The Healthy Human Blood Microbiome: Fact or Fiction?
 Diego J. Castillo
Diego J. Castillo Riaan F. Rifkin
Riaan F. Rifkin Don A. Cowan
Don A. Cowan Marnie Potgieter
Marnie Potgieter- 1Department of Biochemistry, Genetics and Microbiology, Centre for Microbial Ecology and Genomics, University of Pretoria, Pretoria, South Africa
- 2Human Origins and Palaeo Environmental Research Group, Department of Anthropology and Geography, Oxford Brookes University, Oxford, United Kingdom
The blood that flows perpetually through our veins and arteries performs numerous functions essential to our survival. Besides distributing oxygen, this vast circulatory system facilitates nutrient transport, deters infection and dispenses heat throughout our bodies. Since human blood has traditionally been considered to be an entirely sterile environment, comprising only blood-cells, platelets and plasma, the detection of microbes in blood was consistently interpreted as an indication of infection. However, although a contentious concept, evidence for the existence of a healthy human blood-microbiome is steadily accumulating. While the origins, identities and functions of these unanticipated micro-organisms remain to be elucidated, information on blood-borne microbial phylogeny is gradually increasing. Given recent advances in microbial-hematology, we review current literature concerning the composition and origin of the human blood-microbiome, focusing on bacteria and their role in the configuration of both the diseased and healthy human blood-microbiomes. Specifically, we explore the ways in which dysbiosis in the supposedly innocuous blood-borne bacterial microbiome may stimulate pathogenesis. In addition to exploring the relationship between blood-borne bacteria and the development of complex disorders, we also address the matter of contamination, citing the influence of contaminants on the interpretation of blood-derived microbial datasets and urging the routine analysis of laboratory controls to ascertain the taxonomic and metabolic characteristics of environmentally-derived contaminant-taxa.
Introduction
The human microbiome comprises a vast corpus of bacterial, archaeal, viral and fungal microbial taxa. While most of these micro-organisms are commensal, many are mutualistic and some are pathogenic. Regardless of whether their presence is beneficial, inconsequential or detrimental, our lives are inextricably linked to the microbes with which we share our bodies. In fact, despite being 1,000 times smaller than human cells, bacteria comprise ~2% of the adult human body mass (1.5 kg), roughly equivalent in size to the human brain or liver (Molina and DiMaio, 2012). Given our extensive co-evolutionary history with microbes (Moeller et al., 2016), it is not surprising that the estimated number of unique bacterial genes in our “accessory genome” (~3,300,000) exceeds the number of our own genes (~22,000) by a factor of 150 (Qin et al., 2010). Human microbiome research, described as the study of the entire DNA content of micro-organisms inhabiting our bodies, has rapidly evolved over the past decade. As this topic has been reviewed extensively elsewhere (Cho and Blaser, 2012; Morgan et al., 2012; Kim et al., 2013; Khanna and Tosh, 2014; Lloyd-Price et al., 2016), we focus here on current evidence indicative of the existence of a “healthy” human blood-microbiome (HBM).
The exploration of our “microbial-selves” has been facilitated largely by the introduction of Next Generation Sequencing (NGS) and the advent of whole metagenome shotgun sequencing (WMGS) as techniques to study microbial genetic material present in different human body-sites (Segata et al., 2013). For many years, scientists have aimed to establish a taxonomy-based set of core human-associated micro-organisms. However, a more valuable approach involves ascertaining the primary core microbial composition based on functional (metabolic) capacity, since it is easier to correlate pathogenesis with deviations or changes (i.e., dysbiosis) in a “core” microbiome (Turnbaugh et al., 2009). In this regard, several large-scale population-based studies have sequenced the metagenomes of the human intestinal-microbiome (IM), as well as other medically-relevant body-sites including the skin, vagina and mouth. Two notable collaborative projects have been developed to achieve this fundamental aim. As part of the “Metagenomes of the Human Intestinal Tract” project (Qin et al., 2010; Le Chatelier et al., 2013; Li et al., 2014) and the “Human Microbiome Project” (HMP) (Aagaard et al., 2013), more than 2,000 people from across the globe had contributed to the study of the microbiome structure of healthy individuals since 2006 (Lloyd-Price et al., 2016). Although most contemporary research focuses on the human IM, the microbial communities present in the human mouth and eyes, on the skin, lungs and in the placenta and urogenital tracts have also been described (Aagaard et al., 2013; Blekhman et al., 2015; Lloyd-Price et al., 2016).
More recently, the prospect of the existence of a “healthy” HBM has roused much interest in the scientific community (McLaughlin et al., 2002; Bahrani-Mougeot et al., 2008; Païssé et al., 2016). Human blood comprises ~54.3% plasma, ~45% red blood cells (erythrocytes), ~0.7% white blood cells (lymphocytes) and a variable number of platelets (thrombocytes), depending on health status (Alberts et al., 2002). Following the first documented observation of erythrocytes by Antonie van Leeuwenhoek in 1674 (Bessis and Delpech, 1981) (Figure 1), it is now known that blood is the liquid medium that carries and sustains the most basic, but most essential, elements of life. Whereas erythrocytes are responsible primarily for the transport of oxygen, lymphocytes serve as a highly efficient surveillance system that monitors the blood for invasive microbes (Jerne, 1973). The primary function of thrombocytes is to react to bleeding from blood vessel injury by clotting (Blache, 1992). Because blood has traditionally been considered to be a sterile environment, devoid of all other forms of foreign (e.g., bacterial) cells, it is not surprising that the concept of a healthy HBM has been met with criticism (Nikkari et al., 2001; McLaughlin et al., 2002; Païssé et al., 2016). While evidence for the existence of a blood-microbiome in various domesticated mammals and birds do exist (Sze et al., 2014; Mandal et al., 2016; Vientós-Plotts et al., 2017), we focus on the healthy human blood-microbiome. Given that an increasing number of studies are exploring the notion that the presence of “foreign” micro-organisms in human blood does not necessarily equate with infection or with a diseased state, we review evidence concerning the discovery and tentative acceptance of the healthy HBM. In addition, we explore the potential origins and identities of “resident” micro-organisms, their phylogenetic affiliations and the clinical relevance of an allegedly healthy HBM. We also address the adverse influence that contaminants derived from reagents and laboratory environments exert on sequence-based IM and HBM research and the recovery of numerous microbial taxa in DNA extraction and library preparation controls.
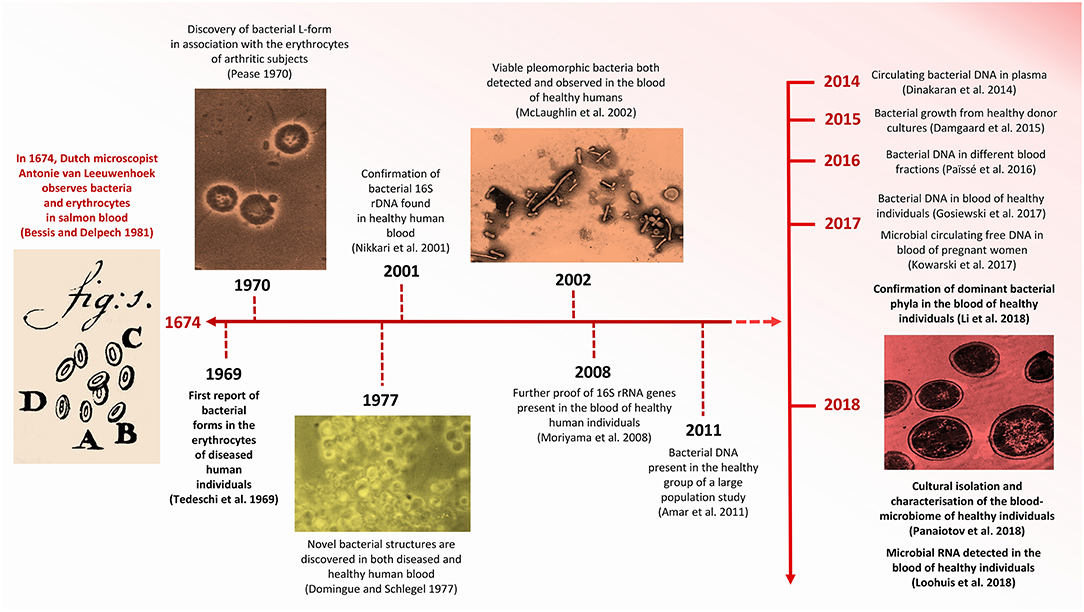
Figure 1. Timeline indicating significant advances concerning HBM research. Images modified from Pease (1970), Domingue and Schlegel (1977), Bessis and Delpech (1981), McLaughlin et al. (2002), and Panaiotov et al. (2018).
