The Long and Short of Next Generation Sequencing for Cryptosporidium Research
 Tapoka T. Mkandawire
Tapoka T. Mkandawire Adam Sateriale
Adam Sateriale- Laboratory of Host-Pathogen Interactions in Cryptosporidiosis, Francis Crick Institute, London, United Kingdom
The intestinal parasite Cryptosporidium is a significant cause of severe diarrhoeal disease that can have long term effects. Therapeutic options remain limited despite a significant impact on public health, partly due to various challenges in the field of Cryptosporidium research, including the availability of genomic and transcriptomic data from environmental and clinical isolates. In this review we explore how long read DNA and RNA sequencing technologies have begun to provide novel insights into the biology of the parasite. The increased deployment of these technologies will help researchers address key gaps in the understanding of Cryptosporidium biology, and ultimately drive translational research and better parasite control.
Introduction
Cryptosporidium is an intracellular parasite that is an important cause of global diarrhoeal disease in animals and humans. Infection in animals, particularly livestock, results in increased agricultural cost and decreased production (Shaw et al., 2020). In humans, children and immunocompromised patients carry a disproportionate burden of disease and early life infections have been shown to be associated with stunted growth (Khalil et al., 2018). Acute disease accounts for 4.22 million disability adjusted life years (DALYs) annually, yet the chronic effects of disease, such as growth stunting, are estimated to account for a further 7.85 million DALYs (Khalil et al., 2018). Despite this substantial impact on public health, there are no fully effective treatments and no available vaccinations against disease (Schneider et al., 2021).
The life cycle of the Cryptosporidium parasite is complex and progresses through several morphologies – the infective form of the parasite is the oocyst, which is an environmentally hardy form that transmits via a faecal-oral route. Oocysts that are ingested by the host release four motile forms of the parasite, sporozoites, that invade epithelial cells that line the host intestinal tract. Once inside the epithelial cell, the parasite undergoes asexual replication, known as schizogony or merogony, before making a sexual commitment and undergoing sexual replication, known as gametogony. These sexual forms then come together to create new infective oocysts that are released into the environment in the host faeces (Current and Reese, 1986).
Cryptosporidium is a prominent infection in vertebrates and there are over 35 recognised species with varying host specificity (Feng et al., 2018). Infection in humans is primarily driven by two species: Cryptosporidium hominis and Cryptosporidium parvum. To date, fifteen unique genome assemblies of Cryptosporidium have been generated and eight genomes annotated (Warrenfeltz et al., 2020). The inconsistent availability and quality of Cryptosporidium reference genomes has not only impacted our understanding of the basic biology of the parasite, but translational advances in surveillance, diagnostics, and therapeutics have also been lagging. For many Cryptosporidium genomes significant time has elapsed since they were initially sequenced and assembled. These references may require updated sequencing with next generation technologies, or genome re–annotation and polishing to ensure that as reference genomes they do not mislead interpretations during genomic or molecular studies (Baptista et al., 2022). A greater understanding of the parasite genome in silico will complement recent in vivo and in vitro advances that have increased our understanding of parasite and infection biology (Marzook and Sateriale, 2020). In this review we describe some of the many challenges facing the Cryptosporidium research community, and we explore the ways recent advances in sequencing, specifically long read technologies, have begun and will continue to address them.
Challenges in Cryptosporidium Research
Historically, Cryptosporidium research has suffered from a lack of suitable models to study infection. However, recent developments have started to turn the tides and expand the capabilities of researchers. Stem-cell based models of infection, such as the organoid (Heo et al., 2018) and air-liquid interface models (Wilke et al., 2019), more closely recapitulate the cellular diversity and architecture of the gut, allowing for more translational studies of infection. A recently developed mouse model of cryptosporidiosis also offers a fully genetically tractable system that replicates human pathology in vivo (Sateriale et al., 2019). Despite these advances, there is still no reliable method for continuous culture of the Cryptosporidium parasite. In vitro culturing is still limited to pre-fertilisation stages as parasites are unable to complete sexual reproduction using traditional cell culture methods (Tandel et al., 2019). These culturing constraints mean that clonal populations of parasites cannot be generated, and this is particularly pertinent for genomic studies.
