone-induced ER Ca2+ release, indicating that D,L-methadone caused Gαi-mediated downregulation of AC activity, resulting in reduced [cAMP]i. While Gβγ in pancreatic acini cells was shown to also directly activate IP3R independent of IP3 production, causing ER Ca2+ release (Zeng et al., 2003), the inability of gallein to inhibit D,L-methadone-induced ER Ca2+ release, and the ability of PTx to completely inhibit D,L-methadone-induced ER Ca2+ release indicate that D,L-methadone-induced ER Ca2+ release in leukemia cells does not involve direct activation of IP3R by Gβγ. The requirement for PKA inhibition in D,L-methadone-induced OPRM1-mediated ER Ca2+ release was proven by our data showing that the 14–22 amide (myr) PKA inhibitor alone evoked ER Ca2+ release, and that subsequent treatment with D,L-methadone did not cause additional ER Ca2+ release. The involvement of Gαi-AC-cAMP-PKA-PLC was further supported by our observations that D,L-methadone triggered immediate downregulation of PKA-mediated PLCβ3 inhibitory phosphorylation at Ser1105, which is required for eliciting IP3/IP3R-mediated ER Ca2+ release (Zhong et al., 2008), and that pre-treatment with forskolin did not affect D,L-methadone-induced PLCβ3 phosphorylation at Ser1105. Potentially, protein phosphatase (PP)1/PP2A (Chiang et al., 2003; Zhong et al., 2008) is involved in the dephosphorylation of PLCβ3 at Ser1105 following D,L-methadone-induced inactivation of PKA. Our finding that D,L-methadone also downregulated BAD phosphorylation at Ser118 is consistent with previously published works showing cytochrome C release and caspase activation in D,L-methadone-induced ALL cell apoptosis (Lee et al., 2021). These findings indicate that PKA serves as a link between the two previously described D,L-methadone-induced OPRM-mediated apoptotic pathways: (i) Gαi-AC-↓[cAMP]i-caspase (Friesen et al., 2013) and ↑[Ca2+]i-calpain-1-Bid-cyt C-caspase-3/1216. Our data showing that the 14–22 amide (myr) PKA inhibitor alone reduced the levels of pSer118-BAD and pSer1105-PLCβ3, which are part of the above two apoptotic pathways, respectively (Figure 7B), led us to examine whether 14–22 amide (myr) induces ALL cell apoptosis. In fact, we found that PKA inhibition using 14–22 amide (myr) effectively killed ALL cells. The lesser extent of apoptosis in cells depleted of HAP1 (#+pRS-shHAP1) compared to control cells (#+pRS) upon treatment with 14–22 amide (myr) or D,L-methadone is likely due to the inability of #+pRS-shHAP1 cells to induce IP3R-mediated ER Ca2+ release. Since 14–22 amide (myr) and D,L-methadone caused a similar extent of ER Ca2+ release in #+pRS cells, our observation that 14–22 amide (myr) causes greater apoptosis compared to D,L-methadone in both #+pRS and #+pRS-shHAP1 cells may be attributed to the greater efficiency of the 14–22 amide (myr) PKA inhibitor in downregulating BAD Ser118 phosphorylation and stimulating cyt C release. While PKA and PLCβ are involved in autophagy, there is no report indicating that D,L-methadone affects autophagy in ALL cells.
While lack of OPRM1 causes ALL cell resistance to asparaginase (Kang et al., 2017), the fact that PKA inhibition can circumvent the upsteam OPRM1-AC-cAMP step suggests that PKA may be targeted for therapy in L-asparaginase-resistant ALL. In addition, although D,L-methadone holds promise as future therapy for ALL (Friesen et al., 2013; Michalska et al., 2017; Onken et al., 2017), it is clear that PKA may also be targeted for therapy in both D,L-methadone-refractory and -sensitive ALL.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
HK: Data curation, Formal Analysis, Investigation, Methodology, Visualization, Writing–original draft. JL: Investigation, Methodology, Supervision, Validation, Visualization, Writing–original draft. K-YL: Conceptualization, Funding acquisition, Resources, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by a grant from CIHR (PJT-174983) to K-YL, which was transferred to Dr. Karl Riabowol due to K-YL’s retirement.
