NMR Structure and Dynamics of TonB Investigated by Scar-Less Segmental Isotopic Labeling Using a Salt-Inducible Split Intein
 Annika Ciragan
Annika Ciragan  Kornelia M. Mikula
Kornelia M. Mikula Hannes M. Beyer
Hannes M. Beyer O. H. Samuli Ollila
O. H. Samuli Ollila Hideo Iwaï
Hideo Iwaï- Institute of Biotechnology, University of Helsinki, Helsinki, Finland
The growing understanding of partially unfolded proteins increasingly points to their biological relevance in allosteric regulation, complex formation, and protein design. However, the structural characterization of disordered proteins remains challenging. NMR methods can access both the dynamics and structures of such proteins, yet suffering from a high degeneracy of NMR signals. Here, we overcame this bottleneck utilizing a salt-inducible split intein to produce segmentally isotope-labeled samples with the native sequence, including the ligation junction. With this technique, we investigated the NMR structure and conformational dynamics of TonB from Helicobacter pylori in the presence of a proline-rich low complexity region. Spin relaxation experiments suggest that the several nano-second time scale dynamics of the C-terminal domain (CTD) is almost independent of the faster pico-to-nanosecond dynamics of the low complexity central region (LCCR). Our results demonstrate the utility of segmental isotopic labeling for proteins with heterogenous dynamics such as TonB and could advance NMR studies of other partially unfolded proteins.
Introduction
Segmental isotopic labeling facilitates nuclear magnetic resonance (NMR) studies of large or multi-domain proteins, and proteins with low complexity regions by alleviating NMR signal overlaps (Yamazaki et al., 1998; Xu et al., 1999; Skrisovska and Allain, 2008; Volkmann and Iwaï, 2010). Segmentally introducing isotopic labels into proteins also allows their NMR spectroscopic investigation by restricting the emerge of signals to a particular protein domain or region in the full-length protein context (Busche et al., 2009; Buchinger et al., 2010; Minato et al., 2012, 2017; Shiraishi et al., 2018; Wiegand et al., 2018). Several methods, including expressed protein ligation (EPL), protein-trans splicing (PTS), fragment ligation using sortases or asparagine endopeptidases have been used for segmental isotopic labeling (Skrisovska and Allain, 2008; Muona et al., 2010; Freiburger et al., 2015; Kwon et al., 2015; Williams et al., 2016; Frederick et al., 2017; Mikula et al., 2017, 2018; Wiegand et al., 2018). Albeit these various available methods, segmental isotopic labeling for structural studies remains challenging due to the requirement of relatively high quantities (>mg scale) and high purity. Moreover, segmentally isotopic-labeled samples prepared by these methods often contain mutations at the ligation site due to the technical requirements. For example, sortase-mediated ligation requires a sequence of “LPXTGG,” remaining as a “scar” in the ligated product (Freiburger et al., 2015; Williams et al., 2016). PTS by the widely used split DnaE intein from Nostoc punctiforme (NpuDnaE) and EPL require a cysteine mutation (remaining as a “scar” in the product) at the ligation site. Introducing such modifications could potentially influence the structure and dynamics of the proteins of interest. The “scar” problem at the ligation site stimulated several protein engineering attempts of inteins, such as converting the nucleophilic cysteine at the so-called +1 position to serine (Lockless and Muir, 2009; Cheriyan et al., 2013). The recent development of the split MCM2 intein from Halorhabdus utahensis (HutMCM2) could alleviate the “scar” problem as well as the solubility issue associated with split inteins (Ciragan et al., 2016). The minimal sequence requirement for PTS using the split HutMCM2 intein is theoretically a Ser at the +1 position, which is more prevalent than cysteine among diverse proteins (Ciragan et al., 2016). Additionally, the halophilic intein is highly soluble even after artificial splitting into trans-reacting fragments, making this intein very attractive for scar-less segmental isotopic labeling (Aranko et al., 2014a,b; Ciragan et al., 2016).
Many multi-domain proteins or regions lack a well-structured three-dimensional fold. These intrinsically disordered proteins (IDPs) and intrinsically disordered protein regions (IDRs) are very prevalent, and their importance has been increasingly recognized in allosteric regulation, complex formation, and protein design (Chen et al., 2013; Papaleo et al., 2016; Berlow et al., 2018; Keul et al., 2018). However, IDRs typically have low complexity sequences, and their NMR signals tend to degenerate due to the lack of a well-defined fold. The higher degeneracy of IDRs compared to highly structured proteins could hinder their detailed NMR analysis, calling for strategies to alleviate signal overlaps when studying IDPs and IDRs (Nabeshima et al., 2014; Frederick et al., 2017). We chose TonB from Gram-negative bacteria as an example of multi-domain proteins where a central proline-rich low complexity region (LCCR) connects a globular C-terminal domain (CTD) to an N-terminal transmembrane (TM) domain (Figures 1, 2A, Figure S1). TonB plays an essential role in the TonB-dependent transport mechanism transducing the cytoplasmic proton motive force (PMF) to the TonB-dependent transporters (TBDT) in the outer membrane (OM) (Figure 1) (Postle and Larsen, 2007). The transmembrane domain of TonB is associated with the inner membrane-bound proteins ExbB and ExdD, and it interacts with TBDTs in the outer-membrane via a globular C-terminal domain (CTD) (Figure 1). The LCCR likely spans across the periplasm for positioning the CTD to contact TBDTs in the outer membrane (Shultis et al., 2006). The functional role of the LCCR in the periplasmic space is poorly understood, although an extended polyproline type II helical conformation has been proposed (Evans et al., 1986; Brewer et al., 1990; Köhler et al., 2010). The LCCR connecting N-terminus and CTD is highly divergent but contains a proline-rich repetitive sequence in nearly every case, presumably having high degeneracy of NMR signals. Thus, the LCCR could hinder the detailed analysis of the full-length TonB by NMR (Figure S1).
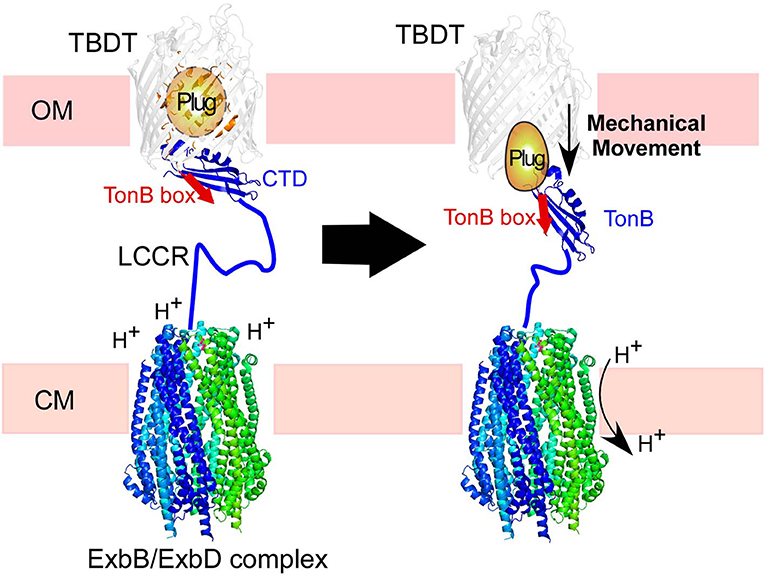
Figure 1. Illustration of TonB-dependent transporters (TBDT) in the cell envelope of Gram-negative bacteria. The C-terminal domain (CTD) of TonB is proposed to interact with the TonB box in the plug domain of TBDT (left). The proton-motive force across the cytoplasmic membrane (CM) is transduced by the ExbB/ExbD complex to induce mechanical force via TonB. The low complexity central region (LCCR) spans in the periplasmic space to interact with TBDTs in the outer-membrane.
