Interaction Between Autophagy and Porphyromonas gingivalis-Induced Inflammation
 Sen Kang
Sen Kang Anna Dai
Anna Dai Huiming Wang
Huiming Wang Pei-Hui Ding
Pei-Hui Ding- Stomatology Hospital, School of Stomatology, Zhejiang University School of Medicine, Clinical Research Center for Oral Diseases of Zhejiang Province, Key Laboratory of Oral Biomedical Research of Zhejiang Province, Zhejiang, China
Autophagy is an immune homeostasis process induced by multiple intracellular and extracellular signals. Inflammation is a protective response to harmful stimuli such as pathogen microbial infection and body tissue damage. Porphyromonas gingivalis infection elicits both autophagy and inflammation, and dysregulation of autophagy and inflammation promotes pathology. This review focuses on the interaction between autophagy and inflammation caused by Porphyromonas gingivalis infection, aiming to elaborate on the possible mechanism involved in the interaction.
Introduction
Periodontal disease (PD) is one of the most common chronic inflammatory oral diseases. It ranks as the sixth most prevalent disease worldwide (Kassebaum et al., 2017). PD can be mainly divided into two major categories: gingivitis and periodontitis. Periodontitis is the primary cause of tooth loss in adults. PD is initiated by gram-negative microorganisms in the sub-gingival plaque biofilms (Socransky et al., 1998). Among these microorganisms, Porphyromonas gingivalis (P. gingivalis) is a keystone pathogenic bacterium (Whitney et al., 1992; How et al., 2016; Ebersole et al., 2020). However, recent reports have suggested that periodontitis might raise the risk of several systematic inflammatory diseases, such as cardiovascular diseases (Humphrey et al., 2008), inflammatory bowel diseases (Jia et al., 2020), rheumatoid arthritis (Rutger Persson, 2012), Alzheimer’s disease (Singhrao et al., 2015), hepatitis (Yoneda et al., 2012) and et al. The links between them probably depends on the P. gingivalis’ ability of adhering and internalizing to the host cells (Hajishengallis and Lamont, 2014).
Autophagy is a self-degradative process. It is important in balancing energy and maintaining homeostasis in response to various stimuli, including bacterial infection (Krause et al., 2018), hypoxia (Cosin-Roger et al., 2017), reactive oxygen species (ROS) (Li et al., 2016), and endoplasmic reticulum (ER) stress (Periyasamy et al., 2016). Cellular contents such as microorganisms or damaged organelles can be delivered into lysosomes for degradation so that nutrients can be recycled by autophagy (Mizushima et al., 2008; Mizushima and Komatsu, 2011).
Autophagy has been linked with immune responses to defense against infection (Randow and Munz, 2012; Thurston et al., 2016). The type of antibacterial autophagy (xenophagy) can specifically target intracellular pathogens to lysosomes, and the involved mechanism relies on the cargo receptors, galectin 8 and ubiquitin (Thurston et al., 2012; Ravenhill et al., 2019). Autophagy plays a protective role in decreasing P. gingivalis-induced inflammation (Bullon et al., 2012; Park et al., 2017). When host cells are stimulated by P. gingivalis or its virulence factors, autophagy can inhibit the activation of inflammasomes such as NLR family pyrin domain containing 3 (NLRP3) and the secretion of pro-inflammatory cytokines such as IL-1β through degrading endogenous stimulus, negatively regulating inflammation and establishing cellular homeostasis (Harris et al., 2011; Takahama et al., 2018).
However, bacteria such as P. gingivalis have evolved a variety of strategies to survive in the host cells. For example, P. gingivalis can enter single membrane vesicles and prevent autophagosome fusion with the lysosome. In this way, P. gingivalis can utilize proteins or other nutrients in the autophagosome to provide energy for its survival (Belanger et al., 2006; Lee et al., 2018). The interaction between autophagy and bacterial infection and how it affects the immune response in host cells is unclear. This review focuses on the studies within the last five years on the interaction between autophagy, P. gingivalis infection, and inflammation and summarizes the possible involved mechanisms.