Controversy and Evolution of a Novel Concept
The controversy concerning the incidence of foreign cells in human blood extends back to the late 1960s, when Tedeschi et al. (1969) reported the presence of metabolically active bacteria in the blood of healthy human subjects (Figure 1). Specifically, the increased absorption of nucleosides and amino acids in erythrocyte suspensions led them to hypothesize that mycoplasm-like or L-phase (cell-wall deficient) bacteria were present in the blood of overtly non-diseased individuals. Nearly a decade later, in 1977, Gerald Domingue and Jorgen Schlegel reported that ~7% of blood samples derived from a healthy human cohort exhibited bacterial growth following osmotic lysis and filtering (Domingue and Schlegel, 1977).
More recent evidence for the hypothetical existence of a healthy HBM derives from Nikkari et al. (2001) who reported the presence of bacterial DNA in the blood of a healthy human cohort. This study, based on qPCR, included the use of rRNA-specific fluorescent probes and 16S rRNA gene-specific primers and identified bacterial taxa belonging to five divisions and seven phylogenetic groups. This study is, however, limited by the fact that all observations were based on the analysis of the blood of only four individuals (Table 1). Shortly thereafter, McLaughlin et al. (2002) described the presence of pleomorphic bacteria in the blood of individuals who did not present any perceptible clinical manifestations of disease. In this study, transmission electron microscopy (TEM), dark-field microscopy (DFM), fluorescence in situ hybridization (FISH) and the sequencing of PCR-amplified 16S rRNA and gyrB genes confirmed the presence of bacterial DNA in the blood of healthy individuals. Correspondingly, Moriyama et al. (2008) contributed to the concept of a “healthy” HBM by confirming the presence of bacterial 16S rRNA genes in the blood of healthy humans.
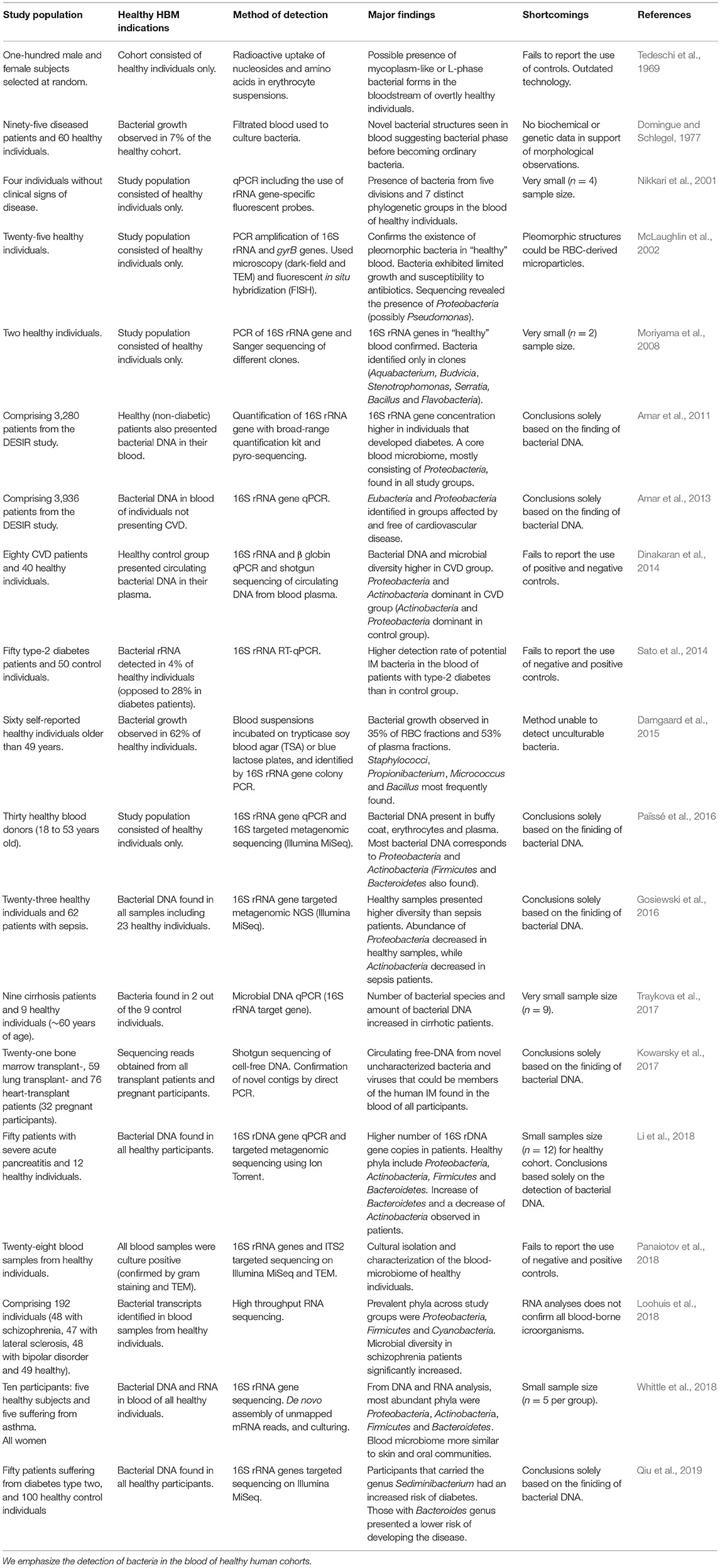
Table 1. HBM studies concerning both healthy and diseased human participants.
As anticipated, challenging the traditional conviction concerning the sterility of blood in healthy humans under normal circumstances has generated considerable controversy. Mitchell et al. (2016) assessed the findings of McLaughlin et al. (2002) and other studies, concluding that the pleomorphic bacteria identified in the blood of healthy humans were, in fact, nothing more than micro-particles derived from disintegrated erythrocytes. Martel et al. (2017) supported this argument with the discovery that bacteria-like structures closely resembled membrane vesicles and that vibrating refringent particles captured by dark-field microscopy were merely aggregates of blood proteins. Although the visual confirmation of micro-organisms present in the blood of healthy individuals requires further examination, evidence confirming the presence of microbial genetic material in the blood-circulatory system is accumulating (McLaughlin et al., 2002; Moriyama et al., 2008; Païssé et al., 2016).
The application of innovative analytical technologies, such as targeted NGS of the 16S rRNA gene, has provided increasingly robust evidence for the existence of a non-diseased HBM (Dinakaran et al., 2014; Damgaard et al., 2015; Gosiewski et al., 2016; Païssé et al., 2016; Kowarsky et al., 2017; Whittle et al., 2018; Qiu et al., 2019). RNA-sequence data have also contributed to this premise, as bacterial transcripts have been identified in non-diseased control groups (Loohuis et al., 2018; Whittle et al., 2018). Researchers characterizing the blood-microbiome in diseased patients, largely through culture-independent methods, have also detected genetic material in their healthy control groups (Table 1). Moreover, the presence of comparable bacterial phyla in different studies appears to lend support for the existence of a healthy HBM (McLaughlin et al., 2002; Amar et al., 2011, 2013; Dinakaran et al., 2014; Païssé et al., 2016; Li et al., 2018; Loohuis et al., 2018; Whittle et al., 2018; Qiu et al., 2019).