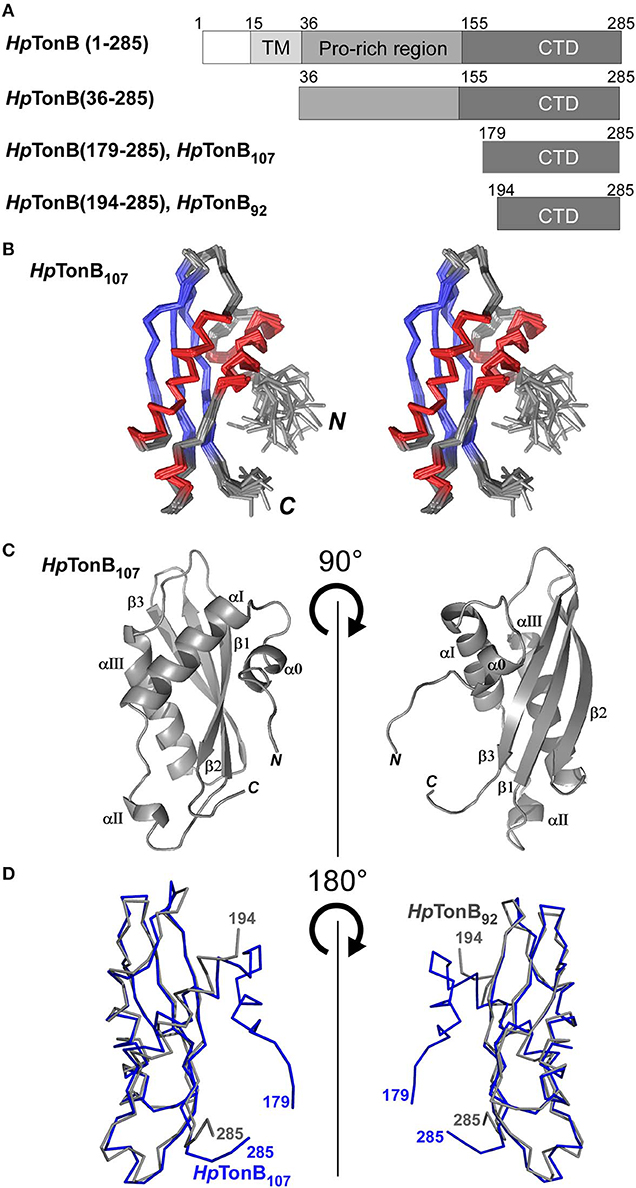
Figure 2. NMR solution structure of HpTonB107 (A) Schematic representations of the domain organization of the full-length TonB and dissected HpTonB variants used in this study. (B) A stereo-view of an ensemble of the 20 NMR-refined conformers of HpTonB107. The regions for α-helices and β-sheets are colored in red and blue, respectively. (C) Cartoon drawings of the HpTonB107 structure with the secondary structure elements labeled. (D) A superposition of the two NMR structures of HpTonB107 (in blue) and HpTonB92 (in gray). The residue numbers for the termini are shown.
Here, we report the NMR structures of the newly dissected CTD of TonB from Helicobacter pylori (HpTonB) and analyzed the effects of the LCCR on the conformational dynamics of TonB by comparison with two differently dissected CTDs. We used a scar-less segmental isotopic labeling approach utilizing a salt-inducible split intein to alleviate the high degeneracy of NMR signals due to the proline-rich low complexity region of TonB.
Results
NMR Solution Structure of TonB (179–285) From Helicobacter pylori (HpTonB107)
First, we determined the NMR solution structure of a 107-residue CTD of HpTonB (179–285) (HpTonB107) for the comparison with HpTonB92 which has been previously reported (Ciragan et al., 2016; Figure 2, Figure S2, Table 1). Three-dimensional structures of variously dissected CTDs of TonB from several Gram-negative bacteria determined by NMR and X-ray crystallography demonstrated variations mainly in the N- and C-terminal regions, suggesting that the dissection positions for the CTD could influence the conformation of the CTD (Chang et al., 2001; Ködding et al., 2005; Peacock et al., 2005; Chu et al., 2010; Oeemig et al., 2018). Notably, the previously determined HpTonB92 indicated notable chemical shift differences over the entire primary structure compared with the chemical shifts of HpTonB (36–285), suggesting possible structural differences of the CTD in the full-length context (Ciragan et al., 2016). The newly determined NMR structure of HpTonB107 with a slightly longer length is very similar to the previously determined NMR structure of HpTonB92 with the root-mean-square deviation (RMSD) of 1.37 Å for the backbone heavy atoms for residues 195–282 (Figures 2A,D). HpTonB107 consists of one β-sheet composed of three anti-parallel β-strands and four α-helices, including an additional short N-terminal helix (α0), which is absent in HpTonB92 (Figure 2C). The extra short N-terminal helix (α0) interacts with the last β-strand (β3). The β3 strand coincides with the region where we observed more substantial chemical shift differences between HpTonB92 and HpTonB (36–285) (Figures 2C, 5A,C).

Table 1. Experimental data for the NMR structure calculation and the structural statistics of the 20 energy-minimized conformers of HpTonB107.
Design for Segmental Isotopic Labeling of HpTonB (36–285)
We believe that the NMR structure of HpTonB107 represents the CTD of TonB in the full-length context better than HpTonB92 because of the higher similarity of the HSQC spectrum to that of HpTonB (36–285). To examine the structure and dynamics of the CTD, we performed segmental isotopic labeling of HpTonB (36–285) lacking the transmembrane region. The presence of a proline-rich repeating sequence in the LCCR of HpTonB suggests that the LCCR is unstructured compared to the CTD, and likely to have very different dynamical properties due to the increased flexibility (Figure S1). For protein dynamics analysis, series of HSQC spectra are typically recorded with various delay times to measure the longitudinal (T1) and transverse (T2) relaxation times. Thus, signal overlaps in the HSQC spectra of the uniformly 15N-labeled HpTonB (36–285) interfere with the 15N relaxation experiments, thereby reducing the reliability of the relaxation data and the number of data points. Therefore, the reduction of the signal degeneracy in the HSQC spectra by segmental isotopic labeling could increase the number of probe residues that could be used for 15N relaxation analysis of the CTD in the presence of the LCCR as well as the LCCR itself.
In this work, we chose a split MCM2 intein from Halorhabdus utahensis (HutMCM2) for the production of segmental isotope-labeled samples, instead of the frequently used naturally split DnaE intein from Nostoc punctiforme (NpuDnaE intein) (Iwai et al., 2006). Whereas, the robust NpuDnaE intein has a Cys residue at the +1 position, the HutMCM2 intein has Ser at the +1 position (Ellilä et al., 2011; Ciragan et al., 2016). HutMCM2 intein thus enables us to make use of a naturally occurring Ser residue in the sequence of HpTonB. Additionally, we decided to perform the protein ligation in vitro to prevent isotopic scrambling, which might interfere with the 15N relaxation analysis (Züger and Iwai, 2005). We split HpTonB between Lys154 and Ser155 and fused each part with the split HutMCM2 intein fragments. To have the native sequence of HpTonB (36–285), we used Lys154 at the −1 position of the N-terminal split HutMCM2 intein, compromising the splicing efficiency (Ciragan et al., 2016). Thus, the split HutMCM2 intein ligation product will retain the native HpTonB sequence at the junction. Such scarless segmental isotopic labeling approaches have been challenging despite the plethora of available methods.
Segmental Isotopic Labeling of HpTonB(36–285)
We used a similar strategy for segmental isotopic labeling of HpTonB with two different labeling schemes for this study, as previously reported and depicted in Figure 3A (Ciragan et al., 2016). Briefly, HpTonB (36–154) was fused with the N-terminal split fragment of the HutMCM2 intein carrying a C-terminal octahistidine tag (HutMCM2ΔC62-H8) (Ciragan et al., 2016). We introduced the octahistidine tag instead of a commonly used hexahistidine tag to account for the highly negatively charged HutMCM2 intein reducing the binding to immobilized metal chelate columns. The C-terminal fragment of HpTonB (155–285) was fused to the C-terminal split HutMCM2 intein with an N-terminal hexahistidine tag (H6-HutMCM2C42). The short names of HutMCM2ΔC62 and HutMCM2C42 for the split fragments are termed for convenience according to the previously reported definition (Aranko et al., 2014b). These two precursor fragments were independently expressed and purified using LB and [20% 13C, 100% 15N]-containing M9 media. We ligated the two independently purified precursors in vitro after mixing at equimolar concentrations with different labeling combinations. To initiate a trans-splicing reaction by the salt-inducible split intein, we elevated the NaCl concentration to 3.5 M to reconstitute an active conformation of the HutMCM2 intein either by rapid dilution or dialysis. The reaction mixture under the high salinity was incubated at room temperature for 1–3 days (Figure 3B). Despite the compromised ligation efficiency due to Lys at the −1 position, the histidine tags incorporated into the split HutMCM2 intein fragments facilitated the efficient removal of both unreacted precursors and reacted split intein fragments (Figure 3A). We obtained 5.8 mg and 47.3 mg of [15N, 36–154]-labeled HpTonB (36–285) and [15N, 155–285]-labeled HpTonB (36–285), respectively.
