PGE2 suppression of innate immunity during mucosal bacterial infection
- Department of Microbiology and Immunology, Tulane University School of Medicine, New Orleans, LA, USA
Prostaglandin E2 (PGE2) is an important lipid mediator in inflammatory and immune responses during acute and chronic infections. Upon stimulation by various proinflammatory stimuli such as lipopolysaccharide (LPS), interleukin (IL)-1β, and tumor necrosis factor (TNF)-α, PGE2 synthesis is upregulated by the expression of cyclooxygenases. Biologically active PGE2 is then able to signal through four primary receptors to elicit a response. PGE2 is a critical molecule that regulates the activation, maturation, migration, and cytokine secretion of several immune cells, particularly those involved in innate immunity such as macrophages, neutrophils, natural killer cells, and dendritic cells. Both Gram-negative and Gram-positive bacteria can induce PGE2 synthesis to regulate immune responses during bacterial pathogenesis. This review will focus on PGE2 in innate immunity and how bacterial pathogens influence PGE2 production during enteric and pulmonary infections. The conserved ability of many bacterial pathogens to promote PGE2 responses during infection suggests a common signaling mechanism to deter protective pro-inflammatory immune responses. Inhibition of PGE2 production and signaling during infection may represent a therapeutic alternative to treat bacterial infections. Further study of the immunosuppressive effects of PGE2 on innate immunity will lead to a better understanding of potential therapeutic targets within the PGE2 pathway.
Introduction
Prostaglandin E2 (PGE2) is an important lipid mediator in inflammatory and immune responses during acute and chronic infections (Phipps et al., 1991; Yu and Chadee, 1998; Harris et al., 2002; Nagamatsu and Schust, 2010). Upon stimulation by various proinflammatory stimuli such as lipopolysaccharide (LPS), interleukin (IL)-1β, and tumor necrosis factor (TNF)-α, PGE2 synthesis is upregulated by the expression of one of three cyclooxygenases (Filion et al., 2001; Kis et al., 2006; Park et al., 2006). Biologically active PGE2 is then able to signal through four primary receptors to elicit a response (Sugimoto et al., 1992; Honda et al., 1993; Nishigaki et al., 1996; Hata and Breyer, 2004). Molecular concentrations of PGE2 and receptor signaling are both influential in regulating proinflammatory and immunosuppressive immune cell phenotypes (Kalinski, 2012). PGE2 is a critical molecule that regulates the activation, maturation, migration, and cytokine secretion of several immune cells, particularly those involved in innate immunity such as macrophages, neutrophils, natural killer cells, and dendritic cells (Bankhurst, 1982; Goto et al., 1983; Kaliński et al., 1997; Yu and Chadee, 1998; Aronoff et al., 2004; Serezani et al., 2007; Nagamatsu and Schust, 2010). Both Gram-negative and Gram-positive bacteria can induce PGE2 synthesis to regulate immune responses during bacterial pathogenesis (Harris et al., 2002; Hessle et al., 2003). This review will focus on PGE2 in innate immunity and how bacterial pathogens influence PGE2 production during enteric and pulmonary infections. Inhibition of PGE2 production, recognition, and signaling may lead to therapeutic alternatives to regulate the innate immune response during bacterial infection. Active mechanisms utilized by bacteria may also promote PGE2 synthesis during pathogenesis. Examination of these mechanisms could elicit a better understanding of disease progression and infection outcome.
PGE2 Production
While PGE2 can be produced by all cell types, immune cells are a primary source of PGE2 production during an inflammatory response (Kalinski, 2012). Within these cells, PGE2 is derived from the release of arachidonic acid (AA) from cell membranes by phospholipase A2 (PLA2) enzymes. While there are multiple members within the PLA2 family, the most utilized enzyme for PGE2 synthesis is the cytosolic calcium-dependent PLA2 (cPLA2) (Lambeau and Lazdunski, 1999). Subsequently, one of two primary cyclooxygenases utilizes AA as a substrate to produce the biological precursor prostaglandin H2 (PGH2). The two cyclooxygenases available for this reaction are COX-1 (constitutively active at basal levels) and COX-2 (highly inducible by inflammatory cytokines and growth factors) (Phipps et al., 1991). PGE2 is then enzymatically produced as an end product of the reaction with the aid of PGE2 synthase (PGES) (Park et al., 2006). Biologically active PGE2 can then readily signal through one of four eicosanoid receptors (EP) (Figure 1). The rate of PGE2 production during an immune response is primarily believed to be dependent upon the expression and activity of COX-2 (Kalinski, 2012), thus it is an important enzyme on which to focus when examining PGE2. PGE2 is relatively stable in vitro, yet is rapidly degraded in tissues by 15-hydroxyprostaglandin dehydrogenase (15-PGDH) (Fitzpatrick et al., 1980; Tai et al., 2002). Accordingly, in order to examine PGE2 under biological conditions, it is necessary to account for its rate of production via COX-2 and PGES and its degradation in response to different stimuli.
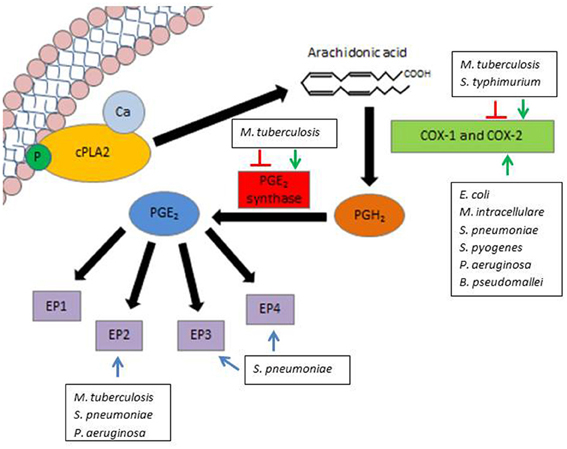
Figure 1. Modulation of PGE2 by pathogenic bacteria. PGE2 synthesis is initiated by the release of AA from the lipid cell membrane by the enzyme cPLA2. Either COX-1 (constitutively expressed) or COX-2 (inducible) can utilize AA as a substrate to produce the precursor PGH2. PGH2 is converted to biologically active PGE2 by means of the enzyme PGE2 synthase. PGE2 can then readily signal through one of four EP, denoted EP1, EP2, EP3, and EP4. Various pathogens influence PGE2 production and signaling along different steps of this pathway. Green arrows indicate activation of COX-2 or PGES, while red lines indicate inhibition of COX-2 or PGES by select pathogens. Blue lines indicate a potential role for EP signaling in pathogen survival.
PGE2 Receptor Signaling
There are four known PGE2 receptors designated EP1, EP2, EP3, and EP4, with at least three splice variants of EP3 recognized as EP3α, EP3β, and EP3γ. This diversity of PGE2 receptors influences the pro-inflammatory and immunosuppressive functions of this molecule within the body under different environmental conditions. EP3 and EP4 are considered to be high-affinity receptors, requiring lower levels of PGE2 for signaling. Conversely, EP1 and EP2 demand higher concentrations of PGE2 for proper signaling. Additionally, the four PGE2 receptors vary in their signal durations (Sugimoto et al., 1992; Honda et al., 1993; Nishigaki et al., 1996; Hata and Breyer, 2004). While the PGE2 receptors have fundamental differences in affinity and signal durations, there are mechanistic similarities between some of these receptors. For example, EP2 and EP4 are both Gs-coupled receptors that signal primarily through the adenylate cyclase-dependent cAMP/PKA/CREB pathways. EP2 and EP4 are the predominant receptors responsible for the anti-inflammatory and immunosuppressive effects of PGE2 (Fujino et al., 2005). Both receptors are primarily thought to function in a cAMP-dependent manner, however EP4 also signals in a phosphatidylinositol 3-kinase (PI3K)-dependent manner to activate the extracellular-signal-regulated kinase 1/2 (ERK1/2) pathway (Fujino et al., 2003). Conversely, EP1 and EP3 do not require cAMP for activation. Few studies have examined the low-affinity EP1, although PGE2 signaling through this receptor leads to an increase in the release of cellular calcium (Hata and Breyer, 2004). Signaling through EP3 primarily involves Gi-coupled receptors that inhibit the activity of adenylate cyclase, and consequently decrease levels of cAMP in the cell. Nevertheless, EP3 splice variants are also able to signal through Gs-coupled receptors, enhancing the diverse signaling ranges among these PGE2 receptors (Sugimoto et al., 1992).
The diversity of receptors, signaling pathways, and signal duration enables PGE2 to act as an adaptable signaling molecule in a wide range of cell types in response to environmental stimuli. The complexities of PGE2 signaling help address its paradoxical ability to elicit both inflammatory and immunosuppressive responses under various concentrations and environmental conditions at early and late stages of bacterial infection (Hessle et al., 2003; Stefanelli et al., 2012). Furthermore, while PGE2-mediated immunoregulation is essential for maintaining homeostasis, the immunosuppressive effects of PGE2 during innate immune responses may be detrimental during bacterial infection, as examined in depth below.
