Tripartite ATP-Independent Periplasmic (TRAP) Transporters and Tripartite Tricarboxylate Transporters (TTT): From Uptake to Pathogenicity
 Leonardo T. Rosa
Leonardo T. Rosa Matheus E. Bianconi
Matheus E. Bianconi Gavin H. Thomas
Gavin H. Thomas David J. Kelly
David J. Kelly- 1Department of Molecular Biology and Biotechnology, University of Sheffield, Sheffield, United Kingdom
- 2Department of Animal and Plant Sciences, University of Sheffield, Sheffield, United Kingdom
- 3Department of Biology, University of York, York, United Kingdom
The ability to efficiently scavenge nutrients in the host is essential for the viability of any pathogen. All catabolic pathways must begin with the transport of substrate from the environment through the cytoplasmic membrane, a role executed by membrane transporters. Although several classes of cytoplasmic membrane transporters are described, high-affinity uptake of substrates occurs through Solute Binding-Protein (SBP) dependent systems. Three families of SBP dependant transporters are known; the primary ATP-binding cassette (ABC) transporters, and the secondary Tripartite ATP-independent periplasmic (TRAP) transporters and Tripartite Tricarboxylate Transporters (TTT). Far less well understood than the ABC family, the TRAP transporters are found to be abundant among bacteria from marine environments, and the TTT transporters are the most abundant family of proteins in many species of β-proteobacteria. In this review, recent knowledge about these families is covered, with emphasis on their physiological and structural mechanisms, relating to several examples of relevant uptake systems in pathogenicity and colonization, using the SiaPQM sialic acid uptake system from Haemophilus influenzae and the TctCBA citrate uptake system of Salmonella typhimurium as the prototypes for the TRAP and TTT transporters, respectively. High-throughput analysis of SBPs has recently expanded considerably the range of putative substrates known for TRAP transporters, while the repertoire for the TTT family has yet to be fully explored but both types of systems most commonly transport carboxylates. Specialized spectroscopic techniques and site-directed mutagenesis have enriched our knowledge of the way TRAP binding proteins capture their substrate, while structural comparisons show conserved regions for substrate coordination in both families. Genomic and protein sequence analyses show TTT SBP genes are strikingly overrepresented in some bacteria, especially in the β-proteobacteria and some α-proteobacteria. The reasons for this are not clear but might be related to a role for these proteins in signaling rather than transport.
Solute Binding-Protein (SBP) Dependant Secondary Transporters: The TRAP and TTT Systems
Solute Binding-Protein (SBP) dependent transport systems contain, in addition to the membrane proteins, a soluble extra-cytoplasmic protein, located either free in the periplasm or anchored to the membrane in the case of Gram-positive bacteria, which binds the substrate with high affinity, and specificity, allowing uptake even in very low concentrations of ligands. Three families of SBP dependant transporters are currently known, the composition of which are summarized in Figure 1. ATP-binding cassette (ABC) transporters use the free energy of ATP binding and hydrolysis to move substrates across the membrane against a concentration gradient. First described in the early 1970‘s (Kalckar, 1971; Willis and Furlong, 1974), this family is by far the best investigated SBP-dependant transporter family, with the maltose and vitamin B12 uptake systems as the most thoroughly studied models, and was subject of several reviews over recent years (Jones and George, 2004; Davidson et al., 2008; Rice et al., 2014; Maqbool et al., 2015; Wilkens, 2015; Locher, 2016).
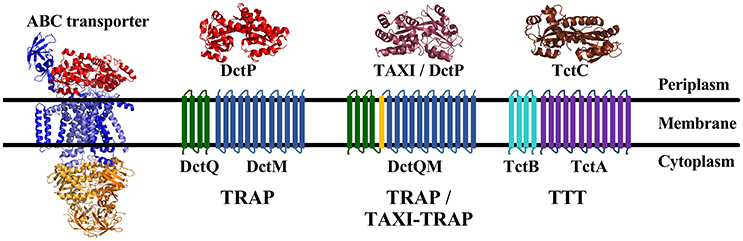
Figure 1. Overall topologies and structures of the different Solute Binding Protein (SBP dependant) transporter families. The ABC transporters are represented by the E. coli maltose transporter MalEFGK2 (PDB 2R6G). It is composed of a solute binding protein (red), two Transmembrane (TM) domains (blue), and two Nucleotide binding domains (NBD) (orange); The secondary Tripartite ATP-independent periplasmic (TRAP) Transporters are composed of a 12 TM domain channel DctM (blue) and a 4 TM domain protein DctQ (green), which can be fused together by an additional TM domain (yellow) in a DctQM protein, and a DctP or TAXI SBP protein, represented respectively by the SiaP from H. influenza (PDB 2CEY) (light red) and the TT1099 from T. thermophilus (PDB 1US4) (dark red). TAXI-TRAP were always found associated with fused DctQM proteins; The Tripartite Tricarboxylate Transporter (TTT), is formed also by a 12 TM channel TctA (purple), a 4 TM protein TctB (cyan) and a TctC solute binding protein, represented by Bug27 from B. pertussis (PDB 2QPQ) (Brown). In some rare cases, TctAB proteins may be also fused. Although sharing similar topology, the TRAP and TTT systems share no sequence similarity.
The Tripartite ATP-independent periplasmic (TRAP) transporters (TC: 2.A.56) and Tripartite Tricarboxylate Transporters (TTT, TC: 2.A.80), on the other hand, use ion-electrochemical gradients to move substrates in a symporter mechanism, thus being defined as secondary transporters. These two families are significantly less well-understood than ABC systems but share a similar overall protein composition and topology, as well as genomic organization. In addition to the SBP‘s (“P” subunit in TRAP systems, “C” subunit in TTT), each system is comprised of two transmembrane proteins, one well-conserved 12 transmembrane (TM) domain protein (“M” subunit in the TRAP systems, “A” subunit in TTT) and one poorly conserved 4 TM domain protein (“Q” subunit in TRAP systems, “B” subunit in the TTT) (Forward et al., 1997; Winnen et al., 2003; Thomas et al., 2006; Hosaka et al., 2013, Figure 1). However, no sequence similarity is found between the corresponding proteins in these families, thus representing either a case of convergent evolution (Fischer et al., 2010) or very ancient orthology and divergence (Winnen et al., 2003).
Regardless of their lack of sequence similarity, the SBPs from these two families show very similar tertiary structures. They are folded in a “Venus fly-trap” shape, with two wings composed of one β-sheet containing four to six strands, surrounded by α-helices and connected by a hinge. Opened in the apo form, the wings close around the substrate in a very specific manner, binding the substrate tightly in a cleft formed between the two domains. The enclosure of the substrate then allows the protein to interact with the transmembrane domains (Herrou et al., 2007). It is suggested that these two wings were generated by a duplication event in early TTT (and other SBP dependent) transporters (Winnen et al., 2003). Classification of SBPs into related clusters has been proposed, based on their secondary and tertiary structural patterns and their substrate specificities, with the first classification into three distinct types proposed by Fukami-Kobayashi et al. (1999). With the exponential increase in new entries for SBPs in genomic databases due to new sequencing capabilities, it became clear that the separation into three types was too simplistic to comprise SBP diversity, and thus a new model was presented by Berntsson et al. (2010) and recently revised by Scheepers et al. (2016). Both TRAP and TTT SBP‘s are contained within the Type II group in the first classification, and inside Cluster E in the latter. This review summarizes the evidence of a relationship between these two classes of secondary high-affinity uptake systems and pathogenicity. Additionally, it adds an evolutionary perspective regarding the expansion of the TTT family in some pathogens.
The TRAP Transporter Family
The first characterization and naming of TRAP transporters was described in Rhodobacter capsulatus by Forward et al. (1997), when a SBP encoding gene was found adjacent to two genes encoding transmembrane proteins of 12 (DctM) and 4 (DctQ) predicted helices. Functional studies showed symport of C4-dicarboxylic acids apparently energized by the proton motive-force. Subsequent studies showed that these systems can transport a variety of substrates under different contexts. Detailed reviews about this family were provided by Kelly and Thomas (2001) and Mulligan et al. (2011), and the following sections will focus on more recent insights.
Substrate Diversity of the TRAP Family and Roles in Pathogenicity
The best studied TRAP system is undoubtedly SiaPQM from Haemophilus influenzae (Figures 1, 2A), discovered by Severi et al. (2005) to be involved in the uptake of sialic acid. Sialic acid is a generic name for a class of 9-carbon sugar acids used by most eukaryotic cells in the form of cell surface glycoproteins. For this reason, many pathogens evolved to mimic these surface structures in their own cell envelope, constituting an important virulence factor which improves evasion of the human immune system (Bouchet et al., 2003). In H. influenzae, absence of SiaPQM causes loss of sialic acid uptake and lack of incorporation in the lipo-oligossacharide (Allen et al., 2005), and a subsequent study showed an increased susceptibility of this pathogen to human serum and decreased virulence in the chinchilla otitis model (Jenkins et al., 2010). Systems homologous to SiaPQM were subsequently found to be involved in the uptake of sialic acid in several pathogens, such as Vibrio cholerae, Fusobacterium nucleatum, and Vibrio vulnificus (Severi et al., 2005), the latter being shown to transport sialic acid in 67 clinical isolates (Lubin et al., 2012). Signature-tagged mutagenesis studies in Pasteurella multocida, an opportunistic pathogen of livestock, showed that disruption of genes related to sialic acid metabolism resulted in decrease of virulence in mice models (Fuller et al., 2000). A subsequent study showed that a SiaP homolog was involved in the uptake of sialic acid in this bacterium (Steenbergen et al., 2005), and that disruption of sialic acid uptake resulted in decreased virulence in a turkey model (Tatum et al., 2009). Severi et al. (2007) provides a review of how sialic acid uptake and metabolism is used as a virulence factor in different pathogens, and Vimr et al. (2004) provides a more general review about sialic acid metabolism. Thomas (2016) gives a recent overview of the different uptake strategies and transport systems used by different pathogens for the uptake of sialic acid.
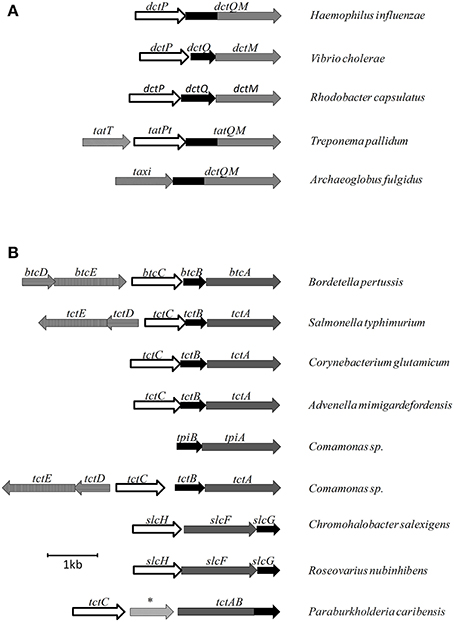
Figure 2. Examples of genetic organization for different secondary SBP dependant transporters. (A) Gene organizations for TRAP systems. (B) Gene organization for TTT systems. * represents an amido-hydrolase gene.
In Bordetella pertussis, the causative agent of whooping cough, two DctP homologs are encoded in the vicinity of virulence-related operons modulated by the BvgA/BvgS two-component system. The two proteins were crystalized by Rucktooa et al. (2007) with a pyroglutamic acid bound in the substrate cleft. One of these proteins is highly expressed in B. pertussis, although the membrane components of the system seem to be mutated and non-functional. Although it is unclear what physiological role pyroglutamic acid would have, this amino-acid is present in the filamentous hemagglutinin produced by B. pertussis, and it was speculated it could serve as a glutamate reserve. In fact, BugE, an abundantly expressed SBP from the TTT family also was shown to bind glutamate (Huvent et al., 2006b), suggesting glutamate metabolism might play an important role in the pathophysiology of this bacterium.
