Synthesis of Ni/NiAlOx Catalysts for Hydrogenation Saturation of Phenanthrene
 Dao-Cheng Liu
Dao-Cheng Liu Yu Chen1,2
Yu Chen1,2  Jie-Ying Jing
Jie-Ying Jing Antony Rajendran
Antony Rajendran Wen-Ying Li
Wen-Ying Li- 1State Key Laboratory of Clean and Efficient Coal Utilization, Taiyuan University of Technology, Taiyuan, China
- 2Key Laboratory of Coal Science and Technology Ministry of Education, Taiyuan University of Technology, Taiyuan, China
- 3State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering, Ningxia University, Yinchuan, China
The saturation of octahydrophenanthrene was the rate-determining step in the hydrogenation process from phenanthrene to perhydrophenanthrene, which was due to the steric hindrance and competitive adsorption of octahydrophenanthrene. In this work, a series of Ni/NiAlOx catalysts with a uniform electron-deficient state of Ni derived from the nickel aluminate structure was synthesized to overcome the disadvantage of noble catalyst and the traditional sulfided catalysts in the saturation hydrogenation process of phenanthrene. Results showed that the catalyst calcinated at 650°C possessed more Ni2+ (∼98%) occupying octahedral sites and exhibited the highest robs (1.53 × 10−3 mol kg−1 s−1) and TOF (14.64 × 10−3 s−1) for phenanthrene hydrogenation. Furthermore, its ability to overcome steric hindrance and promote the rate-determining step was proven by octahydrophenanthrene hydrogenation. Comparing the evolution of hydrogenation activity with the change in the electronic structure of surface Ni sites, it was shown that the increase of metallic electron deficiency hindered the π-back bonding between surface Ni and aromatic rings, which was unfavorable for aromatic adsorption. As a result, the phenanthrene hydrogenation saturation performance can be enhanced by stabilizing the electron-deficient state of surface Ni on an optimal degree.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are predominantly present in coal tar, whose direct discharge and insufficient combustion would cause environmental pollution (Hayakawa et al., 2000; Li and Suzuki 2010). Therefore, the treatment of PAHs has gained undivided attention, and hydrogenation of PAHs is an attractive method as it effectively yields the cycloalkanes which can be blended into the jet fuels to improve the volumetric calorific value and combustion performance (Yoon et al., 1996; Zhang et al., 2018; Jing et al., 2020; Jia et al., 2021). However, it is quite difficult to achieve complete saturation while performing the hydrogenation of PAHs with more aromatic rings, and in particular, the saturation of the final aromatic ring is more difficult (Beltramone et al., 2008). To solve this problem, a variety of catalysts have been attempted to promote the hydrogenation of PAHs using phenanthrene (PHE) as a typical PAH compound. PHE has three aromatic rings, and its hydrogenation process is typically exothermic and consecutive. During the hydrogenation of PHE, the complete saturation, that is, the higher selectivity of perhydrophenanthrene (PHP), is rarely reported, with the exception of very costly noble bimetallic catalyst requiring the higher metal loadings (2.0 wt% Pt and 5.4 wt% Pd) (Qian et al., 1999). Considering the traditional sulfide catalysts (Ni(Co)Mo(W)S), the complete saturation of PHE is still a big challenge despite a list of strategies having been attempted (Fu et al., 2015; Jiang et al., 2017; Luo et al., 2018; Wang D et al., 2019). The complete saturation of PHE is determined by the hydrogenation of symmetric-octahydrophenanthrene (s-OHP) to PHP (the rate-determining step), whose rate is limited by the natural steric hindrance and unfavorable competitive adsorption of s-OHP between partially hydrogenated aromatics (Korre et al., 1995; Beltramone et al., 2008). In this milieu, the catalysts which are low cost without compromising the higher hydrogenation efficiency, especially for the conversion of s-OHP to PHP, have received much attention in the literature, but such kind of catalysts are still scarce.
Being the familiar low-cost hydrogenation catalysts with an excellent H2 dissociation capacity, Ni-based catalysts always seem to be the alternative choice for the group-VIII noble metal catalysts (Pd and Pt) (Hammer and Norskov 1995; De et al., 2016). However, though it accomplishes the excellent conversion of PHE, the Ni-based catalysts still struggle to attain the higher selectivity of PHP during the hydrogenation. For instance, the typical Ni/Al2O3 catalyst displays 97.9% conversion of PHE but achieves a very low selectivity of PHP (20.8%) (Nuzzi and Marcandalli 2003). This is because the adsorption energy of polycyclic aromatic hydrocarbons is higher over Ni (111) surface than that over Pt (111) (Yazdi et al., 2017). In line with the general adsorption mechanism of aromatics over the metallic surfaces, the adsorption of PHE over the Ni surface can be improved through σ bonding (electronic transfer from filled π molecular orbital of aromatics to atomic vacant d orbital of metallic Ni) and through a π-back bonding (electronic back-donation from filled atomic d orbital of Ni to vacant π* molecular orbital of aromatics) (Stanislaus and Cooper 1994). This underlines the importance of electron deficiency in metallic Ni active sites to establish the improved adsorption of PHE through σ bonds, which in turn directs to look for the method to tune the electronic properties of Ni active sites.
The electron transfer driven by differences in the Fermi level of the metal nanoparticles and the support is an important phenomenon in metal-support interaction (MSI). Therefore, enhancing the MSI would favorably influence the electronic properties of metallic active sites (van Deelen et al., 2019). NiAl2O4 spinel-based catalysts seem to be attractive because they offer the enhanced MSI between reduced Ni and oxide support due to the confinement effect of spinel precursors. As a result, the improved metallic Ni dispersion (Srifa et al., 2018), decreased practical size Rubén et al. (2012), and good resistance to coke formation (Salhi et al., 2011; Suffredini et al., 2017) can be envisaged. Deviating from the typical spinel structure, Ni2+ can occupy both the tetrahedral (Nitetra) and octahedral cation sites (Niocta) in NiAl2O4, that is, an inverse spinel structure. Furthermore, Niocta sites are essential as they exhibit strong interaction and better reducibility (Yancheshmeh et al., 2020).
Based on the above facts, in this work, a series of nickel aluminate–based catalysts with more Niocta sites were synthesized by the modified sol–gel method using citric acid. The facile method that varies calcination temperatures was selected to adjust Ni2+ distribution and the electronic state of Ni active sites based on the enhanced MSI. The catalytic activities of the prepared catalysts were evaluated in the hydrogenation of PHE as well as s-OHP, which provided the insights into the potential of present catalysts to overcome the steric hindrance associated with the hydrogenation of PAHs. Moreover, the role of electron deficiency in Ni active sites was emphasized and discussed.