PGE2 and Innate Immunity
Neutrophils
Neutrophils are the first leukocytes recruited to sites of infection during an innate immune response. These cells possess several immune defense mechanisms including phagocytosis, proteolytic enzymes, oxygen-reactive agents, and inflammatory mediators. Accordingly, proper migration as well as signaling between these granulocytes and other immune cells is important to allow for an effective immune response at early stages of infection. Activation and aggregation of human neutrophils is inhibited after exogenous treatment with PGE2 in vitro (Ney and Schrör, 1991; Wheeldon and Vardey, 1993; Talpain et al., 1995). PGE2 also inhibits the activation of rat and guinea pig neutrophils in vitro, suggesting a conserved inhibitory role of PGE2 signaling among mammalian immune responses (Ham et al., 1983; Takenawa et al., 1986; Wise and Jones, 1994; Wise, 1996). Activation of mammalian neutrophils by formylmethionyl-leucyl-phenylalanine (fMLP) is inhibited by PGE2 in an EP2-dependent manner (Takenawa et al., 1986; Burelout et al., 2004, 2007). Inhibition of EP2 signaling improves neutrophil migration to promote bacterial clearing and enhances mouse survival following intratracheal infection with Pseudomonas aeruginosa (Sadikot et al., 2007; Aronoff et al., 2012). Bacterial pathogens and their structural components directly promote PGE2 synthesis by neutrophils during infections. For example, Streptococcus pneumoniae infection induces PGE2 production by human neutrophils and obstructs activation and migration in vitro (Cockeran et al., 2001). Neutrophils also produce increased concentrations of PGE2 after treatment with E. coli LPS or post-infection with P. aeruginosa in rat and mouse models, respectively (He et al., 2001; Alba-Loureiro et al., 2004). Since neutrophils represent a first line of defense against infection, it is important to further elucidate PGE2 production during bacterial infection and examine its immunomodulatory effects on the antimicrobial functions of neutrophils.
Macrophages
Through phagocytosis and the generation of a strong cytokine response, macrophages are important cells in innate immune responses and immunomodulation. While PGE2 is able to locally attract macrophages at early stages of inflammation (Nakayama et al., 2006), macrophage activation can be inhibited by PGE2 through EP2 signaling (Zaslona et al., 2012). The phagocytic properties of alveolar macrophages are inhibited in an EP2-dependent manner during infection with Klebsiella pneumoniae and S. pneumoniae in the rat and mouse models, respectively. Phagocytosis is restored through the inhibition of PGE2 synthesis with non-selective COX inhibitors such as indomethacin (Aronoff et al., 2004; Aronoff, 2012). The phagocytic properties of macrophages are dampened by PGE2 through the induction of immunosuppressive IL-1R-associated kinase-M (IRAK-M), impairing bacterial clearance of P. aeruginosa (Hubbard et al., 2010). PGE2 also affects the inflammatory response of macrophages during infection by altering cell signaling and inhibiting bactericidal mechanisms. Upon PGE2 stimulation, NAPDH oxidase is inhibited inside the macrophage, leading to reduced killing of K. pneumoniae (Serezani et al., 2007). PGE2 also suppresses macrophage activity by inhibiting the production of nitric oxide radicals (Marotta et al., 1992; Asakrah et al., 2013). PGE2 alters the cytokine response of macrophages and promotes an immunosuppressive phenotype. Most notable perhaps is that PGE2 induces the production of immunoregulatory cytokines, such as IL-10 and IL-17 (Kunkel et al., 1986, 1988; Huang et al., 1998; Stolina et al., 2000; Liu et al., 2012). Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a downstream product of PGE2 signaling that negatively regulates alveolar macrophage phagocytosis and bacterial killing during P. aeruginosa infection. Inhibition or genetic knockout of PTEN restores the phagocytic functions of macrophages and enhances bacterial clearance in vivo (Hubbard et al., 2010).
Natural killer cells
Natural killer (NK) cells are potent granulocytes important in controlling infection during innate immune responses. While NK cells are most commonly associated with controlling viral infections, they are also important during bacterial infection. These cells respond to changes in the cytokine profile during infection in order to lyse infected cells, and PGE2 has a negative effect on the cytolytic activities of NK cells by suppressing their responsiveness to cytokines such IL-12 and IL-15 (Bankhurst, 1982; Goto et al., 1983; Joshi et al., 2001; Walker and Rotondo, 2004). In a leukemia rat model, an increase in PGE2 concentration is associated with diminished NK cell cytolysis and decreased animal survival, which is relieved upon COX inhibition by etodolac (Inbar et al., 2011). NK cells also secrete IFN-γ as a signaling mechanism to activate macrophages during the innate immune response and to aid dendritic cells in driving Th1 responses. PGE2 suppresses NK cell-mediated activation of macrophages by inhibiting the production of IFN-γ (Mailliard et al., 2005). Not only does PGE2 have an inhibitory effect on the cytokine response of NK cells, but it also downregulates the expression of receptors important for NK cell effector functions, including CD94/NKG2C, DNAM-1, NKp80, 2B4, and CD161. PGE2 also has a deleterious effect on the homing, migration, and survival of NK cells in humans infected with Human Herpes Virus 8 who have developed Kaposi's sarcoma (Dupuy et al., 2012). This demonstrates similarity to PGE2's ability to negatively affect the aggregation of neutrophils, suggesting that there may be a conserved signaling mechanism across immune cell types. Further research must be conducted in order to elucidate the effect of PGE2 on NK cell activity during bacterial infection.
Dendritic cells
Dendritic cells (DCs) process and present antigen to immune cells during innate and adaptive immune responses and are consequently important in controlling disease progression and outcome. They initiate an adaptive immune response and are key participants in shifting immunity between Th1 and Th2 responses. PGE2 can disrupt DC differentiation at early stages of development (Kaliński et al., 1997). At later stages of DC differentiation, PGE2 can hasten DC maturation in the presence of IL-1β and TNF-α (Rieser et al., 1997; Kaliński et al., 1998). DCs that are fully developed, but functionally immature are prompted by PGE2 to migrate to lymph nodes (Jonuleit et al., 1997). Enhanced DC migration may be due to the ability of PGE2 to induce the expression of the lymphoid-homing chemokine receptor CCR7 on DCs (Luft et al., 2002; Scandella et al., 2002; Kalinski, 2012). However, PGE2 suppresses the production of chemokines, such as the CCR7 ligand CCL19, by DCs and inhibits naïve T cell attraction in the lymph nodes (Muthuswamy et al., 2010). PGE2 also impairs the ability of DCs to induce NK cell-mediated immunity (Gustafsson et al., 2008). These contrasting DC characteristics may be in part due to differences in PGE2 concentrations as well as receptor signaling. While it is generally accepted that DCs matured in the presence of PGE2 promote T cell expansion, these particular DCs suppress Th1 responses and support Th2 responses (Kalinski, 2012). For example, while PGE2-matured DCs effectively prime naïve T cells (Jonuleit et al., 1997), they also suppress the cytotoxic T lymphocyte (CTL) response (Obermajer et al., 2011). This alteration may be in part due to a transformed cytokine profile of PGE2-matured DCs. In particular, DCs matured in the presence of PGE2 display an enhanced production of immunosuppressive cytokines such as IL-10 (Kaliński et al., 1997) and suppress their own production of proinflammatory cytokines such as IL-12p70 (Kaliński et al., 1998). By shifting cytokine profiles away from a Th1 and toward a Th2 response, PGE2 may in fact promote the maturation of DCs that are better-suited to allow for intracellular bacteria to establish infection.
The Role of Pge2 During Bacterial Infection
Multiple bacterial pathogens elicit an increase in PGE2 production upon infection. Both Gram-negative and Gram-positive bacteria are able to induce PGE2 synthesis, yet Gram-negative bacteria elicit a stronger PGE2 response by human monocytes (Hessle et al., 2003). While passive recognition of LPS can contribute to PGE2 production in response to Gram-negative bacteria (Alba-Loureiro et al., 2004), it is becoming apparent that bacteria also actively induce PGE2 production during infection. Levels of PGE2 are highly regulated in the lung and gastrointestinal tract to maintain the integrity of the mucosal barrier (Takeuchi et al., 2010; Bozyk and Moore, 2011), and bacteria may modulate PGE2 biosynthesis to aid colonization of the lung and gut. In fact, enhanced PGE2 synthesis by immune cells appears to be a conserved event during bacterial infections within the mucosa, and this will be discussed in the following sections.