Autophagy Introduction
Autophagy can be divided into macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy. Only macroautophagy involves the formation of another organelle termed autophagosome (Yim and Mizushima, 2020). This review focuses on macroautophagy. Macroautophagy is divided into selective and non-selective autophagy, depending on whether the substrate is specifically targeted to the autophagosome (Kissova et al., 2007).
Non-Selective Autophagy Mechanisms
Non-selective autophagy is a non-specific process during which cytoplasmic components are recycled into autolysosomes for degradation and damaged organelles are removed under stress conditions to maintain cellular homeostasis. This type of autophagy involves a series of steps: initiation, nucleation, formation of the phagophore, phagophore elongation, fusion of the autophagosome with lysosomes, and degradation (Feng et al., 2014) (Figure 1). The formation of these structures is regulated by many signal pathways and related proteins are encoded by autophagy-related genes (ATGs). The process involves two complexes, the unc-51-like kinase (ULK) complex and class III phosphatidylinositol 3-kinase (PI3K) complex. It also includes two ubiquitin-like conjugations (UBL) systems, the Atg16L1 complex and microtubule-associated protein 1 light chain 3 (LC3) (Mizushima and Komatsu, 2011; Mizushima and Levine, 2020).
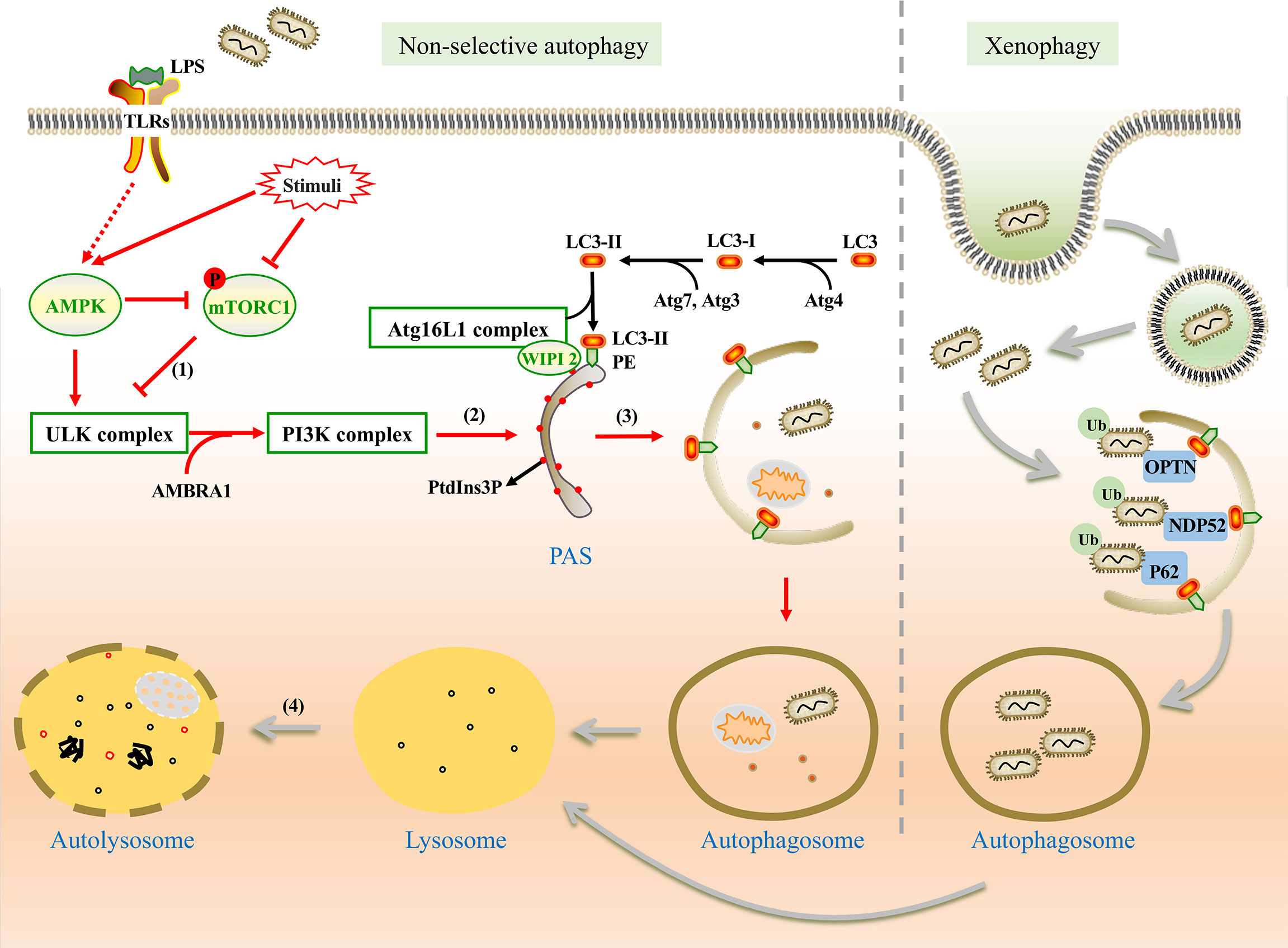
Figure 1 The mechanism of non-selective and xenophagy. Non-selective autophagy (Left) is initiated by the stimulation of LPS. (1) Under the stimulation, AMPK is activated and mTORC1 is inhibited. Thus, the ULK1 complex is activated and phosphorylates the PI3K complex. (2) WIPI2 recruits the Atg16L1 complex to the pre-autophagosomal structure (PAS) by the PtdIns3P binding protein and promotes the conjugation between LC3-II and phosphatidylethanolamine (PE). (3) Autophagic membrane is expanded and completed. Damaged organelles, pathogens, and other components are encapsulated in autophagosomes without selection. (4) Autophagosome fuses with lysosome for degradation of the substances in vesicles. Xenophagy (Right) can capture bacteria selectively. With the help of ubiquitin-binding protein adaptors, such as p62, OPTN, and NDP52, ubiquitinated bacteria are captured by autophagosome for degradation.