In addition to challenging the status quo of the “germ-free” human blood paradigm, methodological obstacles have hindered HBM research. Many micro-organisms found naturally within human blood may in fact be in a dormant state (Potgieter et al., 2015). Accordingly, culture-based methods cannot be reliably employed to support the existence of a HBM. Furthermore, while the concentration of bacterial DNA in the blood is typically very low, increasingly-sensitive analytical techniques, particularly qPCR, and targeted NGS may substantiate current evidence for the presence of “innocuous” bacterial taxa in the blood of healthy humans (Païssé et al., 2016).
However, rigorous experimental controls, which are essential when studying low-biomass microbiomes prone to contamination from external sources, are not routinely included. This is particularly problematic as the detection of >90 microbial genera in DNA extraction and library preparation controls (Salter et al., 2014; Lauder et al., 2016) highlights the influence that contaminants derived from reagents and laboratory environments exert on sequence-based HBM analyses. The analyses of negative DNA extraction controls, as performed by Moriyama et al. (2008), showed significantly less 16S rRNA gene amplification when compared to blood derived from healthy individuals. These controls comprised saline water which had been in contact with povidone iodine sterilized skin (Moriyama et al., 2008). In another study by Dinakaran et al. (2014), the analyses of comparable samples as negative controls for 16S rRNA gene targeted Illumina MiSeq WMGS showed virtually no amplified “contaminant” taxa. Although a number of these samples did yield >10,000 DNA sequence reads, their taxonomic composition differed significantly from that of both the diseased and healthy blood samples (Dinakaran et al., 2014). While investigating the blood-microbiome of cirrhotic patients, Traykova et al. (2017) recovered bacterial DNA from the blood of >20% of the healthy cohort. In this study, sterile water and pan-bacterial assays, which detect a broad range of bacterial taxa, were used as negative and positive controls. The use of controls was also implemented when the blood-microbiome in different blood fractions was characterized (Païssé et al., 2016). In a recent study by Loohuis et al. (2018) the importance of including stringent controls when studying low biomass microbiomes, such as human blood, was clearly demonstrated. While investigating the blood-microbial transcriptomes of both healthy human individuals and of patients affected with brain disorders, RNA obtained from lymphoblast cell lines were used as negative controls, and cells infected with Chlamydia, as positive controls. RNA reads were identified only for the Chlamydiae phylum in the positive controls, and no microbial sequences were detected in the lymphoblast cells.
It is evident that further research is required to establish whether the microbial DNA and RNA found in healthy human blood represents either living or dead, or active or non-active bacterial taxa. Although contamination derived from human sources poses a significant challenge to blood-microbiome research, bacteriological activity in the blood could potentially be studied through viability assay techniques such as propidium monoazide (PMA) treatment and cellular energy measurements (Emerson et al., 2017). However, there is presently no specific and reliable means of detecting living bacteria in human blood.
Despite the fact that evidence for the presence of bacterial taxa comprising a healthy blood-microbiome in humans is accumulating, not much is known about the presence of other micro-organisms, such as viruses, Archaea and lower eukaryotes (i.e., fungi) in the blood of healthy humans. The presence of archaeal DNA is generally not reported (Nikkari et al., 2001; McLaughlin et al., 2002; Moriyama et al., 2008; Dinakaran et al., 2014; Damgaard et al., 2015; Gosiewski et al., 2016; Païssé et al., 2016), presumably due to low abundance or the complete absence of archaea from blood samples. Dinakaran et al. (2014) did, however, document a relative abundance of 0.01% circulating archaeal DNA in the blood plasma of healthy individuals. The fungal microbiomes of the human intestinal tract, mouth, skin, lungs, and other body-sites have been explored (Cui et al., 2013; Huffnagle and Noverr, 2013). As the presence of fungi in the blood of healthy individuals has only recently been reported (Panaiotov et al., 2018), more research concerning the human blood myco-biome, in particular studies including stringent negative controls, is required. Regarding a tentative human blood-virome, and following the exclusion of taxa attributable to contamination, Moustafa et al. (2017) recovered 19 viral taxa from 42% of overtly non-diseased individuals. Previous reports have confirmed the presence of eukaryotic viruses, such as rhabdoviruses (Stremlau et al., 2015), anelloviruses (Furuta et al., 2015), and other families including Herpesviridae and Poxiviridae (Rascovan et al., 2016) in healthy human blood. Additional research concerning the human blood-virome is therefore required to determine whether viruses are resident members of the HBM, or simply remnants of previous infections.
Origin and Location of Blood-Borne Bacteria
It is not known whether blood-borne bacteria are exploiting a viable ecological niche, or whether they are simply transient residents in the blood. Some researchers suggest that the presence of bacteria in blood is a consequence of translocation from other body-sites, particularly the gastro-intestinal tract (Païssé et al., 2016). Indeed, the etiology of diabetes, cardiovascular disease, hematological disorders and cirrhosis has been ascribed to the translocation of bacteria from the intestinal tract, primarily via the intestinal epithelial mucosa (Amar et al., 2013; Dinakaran et al., 2014; Sato et al., 2014; Manzo and Bhatt, 2015; Traykova et al., 2017). Correspondingly, it has been suggested that bacteria derived from the skin- (Cogen et al., 2008) and oral-microbiomes could also diffuse into the blood when the barriers between these environments and the circulatory system are compromised (Forner et al., 2006; Bahrani-Mougeot et al., 2008; Iwai, 2009). Recently, Whittle et al. (2018) compared microbial DNA data derived from healthy individuals to HMP microbiome data. They demonstrated that, whereas the blood-microbiome closely resembles the skin- and oral microbiomes, it differs substantially from the intestinal microbiome. While most studies tend to consider the diffusion of bacteria into the blood-circulatory system as exceptional, this phenomenon may therefore occur rather frequently in healthy individuals, supporting the recent findings of a healthy HBM (Moriyama et al., 2008; Païssé et al., 2016). Even if the intestinal epithelial membrane is not compromised, other mechanisms could facilitate the entry of intestinal bacteria into the circulatory system. Micro-organisms can be absorbed by dendritic cells (antigen or “accessory” cells of the mammalian immune system) and transported through the intestinal epithelium (Rescigno et al., 2001; Niess et al., 2005) or via the assistance of intestinal mucus-secreting goblet cells (McDole et al., 2012). Additionally, intestinal M-cells (specialized epithelial cells of the mucosa-associated lymphoid tissues) could also be involved in the translocation of bacteria from the intestinal lumen to the blood-circulatory system (Vazquez-Torres et al., 1999; Jang et al., 2004; Lelouard et al., 2012).