Experiment
Preparation of Catalysts
Nickel aluminate–based catalysts were prepared by the modified sol–gel method using citric acid. Ni(NO3)2·6H2O and Al(NO3)3·9H2O were taken in a certain molar ratio of Ni:Al (1.45:2). The molar ratio of citric acid and water to metal ions (Ni2+ and Al3+) was 1:1 and 40:1, respectively. Ni(NO3)2·6H2O, Al(NO3)3·9H2O, and citric acid were initially dissolved in deionized water and stirred at room temperature for 3 h. Stirring was further continued but at 80°C for 6 h to ensure the condensation reaction. The resulting solid was kept overnight at room temperature and sequentially dried at two different conditions (100°C for 12 h and 120 °C for 12 h, yielding a green fluffy xerogel). The obtained solids were calcinated at different temperatures (550, 600, 650, 700, and 750°C) for 2 h in air and subsequently reduced at 520 °C for 5 h under H2 flow to produce the five different Ni/NiAlOx-T catalysts (T: calcination temperature). Pure NiO and Al2O3 were also prepared by exactly following the abovementioned sol–gel method and calcination procedure (650°C for 2 h), and mechanically mixed together with the same Ni/Al molar ratio to prepare the catalyst for comparison (NiO + Al2O3-mixed).
Characterization
The powder X-ray diffraction (XRD) patterns of samples were recorded on a Rigaku Ultima IV multifunctional X-ray diffractometer (Rigaku, Japan) in the range of 10–90° at a rate of 4°min−1. For this, the monochromatic Cu Kα radiation was generated at 40 kV and 40 mA. The Scherrer formula was applied on the powder XRD data to calculate the crystallite size. Varian Cary 300 was used to record the diffuse reflectance UV-Vis spectra of powder samples in the range of 300–800 nm. The H2 temperature-programmed reduction (TPR) experiment was performed on Micromeritics AutoChem II 2920 analyzer. 0.1 g of the sample was pretreated at Ar atmosphere (300°C at 2 h) and then heated to 900°C at 10 °C min−1 in the presence of 10 vol% H2/Ar at a flow rate of 25 ml min−1. The transmission electron microscope (TEM) images of the samples were captured using FEI Tecnai G2 F30 to explore the morphologies of the as-synthesized catalysts at an accelerate voltage of 200 kV. For the calculation of particle size distribution, at least 300 different points of Ni particles were considered. The textural properties of the as-synthesized catalysts were analyzed using N2 adsorption–desorption measurements (Quantachrome Autosorb-iQ instrument) in a liquid N2 bath. The specific surface area (SBET) was estimated by Brunauer–Emmett–Teller (BET) equation. The microporous and mesoporous surface area values (Smic and Smeso) were estimated by t-plot. Fourier transform infrared spectra of the samples using CO as a probe molecule (CO-FTIR) were acquired on a Bruker TENSOR 27 spectrophotometer (4,000–400 cm−1, resolution 2 cm−1) equipped with an in situ stainless steel reactor. 10 mg catalyst was pressed into a wafer (Φ = 12.6 mm) and reduced in situ at 520°C for 5 h in H2 flow (50 ml min−1). The sample was cooled to 40°C under Ar, and then the gas flow was changed to 5 vol% CO/Ar that was maintained for 1 h to ensure the maximum CO adsorption. Prior to the analysis, the physically absorbed CO was purged under Ar flow (40 ml min−1). X-ray photoelectron spectra (XPS) of the samples were carried out on a Kratos analytical AXIS SUPRA equipped with an Al Kα (X-ray source). The CO pulse chemisorption was performed on Micromeritics AutoChem II 2920 instrument. 15 mg of the sample was put in a quartz tube and reduced in situ at 520°C for 5 h in H2 flow (50 ml min−1). After stabilizing the temperature of the reactor at 40°C under Ar flow, 5 vol % CO/Ar was pulsed into the reactor and repeated for 40 times with the sampling loop capacity of 0.1 ml. The element content of the as-synthesized samples was detected by inductively coupled plasma optical emission spectrometer (ICP‒OES, Agilent 5110).
Catalytic Activity
The hydrogenation of PHE was carried out in a fixed-bed continuous-flow stainless steel reactor with 10 mm internal diameter and 500 mm length. The dried catalyst (60–80 mush) was diluted with silica sand to 2 mL before being loaded into the reactor. Both the ends of the catalyst bed were filled with additional silica sand of 2 mL. The catalyst was directly reduced in the fixed-bed reactor by heating to 520°C at a rate of 3°Cmin−1 in a H2 flow (50 ml min−1) and kept for 5 h. Then, the catalyst was naturally cooled to reaction temperature in a continuous H2 flow. The operating conditions for hydrogenation of PHE were as follows: total pressure (5.0 MPa), temperature (280°C), feed rate of 1.0 wt% PHE in decalin (6 ml h−1), flow rate of H2 (60 ml min−1), and the weight hourly space velocity (WHSV) of 52 h−1. The effect of external and internal diffusion was checked by adjusting the mass flow rate and particle diameter, respectively. The product was collected and analyzed using Shimadzu GC-2010 installed with RTX-5 column and flame ionization detector (FID). The products were further qualitatively analyzed with GC equipped with a mass spectrometer (Agilent GC-MS).
To investigate the effect of steric hindrance on hydrogenation of PHE, s-OHP was selected as the model compound instead of PHE, and the abovementioned experimental procedure was exactly followed. For the investigation of intrinsic activity of catalysts, the conversion of PHE and s-OHP was kept low by changing the values of WHSV.
Herein, the intrinsic hydrogenation activity of catalysts was observed in terms of the reaction rate (robs, mol s−1kg−1) using Eq. 1.
where F is the reactant molar flow (mol s−1), x is the PHE conversion (%) or the conversion of s-OHP into PHP (%), and W is the catalyst mass (kg).
The hydrogenation turnover frequency (TOF, s−1) values of PHE and s-OHP were calculated from Eq. 2.
where F is the reactant molar flow (mol s−1), x is the PHE conversion (%) or the conversion of s-OHP into PHP (%), NNi is the number of Ni atoms in the catalyst (mol), and fNi is the dispersion of Ni detected by CO pulse adsorption (%).
Results and Discussion
Properties of the As-Synthesized Catalysts
Element Composition and Textural Properties
The actual concentration of nickel and aluminum in the prepared spinel-based catalysts was determined by ICP-OES, and the actual Ni/Al ratio was accordingly calculated (Table 1). All the prepared catalysts displayed the same Ni/Al molar ratio which is equal to the theoretical Ni/Al ratio. This indicated that no loss of Al or Ni ions occurred during the preparation process regardless of calcination temperature. This also led to the assumption that the different physiochemical properties of prepared nickel aluminate samples might be attributed to the variation in the distribution of Niocta and Nitetra with the variation of calcination temperature. The Ni/Al molar ratio (0.73) of the prepared nickel aluminate samples was higher than that of typical nickel aluminate (NiAl2O4, Ni/Al molar ratio was 0.5), indicating that there were additional Ni2+ ions in the present spinel structures. This could be due to the inversion structure of the nickel aluminate spinel structure, in which the additional Ni2+ ions were occupied in the octahedral sites to provide more desirable active sites upon the reduction.