TRAP systems are very important also in environmental organisms and in biotechnologically relevant processes. In Halomonas elongata, Grammann et al. (2002) showed that the TeaABC operon was responsible for accumulation of the compatible solute ectoine, in response to osmolarity stress, and that this transporter was osmoregulated. In R. capsulatus a TRAP system was shown to be involved in the import of several monocarboxylic 2-oxo-acids involved in amino-acid biosynthesis (Thomas et al., 2006). Chae and Zylstra (2006) showed the involvement of TRAP transporters in the degradation of several benzoate derivatives, including toxic chlorinated aromatics. In Rhodopseudomonas palustris the TarPQM system was shown to be involved in the degradation of lignin-derived aromatic compounds, in a redundant function also executed by an ABC transporter in the same gene cluster (Salmon et al., 2013). GaaPQM from Agrobacterium tumefaciens was described to be involved in plant virulence (Zhao and Binns, 2016). Maimanakos et al. (2016) showed that TRAP transporters are found in the vicinity of arylmalonate decarboxylases (AMDases) and recently, Meinert et al. (2017) showed a TRAP system involved with the uptake of five different sugars in Advenella mimigardefordensis, but only after they have been converted to their respective sugar acids in the periplasm (Thomas, 2017).
Vetting et al. (2015) published a highly significant study, which multiplied several times our understanding about substrate specificity in TRAP systems. 8,240 SBP‘s were used to build a sequence similarity network, grouping them into several clusters. From these, 304 representatives of non-characterized groups were then screened, coupling differential scanning fluorescence, crystallography and mass spectrometry of co-purified ligands. The methodology shows the importance of using complementary methods and proposes an efficient strategy for the study of SBP‘s. As a result, 71 of the isofunctional clusters had a ligand assigned; 69 high-resolution crystal structures were obtained; previously known ligands were assigned to non-characterized clusters and several new ligands were found to be captured by TRAP transporters, such as D-glucuronate/D-galacturonate, 6-carbon aldonic acids, cell-wall constituents, lipopolysaccharide components, glycerol-3-phosphate/diglycerol-phosphate, 2-acetolactate, orotic acid, indole acids, pantoate/D-erythonate, and ethanolamine, this last being a particular surprise due to its positive charge in contrast to the typical negatively charged carboxylates of most other TRAP transporter substrates. This work was done as part of the Enzyme Function Initiative (EFI), a network aiming to characterize the biochemical and physiological function of different classes of enzymes, among which are soluble binding proteins, through high-throughput sequence/structure based strategies (http://www.enzymefunction.org/).
The Neglected Group: TAXI-TRAP Transporters
It was observed by Kelly and Thomas (2001) that, in some cases, the SBPs associated with the DctQM subunits in the genome showed very limited sequence similarity to DctP, forming a distinct group, TRAP associated extracytoplasmic immunogenic (TAXI) proteins, named after an immunogenic protein of unknown function from the pathogen Brucella (Mayfield et al., 1988). A previous study by Rabus et al. (1999) had found some similarity between TAXI proteins and the E. coli glutamate binding protein, and the only structure available for a TAXI protein, generated by Takahashi et al. (2004), reinforced these initial findings, as it was described as a glutamate/glutamine binding protein. However, the deletion of a TAXI protein from Psychrobacter arcticus was shown by Bakermans et al. (2009) to affect growth also in other dicarboxylic acids such as acetate, butyrate and fumarate. TAXI-TRAP systems usually have the DctQM subunits fused (Figures 1, 2A) and, because they are found also in many Archaea species, it is believed that this system is an ancient form of TRAP transporter. Although Mulligan et al. (2011) provided a brief speculation about potential function of TAXI-TRAP systems based on their genomic context, a complete characterization of this group is still to be generated.
Two Is Not Enough: The TPAT System
In addition to the classical TRAP and the TAXI-TRAP transporters, a third class of TRAP system was characterized by Deka et al. (2012) in the pathogen Treponema pallidum. T. pallidum is the causative agent of syphilis, a disease which continues to be a challenge in global health. This organism is an obligatory pathogen, which lacks many vital biosynthetic pathways for nucleotides, lipids, and most amino-acids, relying on transport systems to obtain these vital requirements from the human host (Radolf et al., 2016). Deka et al. (2012) observed the existence of a single operon encoding a TRAP system in T. pallidum genome, composed of three genes, as shown in Figure 2A. One dctP and one dctQM homolog, named tatPT and tatQM, and a third gene of unknown function, named tatT. The biochemical and crystallographic characterization of TatT showed this soluble protein was formed by 13 α-helixes and one small helix, structured around a central hydrophobic pore which opened to both ends of the structure. Some of these helices were homologous to a tetratricopeptide motif (TPR), normally involved in protein-protein interactions (D'Andrea and Regan, 2003), which gave the name for this group of TRAP transport systems as TPR-protein associated transporters (TPAT). Using cross-linking, western blotting, analytical ultracentrifugation and computational modeling, it was shown that TatT formed a trimer, which in turn interacted with three subunits of the DctP homolog TatPT (Deka et al., 2012). A later study by Brautigam et al. (2012) confirmed these predictions through crystallization of the TatT and TatPT complex. In these structures, it was shown that the substrate cleft from TatPT was aligned to the C-terminal side of the pore in TatT, with minor structural changes happening upon complexation, the main one being the displacement of one loop from TatPT domain 2 in contact with the binding cleft, called a “cleft-finger.” The hydrophobicity observed both in TatT pore and TatPT cleft, together with the presence of a linear hydrophobic molecule crystalized in the TatT pore, suggested that this system is involved in the uptake of hydrophobic molecules (Deka et al., 2012). As both proteins are found in vivo as lipoproteins, anchored to the membrane, it was suggested as a mechanism that this interaction created a chaperone environment for the transport of lipids through the periplasmic hydrophilic environment, where TatT would receive the lipid from the host, anchored in the outer membrane, and transfer it to TatPT, anchored in the inner membrane, which in turn would deliver it to the TatQM subunit (Brautigam et al., 2012). TPAT systems were found in 35 other species, among other spirochaetes and also among free-living proteobacteria. In this latter group, it was mostly found in species capable of degrading hydrocarbons, reinforcing the potential role in aliphatic transport this distinct group of TRAP transporters might have.
Biochemical and Functional Studies of the DctQM Subunits
Unlike the ABC transporters, no crystal structures have been obtained to date regarding the membrane components of TRAP systems, however some mechanistic information is available particularly regarding energy-coupling. In many systems, such as SiaPQM from H. influenzae, the DctM and DctQ membrane units are not expressed separately, but fused in one only protein containing 17 transmembrane helices, one more than expected due to an additional helix that connects the cytoplasmic C-terminal part of DctQ with the periplasmic N-terminal part of DctM (Figure 2A, Mulligan et al., 2009). Even when expressed separately, DctM and DctQ were shown to form a tight complex with a 1:1 stoichiometry during the folding procedure, and attempts to separate the two proteins resulted in disruption of function (Mulligan et al., 2012). While DctM is believed to form a translocation channel and is a member of the ion transporter superfamily (Rabus et al., 1999), the role of DctQ has not been established yet and has a much more variable sequence. It is known that it is essential for transporter function and it was suggested that DctQ might act to mediate interactions between DctM and DctP, chaperoning DctM and stabilizing it in the membrane or participating in energy coupling (Wyborn et al., 2001). Mulligan et al. (2009) performed a series of experiments showing that the presence of Na+ ions was required for sialic acid transport via SiaPQM in H. influenzae. Replacement of Na+ for Li+ ions did not result in uptake activity, and although neither ΔpH or Δψ alone resulted in transport in absence of Na+, the gradients were able to promote substrate uptake when Na+ was present in equal concentrations in both sides of the membrane. These results show that substrate uptake in TRAP transporters is Na+ dependent and characterized as an eletrogenic process, where at least two Na+ ions are co-transported. Not surprisingly, the TRAP family is widely found in bacteria living in saline environments, using the naturally provided Na+ gradient to provide substrate uptake, as discussed by Mulligan et al. (2007). In addition, Mulligan et al. (2009) showed that in opposition to conventional secondary transporters such as the ones from the MFS family, the transport in the TRAP family is unidirectional. The substrate transporter exposes the binding cavities alternatively in the cytoplasm and the periplasm, but because in TRAP transporters the exposure in the periplasmic side only occurs when in interaction with the SBP, movement in the opposite direction is blocked, even when gradients are inverted. The only condition in which contrary movement was observed was in the presence of an excess of un-liganded SiaP in the periplasm, but these conditions are not physiologically relevant. In addition, it was shown that replacement of the SiaP in H. influenzae (HiSiaP) by an homolog from V. cholerae (VcSiaP) did not complement its function, suggesting that the interactions between DctP proteins and the membrane counterparts are specific in each case, rather than promiscuous among the family (Mulligan et al., 2009). Mulligan et al. (2012) performed these same transport assays and characterization of the SiaQM subunits in the homologous system from V. cholerae, which comprises a true tripartite system instead of the fused subunits. The results from this study were very similar to the H. influenzae fused SiaQM system.
Crystal Structure and Dynamics of TRAP SBP's
The first crystal structure of a TRAP SBP was the SiaP protein from H. influenzae (Müller et al., 2006). TRAP SBP‘s have wings very similar to the Type II proposed structure by Fukami-Kobayashi et al. (1999), but with a remarkably large single β-strand, which connects both domains and participates in both β-sheet domains (Figure 3A). In addition, this family contains a long α-helix, which spans both domains and kinks upon ligand binding. These features characterize the TRAP transporters in Cluster E of the division proposed by Scheepers et al. (2016). The hinge-bending upon ligand-binding was estimated by Müller et al. (2006) to be ~30Å based on comparison between unliganded and ligand protein crystals.
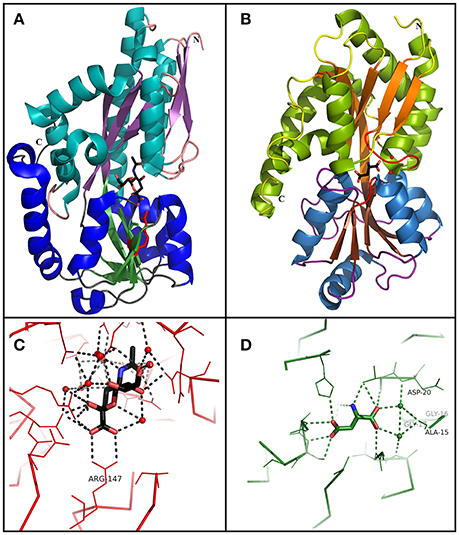
Figure 3. Comparison between TRAP and TTT SBP crystal structures. (A) Overall structure of SiaP, a sialic acid binding SBP from the TRAP family in H. influenzae (PDB 3B50). Domain 1 is represented in cyan (α-helix) and purple (β-sheet), and domain 2 is represented in blue (α-helix) and green (β-sheet). A sialic acid molecule is shown in the binding pocket. Arg147, important to perform a salt bridge with the carboxylic group of the substrate in most SiaP homologs is shown in red. (B) Overall structure of BugD, a aspartate binding SBP from the TTT family in B. pertussis (PDB 2F5X). Domain 1 is represented in green (α-helix) and orange (β-sheet), and domain 2 is represented in blue (α-helix) and brown (β-sheet). An aspartate molecule is shown in the binding pocket. Two loops, between β1 and α1 and between β7 and α6, are involved in the conserved coordination of two water molecules, which bridge hydrogen bonds with the proximal carboxylic group in the substrate. These loops and waters are shown in red (C) Binding pocket of SiaP, showing the coordination of the sialic acid molecule. Arg147 perform a salt bridge with the carboxylic group in the substrate. The remaining of the molecule is coordinated by hydrogen bonds and water molecules which are variable inside the TRAP family. (D) Binding pocket of BugD, showing the coordination of the aspartate molecule. The residues Ala-15, gly-16, gly-17, and Asp-20 participate in the loop between β1 and α1, coordinating two water molecules which bridge hydrogen bonds with the proximal carboxylic group in the substrate. This coordination is very conserved among the TTT family. The remaining substrate coordination occur through non-conserved hydrogen bonds and water bridging.