Enteric Infections
Salmonella
Salmonella is a Gram-negative facultative intracellular bacillus that is able to infect and survive inside several cell types including intestinal epithelial cells and macrophages. Several serotypes of Salmonella including S. enterica, S. dublin, and S. typhimurium induce the expression on PGE2 during infection (Ochman et al., 1996; Eckmann et al., 1997; Uchiya and Nikai, 2004). One of Salmonella's most well-characterized virulence factors is the pathogenicity island 2 (SPI-2). This pathogenicity island is necessary for growth within the macrophage and is an important virulence factor in establishing infection in mice (Ochman et al., 1996; Cirillo et al., 1998; Hensel et al., 1998). SpiC, an important gene product encoded within SPI-2, is necessary for survival of S. typhimurium within macrophages (Uchiya et al., 1999). SpiC activates the ERK1/2 signal transduction pathway to enhance COX-2 expression and PGE2 synthesis in infected macrophages, indicating that Salmonella possesses active mechanisms to alter host cell signaling in intestinal epithelial cells which enhances PGE2 production (Resta-Lenert and Barrett, 2002; Uchiya and Nikai, 2004). Salmonella-induced PGE2 activates the protein kinase A (PKA) pathway and upregulates IL-10 production by macrophages, promoting an immunosuppressive phenotype and impaired killing ability. COX inhibition by indomethacin or SC-58125 restores the bactericidal properties of macrophages during Salmonella infection in vitro (Uchiya and Nikai, 2004). PGE2 production is also dependent upon the expression of Salmonella DNA adenine methylase (dam). Salmonella dam mutants are unable to promote COX-2 expression, leading to reduced PGE2 production in infected murine macrophages (Cristina Cerquetti et al., 2008). Along with the inability to elicit a strong PGE2 response, dam mutants are less cytotoxic to M cells, deficient in cell invasion (García-Del Portillo et al., 1999), and confer cross-protective Salmonella immunity in a mouse model (Heithoff et al., 2001).
During experimental salmonellosis with S. typhimurium, COX-2 expression and PGE2 concentrations in macrophages and dendritic cells within the mesenteric lymph nodes remain elevated 3 days after intragastric infection in the mouse model. At early stages of acute infection in the mouse model, COX-2 inhibition with celecoxib leads to an increase in bacterial loads in the mesenteric lymph nodes; however, at later stages of infection, COX-2 inhibition enhances host survival (Bowman and Bost, 2004). Thus, while PGE2 may have beneficial proinflammatory properties during acute Salmonella infection, prolonged exposure to PGE2 may be detrimental and promote an environment susceptible to chronic disease.
Escherichia coli
Enteropathogenic E. coli (EPEC) and enterohemorrhagic E. coli (EHEC) are Gram-negative bacteria that colonize the intestine and cause diarrheal disease. Both EPEC and EHEC induce PGE2 production by intestinal epithelial cells, however the most potent inducers of PGE2 are invasive strains such as E. coli O29:NM (Eckmann et al., 1997). The Type 3-secreted effector EspT is a guanine nucleotide exchange factor important for EPEC cellular invasion. EPEC strains expressing EspT promote increased COX-2 expression and PGE2 production by infected macrophages (Raymond et al., 2011). This suggests PGE2 increases in part through EspT expression and does not rely entirely on passive immune recognition of LPS or other Toll-like receptor (TLR) agonists, such as flagellin. This also suggests bacteria utilize active signaling mechanisms to exploit PGE2 for intracellular survival. High concentrations of E. coli LPS also induce PGE2 production by macrophages (Kurland and Bockman, 1978; Rosenstreich et al., 1978). E. coli LPS administered at 40 mg/kg is 100% fatal in the normal mouse model, yet COX-2−/− mice demonstrate 100% survival at this dosage and are significantly protected against LPS doses as high as 100 mg/kg (Ejima et al., 2003). Accordingly, COX-2 inhibition may represent a therapeutic strategy in controlling infection with pathogenic E. coli.
Other enteric species
It is not surprising that additional enteric pathogens are able to elicit a PGE2 response upon infection. Vibrio cholerae is an enteric bacterial pathogen whose infection leads to acute watery diarrhea and an increase in PGE2 secretion in infected intestinal tissues. Specifically, jejunal fluids from patients presenting with acute cholera infection contain increased concentrations of PGE2 (Speelman et al., 1985). Both children and adults infected with V. cholerae O1 and V. cholerae O139 demonstrate significantly higher concentrations of PGE2 in stools when compared to healthy controls during the acute stages of infection. However, there is no significant difference in plasma PGE2 levels in these patients, suggesting the PGE2 response is restricted to the infected mucosa (Qadri et al., 2002). Cholera toxin (CT) also influences PGE2 production, as murine macrophages display enhanced PLA2 activity and PGE2 synthesis when stimulated with exogenous CT (Burch et al., 1988). Similarly, stimulation of isolated intestinal rabbit cells with CT leads to an increase in PGE2 concentrations (Peterson et al., 1994).
Other enteric bacterial pathogens demonstrate an ability to induce PGE2 production by infected cells. Both pediatric and adult patients presenting with acute shigellosis exhibit significantly higher concentrations of PGE2 in stool samples when compared to healthy controls (Raqib et al., 2000). Further studies must be conducted in order to determine the mechanisms by which enteric pathogens elicit PGE2 production in infected cells. Moreover, it will be necessary to determine how PGE2 concentrations affect both the host immune response and bacterial pathogenesis at various stages of enteric infection.
Pulmonary Infections
Mycobacteria
Mycobacteria are acid-fast bacilli that cause progressive or latent pulmonary disease after aerosol inhalation (Torrado et al., 2011). Several Mycobacteria species induce PGE2 production during infection. In the mouse model, M. intracellulare induces PGE2 synthesis, inhibiting the production of lymphokines in infected macrophages and suppressing an effective immune response (Edwards et al., 1986). M. bovis bacillus Calmette-Guerin (BCG) also enhances COX-2 expression and PGE2 production in a TLR2-dependent manner in infected macrophages in vitro and in a mouse model (Bansal et al., 2009). In particular, the presence of PGE2 has been noted in the sera and cerebrospinal fluid of tuberculosis patients (Bansal et al., 2009). Mice infected with M. tuberculosis demonstrate a 13-fold increase in lung PGE2 levels at 30 days post-infection compared to uninfected mice (Peres-Buzalaf et al., 2011). Granuloma formation, a hallmark of tuberculosis infection, is comprised of macrophages exhibiting high levels of COX-2 expression and PGE2 synthesis in the mouse model (Rangel Moreno et al., 2002). A gene encoding early secreted antigenic target protein 6 (ESAT-6), present in all pathogenic strains of Mycobacterium, induces COX-2 expression and PGE2 production in a TLR2-dependent manner in infected macrophages in vitro (A et al., 2012). Interestingly, the avirulent M. tuberculosis stain H37Ra was shown to promote macrophage PGE2 production leading to cellular apoptosis, while the virulent strain H37Rv induced significantly less PGE2 and caused macrophage necrosis (Chen et al., 2008; Divangahi et al., 2009). PGES−/− macrophages are unable to control H37Rv replication and PGES−/− mice demonstrate significantly higher bacterial burdens at 5 weeks post-infection with virulent M. tuberculosis, suggesting that PGE2 is necessary to control M. tuberculosis during the early stage of infection (Chen et al., 2008). Similar results were reported by Rangel Moreno et al. (2002) using wild type mice infected with H37Rv. COX-2, PGES, and PGE2 expression were low and relatively stable during the early phase of infection (up to 21 days), and COX-2 inhibition during early infection led to increased bacterial growth and immunopathology. In contrast, COX-2, PGES, and PGE2 expression increased during the chronic phase of infection (60–90 days), and inhibition of COX-2 led to increased iNOS expression with a concomitant reduction in lung bacterial load and granuloma size (Rangel Moreno et al., 2002). Clearly modulation of PGE2 can impact disease outcome during M. tuberculosis infection, and the consequences of PGE2 inhibition may differ between acute and chronic stages of tuberculosis infection. Therapeutic strategies targeting PGE2 may lead to alternative therapies in controlling Mycobacterium infection in the lung.
Streptococcus
Community-acquired pneumonia is one of the leading causes of death worldwide (Finch, 2001), and is most commonly caused by S. pneumoniae (Mandell et al., 2007). In patients suffering from acute pneumonia, COX-2 is expressed in alveolar epithelial cells (AECs). Similarly, AECs, alveolar macrophages, and vascular endothelial cells of human lung tissue in vitro exhibit time-dependent increases in both COX-2 expression and PGE2 production post-infection with S. pneumoniae (Szymanski et al., 2012). Streptococcal toxins also promote PGE2 production in immune cells. Particularly, pneumolysin produced by S. pneumoniae promotes the production of PGE2 in neutrophils and endothelial cells by inducing the expression of PLA2 (Rubins et al., 1994; Cockeran et al., 2001). Enhanced PGE2 production by neutrophils treated with pneumolysin inhibits an effective immune response by obstructing neutrophil activation and migration (Takenawa et al., 1986; Cockeran et al., 2001; Burelout et al., 2004, 2007). Inhibiting PGE2 production during Streptococcus infection enhances macrophage phagocytosis and generation of reactive oxygen species, aiding in bacterial clearance (Stables et al., 2010). PGE2 signaling post-Streptococcus infection relies on both EP2 and EP4 signaling (Aronoff et al., 2012; Szymanski et al., 2012). EP2−/− murine alveolar macrophages demonstrate enhanced phagocytosis, intracellular killing, and increased generation of reactive oxygen in vitro, while EP2−/− mice demonstrate improved bacterial clearance and survival post-infection with S. pneumoniae. Animal survival may be associated with a heightened production of pro-inflammatory cytokines, such as IL-12p40 (Aronoff et al., 2012). EP3 also plays a large role in PGE2 signaling post-infection with Streptococcus both in vitro and in a mouse model. EP3−/− macrophages in vitro have enhanced phagocytic properties and bacterial killing mechanisms, such as nitric oxide production. EP3−/− mice also exhibit greater levels of protection against S. pneumoniae when compared to wildtype mice. Specifically, EP3−/− mice demonstrate heightened bacterial clearance in the lung by alveolar macrophages, with a decrease in infiltrating lung neutrophils and blood leukocytes (Aronoff et al., 2009). The immunosuppressive qualities of PGE2 have characteristically been attributed to EP2 and EP4 signaling, but EP3 signaling also contributes to increased production of PGE2 during pneumococcal infection.