The vertical transmission of the microbiome is an almost universal phenomenon in the animal kingdom (Funkhouser and Bordenstein, 2013) and, as for the human IM, blood-borne bacteria could also have a maternal origin. Although fetal and maternal blood does not mix during gestation, bacteria could colonize the fetal circulatory system even before delivery. Following the isolation of bacterial DNA from the umbilical cords of healthy new-borns delivered by C-section, Jiménez et al. (2005), suggested the tentative existence of a pre-natal blood-microbiome. Blood from 20 neonates was collected and processed in a class II safety cabinet to avoid contamination. Thereafter, 16S rRNA gene sequencing was performed from colonies obtained after culturing in brain and heart infusion broth (Jiménez et al., 2005). Appropriate negative controls were included in the culturing and sequencing steps. The presence of micro-organisms in the blood of new-born humans could be derived from other body-sites, in utero, such as fetal intestinal or oral sites. Thus, while it is widely believed that the initial new-born inoculum stems from contact with the vaginal-, fecal- or skin-microbiota of the mother during labor and that this infant-microbiome is subsequently enriched through breastfeeding (Penders et al., 2006; Biasucci et al., 2008; Dominguez-Bello et al., 2010; Azad et al., 2013), some evidence indicates the existence of a fetal microbiome in utero and the enrichment of that original set of micro-organisms following birth (Romano-Keeler and Weitkamp, 2014). Although controversial, various research groups have suggested the presence of bacteria in the placenta (Aagaard, 2014; Aagaard et al., 2014), the amnion (Hitti et al., 1997; Bearfield et al., 2002; DiGiulio et al., 2010), fetal membranes (Steel et al., 2005) and the meconium (Jiménez et al., 2008; Gosalbes et al., 2013; Moles et al., 2013), favoring this hypothesis (Funkhouser and Bordenstein, 2013). While the mechanisms implicated in the subsequent transmission of bacteria from other body-sites to the fetus are unknown, one possible route of entry into the fetus could involve the ingestion of amniotic fluid during gestation (Romano-Keeler and Weitkamp, 2014). The possible maternal origin of the blood-microbiome requires further investigation.
It is evident that the exact origin of the HBM remains to be fully elucidated. It could, hypothetically, derive from the mother preceding birth, or from the translocation of micro-organisms derived from other sources after birth and during the normal human lifecycle. As with the human IM, and besides the influence on IM taxonomic composition of diet (De Filippo et al., 2010), age (Yatsunenko et al., 2012), seasonal variation (Smits et al., 2017), and host immuno-modulation (Thorburn et al., 2014), the HBM may well comprise an adaptive micro-ecological system prone to environmental influences and exposure to novel microbial taxa. Accordingly, if micro-organisms present in human blood do originate from other body-sites, and if bacterial translocation is not an infrequent event, it appears reasonable to assume that the healthy HBM is highly dynamic. Be that as it may, recent findings based predominantly on cross-sectional studies targeting the 16S rRNA gene show a set of dominant blood-borne bacterial phyla (i.e., Proteobacteria, followed by Actinobacteria, Firmicutes, and Bacteroidetes) (McLaughlin et al., 2002; Amar et al., 2011, 2013; Dinakaran et al., 2014; Gosiewski et al., 2016; Païssé et al., 2016; Loohuis et al., 2018; Whittle et al., 2018; Qiu et al., 2019) and are indicative of longer-term HBM stability.
With regards the precise location of micro-organisms inside human blood, current evidence suggest that bacterial taxa can survive inside both erythrocytes and leukocytes. Chlamydia pneumoniae, an intracellular bacterium and the major causative agent of pneumonia, has been found to inhabit peripheral blood mononuclear cells (PBMCs) in healthy individuals (Yamaguchi et al., 2004). Other bacteria, for example Staphylococcus aureus, can also invade and persist in white blood cells (WBCs). As far back as 2000, Gresham et al. (2000) showed that these bacteria both reside and retain their virulence within neutrophils. Thwaites and Gant (2011) have also suggested that WBCs, and especially neutrophils, could act as “Trojan horses” by offering protection against human antibodies, thereby facilitating the dissemination of S. aureus to different body-sites. Moreover, when Païssé et al. (2016) analyzed the blood-microbiome of healthy individuals, most bacterial DNA (93.74%) was found to be localized within the buffy coat (BC), which consists primarily of WBCs and platelets. A correlation between leukocyte concentration and the number of 16S rRNA gene copies in the BC of study participants was also identified. Similarly, some bacteria can enter RBCs directly, and persist within the nutrient-rich environment; it has been shown that Staphylococcus aureus, a species commonly found in both healthy and diseased human IM (Grice et al., 2009), can utilize iron (Fe) present in RBCs as a nutrient source (Yamaguchi et al., 2013). Yamaguchi et al. (2013) also showed that Streptococcus pneumoniae, a bacterium implicated in the onset of pneumonia and sepsis, became increasingly viable when incubated with erythrocytes. Similarly, it has been reported that Brucella melitensis, the causative agent of ovine brucellosis, and Francisella tularensis, a Gram-negative bacterium that causes tularaemia, also possess the capacity to invade and persist in erythrocytes (Horzempa et al., 2011; Vitry et al., 2014).
Composition of the Putative Healthy Human Blood-Microbiome
Despite the fact that the existence of a blood-microbiome in healthy human individuals appears to be supported by recent studies, knowledge of the phylogenetic diversity of blood-borne bacteria remains limited. In contrast to the dominant bacterial phyla typically observed in the human IM (i.e., Firmicutes and Bacteroidetes), the HBM appears to be dominated by the phyla Proteobacteria followed by Actinobacteria, Firmicutes, and Bacteroidetes (McLaughlin et al., 2002; Amar et al., 2011, 2013; Dinakaran et al., 2014; Gosiewski et al., 2016; Païssé et al., 2016; Li et al., 2018; Loohuis et al., 2018; Whittle et al., 2018; Qiu et al., 2019). The characterization of blood bacterial diversity, however, varies between studies. In 2008, Moriyama et al. (2008) identified a set of bacterial taxa in their study of bacteria from the blood of two healthy individuals comprising mostly Bacillus, Flavobacteria, Stenotrophomas and Serratia. Using a culture-based approach, Damgaard et al. (2015) observed bacterial growth in the blood of ~62% of healthy individuals. The most prominent taxa detected were Propionibacterium acnes and Staphylococcus epidermis, as well as Bacilli and Micrococcus species (Damgaard et al., 2015). In 2016, Païssé et al. (2016) analyzed bacterial DNA present in different fractions of human blood. At class level, Fusobacteria and Flavobacteria were more abundant in RBCs, while members of the Clostridia class were dominant in plasma and erythrocyte fractions. Seven genera were identified in the RBC fraction, including two opportunistic pathogens, namely Acinetobacter baumanni and Stenotrophomonas maltophilia. Problematically, although the incidence of diverse blood taxonomic composition might indeed reflect actual microbial configuration, bacterial DNA found to contaminate DNA extraction kits typically includes Bacillus, Flavobacteria, Fusobacteria, Propionibacterium, and Serratia (Glassing et al., 2016). In addition to a critical awareness of potentially contaminating taxa, there is also a need for much broader metagenomic studies encompassing larger cohorts of both healthy and diseased individuals, as these would provide valuable insights into the composition of the putatively “healthy” HBM as well as its functionality and potential role in maintaining optimal human health and the onset of disease.
The Clinical Relevance of the Healthy HBM
With reference to the role of the human microbiome in pathogenesis, the concept of “dysbiosis,” which refers to a change in the composition of symbiotic or commensal microbial communities (Petersen and Round, 2014), is particularly relevant. Although it is not known whether dysbiosis is a cause, or simply a reflection, of a diseased state (Bäckhed et al., 2012), numerous studies have related changes in human microbial community composition with the onset of disease. Examples include diabetes (Qin et al., 2012), asthma (Teo et al., 2015), inflammatory bowel disease (Morgan et al., 2012), autism (Parracho et al., 2005) and even complex disorders such as Alzheimer's disease (Pistollato et al., 2016). While considerable research has been dedicated to address the relationship between the IM and human health, a limited number of studies have explored dysbiosis of the HBM and its potential role in pathogenesis. Conditions such as diabetes, pancreatitis and also cardiovascular- and liver-disease have, however, been related to changes in the HBM. Using an IM microbial qPCR microarray, the screening for bacterial DNA in the blood of both cirrhotic and healthy individuals by Traykova et al. (2017) resulted in the detection of higher levels of bacterial diversity in patients with cirrhosis, as opposed to a healthy control cohort. They also reported an increase of total bacterial DNA concentration in the blood of the diseased cohort compared to healthy controls. The HBM in patients with severe acute pancreatitis has also been analyzed by 16S rRNA gene amplicon sequencing (Li et al., 2018). Microbial taxonomic diversity was found to be reduced when compared to the healthy human cohort, and an increase in Bacteroidetes and a decrease in Actinobacteria were observed in pancreatitis patients. At class level, Bacteroidia and Clostridia increased in abundance, while Actinobacteria, Flavobacteria, and Bacilli were reduced in the diseased group when compared to healthy controls (Li et al., 2018). These variations in dominant taxa are strongly suggestive of blood-microbiome dysbiosis in pancreatitis patients. The onset of cardiovascular diseases may also be linked to HBM dysbiosis. In 2013, Amar et al. (2013) discovered that the blood of patients who presented an acute cardiovascular event, even years following sample collection, had a significant decrease of total bacterial DNA when compared to a healthy cohort, as well as an increase in taxa assigned to the Proteobacteria. Accordingly, it was concluded that dysbiosis in the HBM could serve as a “marker” for CVD prediction. One year later, Dinakaran et al. (2014) also proposed the likelihood of an increase in microbial diversity and bacterial DNA concentration when analyzing cell-free DNA circulating in the blood of CVD patients. In this study, it was observed that Actinobacteria were dominant over Proteobacteria in CVD patients, while an opposite trend was observed in the healthy cohort.
