 Sobia A. Halim
Sobia A. Halim Shanza Khan
Shanza Khan Ajmal Khan
Ajmal Khan Abdul Wadood4
Abdul Wadood4- 1Department of Biochemistry, Kinnaird College for Women, Lahore, Pakistan
- 2Department of Chemistry, COMSATS Institute of Information Technology, Abbottabad, Pakistan
- 3UoN Chair of Oman Medicinal Plants and Marine Products, University of Nizwa, Nizwa, Oman
- 4Department of Biochemistry, Shankar Campus, Abdul Wali Khan University Mardan, Mardan, Pakistan
- 5Department of Biological Sciences and Chemistry, College of Arts and Sciences, University of Nizwa, Nizwa, Oman
Dengue fever is an emerging public health concern, with several million viral infections occur annually, for which no effective therapy currently exist. Non-structural protein 3 (NS-3) Helicase encoded by the dengue virus (DENV) is considered as a potential drug target to design new and effective drugs against dengue. Helicase is involved in unwinding of dengue RNA. This study was conducted to design new NS-3 Helicase inhibitor by in silico ligand- and structure based approaches. Initially ligand-based pharmacophore model was generated that was used to screen a set of 1201474 compounds collected from ZINC Database. The compounds matched with the pharmacophore model were docked into the active site of NS-3 helicase. Based on docking scores and binding interactions, 25 compounds are suggested to be potential inhibitors of NS3 Helicase. The pharmacokinetic properties of these hits were predicted. The selected hits revealed acceptable ADMET properties. This study identified potential inhibitors of NS-3 Helicase in silico, and can be helpful in the treatment of Dengue.
Introduction
Dengue is one of the most common infections in tropical and subtropical countries, and one of the major diseases in Pakistan since 2005. Over past few years around 48,910 cases appeared with 566 death cases. The first deadly outbreak was reported in Lahore in 2011, where 21,685 cases with 350 deaths were recorded (Lindenbach and Rice, 2003; Mukhtar et al., 2011; Ali et al., 2013; Khan et al., 2013, 2014, 2015, 2016; Rasheed et al., 2013; Raza et al., 2014), while about 50–100 million cases appeared worldwide. Dengue is caused by dengue virus (DENV) which belongs to the Flavivirus genus of the Flaviviridae family (Lindenbach and Rice, 2003). The infected dengue cases show several clinical symptoms including high fever, headache, muscular pain, and nausea/vomiting which can lead to serious conditions such as dengue shock syndrome (DSS) or dengue hemorrhagic fever (DHF). DSS/DHF can eventually cause death within 24 h. Till date no effective drugs are available to cure the disease completely (Kuhn et al., 2002; van Gorp et al., 2002; Seneviratne et al., 2006; Bhatt et al., 2013). Therefore, new and effective inhibitors are needed to be designed as potential therapeutics to cure the disease.
DENV consists of positive stranded RNA composed of a 5′ untranslated region (UTR), a large open reading frame, and a 3′ UTR (Lindenbach and Rice, 2003). The viral genome is translated into three structural proteins: C (Capsid), prM (Membrane precursor), E (Envelop) and seven non-structural proteins (NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5) (Kuhn et al., 2002).
Viral particles appear as smooth surfaces of DENV-E dimers arranged in a head-to-tail fashion parallel to the viral membrane (Modis et al., 2003). The crystal structure of E revealed each monomer consists of three domains that plays different roles in the virus life cycle (Gubler and Clark, 1995; Modis et al., 2005). E is involved in receptor binding, induction of antibody responses, viral fusion and assembly (Gubler and Clark, 1995). The other viral proteins help in RNA packaging and assembly. Capsid protein encapsidates viral RNA and interacts with the genetic material. Membrane glycoprotein is the mature form of pre-membrane (prM) which acts as a chaperone for E. During viral egress through the trans-Golgi network, host furin protein cleaves prM to M. Following cleavage, viral particles are considered mature (Lindenbach and Rice, 2003). The seven DENV non-structural proteins are essential in the virus life cycle. NS1 play role in signaling and replication of virus RNA. NS2A is essential for viral replication and packaging. NS2B is serine protease which acts as a co-factor for NS3. NS3 is a multifunctional protein that posses serine protease, helicase (DENV NS3H), RNA-stimulated nucleoside tri-phosphatase (NTPase/ATPase/helicase), and RNA 5′-triphosphatase (RTPase) activities which are essential for viral RNA replication and capping (Kadaré and Haenni, 1997; Singleton and Wigley, 2002; Wang et al., 2009). These proteins have 67% similarity in four strains of DENV (DENV1-4). These enzymes are important in replication and translation process. NS4A and NS4B are transmembrane proteins responsible for the membrane arrangements leading to the formation of the viral replication complex (Nemésio et al., 2012). Along with NS2A, these proteins have been implicated in interferon antagonism (Muñoz-Jordán et al., 2005). NS5 protein is a RNA-dependent RNA polymerase (Lindenbach and Rice, 2003).
Currently DENV enzymes are targeted to design anti-viral therapies. In this study NS3 helicase was targeted. The helicase domain resides within the region of 170–618 residues of NS3 protein. NS3 helicase unwinds double stranded RNA to release single stranded RNA, which is then used as a template for NS5 protein in replication (Bartelma and Padmanabhan, 2002). Studies showed that the ATPase/helicase and RTPase activities of DENV NS3 share a common active site (Borowski et al., 2001; Benarroch et al., 2004). However, later on, X-ray crystallographic studies proved that ATPase and helicase activities are conferred by distinct sites (Luo et al., 2008). Functional activities of helicase are well-characterized for other species of the flaviviridae family such as Hepatitis C Virus (HCV), and Yellow fever virus (YFV) etc. (Warrener et al., 1993; Utama et al., 2000). DENV has four antigenically distinct serotypes, DENV 1–4. DENV-2 was prominent in outbreaks in 2011. Phylogenetic analysis of partial DENV-2 sequences has revealed that genotype IV or cosmopolitan genotype of DENV-2 is circulating in Pakistan (Fatima et al., 2011). NS3 and NS5 are conserved within the four serotypes (Li et al., 2005), that permit the design of drugs which could be effective against all dengue virus serotypes and other related flaviviruses (Xu et al., 2005; Keller et al., 2006). The structural details of DENV helicase in complex with ssRNA (Luo et al., 2008) and in apo form (Xu et al., 2005) are available at good resolution which opens the opportunities to design novel drugs against dengue.