Although ligand positioning inside the binding pocket is conserved, the hydrogen bonds and hydrophobic interactions for each molecule coordination vary, making substrate prediction difficult for this family. A conserved arginine residue in domain 2, however, turns out to be crucial for ligand interaction (Figure 3C), as discussed by Fischer et al. (2010, 2015). Localized in β-strand 6, which is in a stable β-sheet, the side chain of this highly conserved residue (96.8% of 6,142 sequences searched) points toward the binding cavity and, unusually, is stabilized through a hydrophobic patch and a hydrogen bond, with a bending in Cβ which allows the side chain to reach the pocket (Figures 3A,C). In the presence of ligand, it makes a salt bridge with the ligand carboxylate group, believed to be the first step in ligand coordination. This interaction is believed to be critical for proper functioning of most SBPs from the TRAP family as high-affinity binding proteins, although it is not essential for the coordination of domain closure upon ligand binding (Fischer et al., 2015). The TatPT homologs, believed to be involved in the uptake of aliphatic substrates, mostly lack this residue, having, instead, an alanine (Deka et al., 2012). Mutations of this arginine residue in SiaP were shown by Johnston et al. (2008) to disrupt sialic acid uptake in H. influenzae and recently Fischer et al. (2015) showed that replacing it by a lysine decreased the binding affinity for sialic acid by SiaP from 0.14 to 38.7 μM, and mutating it to an alanine resulted in no binding. Crystallization of these two mutant proteins, however, showed minor differences in ligand coordination, where in place of the missing N atoms, coordinated water molecules bridged the carboxylic group of the ligand to the protein, dissipating the negative charge. Subsequent growth experiments showed that cell growth could be restored in the presence of high external concentrations of sialic acid, as the higher concentration would compensate for the weaker affinity; In the same study, it is shown also that this water coordination enables a higher promiscuity in the binding pocket, allowing it to coordinate an analog ligand containing an amide group in place of the carboxylic acid. In addition, PELDOR spectroscopy analysis recently performed by Glaenzer et al. (2017) showed that no intermediate state of VcSiaP is observed in solution upon ligand binding, which can only be in an open or closed conformation. Moreover, the protein does not alternate to closed conformation unless the ligand is present, a fact that supports the current model in which the SBP will only return to the open conformation upon interaction with the membrane components, avoiding unproductive opening and closing of the binding protein (Mulligan et al., 2011).
In addition, the variable positioning of helix 3 across different proteins seem to be responsible for the adaptation of the binding pocket for different ligand sizes, given by structural changes in regions flanking this helix (Lecher et al., 2009). In some cases, generally for smaller TRAP ligands, cation atoms are also required for ligand coordination. However, as shown by Akiyama et al. (2009), these cations are usually non-specific, and have a structural role to bridge the interaction with the protein chain, and are not necessary when the ligands are capable of filling the respective space and interact directly (Fischer et al., 2010).
Although most TRAP SBPs are found to act as monomers, there is evidence that some of them might require dimerization for function. Gonin et al. (2007) showed that TakP, a pyruvate binding protein from Rhodobacter sphaeroides, crystalized as a dimer, and the functional importance of dimerization was validated by tryptophan fluorescence quenching, gel filtration and cross-linking experiments. The dimerization is believed to occur through a kinked C-terminal helix, which swaps its position with the same portion of the dimer counterpart. Additionally, Akiyama et al. (2009) crystalized a lactate binding protein from Thermus thermophilus which interacts back-to-back in a dimerization process stabilized by hydrophobic interactions in the C-terminal region of the protein. Finally, Cuneo et al. (2008) confirmed also the dimerization state of a TRAP protein from Thermotoga maritima through gel filtration analysis and X-ray scattering. It remains unclear, however, how this dimerization process would promote or interfere with the transport mechanism when interacting with the DctQM subunits.
The TTT Family
As with the TRAP transporters, systems in the TTT family are composed of a conserved 12 TM protein, (TctA homologs) believed to act as a symport protein energized by an electrochemical ion-gradient (although this has not been experimentally determined) and poorly-conserved 4 TM protein (TctB homologs) with unknown function, in combination with an SBP (TctC homologs) which binds the substrate with high affinity (Figure 1, Sweet et al., 1979; Winnen et al., 2003). However, the TTT family has not been subject to many experimental studies and knowledge about this family is still scarce; the topic being last reviewed by Winnen et al. (2003).
Substrate Diversity of the TTT Family: Role of the Solute Binding-Protein and Occurrence in Pathogens
As the prototype for the TTT family, the TctC citrate transporter was first described by Sweet et al. (1979) in the pathogen Salmonella typhimurium, one of the most important causative agents of food-borne gastrointestinal infections and a growing problem due to the recent emergence of multidrug resistant strains (Hur et al., 2012). TctC was found to be involved in the uptake of the tricarboxylic acid citrate with low-μM affinity, with citrate uptake severely reduced in this organism upon disruption of the tctC gene. This function gave the name to the family as tricarboxylate transporters (Sweet et al., 1979; Somers et al., 1981). A series of 36 tricarboxylate and di-carboxylate metabolites were later shown by Sweet et al. (1984) to inhibit citrate binding to TctC to varying extents, suggesting that the substrate range for this protein might not be restricted to citrate. Genetic mapping studies initiated by Somers and Kay (1983) and finished by Widenhorn et al. (1988) showed that downstream of the tctC locus there were two more encoded proteins, of 19 and 45 kDa, corresponding, respectively, to the transmembrane proteins TctB and TctA (Figures 1, 2B). The gene arrangement of tctCBA, is similar to that found for the majority of TRAP transporters (Mulligan et al., 2011). Encoded in the opposite direction, a fourth gene, tctD, was shown by Widenhorn et al. (1989) to encode a transcription regulator of the tctCBA operon, which was found to be repressed when tctD was deleted or in the presence of glucose in the medium. Homologous systems to TctCBA are found in many bacteria, mainly Proteobacteria, and citrate uptake is the commonest identified role for the few other TTT systems experimentally characterized to date (Antoine et al., 2003; Brocker et al., 2009; Hosaka et al., 2013; Graf et al., 2016). Citrate has been shown to act in some cases as an iron chelator for different transport systems (Yancey and Finkelstein, 1981; Braun, 2001; Luck et al., 2001; Banerjee et al., 2016), and although the potential role of TctC acting as an iron transport protein has not been investigated to date, experiments performed with S. typhimurium (Sweet et al., 1979) showed that citrate binding to TctC is improved in the presence of Na+ Ca2+, Mn2+ and Fe2+, while partially inhibited by Mg2+, Ni2+, Zn2+ and Co2+. In addition, growth experiments performed by Brocker et al. (2009) using a homologous TctCBA system from Corynebacterium glutamicum showed that this system was able to uptake citrate in the presence of Ca2+ and Mg2+, but not Sr2+.
After the characterization of TctC, all proteins homologous to the TctCBA systems in newly released genomes were annotated either as unknown proteins or citrate uptake systems, and this family was neglected for over a decade, until one TctC homolog was found by Antoine et al. (2000) to be encoded upstream of the pertussis toxin (PTX) virulence island, one of the most important toxins produced by the causative agent of whooping cough, B. pertussis. This gene was found to be conserved in this locus for different Bordetella species and was named bugT, standing for “Bordetella uptake gene.” Although a relationship between the BugT protein and the production of PTX was not confirmed, regions coding for other virulence factors, such as the adenylate cyclase toxin (AC) and the dermonecrotic toxin (DNT) also contained bugT homologs, while two bug homologs were negatively regulated by the BvgAS two-component system, responsible for the activation of virulence factor production (Antoine et al., 2000, 2003). Recently, a single-nucleotide-polymorphism (SNP) in one bug gene was consistently identified in an Australian epidemic strain of B. pertussis (Safarchi et al., 2016). As discussed in the next sections, further searches in the B. pertussis genome found homologs of Bug proteins to be extensively overrepresented, with 76 genes encoding distinct homologs (Antoine et al., 2000). In contrast, only two sets of genes coding for transmembrane proteins homologous to tctAB were found in B. pertussis, and most of the BugT homologs showed no obvious membrane counterparts encoded in their genomic vicinity, hence the designation of them as “orphan” proteins.
The only complete operonic encoded TTT system in B. pertussis, encoded by bctCBA, contains bug4 as the tctC homolog, and was found to be the equivalent of tctCBA from S. typhimurium, as expression was upregulated by citrate and gene disruption resulted in lower citrate uptake rates (Antoine et al., 2003). As shown by Antoine et al. (2005), upstream of the bctCBA operon is encoded the two-component system bctDE, transcribed in the same direction but forming a separate operon (Figure 2B), which showed a basal expression level independent of citrate. When this two-component system was deleted, expression of bctCBA was not detected, showing that btcDE was in fact activating transporter gene expression. Disruption of the bctBA components, on the other hand, increased operon expression, due to an accumulation of citrate in the periplasm to be directed to signaling purposes (Antoine et al., 2005). Finally, when bctC was deleted, bctBA expression was reduced to basal levels even in high citrate concentrations, implying that the two-component system is enough to maintain a basal level expression, but is not enough to enhance expression in the presence of citrate. Together, those data showed that citrate-bound BctC was required for both transport and signaling, interacting either with BctE or BctA; a model confirmed in the same study by bacterial two-hybrid assays, showing unprecedented evidence that TctC homologs can be involved also in regulatory processes (Antoine et al., 2005). The presence of citrate responsive regulatory genes and two-component systems adjacent to tctCBA operons is not uncommon, as shown for S. typhimurium by Widenhorn et al. (1989), for Comamonas sp. by Hosaka et al. (2013), for A. mimigardefordensis by Wübbeler et al. (2014); and in the genomic searches provided by Antoine et al. (2003). Brocker et al. (2009) also characterized a citrate-responsive two-component system controlling tctCBA expression in C. glutamicum, although in this case the regulatory proteins were adjacent to another transport system. Interestingly, some of the TTT systems are found in the genome with the tctB subunit downstream of tctA, such as the slcHFG systems from Roseovarius nubinhibens (Denger et al., 2009) and Chromohalobacter salexigens (Figure 2B, Denger and Cook, 2010). This feature is also observed in some TRAP systems, and systems with this genomic organization are thought to be more similar among them (Mulligan et al., 2011).