The association between HBM-dysbiosis and the onset of liver-disease has also been explored (Lelouvier et al., 2016; Schierwagen et al., 2018), resulting in the proposal that blood-microbiota could serve as biomarkers for non-alcoholic fatty liver disease (NAFLD) prediction in obese patients (Lelouvier et al., 2016). In this study, qPCR and 16S rRNA gene targeted NGS was employed. Liver fibrosis patients exhibited higher concentrations of 16S rRNA gene in their blood when compared to non-diseased participants. In addition, unique bacterial taxonomic clustering was observed in patients suffering from severe liver fibrosis (Lelouvier et al., 2016). In 2018, Schierwagen et al. (2018) performed 16S rRNA gene NGS analyses on blood samples obtained from the portal vein, central and peripheral venous blood and liver outflow in patients suffering from liver fibrosis. Their findings corroborated those in the NAFLD study (Lelouvier et al., 2016). Furthermore, each of these circulatory compartments exhibited a unique taxonomic composition at genus level (Schierwagen et al., 2018).
In addition to these examples, various other studies have established possible associations between blood-derived bacteria originating from the IM, and the onset of diabetes; whereas IM bacteria have been detected to occur in ~28% of diabetes patients, healthy participants exhibited only ~4% of IM-derived bacterial taxa (Sato et al., 2014). The most abundant taxa identified in the diabetes group included Clostridium coccoides and the Atopobium cluster (Sato et al., 2014). Although Amar et al. (2011) could not convincingly demonstrate a significantly different HBM in patients prone to the development of diabetes, they did observe a higher 16S rRNA gene concentration in the blood of participants. Consequently, high concentrations of blood-derived bacterial DNA could potentially be used as a predictive marker for this condition. Recently, Qiu et al. (2019) did not find a significant difference in terms of blood bacteria diversity between type two diabetes mellitus patients and healthy individuals Nonetheless, participants containing the genus Sediminibacterium in their blood showed a higher risk to develop diabetes, while individuals that carried the genus Bacteroides had a decreased risk of presenting the disease (Qiu et al., 2019).
Concluding Remarks and Future Prospects
The existence of a healthy HBM remains to be challenged, particularly in light of recent criticisms of, for example, the existence of a discrete human placental-microbiome (Lauder et al., 2016). A number of studies on the HBM are furthermore mired by essential shortcomings (Table 1), which casts some doubt over the validity of their results. Moreover, current definitions of “healthy” are vague and consequently problematic in terms of defining a “healthy” HBM. As with the human IM, “healthy” could be defined in terms of ecological stability (the ability to resist community change or rapidly return to a baseline state following stress-related change), an idealized (presumably “health-associated”) composition or by a most desirable functional profile (including metabolic and trophic provisions to the host) (Bäckhed et al., 2012).
Evidence indicative of the presence of a microbial component in the blood of healthy human individuals is, nevertheless, steadily accumulating and recent studies have identified comparable bacterial phyla in the blood of healthy individuals (McLaughlin et al., 2002; Amar et al., 2011, 2013; Dinakaran et al., 2014; Gosiewski et al., 2016; Païssé et al., 2016; Li et al., 2018; Loohuis et al., 2018; Whittle et al., 2018; Qiu et al., 2019). Not discounting the shortcomings of each of the cited studies, a coherent picture of the healthy HBM emerges when one considers the positive aspects introduced by each study. For example, the inclusion of microscopy (McLaughlin et al., 2002) and the application of both DNA (Dinakaran et al., 2014; Païssé et al., 2016; Li et al., 2018; Whittle et al., 2018) and RNA analyses to the blood of healthy individuals (Loohuis et al., 2018; Whittle et al., 2018). From the literature reviewed here, and taking into consideration the trend toward including analytical (positive and negative) controls in HBM-related studies, we conclude that the notion that a healthy (non-diseased) human blood-microbiome exists cannot simply be discarded.
With regards to future studies on the “healthy” HBM, we recommend thorough experimental designs that ensure the reduction of both contamination and technical biases. For this, researchers must improve the protocols implicated in obtaining (drawing blood), processing (using kits and storage), and generating (sequencing protocols and techniques) both healthy and diseased blood microbial data. This could be achieved by studying the blood microbiome together with that of the skin around the puncture site from where the blood was obtained, as well as characterizing microbial DNA derived from potential microorganisms present in the needle, vacutainers, reagents, and other consumable items used during sampling.
We furthermore encourage researchers to investigate this unique microbiome, as it promises to hold the potential to facilitate both the diagnosis and improved understanding of the onset of numerous human diseases. It is still unclear whether the putative healthy HBM comprises a core set of bacterial taxa, or a dynamic and adaptive group of micro-organisms. We recommend performing studies that also take into consideration the “time” element in the HBM. Most of the literature reviewed here deals with snapshots of blood bacterial communities, potentially overlooking important changes of the HBM across time. In addition, the influence of age, geography and socio-economic status (e.g., access to nutritional diet and healthcare services) on healthy HBM composition remains ambiguous. With reference to the location of micro-organisms in the human blood-circulatory system, it is conceivable that bacteria either reside inside, or adhere to blood cells or in the plasma fraction of the blood. Moreover, with regards to the origin of the healthy HBM, it appears that blood-borne micro-organisms, particularly bacteria, may well originate from various body-sites and that many might have a maternal (i.e., vertical) origin. Samples from the skin-, oral- and Intestinal-microbiomes should be analyzed at the same time as blood samples to gain insight into the potential origin of these microbes. Finally, in order to ascertain the potential roles and functions linked to bacteria and other microorganisms in the human blood, studies on the HBM based on WMGS are undoubtedly essential. The increasing recognition of the existence of a healthy HBM stimulates novel and diverse research avenues, some of which may well turn out to be of considerable clinical significance.
Author Contributions
DC initiated and conceived the review, investigated the literature, and wrote the review. RR contributed to the writing and critical revision of the manuscript. DAC contributed to revision of the manuscript. MP directed and contributed to the review design, investigated the literature, and contributed to the critical revision of the manuscript.
Funding
MP is supported by an NRF career advancement fellowship and an MRC self-initiated research grant. RR is funded by a National Geographic Society Scientific Exploration Grant (Nr. NGS-371R-18) and the University of Pretoria.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Marc Van Goethem for insightful discussions and valuable feedback.