The upstream regulators of non-selective autophagy are the mammalian target of rapamycin complex 1 (mTORC1) and adenosine monophosphate-activated protein kinase (AMPK). Under nutrient-rich conditions, mTORC1 inhibits autophagy by phosphorylating ULK1 (Ganley et al., 2009). However, as a positive regulator, AMPK can directly activate autophagy by phosphorylating ULK1 at specific sites or indirectly activate autophagy by impairing mTORC1 (Yang and Klionsky, 2010; Fan et al., 2015).
Under stress conditions, autophagy is initiated by the assembly of the ULK complex which phosphorylates Beclin-1-regulated autophagy protein 1 (AMBRA1) and leads to the activation of the PI3K complex (Li et al., 2016). PI3K complex promotes the formation of phosphatidylinositol-3-phosphate (PtdIns3P) on the endoplasmic reticulum (ER), Golgi intermediate, or other membrane structures to mediate membrane nucleation (Maho et al., 2013). The Atg16L1 complex is recruited to the pre-autophagosomal structure (PAS) by PtdIns3P binding protein such as WIPI2 (Polson et al., 2010; Dooley et al., 2014). LC3 is processed into LC3-II with the assistance of Atg7, Atg4, and Atg3 (Glick et al., 2010), and then LC3-II binds with phosphatidylethanolamine (PE), which is essential for the expansion and completion of the autophagic membrane (Mizushima et al., 1998; Glick et al., 2010). Finally, the autophagosome fuses with the lysosome to form an autolysosome with the involvement of some proteins, including the small GTPase Rab7, syntaxin 17, vesicle-associated membrane protein (VAMP7, VAMP8) (Jager et al., 2004; Itakura et al., 2012; Wang et al., 2016).
Selective Autophagy (Xenophagy Mechanisms)
Evidence indicates that selective autophagy can degrade specific proteins, organelles, and invading bacteria. According to the specific targeted substrates, selective autophagy can be classified as xenophagy (Huang and Brumell, 2014; Castrejon-Jimenez et al., 2015), mitophagy (Zhong et al., 2016), ER-phagy (Bernales et al., 2006), and lipophagy (Singh et al., 2009). Among them, xenophagy acts as anti-bacterial autophagy that plays a crucial role in eliminating intracellular bacterial (Wang and Li, 2020).