Computational tools have a large impact in drug discovery because of its fast and promising results. In silico techniques are categorized as structure- and ligand based. Both structure and ligand based methods are used to predict binding affinities of newly designed compounds. With our interest in computational analysis of several biologically important drug targets (Halim et al., 2013, 2015; Halim and Zaheer-ul-Haq, 2015), we conducted this study to identify novel and effective DENV NS3-helicase inhibitors in silico. The compound which shows potential will be selected for biological testing in future to accelerate the therapeutic process against dengue.
Materials and Methods
All the computation experiments were performed on Windows 8 workstation. LigandScout version 3.12 (Wolber and Langer, 2005) was used for pharmacophore modeling. The X-ray crystallography structure of DENV-2 NS-3 helicase [PDB ID: 2BMF (Xu et al., 2005)] was retrieved from RCSB Protein Data Bank (https://www.rcsb.org) (Berman et al., 2000) for molecular docking. Docking was performed on FRED (Fast Rigid Exhaustive Docking) (McGann, 2012). Protein-ligand interactions were visualized on Chimera software (Pettersen et al., 2004). ADMET properties were calculated on admetSAR server (Cheng et al., 2012). The flow of work is shown in Scheme 1.
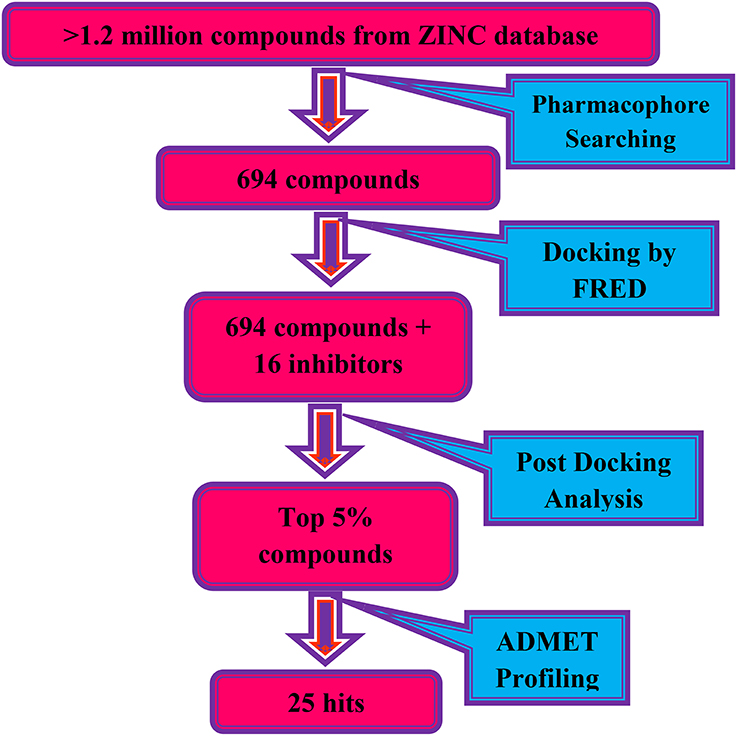
Scheme 1. The virtual screening work flow.
Pharmacophore Modeling and Screening of ZINC Database
The pharmacophore model was generated by LigandScout (Wolber and Langer, 2005) using three most active inhibitors of DENV NS-3 Helicase (Table 1). The known inhibitors were selected from literature (Mastrangelo et al., 2012; Basavannacharya and Vasudevan, 2014; Sweeney et al., 2015). The compounds structures and IC50 values are shown in Table 1. Lig and Scout generates pharmacophore model by using structural data from protein-ligand complex structures or from small compounds. Subsequently protein-ligand interactions are depicted by chemical features including H-bond donors, H-bond acceptors, lipophilic areas, positively and negatively ionizable chemical groups. A pattern-matching based alignment method is used to align the generated pharmacophores. The aligned pharmacophore from different complexes are used to either create “shared feature pharmacophore (SFP)” or “merged feature pharmacophore (MFP).” SFP shares common interactions of several complexes, while MSP comprises of extended pharmacophore. We used SFP method. 1201474 compounds from “Drug Now” category of ZINC database (Irwin et al., 2012) was selected as a screening library. The pharmacophore was validated by adding the known inhibitors of DENV NS-3 Helicase in the screening database. The compounds that matched with pharmacophore model were docked into the protein by FRED docking program.
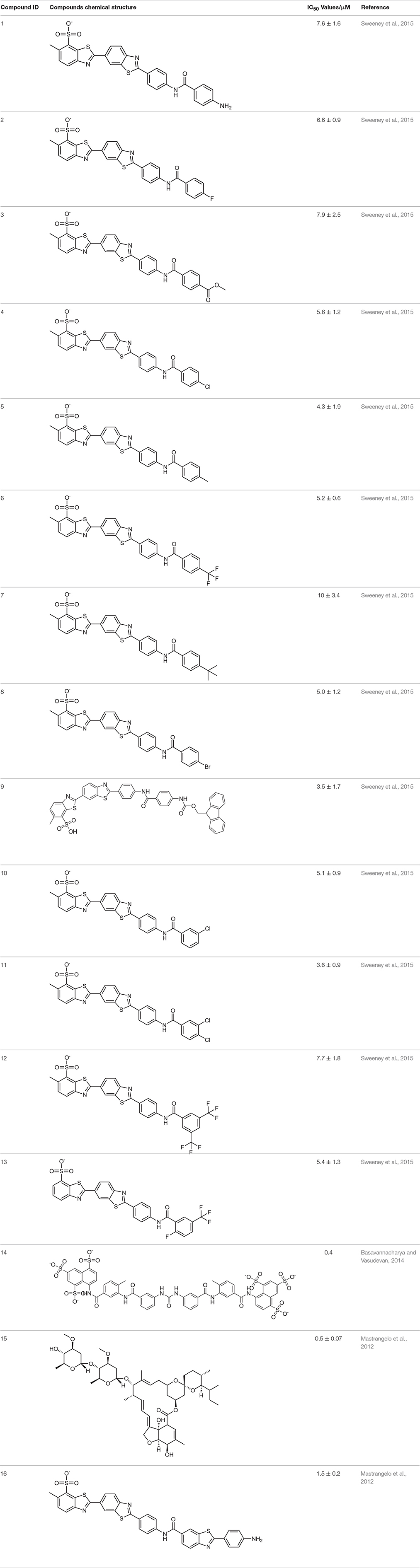
Table 1. The Chemical structures and IC50 values of 16 known inhibitors of NS-3 Helicase.
