Computational pharmacology and computational chemistry of 4-hydroxyisoleucine: Physicochemical, pharmacokinetic, and DFT-based approaches
 Imad Ahmad1
Imad Ahmad1  Aleksey E. Kuznetsov2
Aleksey E. Kuznetsov2  Abdul Saboor Pirzada1
Abdul Saboor Pirzada1  Khalaf F. Alsharif3
Khalaf F. Alsharif3  Maria Daglia4,5
Maria Daglia4,5  Haroon Khan1*
Haroon Khan1*- 1Department of Pharmacy, Abdul Wali Khan University Mardan, Mardan, Pakistan
- 2Department of Chemistry, Universidad Tecnica Federico Santa Maria, Santiago, Chile
- 3Department of Clinical Laboratory, College of Applied Medical Science, Taif University, Taif, Saudi Arabia
- 4Department of Pharmacy, University of Naples Federico II, Naples, Italy
- 5International Research Centre for Food Nutrition and Safety, Jiangsu University, Zhenjiang, China
Computational pharmacology and chemistry of drug-like properties along with pharmacokinetic studies have made it more amenable to decide or predict a potential drug candidate. 4-Hydroxyisoleucine is a pharmacologically active natural product with prominent antidiabetic properties. In this study, ADMETLab 2.0 was used to determine its important drug-related properties. 4-Hydroxyisoleucine is compliant with important drug-like physicochemical properties and pharma giants’ drug-ability rules like Lipinski’s, Pfizer, and GlaxoSmithKline (GSK) rules. Pharmacokinetically, it has been predicted to have satisfactory cell permeability. Blood–brain barrier permeation may add central nervous system (CNS) effects, while a very slight probability of being CYP2C9 substrate exists. None of the well-known toxicities were predicted in silico, being congruent with wet lab results, except for a “very slight risk” for respiratory toxicity predicted. The molecule is non ecotoxic as analyzed with common indicators such as bioconcentration and LC50 for fathead minnow and daphnia magna. The toxicity parameters identified 4-hydroxyisoleucine as non-toxic to androgen receptors, PPAR-γ, mitochondrial membrane receptor, heat shock element, and p53. However, out of seven parameters, not even a single toxicophore was found. The density functional theory (DFT) study provided support to the findings obtained from drug-like property predictions. Hence, it is a very logical approach to proceed further with a detailed pharmacokinetics and drug development process for 4-hydroxyisoleucine.
1 Introduction
4-Hydroxyisoleucine (4HIL) is an amino acid which has been purified from Trigonella foenum-graecum (fenugreek seeds; Fabaceae). Fenugreek is known for its antidiabetic properties in traditional medicine (Fowden et al., 1973; Abou El-Soud et al., 2007). 4-Hydroxyisoleucine increases insulin secretion via a direct effect on isolated islet cells both in rats and humans. The stimulating effect was strictly glucose dependent. Furthermore, it was shown that the insulinotropic effect is biphasic, without associated harm to other islet cells (α and δ-cells) and increase in insulin secretion by increasing glucose concentration (Sauvaire et al., 1998). 4-Hydroxyisoleucine reduces insulin resistance in the muscles and the liver by the activation of insulin receptor substrate-associated phosphoinositide-3 kinase activity. Reduction in body weight and plasma triglyceride and total cholesterol levels resulted from application of 4-hydroxyisoleucine. This plant-derived amino acid offers an attractive package for the treatment of the metabolic syndrome as a whole and also type 2 diabetes, obesity, and dyslipidaemias (Jetté et al., 2000; Narender et al., 2006). Keeping in mind the pharmacological importance of 4HIL, its efficacy needs to be evaluated before it moves to trials and/or is dismissed. Undesirable pharmacokinetic (PK) properties or unacceptable toxicity are the main causes of the failure of drug candidates at the clinical trial stage (Jia et al., 2020).
Since the concept of drug likeness was first proposed, it has become an important consideration in the selection of compounds with desirable bioavailability during the early phases of drug discovery. Over the past decade, online resources have facilitated drug-likeness studies in an economical and time-efficient manner. Michael J. Waring and colleagues compiled and analyzed the attrition of drug candidates from four pharma giants, AstraZeneca, Eli Lilly and Company, GlaxoSmithKline, and Pfizer. They reaffirmed that the control of physicochemical properties during structural optimization helps identify a compound of drug quality. Safety and toxicology are the largest sources of failure within the data set. However, none of the physicochemical descriptors correlate with preclinical toxicology outcomes (Waring et al., 2015).
While considering the various pharmacology effects in different diseased conditions, we calculated various parameters related to drug discovery for 4-hydroxyisoleucine using ADMETLab 2.0. in order to predict the drug-like properties and safety profile and thereby therapeutic potentials.
2 Methodology
Various virtual labs are authorized to share the pharmacokinetic profile of pharmacologically active drug candidates such as SwissADME (Daina et al., 2017), FAF-Drugs4 (Lagorce et al., 2017), ADMETlab (Xiong et al., 2021), admetSAR (Yang et al., 2019), pkCSM (Pires et al., 2015), and ProTox-II (Banerjee et al., 2018). However, ADMETLab was selected because it provides predictive spectrum of multiple drug-likeness parameters and most of ADMET properties. The updated version, ADMETLab 2.0, has overpowered all known shortcomings of the older version while keeping its valuable advantages.
This virtual lab calculates 88 ADMET-related properties, i.e., 17 physicochemical parameters, 13 medicinal chemistry properties, 23 ADME properties, 27 toxicity endpoints, and eight toxicophore rules (751 substructures). ADMETLab 2.0 is freely accessible at https://admetmesh.scbdd.com. The 2D structure of 4-hydroxyleucine was drawn in ADMETLab 2.0 (Table 1). The user interface also offers string-based search. The search string for 4-hydroxyisoleucine is CC(O)C(C)C(N)C (=O)O.

TABLE 1. Structure, IUPAC name, and SMILES notation of 4-hydroxyisoleucine.
An oral bioavailability graph obtained from SwissADMET was calculated using physicochemical characteristics. This graph uses six parameters, i.e., lipophilicity, insolubility, size, unsaturation, polarity, and flexibility, and defines appropriate ranges for oral bioavailability. The GI absorption (HIA), BBB penetration, and being Pgp substrate were also predicted with SwissADMET. The result is depicted as “boiled egg” having two areas: one for HIA (white) and another for BBB penetration (yolk). The background is a gray zone, indicating neither GI absorption nor BBB penetration.
The human ether-a-go-go-related gene (hERG) K+ channels blockade is linked with fatal cardiac arrhythmias. The hERG K+ channel blockade was predicted with both ADMETLab 2.0 and pred-hERG 4.2. The pred-hERG 4.2, http://predherg.labmol.com.br, is a web tool for early detection of presumable hERG blockers and non-blockers, to predict cardiac toxicity.
DFT studies were performed with Gaussian 16 software (Frisch et al., 2016). We optimized the 4-hydroxyisoleucine molecule without any symmetry constraints, checking different possible isomers (vide infra), and then performed frequency calculations to verify that the optimized structures were true energy minima. All calculations were performed using the combination of the hybrid density functional B3LYP (Axel, 1993) with the triple zeta split-valence polarized basis set of Ahlrichs and co-workers Def2TZVP (Weigend and Ahlrichs, 2005; Weigend, 2006); the approach is furthermore referred to as B3LYP/Def2TZVP. We studied the 4-hydroxyisoleucine both in the gas phase (vacuum) and with the implicit effects from water (dielectric constant ε = 78.3553) as a solvent taken into account. In the second type of calculations, we used the self-reliable IEF-PCM approach (Tomasi et al., 2005) using the UFF default model as implemented in Gaussian 16 software, with the electrostatic scaling factor α = 1.0. The charge analysis was performed using the natural bond orbital (NBO) method as implemented in Gaussian 16 program (Reed et al., 1988), using the B3LYP/Def2TZVP approach with the implicit water effects. Frontier molecular orbitals (FMOs) and molecular electrostatic potential (MEP) were computed at the B3LYP/Def2TZVP level with the implicit water effects as well.
We consider the calculated structures of the seven isomers of 4-hydroxyisoleucine, structural parameters for the lowest-lying isomer, its NBO charges, FMOs, and MEP. Furthermore, we used the values of the energies of the lowest-lying isomer, HOMO and LUMO, to compute its global reactivity parameters (GRP) (Geerlings et al., 2003; Chakraborty et al., 2013; Jorio et al., 2019) (Eqs 1–6). Eqs 1, 2 were used to calculate the values of the ionization potential (IP) and electron affinity (EA):
For global hardness η and electronegativity X values, we used Eqs 3, 4:
Also, global electrophilicity ω value was calculated by Eq. 5:
where
Finally, the global softness σ value was computed with Eq. 6:
Open GL version of Molden 5.8.2 visualization program was used for the visualization of the structures and FMOs (Schaftenaar and Noordik, 2000), and Avogadro, version 1.1.1, was used to visualize the MEP maps (Hanwell et al., 2012).
3 Results
ADMETLab 2.0 offers results in different formats, even in a downloadable pdf format. The following parameters were predicted for 4-HIL.
3.1 Physicochemical properties
The widely accepted physicochemical property guidelines are the Lipinski’s rule of five (RO5), which is satisfied by an ideal drug molecule. It offers a prediction of the drug likeness of a chemical compound having a biological activity and designed for oral route of administration. The molecular weight of 4-hydroxyisoleucine is 147.09 g/mol, which reflects its suitability for oral drug development. This provides therapeutic convenience of its formulation as oral dosage form. The optimal range is 100–600 approximately. The molecular volume to van der Waals volume ratio was calculated as 147.06. The number of hydrogen bond acceptors (nHA; optimal: 0–12) is 4. The number of hydrogen bond donors (nHD; optimal: 0∼7) is also 4. 4-Hydroxyisoleucine has three rotatable bonds only (optimal: 0∼11).
There are no rings or cyclic groups in 4-HIL (optimal: 0–6). There are four heteroatoms in 4-hydroxyisoleucine, which satisfy the range 1∼15 given by ADMETLab. There is no formal charge on this molecule. A charge range of −4∼4 is devised as standardized parameter for a drug candidate. The number of rigid bonds may range from 0 to 30; 4HIL has a single rigid bond. This makes the molecule flexible enough to achieve a stable conformation at its target. In this continuity, the flexibility value is 3.0 obtained as ratio of nRot/nRig. The molecule has three stereo centers which is somewhat closer to the optimal value of ≤2. A stereo center may achieve a different conformation at the active site. The topological polar surface area (TPSA) for 4-hydroxyisoleucine is 83.55.
The aqueous solubility (logS) is 0.06, falling within the range of −4∼0.5 log mol/L. LogP value is the Log of octanol/water partition coefficient whose optimal range is 0∼3. A higher logP value implies greater lipophilicity, and this depends upon molecular size, polarity, and hydrogen bonding. LogP value of 4-hydroxyisoleucine is −2.63 which reflects its partition into water compartment. This value is congruent to LogS value described previously. The value of logP at normal physiological pH 7.4 is logD. For 4-hydroxyisoleucine it is calculated as −0.93 just below the optimal range of 1∼3. Physicochemical properties are summarized in Table 2.