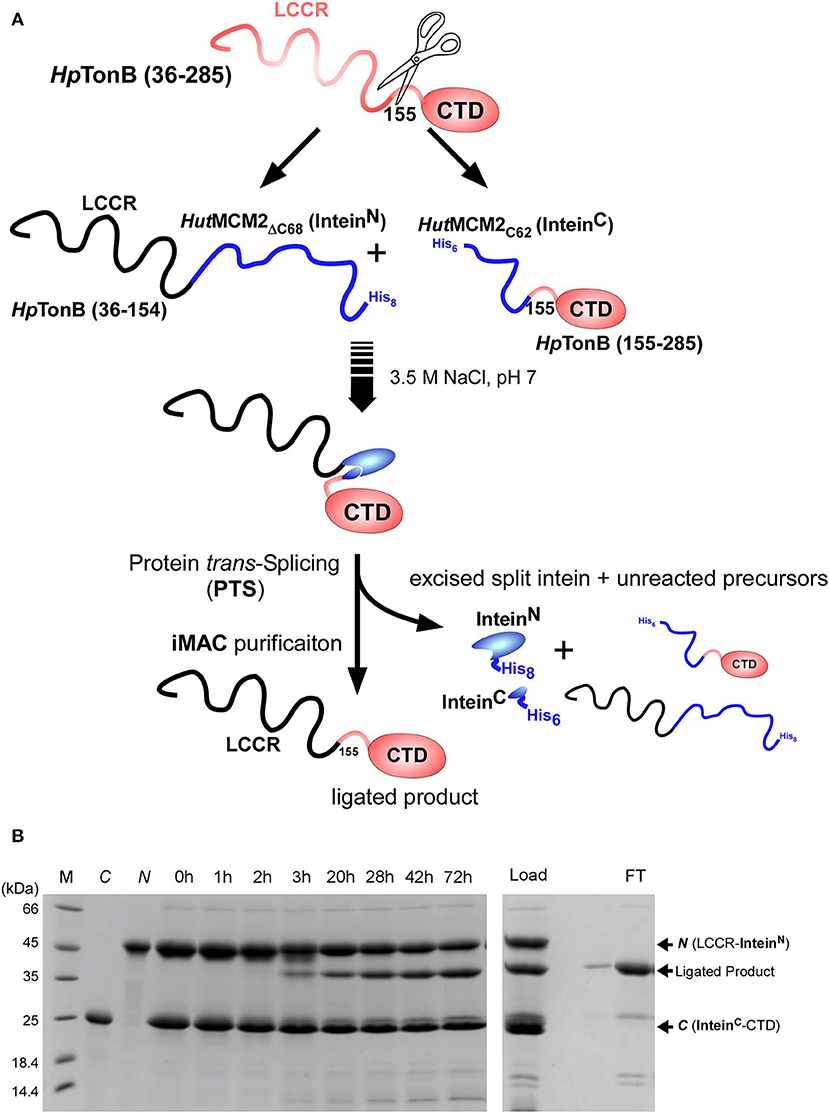
Figure 3. Segmental labeling of HpTonB (36–285). (A) Schematic diagram showing the scheme for segmental labeling either the C-terminal or N-terminal domain of HpTonB (36–285) by the split HutMCM2 intein. (B) In-vitro ligation of segmentally labeled HpTonB (36–285) by PTS using the salt-inducible HutMCM2 intein with Lys at the −1 position. SDS-PAGE analysis shows the time course of the protein ligation reaction. M, C, and N stand for molecular weight marker, C-terminal and N-terminal precursors before mixing, respectively. Lanes 0–72 h indicate the time (hours) of the ligation reaction after mixing the N- and C-terminal precursors in 3.5 M NaCl. The right panel shows the SDS-PAGE analysis of the IMAC purification step, which removes His-tagged precursors and excised intein fragments simultaneously. “Load” and “FT” indicate the sample loaded on the IMAC column and the flow-through fraction, respectively. N and C indicate the N- and C-terminal precursors, respectively.
Backbone Assignments of HpTonB(36–285) Using Segmentally Isotope-Labeled Samples
Segmentally isotope-labeled HpTonB (36–285) samples facilitated more reliable resonance assignments of the crowded regions of the NMR spectrum between 8.2 and 8.5 ppm of the 1H dimension, where NMR signals originate mainly from the N-terminal low complexity region (Figure 4). 71.1% of all backbone resonance assignments (HN, NH, Cα, Cβ, and C′) of HpTonB (36–285) were completed by making use of the two segmentally labeled samples. Of the backbone amide 15N and 1H resonances, 78.6% were assigned. Despite relying on segmental isotopic labeling, NMR signals from the proline-rich repeating amino acid sequences in the N-terminal low complexity region were not entirely resolved in the [1H,15N]-HSQC spectrum (Figure 4C; Figure S1). However, it was possible to unambiguously compare the chemical shifts of the CTD with and without the LCCR. Whereas, the previously reported HpTonB92 indicated notable chemical shift changes when compared with HpTonB (36–285) (Figure 5A), the chemical shift difference was much smaller for HpTonB107 (Figure 5B). This observation confirms that the NMR structure of HpTonB107 likely resembles that of the full-length HpTonB protein. The differences in the chemical shifts of a few N-terminal residues, including Gly181, Ala182, and Thr183, can be attributed to the higher flexibility in the N-terminal end, as observed in the 15N relaxation rates (see below).
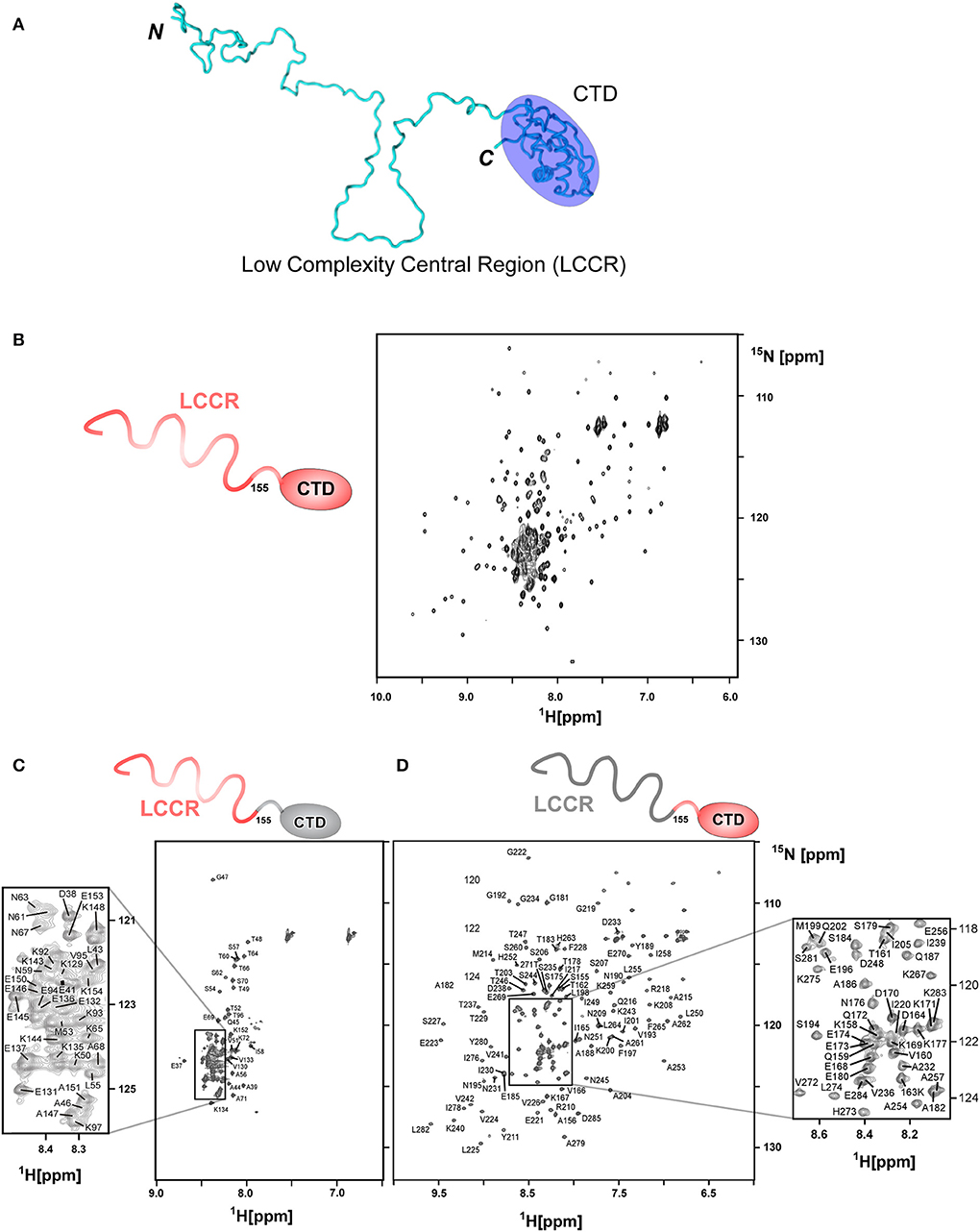
Figure 4. Segmental labeling of HpTonB (36–285) demonstrated by [1H, 15N]-HSQC spectra. (A) A cartoon model of HpTonB (36–285), highlighting the globular CTD and the remaining proline-rich low complexity central region (LCCR). (B) [1H, 15N]-HSQC spectrum of the uniformly 15N-labeled HpTonB (36–285). (C) [1H, 15N]-HSQC spectrum of the segmentally 15N-labeled LCCR (residues 36–154) of HpTonB (36–285). (D) [1H, 15N]-HSQC spectrum of segmentally 15N-labeled CTD (residues 155–285) of HpTonB (36–285).