Fred Docking Protocol
FRED performs rigid docking using exhaustive search algorithm (McGann, 2012) that requires pre-made multiconformer library of ligand. Exhaustive search algorithm rotates and translates each conformation of compound in the protein's binding site to select the best pose that do not clash with the active site or extend far away. Subsequently the best poses are assigned a score. Initially ligand conformations were generated by Omega 2.4.6 (Hawkins et al., 2010). The maximum number of conformations was set as 10 for each ligand along with a dielectric constant of 1.0 and the search force field mmff94s_NoEstat. The 3D structure of DENV NS-3 Helicase (PDB code: 2BMF, resolution: 2.4Å) was retrieved from PDB. The protein file was prepared on FRED make receptor 2.2.5 software. Missing atoms and bonds of protein was checked. Docking box was constructed on the single stranded RNA binding site with a volume of 18869Å3. Inner and outer counter was set as 12Å3 and 6673Å3, respectively. Eight scoring functions (McGann, 2012): Shapegauss (SG), Piecewise Linear Potential (PLP), ChemScore, Chemgauss 2 (CG2), and Chemgauss 3 (CG3), OeChemSscore, ScreenSscore and Zapbind were used in docking. The receptor file was docked with multiple conformer libraries of ligands and top fifty poses of each ligand were saved for further analysis.
Admet Prediction
ADMET properties predict the absorption, distribution, metabolism, excretion, and toxicity of compounds in and through the human body. It estimates pharmacokinetic and pharmacodynamic profiles of drugs, and plays crucial role in drug development. The ADMET properties of the selected ligands were estimated by online server admetSAR (Cheng et al., 2012). admetSAR collects data of diverse compounds associated with ADMET properties from literature, and provides ADMET structure-activity relationship models to predict ADMET properties of drug candidates.
Results
Pharmacophore Modeling and Virtual Screening
ZINC provides chemical molecules repositories that contain millions of diverse compounds. >1.2 million compounds were retrieved from the “Drug Now” category of ZINC database. To reduce the size of the dataset for docking, ligand-based pharmacophore model was constructed via LigandScout based on 3D structure of three most active known inhibitors (Compound ID: 10, 14, and 15). The pharmacophore model was composed of 5 Hydrogen Bond Acceptors (Red spheres), 4 Hydrogen Bond Donors (Green spheres) and 2 hydrophobic features (Yellow spheres) (Figure 1). 694 compounds were matched with the pharmacophore query and 16 known inhibitors were subjected to molecular docking.
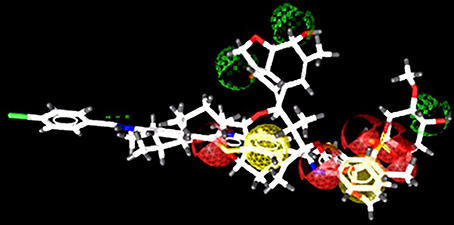
Figure 1. Pharmacophore model consist of 5 H-Bond acceptors (Red spheres), 4 H-Bond donors (Green spheres), and 2 hydrophobic features (Yellow spheres). The known inhibitors are depicted in stick models.
Molecular Docking
The compounds retrieved by pharmacophore based screening were docked into the NS-3 Helicase active site by FRED. After docking, the results of the eight scoring functions were compared. Those scoring functions were selected that ranked all 16 known inhibitors at the top of its ranking list. This retrospective analysis shows that Chemgauss2 (CG2) and Shapegauss (SG) placed all the known inhibitors at the top of their docking results (Table 2). Subsequently consensus strategy was used for the selection of best predicted hits. Based on CG2 and SG ranking, top 5% compounds were selected as hits. The binding interactions analysis of the selected hits showed that 25 compounds acts as potential NS-3 inhibitors. The chemical structures and ZINC codes of selected 25 hits are shown in Table 3, while docking results are tabulated in Table 4.
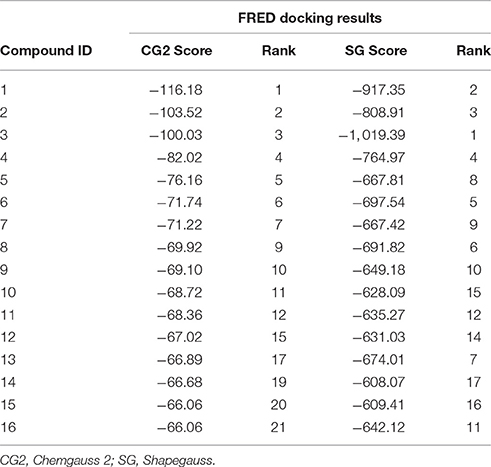
Table 2. FRED docking results of Known inhibitors (scores and rank).

Table 3. The chemical structures of 25 hits selected after post docking analysis.