Other species of Streptococcus induce an increase in PGE2 synthesis during pulmonary infection as well. Group B Streptococcus is a leading cause of neonatal sepsis and pneumonia, and infection with this bacterial pathogen leads to enhanced expression of COX-2 and increased concentrations of PGE2 in A549 human lung epithelial cells (Glibetic et al., 2001; Natarajan et al., 2007). S. pyogenes, a causative agent of pharyngitis, induces the expression of COX-2 and PGE2 synthesis in the macrophages of tissue biopsies from infected patients as well as in infected mice. Pharmacological inhibition of PGE2 synthesis by PKI (14–22) or genetic ablation of COX-2 expression promotes bacterial clearance and improves disease outcome in the mouse model (Goldmann et al., 2010).
Pseudomonas aeruginosa
P. aeruginosa is one of the most virulent opportunistic pathogens and is the leading cause of morbidity and mortality in cystic fibrosis patients (Sato et al., 2003; Sadikot et al., 2005). P. aeruginosa is also a common cause of hospital-acquired pneumonia (Sadikot et al., 2005). In a murine model of P. aeruginosa infection, overproduction of PGE2 in the lung diminishes phagocytosis and TNF-α production by alveolar macrophages (Ballinger et al., 2006; Hubbard et al., 2010). The inhibitory effects of PGE2 appear to partially signal through EP2, as EP2−/− mice demonstrate decreased bacterial loads post-infection (Sadikot et al., 2007). P. aeruginosa induces cPLA2 activity within infected A549 epithelial cells in an ERK 1/2-dependent manner to trigger a four-fold increase in PGE2 production, which can be suppressed with the use of a specific cPLA2 inhibitor (Hurley et al., 2011). COX-2-deficient mice display enhanced bacterial clearance post-infection when compared to wildtype control mice. Recruitment of inflammatory cells in COX-2-deficient mice does not differ from those of control mice post-infection, suggesting bacterial clearance is associated with impaired effector functions of immune cells (Sadikot et al., 2007). Inhibition of COX-2 expression also decreases the severity of P. aeruginosa infection and increases survival rates in mice (Saliba et al., 2005; Sadikot et al., 2007). Murine bone marrow-derived macrophages treated with the selective COX-2 inhibitor NS-398 prior to infection with P. aeruginosa have lower concentrations of PGE2 and show an increase in superoxide production post-infection when compared to mock-treated controls (Sadikot et al., 2005).
Other pulmonary species
Burkholderia pseudomallei is a facultative intracellular Gram- negative bacillus that causes a fatal disease known as melioidosis. Patients acquire the infection through different routes and can present with a wide range of clinical symptoms including debilitating pneumonia and septic shock (Cheng and Currie, 2005). Recent work from our laboratory has demonstrated that PGE2 plays a critical role in the pathogenesis of B. pseudomallei infection in mice (Asakrah et al., 2013). PGE2 promotes B. pseudomallei intracellular survival through the activation of arginase 2 which competes with inducible nitric oxide synthase for the substrate, L-arginine, thereby limiting nitric oxide production. This process is antagonized by blocking PGE2 synthesis with a selective COX-2 inhibitor, NS398 (Asakrah et al., 2013). Treatment of bone marrow-derived macrophages with NS398 reduces endogenous PGE2 production and intracellular survival of B. pseudomallei. Conversely, addition of exogenous PGE2 to NS398-treated macrophages restores B. pseudomallei survival. Administration of NS-398 or Celecoxib significantly enhances mouse survival from lethal pulmonary infection with B. pseudomallei (Asakrah et al., 2013).
Burkholderia cepacia is a Gram-negative bacterium that causes fatal lung infections in cystic fibrosis patients. Approximately 20% of infected patients have severe pulmonary epithelial deterioration that can lead to death within a matter of weeks (Isles et al., 1984). In human lung epithelial cells, B. cepacia promotes enhanced PGE2 synthesis, possibly increasing the severity of disease in immunocompromised individuals (Fink et al., 2003). Bordatella pertussis infections result in a severe pulmonary illness known as pertussis or “whooping cough.” Pertussis toxin (PT) stimulates an increase in PGE2 production in infected murine macrophages in vitro (Burch et al., 1988; Schulze-Specking et al., 1991). Further research is warranted to identify the mechanisms behind which various pulmonary pathogens modulate PGE2 responses in the lung in order to aid infection.
Active Induction of Pge2
When inactivated, many bacteria are unable to elicit a strong PGE2 response by host cells. For example, when compared to live bacteria, UV-irradiated S. typhimurium are unable to induce COX-2 expression in infected macrophages, suggesting that Salmonella uses active mechanisms to alter gene expression in infected tissues for the production of PGE2 (Bowman and Bost, 2004). Similarly, UV-irradiated S. aureus are unable to promote PGE2 biosynthesis in infected osteoblasts (Somayaji et al., 2008). Both live and gamma-irradiated M. avium induce PGE2 production in infected human peripheral blood monocyte-derived macrophages, yet gamma-irradiated M. avium induce significantly lower concentrations of PGE2 (Rastogi et al., 1992). Heat inactivation of B. pseudomallei also led to a significant reduction in COX-2 expression and PGE2 production by murine macrophages (Asakrah et al., 2013). The reduced ability of inactivated bacteria to elicit a strong PGE2 response during infection suggests these bacteria have evolved active mechanisms to alter host cell signaling to promote PGE2 synthesis that may aid infection.
Type three secretion systems (T3SS) are important bacterial secretion systems, some of which stimulate PGE2 production during bacterial pathogenesis (Sato et al., 2003; Saliba et al., 2005; Sadikot et al., 2007; Raymond et al., 2011). ExoU is a T3SS effector molecule associated with P. aeruginosa infections which lead to nosocomial pneumonia and bacteremia (Berthelot et al., 2003; Schulert et al., 2003). This cytotoxin possesses phospholipase activity and induces rapid AA release from the cell wall and enhances PGE2 production during the infection of human epithelial cells (Sato and Frank, 2004; Saliba et al., 2005; Sadikot et al., 2007). Mice infected with ExoU-deficient P. aeruginosa have a significant decrease in COX-2 expression and diminished PGE2 production in the lung and a lower bacterial load in infected tissue, indicating that the secretion of this effector molecule aids in establishing infection (Saliba et al., 2005; Sadikot et al., 2007). E. coli also utilizes a T3SS effector molecule, EspT, to elicit a PGE2 response in infected macrophages (Raymond et al., 2011). Taken together, these studies highlight a conserved mechanism among bacterial T3SSs that induce PGE2 production during infection, and elucidation of these effectors may identify new therapeutic targets.
PGE2 as a Potential Therapeutic Target During Bacterial Infection
COX-2 Inhibition
Since PGE2 production has inhibitory effects on immune cells, particularly those involved in innate immune responses, inhibition of PGE2 may benefit the host during bacterial infection (Goto et al., 1983; Kunkel et al., 1986; Phipps et al., 1991; Strassmann et al., 1994; Kaliński et al., 1997; Harris et al., 2002). In support of this, mice deficient in COX-2 demonstrate enhanced survival post-infection with several bacterial pathogens. For example, COX-2−/− mice exposed intraperitoneally to high doses of E. coli endotoxin exhibit increased survival compared to wildtype mice (Ejima et al., 2003). COX-2-deficient mice also demonstrate greater survival rates and exhibit lower bacterial loads in the liver and spleen after intravenous infection with S. pyogenes (Bowman and Bost, 2004). When compared to wildtype mice, COX-2−/− mice exhibit increased bacterial clearance and enhanced survival at 6 days post-intratracheal infection with P. aeruginosa (Sadikot et al., 2007).
COX inhibitors, which are already widely used in the human population for the relief of pain and inflammation, block the production of PGE2 and other prostaglandins and may offer therapeutic benefit during bacterial infections. For example, non-selective COX inhibitors such as ibuprofen and indomethacin, significantly reduce the bacterial load and PGE2 production in the bronchoalveolar lavage (BAL) after intratracheal P. aeruginosa infection in mice (Saliba et al., 2005). COX-2 inhibition by NS-398 also significantly improves mouse survival post-intratracheal infection with lethal doses of P. aeruginosa (Sadikot et al., 2007). Moreover, NS398 administered post-exposure to mice infected with B. pseudomallei significantly reduces lung PGE2 levels and enhances animal survival (Asakrah et al., 2013). COX-2 inhibition results in higher bacterial loads during acute S. typhimurium and M. tuberculosis infection in mouse models, however administration of a COX-2 inhibitor during chronic infection with S. typhimurium or M. tuberculosis improves host protection (Rangel Moreno et al., 2002; Bowman and Bost, 2004). Similarly, Celecoxib treatment reduces lung levels of PGE2 and enhances the 60-day survival of M. tuberculosis-infected mice (Peres-Buzalaf et al., 2011). Because COX-2 inhibition impairs the production of prostaglandins in addition to PGE2, it is important to consider the potential contribution of other prostaglandins in such studies. Furthermore, additional studies in highly relevant animal models are needed to determine the therapeutic efficacy of COX inhibitors against mucosal bacterial infections.