The study of two abundantly expressed Bug proteins led to the first crystal structures for TTT family SBPs. These proteins, discussed in detail in the next sections, were fortuitously crystalized with substrates in their binding pocket. BugD contained an aspartate molecule (Huvent et al., 2006a) and BugE contained a glutamate molecule (Huvent et al., 2006b). Amino-acids are the most important carbon and nitrogen sources for B. pertussis, which is incapable of metabolism of substrates through the glycolytic pathway (Huvent et al., 2006b); As the Bug proteins are highly expressed, they might play a crucial role in uptake of core metabolic pathways. Herrou et al. (2007) characterized one of the Bug proteins (Bug27), found to be overexpressed in the presence of nicotinic acid, an essential vitamin and a negative modulator of B. pertussis virulence. It was shown that this protein binds, with an affinity lower than 1 μM, not only to nicotinate, but also nicotinamide, citrate, benzoate and quinaldic acid. This protein generated also the first TTT SBP crystal structure in an unliganded conformation (Herrou et al., 2007). The binding of Bug27 to nicotinic acid/nicotinamide might suggest it plays a role in virulence modulation, either by interacting with a membrane signal protein or simply transporting nicotinic acid to the cytoplasm. Interestingly, Brickman et al. (2017) suggested that another Bug protein, Bug69, might also be related to the uptake of nicotinic acid and related compounds.
Although not the focus of this review, the potential of the TTT family as a new source for biotechnology relevant uptake systems was also exposed by the genomic analysis performed by Antoine et al. (2003), where it was observed that in many organisms, the bug homologs were located near operons that conferred specific abilities to each strain, such as catechol degradation, showing that the importance of this family is wider than the suggested so far and that its diversity might correlate with the metabolic versatility and adaptability of an organism. In addition, a genomic search regarding arylmalonate decarboxylases (AMDases) by Maimanakos et al. (2016) found several members of the TTT family in the vicinity of these enzymes for five of the eight predicted AMDase clusters, either as orphan proteins, in the case of β-proteobacteria; or complete systems, in the case of α-proteobacteria, suggesting the TTT proteins might act to import the carboxylated substrates for subsequent catalysis by the AMDases. Other biotechnologically relevant discoveries include sulfolactate metabolic pathways in R. nubinhibens (Denger et al., 2009) and Chromohalobacter salexigens, which contain a TTT uptake system for this substrate (Denger and Cook, 2010) named slcHFG; a TTT system from Comamonas sp., TpiBA and TphC, able to uptake terephthalate (Hosaka et al., 2013); the TctCBA from A. mimigardefordensis able to uptake the synthetic molecule disulfide 3,3′-dithiodipropionic acid (DTDP), a precursor for synthetic polythioesters (Figure 2B, Wübbeler et al., 2014); a tctA homolog genetically proximal to genes coding for esterase enzymes that degrade organophosphates and potentially related to aromatic compound degradation (Batista-García et al., 2014); the TctABC system from Halomonas involved in galactarate/glucarate metabolism (Leyn et al., 2017); and a recent discovery from our group of AdpC, an “orphan” SBP from R. palustris which binds medium chain-length dicarboxylic acids ranging from adipate (C6) to azelate (C9) (Rosa et al., 2017). Searching the Enzyme Function initiative (EFI) database, it was observed that 19 homologs of TctC were in their library, however only one of them, a TctC homolog from Polaromonas sp. was crystalized in the open apo conformation (PDB accession code 4X9T). Table 1 summarizes the known range of characterized TTT systems with their respective ligands. Initially believed to bind exclusively to citrate, the substrate range for the TTT family is clearly much broader and new substrates are continually being found. With the exception of nicotinic acid in Bug27 (Herrou et al., 2007), all substrates characterized so far seem to have two carboxylic groups, or other functional groups such as sulfate and amide, and further studies will show if this is indeed a required property for substrates in the TTT family.
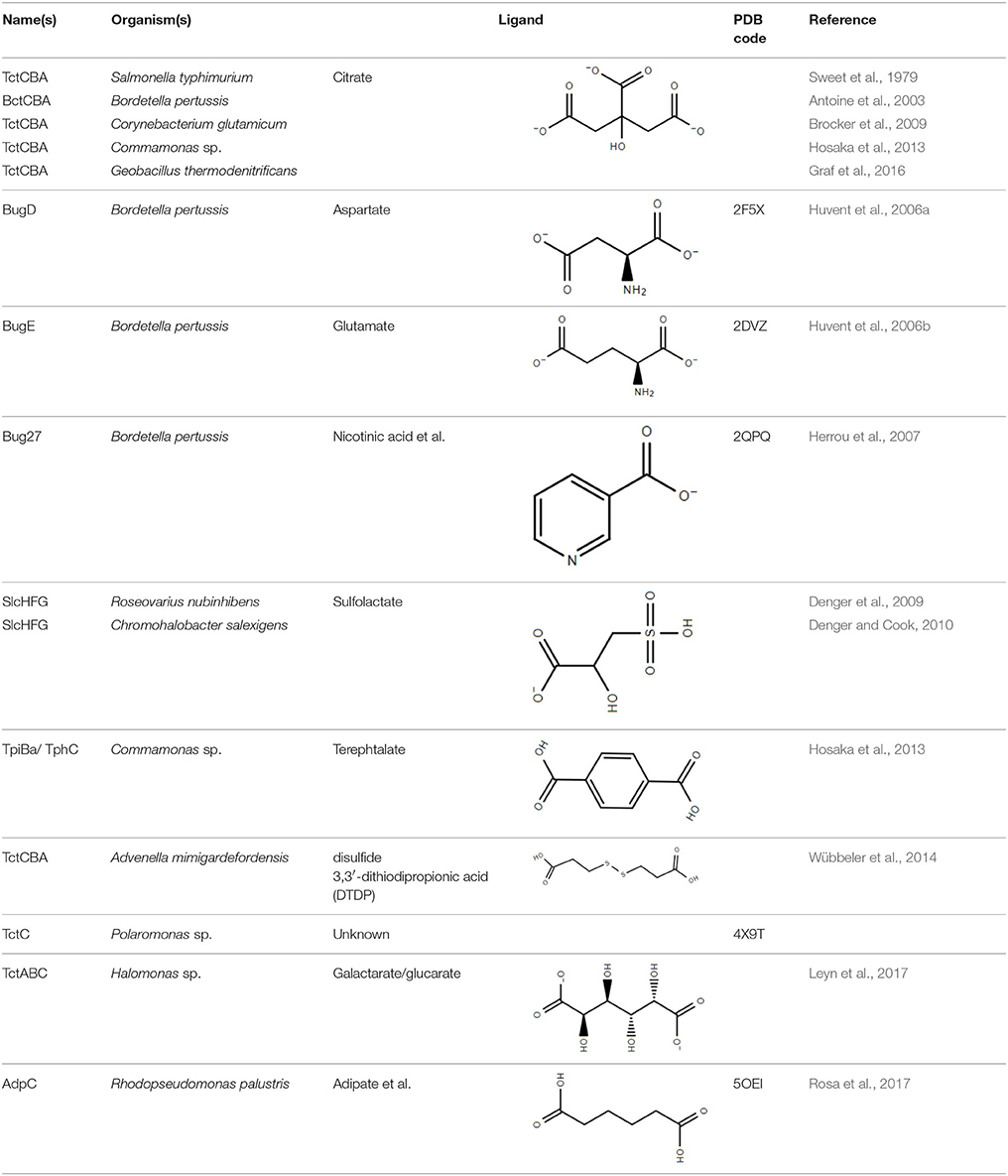
Table 1. Experimentally characterized TTT transporters and SBPs.
Properties and Function of the TctAB Subunits
The TTT systems are predicted to contain two membrane proteins, homologous to TctA and TctB. Although crystal structures of these proteins have not been elucidated, information from the primary and secondary sequences of these subunits were studied, in addition to some physiological characterizations, in an attempt to understand the energetic and structural mechanisms of the TTT family.
Winnen et al. (2003) showed that while TctB and TctC showed only 27 and 31% identity on average between family members, respectively, TctA orthologs suggested 42% identity and 53% similarity in similar comparisons. Topology predictions suggested the number of transmembrane helices in TctA homologs might vary in different systems, ranging from 9 to 12 in bacteria, and 7 to 11 in archaea. In either group, the N-terminal side was predicted to be in the cytoplasm, and large hydrophilic loops between helices 2 and 3 are conserved among all organisms analyzed, suggesting this region must have an important role in protein function. The motif G-Hy3-*G-Hy3-*G-Hy2-*P-G-Hy-G, where Hy is an aliphatic hydrophobic residue and * means a fully conserved residue, are found to be highly conserved both in TM1 and TM7, suggesting the 12-TM protein originated from a duplication in a 6-TM ancestor. TctB homologs were predicted to have between 4 and 5 transmembrane domains, also with predicted cytoplasmic N-termini, and are very poorly conserved among bacteria and were not observed in archaeal sequences (Winnen et al., 2003). In a systems biology study comparing a wild type and a multi-drug resistant strain of Salmonella enterica, Ricci et al. (2012) observed a G109S SNP in TctA. Although shown to be unrelated to the antimicrobial resistance, this mutation compromised growth on several carbon sources, and curiously seemed to confer a delay in the production of Reactive Oxygen Species (ROS) under stressful conditions. Our genome searches revealed for the first time, that in at least one case, in Paraburkholderia caribensis, a fusion between the two membrane subunits can also be observed, forming a single TctAB protein (Figure 2B), similar to what was described for some DctQM proteins from the TRAP family, in particular the ones constituting TAXI-TRAP systems (Mulligan et al., 2011).
Evidence of cation dependence for transport in the TTT family was first provided by Sweet et al. (1979), showing that binding of citrate to TctC was enhanced in the presence of Na+ Ca2+, Mn2+, and Fe2+, while Mg2+, Ni2+, Zn2+, and Co2+ inhibited uptake. Similarly, Brocker et al. (2009) demonstrated that citrate transport by a TctCBA system of C. glutamicum was enhanced by Ca2+ and Mg2+, but not Sr2+. Later studies by Hosaka et al. (2013), however, showed that both the addition of protonophores and an alkaline pH disrupted terephthalate uptake by the TpiBA system in Comamonas sp., while deletion experiments in the same study showed that both subunits were essential for substrate uptake. Furthermore, terephthalate uptake was not disrupted when Comamonas sp. was grown in absence of Na+, suggesting that at least this particular process was more dependent on the proton-motive force rather than a sodium gradient, distinct from SiaPQM sialic acid TRAP transporters (Mulligan et al., 2009).
Discovering a tctA homolog next to esterase genes in metagenomics searches, named tctA_ar, Batista-García et al. (2014) attempted to build a structural model for TctA_ar based on its primary sequence, given that this protein showed no considerable homology to any other secondary transporter available in the database. The TctA homolog of Comamonas sp., characterized in previous studies (Hosaka et al., 2013), was used as a control and called TctA_ct. Two templates were used against each sequence (PDB codes 3VVN and 4K1C), resulting in 4 models in total. In addition to the already described duplicated motif, a high degree of identity was observed between residues 60 and 110 in TctA homologs. The computational models agreed with the predicted 12-TM domain protein, and it was proposed in the 3VVN-based models that the 20 residue conserved motif were in the vicinity of the predicted binding pocket of the protein. In addition, G106, mutated in previous studies (Ricci et al., 2012), would be located between the two repeats, being involved in the beginning of the translocation pathway. In the 4K1C model, the two copies of the domain would be in contact with each other in a helix, which would facilitate conformational changes during the transport cycle. Moreover, given that the TTT transporters were initially known for the transport of citrate, docking of this molecule into the two models was attempted, in addition to the modeling of a Na+ binding pocket, suggested to be necessary for citrate transport (Sweet et al., 1979). It was shown in the 3VVN models that the potential Na+ binding pockets were located in C-terminal variable regions, while the citrate-binding pocket differs in each of the two proteins. The TctA_ar showed a unique pocket for citrate, while TctA-ct showed several along the predicted channel, which might act as different steps in the translocation pathway. In the 4K1C models, several binding pockets were predicted for citrate in both proteins, with at least one positively charged residue to interact with the ligand in each of them. These models are an important step toward understanding of translocation mechanisms in the TTT family, although biological confirmation of the transport mechanisms in this family are clearly essential.