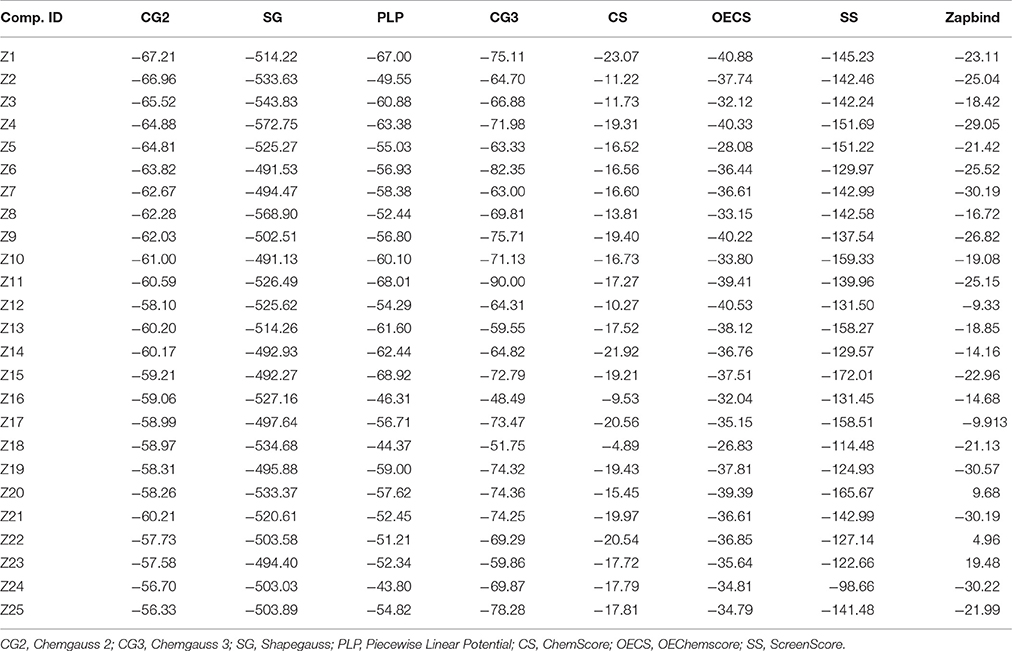
Table 4. FRED Docking Results of the 25 best selected Compounds.
Binding Interaction Analysis
The docked view of the selected compounds are depicted in Figure 2. The binding mode of the compound Z1 showed that the compound forms H-bond with Lys388 and Arg599. The calculated H-bond distance between pyrimidine nitrogen of Z1 and side chain of Lys388 is 3.0Å, while pyrimidine moiety and side chain of Arg599 is 2.3Å. Furthermore, the side chains of surrounding residues stabilize the compound in the active site of NS3. The carbonyl oxygen of compound Z2 formed H-bond with the amino side chain of Arg387 at a distance of 2.3Å. The carbonyl and amino group of compound Z3 mediates H-bond with the amino side chain of Arg599 (2.9Å) and side chain of Arg387 (1.9Å), respectively. The docked view of the compound Z4 shows that this compound is mainly stabilized in the binding site via hydrophobic interactions, while the carbonyl oxygen mediates weak H-bonding with amino side chain of Lys366 (3.1Å). The bromo-pyrimidine nitrogen of compound Z5 is H-bonded to amino side chain of Lys388 (3.0Å). The amino group also formed H-bond with the carbonyl group of Arg599 (2.2Å). The compound Z6 mediates H-bond with Lys388. The pyrrolidine-dione ring accepts H-bonds from amino side chains of Lys388 (2.9Å). The compound Z7 interacts with Arg599. The phenyl nitrogen formed weak H-bond with the amino side chain of Arg599 (3.1Å). The dimethyl-morpholine moiety of compound Z8 found oriented toward Lys388 and the oxygen formed H-bond with its amino side chain at a distance of 3.0Å. The other polar groups of the compound do not interact with surrounding residues as the polar moieties of residues are oriented away from the compound. However, Arg387 provides strong hydrophobic interactions to the methoxy benzene moiety of compound Z8. The oxygen at the cyclopentyl ring of compound Z9 interacted with the amino side chain of Lys388 at a distance of 3.1Å. The methoxyphenyl moiety is oriented toward Arg599 showing strong hydrophobic interactions. The tetrahydrofuran ring of compound Z10 formed bi-dentate interactions with Arg387. The ring is H-bonded to the amino side chain of Arg387 at a distance of 2.7Å and 3.1Å. The compound Z11 is composed of five rings in which the pyrrolidine ring mediates H-bonding with amino side chain of Arg387 at a distance of 2.9Å. The surrounding residues Arg538, Arg599, Lys399, and Arg387 hydrophobically stabilize the compound. The triazole moiety of compound Z12 mediates weak H-bond with the carbonyl side chain of Arg599 at a distance of 3.1Å. The quinolone oxygen of compound Z13 accepts H-bond from the amino group of Lys366. The H-bond distance is 2.8Å. The compound Z14 is H-bonded to the amino side chain of Lys388 2.6Å and the amino group is H-bonded to Arg599 (1.9Å). Similarly compound Z15 is stabilized by two H-bonds with Arg599 and Lys366. The oxadiazole ring accepts H-bond from the amino side chain of Lys366 (2.9Å). The amino group attached with the fluoro-benzene ring donates its H-bond to the carbonyl group of Arg599 (2.6Å). The pyrrolidine moiety of compound Z16 mediates H-bond with the side chain of Lys388 (2.9Å). However, the benzene ring is tilted toward Arg387. The amino nitrogen of compound Z17 formed H-bond with the carbonyl group of Arg387 with the bond length of 1.8Å. The carbonyl moiety of compound Z18 accepts H-bond from the amino side chain of Lys388 (2.7Å). The dimethyl substituted benzene ring found oriented toward Arg387. The compound Z19 mainly interacts with Arg538 and Arg599. The dihydrobenzene substituted amino groups mediates bidentate interactions with carbonyl group of Arg599 with bond length of 2.1Å and 2.0Å. Moreover, compound also interacts with Lys388 via hydrophobic interactions. The quinoxalin moiety of compound Z20 accepts H-bond from the amino side chain of Arg387 (3.0Å). The quinazolin moiety of compound Z21 mediated H-bond with the amino side chain of Lys388 (3.0Å). The cyclopentane ring of the Z21 is tilted toward Arg387 thus stabilizing the compound. The pyrimidine moiety of compound Z22 accepts H-bond from Lys388 and Arg599 at a distance of 2.3Å and 2.7Å, respectively. The fluoro benzene ring is oriented toward Arg387. Compound Z23 is composed of four rings in which the oxo-tetrahydrofuran ring is tilted toward Arg387. Lys388 and Arg599 provide strong hydrophobic interactions to stabilize the compound. The pyrimidine nitrogen of compound Z24 is H-bonded to amino side chain of Lys388 (2.7Å). The phenyl ring formed strong hydrophobic interaction with Arg599. The pyrimidine nitrogen of compound Z25 mediates H-bonding with amino side chain of Lys388 (3.0Å). The binding modes of the selected compounds showed that strong hydrophobic interactions are provided by the surrounding active site residues of the DENV NS-3 Helicase to stabilize these compounds in the active site.