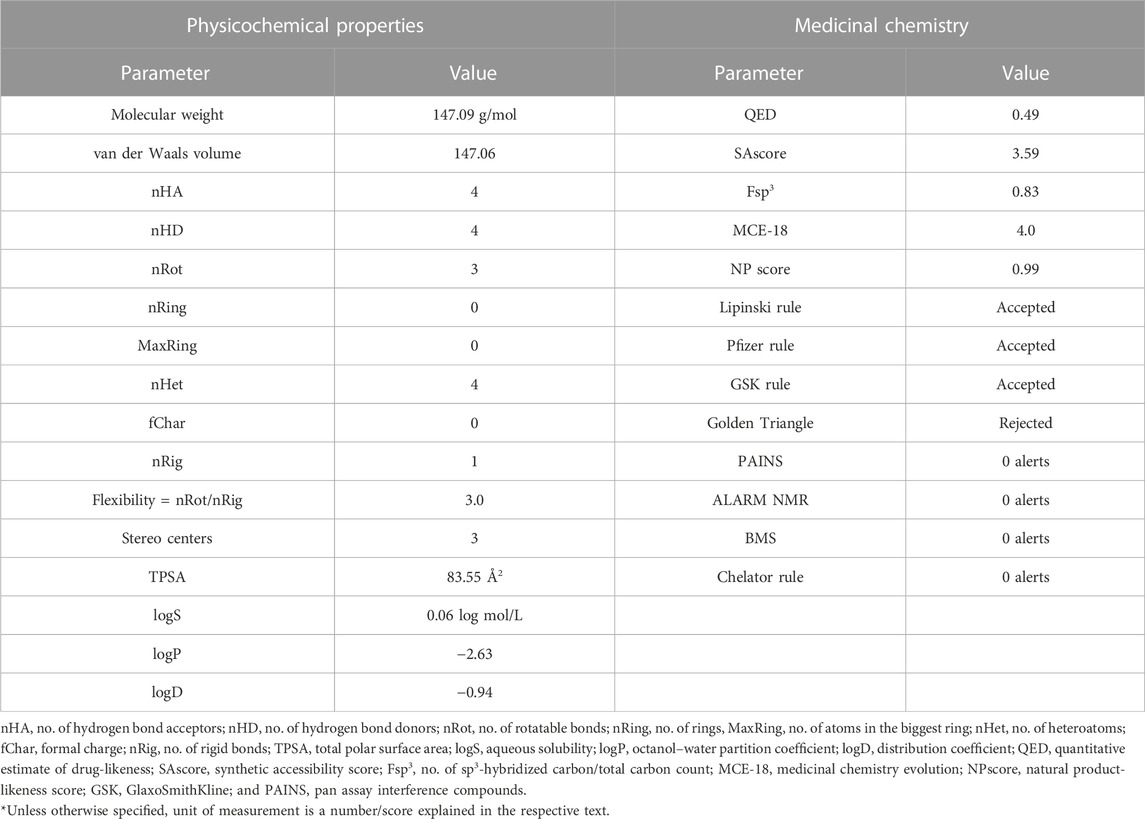
TABLE 2. Physicochemical properties and medicinal chemistry parameters of 4-hydroxy isoleucine calculated with ADMETLab 2.0.
3.2 Medicinal chemistry
The QED score (quantitative estimate of drug-likeness) is 0.49, which implies it is an unattractive molecule. An attractive drug has a QED score of >0.67; for unattractive molecules, the score is 0.49∼0.67, while it is <0.34 for too complex molecules. The QED score is a drug-likeness score based on the calculated physicochemical properties of marketed oral drugs and published human data.
SAscore is implemented based on synthetic accessibility score, which is an estimated ease of synthesis of a drug-like molecule. Based on SAscore (3.59), 4-hydroxyisoleucine is easy to synthesize. Drug-like molecules having SAscore of ≥6 are difficult to synthesize, while those with a score of <6 are easy to synthesize. The Fsp3 value for 4-HIL is 0.83. Fsp3 is the fraction or number of sp3 carbon atoms out of the total carbon count. This parameter reflects the carbon saturation of molecules and characterizes the complexity of their spatial structure. The suitable value defined by ADMETLab for Fsp3 is >0.42, with 4-hydroxyisoleucine being compliant.
The optimum MCE-18 score limit given by ADMETlab 2.0 is ≥45. For 4-HIL MCE-18 score is 4.0, which is extremely low. NP score reflects the natural product-likeness score. This score ranges from −5 to 5; the higher the score, the higher the probability of being a natural product. Natural products are more likely to be safe, with reduced risk of toxicity. NP score for 4-hydroxyisoleucine is 0.99, which is in this safe range.
ADMETLab 2.0 has described Lipinski’s rule as follows: MW ≤ 500, logP ≤5, Hacc ≤10, and Hdon ≤5. These parameters are already described individually under physicochemical properties. All these parameters are met by 4-HIL and thus the widely accepted Lipinski rule is satisfied. In the case where two of these properties are out of range, there is a possibility of poor absorption or permeability. Deviation of only one property is allowed. 4-Hydroxyisoleucine also complies with Pfizer’s rule, i.e., logP >3 and TPSA <75. Compounds which satisfy the GSK rule may have a more favorable ADMET profile. 4-HIL concurs with GSK’s rule as well. The Golden Triangle rule devises the range for two parameters as follows: 200 ≤ MW ≤ 50 and −2 ≤ logD ≤5. Compounds satisfying this Golden Triangle rule are more likely to have favorable ADMET profiles. As per this criterion, 4-hydroxyisoleucine is rejected. However, ADMET profiles are separately described later which possess desirable pharmacokinetics.
4-HIL has zero PAINS alerts, and thus has been excluded from frequent hitters, α-screen artifacts, and reactive compounds. 4-Hydroxyisoleucine has no ALARM NMR alerts and is not a thiol reactive compound. 4-Hydroxyisoleucine has no red alert for BMS and the chelator rule and so is a desirable, non-reactive, and non-chelating compound. Medicinal chemistry parameters are summarized in Table 2.
3.3 Pharmacokinetics
3.3.1 Absorption
Caco-2 cell lines (Caucasian colon adenocarcinoma) are still widely used in absorption studies. The Caco-2 permeability score obtained was −5.99 which is lower than the minimum optimal score of −5.15 log unit. The Madin–Darby canine kidney (MDCK) permeability score was 0.01 cm/s which is in the acceptable limit. The standard scores for low permeability, medium permeability, and high passive permeability are <2 × 10−6 cm/s, 2–20 × 10−6 cm/s, and >20 × 10−6 cm/s, respectively.
4-Hydroxyisoleucine has a very low probability as Pgp inhibitor as per its score of 0.01. Therefore, it is regarded as safe from having significant drug interactions. For an inhibitor, the score is 1, while 0 score denotes a non-inhibitor. This output value exhibits the probability of being a Pgp inhibitor. The probability of being Pgp substrate is the lowest, as indicated by its score 0.05. For a Pgp substrate, the score is 1, while for a non-substrate, the score is 0.
4-HIL is predicted to have low human intestinal absorption (HIA <30%) as indicated by its HIA score, 0.09. The bioavailability is predicted to be greater than 20% and 30% (F20% 0.004, F30% 0.002).
The oral bioavailability graph shown in Figure 1A is predicted with the SwissADMET database. This graph is based on six physicochemical characteristics, i.e., lipophilicity, size, polarity, insolubility, unsaturation, and flexibility. The result of the compound 4-hydroxyisoleucine was within these limits, demonstrating a favorable physio-chemical profile, which is a prerequisite factor that must be monitored in pharmaceutical and clinical studies. The colored zone is the suitable physicochemical space for oral bioavailability.
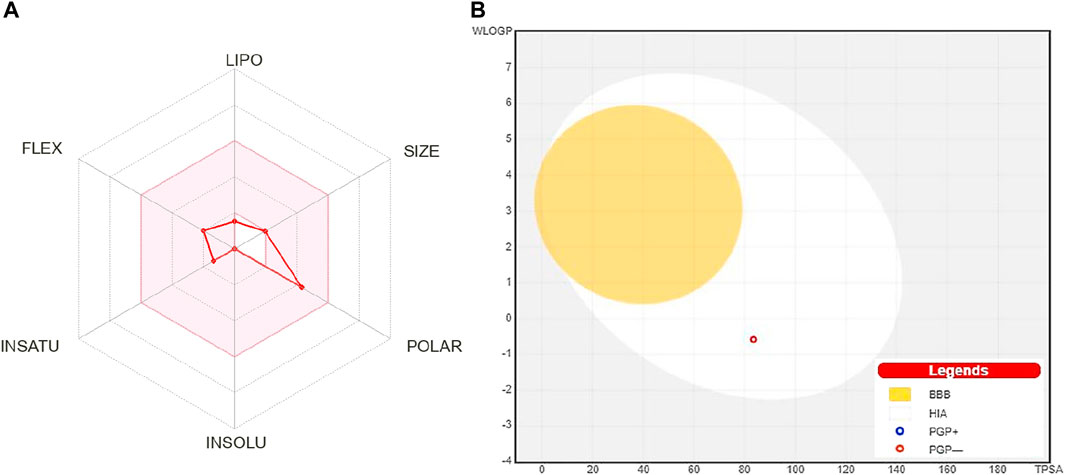
FIGURE 1. (A) Oral bioavailability graph from the SwissADME database; the pink zone is the physicochemical space for oral bioavailability, and the red line defines oral bioavailability properties of 4-hydroxyisoleucine. (B) Predicted boiled-egg plot from the SwissADME database reveals human intestinal absorption and not being Pgp substrate. LIPO: lipophilicity, POLAR: polarity, INSOLU: insolubility, INSATU: unsaturation, FLEX: flexibility, BBB: blood–brain barrier, HIA: human intestinal absorption, PGP+: p-glycoprotein substrate, and PGP-: not a p-glycoprotein substrate.
HIA and central nervous system (CNS) absorption needs to be checked for every biomolecule before its formulation in the pharmaceutical or clinical trials. The blood–brain barrier penetration is essential only if the compound has a target in the CNS, but the inactive compounds on the CNS must not intersect to avoid adverse effects. The boiled-egg plot shows that 4-hydroxyisoleucine has a high gastrointestinal absorption without BBB permeability, indicating low occurrence for CNS side effects (Figure 1B).
3.3.2 Distribution
Plasma protein binding (PPB), unbound fraction in plasma (Fu), volume of distribution (VD), and blood–brain barrier (BBB) permeability were considered to study the distribution (Table 4). The predicted PPB was 10.90%. The optimal range of PPB is <90% because drugs which are high protein-bound may have a low therapeutic index. The volume of distribution (VD) was calculated as 0.39 L/kg. The optimal range for VD is 0.04–20 L/kg. 4-Hydroxyisoleucine has more tendency for blood–brain barrier penetration reflected by the score, 0.73. The output value indicates the probability of being able to cross BBB. The unbound fraction in plasma (Fu) was calculated as 87.84%. The scores for low, middle, and high unbound plasma fraction are <5%, 5∼20%, and >20%, respectively. This implies that more unbound plasma fraction will be available for pharmacological action.
3.3.3 Metabolism
This parameter is important from the drug plasma concentration perspective. The database defines the ligands’ probability to inhibit the enzyme or not by its placement either in category 1 (Inhibitor) or category 0 (non-inhibitor). Similarly, the probability of being substrate for the enzyme is exhibited by the score 1 or 0. Category 1 denotes the molecule is substrate, while category 0 describes it to be non-substrate of the enzyme.
4-Hydroxyisoleucine is likely a non-inhibitor of CYP1A2 as indicated by the assigned score 0.01. The CYP1A2 substrate score obtained as 0.06 implies it is a non-substrate. The probability of CYP2C19 inhibition and being CYP2C19 substrate is extremely low. 4-Hydroxyisoleucine appeared to be non-inhibitor of CYP2C9. However, very low probability exists for being CYP2C9 substrate. Similarly, there is no incidence of CYP2D6 inhibition and being a CYP2D6 substrate. 4-Hydroxyisoleucine is not deemed a CYP3A4 inhibitor nor is it a substrate for CYP3A4.