Receptor Inhibition
Specific targeting of one or more PGE2 receptors may also hold therapeutic promise. EP2 is a major receptor responsible for the immunosuppressive activities of PGE2 signaling (Fujino et al., 2005). EP2−/− alveolar macrophages exhibit improved phagocytosis, increased production of reactive oxygen intermediates and pro-inflammatory cytokines, such as TNF-α and MIP-2, and enhanced killing of P. aeruginosa (Aronoff et al., 2012). Impaired EP2 signaling improves disease outcome in P. aeruginosa-infected mice, as EP2-deficient mice show enhanced survival and bacterial clearance correlated with enhanced neutrophil migration and IL-12 production in the lung (Sadikot et al., 2007; Aronoff et al., 2012). Inhibition of EP3 may also be beneficial in controlling bacterial infections. EP3-deficient alveolar macrophages demonstrate increased phagocytic activity and nitric oxide production, and enhanced bacterial killing during S. pneumoniae infection. EP3−/− mice exhibit greater bacterial clearance and higher survival post-intraperitoneal infection (Aronoff et al., 2009). Specific EP inhibitors or antagonists may aid in therapeutically controlling microbial infection and require further study.
Conclusions and Future Directions
PGE2 is an important lipid mediator that regulates inflammation and immune responses during infection (Phipps et al., 1991; Yu and Chadee, 1998; Harris et al., 2002; Nagamatsu and Schust, 2010). Four principle PGE2 receptors respond to varying concentrations of PGE2 in order to elicit dynamic downstream signaling events during immune responses. It is increasingly evident that PGE2 biosynthesis and its inhibitory actions on innate immune defenses can impact bacterial pathogenesis and disease outcome. For infected macrophages, PGE2 production correlates with diminished phagocytosis, nitric oxide production, and intracellular killing (Marotta et al., 1992; Aronoff et al., 2004; Hubbard et al., 2010), and promotes an immunosuppressive cytokine profile (Kunkel et al., 1986, 1988; Huang et al., 1998; Stolina et al., 2000; Liu et al., 2012). Neutrophil and NK cell activation, migration, and aggregation are inhibited by PGE2 (Bankhurst, 1982; Goto et al., 1983; Takenawa et al., 1986; Joshi et al., 2001; Burelout et al., 2004, 2007; Walker and Rotondo, 2004). PGE2 shifts the immune response away from a Th1 response and toward a Th2 response by promoting the production of anti-inflammatory cytokines and modulating the interactions between DCs and other immune cells (Kaliński et al., 1997, 1998; Gustafsson et al., 2008; Obermajer et al., 2011). The conserved ability of many bacterial pathogens to promote PGE2 responses during infection suggests a common signaling mechanism to deter protective pro-inflammatory immune responses. Inhibition of PGE2 production and signaling during infection may represent a therapeutic alternative to treat certain bacterial infections. Further study of the immunosuppressive effects of PGE2 on innate immunity will lead to a better understanding of potential therapeutic targets within the PGE2 pathway.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
A, S. K., Bansal, K., Holla, S., Verma-Kumar, S., Sharma, P., and Balaji, K. N. (2012). ESAT-6 induced COX-2 expression involves coordinated interplay between PI3K and MAPK signaling. Mol. Immunol. 49, 655–663. doi: 10.1016/j.molimm.2011.11.011
Alba-Loureiro, T. C., Martins, E. F., Miyasaka, C. K., Lopes, L. R., Landgraf, R. G., Jancar, S., et al. (2004). Evidence that arachidonic acid derived from neutrophils and prostaglandin E2 are associated with the induction of acute lung inflammation by lipopolysaccharide of Escherichia coli. Inflamm. Res. 53, 658–663. doi: 10.1007/s00011-004-1308-7
Aronoff, D. M. (2012). Cyclooxygenase inhibition in sepsis: is there life after death? Mediat. Inflamm. 2012:696897. doi: 10.1155/2012/696897
Aronoff, D. M., Bergin, I. L., Lewis, C., Goel, D., O'Brien, E., Peters-Golden, M., et al. (2012). E-prostanoid 2 receptor signaling suppresses lung innate immunity against Streptococcus pneumoniae. Prostaglandins Other Lipid Mediat. 98, 23–30. doi: 10.1016/j.prostaglandins.2012.03.002
Aronoff, D. M., Canetti, C., and Peters-Golden, M. (2004). Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J. Immunol. 173, 559–565.
Aronoff, D. M., Lewis, C., Serezani, C. H., Eaton, K. A., Goel, D., Phipps, J. C., et al. (2009). E-prostanoid 3 receptor deletion improves pulmonary host defense and protects mice from death in severe Streptococcus pneumoniae infection. J. Immunol. 183, 2642–2649. doi: 10.4049/jimmunol.0900129
Asakrah, S., Nieves, W., Mahdi, Z., Agard, M., Zea, A. H., Roy, C. J., et al. (2013). Post-exposure therapeutic efficacy of COX-2 inhibition against Burkholderia pseudomallei. PLoS Negl. Trop Dis. 7:e2212. doi: 10.1371/journal.pntd.0002212
Ballinger, M. N., Aronoff, D. M., McMillan, T. R., Cooke, K. R., Olkiewicz, K., Toews, G. B., et al. (2006). Critical role of prostaglandin E2 overproduction in impaired pulmonary host response following bone marrow transplantation. J. Immunol. 177, 5499–5508.
Bankhurst, A. D. (1982). The modulation of human natural killer cell activity by prostaglandins. J. Clin. Lab. Immunol. 7, 85–91.
Bansal, K., Narayana, Y., Patil, S. A., and Balaji, K. N. (2009). M. bovis BCG induced expression of COX-2 involves nitric oxide-dependent and -independent signaling pathways. J. Leukoc. Biol. 85, 804–816. doi: 10.1189/jlb.0908561
Berthelot, P., Attree, I., Plésiat, P., Chabert, J., de Bentzmann, S., Pozzetto, B., et al. (2003). Genotypic and phenotypic analysis of type III secretion system in a cohort of Pseudomonas aeruginosa bacteremia isolates: evidence for a possible association between O serotypes and exo genes. J. Infect. Dis. 188, 512–518. doi: 10.1086/377000
Bowman, C. C., and Bost, K. L. (2004). Cyclooxygenase-2-mediated prostaglandin E2 production in mesenteric lymph nodes and in cultured macrophages and dendritic cells after infection with Salmonella. J. Immunol. 172, 2469–2475.
Bozyk, P. D., and Moore, B. B. (2011). Prostaglandin E2 and the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 45, 445–452. doi: 10.1165/rcmb.2011-0025RT
Burch, R. M., Jelsema, C., and Axelrod, J. (1988). Cholera toxin and pertussis toxin stimulate prostaglandin E2 synthesis in a murine macrophage cell line. J. Pharmacol. Exp. Ther. 244, 765–773.