References
Aagaard, K., Ma, J., Antony, K. M., Ganu, R., Petrosino, J., and Versalovic, J. (2014). The placenta harbors a unique microbiome. Sci. Transl. Med. 6:237ra265. doi: 10.1126/scitranslmed.3008599
Aagaard, K., Petrosino, J., Keitel, W., Watson, M., Katancik, J., Garcia, N., et al. (2013). The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J. 27, 1012–1022. doi: 10.1096/fj.12-220806
Aagaard, K. M. (2014). Author response to comment on “The placenta harbors a unique microbiome.” Sci. Transl. Med. 6:254lr253. doi: 10.1126/scitranslmed.3010007
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. (2002). Molecular Biology of the Cell, 4th Edn. New York, NY: Garland Science.
Amar, J., Lange, C., Payros, G., Garret, C., Chabo, C., Lantieri, O., et al. (2013). Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: the DESIR study. PLoS ONE 8:e54461. doi: 10.1371/journal.pone.0054461
Amar, J., Serino, M., Lange, C., Chabo, C., Iacovoni, J., Mondot, S., et al. (2011). Involvement of tissue bacteria in the onset of diabetes in humans: evidence for a concept. Diabetologia 54, 3055–3061. doi: 10.1007/s00125-011-2329-8
Azad, M. B., Konya, T., Maughan, H., Guttman, D. S., Field, C. J., Chari, R. S., et al. (2013). Gut microbiota of healthy Canadian infants: profiles by mode of delivery and infant diet at 4 months. CMAJ 185, 385–394. doi: 10.1503/cmaj.121189
Bäckhed, F., Fraser, C. M., Ringel, Y., Sanders, M. E., Sartor, R. B., Sherman, P. M., et al. (2012). Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe 12, 611–622. doi: 10.1016/j.chom.2012.10.012
Bahrani-Mougeot, F. K., Paster, B. J., Coleman, S., Ashar, J., Barbuto, S., and Lockhart, P. B. (2008). Diverse and novel oral bacterial species in blood following dental procedures. J. Clin. Microbiol. 46, 2129–2132. doi: 10.1128/JCM.02004-07
Bearfield, C., Davenport, E. S., Sivapathasundaram, V., and Allaker, R. P. (2002). Possible association between amniotic fluid micro-organism infection and microflora in the mouth. BJOG 109, 527–533. doi: 10.1111/j.1471-0528.2002.01349.x
Bessis, M., and Delpech, G. (1981). Discovery of the red blood cell with notes on priorities and credits of discoveries, past, present and future. Blood Cells. 7, 447–480.
Biasucci, G., Benenati, B., Morelli, L., Bessi, E., and Boehm, G. (2008). Cesarean delivery may affect the early biodiversity of intestinal bacteria. J. Nutr. 138, 1796S−1800S. doi: 10.1093/jn/138.9.1796S
Blache, D. (1992). Structure and function of blood platelets. Arch. Int. Physiol. Biochim. Biophys. 100:A17–24. doi: 10.3109/13813459209000709
Blekhman, R., Goodrich, J. K., Huang, K., Sun, Q., Bukowski, R., Bell, J. T., et al. (2015). Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 16:191. doi: 10.1186/s13059-015-0759-1
Cho, I., and Blaser, M. J. (2012). The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 13:260. doi: 10.1038/nrg3182
Cogen, A., Nizet, V., and Gallo, R. (2008). Skin microbiota: a source of disease or defence? Br. J. Dermatol. 158, 442–455. doi: 10.1111/j.1365-2133.2008.08437.x
Cui, L., Morris, A., and Ghedin, E. (2013). The human mycobiome in health and disease. Genome Med. 5:63. doi: 10.1186/gm467
Damgaard, C., Magnussen, K., Enevold, C., Nilsson, M., Tolker-Nielsen, T., Holmstrup, P., et al. (2015). Viable bacteria associated with red blood cells and plasma in freshly drawn blood donations. PLoS ONE. 10:e0120826. doi: 10.1371/journal.pone.0120826
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696. doi: 10.1073/pnas.1005963107
DiGiulio, D. B., Romero, R., Kusanovic, J. P., Gómez, R., Kim, C. J., Seok, K. S., et al. (2010). Prevalence and diversity of microbes in the amniotic fluid, the fetal inflammatory response, and pregnancy outcome in women with preterm pre-labor rupture of membranes. Am. J. Reprod. Immunol. 64, 38–57. doi: 10.1111/j.1600-0897.2010.00830.x
Dinakaran, V., Rathinavel, A., Pushpanathan, M., Sivakumar, R., Gunasekaran, P., and Rajendhran, J. (2014). Elevated levels of circulating DNA in cardiovascular disease patients: metagenomic profiling of microbiome in the circulation. PLoS ONE. 9:e105221. doi: 10.1371/journal.pone.0105221
Domingue, G., and Schlegel, J. (1977). Novel bacterial structures in human blood: cultural isolation. Infect. Immun. 15, 621–627.
Dominguez-Bello, M. G., Costello, E. K., Contreras, M., Magris, M., Hidalgo, G., Fierer, N., et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U.S.A. 107, 11971–11975. doi: 10.1073/pnas.1002601107
Emerson, J. B., Adams, R. I., Román, C. M. B., Brooks, B., Coil, D. A., Dahlhausen, K., et al. (2017). Schrödinger's microbes: tools for distinguishing the living from the dead in microbial ecosystems. Microbiome 5:86. doi: 10.1186/s40168-017-0285-3
Forner, L., Larsen, T., Kilian, M., and Holmstrup, P. (2006). Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J. Clin. Periodontol. 33, 401–407. doi: 10.1111/j.1600-051X.2006.00924.x
Funkhouser, L. J., and Bordenstein, S. R. (2013). Mom knows best: the universality of maternal microbial transmission. PLoS Biol. 11:e1001631. doi: 10.1371/journal.pbio.1001631
Furuta, R. A., Sakamoto, H., Kuroishi, A., Yasiui, K., Matsukura, H., and Hirayama, F. (2015). Metagenomic profiling of the viromes of plasma collected from blood donors with elevated serum alanine aminotransferase levels. Transfusion 55, 1889–1899. doi: 10.1111/trf.13057
Glassing, A., Dowd, S. E., Galandiuk, S., Davis, B., and Chiodini, R. J. (2016). Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 8:24. doi: 10.1186/s13099-016-0103-7
Gosalbes, M., Llop, S., Valles, Y., Moya, A., Ballester, F., and Francino, M. (2013). Meconium microbiota types dominated by lactic acid or enteric bacteria are differentially associated with maternal eczema and respiratory problems in infants. Clin. Exp. Allergy. 43, 198–211. doi: 10.1111/cea.12063
Gosiewski, T., Ludwig-Galezowska, A., Huminska, K., Sroka-Oleksiak, A., Radkowski, P., Salamon, D., et al. (2016). Comprehensive detection and identification of bacterial DNA in the blood of patients with sepsis and healthy volunteers using next-generation sequencing method-the observation of DNAemia. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 1–8. doi: 10.1007/s10096-016-2805-7
Gresham, H. D., Lowrance, J. H., Caver, T. E., Wilson, B. S., Cheung, A. L., and Lindberg, F. P. (2000). Survival of Staphylococcus aureus inside neutrophils contributes to infection. J. Immunol. 164, 3713–3722. doi: 10.4049/jimmunol.164.7.3713
Grice, E. A., Kong, H. H., Conlan, S., Deming, C. B., Davis, J., Young, A. C., et al. (2009). Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192. doi: 10.1126/science.1171700
Hitti, J., Riley, D. E., Krohn, M. A., Hillier, S. L., Agnew, K. J., Krieger, J. N., et al. (1997). Broad-spectrum bacterial rDNA polymerase chain reaction assay for detecting amniotic fluid infection among women in premature labor. Clin. Infect. Dis. 24, 1228–1232. doi: 10.1086/513669