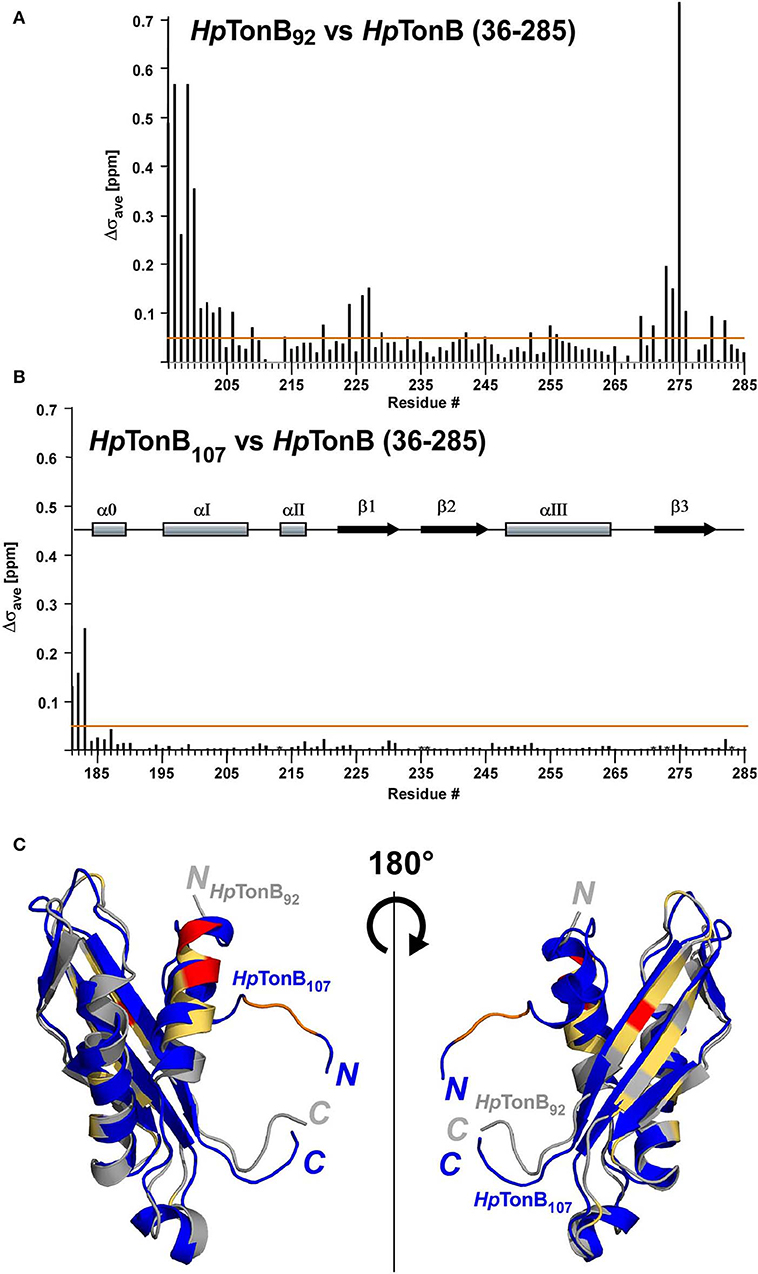
Figure 5. The effects on chemical shifts by dissection of the full-length of HpTonB. (A) Chemical shift differences (Δσave) between HpTonB92 and HpTonB (36–285). (B) Chemical shift difference (Δσave) between HpTonB107 and HpTonB (36–285). The chemical shift difference (Δσave) was derived from the equation: Δσave = ((ΔδH)2 + (0.154 × ΔδN)2)1/2. Orange lines indicate Δσave = 0.05 ppm. The locations of the secondary structural elements of HpTonB107 are indicated above the bar graph. (C) Ribbon drawings of the superimposition of the two structures of HpTonB92 and HpTonB107 shown in gray and blue, respectively. Residues with >0.3 ppm for Δσave are colored in red. Residues above the threshold of Δσav = 0.05 ppm are colored in orange for HpTonB107 and light orange for HpTonB92. The N- and C-termini are indicated by N and C, respectively.
Backbone Dynamics of HpTonB (36–285) Investigated by 15N Relaxation Times Using Segmentally Isotope-Labeled Samples
15N relaxation times have been conveniently used for investigating the internal dynamics of proteins (Palmer, 2004). However, the typical 15N relaxation analysis for N-H bonds used for protein dynamics requires quantifications of peak volumes in a series of [1H, 15N]-HSQC spectra with different relaxation delays. When peaks are not well-resolved in a [1H,15N]-HSQC spectrum, the quantification of the 15N relaxation rate becomes unreliable or even unavailable. Segmental isotopic labeling can alleviate the high signal degeneracy, thereby increasing the number of probes that can be used for NMR analysis. It is thus an attractive method, particularly when no sequence alternation is involved. We combined the 15N relaxation analysis of HpTonB (36–285) from the two differently segmentally isotope-labeled samples (one sample was 15N-labeled for residues 36–154 of HpTonB, and the other had 15N-labeling for residues 155-285) (Figure 4). The segmental isotope-labeled samples indeed increased the number of probes used for the 15N relaxation analysis, thereby enhancing the reliability of the relaxation analysis.
Here, we compared the 15N relaxation rates from three different constructs of HpTonB92, HpTonB107, and HpTonB (36–285) by making use of segmentally labeled samples (Figure 6). The 15N relaxation studies showed that the structured CTD has distinct protein dynamics from the pro-rich LCCR, which has fast dynamics in the picosecond-to-nanosecond time scale as observed from the reduced heteronuclear NOEs (Figure 6). On the other hand, the differences in the spin relaxation times for the different CTDs are small. Assuming that T1 and T2 are independent of internal motions and that the rotation of CTD is spherically isotropic (Kay et al., 1989), the overall correlation times estimated from the T1/T2 ratio of the globular regions were 5.8, 6.9, and 8.2 ns for HpTonB92, HpTonB107, and HpTonB (36–285), respectively (Ollila et al., 2018). Since even small anisotropy could affect the analysis of protein dynamics (Lüginbuhl et al., 1997), the detailed analysis of rotational dynamics taking into account the effect of anisotropy and disordered linker using molecular dynamics simulation is currently under investigation and will be published elsewhere. The overall correlation times for the truncated constructs are only slightly shorter than HpTonB (36–285), even though the molecular size of HpTonB (36–285) is double of HpTonB107. Because the NMR structure of HpTonB107 likely represents the structure in the full-length context, we assume that the dynamical properties of the CTD are mainly independent of the very flexible pro-rich LCCR region. The negative heteronuclear NOE values for the N- and C-termini of HpTonB107 and HpTonB (36–285) are indicative of rapid local motions. The N-terminal region (181–182) of HpTonB107 showed decreased heteronuclear NOEs compared with the corresponding region of HpTonB (36–285). This observation suggests that the truncation of the LCCR in HpTonB107 induced flexibility for this region, which is more restricted in HpTonB (36–285). The change in the local dynamics could account for the observed chemical shift differences between HpTonB107 and HpTonB (36–285) around this region (Figure 5B).