Evolutionary studies on the TTT family performed by Winnen et al. (2003) suggested that the TctA protein is the original core transport protein, and that the TctB homolog worked as an accessory protein. This claim is supported by the presence of TctA homologs in Archaea, added to the absence of TctB or TctC homologs, and reinforced by the model studies performed by Batista-García et al. (2014). However, the findings on the TRAP transporters, where the DctQ subunit is also observed in archaea, and was found essential to function challenge this hypothesis (Mulligan et al., 2011, 2012). Because the archaea harboring TctA homologs were found in extreme environments, amongst groups of methanogens, hyperthermophiles, and halophiles, it was suggested that the TctA homologs would be involved in a range of different specific metabolic niches (Winnen et al., 2003), but experimental evidence is still lacking to show if this is the case.
Crystal Structure and Substrate Coordination in TctC Homologs
The binding proteins from the TTT family, homologous to TctC (Sweet et al., 1979), show a conserved size (ranging from 29 to 33 kDa), topology and secondary structure organization, but differ considerably in the primary sequence, where among them an identity around 30% is observed. Consequently, a big difference in overall pI is also seen, ranging from 5 to 9.6 (Antoine et al., 2000). At the time of writing, only six structures of TTT SBPs have been deposited in the Protein Data Bank (PDB), four of them with a substrate in the binding pocket; it is already possible to identify, however, some common features among them. The average of 300 amino-acid residues comprises the mature form of the proteins (without signal peptide), separated into two globular domains. Domain one is usually formed by residues 1~100 and 230~300 from the N and C termini, forming a β-sheet of five strands, with topological arrangement β2-β1-β3-β9-β4, surrounded by ~6 α-helices. Domain 2 is comprised of residues 100~229, forming also a central β-sheet of 5 strands with topological arrangement β6-β5-β7-β4-β8, surrounded by ~4 helices. Domain 2 sometimes contains a disulphide bridge between cysteine residues located in α5 and β7, but this is not a feature common to all proteins (Huvent et al., 2006a; Rosa et al., 2017). The junction of the two domains is formed by two β-strands, S4 and S9, which are part of domain 1 but extend up to domain 2, and hydrogen bonds between the two domains are scarce. All these features show that TTT SBP characterized so far can be classified into the Type II binding protein group, according to the scheme of Fukami-Kobayashi et al. (1999), or cluster E-II, accordingly to the new division proposed by Scheepers et al. (2016). Figure 3B shows the crystal structure of BugD as a representative for the TctC homologs. Upon binding to the substrate, it was estimated that the two domains close in an angle of 24.7°, based on the structure of the unliganded nicotinic acid binding protein Bug27 (Herrou et al., 2007). Although TctC proteins most commonly seem to bind to molecules containing carboxylic groups, curiously there is usually a slight overall negative charge in the binding pocket, likely dissipated by the water molecules or dipole effects of the surrounding helices (Rosa et al., 2017). Two β-turns, between β1 and α1; and β7 and α7, form a “pincer-like” structure important in substrate coordination, closing around one carboxylic group of the ligand, while the remainder of it is buried in the pocket (Figure 3). The residues present in the loops characterize distinguishing signatures for proteins of this family, with the motif [P*-F-X-A-G*-G*-X-X-D*] in domain 1 being almost ubiquitous among the protein sequences, where X means any residue and * means a very conserved residue. The backbone atoms of residues in this region seem to make hydrogen bonds with two water molecules, present in all substrate-containing structures and also very well-conserved in position. These water molecules bridge hydrogen bonds between the protein main chain and the proximal carboxylic group in the substrate. This pattern is observed in the coordination of adipate by AdpC (Rosa et al., 2017), aspartate in BugD (Huvent et al., 2006a) and glutamate in BugE (Huvent et al., 2006b). In some Bug protein sequences, although these residues are not conserved, they are substituted by others where the side-chain would contain a hydroxyl group, potentially maintaining hydrogen bonds in similar position to what would be expected of the water molecules (Huvent et al., 2006a). As shown by Herrou et al. (2007), the two β-loops which form the “pincer-like” structure and the two water molecules are not well defined when the ligand is not present. The coordination of the ligand's distal carboxylate group, buried in the pocket, is much less conserved, with α3 and α5 helices apparently varying in position to accommodate each substrate (Huvent et al., 2006a; Rosa et al., 2017). Although not conserved in sequence or topology, the involvement of water molecules in the coordination of the distal carboxylate groups was observed in all cases. In some proteins, hydroxyl groups from threonine and serine residues also form hydrogen bonds with the carboxylate in the substrate, but their positions vary. The carbon chain of the substrate, on the other hand, is stabilized by much more conserved hydrophobic interactions, such as Phe14 in AdpC, which seem to act as a docking site for the substrate, and two glycines (Gly18 and Gly163 in AdpC). As a dynamic model, Herrou et al. (2007) suggested that the unliganded form of the SBP would be in an open conformation, with the “pincer-like” structures flexible. Substrate would then bind to domain 1, which would cause a conformational change that would bring the water molecules and domain 2 together. A comparison between the five available TctC homologs available in the PDB (only one AdpC structure was used) is shown in Table 2. The root mean square deviations (RMSD), as expected, show a bigger difference when comparing closed and Apo structures, and smaller RMSDs when comparisons were made between two liganded structures. Taken together, the crystal structures presented to date give a good general mechanism for ligand coordination in the TctC homologs, and further studies will enable us to validate this model and detail the potential differences for substrates containing different functional groups, such as nicotinamide (Herrou et al., 2007) and sulfolactate (Denger and Cook, 2010).
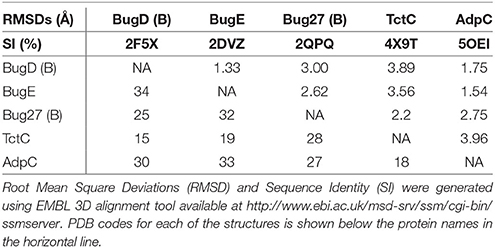
Table 2. Comparison between the TctC homologs structures available in the PDB.
Present in Abundance: The Overrepresentation of TctC Homologs in Some Bacteria
The limited number of sequences available at the time resulted in a bias in the studies performed by Winnen et al. (2003), which suggested that the TTT systems were mostly present in α-proteobacteria, and that most other bacterial groups had few or no homologs of these proteins. Genomic searches following the discovery of BugT by Antoine et al. (2000), however, revealed the bug genes to be very overrepresented in B. pertussis, with 79 BugT homologs, making this family the most abundant in the genome. Following this discovery, Antoine et al. (2003) performed a wider genome analysis, showing that this overrepresentation was extended to several Bordetella species, and that some of the Bug proteins were also among the most abundant in cell protein extracts in B. pertussis. As stated in previous sections, the numbers of TTT transmembrane components did not follow the same process, being found in small numbers and consequently configuring most BugT homologs as “orphan proteins,” with no obvious transmembrane counterparts. The existence of orphan bug homologs was also observed by Antoine et al. (2003) in the genomes of several other bacteria genera, although in that study the only bacterium shown to have as many representatives as the Bordetella species was the β-proteobacterial relative Cupriavidus metallidurans. At the time that search was performed, there were around 200 complete bacterial genomes available in the databases, and more recent genome releases showed that at least two other β-proteobacteria genera, Advenella and Cupriavidus also showed an overrepresentation of tctC homologs (Wübbeler et al., 2014). For this review, we reassessed the distribution of TTT systems using the 8049 fully assembled genomes in Genbank, of 2,323 different species, to provide an updated analysis of the presence of TTT systems in bacterial genomes.
A total of 2,323 complete bacterial genomes retrieved from the NCBI database, one per species, were screened for TctA and TctC homologs using the TBLASTN tool. Searches were performed against the coding sequence (CDS) database of each species using lists of protein sequences of either TctA or TctC homologs as queries rather than single sequences, in order to avoid query bias. For the TctC search, the queries were: TctC from S. enterica (Sweet et al., 1979); BugD, BugE, and Bug27, from B. pertussis (Herrou et al., 2007), TphC fom Comamonas sp. (Hosaka et al., 2013) and AdpC from R. palustris (Rosa et al., 2017). For the TctA search, we used the TctA from S. enterica (Sweet et al., 1979), A. mimigardefordensis (Wübbeler et al., 2014), and C. glutamicum (Brocker et al., 2009); the BctA from B. pertussis (Antoine et al., 2005); the TpiA from Comamonas sp. (Hosaka et al., 2013) and the SlcF from R. nubinhibens (Denger et al., 2009). Due to the poor sequence conservation among the TctB proteins, our searches with this subunit proved to be unsuccessful. TBLASTN reports were obtained for a range of E-values from 1 to 10−15 in order to determine the best threshold to avoid spurious hits, while still retaining distant paralogs. The complete table with number of hits for both proteins with an e-value of 10−15 is presented in Supplementary Table 1.
Our searches revealed that, in accordance with the findings of Antoine et al. (2003) and further reinforced by Wübbeler et al. (2014), the most extreme examples of overrepresentation of this group of proteins are found among β-proteobacteria, especially among Bordetella species, as shown in Table 3. However, this phenomenon is not restricted to this group, but extends also to species in the α-proteobacteria class (Table 3). In fact, the genome of the environmental α-proteobacterium Rhodoplanes sp. encodes 434 TctC homologs in its 8.2 Mbp genome, more than double that of some Bordetella species. A more detailed investigation of the expansion in Rhodoplanes will be reported elsewhere (manuscript in preparation). Although this analysis shows that the overrepresentation of TctC homologs is mostly found in proteobacteria, a deeper phylogenetic analysis is still required in order to clarify whether this feature found in different subgroups originates from duplications in a common ancestor or were independent events resulting from convergent evolution and independent multiplication events. A search for TctA homologs, on the other hand, as shown in Table 4, suggests that the genomes containing the largest numbers of homologs are found among α and γ-proteobacteria, with only two β-proteobacteria showing 8 or more homologs. In this search, the top hits are no higher than 21 per genome, and are usually associated with a similar number of TctC homologs, possibly forming complete tripartite systems. In this case, organisms outside of the class of proteobacteria, such as clostridia, spirochaetes, and bacilli are also observed to harbor these homologs. An overview of the number of genomes encoding different numbers of TctC and TctA homologs are shown in Figure 4, and the full complement of genomes analyzed is shown in Supplementary Table 1.
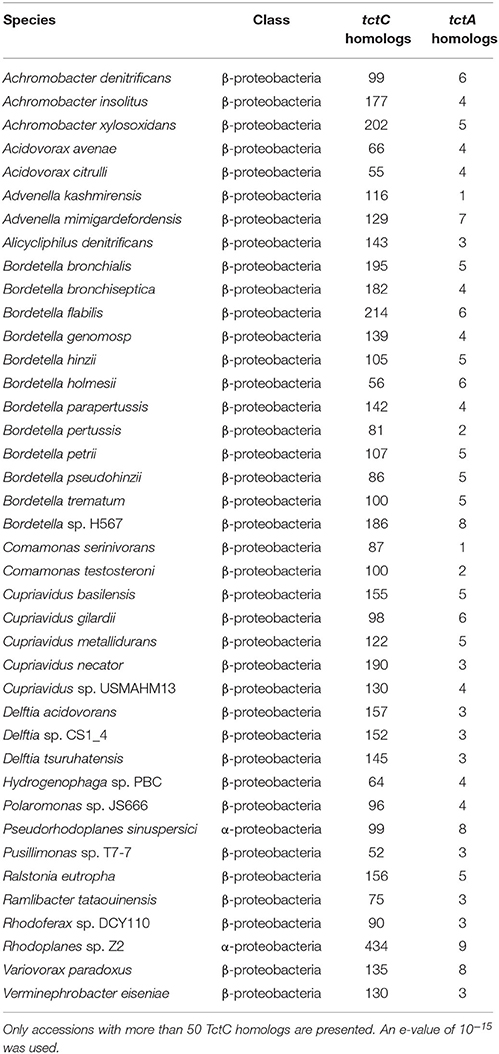
Table 3. Number of TctC and TctA homologs per accession.