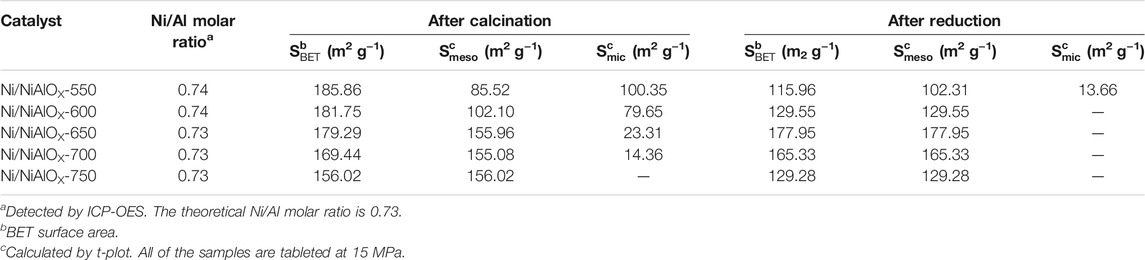
TABLE 1. Ni/Al molar ratio and surface properties of Ni/NiAlOX catalysts with different calcination temperatures.
N2 physisorption was carried out to determine the SBET, Smicro, and Smeso of synthesized catalysts. All the analyzed samples displayed the hysteresis loops corresponding to the mesoporous structure with no significant deviation in the pore diameter values. While increasing the calcination temperature of samples, the SBET value was decreased from 185.86 to 156.02 m2/g, whereas the Smeso value was increased with a loss of Smicro and became stagnant after a certain calcination temperature (650°C) (Table 1). Compared to the reported nickel aluminate, the obtained higher surface area and porosity of the samples could be attributed to the evolution of CO2 from citric acid (Rogers et al., 2016; Miryam et al., 2017; Jing et al., 2018). Considering the textural properties of reduced catalysts, Ni/NiAlOX-650 exhibited the highest mesoporous surface area (177.95 m2/ g) by which Ni/NiAlOX-650 might possess a higher performance in the hydrogenation of PHE with a better mass transfer.
Analysis of Ni Occupation
In order to confirm the Ni occupation in the as-prepared catalyst, we conducted XRD characterization. Figure 1 displayed the powder XRD patterns of the samples before and after reduction. Compared with the standard peaks of NiO (PDF#78-0643) and NiAl2O4 (PDF#78-1601), the peaks of the samples calcined at the lower temperature (<750oC) were shifted (Figure 1a), which was related to the expansion in the nickel aluminate spinel structure. The expansion was derived from the replacement of smaller Al3+ (0.54 Å) by larger Ni2+ (0.69 Å) ions, especially at the octahedral sites. This phenomenon indicated that major Ni2+ ions occupied at the octahedral sites in the spinel structure. The occupancy of Ni2+ ions predominantly at the octahedral sites in the spinel structure can be evidenced by the larger intensity of the peak (45.0°) corresponding to the presence of Niocta sites as well as no intensity of the peak (31.4°) corresponding to the presence of Nitetra sites (Han et al., 2004; Tirsoaga et al., 2011; Miryam et al., 2017). On the other hand, for the sample prepared by calcining at 750oC, the appearance of characteristic powder XRD peaks of NiAl2O4 and NiO species is apparent. Especially, the diffraction peak at 31.4°, which belonged to Nitetra, was obvious. It indicated that the higher calcination temperature promoted the occupancy of tetrahedral sites by Ni2+ ions (O'Neill et al., 1991). Meanwhile, the separation of NiO and Nitetra structures occurred at the higher calcination temperature. DR UV-Vis spectra of the calcined samples (Supplementary Figure S1) further proved this conclusion. The absorption bands of the stoichiometric NiAl2O4 spinel structure at 642 and 597 nm were not seen for the samples calcined less than 700oC. The intensity of these peaks was weak for the sample calcined at 700oC and became relatively strong in the case of the sample calcined at 750oC. This was in accordance with the XRD results and further indicated the formation of the stoichiometric NiAl2O4 spinel structure with Ni2+ occupying Nitetra sites in the samples calcined at temperature (≥700°C) according to the ion-exchange reaction
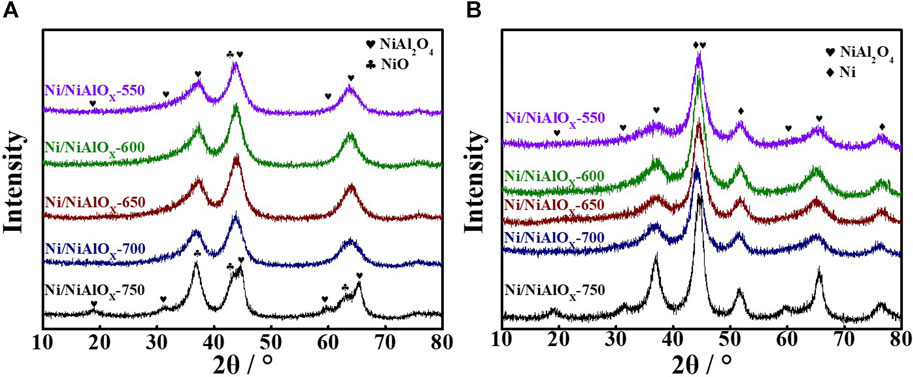
FIGURE 1. Powder XRD spectra of Ni/NiAlOx catalysts after calcination (A) and reduction (B) at different temperatures.
After the reduction of calcined samples at 520oC, the reduced samples were characterized by the powder XRD analysis (Figure 1b). The characteristic peak of metallic Ni species (PDF#65-2865) appeared with the obvious decrease in the intensity of the characteristic peak corresponding to Niocta sites. However, though the obvious decrease in the peak intensity was noticed, there were still the characteristic peaks of the nickel aluminate structure seen in Figure 1b. This suggested that those unreduced Ni2+ ions were present in the bulk nickel aluminate phase and served as the support. The peak corresponding to Ni(200) surface was considered for measuring the crystallite size (3 nm) of metallic Ni species by the Scherrer formula. This small crystallite size can be assigned to the strong MSI experienced by the predominant Niocta sites in the calcined samples, and these Ni2+ ions are supposedly reduced to small Ni0 sites with a higher dispersion.
Quantification Analysis of Different Ni2+ Species
From the H2-TPR (Figure 2) analysis of calcined samples, the quantification of different Ni2+ was accomplished on the basis of Ni–support interaction (reducibility of different Ni2+ species). The H2-TPR data of bare Al2O3 prepared by the same modified sol–gel method was also given to make sure the observed TPR peaks belong to the reduction of Ni2+ species. In general, for the nickel aluminate samples, the reducible Ni2+ ions were categorized into four types by reduction temperatures, such as the bulk or free NiO, NiO bonded to Al2O3, and Ni2+ ions occupying the crystalline spinel structure of NiAl2O4 (Niocta or Nitetra sites) (Zhou et al., 2015; Li et al., 2019). According to the qualitative analysis of XRD and DR UV-Vis data discussed above, the different Ni2+ ions found in the prepared nickel aluminate samples could be free NiO or NiO bonded to nickel aluminate, Niocta and Nitetra of the NiAl2O4 spinel structure. As compared to the literature, the reduction temperature for NiO seemed higher in Figure 2, suggesting the interaction of formed NiO species with nickel aluminate (Yu et al., 2019). In comparison to samples calcined at other temperatures, the reduction peak corresponding to NiO was negligible for the sample calcined at 650oC. Furthermore, this particular sample calcined at 650oC contains more Niocta sites (98%) of the spinel structure according to the intensity of reduction peak observed at 658°C. For the samples calcined at the temperature above 650oC, the reduction peak corresponding to Nitetra appeared. Moreover, the increase in the intensity of reduction peak corresponding to NiO bonded to nickel aluminate was noticed. Thus, H2-TPR analysis proved the decrease of the inversion parameter in the spinel structure of nickel aluminate with the increase of calcination temperature above 650oC (O'Quinn et al., 2017).