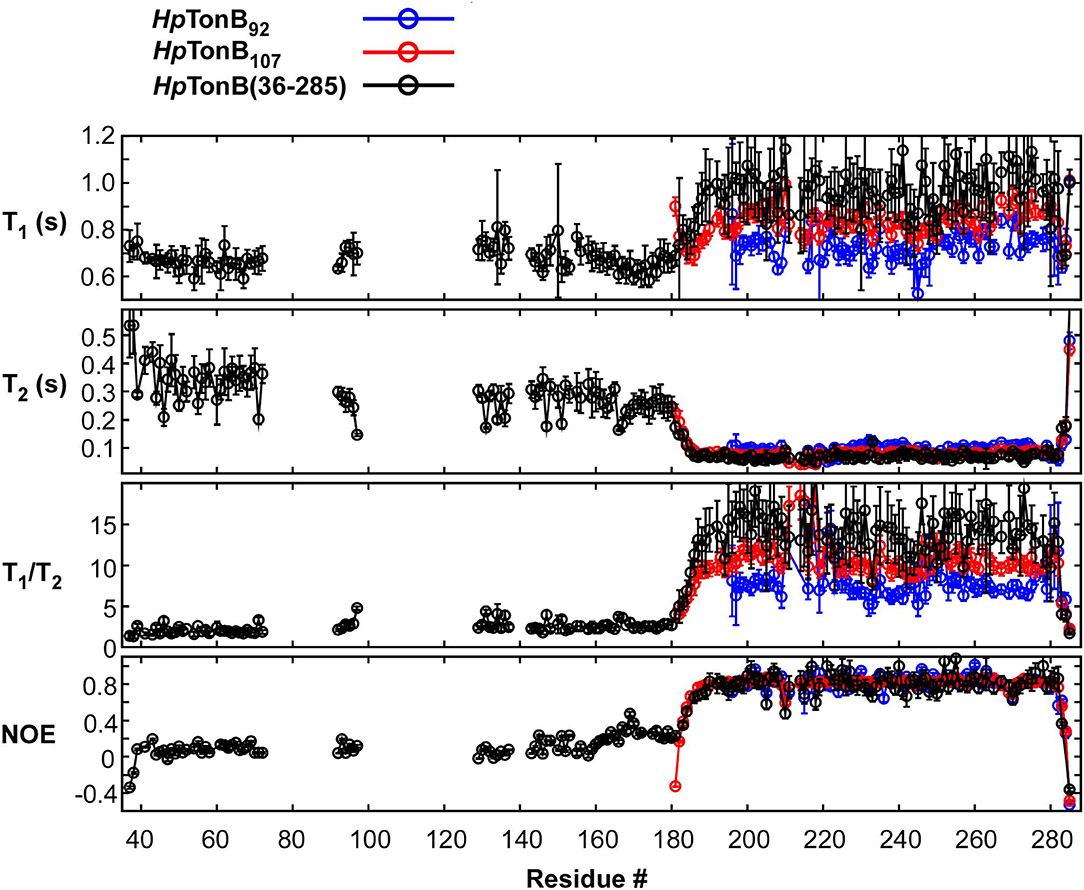
Figure 6. 15N relaxation data obtained for HpTonB92, HpTonB107, and HpTonB (36–285) are shown in blue, red, and black, respectively. The longitudinal relaxation time (T1), transverse relaxation time (T2), T1/T2 ratios, and heteronuclear 15N{1H}-NOE are plotted against the residue number. Residues with overlaps and residues with ambiguous assignments were removed from the plots.
Discussion
It has been a common practice in structural biology to dissect a protein into a globular domain for structural investigation by removing flexible or disordered regions because it often facilitates crystallization and also alleviates the signal overlap problem for NMR analysis. However, IDRs and IDPs are increasingly recognized for their functional importance. Thus, structural investigations of proteins in their full-length contexts, including disordered regions, have become imperative to understand how proteins function in living systems, imposing technical challenges. NMR spectroscopy has the advantage of investigating protein structures with disordered regions because it does not require any crystallization and could be studied under various solution conditions. However, NMR signal assignments could be even more time-consuming and/or difficult when proteins contain repetitive sequences because of the increased complexity and high degeneracy of NMR signals.
Here, we demonstrated scarless segmental isotopic labeling by protein trans-splicing for the structural investigation of HpTonB, resulting in no mutation at the ligation site despite the low ligation efficiency. Segmental labeling of HpTonB indeed alleviated the high degeneracy of NMR spectra originating from the low complexity central region, thereby enabling the detailed comparison of the differently dissected CTDs and a full-length construct without TM region. The comparison between the two structures of HpTonB92 and HpTonB107 demonstrated that a dissected domain does not necessarily represent the structure in the full-length context, which is supported by the more substantial chemical shift differences observed for HpTonB92. In line with this observation, we found the instability of HpTonB92 because it precipitated during extended NMR measurements.
In summary, our study using HpTonB system highlights the importance of performing structural investigations with the full-length protein or at least fragments close to the full-length. The 15N-relaxation analysis by segmentally isotopically labeled sample confirmed that the LCCR of HpTonB (36–285) is flexible with largely independent motions of the CTD. Even though the sequence conservation of LCCR is very weak within the N-terminal and the central regions, the Pro-Lys and Pro-Glu repeats in the LCCR constitute the flexible region, influencing the structural and rotational dynamics of the CTD. It remains elusive how the flexible disordered linker of the LCCR could mediate the biological function in the TonB-dependent transduction system. It would be of remaining interests to investigate the structure and dynamics of TonB inserted into the membrane environment, which might require three-fragment ligation with an additional ligation step. With further improvement of protein ligation by PTS, scarless segmental isotopic labeling by two- and three-fragment ligation is expected to pave the way for NMR studies of unfolded proteins and multi-domain proteins with mixed dynamics as exemplified with HpTonB.
Materials and Methods
Production and Purification of Labeled and Unlabeled Precursors
E. coli strain T7 Express (New England Biolabs) was transformed with the plasmid encoding either the N-terminal precursor encoding HpTonB (36–154)-HutMCM2ΔC62-H8 (pACRSF5) (Ciragan et al., 2016) or the C-terminal precursor H6-HutMCM2C42-HpTonB (155–285) (pBHRSF165) (Ciragan et al., 2016) and grown in 50 mL LB medium supplemented with 25 μg/mL kanamycin at 30°C overnight. The pre-cultures were used to inoculate 1 liter for pACRSF5 or 2 liters for pBHRSF165 of LB medium supplemented with 25 μg/mL kanamycin and incubated at 37°C while shaking at 200 rpm. When OD600 reached 0.6, expression was induced with a final concentration of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG), and induction was continued for 4 h.
For the production of the labeled N-terminal precursor protein, 5 mL of pre-culture containing plasmid pACRSF5 was used to inoculate 50 mL of [20% 13C, 100% 15N]-containing M9 medium with 15NH4Cl (1 g/L) and a mixture of 13C6 D-glucose (0.2 g/L) and unlabeled D-glucose (0.8 g/L) as sole nitrogen and carbon sources, respectively, supplemented with 25 μg/mL kanamycin. The culture was incubated at 30°C with shaking at 200 rpm overnight and then transferred into pre-warmed 1950 mL of [20% 13C, 100% 15N]-containing M9 medium supplemented with 25 μg/mL kanamycin and grown at 37°C under shaking. The protein was induced with a final concentration of 1 mM IPTG when OD600 reached 0.6 and lasted 4–5 h.
Expression cultures for the production of labeled C-terminal precursor protein containing plasmid pBHRSF165 were first grown in 2 L of LB medium until OD600 reached 0.6. The cultures were then harvested by centrifugation at 1,600 g for 12 min at 20°C and gently resuspended in 12 mL of [20% 13C, 100% 15N]-containing M9 medium. The cell suspension was added to 1.8 L of complete M9 medium containing 25 μg/mL kanamycin and grown at 30°C while shaking at 200 rpm for 4 h. After 20 min, expression was induced with 1 mM IPTG. Cells were harvested by centrifugation at 4,000 g, 4°C for 10 min, resuspended in 20 mL of 50 mM sodium phosphate buffer, pH 8.0, 300 mM NaCl, frozen in liquid nitrogen, and stored for further use at −74°C. The cell pellets were thawed and lyzed at 15,000 psi for 10 min, 4°C using an Emulsiflex C3 homogenizer (Avestin). Cell debris was removed by centrifugation at 38,000 g, 4°C for 60 min, the supernatant was passed through a 0.45 μm filter and followed by loading on a 5 mL HisTrap HP column (GE Healthcare Life Sciences), which was pre-equilibrated with 50 mM sodium phosphate buffer, pH 8.0, 300 mM NaCl, 10 mM imidazole. After washing with 50 mM sodium phosphate buffer, pH 8.0, 300 mM NaCl, 30 mM imidazole, the protein was eluted with a linear gradient of 30–250 mM imidazole in 50 mM sodium phosphate buffer, pH 8.0, 300 mM NaCl. The elution was dialyzed overnight against phosphate-buffered saline (PBS) at 8°C. N- and C-terminal precursors for segmental labeling were concentrated using ultra-centrifugal devices and frozen in liquid nitrogen for storage at −74°C.