3.3.4 Excretion
The clearance (CL) for 4-hydroxyisoleucine is calculated as 7.27 mL/min/kg which is a moderate clearance rate. If a drug has a high clearance rate, its score will be > 15 mL/min/kg; for a moderate clearance rate, it will be 5–15 mL/min/kg; and for a low clearance rate, it will be < 5 mL/min/kg. The half-life (T1/2) value is 0.62, which reflects the probability of long half-life. The database output value may be from category 1 (long half-life, >3 h) or category 0 (short half-life, <3 h). All the pharmacokinetic parameters (absorption, distribution, metabolism, and excretion) are summarized in Table 3.
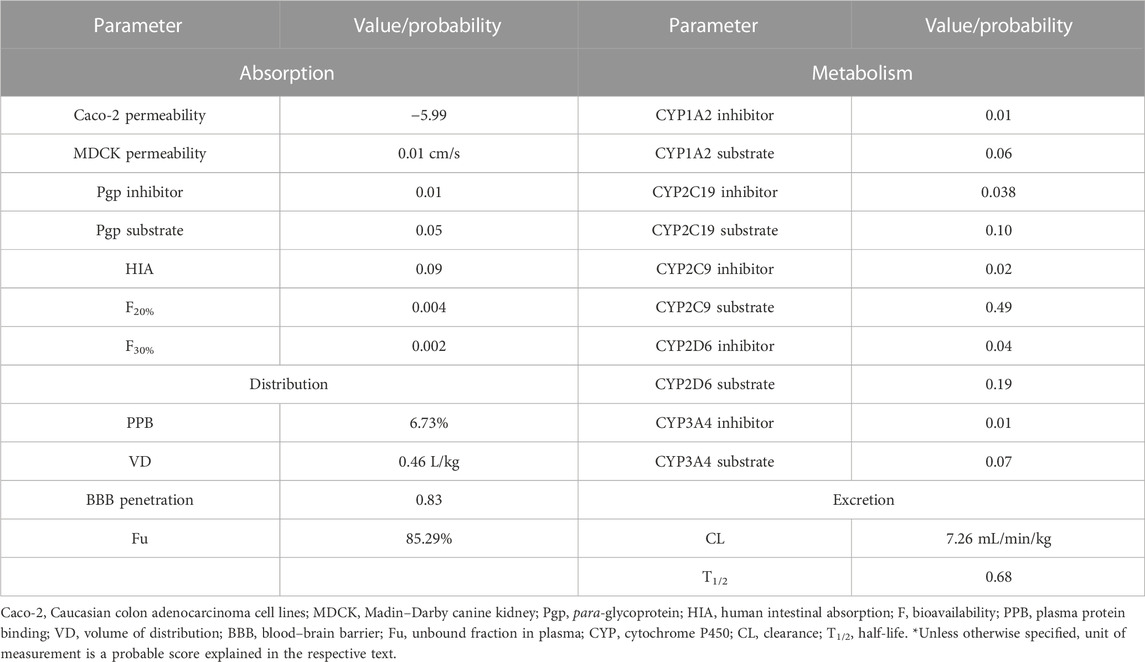
TABLE 3. Pharmacokinetics of 4-Hydroxy isoleucine calculated with ADMETLab 2.0.
3.4 Toxicity
The toxicity parameters verified by ADMETlab include hERG blockade, human hepatotoxicity, drug-induced liver injury, AMES toxicity, rat oral acute toxicity, FDA maximum daily dose, carcinogenicity, mutagenicity, skin sensitization, eye corrosion, eye irritation, and respiratory toxicity (Table 4).
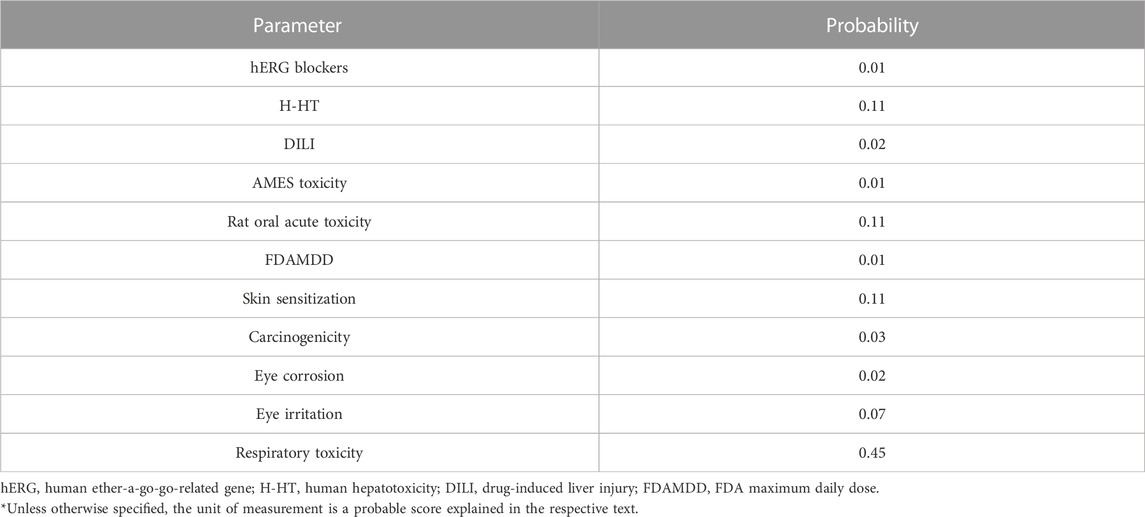
TABLE 4. Toxicity profile of 4-hydroxy isoleucine calculated with ADMETLab 2.0.
The ADMETLab 2.0 score for hERG blocker is 0.01 which shows the safety of 4-hydroxyisoleucine. The result was further in line with Pred-hERG which defined 4-hydroxyisoleucine as non-cardiotoxic with 90% confidence, as shown in Figure 2. The red dotted lines defined the area which negatively contributes to hERG blockade, while contour lines with intense green color represent a higher positive contribution of an atom to hERG blockade. 4-Hydroxyisoleucine is not human hepatotoxic (H-HT) as its score is 0.11. Molecules falling in category 1 are hepatotoxic (H-HT positive, +), while category 0 reflects non-hepatotoxic (H-HT negative, −). Similarly, there is low risk of drug-induced liver injury (DILI), 0.02. Commonly, category 1 drugs have high risk of DILI, while category 0 drugs have no risk of DILI. Furthermore, Ames toxicity score also indicated safety of 4-hydroxyisoleucine, evident from its score of 0.01. Category 1 drugs are classified as Ames positive (+, toxic), while category 0 represents Ames negative (−, non-toxic). Finally, the risk of oral toxicity is low. The rat oral acute toxicity score is 0.11, which classifies 4-hydroxyisoleucine in category 0 (low-toxicity), as category 1 indicates high-toxicity.
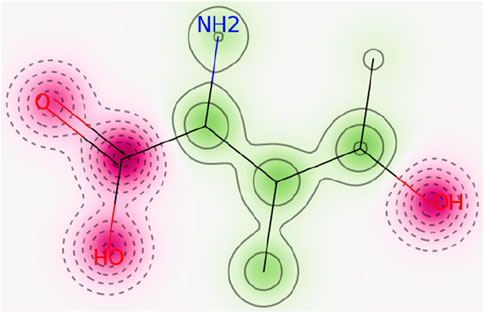
FIGURE 2. In the probability map of hERG blockage (Pred-hERG web tool), the red dotted lines reflect non-hERG blocker. Contour lines with intense green color indicate a higher positive contribution of an atom to hERG blockade.
4-Hydroxyisoleucine has the probability of being skin non-sensitizer with a value of 0.11. Category 1 drugs are skin sensitizers, while category 0 are non-sensitizers. The risk of carcinogenicity (0.03) is lower, and the molecule is safe from adverse reactions. Category 1 drugs are carcinogens, while category 0 are non-carcinogens. A negligible risk of eye corrosion is predicted as exhibited by its score, 0.02. Category 1 drugs are corrosives, while category 0 drugs are non-corrosive to the eye. The output value indicates the probability of being non-irritant to the eye. The eye irritation score is 0.07. Category 1 drugs are irritants, while category 0 drugs are non-irritants. Finally, the respiratory toxicity score (0.45) declares borderline low probability to be free from respiratory toxicity. Category 1 indicates respiratory toxicants, and category 0 drugs are believed to be free from respiratory toxicity.
3.5 Environmental toxicity
For a drug, it is also important to be non-toxic to the environment. Therefore, ADMETLab defines some of the environmental toxicity indicators which are provided in Table 5. The bioconcentration factor, 0.17, is in favor of 4-hydroxyisoleucine non-ecotoxicity. The 50% growth inhibitory concentration (IGC50) for Tetrahymena pyriformis is 2.43. For 96-h fathead minnow (Pimephales promelas), the 50% lethal concentration LC50FM of 4-hydroxyisoleucine is 2.92 (Dionne et al., 2021). The score for a 48-h daphnia magna 50 percent lethal concentration (LC50DM) for 4-hydroxyisoleucine is 2.36.
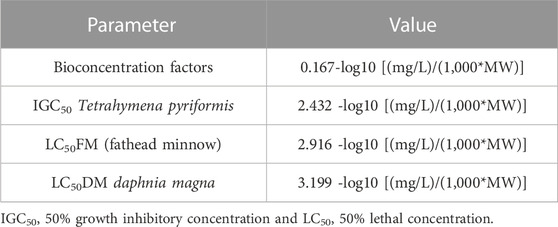
TABLE 5. Environmental toxicity of 4-hydroxyisoleucine calculated with ADMETLab 2.0.
3.6 Tox21 pathway
There is probability, to a negligible extent, of 4-hydroxyisoleucine binding to androgen receptor (NR-AR). The output value is 0.05, indicating its safety toward genetic instability. In another approach to test its safety, it is predicted that 4-hydroxyisoleucine will not bind to androgen receptor ligand-binding domain (NR-AR-LBD). Such a reduced probability is exhibited from its lowest value 0.01.
With such a reduced probability (0.01), inactivity at aryl hydrocarbon receptor is evidenced. No activity at NR-Aromatase is predicted, as 0.01 is near to zero, which means inactive. However, 4-hydroxyisoleucine has a low probability to remain inactive at binding to the estrogen receptor (0.42), while reduced affinity for estrogen receptor ligand-binding domain exists (0.01). The peroxisome proliferator-activated receptor gamma is also unlikely to be affected by 4-hydroxyisoleucine, as the score is 0.01. 4-Hydroxyisoleucine is not predicted to be an antioxidant response element since the probability is negligible at 0.02. It is also unlikely to interfere with the activity of ATPase family AAA domain-containing protein 5 as predicted score is 0.01. Furthermore, it is unlikely to activate the heat shock factor response element as the score is 0.01. The score for non-modification of mitochondrial membrane potential is 0.01. Finally, 4-hydroxyisoleucine also has low probability (0.01) of affecting p53 (see Table 6).