The Cryptosporidium genome is very compact at ~9 Mb and the parasite scavenges nutrients from the host, obscuring metabolic pathways. The parasite also uses alternative splice forms, making it a challenge to validate and annotate genes and their functions (Baptista et al., 2022). As a result, many genome assemblies contain large percentages of hypothetical or uncharacterised proteins (Table 1). Oocysts are often sampled as the most readily available life cycle stage, however they only display minute morphological differences at the species level. Individuals in endemic regions are often co-infected with multiple strains or species and these differences can only be observed genetically (Ryan et al., 2021). Importantly, as each oocyst contains four sporozoites, an individual oocyst is considered a mixed population. Even single cell sequencing only partially addresses concerns around mixed populations, but does facilitate the detection of low frequency variation (Troell et al., 2016; Baptista et al., 2021). Genetic recombination occurs during sexual replication generating diversity that influences virulence and transmissibility. Establishing clonal cultures of isolates would address some of the challenges in deconvoluting this diversity.
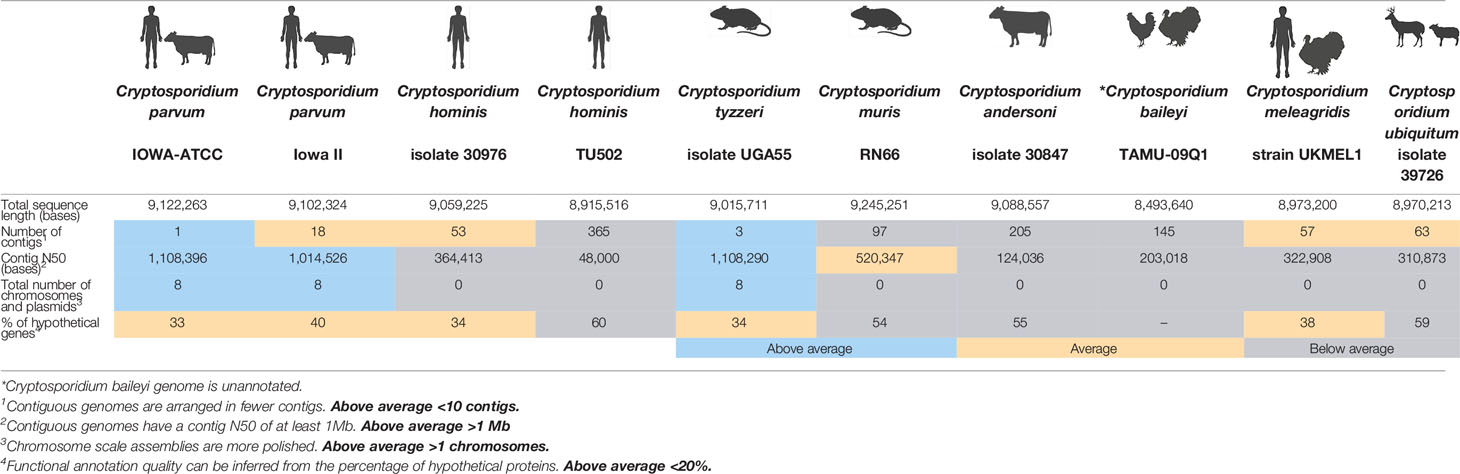
Table 1 Genome assembly statistics for Cryptosporidium species.
Long Read Genomics to Improve Our Understanding of Parasite Virulence and Transmission
Despite the decreasing cost and increasing relative ease of sequencing it is an underutilised tool in Cryptosporidium research; and for many years, the field has relied on the C. parvum and C. hominis genomes generated in 2004 through whole genome shotgun Sanger sequencing and HAPPY mapping (Abrahamsen et al., 2004; Xu et al., 2004). Annotation remains a resource and labour intensive process and while there have been several re-annotations of the C. parvum genome, it was only recently updated using a combination of Pacific Biosciences long reads and Illumina DNA and RNA short reads. This combined approach significantly improved the genome assembly quality across key metrics– including contiguity, completeness, and correctness, and generated a high quality C. parvum IOWA-ATCC reference genome (Baptista et al., 2022). This revised genome has already revealed several new insights into the parasite’s biology, including the identification of new parasite transporters. One surprising find from this reannotation is the amount of copy number variation in the Cryptosporidium genome (Figure 1). Variations in copy number have been shown to affect gene expression and increase phenotypic variation (Freeman et al., 2006), and in C. parvum it is thought to contribute to phenotypes such as host specificity and sporozoite invasion (Zhang et al., 2019). DNA long reads– especially through intergenic and repeat regions in the genome, can help resolve artificial compression of the genome and reveal variations in copy number (Logsdon et al., 2020). The recent updates to the C. parvum genome revealed at least 13 genes with variations in copy number, including the recently identified MEDLE family of host-exported virulence factors (Dumaine et al., 2021). This reannotation of the C. parvum genome is an exemplary case and the quality of the Cryptosporidium reference genomes varies greatly across species, therefore more concerted efforts are needed to generate high quality references of the other species (Table 1). In particular, C. hominis as the other prominent causative agent of human disease and C. tyzzeri as a murine model of human infection, are both desperately in need of a reannotation. The C. tyzzeri genome specifically, illustrates how current strategies using the limited genomic data available can present a false confidence in the quality of a genome assembly. The current C. tyzzeri assembly (Sateriale et al., 2019) relies heavily on C. parvum data to scaffold the genome and this cross species assembly can lead to a loss of information, especially translocations and inversions. Increased use of long read sequencing across Cryptosporidium species will address these inaccuracies and lead to more informative assemblies.