Salt-Induced Protein Ligation by Protein Trans-Splicing (PTS)
Ligation of HpTonB(36–154)-HutMCM2ΔC62-H8 with H6-HutMCM2C42-HpTonB (155–285) was performed according to the protocol published previously with slight modifications (Ciragan et al., 2016). Purified 15N-labeled C-terminal precursor and the unlabeled N-terminal precursor were mixed in an equimolar ratio at a final concentration of 0.25 mM each, in a total volume of 11 mL with 0.5 M sodium phosphate buffer, pH 7.0, 3.5 M NaCl and 0.5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and incubated at room temperature for 3 days. The ligation mixture was dialyzed at room temperature against PBS and then against 50 mM sodium phosphate buffer, pH 8.0, 350 mM NaCl. The sample was further diluted twice with fresh buffer before loading onto a 5 mL HisTrap HP column to remove the unreacted precursors and excised intein fragments. For producing HpTonB (36–285) using a 15N-labeled N-terminal precursor instead, the two precursor proteins were mixed in an equimolar ratio at 0.325 mM each, in 4 mL PBS and dialyzed overnight at room temperature against 350 mM sodium phosphate buffer, pH 7.0, 3.5 M NaCl, 0.5 mM TCEP. After 24 h, the dialysis buffer was exchanged with PBS for 2 h. The sample was diluted 5-fold with 50 mM sodium phosphate buffer, pH 8.0, 300 mM NaCl, and purified using a 5 mL HisTrap HP column as described above. The flow-through fractions containing ligated segmentally labeled HpTonB (36–285) protein were dialyzed overnight at 8°C against 20 mM sodium phosphate buffer, pH 6.0 for NMR measurements.
Molecular Cloning and Production of Uniformly Labeled HpTonB107
The coding sequence of the 107-residue C-terminal domain of HpTonB (HpTonB107) was PCR-amplified from Helicobacter pylori genomic DNA using the oligonucleotides I472: TTAAAGCTTAGTCTTCTTTCAAGCTATAAGCGATAG and I812: AAAGGATCCGAGGGGGCCACTTCCGAAGCTCAGGCTTATAACC. The PCR product was digested with BamHI and HindIII and ligated into pHYRSF53 (Addgene #64696), resulting in pBHRSF159, encoding HpTonB with N-terminal hexahistidine tag and SUMO fusion (H6-SUMO-HpTonB107) (Guerrero et al., 2015). For protein production, the plasmid was transformed into E. coli T7 Express cells (New England Biolabs). The cells were grown overnight at 30°C in 50 ml LB medium supplemented with 25 μg/ml kanamycin. The cells were spun down at 900 g for 15 min and gently resuspended in 2 L pre-warmed M9 medium with 25 μg/ml kanamycin, containing 15NH4Cl and 13C-glucose as the sole nitrogen and carbon source, respectively. Further protein expression and purification were performed as described for the N-terminal labeled HpTonB (36–285) precursor, except that the expression culture was grown at 30°C for 5 h. Cells were harvested, lyzed, and purified, as described above. The N-terminal H6-SUMO fusion was removed using the SUMO-specific protease Ulp1 and the final [100% 13C, 100% 15N]-labeled HpTonB107 protein was recovered in the flow-through after passage through a 5 mL HisTrap HP column as previously described (Guerrero et al., 2015). The purified protein was dialyzed against 20 mM sodium phosphate buffer, pH 6.0 for NMR analysis, and concentrated using ultrafiltration.
Sample Preparation for NMR Measurements
Segmentally labeled HpTonB (36–285) or uniformly labeled HpTonB107 protein samples in 20 mM sodium phosphate buffer, pH 6.0 were mixed at the given concentrations with the following additives: HpTonB (36–285) with N- or C-terminal segmental labeling, 300 μM, 150 mM NaCl, 5% D2O in 220 μL; uniformly labeled HpTonB107, 450–500 μM in 250 μL with 10% D2O.
Determination of NMR Solution Structures
For the sequential resonance assignment, a standard set of double and triple resonance spectra was recorded: [1H, 15N]-HSQC, [1H, 13C]-HSQC, HNCO, HNCA, HNCACB, HN(CO)CA, HN(CA)CO, CBCA(CO)NH, HCCH-COSY, H(CCCO)NH, (H)CC(CO)NH (Sattler et al., 1999). NMR measurements were performed on Bruker Avance IIIHD 850 MHz spectrometer equipped with a cryogenically cooled probe head at 303 K at the Institute of Biotechnology, University of Helsinki. Resonance assignment was done using CcpNmr Analysis (Vranken et al., 2005). Automated NMR structure calculation was performed by CYANA 3.0, which is based on automated NOESY cross-peaks assignment using previously determined chemical shift values (Güntert et al., 1997; Herrmann et al., 2002). For the structure calculation, triple resonance 15N-edited NOESY-HSQC and 13C-edited NOESY-HSQC spectra with mixing times of 70 and 90 ms, respectively, were used for HpTonB107. The chemical shift values from the sequential resonance assignment were used together with the NOE peak lists for the structure calculation (Güntert et al., 1997; Herrmann et al., 2002). Energy minimization of the structure was done using the 20 best conformers based on the lowest values of the CYANA target function by AMBER 14 (Cornell et al., 1995; Pearlman et al., 1995). Quality analysis of the structure was performed using PSVS 1.5 validation software suite (Bhattacharya et al., 2007), PROCHECK-NMR (Laskowski et al., 1996), and WHAT IF (Vriend, 1990).
Backbone Dynamics of HpTonB
The spin relaxation times of HpTonB92 were published previously (Ollila et al., 2018), and HpTonB107 relaxation times were measured with the identical setup described in that publication. For HpTonB (36–285) with residues 36–154 or 155–285 labeled, the same pulse sequence (Kay et al., 1989; Barbato et al., 1992; Farrow et al., 1994) was used with slightly modified delay times. In HpTonB (36–285) experiments with residues 36–154 labeled, the delay times of 50, 100, 200, 300, 500, 700, and 900 ms were used for T1, and delay times of 51, 68, 85, 119, 153, 187, 220, and 254 ms were used for T2. The recycling delay of 3.5 s was used for both T1 and T2, and 5.0 s for heteronuclear NOE. The same settings were used for the segmentally [15N, 154–285]-labeled sample of the HpTonB (36–285), except for the T2 delays, which were 51, 68, 85, 102, 136, 170, 204, and 237 ms, and three-second recycling delays for T1 and T2. The temperature in these experiments was 298 K. The T1 and T2 relaxation data were processed and analyzed using Bruker Dynamic Center software (version 2.5.5) and the NOE relaxation times were processed and analyzed using CcpNmr Analysis software (version 2.4.2) (Vranken et al., 2005). The approximate time-scales for the CTD rotation were estimated, minimizing the equation (11) from Ollila et al. (2018) with respect to the experimentally measured T1/T2 ratios as originally proposed by Kay et al. (1989).
Data Availability Statement
The coordinates and the chemical shifts for this study can be found in the protein data bank (http://www.rcsb.org/pdb/home/home.do) with PDB ID code: 6SLY and in BMRB (http://www.bmrb.wisc.edu/) with the accession number: 34425.
Author Contributions
HI and OS designed and supervised the project. AC, HB, and KM have contributed to the production of NMR samples. AC and SB performed the NMR analysis and structure determination. OS contributed to the relaxation studies. All authors contributed to writing of the manuscript.