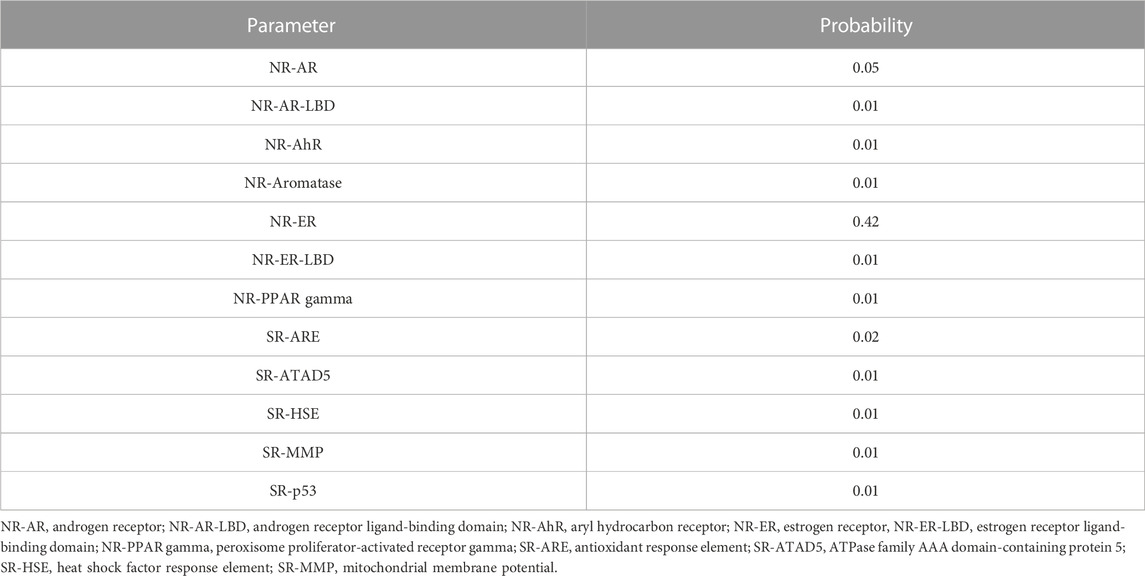
TABLE 6. Toxicity profile of 4-hydroxyisoleucine calculated with ADMETLab 2.0.
3.7 Toxicophore rules
This parameter rules out the presence of a toxicophore, causing severe adverse effects. The acute toxicity rule exhibit zero alerts for acute toxicity during oral administration. The molecule is safe from genotoxic carcinogenicity or mutagenicity. Furthermore, 4-hydroxyisoleucine is clear of a non-genotoxic carcinogenic mechanism as well, with zero alerts. No toxicophore was found for skin sensitization, and 4-hydroxyisoleucine is expected to be free from skin irritation. The molecule is not toxic to aquatic life, with zero alerts for the aquatic toxicity rule (water toxicity). 4-Hydroxyisoleucine also appears biodegradable as ADMETlab revealed zero alerts for the non-biodegradable rule. 4-Hydroxyisoleucine has no enlistment in SureChEMBL database, so it has zero alerts for the SureChEMBL rule and is not classified as MedChem unfriendly status (see Table 7).
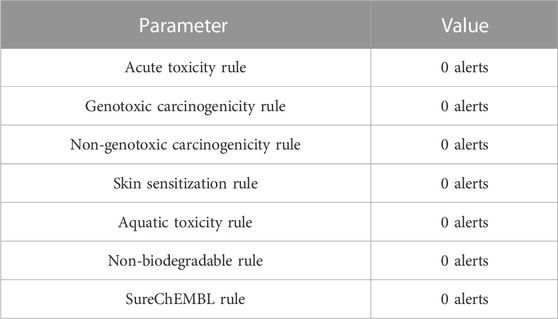
TABLE 7. Toxicophore based profile of 4-hydroxy isoleucine calculated with ADMETLab 2.0.
3.8 DFT study results
In Figure 3, the optimized structures of seven isomers studied for 4-hydroxyisoleucine are shown, and Table 8 summarizes the energies, relative energies, HOMO/LUMO energies, and HOMO/LUMO energy gaps of these seven isomers. As can be seen from Figure 3, the isomers differ from each other by orientations of 4-hydroxy and amino groups and hydrogens of the carbonyl, hydroxyl, and amino groups. The lowest-lying isomer 5 (Figure 3) has the carbonyl oxygen of the carboxyl group and amino group located on the same molecule side and the 4-hydroxy group located in the plane approximately perpendicular to the carboxyl group, with hydrogen bonds formed between the 4-hydroxy and amino groups and between the amino group and the carbonyl oxygen of the carboxyl group (see more detailed discussion later). Furthermore, from Table 8, it follows that all six higher-lying 4-hydroxyisoleucine isomers are quite close in energy to the global minimum isomer 5, being within 0.09 (isomer 3)–2.89 (isomer 7) kcal/mol higher in energy. We focus on the more detailed consideration of only the lowest-lying isomer 5. Further consideration of Table 4 data shows that all seven isomers have significant HOMO/LUMO gaps, 6.51–7.07 eV, with the isomer 5 having the HOMO/LUMO gap value 6.86 eV, the highest in energy isomer 7 having the smallest HOMO/LUMO gap value, 6.51 eV, and the second lowest-lying isomer 3 having the largest HOMO/LUMO gap value, 7.07 eV.
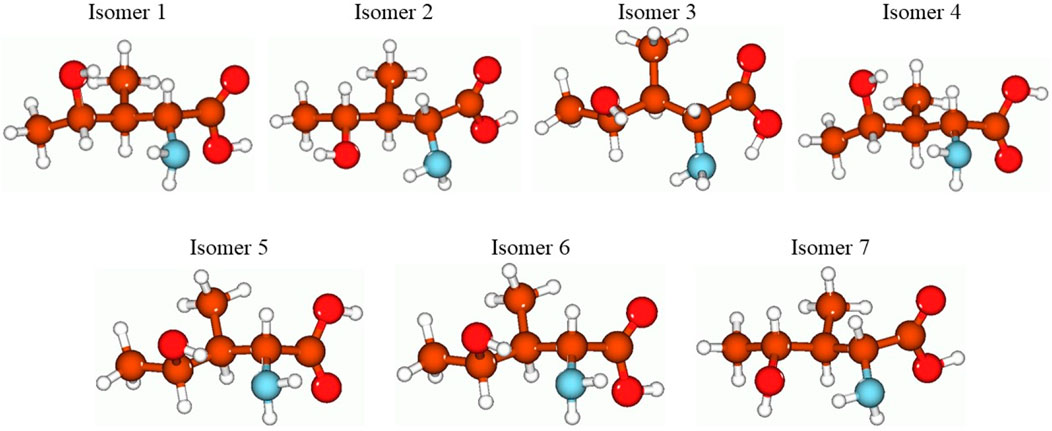
FIGURE 3. Structures of the seven studied isomers of the 4-hydroxyisoleucine optimized using the B3LYP/Def2TZVP method with the implicit effects of water. Color coding: brown for C, red for O, light blue for N, and light gray for H.
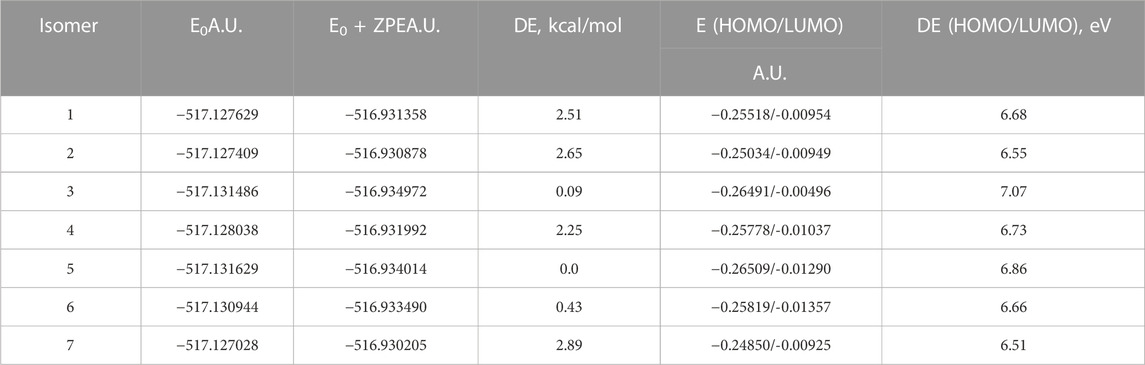
TABLE 8. Energetics of the seven optimized isomers of 4-hydroxyisoleucine calculated using the B3LYP/Def2TZVP method with the implicit effects of water.
Figure 4 presents the selected bond distances, Å (Figure 4A), NBO charges on the selected atoms (Figure 4B), FMOs (Figure 4C), and MEP plot (Figure 4D) for the lowest-lying isomer 5. As aforementioned, there are several intermolecular hydrogen bonds in this isomer, between the 4-hydroxy group hydrogen and amino group nitrogen (bond distance, 2.166 Å), between the amino group hydrogens and the carbonyl oxygen of the carboxyl group (bond distances, 2.652 and 3.097 Å), and within the carboxyl group, 2.307 Å. Thus, as can be seen, intra- and also intermolecular hydrogen bonds can be formed by 4-hydroxyisoleucine, which might explain the presence of several isomers close in energy. Furthermore, NBO charge analysis (Figure 4B) shows noticeable negative charges on the oxygens and nitrogen of the molecular functional groups, −0.625 to −0.835 e, along with noticeable positive charges on the hydrogens of these functional groups, 0.372–0.503 e. These charges would explain the formation of intramolecular hydrogen bonds along with intermolecular interactions. The HOMO and LUMO plots (Figure 4C) show that essentially the whole molecule contributes to FMOs, thus being able to participate in potential processes of donating/accepting electrons. Finally, analysis of the MEP plot (Figure 4D) shows accumulation of both negative (as indicated by red color) and positive (as indicated by blue color) electrostatic potential in the molecule. Negative MEP is accumulated on the carboxyl oxygens and on the 4-hydroxy oxygen, whereas positive MEP is accumulated on the amino group, carboxyl group hydrogen, and 4-hydroxy group hydrogen. This implies that the 4-hydroxyisoleucine molecule can behave both as a nucleophilic and electrophilic agent in chemical reactions. Also, these MEP accumulations imply possibility of intermolecular interactions for this compound.
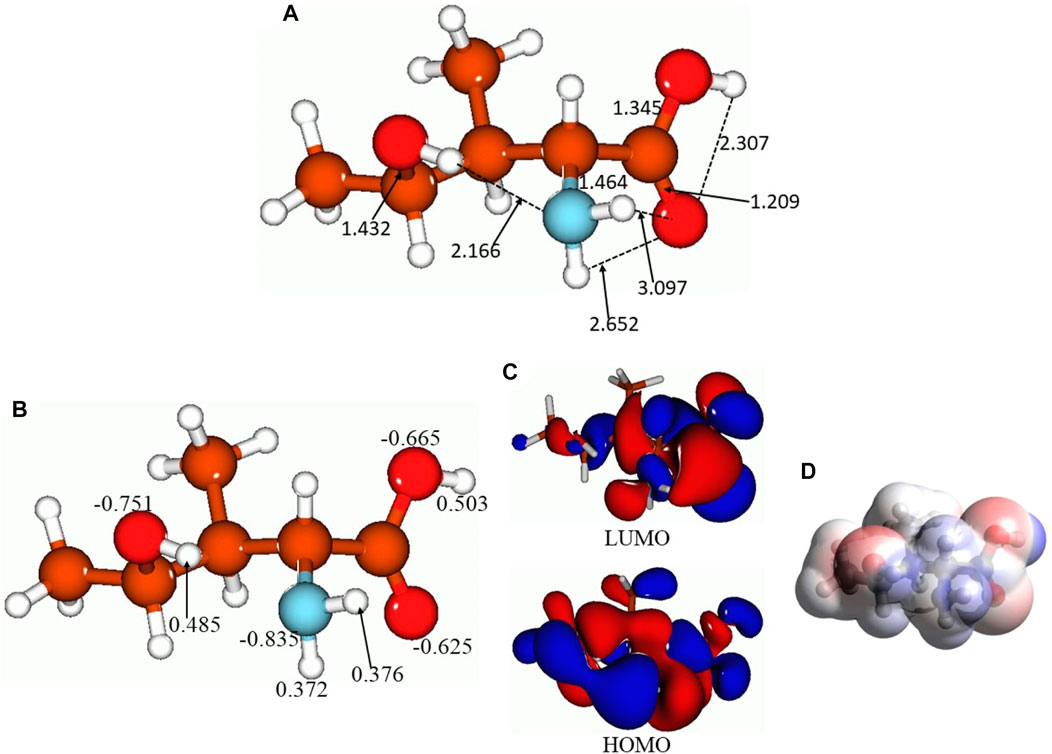
FIGURE 4. Lowest-lying isomer 5 of 4-hydroxyisoleucine: selected bond distances, Å (A), NBO charges on the selected atoms (B), FMOs (C), and MEP plot (D).