Unlike nutrient starvation-induced non-selective autophagy, ubiquitination is more important in xenophagy rather than the regulation of mTORC1 and AMPK (Shaid et al., 2013; Losier et al., 2019). Ubiquitinated bacteria could be captured by ubiquitin-binding protein adaptors such as sequestosome 1 (SQSTM1/p62) (Zheng et al., 2009), nuclear domain 10 protein 52 (NDP52) (Li et al., 2013), optineurin (OPTN) (Wild et al., 2011), and et al. These adaptors interact with LC3, which promotes the formation of autophagosome (Sharma et al., 2018; Wu and Li, 2019). Similar to non-selective autophagy, the fate of autophagosomes is to fuse with lysosomes for degradation (Figure 1).
It is supposed that P. gingivalis and its virulence factors are tagged with ubiquitin, which allows them to bind to the adaptor proteins NDP52 and p62 (Lee et al., 2018). These adaptor proteins bind to LC3, thereby ensuring the bacteria be tethered and trapped in the autolysosome.
P. gingivalis Introduction
P. gingivalis expresses diverse virulence factors, such as lipopolysaccharide (LPS), fimbriae, capsule, gingipains, haemagglutinins, and other surface structures (How et al., 2016). Among them, P. gingivalis LPS and fimbriae are two special virulence factors in triggering inflammation, for their structure is variable. LPS consists of three parts: lipid A, core oligosaccharide, and O-antigen. Among them, lipid A is the core component and effector of LPS (Ogawa and Yagi, 2010). P. gingivalis expresses two forms of fimbriae, including long fimbriae, and short fimbriae. Long fimbriae contain FimA subunits protein and short fimbriae are comprised of Mfa1 subunits (Umemoto and Hamada, 2003). Considering the heterogeneity of lipid A and fimbriae, P. gingivalis can induce different immune responses and even escape from immune surveillance and attack (Bostanci and Belibasakis, 2012).
The Interaction Between Autophagy and Infection
Autophagy majorly plays a protective role in the elimination of intracellular bacteria. Upon infection, p62 or other autophagy receptors are recruited to invading ubiquitinated bacteria and delivered them to autophagosome (Zheng et al., 2009). Thurston found that pattern recognition or danger receptors, such as galectin 8 could label invading Salmonella as autophagy cargo and activate xenophagy by recruiting NDP52 (Thurston et al., 2012). Both autophagy receptor p62 and NDP52 could detect ubiquitin-coated bacteria in the cytosol and deliver them into autophagosomes for degradation (Thurston et al., 2009; Randow, 2011).
However, bacteria have evolved to manipulate the autophagic way to escape from immune attacks and utilize autophagy for nutrients (Randow and Munz, 2012; Bah and Vergne, 2017). For example, L. pneumophila could inhibit autophagy flux by cleaving syntaxin-17, a key SNARE protein implicated in autophagosome formation (Arasaki et al., 2017). Besides L. pneumophila could also inhibit the formation of autophagosomes by cleaving phosphatidylethanolamine-conjugated LC3 (Choy et al., 2012). The two mechanisms involved in the cleavage of autophagy machinery were respectively achieved by two specific proteases, Lpg1137 and RavZ (Choy et al., 2012; Arasaki et al., 2017). Canonical macroautophagy limits the growth of bacterial replication. But Steele et al. (Steele et al., 2013) found that F. tularensis could induce an Atg5-independent autophagy pathway which was beneficial for cytoplasmic F. tularensis growth by supplying nutrients in primary human monocytes. F. tularensis growth was significantly decreased by treatment with 3-MA and it was rescued by supplementing excess amino acids. Therefore, autophagy could provide amino acids that support F. tularensis growth (Steele et al., 2013).
In summary, the interaction between bacterial pathogens and the infected host cell determines the former’s survival or elimination. The involved mechanism varies in different bacterial and host cells, which are listed in Table 1.