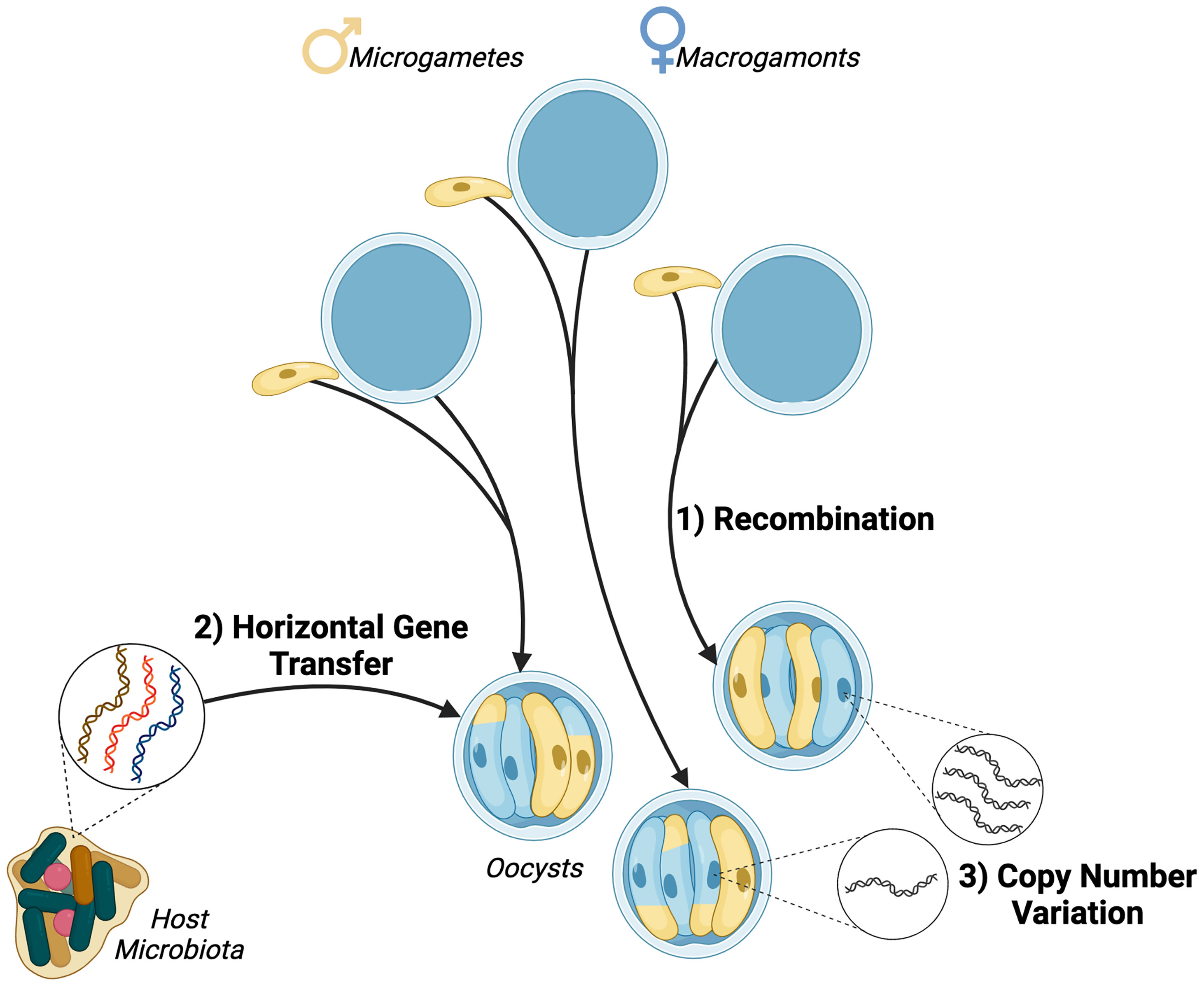
Figure 1 Sources of genetic diversity in Cryptosporidium parasites. 1) Recombination occurs during sexual replication and meiotic divisions distribute parental alleles, resulting in recombinant progeny. 2) Horizontal gene transfer of bacterial genes has been observed in Cryptosporidium species suggesting a link with the host gastrointestinal microbiota. 3) Copy number variation has been observed in at least 13 Cryptosporidium parvum genes with implications for parasite host range and invasion.