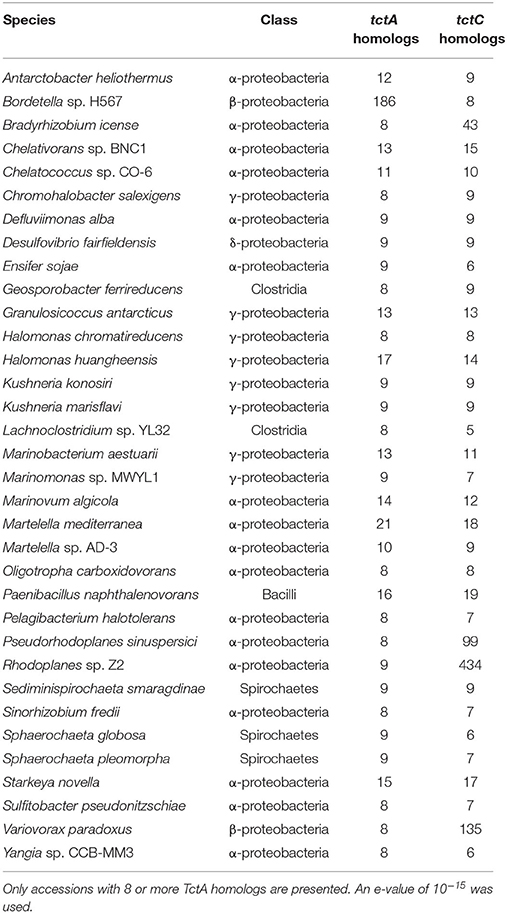
Table 4. Number of TctC and TctA homolog per accession.
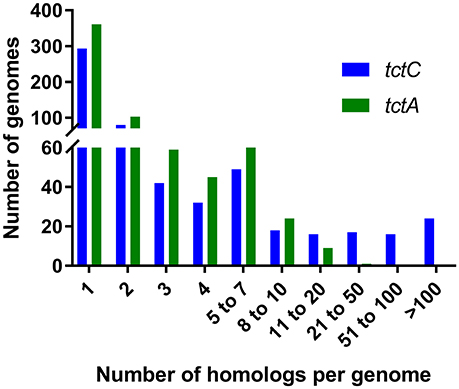
Figure 4. Numbers of genomes containing different ranges of homologs for tctC and tctA.
At an e-value of 10−15 in BLAST searches, the TctC homologs outnumber the TctA homologs in 176 genomes, as shown in Table 5. As already discussed, in these cases it might be that one TctA interacts with more than one TctC, or that the latter are involved in processes other than transport, such as signaling and chemotaxis (Antoine et al., 2003; Piepenbreier et al., 2017). These latter suggestions are reinforced by the fact that in 36 genomes, one tctC homolog was found, but no tctA homologs, although the hypothesis that the binding proteins might interact with transmembrane domains of other transporter classes cannot be excluded. In our initial searches, we found that 210 genomes showed an excess of tctA homologs in relation to tctC, an unprecedented observation to the best of our knowledge. In order to see whether these observations were due to too strict threshold, our searches were repeated with different e-values, shown in Table 5. As shown, using an e-value of 10−9, the number of genomes where this situation occurs is reduced to 66, and to 39 in 10−6. At the latter threshold, 6 genomes indicated the presence of TctA homologs, but no TctC homologs. Investigating these 6 genomes individually, we found that 4 of them contained a truncated tctC homolog in the vicinity of the tctA gene and in one the tctA gene was clearly mutated. The single remaining genome, Mageeibacillus indolicus, indeed seems to have no indication of any periplasmic binding proteins in the vicinity of the tctA gene. If the existence of a tctA gene without any tctC is not a search artifact, one possible explanation would be that SBPs of other types of transport systems could be capable of interacting with the TTT transmembrane subunits. An alternative is that such rare orphan TctA proteins in bacteria are, like in archaea, capable of functioning without the involvement of an SBP (Winnen et al., 2003), although both the TTT systems characterized so far and the experiments with the TRAP transporters suggest otherwise (Brocker et al., 2009; Mulligan et al., 2009; Hosaka et al., 2013). In another 316 genomes, the numbers of tctC and tctA genes match exactly, suggesting all proteins would be involved in transport through tripartite systems. Finally, about half of the genomes searched (1621) contained no homologous proteins to any of the queries used, suggesting, given TctA homologs are also found in archaea (Winnen et al., 2003), that TTT systems were lost during evolution in these phylogenetic branches.
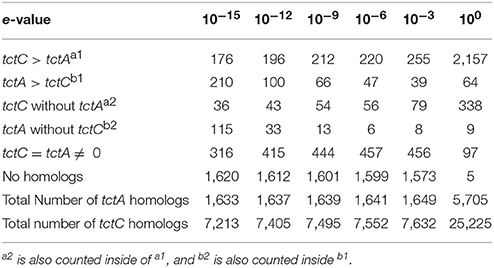
Table 5. Number of genomes showing different patterns in terms of numbers of tctC and tctA homologs, using different e-values as thresholds.
The reason for the overrepresentation of the Bug proteins in Bordetella species and other Proteobacteria remains unclear. Antoine et al. (2003) suggested that the few transmembrane domains of TTT systems evolved to be poorly specific, being able to interact with several TctC homologs and thus be required for the uptake of different substrates. This hypothesis was also suggested by Hosaka et al. (2013), but no evidence for this mechanism is yet available. One could hypothesize that perhaps many of these proteins have similar binding functions, but are expressed differentially during the infection cycle in Bordetella species in order to evade the immune system more efficiently. However, the fact that many environmental Proteobacteria also have this expansion of Bug proteins suggests instead that it is an earlier evolutionary trait. The genome of Ralstonia eutropha containing 154 homologs of tctC, reveals that the majority of them (64.1%) have in their vicinity a regulatory protein, suggesting that most of these proteins are associated with regulatory mechanisms rather than transport (Pohlmann et al., 2006). In this sense, the nomenclature of these SBPs as “uptake genes” might not reflect their actual role in the cell. Piepenbreier et al. (2017) provides a good review of how transporters from different classes can act as first agents in signaling pathways, and further studies will enrich our understanding to whether this is the case for the TTT family.
Concluding Remarks
In this review, we showed how transport systems from the TRAP and TTT families can play important roles in bacteria with a focus on pathogenicity and colonization. Recent high-throughput studies increased substantially the range of substrates known for the TRAP family, while the TTT family is still understudied with a more limited known substrate range, being unraveled in individual studies. In addition, while a lot has been elucidated regarding the binding mechanisms, energetics, and kinetics of the TRAP family, very few equivalent studies exist for the TTT family, where especially the energy-coupling mechanisms are yet to be elucidated properly. For both families, a crystal structure of the complete tripartite systems would greatly increase our understanding of the transport process across the membrane, perhaps with potential applications as new drug targets in pathogenic bacteria, given the absence of these transporters in eukaryotic cells.
Author Contributions
LR reviewed the literature, co-wrote the manuscript and co-analyzed the bioinformatic data. MB generated the bioinformatics data and co-analyzed it. GT edited and commented on the drafts. DK conceived the idea and focus of the review, co-wrote and edited the paper, and provided supervisory guidance.
Funding
The present work was accomplished with funding from Brazilian funding agency CNPQ (National Council for Scientific and Technological Development), though PhD studentships in the remit of “Science Without Borders” program to LR (248597/2013-2) and MB (201873/2014-1). May this manuscript reinforce the importance of prioritizing investment in science and research by the Brazilian government.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2018.00033/full#supplementary-material
Supplementary Table 1. Number of genes homologous to tctC and tctA in 2,323 bacterial genomes, obtained using TBLASTN.
Abbreviations
SBP, Solute Binding-Proteins; TTT, Tripartite Tricarboxylate Transporters; TRAP, Tripartite ATP-independent periplasmic transporter.