Horzempa, J., O'dee, D. M., Stolz, D. B., Franks, J. M., Clay, D., and Nau, G. J. (2011). Invasion of erythrocytes by Francisella tularensis. J. Infect. Dis. 204, 51–59. doi: 10.1093/infdis/jir221
Huffnagle, G. B., and Noverr, M. C. (2013). The emerging world of the fungal microbiome. Trends Microbiol. 21, 334–341. doi: 10.1016/j.tim.2013.04.002
Iwai, T. (2009). Periodontal bacteremia and various vascular diseases. J. Clin. Periodontol. 44, 689–694. doi: 10.1111/j.1600-0765.2008.01165.x
Jang, M. H., Kweon, M.-N., Iwatani, K., Yamamoto, M., Terahara, K., Sasakawa, C., et al. (2004). Intestinal villous M cells: an antigen entry site in the mucosal epithelium. Proc. Natl. Acad. Sci. U.S.A. 101, 6110–6115. doi: 10.1073/pnas.0400969101
Jiménez, E., Fernández, L., Marín, M. L., Martín, R., Odriozola, J. M., Nueno-Palop, C., et al. (2005). Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr. Microbiol. 51, 270–274. doi: 10.1007/s00284-005-0020-3
Jiménez, E., Marín, M. L., Martín, R., Odriozola, J. M., Olivares, M., Xaus, J., et al. (2008). Is meconium from healthy newborns actually sterile? Res. Microbiol. 159, 187–193. doi: 10.1016/j.resmic.2007.12.007
Khanna, S., and Tosh, P. K. (2014). A clinician's primer on the role of the microbiome in human health and disease. Mayo. Clin. Proc. 89, 107–114. doi: 10.1016/j.mayocp.2013.10.011
Kim, B.-S., Jeon, Y.-S., and Chun, J. (2013). Current status and future promise of the human microbiome. J. Pediatr. Gastroenterol. Nutr. 16, 71–79. doi: 10.5223/pghn.2013.16.2.71
Kowarsky, M., Camunas-Soler, J., Kertesz, M., De Vlaminck, I., Koh, W., Pan, W., et al. (2017). Numerous uncharacterized and highly divergent microbes which colonize humans are revealed by circulating cell-free DNA. Proc Natl Acad Sci U S A. 114, 9623–9628. doi: 10.1073/pnas.1707009114
Lauder, A. P., Roche, A. M., Sherrill-Mix, S., Bailey, A., Laughlin, A. L., Bittinger, K., et al. (2016). Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 4:29. doi: 10.1186/s40168-016-0172-3
Le Chatelier, E., Nielsen, T., Qin, J., Prifti, E., Hildebrand, F., Falony, G., et al. (2013). Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546. doi: 10.1038/nature12506
Lelouard, H., Fallet, M., de Bovis, B., Méresse, S., and Gorvel, J. P. (2012). Peyer's patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology 142, 592–601. e593. doi: 10.1053/j.gastro.2011.11.039
Lelouvier, B., Servant, F., Païssé, S., Brunet, A. C., Benyahya, S., Serino, M., et al. (2016). Changes in blood microbiota profiles associated with liver fibrosis in obese patients: a pilot analysis. Hepatology 64, 2015–2027. doi: 10.1002/hep.28829
Li, J., Jia, H., Cai, X., Zhong, H., Feng, Q., Sunagawa, S., et al. (2014). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 32, 834–841. doi: 10.1038/nbt.2942
Li, Q., Wang, C., Tang, C., Zhao, X., He, Q., and Li, J. (2018). Identification and characterization of blood and neutrophil-associated microbiomes in patients with severe acute pancreatitis using next-generation sequencing. Front. Cell Infect. Microbiol. 8:5. doi: 10.3389/fcimb.2018.00005
Lloyd-Price, J., Abu-Ali, G., and Huttenhower, C. (2016). The healthy human microbiome. Genome Med. 8:51. doi: 10.1186/s13073-016-0307-y
Loohuis, L. M. O., Mangul, S., Ori, A. P., Jospin, G., Koslicki, D., Yang, H. T., et al. (2018). Transcriptome analysis in whole blood reveals increased microbial diversity in schizophrenia. Transl. Psychiatry. 8:96. doi: 10.1038/s41398-018-0107-9
Mandal, R. K., Jian, T., Al-Rubaye, A. A., Rhoads, D. D., Wideman, R. F., Zhao, J., et al. (2016). An investigation into blood microbiota and its potential association with Bacterial Chondronecrosis with Osteomyelitis (BCO) in broilers. Sci. Rep. 6:25882. doi: 10.1038/srep25882
Manzo, V. E., and Bhatt, A. S. (2015). The human microbiome in hematopoiesis and hematologic disorders. Blood 126, 311–318. doi: 10.1182/blood-2015-04-574392
Martel, J., Wu, C.-Y., Huang, P.-R., Cheng, W.-Y., and Young, J. D. (2017). Pleomorphic bacteria-like structures in human blood represent non-living membrane vesicles and protein particles. Sci. Rep. 7:10650. doi: 10.1038/s41598-017-10479-8
McDole, J. R., Wheeler, L. W., McDonald, K. G., Wang, B., Konjufca, V., Knoop, K. A., et al. (2012). Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 483, 345–349. doi: 10.1038/nature10863
McLaughlin, R. W., Vali, H., Lau, P. C., Palfree, R. G., De Ciccio, A., Sirois, M., et al. (2002). Are there naturally occurring pleomorphic bacteria in the blood of healthy humans? J. Clin. Microbiol. 40, 4771–4775. doi: 10.1128/JCM.40.12.4771-4775.2002
Mitchell, A. J., Gray, W. D., Schroeder, M., Yi, H., Taylor, J. V., Dillard, R. S., et al. (2016). Pleomorphic structures in human blood are red blood cell-derived microparticles, not bacteria. PLoS ONE 11:e0163582. doi: 10.1371/journal.pone.0163582
Moeller, A. H., Caro-Quintero, A., Mjungu, D., Georgiev, A. V., Lonsdorf, E. V., Muller, M. N., et al. (2016). Cospeciation of gut microbiota with hominids. Science 353, 380–382. doi: 10.1126/science.aaf3951
Moles, L., Gómez, M., Heilig, H., Bustos, G., Fuentes, S., de Vos, W., et al. (2013). Bacterial diversity in meconium of preterm neonates and evolution of their fecal microbiota during the first month of life. PLoS ONE 8:e66986. doi: 10.1371/journal.pone.0066986
Molina, D. K., and DiMaio, V. J. (2012). Normal organ weights in men: part I-the heart. Am. J. Forensic Med. Pathol. 33, 362–367. doi: 10.1097/PAF.0b013e31823d298b
Morgan, X. C., Tickle, T. L., Sokol, H., Gevers, D., Devaney, K. L., Ward, D. V., et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13:R79. doi: 10.1186/gb-2012-13-9-r79
Moriyama, K., Ando, C., Tashiro, K., Kuhara, S., Okamura, S., Nakano, S., et al. (2008). Polymerase chain reaction detection of bacterial 16S rRNA gene in human blood. Microbiol. Immunol. 52, 375–382. doi: 10.1111/j.1348-0421.2008.00048.x
Moustafa, A., Xie, C., Kirkness, E., Biggs, W., Wong, E., Turpaz, Y., et al. (2017). The blood DNA virome in 8,000 humans. PLoS Pathog. 13:e1006292. doi: 10.1371/journal.ppat.1006292
Niess, J. H., Brand, S., Gu, X., Landsman, L., Jung, S., McCormick, B. A., et al. (2005). CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307, 254–258. doi: 10.1126/science.1102901
Nikkari, S., McLaughlin, I. J., Bi, W., Dodge, D. E., and Relman, D. A. (2001). Does blood of healthy subjects contain bacterial ribosomal DNA? J. Clin. Microbiol. 39, 1956–1959. doi: 10.1128/JCM.39.5.1956-1959.2001
Païssé, S., Valle, C., Servant, F., Courtney, M., Burcelin, R., Amar, J., et al. (2016). Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 56, 1138–1147. doi: 10.1111/trf.13477