Understanding transmission hinges on the ability to identify and differentiate different genotypes during surveillance. Currently, genes such as gp60 and 18S rRNA are used for single locus typing (SLT) and a variety of markers are selected on a study by study basis for multilocus sequence typing (MLST) (Robinson and Chalmers, 2012). SLT is not a robust strategy that can account for the effects of recombination during sexual stages. Additionally, the gene currently used for much of single locus typing– gp60, is a virulence gene and therefore under selective pressure which may affect its suitability as a marker gene (Morris et al., 2019). Indeed the lack of global geographical sub structuring observed when comparing gp60 alleles in isolates from different locations demonstrates that the selection pressures driving gp60 evolution mean it cannot be used for universal single locus typing (Widmer, 2009; Robinson and Chalmers, 2012). In order to perform MLST markers are selected and used in single studies, because when applied to isolates from different geographical regions the performance of these panels declines significantly (Robinson and Chalmers, 2012). Furthermore, isolates identified as the same species through 18S rRNA typing can display differing phenotypes and even different host specificity; together with the knowledge that multiple copies of this gene are present across the genome, this suggests that copy number variation (CNV) or variation at other loci will prove to be more informative (Nader et al., 2019; Baptista et al., 2022). It is important to address these incongruencies moving past our reliance on outdated strategies such as SLT, and developing more broadly applicable MLST panels. Improved genomic sequencing will provide the greater sequence coverage of multiple loci required to fully resolve and understand population.
Another major challenge for the surveillance and population genomics of Cryptosporidium is deconvolution. Individuals in endemic regions can present mixed strain and species infections (Ryan et al., 2021) and, without a consensus of marker genes, current strategies struggle to resolve the structure of subpopulations of the same species (Robinson and Chalmers, 2012; Baptista et al., 2021). When investigating subpopulations, the diversity generated by recombination events during sexual replication means that mixed populations of sporozoites can exist in a single oocyst (Figure 1) (Dettwiler et al., 2021). Additionally, Cryptosporidium spp. have acquired large quantities of genes through horizontal gene transfer (Figure 1) (Huang et al., 2004). In order to fully understand the movement of genes into and within parasite populations more high-quality sequences are needed.
Long Read Transcriptomics to Improve Our Understanding of Parasite Biology and Gene Regulation
Advances in genomics and transcriptomics go hand in hand, and combining long and short read DNA sequencing with short read RNA sequencing (RNAseq) has already improved the annotation of the C. parvum, C. hominis, and C. tyzzeri genomes (Baptista et al., 2022). Available strand specific RNAseq data has revealed some information regarding splicing mechanisms and isoforms in Cryptosporidium (Li et al., 2020). The long non-coding RNAs (lncRNA) identified were predominantly antisense sense transcripts (91.7%) that covered nearly 10% of predicted mRNA transcripts and were primarily located at the 3’ end of the sense mRNA (Li et al., 2020). Additionally, these data provided evidence of splicing in lncRNA and suggested the presence of bidirectional promoters (Li et al., 2020), indicating that the parasite has mechanisms for complex control of gene expression (Gil and Ulitsky, 2018). In organisms with compact genomes, like Cryptosporidium, which is only 20% intergenic, utilising RNAseq to annotate and validate genes can be challenging (Baptista et al., 2022). Resolving genes that are in close proximity through transcriptome assembly using short reads can often result in the artificial fusion of exons (Xie et al., 2016). Furthermore, short read RNAseq cannot provide information regarding concurrent alternative or rare splicing isoforms (Lee et al., 2021). There are six modes of alternative splicing (exon skipping, intron retention, constitutive splicing, mutually exclusive exons & alternative 3′ or 5’ splice sites) that are observed at varied frequencies in organisms, with intron retention most frequently detected in protozoa and lower metazoans, and exon skipping in higher metazoans (Wang et al., 2015; Yeoh et al., 2019). Long-read technologies such as Pacific Biosciences (PacBio) IsoSeq and Oxford Nanopore (ONT) direct RNA sequencing could be used to examine alternative splicing aiding in the verification and annotation of genes, and identification novel splice forms. In addition to limited information regarding splicing there are currently, no models for nucleotide modification in Cryptosporidium species and there are mixed reports on the evidence of methylation sites in the genome (Gissot et al., 2008; Aliaga et al., 2019). However, with the discovery of alternative methylation models in other apicomplexans (Baumgarten et al., 2019) it is important to reinvestigate and identify these models in Cryptosporidium species and ONT direct RNAseq will allow researchers to do this.