Figure 2. The DENV NS-3 Helicase structure is depicted in Golden ribbon. The residues of active site are depicted as purple sticks. The predicted inhibitors are displayed as blue stick model.
Admet Prediction
ADMET properties were predicted by online admetSAR server. The results are presented in Table 5. The results revealed that all compounds possess good HIA (Human Intestinal Absorption) values. Greater HIA denotes that the compound could be better absorbed from the intestinal tract upon oral administration. Out of 25 compounds, only four compounds showed low HIA. Moreover, all the compounds displayed negative penetration through the Blood-Brain Barrier (BBB), means these compounds do not cross BBB. In terms of metabolism, we found that some compounds inhibit the members of the cytochrome P450 superfamily of enzymes and some are non-inhibitors. A non-inhibitor of CYP450 means that the molecule will not restrict the biotransformation of drugs metabolized by CYP450 enzyme. AMES toxicity test is employed to predict whether a compound is mutagenic or not. All of the compounds are non-mutagenic. Carcinogenic profile also revealed that all the ligands were non-carcinogenic. Acute oral toxicity test showed that the predicted LD50 values of these compounds are >500 mg/kg but less that 5,000 mg/kg, suggesting that these compounds do not posses acute oral toxicity at lower doses. Important information obtained from admetSAR server was the computed LD50 dose in rat model. Comparing the LD50 doses, a compound with lower dose is more lethal than the compound having higher LD50. From our observation, it was seen that all compounds possess higher LD50 values. The physicochemical properties (Table 5) showed that these compounds acquire drug like properties and can be good inhibitors of DENV NS3 protein when tested in vitro.
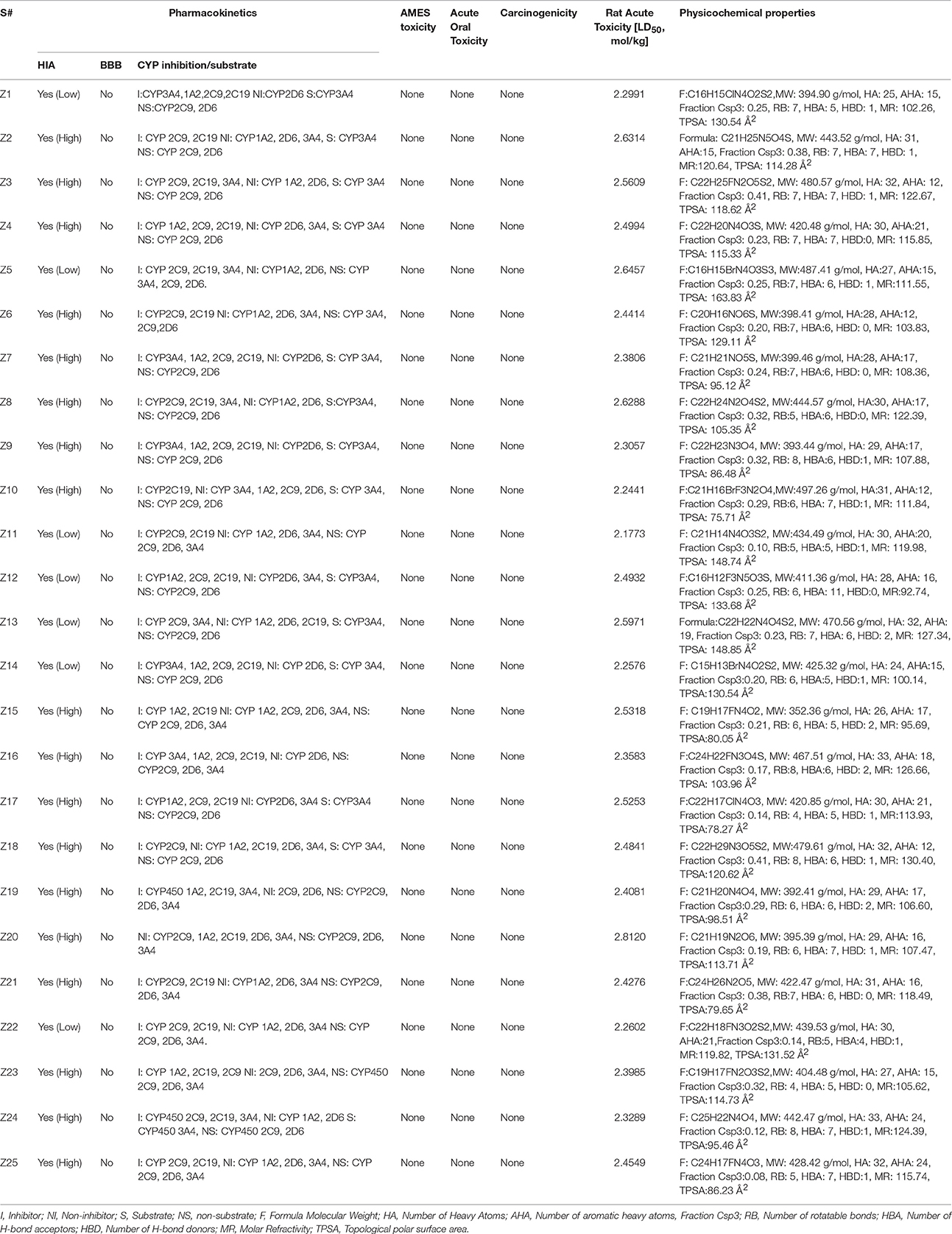
Table 5. Pharmacokinetic and Physiological Properties of 25 selected hits.
Discussion
Computational methods are extensively used in medicinal and pharmaceutical chemistry researches to foster drug discovery process. In silico drug design has given several novel molecules that are in clinical trials. Hence considering the importance of computational drug discovery methods, this study was conducted to discover potential Dengue Virus non-structural protein (DENV NS-3) Helicase inhibitors. DENV NS-3 Helicase is an important drug target to design novel antiviral compounds for the treatment of Dengue.