Panaiotov, S., Filevski, G., Equestre, M., Nikolova, E., and Kalfin, R. (2018). Cultural isolation and characteristics of the blood microbiome of healthy individuals. Adv. Microbiol. 8:406. doi: 10.4236/aim.2018.85027
Parracho, H. M., Bingham, M. O., Gibson, G. R., and McCartney, A. L. (2005). Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J. Med. Microbiol. 54, 987–991. doi: 10.1099/jmm.0.46101-0
Pease, P. (1970). Morphological appearances of a bacterial L-form growing in association with the erythrocytes of arthritic subjects. Ann. Rheum. Dis. 29:439. doi: 10.1136/ard.29.4.439
Penders, J., Thijs, C., Vink, C., Stelma, F. F., Snijders, B., Kummeling, I., et al. (2006). Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521. doi: 10.1542/peds.2005-2824
Petersen, C., and Round, J. L. (2014). Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 16, 1024–1033. doi: 10.1111/cmi.12308
Pistollato, F., Cano, S. S., Elio, I., Vergara, M. M., Giampieri, F., and Battino, M. (2016). Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 74, 624–634. doi: 10.1093/nutrit/nuw023
Potgieter, M., Bester, J., Kell, D. B., and Pretorius, E. (2015). The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol. Rev. 39, 567–591. doi: 10.1093/femsre/fuv013
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59. doi: 10.1038/nature08821
Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F., et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. doi: 10.1038/nature11450
Qiu, J., Zhou, H., Jing, Y., and Dong, C. (2019). Association between blood microbiome and type 2 diabetes mellitus: a nested case-control study. J. Clin. Lab. Anal. e22842. doi: 10.1002/jcla.22842
Rascovan, N., Duraisamy, R., and Desnues, C. (2016). Metagenomics and the human virome in asymptomatic individuals. Annu. Rev. Microbiol. 70, 125–141. doi: 10.1146/annurev-micro-102215-095431
Rescigno, M., Urbano, M., Valzasina, B., Francolini, M., Rotta, G., Bonasio, R., et al. (2001). Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2:86373. doi: 10.1038/86373
Romano-Keeler, J., and Weitkamp, J.-H. (2014). Maternal influences on fetal microbial colonization and immune development. Pediatr. Res. 77, 189–195. doi: 10.1038/pr.2014.163
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Sato, J., Kanazawa, A., Ikeda, F., Yoshihara, T., Goto, H., Abe, H., et al. (2014). Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diabetes Care. 37, 2343–2350. doi: 10.2337/dc13-2817
Schierwagen, R., Alvarez-Silva, C., Madsen, M. S. A., Kolbe, C. C., Meyer, C., Thomas, D., et al. (2018). Circulating microbiome in blood of different circulatory compartments. Gut. 2018:gutjnl-2018-316227. doi: 10.1136/gutjnl-2018-316227
Segata, N., Boernigen, D., Tickle, T. L., Morgan, X. C., Garrett, W. S., and Huttenhower, C. (2013). Computational meta'omics for microbial community studies. Mol. Syst. Biol. 9:666. doi: 10.1038/msb.2013.22
Smits, S. A., Leach, J., Sonnenburg, E. D., Gonzalez, C. G., Lichtman, J. S., Reid, G., et al. (2017). Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 357, 802–806. doi: 10.1126/science.aan4834
Steel, J. H., Malatos, S., Kennea, N., Edwards, A. D., Miles, L., Duggan, P., et al. (2005). Bacteria and inflammatory cells in fetal membranes do not always cause preterm labor. Pediatr. Res. 57, 404–411. doi: 10.1203/01.PDR.0000153869.96337.90
Stremlau, M. H., Andersen, K. G., Folarin, O. A., Grove, J. N., Odia, I., Ehiane, P. E., et al. (2015). Discovery of novel rhabdoviruses in the blood of healthy individuals from West Africa. PLoS Negl. Trop. Dis. 9:e0003631. doi: 10.1371/journal.pntd.0003631
Sze, M. A., Tsuruta, M., Yang, S. W., Oh, Y., Man, S. F., Hogg, J. C., et al. (2014). Changes in the bacterial microbiota in gut, blood, and lungs following acute LPS instillation into mice lungs. PLoS ONE 9:e111228. doi: 10.1371/journal.pone.0111228
Tedeschi, G., Amici, D., and Paparelli, M. (1969). Incorporation of nucleosides and amino-acids in human erythrocyte suspensions: possible relation with a diffuse infection of mycoplasms or bacteria in the L form. Nature 222, 1285–1286. doi: 10.1038/2221285a0
Teo, S. M., Mok, D., Pham, K., Kusel, M., Serralha, M., Troy, N., et al. (2015). The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe 17, 704–715. doi: 10.1016/j.chom.2015.03.008
Thorburn, A. N., Macia, L., and Mackay, C. R. (2014). Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity 40, 833–842. doi: 10.1016/j.immuni.2014.05.014
Thwaites, G. E., and Gant, V. (2011). Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat. Rev. Microbiol. 9:215. doi: 10.1038/nrmicro2508
Traykova, D., Schneider, B., Chojkier, M., and Buck, M. (2017). Blood microbiome quantity and the hyperdynamic circulation in decompensated cirrhotic patients. PLoS ONE. 12:e0169310. doi: 10.1371/journal.pone.0169310
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
Vazquez-Torres, A., Jones-Carson, J., Bäumler, A. J., Falkow, S., Valdivia, R., Brown, W., et al. (1999). Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature 401, 804–808. doi: 10.1038/44593
Vientós-Plotts, A. I., Ericsson, A. C., Rindt, H., Grobman, M. E., Graham, A., Bishop, K., et al. (2017). Dynamic changes of the respiratory microbiota and its relationship to fecal and blood microbiota in healthy young cats. PLoS ONE. 12:e0173818. doi: 10.1371/journal.pone.0173818
Vitry, M. A., Mambres, D. H., Deghelt, M., Hack, K., Machelart, A., Lhomme, F., et al. (2014). Brucella melitensis invades murine erythrocytes during infection. Infect. Immun. 82, 3927–3938. doi: 10.1128/IAI.01779-14
Whittle, E., Leonard, M. O., Harrison, R., Gant, T. W., and Tonge, D. P. (2018). Multi-method characterization of the human circulating microbiome. Front. Microbiol. 9:3266. doi: 10.3389/fmicb.2018.03266
Yamaguchi, H., Yamada, M., Uruma, T., Kanamori, M., Goto, H., Yamamoto, Y., et al. (2004). Prevalence of viable Chlamydia pneumoniae in peripheral blood mononuclear cells of healthy blood donors. Transfusion 44, 1072–1078. doi: 10.1111/j.1537-2995.2004.04005.x
Yamaguchi, M., Terao, Y., Mori-Yamaguchi, Y., Domon, H., Sakaue, Y., Yagi, T., et al. (2013). Streptococcus pneumoniae invades erythrocytes and utilizes them to evade human innate immunity. PLoS ONE 8:e77282. doi: 10.1371/journal.pone.0077282
Keywords: human blood microbiome, bacteria, dysbiosis, disease, contamination
Citation: Castillo DJ, Rifkin RF, Cowan DA and Potgieter M (2019) The Healthy Human Blood Microbiome: Fact or Fiction? Front. Cell. Infect. Microbiol. 9:148. doi: 10.3389/fcimb.2019.00148
Received: 18 January 2019; Accepted: 23 April 2019;
Published: 08 May 2019.
Edited by:
Benoit Chassaing, Georgia State University, United StatesReviewed by:
Barbara Methe, University of Pittsburgh, United StatesKarl M. Thompson, Howard University, United States
Copyright © 2019 Castillo, Rifkin, Cowan and Potgieter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marnie Potgieter, marniepotgieter@yahoo.com