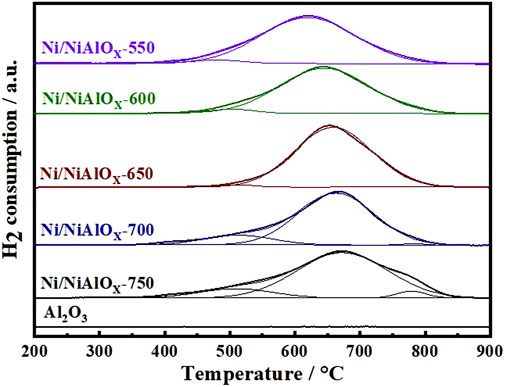
FIGURE 2. H2-TPR profiles of Ni/NiAlOy catalysts with different calcination temperatures.
Electronic Structure
X-ray photoelectron spectroscopy analyses were carried out to identify the electronic state of Ni on the surface of reduced catalysts (Figure 3). The characteristic Ni0 2p3/2, Ni2+ 2p3/2, and the satellite peak of Ni2+ 2p3/2 usually appeared at the binding energy of 852.5, 856.0, and 861.8 eV, respectively (Yancheshmeh et al., 2020). These three peaks were observed in the XPS analysis of the present catalysts. Compared to the sample prepared by mechanical mixing (852.32 eV), the distinctive peak of Ni0 2p3/2 appeared at the higher binding energy for Ni/NiAlOx catalysts due to their electron-deficient Ni sites. As was discussed above, the electron-deficient state of Ni sites in the prepared catalysts could be advantageous for promoting the activation of aromatics through adsorption.
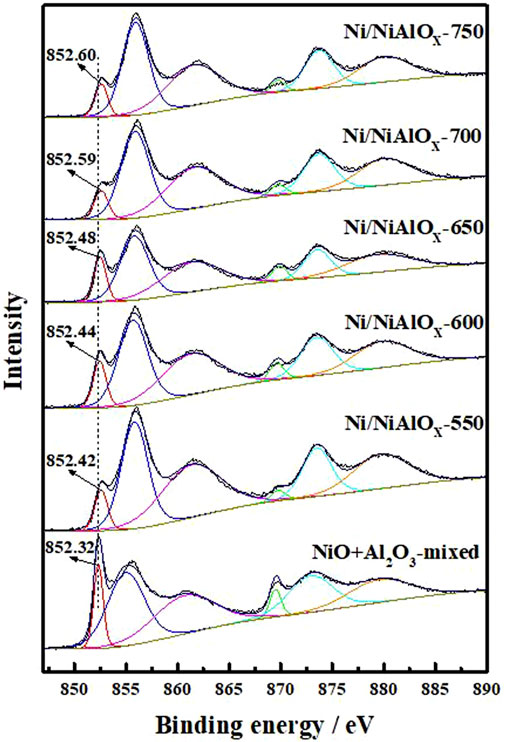
FIGURE 3. XPS spectra in the Ni
Catalytic Hydrogenation Performance
Hydrogenation Activity
Hydrogenation of PHE was performed under optimal reaction conditions (300°C, 5 MPa, a H2/oil volume ratio of 600, and WHSV of 52 h−1) to find out the best catalyst among the as-synthesized catalysts. The change in selectivity of PHP with reaction time during the hydrogenation of PHE over the attempted catalysts is shown in Figure 4, indicating PHP as the major product without the formation of any gas and ring opening products. All the Ni/NiAlOX catalysts presented remarkable initial activity both in terms of PHE conversion (∼99%) and PHP selectivity (∼97%) even under high WHSV (52 h−1) due to the improved adsorption of aromatics that was promoted by the electron-deficient Ni sites. Ni/NiAlOX-650 reveals a comparatively higher hydrogenation activity than the other Ni/NiAlOX-T catalysts due to its metallic Ni sites predominantly produced from Niocta sites of the calcined form which expose the strong MSI. As evidenced by the CO-pulse adsorption data (Table 2), the strong MSI advantageously provided the higher Ni dispersion (1.99%) in Ni/NiAlOx-650 catalysts. The relatively higher Smeso of Ni/NiAlOX-650 (Table 1) could also contribute to the higher performance Ni/NiAlOX-650 in the hydrogenation of PHE.
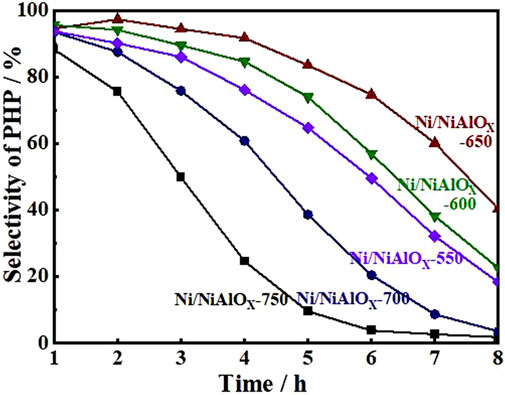
FIGURE 4. Selectivity of perhydro-phenanthrene over Ni/NiAlOx catalysts calcinated at different temperatures.

TABLE 2. Metal dispersion and reactivity of Ni/NiAlOX catalysts with different calcination temperatures.
Function of the Electron-Deficient State
To explain the gradual decrease of PHP selectivity (Figure 4) with an increase of s-OHP selectivity during the PHE hydrogenation, the characterization of spent catalyst was important. The comparative of XPS analysis of fresh and spent Ni/NiAlOX-650 was shown in Figure 5. The characteristic binding energy (Ni0 2p3/2) corresponding to metallic Ni species has been shifted to higher binding energy for the spent Ni/NiAlOX-650 catalyst, which ranges from 852.4 to 852.7 eV, indicating the increase of electron deficiency of Ni0 sites during the PHE hydrogenation. For further insights about the role of the electronic structure of Ni0 species in PHE hydrogenation, the change in the electronic structure of surface Ni0 species after the PHE hydrogenation was studied by FT-IR analysis using CO as a probe molecule (Figure 6). In comparison, the upward shift, that is, 2050 cm−1 to 2064 cm−1 (spent catalyst at 4 h) and 2067 cm−1 (spent catalyst at 8 h), is observed in the characteristic stretch of linear CO absorption after PHE hydrogenation due to the weakening of interaction between surface Ni0 sites and CO (Yang et al., 2017). This can be attributed to the enhancing trend of electron deficiency of Ni0 sites during the PHE hydrogenation progress, which hampered the π-back bonding from Ni0 to CO (Lee and Oyama 2006), as supported by XPS results. Contrary to linear CO adsorption, no obvious shift was noticed in the peaks corresponding to the bridged CO adsorption (1936 cm−1 and 1874 cm−1) but the intensity of such peaks is apparently decreased. In particular, the bridged CO adsorption peak completely disappeared in the CO FT-IR spectrum of spent Ni/NiAlOX-650 catalysts for 8 h. This suggested that the Ni0 sites in the spent Ni/NiAlOX-650 catalysts became completely unsuitable to establish the CO absorption through bridging mode.