Successful docking studies have been performed in the recent years (Zaheer-ul-Haq et al., 2010; Halim et al., 2015). Recently several novel immunomodulators were designed using in silico approaches when screened against Interleukin-2 (Halim et al., 2013). Knowing the importance of computational drug designing methods, in this research, ligand-based pharmacophore modeling was carried out. The pharmacophore model was validated by screening known inhibitors embedded in compounds library collected from ZINC dataset. Furthermore, 1201474 compounds selected from drug-like category of ZINC database was screened by pharmacophore model which led to identify 694 molecules that were subjected to docking by FRED docking suit. The compounds were scored by eight scoring functions (Shapegauss (SG), Piecewise Linear Potential (PLP), ChemScore, Chemgauss 2 (CG2), and Chemgauss 3 (CG3), OeChemScore, ScreenSscore, and Zapbind) to evaluate the performance of selected scoring functions. Shapegauss is a shape based scoring function that select the best pose based on its shape complementarity with active site, but lack estimation of protein-ligand interactions. PLP estimates shape and protein-ligand interactions specifically hydrogen bonding. Chemscore estimates lipophilic interactions, H-bonding, metal/ligand interactions as well as any rotatable bonds in a pose and clash between protein-ligand. CG2 and CG3 are Gaussian scoring functions that calculate shape complementarity, however CG3 also calculate H-bonding between protein and ligand, between ligand and solvent and metallic interactions. OEChemscore is a variant of chemscore, but it is unable to calculate entropy penalty upon complex formation. Screenscore is a hybrid of PLP and FlexX scoring functions; it calculates interactions between polar and non-polar atoms. Zapbind is the most computationally expensive scoring function among all the ones integrated in FRED. It sum up surface area contact term (calculated by Gaussian-based method) and an electrostatic interaction term calculated using the Poisson–Boltzmann (PB) solvent approximation which is calculated by ZAP. Prior to virtual screening experiments the comparison of available scoring functions must be conducted to ensure the suitable scoring function for the target of interest (Zaheer-ul-Haq et al., 2010). For this purpose, retrospective docking analysis gives good idea of which scoring function is best for the protein under observation. Among eight scoring functions, CG2 and SG showed excellent results and predicted all the 16 actives as top ranked inhibitors. Hence the CG2 and SG top ranked ZINC compounds were predicted as efficient inhibitors of DENV NS-3 Helicase. Interaction analysis revealed that 25 compounds significantly showed good interaction with the NS3 Helicase active site. The compounds showed strong hydrogen bonding interactions with the target protein. A model of nucleic acid binding site of NS3 helicase was generated by (Xu et al., 2005). The model revealed interaction between the NS3 nucleotide binding-site with a deoxyuridylate octamer oligonucleotide (single stranded RNA). The oligonucleotide interacted with residues from motifs Ia, IV, and V, and residues Arg-225 (motif Ia), Lys-366 (motif IV), Arg-387, Lys-388, and Arg-538 and Arg-599 from domain III interacted with the phosphodiester backbone. These residues (Arg-225, Lys-366, Arg-387, Lys-388, Arg-538, and Arg-599) binds with the single stranded RNA. However, single stranded RNA binding site was elucidated by Luo et al. (2008). The ssRNA binds with domain I, II and III. The residues Pro223, Arg225, Asp290, Gln243, Thr244, Cys261, Thr264, Thr267 from domain I, Pro363, Ile365, Lys366, Arg387, Thr408, Asp409, Leu429 from domain II, Arg538, and Arg599 and Asp603 from domain III makes a tunnel to accommodate ssRNA. The crystallographic structures of DENV NS-3 Helicase in complex with ssRNA (PDB ID: 2JLU), with ADP (PDB ID: 2JLS), with ligand ANP (phosphoaminophosphonic acid adenylate ester, PDB ID: 2JLR) and with ANP and ssRNA (PDB ID: 2JLV) showed that NTPase/ATPase and helicase active site are distinct (Luo et al., 2008). Our docking results showed that most of the compounds interacted with Arg599, Lys388, and Arg387. Hence the compounds are accommodated in a groove between domain II and III. The interactions are depicted in Figure 3.
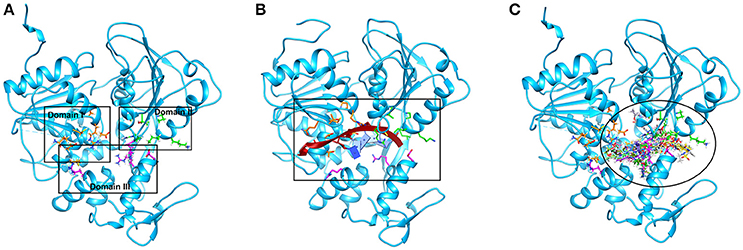
Figure 3. The single stranded RNA binding site is shown (A,B). The residues of domain I, II, and III are shown in orange, green, and magenta color respectively. The docked view of 25 selected hits shows the compounds are fitted between domain II and III (C).
Computational tools also aid in prediction of ADMET properties of compounds. ADMET prediction is essential to remove compounds that show toxicity in biological system. Thus, prior to in vivo trial ADMET properties of selected compounds must be calculated to remove any toxic compound. These assessments further increase the efficacy of drug candidate and reduce the chance of its failure in in vivo trials. Thus, ADMET profiling was conducted via admetSAR and all the compounds revealed good pharmacokinetic properties. These results suggest that these compounds can be considered as potent inhibitors of DENV NS3 Helicase by hindering its active site.
Conclusion
Treatment of Dengue is one of the main public concerns nowadays therefore novel inhibitors need to be urgently designed to cure this disease. DENV NS-3 Helicase is a potential drug target. We employed computational modeling techniques including ligand based pharmacophore modeling and structure based virtual screening to identify novel and potential DENV NS-3 Helicase inhibitors from ZINC database. The in silico results demonstrated 25 hits compatible with active site of NS-3 Helicase and are predicted to block its activity in silico. The current computational results will be validated in wet lab by both in vitro and in vivo testing.
Author Contributions
SH outline the research strategy and idea. SK carried out the literature search, and performed computational experiments. SH drafted and revised the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, TM, and handling Editor declared their shared affiliation.
Acknowledgments
The authors are thankful to Inte:Ligand GmbH and OpenEye Scientific Inc. for providing us free academic license of LigandScout and FRED softwares, respectively for this study.