Burelout, C., Thibault, N., Harbour, D., Naccache, P. H., and Bourgoin, S. G. (2007). The PGE2-induced inhibition of the PLD activation pathway stimulated by fMLP in human neutrophils is mediated by PKA at the PI3-Kgamma level. Biochem. Pharmacol. 74, 730–741. doi: 10.1016/j.bcp.2007.06.013
Burelout, C., Thibault, N., Levasseur, S., Simard, S., Naccache, P. H., and Bourgoin, S. G. (2004). Prostaglandin E2 inhibits the phospholipase D pathway stimulated by formyl-methionyl-leucyl-phenylalanine in human neutrophils. Involvement of EP2 receptors and phosphatidylinositol 3-kinase gamma. Mol. Pharmacol. 66, 293–301. doi: 10.1124/mol.66.2.293
Chen, M., Divangahi, M., Gan, H., Shin, D. S., Hong, S., Lee, D. M., et al. (2008). Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 205, 2791–2801. doi: 10.1084/jem.20080767
Cheng, A. C., and Currie, B. J. (2005). Melioidosis: epidemiology, pathophysiology, and management. Clin. Microbiol. Rev. 18, 383–416. doi: 10.1128/CMR.18.2.383-416.2005
Cirillo, D. M., Valdivia, R. H., Monack, D. M., and Falkow, S. (1998). Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol. Microbiol. 30, 175–188. doi: 10.1046/j.1365-2958.1998.01048.x
Cockeran, R., Steel, H. C., Mitchell, T. J., Feldman, C., and Anderson, R. (2001). Pneumolysin potentiates production of prostaglandin E(2) and leukotriene B(4) by human neutrophils. Infect. Immun. 69, 3494–3496. doi: 10.1128/IAI.69.5.3494-3496.2001
Cockeran, R., Theron, A. J., Steel, H. C., Matlola, N. M., Mitchell, T. J., Feldman, C., et al. (2001). Proinflammatory interactions of pneumolysin with human neutrophils. J. Infect. Dis. 183, 604–611. doi: 10.1086/318536
Cristina Cerquetti, M., Hovsepian, E., Sarnacki, S. H., and Goren, N. B. (2008). Salmonella enterica serovar Enteritidis dam mutant induces low NOS-2 and COX-2 expression in macrophages via attenuation of MAPK and NF-kappaB pathways. Microbes Infect. 10, 1431–1439. doi: 10.1016/j.micinf.2008.08.008
Divangahi, M., Chen, M., Gan, H., Desjardins, D., Hickman, T. T., Lee, D. M., et al. (2009). Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 10, 899–906. doi: 10.1038/ni.1758
Dupuy, S., Lambert, M., Zucman, D., Choukem, S. P., Tognarelli, S., Pages, C., et al. (2012). Human Herpesvirus 8 (HHV8) sequentially shapes the NK cell repertoire during the course of asymptomatic infection and Kaposi sarcoma. PLoS Pathog. 8:e1002486. doi: 10.1371/journal.ppat.1002486
Eckmann, L., Stenson, W. F., Savidge, T. C., Lowe, D. C., Barrett, K. E., Fierer, J., et al. (1997). Role of intestinal epithelial cells in the host secretory response to infection by invasive bacteria. Bacterial entry induces epithelial prostaglandin h synthase-2 expression and prostaglandin E2 and F2alpha production. J. Clin. Invest. 100, 296–309. doi: 10.1172/JCI119535
Edwards, C. K., Hedegaard, H. B., Zlotnik, A., Gangadharam, P. R., Johnston, R. B., and Pabst, M. J. (1986). Chronic infection due to Mycobacterium intracellulare in mice: association with macrophage release of prostaglandin E2 and reversal by injection of indomethacin, muramyl dipeptide, or interferon-gamma. J. Immunol. 136, 1820–1827.
Ejima, K., Layne, M. D., Carvajal, I. M., Kritek, P. A., Baron, R. M., Chen, Y. H., et al. (2003). Cyclooxygenase-2-deficient mice are resistant to endotoxin-induced inflammation and death. FASEB J. 17, 1325–1327. doi: 10.1096/fj.02-1078fje
Filion, F., Bouchard, N., Goff, A. K., Lussier, J. G., and Sirois, J. (2001). Molecular cloning and induction of bovine prostaglandin E synthase by gonadotropins in ovarian follicles prior to ovulation in vivo. J. Biol. Chem. 276, 34323–34330. doi: 10.1074/jbc.M103709200
Finch, R. (2001). Community-acquired pneumonia: the evolving challenge. Clin. Microbiol. Infect. 7(Suppl. 3), 30–38.
Fink, J., Steer, J. H., Joyce, D. A., McWilliam, A. S., and Stewart, G. A. (2003). Pro-inflammatory effects of Burkholderia cepacia on cystic fibrosis respiratory epithelium. FEMS Immunol. Med. Microbiol. 38, 273–282. doi: 10.1016/S0928-8244(03)00169-X
Fitzpatrick, F. A., Aguirre, R., Pike, J. E., and Lincoln, F. H. (1980). The stability of 13, 14-dihydro-15 keto-PGE2. Prostaglandins 19, 917–931. doi: 10.1016/0090-6980(80)90126-4
Fujino, H., Salvi, S., and Regan, J. W. (2005). Differential regulation of phosphorylation of the cAMP response element-binding protein after activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. Mol. Pharmacol. 68, 251–259.
Fujino, H., Xu, W., and Regan, J. W. (2003). Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J. Biol. Chem. 278, 12151–12156. doi: 10.1074/jbc.M212665200
García-Del Portillo, F., Pucciarelli, M. G., and Casadesús, J. (1999). DNA adenine methylase mutants of Salmonella typhimurium show defects in protein secretion, cell invasion, and M cell cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 96, 11578–11583. doi: 10.1073/pnas.96.20.11578
Glibetic, M., Samlalsingh-Parker, J., Raykova, V., Ofenstein, J., and Aranda, J. V. (2001). Group B Streptococci and inducible nitric oxide synthase: modulation by nuclear factor kappa B and ibuprofen. Semin. Perinatol. 25, 65–69. doi: 10.1053/sper.2001.23181
Goldmann, O., Hertzén, E., Hecht, A., Schmidt, H., Lehne, S., Norrby-Teglund, A., et al. (2010). Inducible cyclooxygenase released prostaglandin E2 modulates the severity of infection caused by Streptococcus pyogenes. J. Immunol. 185, 2372–2381. doi: 10.4049/jimmunol.1000838
Goto, T., Herberman, R. B., Maluish, A., and Strong, D. M. (1983). Cyclic AMP as a mediator of prostaglandin E-induced suppression of human natural killer cell activity. J. Immunol. 130, 1350–1355.
Gustafsson, K., Ingelsten, M., Bergqvist, L., Nyström, J., Andersson, B., and Karlsson-Parra, A. (2008). Recruitment and activation of natural killer cells in vitro by a human dendritic cell vaccine. Cancer Res. 68, 5965–5971. doi: 10.1158/0008-5472.CAN-07-6494
Ham, E. A., Soderman, D. D., Zanetti, M. E., Dougherty, H. W., McCauley, E., and Kuehl, F. A. (1983). Inhibition by prostaglandins of leukotriene B4 release from activated neutrophils. Proc. Natl. Acad. Sci. U.S.A. 80, 4349–4353. doi: 10.1073/pnas.80.14.4349
Harris, S. G., Padilla, J., Koumas, L., Ray, D., and Phipps, R. P. (2002). Prostaglandins as modulators of immunity. Trends Immunol. 23, 144–150. doi: 10.1016/S1471-4906(01)02154-8
Hata, A. N., and Breyer, R. M. (2004). Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol. Ther. 103, 147–166. doi: 10.1016/j.pharmthera.2004.06.003
He, L. K., Liu, L. H., Hahn, E., and Gamelli, R. L. (2001). The expression of cyclooxygenase and the production of prostaglandin E2 in neutrophils after burn injury and infection. J. Burn Care Rehabil. 22, 58–64. doi: 10.1097/00004630-200101000-00012
Heithoff, D. M., Enioutina, E. Y., Daynes, R. A., Sinsheimer, R. L., Low, D. A., and Mahan, M. J. (2001). Salmonella DNA adenine methylase mutants confer cross-protective immunity. Infect. Immun. 69, 6725–6730. doi: 10.1128/IAI.69.11.6725-6730.2001
Hensel, M., Shea, J. E., Waterman, S. R., Mundy, R., Nikolaus, T., Banks, G., et al. (1998). Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 30, 163–174. doi: 10.1046/j.1365-2958.1998.01047.x
Hessle, C. C., Andersson, B., and Wold, A. E. (2003). Gram-negative, but not Gram-positive, bacteria elicit strong PGE2 production in human monocytes. Inflammation 27, 329–332. doi: 10.1023/B:IFLA.0000006700.41614.21
Honda, A., Sugimoto, Y., Namba, T., Watabe, A., Irie, A., Negishi, M., et al. (1993). Cloning and expression of a cDNA for mouse prostaglandin E receptor EP2 subtype. J. Biol. Chem. 268, 7759–7762.
Huang, M., Stolina, M., Sharma, S., Mao, J. T., Zhu, L., Miller, P. W., et al. (1998). Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res. 58, 1208–1216.
Hubbard, L. L., Ballinger, M. N., Thomas, P. E., Wilke, C. A., Standiford, T. J., Kobayashi, K. S., et al. (2010). A role for IL-1 receptor-associated kinase-M in prostaglandin E2-induced immunosuppression post-bone marrow transplantation. J. Immunol. 184, 6299–6308. doi: 10.4049/jimmunol.0902828
Hurley, B. P., Pirzai, W., Mumy, K. L., Gronert, K., and McCormick, B. A. (2011). Selective eicosanoid-generating capacity of cytoplasmic phospholipase A2 in Pseudomonas aeruginosa-infected epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L286–L294. doi: 10.1152/ajplung.00147.2010
Inbar, S., Neeman, E., Avraham, R., Benish, M., Rosenne, E., and Ben-Eliyahu, S. (2011). Do stress responses promote leukemia progression? An animal study suggesting a role for epinephrine and prostaglandin-E2 through reduced NK activity. PLoS ONE 6:e19246. doi: 10.1371/journal.pone.0019246
Isles, A., Maclusky, I., Corey, M., Gold, R., Prober, C., Fleming, P., et al. (1984). Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J. Pediatr. 104, 206–210. doi: 10.1016/S0022-3476(84)80993-2
Jonuleit, H., Kühn, U., Müller, G., Steinbrink, K., Paragnik, L., Schmitt, E., et al. (1997). Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol. 27, 3135–3142. doi: 10.1002/eji.1830271209
Joshi, P. C., Zhou, X., Cuchens, M., and Jones, Q. (2001). Prostaglandin E2 suppressed IL-15-mediated human NK cell function through down-regulation of common gamma-chain. J. Immunol. 166, 885–891.