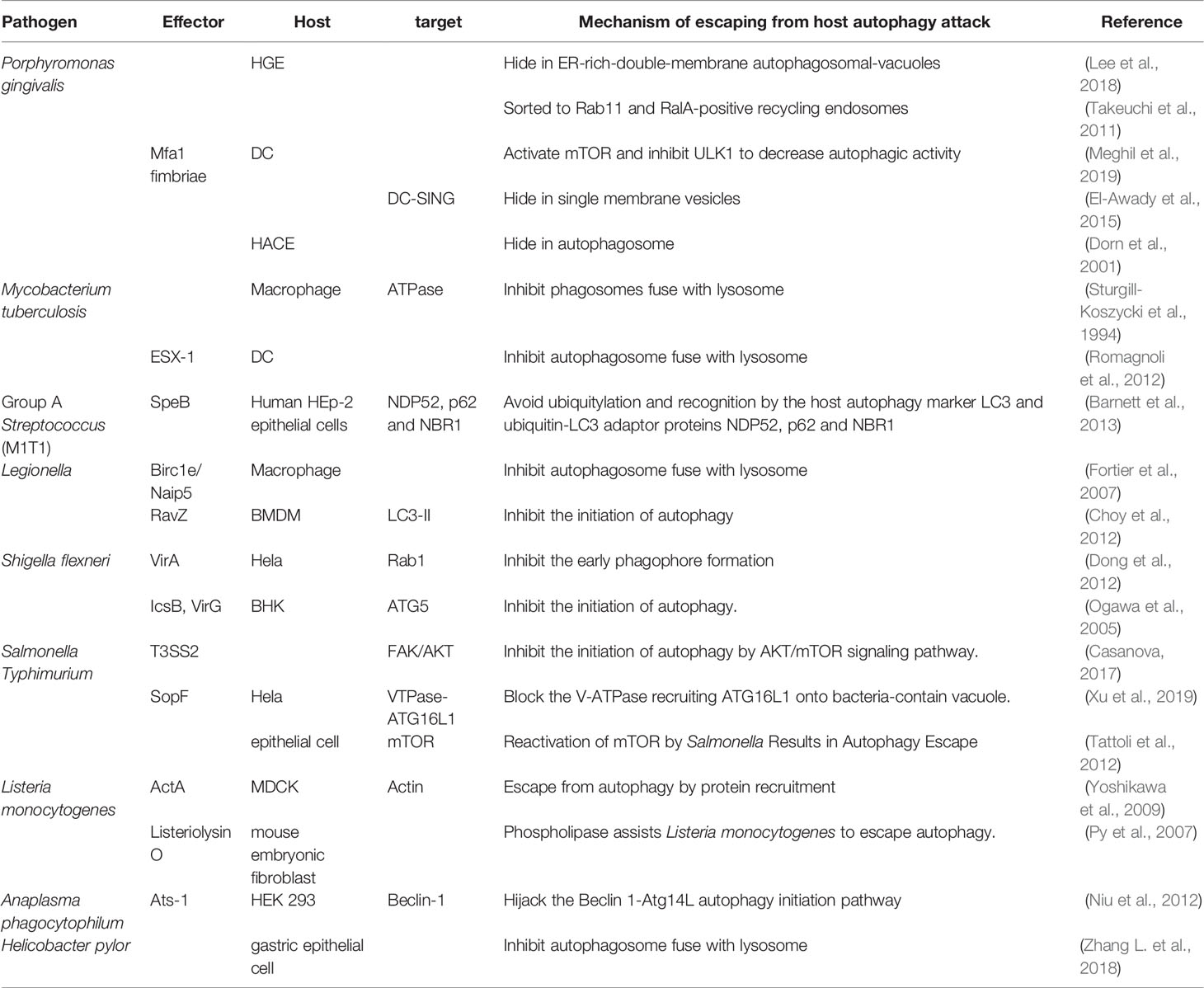
Table 1 Summary of the mechanisms how the bacteria interact with autophagy.