Funding
This work was supported in part by the Academy of Finland (137995, 277335), Sigrid Jusélius Foundation, and Novo Nordisk Foundation (NNF17OC0025402). The NMR facility has been supported by Biocenter Finland and HiLIFE-INFRA.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
CSC-IT center for Science Ltd., Finland, is acknowledged for the allocation of computational resources. H. A. Heikkinen and Tuuli M. Kauppala are acknowledged for their help with NMR spectra measurements and 15N relaxation analysis. We thank S. Jääskeläinen for her help with protein production.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00136/full#supplementary-material
Figure S1. Sequence alignment of TonB protein from four gram-negative bacteria (HP, Helicobacter pylori; EC, Escherichia coli; ST, Salmonella typhimurium; PA, Pseudomonas aeruginosa). Black color indicates identical residues, and gray color indicates residues with similar chemical characteristics. The asterisk (*) indicates the position of the conserved histidine (except for HP) in the transmembrane domain. The domain arrangement of HpTonB is shown above the amino-acid sequence.
Figure S2. Comparison of the two [1H, 15N]-HSQC spectra of (A) HpTonB92 and (B) HpTonB107 Sequence-specific assignments are marked for each peak with one-letter codes for amino-acid types.
Abbreviations
CTD, C-terminal domain; DTT, dithiothreitol; EPL, expressed protein ligation; GB1, B1 domain of IgG binding protein G; HSQC, Heteronuclear Single Quantum Coherence; Hp, Helicobacter pylori; Hut, Halorhabdus utahensis; IDP, intrinsically disordered protein; IDR, intrinsically disordered protein region; IMAC, immobilized metal affinity chromatography; IPTG, isopropyl β-d-1-thiogalactopyranoside; LCCR, low complexity central region; PMF, proton motive force; NMR, nuclear magnetic resonance; Npu, Nostoc punctiforme; OM, outer membrane; PBS, phosphate-buffered saline; PTS, protein trans-splicing; TCEP, tris(2-carboxyethyl)phosphine; TBDT, TonB-dependent transporter; TM, transmembrane.
References
Aranko, A. S., Oeemig, J. S., Zhou, D., Kajander, T., Wlodawer, A., and Iwaï, H. (2014a). Structure-based engineering and comparison of novel split inteins for protein ligation. Mol. Biosyst. 10, 1023–1034. doi: 10.1039/C4MB00021H
Aranko, A. S., Wlodawer, A., and Iwaï, H. (2014b). Nature's recipe for splitting inteins. Protein Eng. Des. Sel. 27, 263–271. doi: 10.1093/protein/gzu028
Barbato, G., Ikura, M., Kay, L. E., Pastor, R. W., and Bax, A. (1992). Backbone dynamics of calmodulin studied by nitrogen-15 relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry 31, 5269–5278. doi: 10.1021/bi00138a005
Berlow, R. B., Dyson, H. J., and Wright, P. E. (2018). Expanding the paradigm: intrinsically disordered proteins and allosteric regulation. J. Mol. Biol. 430, 2309–2320. doi: 10.1016/j.jmb.2018.04.003
Bhattacharya, A., Tejero, R., and Montelione, G. T. (2007). Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795. doi: 10.1002/prot.21165
Brewer, S., Tolley, M., Trayer, I. P., Barr, G. C., Dorman, C. J., Hannavy, K., et al. (1990). Structure and function of X-Pro dipeptide repeats in the TonB proteins of Salmonella typhimurium and Escherichia coli. J. Mol. Biol. 216, 883–895. doi: 10.1016/S0022-2836(99)80008-4
Buchinger, E., Aachmann, F. L., Aranko, A. S., Valla, S., Sjåk-Bræk, G., Iwaï, H., et al. (2010). Use of Protein trans-splicing to produce active and segmentally 2H, 15N labeled mannuronan C5-epimerase AlgE4. Prot. Sci. 19, 1534–1543. doi: 10.1002/pro.432
Busche, A. E. L., Aranko, A. S., Talebzadeh-Farooji, M., Bernhard, F., Dötsch, V., and Iwaï, H. (2009). Segmental isotopic labeling of a central domain in a multi-domain protein by protein trans-splicing using only one robust DnaE intein. Angew. Chem. Int. Edit. 48, 6128–6131. doi: 10.1002/anie.200901488
Chang, C., Mooser, A., Plückthun, A., and Wlodawer, A. (2001). Crystal structure of the dimeric C-terminal domain of TonB reveals a novel fold. J. Biol. Chem. 276, 27535–27540. doi: 10.1074/jbc.M102778200
Chen, X., Zaro, J. L., and Shen, W.-C. (2013). Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 65, 1357–1369. doi: 10.1016/j.addr.2012.09.039
Cheriyan, M., Pedamallu, C. S., Tori, K., and Perler, F. (2013). Faster protein splicing with the Nostoc punctiforme DnaE intein using non-native extein residues. J. Biol. Chem. 288, 6202–6211. doi: 10.1074/jbc.M112.433094
Chu, B. C., Garcia-Herrero, A., Johanson, T. H., Krewulak, K. D., Lau, C. K., Peacock, R. S., et al. (2010). Siderophore uptake in bacteria and the battle for iron with the host; a bird's eye view. Biometals 23, 601–611. doi: 10.1007/s10534-010-9361-x
Ciragan, A., Aranko, A. S., Tascon, I., and Iwaï, H. (2016). Salt inducible protein splicing in cis and trans by inteins from extremely halophilic archaea as a novel protein engineering tool. J. Mol. Biol. 428, 4573–4588. doi: 10.1016/j.jmb.2016.10.006
Cornell, W. D., Cieplack, P., Bayly, C. I., Gould, I. R., Merz, K. M., Ferguson, D. M., et al. (1995). A second generation force-field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117, 5179–5197. doi: 10.1021/ja00124a002
Ellilä, S., Jurvansuu, J. M., and Iwaï, H. (2011). Evaluation and comparison of protein splicing by exogenous inteins with foreign exteins in Escherichia coli. FEBS lett. 585, 3471–3477. doi: 10.1016/j.febslet.2011.10.005
Evans, J. S., Levine, B. A., Trayer, I. P., Dorman, C. J., and Higgins, C. F. (1986). Sequence-imposed structural constraints in the TonB protein of E. coli. FEBS Lett. 208, 211–216. doi: 10.1016/0014-5793(86)81020-1
Farrow, N. A., Muhandiram, R., Singer, A. U., Pascal, S. M., Kay, C. M., Gish, G., et al. (1994). Backbone dynamics of a free and a phosphopeptide complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003. doi: 10.1021/bi00185a040
Frederick, K. K., Michaelis, V. K., Caporini, M. A., Andreas, L. B., Debelouchina, G. T., Griffin, R. G., et al. (2017). Combining DNP NMR with segmental and specific labeling to study a yeast prion protein strain that is not parallel in-register. Proc. Natl. Acad. Sci.U.S.A. 114, 3642–3647. doi: 10.1073/pnas.1619051114
Freiburger, L., Sonntag, M., Hennig, J., Li, J., Zou, P., and Sattler, M. (2015). Efficient segmental isotope labeling of multi-domain proteins using Sortase A. J. Biomol. NMR 63, 1–8. doi: 10.1007/s10858-015-9981-0
Guerrero, F., Ciragan, A., and Iwaï, H. (2015). Tandem SUMO fusion vectors for improving soluble protein expression and purification. Protein Expr. Purif. 116, 42–49. doi: 10.1016/j.pep.2015.08.019
Güntert, P., Mumenthaler, C., and Wüthrich, K. (1997). Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298. doi: 10.1006/jmbi.1997.1284
Herrmann, T., Güntert, P., and Wüthrich, K. (2002). Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227. doi: 10.1016/s0022-2836(02)00241-3
Iwai, H., Züger, S., Jin, J., and Tam, P. H. (2006) Highly efficient protein trans-splicing by a naturally split DnaE intein from nostoc punctiforme. FEBS lett. 580, 1853–1858. doi: 10.1016/j.febslet.2006.02.045
Kay, L. E., Torchia, D. A., and Bax, A. (1989). Backbone dynamics of proteins as studied by nitrogen-15 inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 128, 8972–8979. doi: 10.1021/bi00449a003
Keul, N. D., Oruganty, K., Schaper Bergman, E. T., Beattie, N. R., McDonald, W. E., Kadirvelraj, R., et al. (2018). The entropic force generated by intrinsically disordered segments tunes protein function. Nature 563, 584–588. doi: 10.1038/s41586-018-0699-5