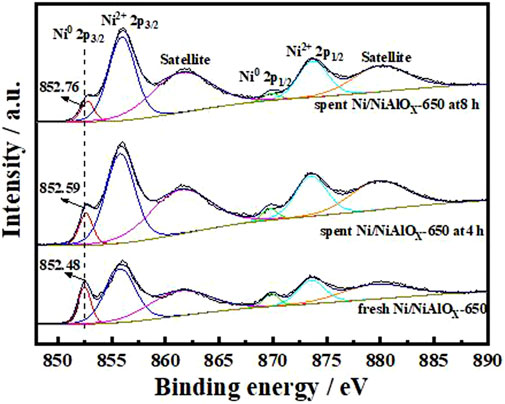
FIGURE 5. XPS spectra in the Ni
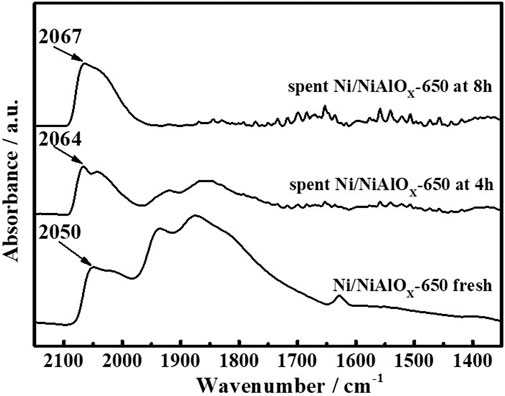
FIGURE 6. CO-FTIR of Ni/NiAlOx-650 catalysts with different reaction times in the PHE hydrogenation process.
To ensure that the decrease in activity during the progress of PHE hydrogenation occurred primarily by the change in the electronic structure, TEM analysis of the spent catalysts have been performed (Figure 7; Yang et al., 2017). According to TEM results, the fresh and spent Ni/NiAlOX-650 catalysts displayed the same pattern of lattice fringes with 0.204 nm lattice spacing that corresponds to Ni(111) plane, indicating that the geometric structure had no impact in inhibiting the PHE hydrogenation with time. Expecting the same adsorption mechanism for aromatics like CO over Ni0 sites (Lee and Oyama 2006), the charge transfer between surface Ni and aromatic rings could be reduced by the enhanced electron-deficiency of Ni0 sites, which was unfavorable for aromatic activation.
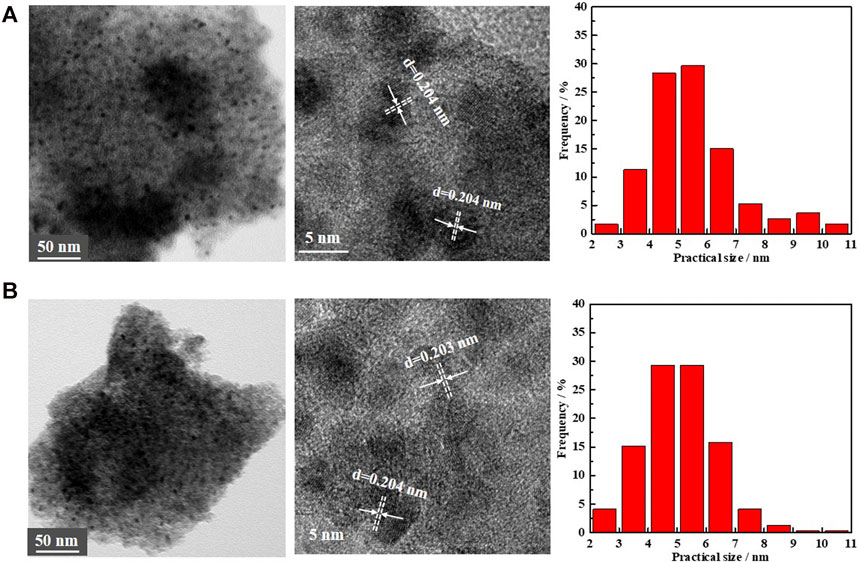
FIGURE 7. TEM images and particles size distribution of the (A) fresh Ni/NiAlOx-650 catalysts and (B) spent Ni/NiAlOx-650 catalyst.
Hydrogenation Mechanism Exploration
To correlate the electronic properties of Ni0 sites with the PHP selectivity at different reaction times, a plot is made between the binding energy of Ni0 2p3/2 in different catalysts (Figure 8). To support this study, a catalyst prepared from the reduction of a mechanical mixture containing NiO and Al2O3 (NiO + Al2O3-mixed) was taken as reference, whose binding energy (852.38 eV) was considered as a null point for the comparison to derive the shift in binding energy of Ni0 2p3/2. As expected, the PHP selectivity was zero while using the NiO + Al2O3-mixed catalyst evaluated at the same hydrogenation conditions. For the comparison, the instant selectivity (1 h) as well as the end selectivity (8 h) of PHP while using different catalysts have been considered. According to Figure 8, the binding energy of Ni0 2p3/2 in the range of 852.4–852.6 eV, that is, 0.1–0.3 eV shift from the actual binding energy of NiO + Al2O3-mixed, was essential, and that provided the favorable electron deficiency of Ni0 sites to enable better PHP selectivity (>90%). This big difference in activity caused by small changes in the electron state could be understood to seek the match of orbital energy levels. The electron-deficient state could improve the electronic donation from aromatics to Ni atom for σ bonding. However, an excessive deficient degree would limit the filling of electrons in antibonding orbitals to form π-back bonding.
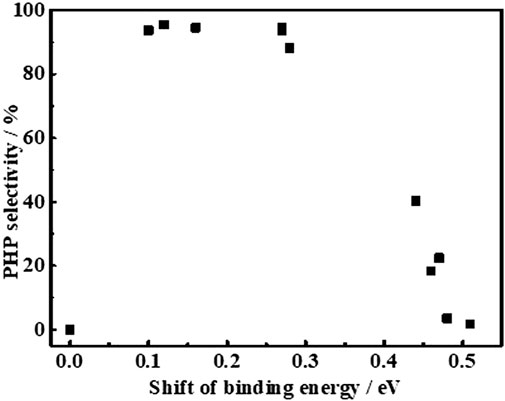
FIGURE 8. Relationship between shifts of Ni°
The TEM image of spent Ni/NiAlOX-650 (Figure 7) indicated the nondisruptive dispersion of Ni0 active sites during the PHE hydrogenation, indicating the absence of sintering of Ni0 active sites. Meanwhile, the Raman spectrum of spent Ni/NiAlOX-650 catalysts for 8 h (Supplementary Figure S2) rules out the formation of carbon deposition on the catalyst surface during PHE hydrogenation. Therefore, it was confirmed that the decrease in PHE hydrogenation capacity of studied catalysts occurred primarily due to the unfavorable increase of electron deficiency in Ni0 active sites.