Table 8 summarizes the values of the GRPs calculated according to Eqs 1–6 (see Materials and Methods section).
Analysis of the GRP values in Table 9 shows the following. 1) The 4-hydroxyisoleucine compound has quite a high IP value, 7.21 eV, but low EA value, 0.35 eV. This implies that this compound should be a poor electron donor and a relatively poor electron acceptor, or, in other words, should be relatively stable in redox processes, which is also supported by its significant HOMO/LUMO gap value, 6.86 eV. 2) The global electronegativity X and global hardness η values of this compound are quite noticeable, 3.78 and 3.43 eV, respectively, which would imply that this compound might show electrophilic behavior in chemical processes but also should be relatively non-reactive and thus thermodynamically stable, which is also supported by its large HOMO/LUMO gap and noticeable value of its chemical potential, −3.78 eV. 3) Furthermore, the global softness of the compound has low value, 0.146 eV, which supports the relatively low reactivity of the compound, and global electrophilicity of the compound can be considered as relatively noticeable, 2.083 eV, which would imply that this compound might show electrophilic behavior in chemical processes.
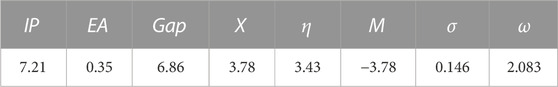
TABLE 9. Calculated GRPs for the isomer 5 (eV).
4 Discussion
The original rule of five (Lipinski’s RO5) dealt with orally active compounds and had defined four simple physicochemical parameters, i.e., molecular weight ≤ 500, log p ≤ 5 representing hydrophobicity, H-bond donors ≤5, and H-bond acceptors ≤10. These properties have been associated with 90% of the orally active drugs that have achieved phase II clinical status (Lipinski, 2004). 4-Hydroxyisoleucine is compliant with one of the most acceptable drug-like parameters. Passing RO5 means acceptable aqueous solubility along with intestinal permeability, which are the first steps in oral bioavailability. The RO5 has found its importance where medicinal and combinatorial chemistry produced thousands of compounds with very poor physicochemical properties. However, passing RO5 never guaranteed that a compound will be drug-like. But if a compound fails, then a high probability of oral activity problems exists.
The molecular weight of 4-hydroxyisoleucine is 147.09, which is ideal for oral drug designing. The intrinsic molecular volume is considered as a molecular descriptor in modeling physicochemical properties and the biological activity. The molecular volume is one of the most common descriptors in quantitative structure–activity relationship (QSAR) studies (Leahy, 1986; McGowan and Mellors, 1986; Consonni et al., 2002; Zhao et al., 2003a). It is related to different physicochemical properties and biological processes including intestinal absorption and BBB penetration, to name a few (Zhao et al., 2003b). The number of hydrogen bond acceptors and donors are within the required strict limits. Oral drugs usually have fewer H-bond acceptors, H-bond donors, and rotatable bonds (Bitew et al., 2021).
The parameter of rotatable bond count is used as “drug filter.” More than 10 rotatable bonds are associated with decreased rat oral bioavailability (Veber et al., 2002). The mechanistic basis for this “rotatable bond filter” is not clear, since its count does not relate to the in vivo clearance rate in rats. However, the filter still remains reasonable from a viewpoint of in vitro screening as ligand affinity decreases at an average of 0.5 kcal for each two rotatable bonds (Dashti et al., 2014). The number of aromatic and heteroaromatic rings in a molecule has a relation with aqueous solubility, lipophilicity, oral bioavailability, serum albumin binding, CYP450 inhibition, and hERG inhibition. The fewer the aromatic rings in an oral drug candidate, the greater is the developable probability of a drug candidate. However, >3 aromatic rings in a molecule are linked with poorer compound developability and an increased risk of knock out. Even within the defined lipophilicity range, increasing aromatic rings lead to decreased aqueous solubility (Ritchie and Macdonald, 2009).
Saturated heterocycles are metabolized at the position of heteroatom or adjacent to it (Jean and Fotsch, 2012). The heteroatom is sometimes beneficial in drug metabolism like nitrogen in 4-hydroxyisoleucine. Redox reactions of drug metabolism such as heteroatom dealkylation, hydroxylation, heteroatom oxygenation, reduction, and dehydrogenation can yield active metabolites. Rarely, even conjugation reactions can give rise to an active metabolite (Obach, 2013). 4-Hydroxyisoleucine has a single rigid bond. This makes the molecule flexible enough to achieve a stable conformation at its target. The optimal range for drug-like property, TPSA, is 0–140 Å2, that of 4-Hydroxyisoleucine within accurate limit (83.55 Å2). Molecules with a TPSA of ≥140 Å2 would be poorly absorbed (<10% of the fraction absorbed), while a TPSA of 60 Å2 favors >90% absorption (Clark, 1999).
The calculation of QED score uses eight important properties that were previously used to assess drug-likeness, i.e., molecular weight, octanol–water partition coefficient (ALOGP), number of H-bond donors, number of H-bond acceptors, molecular polar surface area, number of rotatable bonds, number of aromatic rings, and number of structural alerts (ALERTS). ALERTS is a list of undesirable chemical features such as chemical reactivity or perceived toxicity. Drugs with high QED scores achieve higher absorption and bioavailability, have lower doses, fewer drug–drug interaction warnings and P-glycoprotein interactions, and lower absorption issues due to a food-related effect. Nonetheless, it is surprising that high QED-scoring drugs have similarities to low-scoring drugs with respect to plasma free fraction, the extent of gut-wall metabolism, first-pass hepatic extraction/metabolism, volume of distribution, clearance, elimination half-life, and frequency of dosing (Ritchie and Macdonald, 2014). In this context, 4-hydroxyisoleucine lies at the borderline of attractive and unattractive molecules. Thus, this negligible amount of difference may not provide an impetus for rejection.
Fsp3 is the fraction of sp3 carbon atoms. It is the number of sp3-hybridized carbons out of the total carbon count which tells about carbon saturation of molecules and characterizes the complexity of their spatial structure (Lovering et al., 2009). Natural products generally possess a higher fraction of sp3 than synthetic compounds and so become a rich source of drugs (Jia et al., 2020). Yan and Gesteiger (2003) used it to characterize aliphatic degrees of molecules and ultimately predict solubility (Yan and Gasteiger, 2003). The optimum value of ≥0.42 for Fsp3 is considered a suitable value, and about 84% of marketed drugs meet this criterion (Kombo et al., 2013). However, sp3 content should be increased within an appropriate range; higher Fsp3 score does not always assure higher performance and can increase the difficulty of synthesis (Gerlach et al., 2019). However, critical discussions of Fsp3 scoring have helped in generating new potential descriptors, such as MCE-18, which is discussed in the following paragraphs, and calculated using the following equation (Wei et al., 2020).
A simple sp3 index (number of sp3-hybridized carbon atoms) can lead to a high rate of false-positive results, while the issue can be solved with MCE-18 descriptor. The composition, spatial geometry, and complexity of chemical structures especially for state-of-the-art molecules are not considered in sp3 index, as these special features cannot be reflected using only the portion of sp3-hybridized carbons. They focus on the nature and quality of sp3-rich frameworks rather than sp3 counts only (Ivanenkov et al., 2019).
4-Hydroxyisoleucine is a naturally derived molecule, and therefore, MCE-18 may not be the only guide toward drug discovery. MCE-18 is applied for the assessment of pharmacological novelty and may help in designing new potential chemical entities in modern drug design. This index is also used for profiling HTS-focused libraries and prioritization of molecules. Presumably, compounds with optimal MCE-18 values have greater reliable IP positions. It is a tracer of medicinal chemistry evolution and is used to assess the “distance” between old and new chemistry. During the past 10 years, two of the top four pharmaceutical companies have primarily focused on molecules with MCE-18 values over 60, while the remaining two companies have demonstrated less drug productivity but have reached similar scores (Ivanenkov et al., 2019). The natural product score is congruent with the real-time natural source of 4-hydroxyisoleucine, namely, fenugreek seeds (Fowden et al., 1973).
4-Hydroxyisoleucine is compliant with the Lipinski rule, Pfizer rule, and GSK rule. The parameters for the Pfizer rule are logP >3 and TPSA <75. Compounds with higher log P (>3) and low TPSA (<75) are more likely to be toxic. The most significant work which describes the influence of molecular properties on toxicological outcomes has led to the “3/75 rule.” This has been devised by Pfizer from an analysis of exploratory or dose-finding toxicology studies of 245 compounds. This approach defined that compound with logP <3 and tPSA >75Å2 are expected to be non-toxic (Hughes et al., 2008; Waring et al., 2015). However, 4-hydroxyisoleucine is non-toxic and classified as safe with the Pfizer rule. The GSK rule suggests MW ≤ 400 and logP ≤4 (Hann, 2011) which are well qualified by 4-hydroxyisoleucine. The Golden Triangle aids in achieving metabolically stable and permeable drug candidates. It is a visualization tool developed with three parameters i.e., in vitro permeability, in vitro clearance, and computational data (Johnson et al., 2009). However, the stability and permeation of 4-hydroxyisoleucine is already demonstrated (Gowtham et al., 2022). Thus, the predicted Golden Triangle rejection may be ruled out here.
PAINS, ALARM NMR, BMS, and chelator rule are in favor of 4-hydroxyisoleucine. Pan assay interference compounds have a tendency to produce false-positive hits in high-throughput screening. The mechanism of PAINS activity is poorly understood. However, PAINS are associated with protein reactivity and non-covalent interactions (Bolz et al., 2021). ALARM NMR is critical to recognize reactive compounds that should be prioritized for lead optimization. This has emerged as a new filtering tool that can identify nuisance compounds and ultimately improve the process of hit triage. It is a La assay to detect reactive molecules by NMR by monitoring dithiothreitol-dependent 13C chemical shift changes brought in the human La antigen (Huth et al., 2005; Zega, 2017; Dahlin et al., 2018). Thiol reactive compounds oxidize or form covalent adducts with thiol groups of proteins. Therefore, ALARM NMR is a sensitive tool to rapidly identify compounds with increased risk of side effects in humans, such as alcohol intolerance, reactive oxygen species generation, and drug–drug interactions (Huth et al., 2007). The BMS is an alert for mapping molecular promiscuity and identification of undesirable and reactive compounds, while the chelator rule reflects chelating compounds (Agrawal et al., 2010; Yang et al., 2021).