Kalinski, P. (2012). Regulation of immune responses by prostaglandin E2. J. Immunol. 188, 21–28. doi: 10.4049/jimmunol.1101029
Kaliński, P., Hilkens, C. M., Snijders, A., Snijdewint, F. G., and Kapsenberg, M. L. (1997). IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J. Immunol. 159, 28–35.
Kaliński, P., Schuitemaker, J. H., Hilkens, C. M., and Kapsenberg, M. L. (1998). Prostaglandin E2 induces the final maturation of IL-12-deficient CD1a+CD83+ dendritic cells: the levels of IL-12 are determined during the final dendritic cell maturation and are resistant to further modulation. J. Immunol. 161, 2804–2809.
Kis, B., Snipes, J. A., Gaspar, T., Lenzser, G., Tulbert, C. D., and Busija, D. W. (2006). Cloning of cyclooxygenase-1b (putative COX-3) in mouse. Inflamm. Res. 55, 274–278. doi: 10.1007/s00011-006-0083-z
Kunkel, S. L., Spengler, M., May, M. A., Spengler, R., Larrick, J., and Remick, D. (1988). Prostaglandin E2 regulates macrophage-derived tumor necrosis factor gene expression. J. Biol. Chem. 263, 5380–5384.
Kunkel, S. L., Wiggins, R. C., Chensue, S. W., and Larrick, J. (1986). Regulation of macrophage tumor necrosis factor production by prostaglandin E2. Biochem. Biophys. Res. Commun. 137, 404–410. doi: 10.1016/0006-291X(86)91224-6
Kurland, J. I., and Bockman, R. (1978). Prostaglandin E production by human blood monocytes and mouse peritoneal macrophages. J. Exp. Med. 147, 952–957. doi: 10.1084/jem.147.3.952
Lambeau, G., and Lazdunski, M. (1999). Receptors for a growing family of secreted phospholipases A2. Trends Pharmacol. Sci. 20, 162–170. doi: 10.1016/S0165-6147(99)01300-0
Liu, L., Ge, D., Ma, L., Mei, J., Liu, S., Zhang, Q., et al. (2012). Interleukin-17 and prostaglandin E2 are involved in formation of an M2 macrophage-dominant microenvironment in lung cancer. J. Thorac. Oncol. 7, 1091–1100. doi: 10.1097/JTO.0b013e3182542752
Luft, T., Jefford, M., Luetjens, P., Toy, T., Hochrein, H., Masterman, K. A., et al. (2002). Functionally distinct dendritic cell (DC) populations induced by physiologic stimuli: prostaglandin E(2) regulates the migratory capacity of specific DC subsets. Blood 100, 1362–1372. doi: 10.1182/blood-2001-12-0360
Mailliard, R. B., Alber, S. M., Shen, H., Watkins, S. C., Kirkwood, J. M., Herberman, R. B., et al. (2005). IL-18-induced CD83+CCR7+ NK helper cells. J. Exp. Med. 202, 941–953. doi: 10.1084/jem.20050128
Mandell, L. A., Wunderink, R. G., Anzueto, A., Bartlett, J. G., Campbell, G. D., Dean, N. C., et al. (2007). Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin. Infect. Dis. 44(Suppl. 2), S27–S72. doi: 10.1086/511159
Marotta, P., Sautebin, L., and Di Rosa, M. (1992). Modulation of the induction of nitric oxide synthase by eicosanoids in the murine macrophage cell line J774. Br. J. Pharmacol. 107, 640–641. doi: 10.1111/j.1476-5381.1992.tb14499.x
Muthuswamy, R., Mueller-Berghaus, J., Haberkorn, U., Reinhart, T. A., Schadendorf, D., and Kalinski, P. (2010). PGE(2) transiently enhances DC expression of CCR7 but inhibits the ability of DCs to produce CCL19 and attract naive T cells. Blood 116, 1454–1459. doi: 10.1182/blood-2009-12-258038
Nagamatsu, T., and Schust, D. J. (2010). The immunomodulatory roles of macrophages at the maternal-fetal interface. Reprod. Sci. 17, 209–218. doi: 10.1177/1933719109349962
Nakayama, T., Mutsuga, N., Yao, L., and Tosato, G. (2006). Prostaglandin E2 promotes degranulation-independent release of MCP-1 from mast cells. J. Leukoc. Biol. 79, 95–104. doi: 10.1189/jlb.0405226
Natarajan, G., Glibetic, M., Raykova, V., Ofenstein, J. P., Thomas, R. L., and Aranda, J. V. (2007). Nitric oxide and prostaglandin response to group B streptococcal infection in the lung. Ann. Clin. Lab. Sci. 37, 170–176.
Ney, P., and Schrör, K. (1991). PGD2 and its mimetic ZK 110.841 are potent inhibitors of receptor-mediated activation of human neutrophils. Eicosanoids 4, 21–28.
Nishigaki, N., Negishi, M., and Ichikawa, A. (1996). Two Gs-coupled prostaglandin E receptor subtypes, EP2 and EP4, differ in desensitization and sensitivity to the metabolic inactivation of the agonist. Mol. Pharmacol. 50, 1031–1037.
Obermajer, N., Muthuswamy, R., Lesnock, J., Edwards, R. P., and Kalinski, P. (2011). Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 118, 5498–5505. doi: 10.1182/blood-2011-07-365825
Ochman, H., Soncini, F. C., Solomon, F., and Groisman, E. A. (1996). Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. U.S.A. 93, 7800–7804. doi: 10.1073/pnas.93.15.7800
Park, J. Y., Pillinger, M. H., and Abramson, S. B. (2006). Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin. Immunol. 119, 229–240. doi: 10.1016/j.clim.2006.01.016
Peres-Buzalaf, C., de Paula, L., Frantz, F. G., Soares, E. M., Medeiros, A. I., Peters-Golden, M., et al. (2011). Control of experimental pulmonary tuberculosis depends more on immunostimulatory leukotrienes than on the absence of immunosuppressive prostaglandins. Prostaglandins Leukot. Essent. Fatty Acids 85, 75–81. doi: 10.1016/j.plefa.2011.04.024
Peterson, J. W., Lu, Y., Duncan, S., Cantu, J., and Chopra, A. K. (1994). Interactions of intestinal mediators in the mode of action of cholera toxin. J. Med. Microbiol. 41, 3–9. doi: 10.1099/00222615-41-1-3
Phipps, R. P., Stein, S. H., and Roper, R. L. (1991). A new view of prostaglandin E regulation of the immune response. Immunol. Today 12, 349–352. doi: 10.1016/0167-5699(91)90064-Z
Qadri, F., Raqib, R., Ahmed, F., Rahman, T., Wenneras, C., Das, S. K., et al. (2002). Increased levels of inflammatory mediators in children and adults infected with Vibrio cholerae O1 and O139. Clin. Diagn. Lab. Immunol. 9, 221–229. doi: 10.1128/CDLI.9.2.221-229.2002
Rangel Moreno, J., Estrada García, I., De La Luz García Hernández, M., Aguilar Leon, D., Marquez, R., and Hernández Pando, R. (2002). The role of prostaglandin E2 in the immunopathogenesis of experimental pulmonary tuberculosis. Immunology 106, 257–266. doi: 10.1046/j.1365-2567.2002.01403.x
Raqib, R., Mia, S. M., Qadri, F., Alam, T. I., Alam, N. H., Chowdhury, A. K., et al. (2000). Innate immune responses in children and adults with Shigellosis. Infect. Immun. 68, 3620–3629. doi: 10.1128/IAI.68.6.3620-3629.2000
Rastogi, N., Bachelet, M., and Carvalho de Sousa, J. P. (1992). Intracellular growth of Mycobacterium avium in human macrophages is linked to the increased synthesis of prostaglandin E2 and inhibition of the phagosome-lysosome fusions. FEMS Microbiol. Immunol. 4, 273–279. doi: 10.1111/j.1574-6968.1992.tb05006.x
Raymond, B., Crepin, V. F., Collins, J. W., and Frankel, G. (2011). The WxxxE effector EspT triggers expression of immune mediators in an Erk/JNK and NF-κ B-dependent manner. Cell. Microbiol. 13, 1881–1893. doi: 10.1111/j.1462-5822.2011.01666.x
Resta-Lenert, S., and Barrett, K. E. (2002). Enteroinvasive bacteria alter barrier and transport properties of human intestinal epithelium: role of iNOS and COX-2. Gastroenterology 122, 1070–1087. doi: 10.1053/gast.2002.32372
Rieser, C., Böck, G., Klocker, H., Bartsch, G., and Thurnher, M. (1997). Prostaglandin E2 and tumor necrosis factor alpha cooperate to activate human dendritic cells: synergistic activation of interleukin 12 production. J. Exp. Med. 186, 1603–1608. doi: 10.1084/jem.186.9.1603
Rosenstreich, D. L., Vogel, S. N., Jacques, A. R., Wahl, L. M., and Oppenheim, J. J. (1978). Macrophage sensitivity to endotoxin: genetic control by a single codominant gene. J. Immunol. 121, 1664–1670.