The steric hindrance and competitive adsorption associated with the hydrogenation of s-OHP made it a major product in the main product of PHE hydrogenation Korre et al. (1995), Beltramone et al. (2008), Fu et al. (2015), Wang M et al. (2019), and thus, s-OHP conversion to PHP is regarded as the rate-determining step. Using s-OHP as the model compound, the real benefit of adequate electron deficiency in Ni0 sites in the hydrogenation reactions was highlighted. For the hydrogenation of s-OHP to PHP, the employed reaction conditions were as follows: 300°C, 5 MPa (H2), and H2/oil ratio of 600. The effects of internal and external diffusion were excluded by changing the mass flow rate and grain size, respectively. A carbon balance is attained over 96%. Figure 9 exhibited the change of s-OHP conversion and product selectivity with WHSV over Ni/NiAlOX-650 catalysts, representatively. Three simultaneous reaction processes of s-OHP over the Ni/NiAlOX-650 catalysts were predicted: saturation to PHP, isomerization to as-OHP, and dehydrogenation to THP and DHP. With the decrease of s-OHP, the content of dehydrogenation and isomerization yields in the product mixture was basically unchanged, but the content of PHP was gradually increased, which was almost equal to the conversion rate of s-OHP. These results proved that the conversion of s-OHP to PHP occurred through the hydrogenation route over Ni/NiAlOX-650 catalyst.
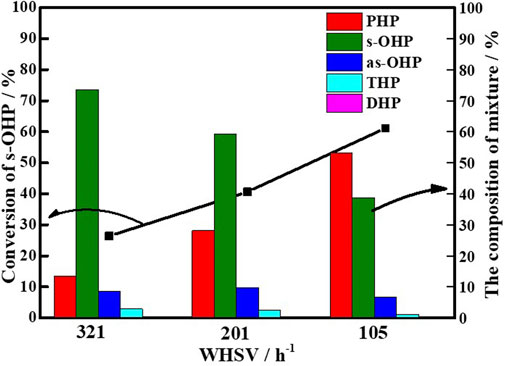
FIGURE 9. Hydrogenation of s-OHP over Ni/NiAlOx-650 catalysts with different weight hourly space velocity (WHSV).
The obvious reaction rate (robs) and the turnover frequency (TOF) in the performed s-OHP were calculated at the lower conversion (<20%) and given in Table 3. Ni/NiAlOX-650 showed a higher robs (0.64 × 10−3 mol kg−1 s−1) and TOF (4.65 × 10−3 s−1) than the other as-synthesized catalysts, and the activity trend of different catalysts was similar to that observed in the PHE hydrogenation. The higher efficiency of Ni/NiAlOX-650 could be assigned to the abundance of Niocta sites after its calcination, which promoted the dispersion of Ni0 sites and provided the more suitable electron deficiency.
The TOF of Ni/NiAlOx-650 in PHE hydrogenation was calculated and compared with that of the catalysts already reported in the literature (Table 3). Comparatively, Ni/NiAlOX-650 (WHSV of 52 h−1) accomplished a much higher average PHP selectivity (79.6%) than the other catalysts. For the robs and TOF calculation, WHSV was increased to 520 h−1 to obtain the lower PHE conversion (<20%). At this condition, the calculated value of robs and TOF is 1.53 × 10−3 mol kg−1 s−1 and 14.64 × 10− s−1, respectively. These values were relatively higher than that reported for various catalysts, indicating the significance of Ni/NiAlOX-650 possessing the suitable electron-deficient Ni0 sites.
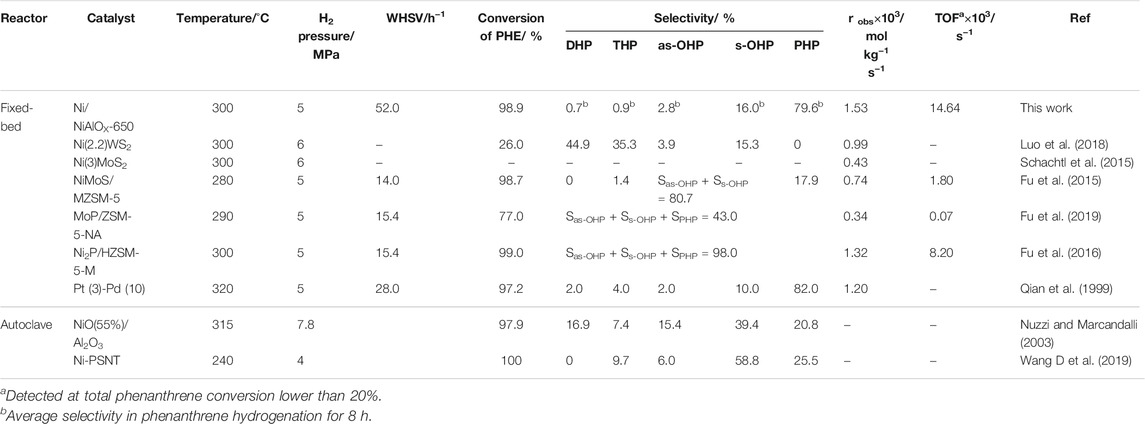
TABLE 3. Comparison of the catalytic performance of Ni/NiAlOX-650 with the literature in the deep hydrogenation of phenanthrene.
Conclusion
The electronic properties of relatively low-cost metallic active site (Ni) were tuned by the facile calcination method to attain the improved activity with a better selectivity of perhydro-products during phenanthrene hydrogenation. It was found that nickel occupation in the nickel aluminate–based catalyst could be regulated by varying the calcination temperatures (550-750oC) of the catalyst’s precursor. When the calcination temperature was 650°C, around 98% octahedral Ni2+ sites were formed, which were relatively easily reduced and constructed the favorable metal–support interaction that improved the electron deficiency degree of Ni active sites in the catalyst (Ni/NiAlOx-650) obtained after reduction. Phenanthrene hydrogenation evaluation showed that Ni/NiAlOx-650 displayed the improved initial selectivity of perhydrophenanthrene (98%) along with a higher TOF (1.53 × 10−3 mol kg−1 s−1) at 300°C, 5 MPa (H2). The characterization of the spent catalysts indicated that electron-deficient degree of Ni active sites played an important role in phenanthrene hydrogenation, which overcomes the steric hindrance and competitive adsorption of octahydrophenanthrene.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Author Contributions
D-CL, J-YJ, and W-YL conceived the research and supervised the whole work. D-CL and YC prepared the catalysts and characterized the samples. AR and H-CB contributed to the analysis of the hydrogenation mechanism. All authors contributed to the manuscript writing and editing, and approved the submitted version.