Caco-2 cell lines are still widely used in absorption studies. Sometime regarded as the “gold standard” technique, Caco-2 cells are used to validate other absorption assays (Artursson et al., 2001; Hidalgo, 2001; Ding et al., 2021; Murador et al., 2021). 4-Hydroxyisoleucine has a lower Caco-2 cell-predicted permeability. However, a considerable chance of MDCK permeability exists. The MDCK cell line is also used for permeability screening in drug discovery (BOKULIĆ et al., 2022). However, both ADMETLab 2.0 and SwissADME are in favor of better gastrointestinal absorption and bioavailability, indicated by the boiled-egg plot as well. However, there are no permeability results available from wet labs. 4-Hydroxyisoleucine is safe from clinically significant drug interactions. The absorption and excretion of Pgp substrate can be modified by its inducer/inhibitor, ultimately paving the way for drug interaction (Yamazaki et al., 2019). A Pgp inhibitor is more likely to have interactions (Telbisz et al., 2021; Terrier et al., 2021). ADMETLab 2.0 and SwissADME declared it as a non-Pgp substrate, diminishing the risk of potential drug interactions and drug resistance mechanisms. Metabolic enzyme inducers or inhibitors can increase or decrease the blood level of another drug which is a substrate for the target enzyme. This can lead to several pharmacokinetic drug interactions with CYP inhibitors or inducers (Bolleddula, 2021; Lang et al., 2021). Only a marginal chance of being CYP2CP substrate is predicted for 4-hydroxyisoleucine. However, no other risk exists for a significant drug interaction. To date, there are no studies available on 4-hydroxyisoleucine–Pgp interactions. Studies on the metabolism of 4-hydroxy isoleucine are also desirable to establish a complete pharmacokinetic profile.
Some of the pharmacokinetic parameters of 4-hydroxyisoleucine (10 mg/kg) were estimated after oral administration in Wistar rats using the ultra-performance liquid chromatography–tandem mass spectrometry-based method. The Vd was estimated as 3,123.59 ± 355.86 mL/kg (Wadhwa et al., 2020), which is lower than that calculated by ADMETLab. The clearance of 4-hydroxyisoleucine appeared to be 204.95 ± 23.97 mL/h/kg which is less than the rate determined by ADMETLab. However, the t1/2 10.83 ± 1.96 h is almost equivalent to that determined by ADMETLab (Wadhwa et al., 2020). The long half-life is congruent with the low clearance rate of 4-hydroxyisoleucine.
A drug with a moderate clearance rate and a better half-life may provide dose flexibility and better patient compliance (Sleep et al., 2013). Also, 4-hydroxy isoleucine is assumed to have a desirable clearance rate and half-life. However, a risk of CNS unwanted effects can be expected as 4-hydroxyisoleucine has the probability to be BBB permeant according to ADMETLab 2.0. Contrary to this, the boiled-egg plot predicted with SwissADME showed that it is non-permeant across the BBB (Daina and Zoete, 2016).
While evaluating toxicity, not even a single risk was found for hERG blockade, H-HT, DILI, Ames toxicity, rat oral acute toxicity, skin sensitization, carcinogenicity, and ophthalmic toxicity. Cardiac arrhythmia is one of the most frequently encountered side effects during drug discovery. This effect is related to inhibition of the hERG cardiac potassium channel. Also, this is the reason regulatory agencies such as US Food and Drug Administration (FDA) and European Medicines Agency recommend early evaluation of hERG toxicity (Garrido et al., 2020). Both ADMETLab 2.0 and Pred-hERG defined 4-hydroxyisoleucine as non-hERG blocker, being safe from cardiotoxicity. Hepatotoxicity is also highly responsible for withdrawal of an approved drug from the market. Hepatotoxicity accounts for >50% of acute liver failure cases as > 600 drugs are associated with it (Williams, 2018). Drug-induced liver injury is one of the leading factors of drug failure in clinical trials. Therefore, its assessment of drug candidates is necessary in advance as an effective strategy to decrease the drug attrition rate (He et al., 2019; Walker et al., 2020). The Ames toxicity test is an indicator for assessing mutagenic compounds’ potential and use bacteria for this assay. 4-HIL is likely to be safe from these types of toxicities (Petkov et al., 2021). However, the risk of respiratory toxicity has yet to be determined.
Toxicity studies on 4-hydroxyisoleucine are scant; however, studies on standardized extracts of its natural origin exist. Singh et al. (2022) evaluated the toxicity of 4-hydroxyisoleucine using human embryonic kidney cells (HEK-293) and healthy rats. The viability of HEK-293 cells was not affected by 4-hydroxyisoleucine, and no signs of toxicity were observed in healthy rats. Another study by Swaroop et al. identified Trigonella foenum-graecum seed extract (source of 4-Hydroxyisoleucine) as safe and efficacious. After extensive acute oral toxicity, 28 days’ sub-chronic toxicity and Ames’ bacterial reverse mutation assay, no toxicity was detected (Swaroop et al., 2014).
Deshpande et al. observed IDM01 (4-hydroxyisoleucine and trigonelline-based standardized fenugreek seed extract) as safe during acute and sub-chronic preclinical toxicity in rats without mutagenicity or genotoxicity. Not a single mortality or treatment-related adverse sign was noted during acute (≤2000 mg/kg) and sub-chronic (90-day repeated doses of 250/500/1,000 mg per kg body weight with 28 days of recovery period). The oral median lethal dose (LD50) was >2,000 mg/kg during the acute oral toxicity study. A non-observed adverse effect level was estimated at 500 mg/kg. The Ames test did not exhibit any mutagenicity up to 5,000 μg/plate and did not induce any structural chromosomal aberrations at < 50 mg/culture (Deshpande et al., 2017).
A genotoxicity study of the fenugreek seed extract (≤40% 4-hydroxyisoleucine) was carried out via standard battery of tests such as reverse mutation assay, mouse lymphoma forward mutation assay, and mouse micronucleus assay. There was no genotoxicity exhibited under the tested conditions (Flammang et al., 2004).
Bioconcentration represents the accumulation of chemicals in organisms at higher concentration than its surrounding (Jambor and Weisener, 2005). Bioconcentration factors are considered to assess secondary poisoning potential and risks to human health via the food chain. The factor is an estimate of the residual organic chemicals used for ranking chemicals as possible hazards to the environment (Gramatica and Papa, 2003). Similarly, the toxicity of organic aromatic compounds is based on logarithm of 50% growth inhibitory concentration (log (IGC50-1)) of Tetrahymena pyriformis, and fathead minnow which have extensive use in ecotoxicology and environmental safety applications (Keshavarz et al., 2021). The environmental toxicity of 4-hydroxyisoleucine exhibited a higher LC50, favoring the evidence of being non-ecotoxic. But the most acceptable evidence can be its origin as natural product, being part of nature chemistry (Avalos-Soriano et al., 2016). In the same manner, no risk of toxicity is predicted for androgen and estrogen receptors, PPAR-γ, heat shock element, mitochondrial membrane potential, and p53 (Bova et al., 2005; Ciocca et al., 2013; Khoo et al., 2014; Barna et al., 2018; Wahl and Smieško, 2018; Xi et al., 2020; Ciallella et al., 2021). Androgen-disruptors include all those chemicals which interfere with the biosynthesis, metabolism, or its physiological action. This results in an abnormal male development characteristic and poor growth and function of reproductive tract. Since androgen is the main regulator, its disruptors can harm reproductive developmental processes (Luccio-Camelo and Prins, 2011). In the same manner, estrogen disruptors can harm the female reproductive programming as the estrogen receptor is the primary determinant of female characteristics (Tsakovska et al., 2011). However, 4-hydroxyisoleucine is devoid of such toxicities and is considered non-toxic.
Mitochondrial membrane potential is generated by proton pumps and one of the essential components in the energy storage process. It is kept stable, and little variations are allowed; however, long-lasting disturbance may lead to adverse pathologies and loss of cell viability (Zorova et al., 2018). 4-Hydroxyisoleucine is free from such disruption of mitochondrial membrane potential. Similarly, genetic toxicology is important for long-term carcinogenicity during the early drug development process. This genotoxicity testing can identify the potentially hazardous drug candidates which may be knocked out at later stages of the drug development process (Custer and Sweder, 2008). However, ADMETLab recommends 4-hydroxyisoleucine as non-carcinogenic and non-genotoxic.
As all these parameters are modifiers of normal human physiology, and their disturbance leads to a red alert toxicity. Furthermore, none of the toxicophores were found in 4-hydroxyisoleucine which may have led to aquatic toxicity, skin sensitization, or created an acute toxicity response. The molecule has proven to be biodegradable and is not enlisted in the SureChEMBL database, implying medicinal chemistry friendly status (Papadatos et al., 2016; Falaguera and Mestres, 2021). The DFT study results provide certain support for the aforementioned discussion. Thus, formation of intra- and intermolecular hydrogen bonds by the 4-hydroxyisoleucine molecules, along with negative and positive MEP accumulation and significant NBO charges, would imply strong interactions with polar solvent molecules, including water, and thus acceptable aqueous solubility of the compound. Also, this would imply interaction of the 4-hydroxyisoleucine molecules with polar parts of large organic molecules. Furthermore, the shown presence of a few H-bond donors and acceptors would support the suggestion of the potential of the compound as an oral drug. Moreover, formation of several low-lying structural isomers confirms that the molecule is flexible enough to achieve a stable conformation at its target. The 4-hydroxyisoleucine stability shown by the GRP analysis supports its long half-life and stability findings achieved by other approaches.
5 Conclusion
To determine the knockout of 4-hydroxyisoleucine, a detailed ADMET profile was predicted via the ADMETLab 2.0 database. This molecule has satisfactory physicochemical properties and medicinal chemistry. The pharmacokinetic profile is in the favor of oral bioavailability, efficient distribution to all tissues including the brain, and non-interruptive drug metabolism, with moderate clearance and half-life. Some of the actual PK parameters are almost comparable to in silico values, but further detailed studies are desirable in the lab. There is no risk of any toxicity predicted both in silico and inside lab, except with low probability of respiratory toxicity predicted. Even it is undesirable for this molecule to disturb some of the most important receptors/targets such as gonadal receptors, p53, heat shock factor, mitochondrial membrane potential, or PPAR-γ. The molecule is safe for the environment, with agreeable lethal concentrations for some of the most common indicators. Finally, not a single toxicophore alert was found for 4-hydroxyisoleucine. All these properties suggest the drug ability of 4-hydroxy isoleucine. DFT study results are in support of the findings obtained by other methods.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Author contributions
IA and AK have written a major part of the article, AP and KA supported in writing the draft, MD and HK supervised the overall article and finalized the submission.
Acknowledgments
This work was partially supported by Taif University Researchers Supporting Program (project number: TURSP-2020/153), Taif Univer Saudi and by the supercomputing infrastructure of the NLHPC (ECM-02).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abou El-Soud, N. H., Khalil, M. Y., Hussein, J. S., Oraby, F. S. H., Farrag, A. R. H., et al. (2007). Antidiabetic effects of fenugreek alkaliod extract in streptozotocin induced hyperglycemic rats. J. Appl. Sci. Res. 3 (10), 1073–1083.