Ködding, J., Killig, F., Polzer, P., Howard, S. P., Diederichs, K., and Welte, W. (2005). Crystal structure of a 92-residue C-terminal fragment of TonB from Escherichia coli reveals significant conformational changes compared to structures of smaller TonB fragments. J. Biol. Chem. 4, 3022–3028. doi: 10.1074/jbc.M411155200
Köhler, S. D., Weber, A., Howard, S. P., Welte, W., and Drescher, M. (2010). The proline-rich domain of TonB possesses an extended polyproline II-like conformation of sufficient length to span the periplasm of Gram-negative bacteria. Protein Sci. 19, 625–630. doi: 10.1002/pro.345
Koradi, R., Billeter, M., and Wüthrich, K. (1996). MOLMOL: a program for display and analysis of macromolecular structures J. Mol. Graph. 14, 51–55. doi: 10.1016/0263-7855(96)00009-4
Kwon, B., Tietze, D., White, P. B., SLiao, S. Y., and Hong, M. (2015). Chemical ligation of the in?uenza M2 protein for solid-state NMR characterization of the cytoplasmic domain. Protein Sci. 24, 1087–1099. doi: 10.1002/pro.2690
Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R., and Thornton, J. M. (1996). AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 8, 477–486. doi: 10.1007/bf00228148
Lockless, S. W., and Muir, T. W. (2009). Traceless protein splicing utilizing evolved split inteins. Proc. Natl. Acad. Sci. U.S.A. 106, 10999–11004. doi: 10.1073/pnas.0902964106
Lüginbuhl, P., Pervushin, K. V., Iwai, H., and Wüthrich, K. (1997). Anisotropic molecular rotational diffusion in 15N spin relaxation studies of protein mobility. Biochemistry 36, 7305–7312. doi: 10.1021/bi963161h
Mikula, K. M., Krumwiede, L., Plückthun, A., and Iwaï, H. (2018). Segmental isotopic labeling by asparaginyl endopeptidase-mediated protein ligation. J. Biomol. NMR 71, 225–235. doi: 10.1007/s10858-018-0175-4
Mikula, K. M., Tascón, I., Tommila, J. J., and Iwaï, H. (2017). Segmental isotopic labeling of a single-domain globular protein without any refolding step by an asparaginyl endopeptidase. FEBS lett. 591, 1285–1294. doi: 10.1002/1873-3468.12640
Minato, Y., Ueda, T., Machiyama, A., Iwaï, H., and Shimada, I. (2017). Dynamic domain arrangement of CheA-CheY complex regulates bacterial thermotaxis, as revealed by NMR. Sci. Rep. 7:16462. doi: 10.1038/s41598-017-16755-x
Minato, Y., Ueda, T., Shimada, I., and Iwaï, H. (2012) Segmental isotopic labeling of a 140 kD dimeric multi-domain protein CheA from Escherichia coli by expressed protein ligation protein Trans-splicing. J. Biomol. NMR 53, 191–207. doi: 10.1007/s10858-012-9628-3
Muona, M., Aranko, A. S., Raulinaitis, V., and Iwaï, H. (2010). Segmental isotopic labeling of multi-domain and fusion proteins by protein trans-splicing in vivo and in vitro. Nat. Protoc. 5, 574–587. doi: 10.1038/nprot.2009.240
Nabeshima, Y., Mizuguchi, M., Kajiyama, A., and Okazawa, H. (2014). Segmental isotope-labeling of the intrinsically disordered PQBP1. FEBS lett. 588, 4583–4589. doi: 10.1016/j.febslet.2014.10.028
Oeemig, J. S., Ollila, O. H. S., and Iwaï, H. (2018). NMR structure of the C-terminal domain of TonB protein from Pseudomonas aeruginosa. PeerJ. 6:e5412. doi: 10.7717/peerj.5412
Ollila, O. H. S., Heikkinen, H. A., and Iwaï, H. (2018). Rotational dynamics of proteins from spin relaxation times and molecular dynamics simulations. J. Phys. Chem. B. 122, 6559–6569. doi: 10.1021/acs.jpcb.8b02250
Palmer, A. G. (2004). NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 104, 3623–3640. doi: 10.1021/cr030413t
Papaleo, E., Saladino, G., Lambrughi, M., Lindorff-Larsen, K., Gervasio, F. L., and Nussinov, R. (2016). The role of protein loops and linkers in conformational dynamics and allostery. Chem. Rev. 116, 6391–6423. doi: 10.1021/acs.chemrev.5b00623
Peacock, R. S., Weljie, A. M., Howard, S. P., Price, F. D., and Vogel, H. J. (2005). The solution structure of the C-terminal domain of TonB and interaction studies with TonB box peptides. J. Mol. Biol. 345, 1185–1197. doi: 10.1016/j.jmb.2004.11.026
Pearlman, D. A., Case, D. A., Caldwell, J. W., Ross, W. S., Cheatham, T. E., DeBolt, S., et al. (1995). AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91, 1–41. doi: 10.1016/0010-4655(95)00041-D
Postle, K., and Larsen, R. A. (2007). TonB-dependent energy transduction between outer and cytoplasmic membranes. BioMetals 20, 453–465. doi: 10.1007/s10534-006-9071-6
Sattler, M., Schleucher, J., and Griesinger, C. (1999). Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectr. 34, 93–158. doi: 10.1016/S0079-6565(98)00025-9
Shiraishi, Y., Natsume, M., Kofuku, Y., Ueda, T., Nakata, K., Mizukoshi, T., et al. (2018). Phosphorylation-induced conformation of ß2-adrenoceptor related to arrestin recruitment revealed by NMR. Nat. Commun. 9, 194. doi: 10.1038/s41467-017-02632-8
Shultis, D. D., Purdy, M. D., Banchs, C. N., and Wiener, M. C. (2006). Outer membrane active transport: structure of the BtuB:TonB complex. Science 312, 1396–1399. doi: 10.1126/science.1127694
Skrisovska, L., and Allain, F. H.-T. (2008). Improved segmental isotope labeling methods for the NMR study of multidomain or large proteins: application to the RRMs of Npl3p and hnRNP L. J Mol. Biol. 375, 151–164. doi: 10.1016/j.jmb.2007.09.030
Volkmann, G., and Iwaï, H. (2010) Protein trans-splicing its use in structural biology: opportunities limitations. Mol. Biosyst. 6, 2110–2121. doi: 10.1039/c0mb00034e
Vranken, W. F., Boucher, W., Stevens, T. J., Fogh, R. H., Pajon, A., Llinas, M., et al. (2005). The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696. doi: 10.1002/prot.20449
Vriend, G. (1990). WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 8, 52–56. doi: 10.1016/0263-7855(90)80070-V
Wiegand, T., Cadalbert, R., von Schroetter, C., Allain, F. H., and Meier, B. H. (2018). Segmental isotope labeling and solidstate NMR of a 12 x 59 kDa motor protein: identification of structural variability. J. Biomol. NMR 71, 237–245. doi: 10.1007/s10858-018-0196-z
Williams, F. P., Milbradt, A. G., Embrey, K. J., and Bobby, R. (2016). Segmental isotope labeling of an individual bromodomain of a tandem domain BRD4 using Sortase A. PLoS ONE. 11:e0154607. doi: 10.1371/journal.pone.0154607
Xu, R., Ayers, B., Cowburn, D., and Muir, T. W. (1999). Chemical ligation of folded recombinant proteins: segmental isotopic labeling of domains for NMR studies. Proc. Natl. Acad. Sci. U.S.A. 96, 388–393. doi: 10.1073/pnas.96.2.388
Yamazaki, T., Otomo, T., Oda, N., Kyogoku, Y., Uegaki, K., Ito, N., et al. (1998). Segmental isotope labeling for protein NMR using peptide splicing. J. Am. Chem. Soc. 120, 5591–5592. doi: 10.1021/ja980776o
Keywords: inteins, protein ligation, NMR spectroscopy, segmental isotopic labeling, TonB, intrinsically disordered protein, multi-domain proteins, protein dynamics
Citation: Ciragan A, Backlund SM, Mikula KM, Beyer HM, Samuli Ollila OH and Iwaï H (2020) NMR Structure and Dynamics of TonB Investigated by Scar-Less Segmental Isotopic Labeling Using a Salt-Inducible Split Intein. Front. Chem. 8:136. doi: 10.3389/fchem.2020.00136
Received: 18 November 2019; Accepted: 13 February 2020;
Published: 19 March 2020.
Edited by:
Julio A. Camarero, University of Southern California, United StatesReviewed by:
Irina Bakunina, G. B. Elyakov Pacific Institute of Bioorganic Chemistry, Far Eastern Branch (RAS), RussiaDavid Cowburn, Albert Einstein College of Medicine, United States
Alexander Shekhtman, University at Albany, United States
Copyright © 2020 Ciragan, Backlund, Mikula, Beyer, Samuli Ollila and Iwaï. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hideo Iwaï, hideo.iwai@helsinki.fi