Funding
We acknowledge the financial support obtained from the National Natural Science Foundation of China (21978190, 22038008), and the Fund for Shanxi 1331 Project and Foundation of State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering (2020-KF-08).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.757908/full#supplementary-material
References
Beltramone, A. R., Resasco, D. E., Alvarez, W. E., and Choudhary, T. V. (2008). Simultaneous Hydrogenation of Multiring Aromatic Compounds over NiMo Catalyst. Ind. Eng. Chem. Res. 47, 7161–7166. doi:10.1021/ie8004258
De, S., Zhang, J., Luque, R., and Yan, N. (2016). Ni-based Bimetallic Heterogeneous Catalysts for Energy and Environmental Applications. Energy Environ. Sci. 9, 3314–3347. doi:10.1039/c6ee02002j
Elias, I., Soon, A., Huang, J., S. Haynes, B., and Montoya, A. (2019). Atomic Order, Electronic Structure and Thermodynamic Stability of Nickel Aluminate. Phys. Chem. Chem. Phys. 21, 25952–25961. doi:10.1039/c9cp04325j
Fu, W., Zhang, L., Wu, D., Xiang, M., Zhuo, Q., Huang, K., et al. (2015). Mesoporous Zeolite-Supported Metal Sulfide Catalysts with High Activities in the Deep Hydrogenation of Phenanthrene. J. Catal. 330, 423–433. doi:10.1016/j.jcat.2015.07.026
Fu, W., Zhang, L., Wu, D., Yu, Q., Tang, T., and Tang, T. (2016). Mesoporous Zeolite ZSM-5 Supported Ni2P Catalysts with High Activity in the Hydrogenation of Phenanthrene and 4,6-Dimethyldibenzothiophene. Ind. Eng. Chem. Res. 55, 7085–7095. doi:10.1021/acs.iecr.6b01583
Fu, W., Zhao, W., Zhang, L., Zhang, T., Tang, T., and Chen, Q. (2019). ZSM-5 Microspheres Consisting of Nanocrystals for Preparing Highly Dispersed MoP Clusters with Good Activity in Phenanthrene Hydrogenation. Ind. Eng. Chem. Res. 58, 17289–17299. doi:10.1021/acs.iecr.9b03477
Hammer, B., and Norskov, J. K. (1995). Why Gold Is the Noblest of All the Metals. Nature 376, 238–240. doi:10.1038/376238a0
Han, Y. S., Li, J. B., Ning, X. S., and Chi, B. (2004). Effect of Preparation Temperature on the Lattice Parameter of Nickel Aluminate Spinel. J. Am. Ceram. Soc. 87, 1347–1349. doi:10.1111/j.1151-2916.2004.tb07733.x
Hayakawa, K., Murahashi, T., Akutsu, K., Kanda, T., Tang, N., Kakimoto, H., et al. (2000). Comparison of Polycyclic Aromatic Hydrocarbons and Nitropolycyclic Aromatic Hydrocarbons in Airborne and Automobile Exhaust Particulates. Polycyclic Aromatic Comp. 20, 179–190. doi:10.1080/10406630008034784
Jia, T., Zhang, X., Liu, Y., Gong, S., Deng, C., Pan, L., et al. (2021). A Comprehensive Review of the thermal Oxidation Stability of Jet Fuels. Chem. Eng. Sci. 229, 116157. doi:10.1016/j.ces.2020.116157
Jiang, Y., Wang, D., Li, J., Li, M., Pan, Z., Ma, H., et al. (2017). Designing MoS2 Nanocatalysts with Increased Exposure of Active Edge Sites for Anthracene Hydrogenation Reaction. Catal. Sci. Technol. 7, 2998–3007. doi:10.1039/c7cy01026e
Jing, J.-y., Zhang, X.-w., Li, Q., Li, T.-y., and Li, W.-y. (2018). Self-activation of CaO/Ca3Al2O6 Sorbents by Thermally Pretreated in CO2 Atmosphere. Appl. Energ. 220, 419–425. doi:10.1016/j.apenergy.2018.03.069
Jing, J.-Y., Wang, J.-Z., Liu, D.-C., Qie, Z.-Q., Bai, H.-C., and Li, W.-Y. (2020). Naphthalene Hydrogenation Saturation over Ni2P/Al2O3 Catalysts Synthesized by Thermal Decomposition of Hypophosphite. ACS Omega 5, 31423–31431. doi:10.1021/acsomega.0c05019
Korre, S. C., Klein, M. T., and Quann, R. J. (1995). Polynuclear Aromatic Hydrocarbons Hydrogenation. 1. Experimental Reaction Pathways and Kinetics. Ind. Eng. Chem. Res. 34, 101–117. doi:10.1021/ie00040a008
Lee, Y., and Oyama, S. (2006). Bifunctional Nature of a SiO2-Supported Ni2P Catalyst for Hydrotreating: EXAFS and FTIR Studies. J. Catal. 239, 376–389. doi:10.1016/j.jcat.2005.12.029
Li, C., and Suzuki, K. (2010). Resources, Properties and Utilization of Tar. Resour. Conservation Recycling 54, 905–915. doi:10.1016/j.resconrec.2010.01.009
Li, S., Wang, Q., Lu, J., Deng, X., Bi, S., Song, Z., et al. (2019). Precise Control of the Growth and Size of Ni Nanoparticles on Al2O3 by a MOF-Derived Strategy. CrystEngComm 21, 6709–6718. doi:10.1039/c9ce01127g
Luo, W., Shi, H., Schachtl, E., Gutiérrez, O. Y., and Lercher, J. A. (2018). Active Sites on Nickel-Promoted Transition-Metal Sulfides that Catalyze Hydrogenation of Aromatic Compounds. Angew. Chem. Int. Ed. 57, 14555–14559. doi:10.1002/anie.201808428
Miryam, G. C., Cristina, J. G., Beatriz, d. R., Ignacio, G. O. J., and Rubén, L. F. (2017). Effect of Ni/Al Molar Ratio on the Performance of Substoichiometric NiAl2O4 Spinel-Based Catalysts for Partial Oxidation of Methane. Appl. Catal. B: Environ. 209, 128–138. doi:10.1016/j.apcatb.2017.02.063
Nuzzi, M., and Marcandalli, B. (2003). Hydrogenation of Phenanthrene in the Presence of Ni Catalyst. Thermal Dehydrogenation of Hydrophenanthrenes and Role of Individual Species in Hydrogen Transfers for Coal Liquefaction. Fuel Process. Tech. 80, 35–45. doi:10.1016/s0378-3820(02)00189-3
O'Neill, H. S. C., Dollase, W. A., and Ross, C. R. (1991). Temperature Dependence of the Cation Distribution in Nickel Aluminate (NiAl2O4) Spinel: a Powder XRD Study. Phys. Chem. Minerals 18, 302–319. doi:10.1007/bf00200188
O'Quinn, E. C., Shamblin, J., Perlov, B., Ewing, R. C., Neuefeind, J., Feygenson, M., et al. (2017). Inversion in Mg1-xNixAl2O4 Spinel: New Insight into Local Structure. J. Am. Chem. Soc. 139, 10395–10402. doi:10.1021/jacs.7b04370