Long-RNA reads have already revealed several insights into the biology of related apicomplexan parasites, Toxoplasma gondii and Plasmodium falciparum. Transcripts from both T. gondii and P. falciparum retain large quantities of intronic sequences (Lee et al., 2021). Interestingly, in these parasites, intron retention rarely corresponds to expanded protein expression through alternative protein products, instead retention of introns primarily renders transcripts untranslatable and marked for nonsense mediated decay (NMD) (Lee et al., 2021). A detailed understanding of the transcriptional landscape in any Cryptosporidium species is outstanding. Putative alternatively spliced genes have been identified in C. parvum and alternative splicing regulators identified in Cryptosporidium muris, yet these splice forms and mechanisms are yet to be experimentally validated (Yeoh et al., 2019; Baptista et al., 2022).
Discussion
The recent advances in sequencing technologies have expanded the capability of genomics and transcriptomics, and this can only be expected to increase as long-read sequencing is used on a population scale. Long read technologies not only increase resolution for haplotype collapsed reference genomes they can provide individual variant validation (Logsdon et al., 2020) and increased use of long read technologies on both these fronts will significantly aid Cryptosporidium research. Generating high quality reference genomes of more Cryptosporidium species will facilitate the identification of robust marker genes that can be used for MLST of isolates during general surveillance and clinical diagnosis (Morris et al., 2019). Individual variant resolution using long-read sequencing will help us characterise isolates, identifying genes and structural variants that drive pathogenicity. One example of where this will be a particularly useful tool is in the investigations of C. tyzzeri. The study by Sateriale et al. isolated a wild strain of the parasite that causes disease in laboratory mice that recapitulates human pathology (Sateriale et al., 2019). In contrast, Russler-Germain et al. identified a strain of C. tyzzeri that occurs commensally in laboratory mice (Russler-Germain et al., 2021). Genomic analysis of these variants (and others) may provide a genetic explanation for the observed differences in pathology.
In addition to facilitating better identification and characterisation of acute infections that can inform and improve patient care, sequencing can inform epidemiological policy more broadly through better parasite surveillance. Generating tools and methods for deconvoluting inter- and intra- specific Cryptosporidium population structure is challenging due to its compact genome, sexual recombination, CNV, and horizontal gene transfer. However, it is imperative that we continue to explore these questions in order to identify the mechanisms behind observations like increased transmissibility and virulence, reinfection, and seasonality, learning from long read sequencing studies in Plasmodium (Lin et al., 2018; Runtuwene et al., 2018; Yang et al., 2021). Studies in other Apicomplexa species should inform our increased use of long-read DNA sequencing in Cryptosporidium and subsequent data sets will facilitate the development of enhanced diagnostics arrays and accurate genotyping.
Long-read RNAseq technologies have and will continue to reveal important insights into the transcriptional landscape of the genome and improve our annotation and validation of genes (Lee et al., 2021; Baptista et al., 2022). Studying complete transcripts can help increase our understanding of the contents of the UTRs; for example, helping us identify promoter sequences that can be used for genetic studies, and may form potential therapeutic targets. Long-read transcriptomics can also shed light on the abundance of splice isoforms across the life cycle, thereby increasing our understanding of what defines each stage and the transitions between them. For example, NMD splicing was found to be prevalent in T. gondii and P. falciparum (Lee et al., 2021), and while NMD is typically thought to be stochastic (Saudemont et al., 2017), it can be regulatory, and in Plasmodium and Toxoplasma alternative splicing modulates transitions between life cycle stages (Yeoh et al., 2019; Lee et al., 2021). Long-read transcriptomics across the different stages of the Cryptosporidium life cycle will likely help us understand the mechanisms that drive sexual commitment, possibly unlocking methods to continuously propagate the parasite, in vitro.
Cryptosporidium research has been hindered in the past by the inconsistent availability and quality of genomic and transcriptomic data. The continuing advances in sequencing technologies, particularly with regards to long-read DNA and RNA sequencing, are perfectly positioned to drive forward Cryptosporidium research. The wider use of such technologies will increase our understanding of the parasite biology and facilitating translational advances in surveillance, diagnostics, and therapeutics.