Acknowledgments
We express our gratitude to Drs. Jesusa L. Rosales, Andrew Braun and Faisal Khan for their suggestion and critical comments for the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2024.1388745/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Gαi-dependent D,L-methadone-induced ER Ca2+ release in POETIC2 cells. POETIC2 cells loaded with Mag-Fluo-4 AM were subjected to Ca2+ tracing by single-cell Ca2+ imaging. After obtaining stable baseline ER Ca2+ levels, cells were pretreated (or not pre-treated, (A) with gallein (B) or PTx (C) for 2 min, then treated with D,L-methadone for 2 min to analyze ER Ca2+ release. The left panels show the average Ca2+ tracing measured per sec in 12 individual cells after gallein (B) or PTx (C) treatment. Data are from one of three independent experiments (n = 3) showing similar results. The charts on the right show the difference in ER Ca2+ release following treatment with D,L-methadone in cells pretreated (or not pretreated, (A) with gallein (B) or PTx (C). F/F0 value of 40 s after D,L-methadone addition (left panel) was used to determine F/F0 reduction. Values are means ± SEM of the three independent experiments. *p < 0.025. N.S., not significant.
SUPPLEMENTARY FIGURE S2 | Inhibition of D,L-methadone-induced ER Ca2+ release by forskolin or 8-CPT-cAMP in POETIC2 cells. POETIC2 leukemic cells loaded with Mag-Fluo-4 AM were subjected to Ca2+ tracings by single-cell Ca2+ imaging. After obtaining stable baseline ER Ca2+ levels, cells were pre-treated (or not pre-treated) with forskolin (A) or 8-CPT-cAMP (B) for 40 s then treated with 1 µg/ml D,L-methadone for 70 s, to analyze ER Ca2+ release. The left panels show the average Ca2+ tracing measured per sec in 12 individual cells after forskolin (A) or 8-CPT-cAMP (B) treatment. Data are from one of three independent experiments (n = 3) showing similar results. The charts on the right show the difference in ER Ca2+ release following treatment with D,L-methadone pretreated (or not pretreated) with forskolin (A) or 8-CPT-cAMP (B). F/F0 value of 30 s after D,L-methadone addition (left panel) was used to determine F/F0 reduction. Values are means ± SEM of the three independent experiments. *p < 0.05.
SUPPLEMENTARY FIGURE S3 | ER Ca2+ release by 14-22 amide (myr) alone in POETIC2 cells. POETIC2 leukemic cells loaded with Mag-Fluo-4 AM were subjected to Ca2+ tracing by single-cell Ca2+ imaging. After obtaining stable baseline ER Ca2+ levels, cells were pretreated (or not pretreated) with 14-22 amide (myr) for 30 s then treated with D,L-methadone or TBHQ for 100 s, to analyze ER Ca2+ release. The left panels show the average Ca2+ tracing measured per sec in 10 individual cells after 14-22 amide (myr) treatment. Treatment with TBHQ, an ER Ca2+ pump inhibitor, caused further ER Ca2+ release, indicating that these cells were viable during analysis. Data are from one of three independent experiments (n = 3) showing similar results. The chart on the bottom shows the difference in ER Ca2+ release following treatment with D,L-methadone pretreated (or not pretreated) with 14-22 amide (myr). F/F0 value of 30 s after D,L-methadone and/or 14-22 amide (myr) addition (left panel) was used to determine F/F0 reduction. Values are means ± SEM of the three independent experiments. *p < 0.025. N.S., not significant.
SUPPLEMENTARY FIGURE S4 | The bar graphs in (A,B) represent the levels of pSer1105-PLCβ3 vs total PLCβ3 ratios in Figure 4A (left upper two panels) and 4B (right upper two panels) and the levels of pSer118-BAD vs total BAD ratios in Figure 4A (left lower two panels) and 4B (right lower two panels), respectively. Values are means ± SEM of the three independent experiments (n = 3). *p < 0.05. N.S., not significant.