Qian, W., Yoda, Y., Hirai, Y., Ishihara, A., and Kabe, T. (1999). Hydrodesulfurization of Dibenzothiophene and Hydrogenation of Phenanthrene on Alumina-Supported Pt and Pd Catalysts. Appl. Catal. A: Gen. 184, 81–88. doi:10.1016/s0926-860x(99)00083-6
Rogers, J. L., Mangarella, M. C., D’Amico, A. D., Gallagher, J. R., Dutzer, M. R., Stavitski, E., et al. (2016). Differences in the Nature of Active Sites for Methane Dry Reforming and Methane Steam Reforming over Nickel Aluminate Catalysts. ACS Catal. 6, 5873–5886. doi:10.1021/acscatal.6b01133
Rubén, L. F., Cristina, J. G., Beatriz, d. R., and José, I. G. O. (2012). Partial Oxidation of Methane to Syngas on Bulk NiAl2O4 Catalyst. Comparison with Alumina Supported Nickel, Platinum and Rhodium Catalysts. Appl. Catal. A: Gen. 437-438, 53–62. doi:10.1016/j.apcata.2012.06.014
Salhi, N., Boulahouache, A., Petit, C., Kiennemann, A., and Rabia, C. (2011). Steam Reforming of Methane to Syngas over NiAl2O4 Spinel Catalysts. Int. J. Hydrogen Energ. 36, 11433–11439. doi:10.1016/j.ijhydene.2010.11.071
Schachtl, E., Zhong, L., Kondratieva, E., Hein, J., Gutiérrez, O. Y., Jentys, A., et al. (2015). Understanding Ni Promotion of MoS2/γ-Al2O3and its Implications for the Hydrogenation of Phenanthrene. ChemCatChem 7, 4118–4130. doi:10.1002/cctc.201500706
Srifa, A., Kaewmeesri, R., Fang, C., Itthibenchapong, V., and Faungnawakij, K. (2018). NiAl2O4 Spinel-type Catalysts for Deoxygenation of palm Oil to green Diesel. Chem. Eng. J. 345, 107–113. doi:10.1016/j.cej.2018.03.118
Stanislaus, A., and Cooper, B. H. (1994). Aromatic Hydrogenation Catalysis: a Review. Catal. Rev. 36, 75–123. doi:10.1080/01614949408013921
Suffredini, D. F. P., Thyssen, V. V., de Almeida, P. M. M., Gomes, R. S., Borges, M. C., Duarte de Farias, A. M., et al. (2017). Renewable Hydrogen from Glycerol Reforming over Nickel Aluminate-Based Catalysts. Catal. Today 289, 96–104. doi:10.1016/j.cattod.2016.07.027
Tirsoaga, A., Visinescu, D., Jurca, B., Ianculescu, A., and Carp, O. (2011). Eco-friendly Combustion-Based Synthesis of Metal Aluminates MAl2O4 (M = Ni, Co). J. Nanopart Res. 13, 6397–6408. doi:10.1007/s11051-011-0392-1
van Deelen, T. W., Hernández Mejía, C., and de Jong, K. P. (2019). Control of Metal-Support Interactions in Heterogeneous Catalysts to Enhance Activity and Selectivity. Nat. Catal. 2, 955–970. doi:10.1038/s41929-019-0364-x
Wang, D., Li, J., Zheng, A., Ma, H., Pan, Z., Qu, W., et al. (2019). Quasi-Single-Layer MoS2 on MoS2/TiO2 Nanoparticles for Anthracene Hydrogenation. ACS Appl. Nano Mater. 2, 5096–5107. doi:10.1021/acsanm.9b01001
Wang, M., Qian, X., Xie, L., Fang, H., Ye, L., Duan, X., et al. (2019). Synthesis of a Ni Phyllosilicate with Controlled Morphology for Deep Hydrogenation of Polycyclic Aromatic Hydrocarbons. ACS Sust. Chem. Eng. 7, 1989–1997. doi:10.1021/acssuschemeng.8b04256
Shokrollahi Yancheshmeh, M., Alizadeh Sahraei, O., Aissaoui, M., and Iliuta, M. C. (2020). A Novel Synthesis of NiAl2O4 Spinel from a Ni-Al Mixed-Metal Alkoxide as a Highly Efficient Catalyst for Hydrogen Production by Glycerol Steam Reforming. Appl. Catal. B: Environ. 265, 118535. doi:10.1016/j.apcatb.2019.118535
Yang, F., Liu, D., Wang, H., Liu, X., Han, J., Ge, Q., et al. (2017). Geometric and Electronic Effects of Bimetallic Ni-Re Catalysts for Selective Deoxygenation of M-Cresol to Toluene. J. Catal. 349, 84–97. doi:10.1016/j.jcat.2017.01.001
Ghadami Yazdi, M., Moud, P. H., Marks, K., Piskorz, W., Öström, H., Hansson, T., et al. (2017). Naphthalene on Ni(111): Experimental and Theoretical Insights into Adsorption, Dehydrogenation, and Carbon Passivation. J. Phys. Chem. C. 121, 22199–22207. doi:10.1021/acs.jpcc.7b07757
Yoon, E. M., Selvaraj, L., Song, C., Stallman, J. B., and Coleman, M. M. (1996). High-temperature Stabilizers for Jet Fuels and Similar Hydrocarbon Mixtures. 1. Comparative Studies of Hydrogen Donors. Energy Fuels 10, 806–811. doi:10.1021/ef950228l
Yu, L., Song, M., Williams, P. T., and Wei, Y. (2019). Alumina-Supported Spinel NiAl2O4 as a Catalyst for Re-forming Pyrolysis Gas. Ind. Eng. Chem. Res. 58, 11770–11778. doi:10.1021/acs.iecr.9b01006
Zhang, X., Pan, L., Wang, L., and Zou, J.-J. (2018). Review on Synthesis and Properties of High-Energy-Density Liquid Fuels: Hydrocarbons, Nanofluids and Energetic Ionic Liquids. Chem. Eng. Sci. 180, 95–125. doi:10.1016/j.ces.2017.11.044
Keywords: phenanthrene hydrogenation, metallic electron-deficient state, nickel aluminate, calcination temperature, steric hindrance, competitive adsorption
Citation: Liu D-C, Chen Y, Jing J-Y, Rajendran A, Bai H-C and Li W-Y (2021) Synthesis of Ni/NiAlOx Catalysts for Hydrogenation Saturation of Phenanthrene. Front. Chem. 9:757908. doi: 10.3389/fchem.2021.757908
Received: 13 August 2021; Accepted: 06 September 2021;
Published: 08 October 2021.
Edited by:
Changhai Liang, Dalian University of Technology, ChinaReviewed by:
Yuefeng Liu, Dalian Institute of Chemical Physics (CAS), ChinaXiangwen Zhang, Tianjin University, China
Copyright © 2021 Liu, Chen, Jing, Rajendran, Bai and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie-Ying Jing, jingjieying@tyut.edu.cn; Wen-Ying Li, ying@tyut.edu.cn