Author Contributions
TM and AS wrote this manuscript together. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from The Francis Crick Institute (https://www.crick.ac.uk/), which receives its core funding from Cancer Research UK, the UK Medical Research Council and the Wellcome Trust.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
All images created with BioRender.com.
References
Abrahamsen, M. S., Templeton, T. J., Enomoto, S., Abrahante, J. E., Zhu, G., Lancto, C. A., et al. (2004). Complete Genome Sequence of the Apicomplexan, Cryptosporidium Parvum. Science 304, 441–445. doi: 10.1126/science.1094786
Aliaga, B., Bulla, I., Mouahid, G., Duval, D., Grunau, C. (2019). Universality of the DNA Methylation Codes in Eucaryotes. Sci. Rep. 9, 173. doi: 10.1038/s41598-018-37407-8
Baptista, R. P., Cooper, G. W., Kissinger, J. C. (2021). Challenges for Cryptosporidium Population Studies. Genes 12, 1–12. doi: 10.3390/genes12060894
Baptista, R. P., Li, Y., Sateriale, A., Sanders, M. J., Brooks, K. L., Tracey, A., et al. (2022). Long-Read Assembly and Comparative Evidence-Based Reanalysis of Cryptosporidium Genome Sequences Reveal Expanded Transporter Repertoire and Duplication of Entire Chromosome Ends Including Subtelomeric Regions. Genome Res. 32, 203–213. doi: 10.1101/gr.275325.121
Baumgarten, S., Bryant, J. M., Sinha, A., Reyser, T., Preiser, P. R., Dedon, P. C., et al. (2019). Transcriptome-Wide Dynamics of Extensive M6a mRNA Methylation During Plasmodium Falciparum Blood-Stage Development. Nat. Microbiol. 4, 2246–2259. doi: 10.1038/s41564-019-0521-7
Current, W. L., Reese, N. C. (1986). A Comparison of Endogenous Development of Three Isolates of Cryptosporidium in Suckling Mice. J. Protozool. 33, 98–108. doi: 10.1111/j.1550-7408.1986.tb05567.x
Dettwiler, I., Troell, K., Robinson, G., Chalmers, R. M., Basso, W., Rentería-Solís, Z. M., et al. (2021). TIDE Analysis of Cryptosporidium Infections by Gp60 Typing Reveals Obscured Mixed Infections. J. Infect. Dis. 225, 685–695. doi: 10.1093/infdis/jiab417
Dumaine, J. E., Sateriale, A., Gibson, A. R., Reddy, A. G., Gullicksrud, J. A., Hunter, E. N., et al. (2021). The Enteric Pathogen Cryptosporidium Parvum Exports Proteins Into the Cytosol of the Infected Host Cell. Elife 10, 1–31. doi: 10.7554/eLife.70451
Feng, Y., Ryan, U. M., Xiao, L. (2018). Genetic Diversity and Population Structure of Cryptosporidium. Trends Parasitol. 34, 997–1011. doi: 10.1016/j.pt.2018.07.009
Freeman, J. L., Perry, G. H., Feuk, L., Redon, R., McCarroll, S. A., Altshuler, D. M., et al. (2006). Copy Number Variation: New Insights in Genome Diversity. Genome Res. 16, 949–961. doi: 10.1101/gr.3677206
Gil, N., Ulitsky, I. (2018). Production of Spliced Long Noncoding RNAs Specifies Regions With Increased Enhancer Activity. Cell Syst. 7, 537–547.e3. doi: 10.1016/j.cels.2018.10.009
Gissot, M., Choi, S.-W., Thompson, R. F., Greally, J. M., Kim, K. (2008). Toxoplasma Gondii and Cryptosporidium Parvum Lack Detectable DNA Cytosine Methylation. Eukaryot. Cell 7, 537–540. doi: 10.1128/EC.00448-07
Heo, I., Dutta, D., Schaefer, D. A., Iakobachvili, N., Artegiani, B., Sachs, N., et al. (2018). Modelling Cryptosporidium Infection in Human Small Intestinal and Lung Organoids. Nat. Microbiol. 3, 814–823. doi: 10.1038/s41564-018-0177-8
Huang, J., Mullapudi, N., Lancto, C. A., Scott, M., Abrahamsen, M. S., Kissinger, J. C. (2004). Phylogenomic Evidence Supports Past Endosymbiosis, Intracellular and Horizontal Gene Transfer in Cryptosporidium Parvum. Genome Biol. 5, R88. doi: 10.1186/gb-2004-5-11-r88