The Interaction Between Autophagy and P. gingivalis Infection Affects the Inflammation
As a major pathogen of periodontitis, P. gingivalis can manipulate the host’s innate immune response through different mechanisms, ultimately leading to the loss of periodontal supporting tissues, including the periodontal ligament and alveolar bone (Kinane et al., 2017). P. gingivalis and its virulence factors are recognized by pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and nod-like receptors (NLRs) (Fan et al., 2015; Watanabe et al., 2020). This process can not only trigger an inflammatory response but also induce autophagy to achieve the purpose of clearing P. gingivalis. However, P. gingivalis has developed diverse mechanisms to avoid recognition, inhibit the autophagic pathway, or may hijack the autophagosome (Wu and Li, 2019). The aberrant of autophagy leads to the imbalance between the pro-inflammatory and anti-inflammatory processes. Consequently, whether autophagy plays a protective role in the P. gingivalis-induced inflammation is determined by the interaction between autophagy and P. gingivalis infection.
Autophagy Defends Against P. gingivalis to Limit Inflammation
P. gingivalis invades the host cells by cytoskeletal rearrangement to form phagocytose, after which autophagy starts up and promotes bacteria elimination by fusing with lysosomes (Hu et al., 2020). In addition, autophagy acts as an innate immune response and plays a protective role in combating P. gingivalis infection and downregulating inflammatory response (Park et al., 2017; Liu et al., 2018).
Autophagy Plays a Protective Role in Periodontal Tissue Cells
Autophagy can protect host cells from apoptosis when periodontal tissues are subjected to P. gingivalis and its virulence factors. The possible involved mechanism is that the activation of autophagy may suppress apoptosis. For example, compared with healthy people, the expression levels of ATGs such as LC3, Beclin-1, Atg7, and Atg12 are higher in periodontal ligament tissue cells (PDLSCs) of periodontitis patients. However, the apoptotic protein caspase-8 expresses less (An et al., 2016). Coincidentally, another research found that the expression of Atg12 and the conversion of LC3-I to L3-II were increased in human gingival fibroblasts (HGFs) treated with P. gingivalis LPS (Bullon et al., 2012). Pro-inflammatory cytokines, such as TNF-α and IL-1β secretion could be limited by P. gingivalis-induced autophagy (Liu et al., 2018). Inhibiting autophagy with 3-Methyladenine (3-MA) increased the apoptosis rate in infected cells (Bullon et al., 2012), which indicated that autophagy might protect host cells from apoptosis in inflammation (Bullon et al., 2012; Liu et al., 2018).
Besides, autophagy can also reduce the secretion or increases the degradation of inflammatory factors. Chang et al. (Chang et al., 2013) found that butyrate, a kind of metabolic production of P. gingivalis, could impair cell cycle progression and induce inflammation via the generation of ROS in HGFs. However, P. gingivalis LPS treatment could also induce an autophagic response in human cultured keratinocyte cells (HaCaT) via increased ROS (Hagio-Izaki et al., 2018). The expression of LPS binding protein and TLRs increased, resulting in the co-localization of P. gingivalis with autophagosomes (Hagio-Izaki et al., 2018). Antibacterial peptide LL-37 could reduce the number of live P. gingivalis in HaCaT in a dose-dependent manner via xenophagy. Treatment with 3-MA weakened the inhibitory effect of LL-37 on the intracellular P. gingivalis (Yang et al., 2020). The findings indicated that autophagy was beneficial to the defense against the P. gingivalis internalized in HaCaT. Furthermore, inhibiting autophagy could enhance inflammation. Huang et al. (Huang et al., 2020) found high glucose environment could disrupt the autophagy lysosomal pathway (ALP) via a special subunit on the ATPase transmembrane (ATP6V0C), consequently enhancing the expression of IL-1β in HGECs.
Moreover, autophagy also plays a protective role by modulating the proangiogenesis ability to protect cells under the inflammatory environment. Wei et al. (Wei et al., 2018) found that proangiogenesis cytokine, angiogenin (Ang) expression is higher in inflammatory human periodontal ligament stem tissues (PDLSCs) and more tube formation was observed in endothelial cells pretreated with TNF-α and IL-1β. Rapamycin or Beclin-1 overexpression-induced autophagy can upregulate the expression level of Ang in PDLSCs, while knockdown of Beclin-1 downregulates its expression.