SUPPLEMENTARY FIGURE S5 | The bar graphs in the left and right represent the levels of pSer1105-PLCβ3 vs total PLCβ3 ratios in Figure 4C (upper two panels) and the levels of pSer118-BAD vs total BAD ratios in Figure 4C (lower two panels), respectively. Values are means ± SEM of the three independent experiments (n = 3). *p < 0.05. N.S., not significant.
SUPPLEMENTARY FIGURE S6 | Downregulation of PLCβ3 Ser1105 inhibitory phosphorylation and BAD phosphorylation at Ser118 in POETIC2 leukemic cells that induce ER Ca2+ release upon D,L-methadone treatment. (A–C) Lysates of POETIC2 cells pre-treated or not pre-treated; (A) with forskolin (B) or 14-22 amide (myr) (C) then treated with D,L-methadone were subjected to SDS-PAGE and immunoblotting for pSer1105-PLCβ3 and total PLCβ3, and/or pSer118-BAD and total BAD. In (C), lysates of cells treated with D,L-methadone for 120 s were used. The numbers under the pSer1105-PLCβ3 and pSer118-BAD bands represent the relative intensity ratios of pSer1105-PLCβ3 or pSer118-BAD vs total PLCβ3 or BAD bands, respectively, with values at time 0 normalized to 1. Data are from one of two independent experiments (n = 2) showing similar results.
SUPPLEMENTARY FIGURE S7 | PKA inhibition-induced ALL cell apoptosis analyzed by flow cytometry. #+pRS and #+pRS-shHAP1 cells pre-treated (or not pre-treated) with 14-22 amide (myr) then treated (not treated) with D,L-methadone for 12 h were stained with PI and FITC-annexin V and subjected to flow cytometry as described in Materials and Methods. Values are means ± SEM from three independent experiments (n = 3). *p < 0.05. N.S., not significant.
References
Al-Hasani, R., and Bruchas, M. R. (2011). Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115, 1363–1381. doi:10.1097/ALN.0b013e318238bba6
Ali, H., Sozzani, S., Fisher, I., Barr, A. J., Richardson, R. M., Haribabu, B., et al. (1998). Differential regulation of formyl peptide and platelet-activating factor receptors. Role of phospholipase Cbeta3 phosphorylation by protein kinase A. J. Biol. Chem. 273, 11012–11016. doi:10.1074/jbc.273.18.11012
Ali, S., Tahir, B., Jabeen, S., and Malik, M. (2017). Methadone treatment of opiate addiction: a systematic review of comparative studies. Innov. Clin. Neurosci. 14, 8–19.
Bidlack, J. M. (2000). Detection and function of opioid receptors on cells from the immune system. Clin. Diagn Lab. Immunol. 7, 719–723. doi:10.1128/CDLI.7.5.719-723.2000
Capria, S., Molica, M., Mohamed, S., Bianchi, S., Moleti, M. L., Trisolini, S. M., et al. (2020). A review of current induction strategies and emerging prognostic factors in the management of children and adolescents with acute lymphoblastic leukemia. Expert Rev. Hematol. 13, 755–769. doi:10.1080/17474086.2020.1770591
Celik, M. O., Labuz, D., Henning, K., Busch-Dienstfertig, M., Gaveriaux-Ruff, C., Kieffer, B. L., et al. (2016). Leukocyte opioid receptors mediate analgesia via Ca(2+)-regulated release of opioid peptides. Brain Behav. Immun. 57, 227–242. doi:10.1016/j.bbi.2016.04.018
Chiang, C. W., Kanies, C., Kim, K. W., Fang, W. B., Parkhurst, C., Xie, M., et al. (2003). Protein phosphatase 2A dephosphorylation of phosphoserine 112 plays the gatekeeper role for BAD-mediated apoptosis. Mol. Cell Biol. 23, 6350–6362. doi:10.1128/MCB.23.18.6350-6362.2003
Dalton, G. D., Smith, F. L., Smith, P. A., and Dewey, W. L. (2005). Alterations in brain Protein Kinase A activity and reversal of morphine tolerance by two fragments of native Protein Kinase A inhibitor peptide (PKI). Neuropharmacology 48, 648–657. doi:10.1016/j.neuropharm.2004.12.006