Khalil, I. A., Troeger, C., Rao, P. C., Blacker, B. F., Brown, A., Brewer, T. G., et al. (2018). Morbidity, Mortality, and Long-Term Consequences Associated With Diarrhoea From Cryptosporidium Infection in Children Younger Than 5 Years: A Meta-Analyses Study. Lancet Glob Health 6, e758–e768. doi: 10.1016/S2214-109X(18)30283-3
Lee, V. V., Judd, L. M., Jex, A. R., Holt, K. E., Tonkin, C. J., Ralph, S. A. (2021). Direct Nanopore Sequencing of mRNA Reveals Landscape of Transcript Isoforms in Apicomplexan Parasites. mSystems 6, 1–20. doi: 10.1128/mSystems.01081-20
Li, Y., Baptista, R. P., Sateriale, A., Striepen, B., Kissinger, J. C. (2020). Analysis of Long Non-Coding RNA in Cryptosporidium Parvum Reveals Significant Stage-Specific Antisense Transcription. Front. Cell. Infect. Microbiol. 10, 608298. doi: 10.3389/fcimb.2020.608298
Lin, J.-W., Reid, A. J., Cunningham, D., Böhme, U., Tumwine, I., Keller-Mclaughlin, S., et al. (2018). Genomic and Transcriptomic Comparisons of Closely Related Malaria Parasites Differing in Virulence and Sequestration Pattern. Wellcome Open Res. 3, 142. doi: 10.12688/wellcomeopenres.14797.1
Logsdon, G. A., Vollger, M. R., Eichler, E. E. (2020). Long-Read Human Genome Sequencing and its Applications. Nat. Rev. Genet. 21, 597–614. doi: 10.1038/s41576-020-0236-x
Marzook, N. B., Sateriale, A. (2020). Crypto-Currency: Investing in New Models to Advance the Study of Cryptosporidium Infection and Immunity. Front. Cell. Infect. Microbiol. 10, 587296. doi: 10.3389/fcimb.2020.587296
Morris, A., Robinson, G., Swain, M. T., Chalmers, R. M. (2019). Direct Sequencing of Cryptosporidium in Stool Samples for Public Health. Front. Public Health 7, 360. doi: 10.3389/fpubh.2019.00360
Nader, J. L., Mathers, T. C., Ward, B. J., Pachebat, J. A., Swain, M. T., Robinson, G., et al. (2019). Evolutionary Genomics of Anthroponosis in Cryptosporidium. Nat. Microbiol. 4, 826–836. doi: 10.1038/s41564-019-0377-x
Robinson, G., Chalmers, R. M. (2012). Assessment of Polymorphic Genetic Markers for Multi-Locus Typing of Cryptosporidium Parvum and Cryptosporidium Hominis. Exp. Parasitol. 132, 200–215. doi: 10.1016/j.exppara.2012.06.016
Runtuwene, L. R., Tuda, J. S. B., Mongan, A. E., Makalowski, W., Frith, M. C., Imwong, M., et al. (2018). Nanopore Sequencing of Drug-Resistance-Associated Genes in Malaria Parasites, Plasmodium Falciparum. Sci. Rep. 88286, 1–13. doi: 10.1038/s41598-018-26334-3
Russler-Germain, E. V., Jung, J., Miller, A. T., Young, S., Yi, J., Wehmeier, A., et al. (2021). Commensal Cryptosporidium Colonization Elicits a Cdc1-Dependent Th1 Response That Promotes Intestinal Homeostasis and Limits Other Infections. Immunity. 54, 2547–2564. doi: 10.1016/j.immuni.2021.10.002
Ryan, U., Zahedi, A., Feng, Y., Xiao, L. (2021). An Update on Zoonotic Cryptosporidium Species and Genotypes in Humans. Anim. (Basel) 11, 1–22. doi: 10.3390/ani11113307
Sateriale, A., Šlapeta, J., Baptista, R., Engiles, J. B., Gullicksrud, J. A., Herbert, G. T., et al. (2019). A Genetically Tractable, Natural Mouse Model of Cryptosporidiosis Offers Insights Into Host Protective Immunity. Cell Host Microbe 26, 135–146.e5. doi: 10.1016/j.chom.2019.05.006
Saudemont, B., Popa, A., Parmley, J. L., Rocher, V., Blugeon, C., Necsulea, A., et al. (2017). The Fitness Cost of Mis-Splicing is the Main Determinant of Alternative Splicing Patterns. Genome Biol. 18, 208. doi: 10.1186/s13059-017-1344-6