References
Akiyama, N., Takeda, K., and Miki, K. (2009). Crystal structure of a periplasmic substrate-binding protein in complex with calcium lactate. J. Mol. Biol. 392, 559–565. doi: 10.1016/j.jmb.2009.07.043
Allen, S., Zaleski, A., Johnston, J. W., Gibson, B. W., and Apicella, M. A. (2005). Novel sialic acid transporter of Haemophilus influenzae. Infect. Immun. 73, 5291–5300. doi: 10.1128/IAI.73.9.5291-5300.2005
Antoine, R., Huvent, I., Chemlal, K., Deray, I., Raze, D., Locht, C., et al. (2005). The periplasmic binding protein of a tripartite tricarboxylate transporter is involved in signal transduction. J. Mol. Biol. 351, 799–809. doi: 10.1016/j.jmb.2005.05.071
Antoine, R., Jacob-Dubuisson, F., Drobecq, H., Willery, E., Lesjean, S., and Locht, C. (2003). Overrepresentation of a gene family encoding extracytoplasmic solute receptors in Bordetella. J. Bacteriol. 185, 1470–1474. doi: 10.1128/JB.185.4.1470-1474.2003
Antoine, R., Raze, D., and Locht, C. (2000). Genomics of Bordetella pertussis toxins. Int. J. Med. Microbiol. 290, 301–305. doi: 10.1016/S1438-4221(00)80026-0
Bakermans, C., Sloup, R. E., Zarka, D. G., Tiedje, J. M., and Thomashow, M. F. (2009). Development and use of genetic system to identify genes required for efficient low-temperature growth of Psychrobacter arcticus 273-4. Extremophiles 13, 21–30. doi: 10.1007/s00792-008-0193-3
Banerjee, S., Paul, S., Nguyen, L. T., Chu, B. C. H., and Vogel, H. J. (2016). FecB, a periplasmic ferric-citrate transporter from E. coli, can bind different forms of ferric-citrate as well as a wide variety of metal-free and metal-loaded tricarboxylic acids. Metallomics 8, 125–133. doi: 10.1039/C5MT00218D
Batista-García, R. A., Sánchez-Reyes, A., Millán-Pacheco, C., González-Zuñiga, V. M., Juárez, S., Folch-Mallol, J. L., et al. (2014). A novel TctA citrate transporter from an activated sludge metagenome: structural and mechanistic predictions for the TTT family. Proteins 82, 1756–1764. doi: 10.1002/prot.24529
Berntsson, R. P., Smits, S. H., Schmitt, L., Slotboom, D. J., and Poolman, B. (2010). A structural classification of substrate-binding proteins. FEBS Lett. 584, 2606–2617. doi: 10.1016/j.febslet.2010.04.043
Bouchet, V., Hood, D. W., Li, J., Brisson, J. R., Randle, G. A., Martin, A., et al. (2003). Host-derived sialic acid is incorporated into Haemophilus influenzae lipopolysaccharide and is a major virulence factor in experimental otitis media. Proc. Natl. Acad. Sci. U.S.A. 100, 8898–8903. doi: 10.1073/pnas.1432026100
Braun, V. (2001). Iron uptake mechanisms and their regulation in pathogenic bacteria. Int. J. Med. Microbiol. 291, 67–79. doi: 10.1078/1438-4221-00103
Brautigam, C. A., Deka, R. K., Schuck, P., Tomchick, D. R., and Norgard, M. V. (2012). Structural and thermodynamic characterization of the interaction between two periplasmic Treponema pallidum lipoproteins that are components of a TPR-protein-associated TRAP transporter (TPAT). J. Mol. Biol. 420, 70–86. doi: 10.1016/j.jmb.2012.04.001
Brickman, T. J., Suhadolc, R. J., McKelvey, P. J., and Armstrong, S. K. (2017). Essential role of Bordetella NadC in a quinolinate salvage pathway for NAD biosynthesis. Mol. Microbiol. 103, 423–438. doi: 10.1111/mmi.13566
Brocker, M., Schaffer, S., Mack, C., and Bott, M. (2009). Citrate utilization by Corynebacterium glutamicum is controlled by the CitAB two-component system through positive regulation of the citrate transport genes citH and tctCBA. J. Bacteriol. 191, 3869–3880. doi: 10.1128/JB.00113-09
Chae, J. C., and Zylstra, G. (2006). 4-Chlorobenzoate uptake in Comamonas sp. strain DJ-12 is mediated by a tripartite ATP-independent periplasmic transporter. J. Bacteriol. 188, 8407–8412. doi: 10.1128/JB.00880-06
Cuneo, M. J., Changela, A., Miklos, A. E., Beese, L. S., Krueger, J. K., and Hellinga, H. W. (2008). Structural analysis of a periplasmic binding protein in the tripartite ATP-independent transporter family reveals a tetrameric assembly that may have a role in Ligand transport. J. Biol. Chem. 283, 32812–32820. doi: 10.1074/jbc.M803595200
D'Andrea, L. D., and Regan, L. (2003). TPR proteins: the versatile helix. Trends Biochem. Sci. 28, 655–662. doi: 10.1016/j.tibs.2003.10.007
Davidson, A. L., Dassa, E., Orelle, C., and Chen, J. (2008). Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 72, 317–364. doi: 10.1128/MMBR.00031-07
Deka, R. K., Brautigam, C. A., Goldberg, M., Schuck, P., Tomchick, D. R., and Norgard, M. V. (2012). Structural, bioinformatic, and in vivo analyses of two Treponema pallidum lipoproteins reveal a unique TRAP transporter. J. Mol. Biol. 416, 678–696. doi: 10.1016/j.jmb.2012.01.015
Denger, K., and Cook, A. M. (2010). Racemase activity effected by two dehydrogenases in sulfolactate degradation by Chromohalobacter salexigens: purification of (S)-sulfolactate dehydrogenase. Microbiology 156(Pt 3), 967–974. doi: 10.1099/mic.0.034736-0
Denger, K., Mayer, J., Buhmann, M., Weinitschke, S., Smits, T. H., and Cook, A. M. (2009). Bifurcated degradative pathway of 3-sulfolactate in Roseovarius nubinhibens ISM via sulfoacetaldehyde acetyltransferase and (S)-cysteate sulfolyase. J. Bacteriol. 191, 5648–5656. doi: 10.1128/JB.00569-09
Fischer, M., Hopkins, A. P., Severi, E., Hawkhead, J., Bawdon, D., Watts, A. G., et al. (2015). Tripartite ATP-independent Periplasmic (TRAP) Transporters Use an Arginine-mediated Selectivity Filter for High Affinity Substrate Binding. J. Biol. Chem. 290, 27113–27123. doi: 10.1074/jbc.M115.656603
Fischer, M., Zhang, Q. Y., Hubbard, R. E., and Thomas, G. H. (2010). Caught in a TRAP: substrate-binding proteins in secondary transport. Trends Microbiol. 18, 471–478. doi: 10.1016/j.tim.2010.06.009
Forward, J. A., Behrendt, M. C., Wyborn, N. R., Cross, R., and Kelly, D. J. (1997). TRAP transporters: a new family of periplasmic solute transport systems encoded by the dctPQM genes of Rhodobacter capsulatus and by homologs in diverse gram-negative bacteria. J. Bacteriol. 179, 5482–5493. doi: 10.1128/jb.179.17.5482-5493.1997
Fukami-Kobayashi, K., Tateno, Y., and Nishikawa, K. (1999). Domain dislocation: a change of core structure in periplasmic binding proteins in their evolutionary history. J. Mol. Biol. 286, 279–290. doi: 10.1006/jmbi.1998.2454
Fuller, T. E., Kennedy, M. J., and Lowery, D. E. (2000). Identification of Pasteurella multocida virulence genes in a septicemic mouse model using signature-tagged mutagenesis. Microb. Pathog. 29, 25–38. doi: 10.1006/mpat.2000.0365
Glaenzer, J., Peter, M. F., Thomas, G. H., and Hagelueken, G. (2017). PELDOR Spectroscopy Reveals Two Defined States of a Sialic Acid TRAP Transporter SBP in Solution. Biophys. J. 112, 109–120. doi: 10.1016/j.bpj.2016.12.010
Gonin, S., Arnoux, P., Pierru, B., Lavergne, J., Alonso, B., Sabaty, M., et al. (2007). Crystal structures of an extracytoplasmic solute receptor from a TRAP transporter in its open and closed forms reveal a helix-swapped dimer requiring a cation for alpha-keto acid binding. BMC Struct. Biol. 7:11. doi: 10.1186/1472-6807-7-11
Graf, S., Broll, C., Wissig, J., Strecker, A., Parowatkin, M., and Unden, G. (2016). CitA (citrate) and DcuS (C4-dicarboxylate) sensor kinases in thermophilic Geobacillus kaustophilus and Geobacillus thermodenitrificans. Microbiology 162, 127–137. doi: 10.1099/mic.0.000171
Grammann, K., Volke, A., and Kunte, H. J. (2002). New type of osmoregulated solute transporter identified in halophilic members of the bacteria domain: TRAP transporter TeaABC mediates uptake of ectoine and hydroxyectoine in Halomonas elongata DSM 2581(T). J. Bacteriol. 184, 3078–3085. doi: 10.1128/JB.184.11.3078-3085.2002
Herrou, J., Bompard, C., Antoine, R., Leroy, A., Rucktooa, P., Hot, D., et al. (2007). Structure-based mechanism of ligand binding for periplasmic solute-binding protein of the Bug family. J. Mol. Biol. 373, 954–964. doi: 10.1016/j.jmb.2007.08.006
Hosaka, M., Kamimura, N., Toribami, S., Mori, K., Kasai, D., Fukuda, M., et al. (2013). Novel tripartite aromatic acid transporter essential for terephthalate uptake in Comamonas sp. strain E6. Appl. Environ. Microbiol. 79, 6148–6155. doi: 10.1128/AEM.01600-13
Hur, J., Jawale, C., and Lee, J. H. (2012). Antimicrobial resistance of Salmonella isolated from food animals: A review. Food Res. Int. 45, 819–830. doi: 10.1016/j.foodres.2011.05.014
Huvent, I., Belrhali, H., Antoine, R., Bompard, C., Locht, C., Jacob-Dubuisson, F., et al. (2006a). Crystal structure of Bordetella pertussis BugD solute receptor unveils the basis of ligand binding in a new family of periplasmic binding proteins. J. Mol. Biol. 356, 1014–1026. doi: 10.1016/j.jmb.2005.11.096
Huvent, I., Belrhali, H., Antoine, R., Bompard, C., Locht, C., Jacob-Dubuisson, F., et al. (2006b). Structural analysis of Bordetella pertussis BugE solute receptor in a bound conformation. Acta Crystallogr. D Biol. Crystallogr. 62(Pt 11), 1375–1381. doi: 10.1107/S0907444906032653
Jenkins, G. A., Figueira, M., Kumar, G. A., Sweetman, W. A., Makepeace, K., Pelton, S. I., et al. (2010). Sialic acid mediated transcriptional modulation of a highly conserved sialometabolism gene cluster in Haemophilus influenzae and its effect on virulence. BMC Microbiol. 10:48. doi: 10.1186/1471-2180-10-48
Johnston, J. W., Coussens, N. P., Allen, S., Houtman, J. C. D., Turner, K. H., Zaleski, A., et al. (2008). Characterization of the N-Acetyl-5-neuraminic Acid-binding Site of the Extracytoplasmic Solute Receptor (SiaP) of Nontypeable Haemophilus influenzae Strain 2019. J. Biol. Chem. 283, 855–865. doi: 10.1074/jbc.M706603200
Jones, P. M., and George, A. M. (2004). The ABC transporter structure and mechanism: perspectives on recent research. Cell. Mol. Life Sci. 61, 682–699. doi: 10.1007/s00018-003-3336-9
Kalckar, H. M. (1971). The Periplasmic Galactose Binding Protein of Escherichia coli. Science 174, 557–565.
Kelly, D. J., and Thomas, G. H. (2001). The tripartite ATP-independent periplasmic (TRAP) transporters of bacteria and archaea. FEMS Microbiol. Rev. 25, 405–424. doi: 10.1111/j.1574-6976.2001.tb00584.x
Lecher, J., Pittelkow, M., Zobel, S., Bursy, J., Bönig, T., Smits, S. H., et al. (2009). The Crystal Structure of UehA in Complex with Ectoine—A Comparison with Other TRAP-T Binding Proteins. J. Mol. Biol. 389, 58–73. doi: 10.1016/j.jmb.2009.03.077
Leyn, S. A., Maezato, Y., Romine, M. F., and Rodionov, D. A. (2017). Genomic reconstruction of carbohydrate utilization capacities in microbial-mat derived consortia. Front. Microbiol. 8:1304. doi: 10.3389/fmicb.2017.01304
Locher, K. P. (2016). Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 23, 487–493. doi: 10.1038/nsmb.3216
Lubin, J.-B., Kingston, J. J., Chowdhury, N., and Boyd, E. F. (2012). Sialic acid catabolism and transport gene clusters are lineage specific in Vibrio vulnificus. Appl. Environ. Microbiol. 78, 3407–3415. doi: 10.1128/AEM.07395-11
Luck, S. N., Turner, S. A., Rajakumar, K., Sakellaris, H., and Adler, B. (2001). Ferric Dicitrate Transport System (Fec) of Shigella flexneri 2a YSH6000 is encoded on a novel pathogenicity island carrying multiple antibiotic resistance genes. Infect. Immun. 69, 6012–6021. doi: 10.1128/IAI.69.10.6012-6021.2001
Maimanakos, J., Chow, J., Gaßmeyer, S. K., Güllert, S., Busch, F., Kourist, R., et al. (2016). Sequence-based screening for rare enzymes: new insights into the world of AMDases reveal a conserved motif and 58 novel enzymes clustering in eight distinct families. Front. Microbiol. 7:1332. doi: 10.3389/fmicb.2016.01332
Maqbool, A., Horler, R. S., Muller, A., Wilkinson, A. J., Wilson, K. S., and Thomas, G. H. (2015). The substrate-binding protein in bacterial ABC transporters: dissecting roles in the evolution of substrate specificity. Biochem. Soc. Trans. 43, 1011–1017. doi: 10.1042/BST20150135
Mayfield, J. E., Bricker, B. J., Godfrey, H., Crosby, R. M., Knight, D. J., Halling, S. M., et al. (1988). The cloning, expression, and nucleotide sequence of a gene coding for an immunogenic Brucella abortus protein. Gene 63, 1–9. doi: 10.1016/0378-1119(88)90540-9
Meinert, C., Senger, J., Witthohn, M., Wübbeler, J. H., and Steinbüchel, A. (2017). Carbohydrate uptake in Advenella mimigardefordensis strain DPN7T is mediated by periplasmic sugar oxidation and a TRAP-transport system. Mol. Microbiol. 104, 916–930. doi: 10.1111/mmi.13692
Müller, A., Severi, E., Mulligan, C., Watts, A. G., Kelly, D. J., Wilson, K. S., et al. (2006). Conservation of structure and mechanism in primary and secondary transporters exemplified by SiaP, a sialic acid binding virulence factor from Haemophilus influenzae. J. Biol. Chem. 281, 22212–22222. doi: 10.1074/jbc.M603463200
Mulligan, C., Fischer, M., and Thomas, G. H. (2011). Tripartite ATP-independent periplasmic (TRAP) transporters in bacteria and archaea. FEMS Microbiol. Rev. 35, 68–86. doi: 10.1111/j.1574-6976.2010.00236.x
Mulligan, C., Geertsma, E. R., Severi, E., Kelly, D. J., Poolman, B., and Thomas, G. H. (2009). The substrate-binding protein imposes directionality on an electrochemical sodium gradient-driven TRAP transporter. Proc. Natl. Acad. Sci. U.S.A. 106, 1778–1783. doi: 10.1073/pnas.0809979106
Mulligan, C., Kelly, D. J., and Thomas, G. H. (2007). Tripartite ATP-independent periplasmic transporters: application of a relational database for genome-wide analysis of transporter gene frequency and organization. J. Mol. Microbiol. Biotechnol. 12, 218–226. doi: 10.1159/000099643
Mulligan, C., Leech, A. P., Kelly, D. J., and Thomas, G. H. (2012). The Membrane Proteins SiaQ and SiaM Form an Essential Stoichiometric Complex in the Sialic Acid Tripartite ATP-independent Periplasmic (TRAP) Transporter SiaPQM (VC1777–1779) from Vibrio cholerae. J. Biol. Chem. 287, 3598–3608. doi: 10.1074/jbc.M111.281030
Piepenbreier, H., Fritz, G., and Gebhard, S. (2017). Transporters as information processors in bacterial signalling pathways. Mol. Microbiol. 104, 1–15. doi: 10.1111/mmi.13633
Pohlmann, A., Fricke, W. F., Reinecke, F., Kusian, B., Liesegang, H., Cramm, R., et al. (2006). Genome sequence of the bioplastic-producing Knallgas bacterium Ralstonia eutropha H16. Nat. Biotechnol. 24, 1257–1262. doi: 10.1038/nbt1244
Rabus, R., Jack, D. L., Kelly, D. J., and Saier, M. H. Jr (1999). TRAP transporters: an ancient family of extracytoplasmic solute-receptor-dependent secondary active transporters. Microbiology 145 (Pt 12), 3431–3445.