Friesen, C., Roscher, M., Alt, A., and Miltner, E. (2008). Methadone, commonly used as maintenance medication for outpatient treatment of opioid dependence, kills leukemia cells and overcomes chemoresistance. Cancer Res. 68, 6059–6064. doi:10.1158/0008-5472.CAN-08-1227
Friesen, C., Roscher, M., Hormann, I., Fichtner, I., Alt, A., Hilger, R. A., et al. (2013). Cell death sensitization of leukemia cells by opioid receptor activation. Oncotarget 4, 677–690. doi:10.18632/oncotarget.952
Gavathiotis, E., Suzuki, M., Davis, M. L., Pitter, K., Bird, G. H., Katz, S. G., et al. (2008). BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081. doi:10.1038/nature07396
Insel, P. A., and Ostrom, R. S. (2003). Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell Mol. Neurobiol. 23, 305–314. doi:10.1023/A:1023684503883
Kang, S. M., Rosales, J. L., Meier-Stephenson, V., Kim, S., Lee, K. Y., and Narendran, A. (2017). Genome-wide loss-of-function genetic screening identifies opioid receptor μ1 as a key regulator of L-asparaginase resistance in pediatric acute lymphoblastic leukemia. Oncogene 36, 5910–5913. doi:10.1038/onc.2017.211
Labuz, D., Schmidt, Y., Schreiter, A., Rittner, H. L., Mousa, S. A., and Machelska, H. (2009). Immune cell-derived opioids protect against neuropathic pain in mice. J. Clin. Invest. 119, 278–286. doi:10.1172/JCI36246
Labuz, D., Schreiter, A., Schmidt, Y., Brack, A., and Machelska, H. (2010). T lymphocytes containing β-endorphin ameliorate mechanical hypersensitivity following nerve injury. Brain Behav. Immun. 24, 1045–1053. doi:10.1016/j.bbi.2010.04.001
Lee, J., Rosales, J. L., Byun, H. G., and Lee, K. Y. D. (2021). D,L-Methadone causes leukemic cell apoptosis via an OPRM1-triggered increase in IP3R-mediated ER Ca2+ release and decrease in Ca2+ efflux, elevating [Ca2+]i. Sci. Rep. 11, 1009. doi:10.1038/s41598-020-80520-w
Lee, J. K., Kang, S., Wang, X., Rosales, J. L., Gao, X., Byun, H. G., et al. (2019). HAP1 loss confers l-asparaginase resistance in ALL by downregulating the calpain-1-Bid-caspase-3/12 pathway. Blood 133, 2222–2232. doi:10.1182/blood-2018-12-890236
Liou, J. T., Liu, F. C., Mao, C. C., Lai, Y. S., and Day, Y. J. (2011). Inflammation confers dual effects on nociceptive processing in chronic neuropathic pain model. Anesthesiology 114, 660–672. doi:10.1097/ALN.0b013e31820b8b1e
Liu, M., and Simon, M. I. (1996). Regulation by cAMP-dependent protein kinease of a G-protein-mediated phospholipase C. Nature 382, 83–87. doi:10.1038/382083a0
Lizcano, J. M., Morrice, N., and Cohen, P. (2000). Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem. J. 349, 547–557. doi:10.1042/0264-6021:3490547
Machelska, H., and Celik, M. Ö. (2018). Advances in achieving opioid analgesia without side effects. Front. Pharmacol. 9, 1388. doi:10.3389/fphar.2018.01388
Michalska, M., Katzenwadel, A., and Wolf, P. (2017). Methadone as a "tumor theralgesic" against cancer. Front. Pharmacol. 8, ARTN 733. doi:10.3389/fphar.2017.00733
Midic, D., Rinke, J., Perner, F., Müller, V., Hinze, A., Pester, F., et al. (2020). Prevalence and dynamics of clonal hematopoiesis caused by leukemia-associated mutations in elderly individuals without hematologic disorders. Leukemia 34, 2198–2205. doi:10.1038/s41375-020-0869-y
Ninkovic, J., and Roy, S. (2013). Role of the mu-opioid receptor in opioid modulation of immune function. Amino Acids 45, 9–24. doi:10.1007/s00726-011-1163-0