Schneider, A., Wendt, S., Lübbert, C., Trawinski, H. (2021). Current Pharmacotherapy of Cryptosporidiosis: An Update of the State-of-the-Art. Expert Opin. Pharmacother. 22, 2337–2342. doi: 10.1080/14656566.2021.1957097
Shaw, H. J., Innes, E. A., Morrison, L. J., Katzer, F., Wells, B. (2020). Long-Term Production Effects of Clinical Cryptosporidiosis in Neonatal Calves. Int. J. Parasitol. 50, 371–376. doi: 10.1016/j.ijpara.2020.03.002
Tandel, J., English, E. D., Sateriale, A., Gullicksrud, J. A., Beiting, D. P., Sullivan, M. C., et al. (2019). Life Cycle Progression and Sexual Development of the Apicomplexan Parasite Cryptosporidium Parvum. Nat. Microbiol. 4, 2226–2236. doi: 10.1038/s41564-019-0539-x
Troell, K., Hallström, B., Divne, A.-M., Alsmark, C., Arrighi, R., Huss, M., et al. (2016). Cryptosporidium as a Testbed for Single Cell Genome Characterization of Unicellular Eukaryotes. BMC Genomics 17, 471. doi: 10.1186/s12864-016-2815-y
Wang, Y., Liu, J., Huang, B. O., Xu, Y.-M., Li, J., Huang, L.-F., et al. (2015). Mechanism of Alternative Splicing and its Regulation. Biomed. Rep. 3, 152–158. doi: 10.3892/br.2014.407
Warrenfeltz, S., Kissinger, J. C., EuPathDB Team (2020). Accessing Cryptosporidium Omic and Isolate Data via CryptoDB.Org. Methods Mol. Biol. 2052, 139–192. doi: 10.1007/978-1-4939-9748-0_10
Widmer, G. (2009). Meta-Analysis of a Polymorphic Surface Glycoprotein of the Parasitic Protozoa Cryptosporidium Parvum and Cryptosporidium Hominis. Epidemiol. Infect. 137, 1800–1808. doi: 10.1017/s0950268809990215
Wilke, G., Funkhouser-Jones, L. J., Wang, Y., Ravindran, S., Wang, Q., Beatty, W. L., et al. (2019). A Stem-Cell-Derived Platform Enables Complete Cryptosporidium Development In Vitro and Genetic Tractability. Cell Host Microbe 26, 123–134.e8. doi: 10.1016/j.chom.2019.05.007
Xie, B., Yang, W., Ouyang, Y., Chen, L., Jiang, H., Liao, Y., et al. (2016). Two RNAs or DNAs may Artificially Fuse Together at a Short Homologous Sequence (SHS) During Reverse Transcription or Polymerase Chain Reactions, and Thus Reporting an SHS-Containing Chimeric RNA Requires Extra Caution. PloS One 11, e0154855. doi: 10.1371/journal.pone.0154855
Xu, P., Widmer, G., Wang, Y., Ozaki, L. S., Alves, J. M., Serrano, M. G., et al. (2004). The Genome of Cryptosporidium Hominis. Nature 431, 1107–1112. doi: 10.1038/nature02977
Yang, M., Shang, X., Zhou, Y., Wang, C., Wei, G., Tang, J., et al. (2021). Full-Length Transcriptome Analysis of Plasmodium Falciparum by Single-Molecule Long-Read Sequencing. Front. Cell. Infect. Microbiol. 11, 631545. doi: 10.3389/fcimb.2021.631545
Yeoh, L. M., Lee, V. V., McFadden, G. I., Ralph, S. A. (2019). Alternative Splicing in Apicomplexan Parasites. MBio 10, 1–15. doi: 10.1128/mBio.02866-18
Keywords: apicomplexa, long read sequencing, population genomics, gene expression, parasite control
Citation: Mkandawire TT and Sateriale A (2022) The Long and Short of Next Generation Sequencing for Cryptosporidium Research. Front. Cell. Infect. Microbiol. 12:871860. doi: 10.3389/fcimb.2022.871860
Received: 08 February 2022; Accepted: 04 March 2022;
Published: 28 March 2022.
Edited by:
William Harold Witola, University of Illinois at Urbana–Champaign, United StatesReviewed by:
Kun Li, Nanjing Agricultural University, ChinaTauqeer Alam, University of Illinois at Urbana-Champaign, United States
Jan Slapeta, The University of Sydney, Australia
Copyright © 2022 Mkandawire and Sateriale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adam Sateriale, adam.sateriale@crick.ac.uk