References
Ali, A., Nasim, Z., Rehman, R. U., Farzana, A., and Zahir, F. (2013). Dengue virus serotype 2 and 3 causing high morbidity and mortality in Swat, Pakistan. Biohelikon. Immun. Dis. 1:a6. doi: 10.1186/s12985-016-0603-6
Bartelma, G., and Padmanabhan, R. (2002). Expression, purification, and characterization of the RNA 5′-triphosphatase activity of dengue virus type 2 nonstructural protein 3. Virology 299, 122–132. doi: 10.1006/viro.2002.1504
Basavannacharya, C., and Vasudevan, S. G. (2014). Suramin inhibits helicase activity of NS3 protein of dengue virus in a fluorescence-based high throughput assay format. Biochem. Biophys. Res. Comm. 453, 539–544. doi: 10.1016/j.bbrc.2014.09.113
Benarroch, D., Selisko, B., Locatelli, G. A., Maga, G., Romette, J. L., and Canard, B. (2004). The RNA helicase, nucleotide 5′-triphosphatase, and RNA 5′-triphosphatase activities of Dengue virus protein NS3 are Mg2+-dependent and require a functional Walker B motif in the helicase catalytic core. Virology 328, 208–218. doi: 10.1016/j.virol.2004.07.004
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nuc. Acids Res. 28, 235–242. doi: 10.1093/nar/28.1.235
Bhatt, S., Gething, P. W., Brady, O. J., Messina, J. P., Farlow, A. W., Moyes, C. L., et al. (2013). The global distribution and burden of dengue. Nature 496, 504–507. doi: 10.1038/nature12060
Borowski, P., Niebuhr, A., Mueller, O., Bretner, M., Felczak, K., Kulikowski, T., et al. (2001). Purification and characterization of West Nile virus nucleoside triphosphatase (NTPase)/helicase: evidence for dissociation of the NTPase and helicase activities of the enzyme. J. Virol. 75, 3220–3229. doi: 10.1128/JVI.75.7.3220-3229.2001
Cheng, F., Li, W., Zhou, Y., Shen, J., Wu, Z., Liu, G., et al. (2012). Admetsar: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Infor. Model. 52, 3099–3105. doi: 10.1021/ci300367a
Fatima, Z., Idrees, M., Bajwa, M. A., Tahir, Z., Ullah, O., Zia, M. Q., et al. (2011). Serotype and genotype analysis of dengue virus by sequencing followed by phylogenetic analysis using samples from three mini outbreaks-2007-2009 in Pakistan. BMC Microbiol. 11:200. doi: 10.1186/1471-2180-11-200
Gubler, D. J., and Clark, G. G. (1995). Dengue/dengue hemorrhagic fever: the emergence of a global health problem. Emer. Inf. Dis. 1, 55–57. doi: 10.3201/eid0102.952004
Halim, S. A., Abdalla, O. M., Mesaik, M. A., Wadood, A., ul-Haq, Z., and Kontoyianni, M. (2013). Identification of novel Interleukin-2 inhibitors through computational approaches. Mol. Div. 17, 345–355. doi: 10.1007/s11030-013-9431-4
Halim, S. A., and Zaheer-ul-Haq (2015). Structure based 3D-QSAR studies of Interleukin-2 inhibitors: comparing the quality and predictivity of 3D-QSAR models obtained from different alignment methods and charge calculations. Chem. Biol. Int. 238, 9–24. doi: 10.1016/j.cbi.2015.05.018
Halim, S. A., Jawad, M., Ilyas, M., Mir, Z., Mirza, A. A., and Husnain, T. (2015). In silico identification of novel IL-1β inhibitors to target protein–protein interfaces. Comp. Biol. Chem. 58, 158–166. doi: 10.1016/j.compbiolchem.2015.06.004
Hawkins, P. C., Skillman, A. G., Warren, G. L., Ellingson, B. A., and Stahl, M. T. (2010). Conformer generation with OMEGA: algorithm and validation using high quality structures from the protein databank and Cambridge structural database. J. Chem. Inf. Model. 50, 572–584. doi: 10.1021/ci100031x
Irwin, J. J., Sterling, T., Mysinger, M. M., Bolstad, E. S., and Coleman, R. G. (2012). ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768. doi: 10.1021/ci3001277
Keller, T. H., Chen, Y. L., Knox, J. E., Lim, S. P., Ma, N. L., Patel, S. J., et al. (2006). Finding New Medicines for Flaviviral Targets. Chichester; New York, NY: John Wiley.
Khan, A., Mehr, M. T., and Shaukat, A. A. (2014). Presentation and prevention of dengue fever. J. Med. Sci. 22, 92–95.
Khan, J., Khan, I., and Amin, I. (2016). A comprehensive entomological, serological and molecular study of 2013 dengue outbreak of Swat, Khyber Pakhtunkhwa, Pakistan. PLoS ONE. 11:e0147416. doi: 10.1371/journal.pone.0147416
Khan, J., Munir, W., Khan, B. T., Ahmed, Z., Shams, W. A., and Khan, A. (2015). Dengue outbreak 2013: Clinical profile of patients presenting at DHQ Bunner and THQ Shangla, Khyber Pakhtunkhwa, Pakistan. Biohelikon. Immun. Dis. 3:a11. doi: 10.5281/zenodo.816342
Khan, M. A., Ellis, E. M., Tissera, H. A., Alvi, M. Y., Rahman, F. F., Masud, F., et al. (2013). Emergence and diversification of dengue 2 cosmopolitan genotype in Pakistan. (2011). PLoS ONE 8:e56391. doi: 10.1371/journal.pone.0056391
Kuhn, R. J., Zhang, W., Rossmann, M. G., Pletnev, S. V., Corver, J., Lenches, E., et al. (2002). Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108, 717–725. doi: 10.1016/S0092-8674(02)00660-8
Li, J., Lim, S. P., Beer, D., Patel, V., Wen, D., Tumanut, C., et al. (2005). Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J. Biol. Chem. 280, 28766–28774. doi: 10.1074/jbc.M500588200
Lindenbach, B. D., and Rice, C. M. (2003). Molecular biology of flaviviruses. Adv. Virus Res. 59, 23–61. doi: 10.1016/S0065-3527(03)59002-9
Luo, D., Xu, T., Watson, R. P., Scherer-Becker, D., Sampath, A., Jahnke, W., et al. (2008). Insights into RNA unwinding and ATP hydrolysis by the flavivirus NS3 protein. EMBO J. 27, 3209–3219. doi: 10.1038/emboj.2008.232
Mastrangelo, E., Pezzullo, M., De Burghgraeve, T., Kaptein, S., Pastorino, B., Dallmeier, K., et al. (2012). Ivermectin is a potent inhibitor of flavivirus replication specifically targeting NS3 helicase activity: new prospects for an old drug. J. Antimicrob. Chemother. 67, 1884–1894. doi: 10.1093/jac/dks147
McGann, M. (2012). FRED and HYBRID docking performance on standardized datasets. J. Comp. Aid. Mol. Des. 26, 897–906. doi: 10.1007/s10822-012-9584-8
Modis, Y., Ogata, S., Clements, D., and Harrison, S. C. (2003). A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 100, 6986–6991. doi: 10.1073/pnas.0832193100
Modis, Y., Ogata, S., Clements, D., and Harrison, S. C. (2005). Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J. Virol. 79, 1223–1231. doi: 10.1128/JVI.79.2.1223-1231.2005
Mukhtar, M., Tahir, Z., Baloch, T. M., Mansoor, F., and Kamran, J. (2011). Entomological investigations of dengue vectors in epidemic-prone districts of Pakistan during 2006–2010. Dengue Bull. 35, 99–115.