Radolf, J. D., Deka, R. K., Anand, A., Šmajs, D., Norgard, M. V., and Yang, X. F. (2016). Treponema pallidum, the syphilis spirochete: making a living as a stealth pathogen. Nat. Rev. Microbiol. 14, 744–759. doi: 10.1038/nrmicro.2016.141
Ricci, V., Loman, N., Pallen, M., Ivens, A., Fookes, M., Langridge, G. C., et al. (2012). The TCA cycle is not required for selection or survival of multidrug-resistant Salmonella. J. Antimicrob. Chemother. 67, 589–599. doi: 10.1093/jac/dkr515
Rice, A. J., Park, A., and Pinkett, H. W. (2014). Diversity in ABC transporters: type, I., II and III importers. Crit. Rev. Biochem. Mol. Biol. 49, 426–437. doi: 10.3109/10409238.2014.953626
Rosa, L. T., Dix, S. R., Rafferty, J. B., and Kelly, D. (2017). Structural basis for high-affinity adipate binding to AdpC (RPA4515), an orphan periplasmic binding protein from the Tripartite Tricarboxylate Transporter (TTT) family in Rhodopseudomonas palustris. FEBS J. 284, 4262–4277. doi: 10.1111/febs.14304
Rucktooa, P., Antoine, R., Herrou, J., Huvent, I., Locht, C., Jacob-Dubuisson, F., et al. (2007). Crystal Structures of two Bordetella pertussis periplasmic receptors contribute to defining a novel pyroglutamic acid binding DctP Subfamily. J. Mol. Biol. 370, 93–106. doi: 10.1016/j.jmb.2007.04.047
Safarchi, A., Octavia, S., Wu, S. Z., Kaur, S., Sintchenko, V., Gilbert, G. L., et al. (2016). Genomic dissection of Australian Bordetella pertussis isolates from the 2008–2012 epidemic. J. Infect. 72, 468–477. doi: 10.1016/j.jinf.2016.01.005
Salmon, R. C., Cliff, M. J., Rafferty, J. B., and Kelly, D. J. (2013). The CouPSTU and TarPQM transporters in Rhodopseudomonas palustris: redundant, promiscuous uptake systems for lignin-derived aromatic substrates. PLoS ONE 8:e59844. doi: 10.1371/journal.pone.0059844
Scheepers, G. H., Lycklama a Nijeholt, J. A., and Poolman, B. (2016). An updated structural classification of substrate-binding proteins. FEBS Lett. 590, 4393–4401. doi: 10.1002/1873-3468.12445
Severi, E., Hood, D. W., and Thomas, G. H. (2007). Sialic acid utilization by bacterial pathogens. Microbiology 153, 2817–2822. doi: 10.1099/mic.0.2007/009480-0
Severi, E., Randle, G., Kivlin, P., Whitfield, K., Young, R., Moxon, R., et al. (2005). Sialic acid transport in Haemophilus influenzae is essential for lipopolysaccharide sialylation and serum resistance and is dependent on a novel tripartite ATP-independent periplasmic transporter. Mol. Microbiol. 58, 1173–1185. doi: 10.1111/j.1365-2958.2005.04901.x
Somers, J. M., and Kay, W. W. (1983). Genetic fine structure of the tricarboxylate transport (tct) locus of Salmonella typhimurium. Mol. Gen. Genet. 190, 20–26. doi: 10.1007/BF00330319
Somers, J. M., Sweet, G. D., and Kay, W. W. (1981). Fluorocitrate resistant tricarboxylate transport mutants of Salmonella typhimurium. Mol. Gen. Genet. MGG 181, 338–345. doi: 10.1007/BF00425608
Steenbergen, S. M., Lichtensteiger, C. A., Caughlan, R., Garfinkle, J., Fuller, T. E., and Vimr, E. R. (2005). Sialic acid metabolism and systemic pasteurellosis. Infect. Immun. 73, 1284–1294. doi: 10.1128/IAI.73.3.1284-1294.2005
Sweet, G. D., Kay, C. M., and Kay, W. W. (1984). Tricarboxylate-binding proteins of Salmonella typhimurium. Purification, crystallization, and physical properties. J. Biol. Chem. 259, 1586–1592.
Sweet, G. D., Somers, J. M., and Kay, W. W. (1979). Purification and properties of a citrate-binding transport component, the C protein of Salmonella typhimurium. Can. J. Biochem. 57, 710–715. doi: 10.1139/o79-089
Takahashi, H., Inagaki, E., Kuroishi, C., and Tahirov, T. H. (2004). Structure of the Thermus thermophilus putative periplasmic glutamate/glutamine-binding protein. Acta Crystallogr. D Biol. Crystallogr. 60(Pt 10), 1846–1854. doi: 10.1107/S0907444904019420
Tatum, F. M., Tabatabai, L. B., and Briggs, R. E. (2009). Sialic acid uptake is necessary for virulence of Pasteurella multocida in turkeys. Microb. Pathog. 46, 337–344. doi: 10.1016/j.micpath.2009.04.003
Thomas, G. H. (2016). Sialic acid acquisition in bacteria–one substrate, many transporters. Biochem. Soc. Trans. 44, 760–765. doi: 10.1042/BST20160056
Thomas, G. H. (2017). On the pull: periplasmic trapping of sugars before transport. Mol. Microbiol. 104, 883–888. doi: 10.1111/mmi.13691
Thomas, G. H., Southworth, T., León-Kempis, M. R., Leech, A., and Kelly, D. J. (2006). Novel ligands for the extracellular solute receptors of two bacterial TRAP transporters. Microbiology 152(Pt 1), 187–198. doi: 10.1099/mic.0.28334-0
Vetting, M. W., Al-Obaidi, N., Zhao, S., San Francisco, B., Kim, J., Wichelecki, D. J., et al. (2015). Experimental strategies for functional annotation and metabolism discovery: targeted screening of solute binding proteins and unbiased panning of metabolomes. Biochemistry 54, 909–931. doi: 10.1021/bi501388y
Vimr, E. R., Kalivoda, K. A., Deszo, E. L., and Steenbergen, S. M. (2004). Diversity of microbial sialic acid metabolism. Microbiol. Mol. Biol. Rev. 68, 132–153. doi: 10.1128/MMBR.68.1.132-153.2004
Widenhorn, K. A., Somers, J. M., and Kay, W. W. (1988). Expression of the divergent tricarboxylate transport operon (tctI) of Salmonella typhimurium. J. Bacteriol. 170, 3223–3227. doi: 10.1128/jb.170.7.3223-3227.1988
Widenhorn, K. A., Somers, J. M., and Kay, W. W. (1989). Genetic regulation of the tricarboxylate transport operon (tctI) of Salmonella typhimurium. J. Bacteriol. 171, 4436–4441. doi: 10.1128/jb.171.8.4436-4441.1989
Wilkens, S. (2015). Structure and mechanism of ABC transporters. F1000Prime Rep. 7:14. doi: 10.12703/P7-14
Willis, R. C., and Furlong, C. E. (1974). Purification and properties of a ribose-binding protein from Escherichia coli. J. Biol. Chem. 249, 6926–6929.
Winnen, B., Hvorup, R. N., and Saier, M. H. Jr. (2003). The tripartite tricarboxylate transporter (TTT) family. Res. Microbiol. 154, 457–465. doi: 10.1016/S0923-2508(03)00126-8
Wübbeler, J. H., Hiessl, S., Schuldes, J., Thürmer, A., Daniel, R., and Steinbüchel, A. (2014). Unravelling the complete genome sequence of Advenella mimigardefordensis strain DPN7T and novel insights in the catabolism of the xenobiotic polythioester precursor 3,3′-dithiodipropionate. Microbiology 160(Pt 7), 1401–1416. doi: 10.1099/mic.0.078279-0
Wyborn, N. R., Alderson, J., Andrews, S. C., and Kelly, D. J. (2001). Topological analysis of DctQ, the small integral membrane protein of the C4-dicarboxylate TRAP transporter of Rhodobacter capsulatus. FEMS Microbiol. Lett. 194, 13–17. doi: 10.1111/j.1574-6968.2001.tb09439.x
Yancey, R. J., and Finkelstein, R. A. (1981). Assmilation of iron by pathogenic Neisseria sp. Infect. Immun. 32, 592–599.
Keywords: solute transport, periplasmic binding-proteins, secondary transporter, high-affinity, carboxylic acids
Citation: Rosa LT, Bianconi ME, Thomas GH and Kelly DJ (2018) Tripartite ATP-Independent Periplasmic (TRAP) Transporters and Tripartite Tricarboxylate Transporters (TTT): From Uptake to Pathogenicity. Front. Cell. Infect. Microbiol. 8:33. doi: 10.3389/fcimb.2018.00033
Received: 29 October 2017; Accepted: 25 January 2018;
Published: 12 February 2018.
Edited by:
Kalai Mathee, Florida International University, United StatesReviewed by:
Todd Kitten, Virginia Commonwealth University, United StatesFranz Narberhaus, Ruhr University Bochum, Germany
Copyright © 2018 Rosa, Bianconi, Thomas and Kelly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David J. Kelly, d.kelly@sheffield.ac.uk