Onken, J., Friesen, C., Vajkoczy, P., and Misch, M. (2017). Safety and tolerance of D,L-Methadone in combination with chemotherapy in patients with glioma. Anticancer Res. 37, 1227–1235. doi:10.21873/anticanres.11438
Rossi, A. M., and Taylor, C. W. (2020). Reliable measurement of free Ca(2+) concentrations in the ER lumen using Mag-Fluo-4. Cell Calcium 87, 102188. doi:10.1016/j.ceca.2020.102188
Sanz, G., Leray, I., Muscat, A., Acquistapace, A., Cui, T., Rivière, J., et al. (2017). Gallein, a Gβγ subunit signalling inhibitor, inhibits metastatic spread of tumour cells expressing OR51E2 and exposed to its odorant ligand. BMC Res. Notes 10, 541. doi:10.1186/s13104-017-2879-z
Sharp, B. M. (2006). Multiple opioid receptors on immune cells modulate intracellular signaling. Brain Behav. Immun. 20, 9–14. doi:10.1016/j.bbi.2005.02.002
Tang, T. S., Tu, H., Chan, E. Y. W., Maximov, A., Wang, Z., Wellington, C. L., et al. (2003). Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239. doi:10.1016/s0896-6273(03)00366-0
Terwilliger, T., and Abdul-Hay, M. (2017). Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 7, e577. doi:10.1038/bcj.2017.53
Virdee, K., Parone, P. A., and Tolkovsky, A. M. (2000). Phosphorylation of the pro-apoptotic protein BAD on serine 155, a novel site, contributes to cell survival. Curr. Biol. 10, R883. doi:10.1016/s0960-9822(00)00843-5
Westphal, D., Dewson, G., Czabotar, P. E., and Kluck, R. M. (2011). Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta 1813, 521–531. doi:10.1016/j.bbamcr.2010.12.019
Wu, E. H., and Wong, Y. H. (2005). Pertussis toxin-sensitive Gi/o proteins are involved in nerve growth factor-induced pro-survival Akt signaling cascade in PC12 cells. Cell Signal 17, 881–890. doi:10.1016/j.cellsig.2004.11.008
Yoon, S. H., Lo, T. M., Loh, H. H., and Thayer, S. A. (1999). Delta-opioid-induced liberation of Gbetagamma mobilizes Ca2+ stores in NG108-15 cells. Mol. Pharmacol. 56, 902–908. doi:10.1124/mol.56.5.902
Yue, C., Dodge, K. L., Weber, G., and Sanborn, B. M. (1998). Phosphorylation of serine 1105 by protein kinase A inhibits phospholipase Cbeta3 stimulation by Galphaq. J. Biol. Chem. 273, 18023–18027. doi:10.1074/jbc.273.29.18023
Zeng, W., Mak, D. O. D., Li, Q., Shin, D. M., Foskett, J. K., and Muallem, S. (2003). A new mode of Ca2+ signaling by G protein-coupled receptors: gating of IP3 receptor Ca2+ release channels by Gbetagamma. Curr. Biol. 13, 872–876. doi:10.1016/s0960-9822(03)00330-0
Zhang, J., Gu, Y., and Chen, B. (2019). Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. 12, 1937–1945. doi:10.2147/OTT.S191621
Keywords: signaling, calcium, apoptosis, leukemia, cancers
Citation: Kamran H, Lee JK and Lee K-Y (2024) PKA inhibition is a central step in D,L-methadone-induced ER Ca2+ release and subsequent apoptosis in acute lymphoblastic leukemia. Front. Cell Dev. Biol. 12:1388745. doi: 10.3389/fcell.2024.1388745
Received: 20 February 2024; Accepted: 03 April 2024;
Published: 24 April 2024.
Edited by:
Elaine Maria Souza-Fagundes, Federal University of Minas Gerais, BrazilReviewed by:
Felipe Henrique de Souza Silva, Federal University of Minas Gerais, BrazilMauro Cunha Xavier Pinto, Universidade Federal de Goiás, Brazil
Copyright © 2024 Kamran, Lee and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ki-Young Lee, kyleecal1957@gmail.com
†These authors have contributed equally to this work