Rubins, J. B., Mitchell, T. J., Andrew, P. W., and Niewoehner, D. E. (1994). Pneumolysin activates phospholipase A in pulmonary artery endothelial cells. Infect. Immun. 62, 3829–3836.
Sadikot, R. T., Blackwell, T. S., Christman, J. W., and Prince, A. S. (2005). Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 171, 1209–1223. doi: 10.1164/rccm.200408-1044SO
Sadikot, R. T., Zeng, H., Azim, A. C., Joo, M., Dey, S. K., Breyer, R. M., et al. (2007). Bacterial clearance of Pseudomonas aeruginosa is enhanced by the inhibition of COX-2. Eur. J. Immunol. 37, 1001–1009. doi: 10.1002/eji.200636636
Saliba, A. M., Nascimento, D. O., Silva, M. C., Assis, M. C., Gayer, C. R., Raymond, B., et al. (2005). Eicosanoid-mediated proinflammatory activity of Pseudomonas aeruginosa ExoU. Cell. Microbiol. 7, 1811–1822. doi: 10.1111/j.1462-5822.2005.00635.x
Sato, H., and Frank, D. W. (2004). ExoU is a potent intracellular phospholipase. Mol. Microbiol. 53, 1279–1290. doi: 10.1111/j.1365-2958.2004.04194.x
Sato, H., Frank, D. W., Hillard, C. J., Feix, J. B., Pankhaniya, R. R., Moriyama, K., et al. (2003). The mechanism of action of the Pseudomonas aeruginosa-encoded type III cytotoxin, ExoU. EMBO J. 22, 2959–2969. doi: 10.1093/emboj/cdg290
Scandella, E., Men, Y., Gillessen, S., Förster, R., and Groettrup, M. (2002). Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood 100, 1354–1361. doi: 10.1182/blood-2001-11-0017
Schulert, G. S., Feltman, H., Rabin, S. D., Martin, C. G., Battle, S. E., Rello, J., et al. (2003). Secretion of the toxin ExoU is a marker for highly virulent Pseudomonas aeruginosa isolates obtained from patients with hospital-acquired pneumonia. J. Infect. Dis. 188, 1695–1706. doi: 10.1086/379372
Schulze-Specking, A., Duyster, J., Gebicke-Haerter, P. J., Wurster, S., and Dieter, P. (1991). Effect of fluoride, pertussis and cholera toxin on the release of arachidonic acid and the formation of prostaglandin E2, D2, superoxide and inositol phosphates in rat liver macrophages. Cell. Signal. 3, 599–606. doi: 10.1016/0898-6568(91)90036-T
Serezani, C. H., Chung, J., Ballinger, M. N., Moore, B. B., Aronoff, D. M., and Peters-Golden, M. (2007). Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am. J. Respir. Cell Mol. Biol. 37, 562–570. doi: 10.1165/rcmb.2007-0153OC
Somayaji, S. N., Ritchie, S., Sahraei, M., Marriott, I., and Hudson, M. C. (2008). Staphylococcus aureus induces expression of receptor activator of NF-kappaB ligand and prostaglandin E2 in infected murine osteoblasts. Infect. Immun. 76, 5120–5126. doi: 10.1128/IAI.00228-08
Speelman, P., Rabbani, G. H., Bukhave, K., and Rask-Madsen, J. (1985). Increased jejunal prostaglandin E2 concentrations in patients with acute cholera. Gut 26, 188–193. doi: 10.1136/gut.26.2.188
Stables, M. J., Newson, J., Ayoub, S. S., Brown, J., Hyams, C. J., and Gilroy, D. W. (2010). Priming innate immune responses to infection by cyclooxygenase inhibition kills antibiotic-susceptible and -resistant bacteria. Blood 116, 2950–2959. doi: 10.1182/blood-2010-05-284844
Stefanelli, P., Teloni, R., Carannante, A., Mariotti, S., Nisini, R., and Gagliardi, M. C. (2012). Neisseria gonorrhoeae triggers the PGE2/IL-23 pathway and promotes IL-17 production by human memory T cells. Prostaglandins Other Lipid Mediat. 99, 24–29. doi: 10.1016/j.prostaglandins.2012.04.002
Stolina, M., Sharma, S., Lin, Y., Dohadwala, M., Gardner, B., Luo, J., et al. (2000). Specific inhibition of cyclooxygenase 2 restores antitumor reactivity by altering the balance of IL-10 and IL-12 synthesis. J. Immunol. 164, 361–370.
Strassmann, G., Patil-Koota, V., Finkelman, F., Fong, M., and Kambayashi, T. (1994). Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J. Exp. Med. 180, 2365–2370. doi: 10.1084/jem.180.6.2365
Sugimoto, Y., Namba, T., Honda, A., Hayashi, Y., Negishi, M., Ichikawa, A., et al. (1992). Cloning and expression of a cDNA for mouse prostaglandin E receptor EP3 subtype. J. Biol. Chem. 267, 6463–6466.
Szymanski, K. V., Toennies, M., Becher, A., Fatykhova, D., N'Guessan, P. D., Gutbier, B., et al. (2012). Streptococcus pneumoniae induced regulation of cyclooxygenase-2 in human lung tissue. Eur. Respir. J. 40, 1458–1467. doi: 10.1183/09031936.00186911
Tai, H. H., Ensor, C. M., Tong, M., Zhou, H., and Yan, F. (2002). Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 68–69, 483–493. doi: 10.1016/S0090-6980(02)00050-3
Takenawa, T., Ishitoya, J., and Nagai, Y. (1986). Inhibitory effect of prostaglandin E2, forskolin, and dibutyryl cAMP on arachidonic acid release and inositol phospholipid metabolism in guinea pig neutrophils. J. Biol. Chem. 261, 1092–1098.
Takeuchi, K., Kato, S., and Amagase, K. (2010). Prostaglandin EP receptors involved in modulating gastrointestinal mucosal integrity. J. Pharmacol. Sci. 114, 248–261. doi: 10.1254/jphs.10R06CR
Talpain, E., Armstrong, R. A., Coleman, R. A., and Vardey, C. J. (1995). Characterization of the PGE receptor subtype mediating inhibition of superoxide production in human neutrophils. Br. J. Pharmacol. 114, 1459–1465. doi: 10.1111/j.1476-5381.1995.tb13370.x
Torrado, E., Robinson, R. T., and Cooper, A. M. (2011). Cellular response to mycobacteria: balancing protection and pathology.Trends Immunol. 32, 66–72. doi: 10.1016/j.it.2010.12.001
Uchiya, K., Barbieri, M. A., Funato, K., Shah, A. H., Stahl, P. D., and Groisman, E. A. (1999). A Salmonella virulence protein that inhibits cellular trafficking. EMBO J. 18, 3924–3933. doi: 10.1093/emboj/18.14.3924
Uchiya, K., and Nikai, T. (2004). Salmonella enterica serovar Typhimurium infection induces cyclooxygenase 2 expression in macrophages: involvement of Salmonella pathogenicity island 2. Infect. Immun. 72, 6860–6869. doi: 10.1128/IAI.72.12.6860-6869.2004
Walker, W., and Rotondo, D. (2004). Prostaglandin E2 is a potent regulator of interleukin-12- and interleukin-18-induced natural killer cell interferon-gamma synthesis. Immunology 111, 298–305. doi: 10.1111/j.1365-2567.2004.01810.x
Wheeldon, A., and Vardey, C. J. (1993). Characterization of the inhibitory prostanoid receptors on human neutrophils. Br. J. Pharmacol. 108, 1051–1054. doi: 10.1111/j.1476-5381.1993.tb13504.x
Wise, H. (1996). The inhibitory effect of prostaglandin E2 on rat neutrophil aggregation. J. Leukoc. Biol. 60, 480–486.
Wise, H., and Jones, R. L. (1994). Characterization of prostanoid receptors on rat neutrophils. Br. J. Pharmacol. 113, 581–587. doi: 10.1111/j.1476-5381.1994.tb17029.x
Yu, Y., and Chadee, K. (1998). Prostaglandin E2 stimulates IL-8 gene expression in human colonic epithelial cells by a posttranscriptional mechanism. J. Immunol. 161, 3746–3752.
Keywords: bacteria, prostaglandin, COX, immunotherapeutic, mucosal, infection
Citation: Agard M, Asakrah S and Morici LA (2013) PGE2 suppression of innate immunity during mucosal bacterial infection. Front. Cell. Infect. Microbiol. 3:45. doi: 10.3389/fcimb.2013.00045
Received: 07 June 2013; Accepted: 30 July 2013;
Published online: 21 August 2013.
Edited by:
Alfredo G. Torres, University of Texas Medical Branch, USAReviewed by:
Maziar Divangahi, McGill University, CanadaNemani V. Prasadarao, Children's Hospital Los Angeles and University of Southern California, USA
Copyright © 2013 Agard, Asakrah and Morici. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa A. Morici, Department of Microbiology and Immunology, Tulane University School of Medicine, 1430 Tulane Ave, New Orleans, LA 70119, USA e-mail: lmorici@tulane.edu