Agrawal, A., Johnson, S., Jacobsen, J., Miller, M., Chen, L. H., Pellecchia, M., et al. (2010). Chelator fragment libraries for targeting metalloproteinases. ChemMedChem 5 (2), 195–199. doi:10.1002/cmdc.200900516
Artursson, P., Palm, K., and Luthman, K. (2001). Caco-2 monolayers in experimental and theoretical predictions of drug transport1PII of original article: S0169-409X(96)00415-2. The article was originally published in advanced drug delivery reviews 22 (1996) 67–84.1. Adv. drug Deliv. Rev. 46 (1-3), 27–43. doi:10.1016/s0169-409x(00)00128-9
Avalos-Soriano, A., De la Cruz-Cordero, R., Rosado, J., and Garcia-Gasca, T. (2016). 4-Hydroxyisoleucine from fenugreek (Trigonella foenum-graecum): Effects on insulin resistance associated with obesity. Molecules 21 (11), 1596. doi:10.3390/molecules21111596
Axel, D. B. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98 (7), 5648–5652. doi:10.1063/1.464913
Banerjee, P., Eckert, A. O., Schrey, A. K., and Preissner, R. (2018). ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic acids Res. 46 (1), W257–W263. doi:10.1093/nar/gky318
Barna, J., Csermely, P., and Vellai, T. (2018). Roles of heat shock factor 1 beyond the heat shock response. Cell. Mol. Life Sci. 75 (16), 2897–2916. doi:10.1007/s00018-018-2836-6
Bitew, M., Desalegn, T., Demissie, T. B., Belayneh, A., Endale, M., and Eswaramoorthy, R. (2021). Pharmacokinetics and drug-likeness of antidiabetic flavonoids: Molecular docking and DFT study. Plos one 16 (12), e0260853. doi:10.1371/journal.pone.0260853
Bokulić, A., Padovan, J., Stupin-Polancec, D., and Milic, A. (2022). Isolation of MDCK cells with low expression of mdr1 gene and their use inmembrane permeability screening. Acta Pharm. 72 (2), 275–288. doi:10.2478/acph-2022-0003
Bolleddula, J., (2021). PBPK modeling to predict drug-drug interactions of ivosidenib as a perpetrator in cancer patients and qualification of the Simcyp platform for CYP3A4 induction. CPT: Pharmacometrics and Systems Pharmacology.
Bolz, S. N., Adasme, M. F., and Schroeder, M. (2021). Toward an understanding of pan-assay interference compounds and promiscuity: A structural perspective on binding modes. J. Chem. Inf. Model. 61 (5), 2248–2262. doi:10.1021/acs.jcim.0c01227
Bova, M. P., Tam, D., McMahon, G., and Mattson, M. N. (2005). Troglitazone induces a rapid drop of mitochondrial membrane potential in liver HepG2 cells. Toxicol. Lett. 155 (1), 41–50. doi:10.1016/j.toxlet.2004.08.009
Chakraborty, A., Pan, S., and Chattaraj, P. K. (2013). “Biological activity and toxicity: A conceptual DFT approach,” in Applications of density functional theory to biological and bioinorganic chemistry, 143–179.
Ciallella, H. L., Russo, D. P., Aleksunes, L. M., Grimm, F. A., and Zhu, H. (2021). Predictive modeling of estrogen receptor agonism, antagonism, and binding activities using machine-and deep-learning approaches. Lab. Investig. 101 (4), 490–502. doi:10.1038/s41374-020-00477-2
Ciocca, D. R., Arrigo, A. P., and Calderwood, S. K. (2013). Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: An update. Archives Toxicol. 87 (1), 19–48. doi:10.1007/s00204-012-0918-z
Clark, D. E. (1999). Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J. Pharm. Sci. 88 (8), 807–814. doi:10.1021/js9804011
Consonni, V., Todeschini, R., Pavan, M., and Gramatica, P. (2002). Structure/response correlations and similarity/diversity analysis by GETAWAY descriptors. 2. Application of the novel 3D molecular descriptors to QSAR/QSPR studies. J. Chem. Inf. Comput. Sci. 42 (3), 693–705. doi:10.1021/ci0155053
Custer, L., and Sweder, K. (2008). The role of genetic toxicology in drug discovery and optimization. Curr. drug Metab. 9 (9), 978–985. doi:10.2174/138920008786485191
Dahlin, J. L., Cuellar, M., Singh, G., Nelson, K. M., Strasser, J. M., Rappe, T., et al. (2018). ALARM NMR for HTS triage and chemical probe validation. Curr. Protoc. Chem. Biol. 10 (1), 91–117. doi:10.1002/cpch.35
Daina, A., Michielin, O., and Zoete, V. (2017). SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7 (1), 42717. doi:10.1038/srep42717
Daina, A., and Zoete, V. (2016). A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 11 (11), 1117–1121. doi:10.1002/cmdc.201600182
Dashti, Y., Grkovic, T., and Quinn, R. J. (2014). Predicting natural product value, an exploration of anti-TB drug space. Nat. Product. Rep. 31 (8), 990–998. doi:10.1039/c4np00021h
Deshpande, P. O., Mohan, V., and Thakurdesai, P. A. (2017). Preclinical toxicological evaluation of IDM01: The botanical composition of 4-hydroxyisoleucine-and trigonelline-based standardized fenugreek seed extract. Pharmacogn. Res. 9 (2), 138–150. doi:10.4103/0974-8490.204649
Ding, X., Hu, X., Chen, Y., Xie, J., Ying, M., Wang, Y., et al. (2021). Differentiated Caco-2 cell models in food-intestine interaction study: Current applications and future trends. Trends Food Sci. Technol. 107, 455–465. doi:10.1016/j.tifs.2020.11.015
Dionne, E., Hanson, M. L., Anderson, J. C., and Brain, R. A. (2021). Chronic toxicity of technical atrazine to the fathead minnow (Pimephales promelas) during a full life-cycle exposure and an evaluation of the consistency of responses. Sci. Total Environ. 755, 142589. doi:10.1016/j.scitotenv.2020.142589
Falaguera, M. J., and Mestres, J. (2021). Identification of the core chemical structure in SureChEMBL patents. J. Chem. Inf. Model. 61 (5), 2241–2247. doi:10.1021/acs.jcim.1c00151
Flammang, A., Cifone, M., Erexson, G., and Stankowski, L. (2004). Genotoxicity testing of a fenugreek extract. Food Chem. Toxicol. 42 (11), 1769–1775. doi:10.1016/j.fct.2004.07.003
Fowden, L., Pratt, H. M., and Smith, A. (1973). 4-Hydroxyisoleucine from seed of Trigonella foenum-graecum. Phytochemistry 12 (7), 1707–1711. doi:10.1016/0031-9422(73)80391-7
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16, revision B. 01. Wallingford, CT: Gaussian.
Garrido, A., Lepailleur, A., Mignani, S. M., Dallemagne, P., and Rochais, C. (2020). hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem. 195, 112290. doi:10.1016/j.ejmech.2020.112290
Geerlings, P., De Proft, F., and Langenaeker, W. (2003). Conceptual density functional theory. Chem. Rev. 103 (5), 1793–1874. doi:10.1021/cr990029p
Gerlach, E. M., Korkmaz, M. A., Pavlinov, I., Gao, Q., and Aldrich, L. N. (2019). Systematic diversity-oriented synthesis of reduced flavones from γ-pyrones to probe biological performance diversity. ACS Chem. Biol. 14 (7), 1536–1545. doi:10.1021/acschembio.9b00294
Gowtham, L., Ujjalkumar, D., Moksha, L., Roy, T. K., Halder, N., and Velpandian, T. (2022). Hydrophilic interaction LC–MS/MS method to avoid endogenous interference in the analysis of 4-hydroxy isoleucine from dietary supplementation of fenugreek. J. Sep. Sci. 45, 1210–1221. doi:10.1002/jssc.202100894
Gramatica, P., and Papa, E. (2003). QSAR modeling of bioconcentration factor by theoretical molecular descriptors. QSAR Comb. Sci. 22 (3), 374–385. doi:10.1002/qsar.200390027
Hann, M. M. (2011). Molecular obesity, potency and other addictions in drug discovery. MedChemComm 2 (5), 349–355. doi:10.1039/c1md00017a
Hanwell, M. D., Curtis, D. E., Lonie, D. C., Vandermeersch, T., Zurek, E., and Hutchison, G. R. (2012). Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. cheminformatics 4 (1), 17. doi:10.1186/1758-2946-4-17
He, S., Ye, T., Wang, R., Zhang, C., Zhang, X., Sun, G., et al. (2019). An in silico model for predicting drug-induced hepatotoxicity. Int. J. Mol. Sci. 20 (8), 1897. doi:10.3390/ijms20081897
Hidalgo, I. J. (2001). Assessing the absorption of new pharmaceuticals. Curr. Top. Med. Chem. 1 (5), 385–401. doi:10.2174/1568026013395010
Hughes, J. D., Blagg, J., Price, D. A., Bailey, S., DeCrescenzo, G. A., Devraj, R. V., et al. (2008). Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 18 (17), 4872–4875. doi:10.1016/j.bmcl.2008.07.071
Huth, J. R., Mendoza, R., Olejniczak, E. T., Johnson, R. W., Cothron, D. A., Liu, Y., et al. (2005). Alarm NMR: A rapid and robust experimental method to detect reactive false positives in biochemical screens. J. Am. Chem. Soc. 127 (1), 217–224. doi:10.1021/ja0455547
Huth, J. R., Song, D., Mendoza, R. R., Black-Schaefer, C. L., Mack, J. C., Dorwin, S. A., et al. (2007). Toxicological evaluation of thiol-reactive compounds identified using a la assay to detect reactive molecules by nuclear magnetic resonance. Chem. Res. Toxicol. 20 (12), 1752–1759. doi:10.1021/tx700319t
Ivanenkov, Y. A., Zagribelnyy, B. A., and Aladinskiy, V. A. (2019). Are we opening the door to a new era of medicinal chemistry or being collapsed to a chemical singularity? Perspective. J. Med. Chem. 62 (22), 10026–10043. doi:10.1021/acs.jmedchem.9b00004
Jambor, J., and Weisener, C. (2005). The geochemistry of acid mine drainage. Environ. Geochem. 9, 149.
Jean, St.D. J., and Fotsch, C. (2012). Mitigating heterocycle metabolism in drug discovery. J. Med. Chem. 55 (13), 6002–6020. doi:10.1021/jm300343m
Jetté, L., Harvey, L., Eugeni, K., and Levens, N. (2000). 4-Hydroxyisoleucine: A plant-derived treatment for metabolic syndrome. Curr. Opin. investigational drugs 10(4). 353–358.