Muñoz-Jordán, J. L., Laurent-Rolle, M., Ashour, J., Martínez-Sobrido, L., Ashok, M., Lipkin, W. I., et al. (2005). Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 79, 8004–8013. doi: 10.1128/JVI.79.13.8004-8013.2005
Nemésio, H., Palomares-Jerez, F., and Villalaín, J. (2012). NS4A and NS4B proteins from dengue virus: membranotropic regions. Biochim. Biophys. Biomemb 1818, 2818–2830. doi: 10.1016/j.bbamem.2012.06.022
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. et al. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Rasheed, S., Butlin, R., and Boots, M. (2013). A review of dengue as an emerging disease in Pakistan. Pub. Heal. 127, 11–17. doi: 10.1016/j.puhe.2012.09.006
Raza, F. A., Rehman, S. U., Khalid, R., Ahmad, J., Ashraf, S., Iqbal, M., et al. (2014). Demographic and clinico-epidemiological features of dengue fever in Faisalabad, Pakistan. PLoS ONE 9:e89868. doi: 10.1371/journal.pone.0089868
Seneviratne, S. L., Malavige, G. N., and de Silva, H. J. (2006). Pathogenesis of liver involvement during dengue viral infections. Trans. Roy. Soc. Trop. Med. Hyg. 100, 608–614. doi: 10.1016/j.trstmh.2005.10.007
Singleton, M. R., and Wigley, D. B. (2002). Modularity and specialization in superfamily 1 and 2 helicases. J. Bacteriol. 184, 1819–1826. doi: 10.1128/JB.184.7.1819-1826.2002
Sweeney, N. L., Hanson, A. M., Mukherjee, S., Ndjomou, J., Geiss, B. J., Steel, J. J., et al. (2015). Benzothiazole and pyrrolone flavivirus inhibitors targeting the viral helicase. ACS Inf. Dis. 1, 140–148. doi: 10.1021/id5000458
Utama, A., Shimizu, H., Morikawa, S., Hasebe, F., Morita, K., Igarashi, A., et al. (2000). Identification and characterization of the RNA helicase activity of Japanese encephalitis virus NS3 protein. FEBS Lett. 465, 74–78. doi: 10.1016/S0014-5793(99)01705-6
van Gorp, E. C., Suharti, C., Mairuhu, A. T., Dolmans, W. M., van Der Ven, J., Demacker, P. N. et al. (2002). Changes in the plasma lipid profile as a potential predictor of clinical outcome in dengue hemorrhagic fever. Clin. Inf. Dis. 34, 1150–1153. doi: 10.1086/339539
Wang, C.-C., Huang, Z.-S., Chiang, P.-L., Chen, C.-T., and Wu, H.-N. (2009). Analysis of the nucleoside triphosphatase, RNA triphosphatase, and unwinding activities of the helicase domain of dengue virus NS3 protein. FEBS Lett. 583, 691–696. doi: 10.1016/j.febslet.2009.01.008
Warrener, P., Tamura, J. K., and Collett, M. S. (1993). RNA-stimulated NTPase activity associated with yellow fever virus NS3 protein expressed in bacteria. J. Virol. 67, 989–996.
Wolber, G., and Langer, T. (2005). LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inform. Model. 45, 160–169. doi: 10.1021/ci049885e
Xu, T., Sampath, A., Chao, A., Wen, D., Nanao, M., Chene, P., et al. (2005). Structure of the dengue virus helicase/nucleoside triphosphatase catalytic domain at a resolution of 2.4 Å. J. Virol. 79, 10278–10288. doi: 10.1128/JVI.79.16.10278-10288.2005
Keywords: dengue virus non-structural protein-3 Helicase, virtual screening, pharmacophore modeling, molecular docking, ADMET, FRED
Citation: Halim SA, Khan S, Khan A, Wadood A, Mabood F, Hussain J and Al-Harrasi A (2017) Targeting Dengue Virus NS-3 Helicase by Ligand based Pharmacophore Modeling and Structure based Virtual Screening. Front. Chem. 5:88. doi: 10.3389/fchem.2017.00088
Received: 12 July 2017; Accepted: 16 October 2017;
Published: 31 October 2017.
Edited by:
Nino Russo, Dipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, ItalyReviewed by:
Tiziana Marino, University of Calabria, ItalyJitrayut Jitonnom, University of Phayao, Thailand
Copyright © 2017 Halim, Khan, Khan, Wadood, Mabood, Hussain and Al-Harrasi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sobia A. Halim, c29iaWEuYWhzYW5Aa2lubmFpcmQuZWR1LnBr; c29iaWFoYWxAZ21haWwuY29t
Ajmal Khan, YWptYWxjaGVtaXN0QHlhaG9vLmNvbQ==