Jia, C.-Y., Li, J. Y., Hao, G. F., and Yang, G. F. (2020). A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today 25 (1), 248–258. doi:10.1016/j.drudis.2019.10.014
Johnson, T. W., Dress, K. R., and Edwards, M. (2009). Using the Golden Triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett. 19 (19), 5560–5564. doi:10.1016/j.bmcl.2009.08.045
Jorio, S., Salah, M., Abou El Makarim, H., and Tabyaoui, M. (2019). Reactivity indices related to DFT theory, the electron localization function (ELF) and non-covalent interactions (NCI) calculations in the formation of the non-halogenated pyruvic esters in solution. Mediterr. J. Chem. 8 (6), 476–485. doi:10.13171/mjc8619072612sj
Keshavarz, M. H., Shirazi, Z., and Sheikhabadi, P. K. (2021). Risk assessment of organic aromatic compounds to Tetrahymena pyriformis in environmental protection by a simple QSAR model. Process Saf. Environ. Prot. 150, 137–147. doi:10.1016/j.psep.2021.04.011
Khoo, K. H., Verma, C. S., and Lane, D. P. (2014). Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 13 (3), 217–236. doi:10.1038/nrd4236
Kombo, D. C., Tallapragada, K., Jain, R., Chewning, J., Mazurov, A. A., Speake, J. D., et al. (2013). 3D molecular descriptors important for clinical success. J. Chem. Inf. Model. 53 (2), 327–342. doi:10.1021/ci300445e
Lagorce, D., Bouslama, L., Becot, J., Miteva, M. A., and Villoutreix, B. O. (2017). FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 33 (22), 3658–3660. doi:10.1093/bioinformatics/btx491
Lang, J., Vincent, L., Chenel, M., Ogungbenro, K., and Galetin, A. (2021). Impact of hepatic CYP3A4 ontogeny functions on drug–drug interaction risk in pediatric physiologically-based pharmacokinetic/pharmacodynamic modeling: Critical literature review and ivabradine case study. Clin. Pharmacol. Ther. 109 (6), 1618–1630. doi:10.1002/cpt.2134
Leahy, D. E. (1986). Intrinsic molecular volume as a measure of the cavity term in linear solvation energy relationships: Octanol-water partition coefficients and aqueous solubilities. J. Pharm. Sci. 75 (7), 629–636. doi:10.1002/jps.2600750702
Lipinski, C. A. (2004). Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 1 (4), 337–341. doi:10.1016/j.ddtec.2004.11.007
Lovering, F., Bikker, J., and Humblet, C. (2009). Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 52 (21), 6752–6756. doi:10.1021/jm901241e
Luccio-Camelo, D. C., and Prins, G. S. (2011). Disruption of androgen receptor signaling in males by environmental chemicals. J. steroid Biochem. Mol. Biol. 127 (1-2), 74–82. doi:10.1016/j.jsbmb.2011.04.004
McGowan, J. C., and Mellors, A. (1986). Molecular volumes in chemistry and biology. Chichester: E. Horwood.
Murador, D. C., De Souza Mesquita, L. M., Neves, B. V., Braga, A. R., Martins, P. L., Zepka, L. Q., et al. (2021). Bioaccessibility and cellular uptake by caco-2 cells of carotenoids and chlorophylls from orange peels: A comparison between conventional and ionic liquid mediated extractions. Food Chem. 339, 127818. doi:10.1016/j.foodchem.2020.127818
Narender, T., Puri, A., Khaliq, T., Saxena, R., Bhatia, G., Chandra, R., et al. (2006). 4-Hydroxyisoleucine an unusual amino acid as antidyslipidemic and antihyperglycemic agent. Bioorg. Med. Chem. Lett. 16 (2), 293–296. doi:10.1016/j.bmcl.2005.10.003
Obach, R. S. (2013). Pharmacologically active drug metabolites: Impact on drug discovery and pharmacotherapy. Pharmacol. Rev. 65 (2), 578–640. doi:10.1124/pr.111.005439
Papadatos, G., Davies, M., Dedman, N., Chambers, J., Gaulton, A., Siddle, J., et al. (2016). SureChEMBL: A large-scale, chemically annotated patent document database. Nucleic acids Res. 44 (1), D1220–D1228. doi:10.1093/nar/gkv1253
Petkov, P., Ivanova, H., Schultz, T., and Mekenyan, O. (2021). Criteria for assessing the reliability of toxicity predictions: I. TIMES ames mutagenicity model. Comput. Toxicol. 17, 100143. doi:10.1016/j.comtox.2020.100143
Pires, D. E., Blundell, T. L., and Ascher, D. B. (2015). pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 58 (9), 4066–4072. doi:10.1021/acs.jmedchem.5b00104
Reed, A. E., Curtiss, L. A., and Weinhold, F. (1988). Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 88 (6), 899–926. doi:10.1021/cr00088a005
Ritchie, T. J., and Macdonald, S. J. (2014). How drug-like are ‘ugly’drugs: Do drug-likeness metrics predict ADME behaviour in humans? Drug Discov. Today 19 (4), 489–495. doi:10.1016/j.drudis.2014.01.007
Ritchie, T. J., and Macdonald, S. J. (2009). The impact of aromatic ring count on compound developability–are too many aromatic rings a liability in drug design? Drug Discov. today 14 (21-22), 1011–1020. doi:10.1016/j.drudis.2009.07.014
Sauvaire, Y., Petit, P., Broca, C., Manteghetti, M., Baissac, Y., Fernandez-Alvarez, J., et al. (1998). 4-Hydroxyisoleucine: A novel amino acid potentiator of insulin secretion. Diabetes 47 (2), 206–210. doi:10.2337/diab.47.2.206
Schaftenaar, G., and Noordik, J. H. (2000). Molden: A pre-and post-processing program for molecular and electronic structures. J. computer-aided Mol. Des. 14 (2), 123–134. doi:10.1023/a:1008193805436
Singh, P., Ishteyaque, S., Prajapati, R., Yadav, K. S., Singh, R., Kumar, A., et al. (2022). Assessment of antidiabetic effect of 4-HIL in type 2 diabetic and healthy Sprague Dawley rats. Hum. Exp. Toxicol. 41, 096032712110618. doi:10.1177/09603271211061873
Sleep, D., Cameron, J., and Evans, L. R. (2013). Albumin as a versatile platform for drug half-life extension. Biochimica Biophysica Acta (BBA)-General Subj. 1830 (12), 5526–5534. doi:10.1016/j.bbagen.2013.04.023
Swaroop, A., Bagchi, M., Kumar, P., Preuss, H. G., Tiwari, K., Marone, P. A., et al. (2014). Safety, efficacy and toxicological evaluation of a novel, patented anti-diabetic extract of Trigonella foenum-graecum Seed Extract (Fenfuro). Toxicol. Mech. methods 24 (7), 495–503. doi:10.3109/15376516.2014.943443
Telbisz, Á., Ambrus, C., Mozner, O., Szabo, E., Varady, G., Bakos, E., et al. (2021). Interactions of potential anti-COVID-19 compounds with multispecific ABC and OATP drug transporters. Pharmaceutics 13 (1), 81. doi:10.3390/pharmaceutics13010081
Terrier, J., Gaspar, F., Fontana, P., Daali, Y., Reny, J. L., Csajka, C., et al. (2021). Drug-drug interactions with direct oral anticoagulants: Practical recommendations for clinicians. Am. J. Med. 134 (8), 939–942. doi:10.1016/j.amjmed.2021.04.003
Tomasi, J., Mennucci, B., and Cammi, R. (2005). Quantum mechanical continuum solvation models. Chem. Rev. 105 (8), 2999–3094. doi:10.1021/cr9904009
Tsakovska, I., Pajeva, I., Alov, P., and Worth, A. (2011). Recent advances in the molecular modeling of estrogen receptor-mediated toxicity. Adv. Protein Chem. Struct. Biol. 85, 217–251. doi:10.1016/B978-0-12-386485-7.00006-5
Veber, D. F., Johnson, S. R., Cheng, H. Y., Smith, B. R., Ward, K. W., and Kopple, K. D. (2002). Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45 (12), 2615–2623. doi:10.1021/jm020017n
Wadhwa, G., Krishna, K. V., Taliyan, R., Tandon, N., Yadav, S. S., Katiyar, C., et al. (2020). Pre-clinical pharmacokinetic and pharmacodynamic modelling study of 4-hydroxyisoleucine using validated ultra-performance liquid chromatography-tandem mass spectrometry. RSC Adv. 10 (10), 5525–5532. doi:10.1039/c9ra08121f
Wahl, J., and Smieško, M. (2018). Endocrine disruption at the androgen receptor: Employing molecular dynamics and docking for improved virtual screening and toxicity prediction. Int. J. Mol. Sci. 19 (6), 1784. doi:10.3390/ijms19061784
Walker, P. A., Ryder, S., Lavado, A., Dilworth, C., and Riley, R. J. (2020). The evolution of strategies to minimise the risk of human drug-induced liver injury (DILI) in drug discovery and development. Archives Toxicol. 94 (8), 2559–2585. doi:10.1007/s00204-020-02763-w
Waring, M. J., Arrowsmith, J., Leach, A. R., Leeson, P. D., Mandrell, S., Owen, R. M., et al. (2015). An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 14 (7), 475–486. doi:10.1038/nrd4609
Wei, W., Cherukupalli, S., Jing, L., Liu, X., Zhan, P., et al. (2020). Fsp3: A new parameter for drug-likeness. Amsterdam: Drug Discovery Today.
Weigend, F. (2006). Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 8 (9), 1057–1065. doi:10.1039/b515623h
Weigend, F., and Ahlrichs, R. (2005). Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7 (18), 3297–3305. doi:10.1039/b508541a
Williams, D. P. (2018). Application of hepatocyte-like cells to enhance hepatic safety risk assessment in drug discovery. Philosophical Trans. R. Soc. B Biol. Sci. 373 (1750), 20170228. doi:10.1098/rstb.2017.0228
Xi, Y., Zhang, Y., Zhu, S., Luo, Y., Xu, P., and Huang, Z. (2020). PPAR-mediated toxicology and applied pharmacology. Cells 9 (2), 352. doi:10.3390/cells9020352
Xiong, G., Wu, Z., Yi, J., Fu, L., Yang, Z., Hsieh, C., et al. (2021). ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res.
Yamazaki, S., Costales, C., Lazzaro, S., Eatemadpour, S., Kimoto, E., and Varma, M. V. (2019). Physiologically-based pharmacokinetic modeling approach to predict rifampin-mediated intestinal p-glycoprotein induction. CPT pharmacometrics Syst. Pharmacol. 8 (9), 634–642. doi:10.1002/psp4.12458
Yan, A., and Gasteiger, J. (2003). Prediction of aqueous solubility of organic compounds based on a 3D structure representation. J. Chem. Inf. Comput. Sci. 43 (2), 429–434. doi:10.1021/ci025590u
Yang, H., Lou, C., Sun, L., Li, J., Cai, Y., Wang, Z., et al. (2019). admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 35 (6), 1067–1069. doi:10.1093/bioinformatics/bty707
Yang, Z.-Y., Lu, A. P., Hou, T. J., and Cao, D. S. (2021). Scopy: An integrated negative design python library for desirable HTS/VS database design. Briefings Bioinforma. 22 (3), bbaa194. doi:10.1093/bib/bbaa194
Zega, A. (2017). NMR methods for identification of false positives in biochemical screens: Miniperspective. J. Med. Chem. 60 (23), 9437–9447. doi:10.1021/acs.jmedchem.6b01520
Zhao, Y. H., Abraham, M. H., Hersey, A., and Luscombe, C. N. (2003). Quantitative relationship between rat intestinal absorption and Abraham descriptors. Eur. J. Med. Chem. 38 (11-12), 939–947. doi:10.1016/j.ejmech.2003.07.005
Zhao, Y. H., Abraham, M. H., and Zissimos, A. M. (2003). Fast calculation of van der Waals volume as a sum of atomic and bond contributions and its application to drug compounds. J. Org. Chem. 68 (19), 7368–7373. doi:10.1021/jo034808o
Keywords: 4-hydroxyisoleucine, pharmacokinetics, computational chemistry, drug likeness, DFT
Citation: Ahmad I, Kuznetsov AE, Pirzada AS, Alsharif KF, Daglia M and Khan H (2023) Computational pharmacology and computational chemistry of 4-hydroxyisoleucine: Physicochemical, pharmacokinetic, and DFT-based approaches. Front. Chem. 11:1145974. doi: 10.3389/fchem.2023.1145974
Received: 20 February 2023; Accepted: 21 March 2023;
Published: 13 April 2023.
Edited by:
Nino Russo, University of Calabria, ItalyReviewed by:
Isabella Romeo, Magna Græcia University, ItalyZoran S. Markovic, University of Kragujevac, Serbia
Copyright © 2023 Ahmad, Kuznetsov, Pirzada, Alsharif, Daglia and Khan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haroon Khan, hkdr2006@gmail.com