Consequently, when periodontal tissue cells are stimulated by P. gingivalis or its virulence factors, autophagy plays a protective role in fighting against the pathogens and decreasing the inflammatory injury. The involved mechanism may be that autophagy can decrease the intracellular P. gingivalis, reduce the secretion of inflammatory factors, inhibit host cell apoptosis death, and promote angiogenesis ability.
Autophagy Plays a Dual Role in Osteoclasts Differentiation and Bone Resorption
As an important indicator for the diagnosis of periodontitis, alveolar bone resorption is largely irreversible (Armitage, 2004). P. gingivalis infection could accelerate the alveolar bone pathological resorption by ROS generation and autophagic activity (Waddington et al., 2000; Tokutomi et al., 2015; Huck et al., 2020). Autophagy plays a dual role in osteoclast differentiation and bone resorption during periodontitis. DeSelm et al. (DeSelm et al., 2011) elucidated that osteoclasts degrade bone by directionally secreting lysosomal enzymes to a complex structure named ruffled border. The ruffled border is the product of the fusion of secretory vesicles to the bone-apposed plasma membrane. Lysosomal fusion with the plasmalemma results in the release of matrix-degrading molecules such as cathepsin K into the extracellular space to digest the bone matrix (DeSelm et al., 2011). However, PE-conjugated LC3 system and ubiquitin systems are essential for generating the osteoclast ruffled border, the secretory function of osteoclasts, and bone resorption (DeSelm et al., 2011). The research found that Syndecan 4 (SDC4), a member of the syndecan family, was highly expressed in the experimental periodontitis rat model (Li J. et al., 2021). While SDC4 silencing inhibits osteoclast differentiation and SDC4 overexpression enhanced osteoclast differentiation and autophagy induced by receptor activator of NF-κB ligand (RANKL). However, inhibiting autophagy with 3-MA abolished osteoclast differentiation which was enhanced by SDC4, indicating that autophagy might promote the osteoclast differentiation during periodontitis (Li J. et al., 2021).
On the other hand, autophagy also plays a negative role in osteoclast differentiation. Song et al. (Song et al., 2019) found that osteoclast differentiation and mRNA expression of osteoclast−specific genes were enhanced in bone marrow macrophages stimulated by IL-17A. Inhibition of autophagy with 3−MA attenuated the IL−17A−induced osteoclast differentiation. Tang et al. (Tong et al., 2018) found that autophagy plays an essential role in the process via AMPK/mTOR/p70S6K signaling pathway. Inhibiting autophagy with chloroquine (CQ) attenuated the OPG’s inhibition of osteoclast differentiation and bone resorption. Besides, autophagy also plays an important role in orthodontic tooth movement (Li Y. et al., 2021). Autophagy activity enhanced on the compression side of the tooth, which was associated with osteoclast recruitment and inflammatory cytokine expression (Li Y. et al., 2021). Treatment with rapamycin, an autophagy promotor, resulted in less bone resorption and tooth movement, suggesting that autophagy downregulated the inflammatory response and osteoclast differentiation during orthodontic tooth movement (Li Y. et al., 2021). To sum up, regulating autophagy activity may be an effective way to protect alveolar bone from excessive resorption in periodontitis.
Autophagy Plays a Protective Role in Antigen-Presenting Cells
P. gingivalis can invade antigen-presenting cells (APCs) such as macrophage and dendritic cells (DCs) and elevate autophagy activity. Autophagy can also inhibit inflammation by the elimination of P. gingivalis and degradation of inflammatory factors in APCs.
As for macrophages, Park et al.. (Park et al., 2017) preliminarily elucidated the link among bacteria, autophagy, and inflammation in the model of P. gingivalis infection in THP-1 cells. The infection promoted the secretion of mature IL-1β through activation of NLRP3 inflammasomes and downstream caspase-1. Similarly, Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans), another important pathogen of periodontitis, can also promote IL-1β maturation through the activation of AIM2 (absent in melanoma 2) inflammasome (Lee et al., 2020). The induction of AIM2 or NLRP3 inflammasomes could trigger autophagosome formation, which is dependent on the presence of the inflammasome sensor (Shi et al., 2012). The inflammasomes could be ubiquitinated and then recruit the ubiquitin-binding protein adaptor p62. With its assistance, the inflammasomes were delivered into autolysosome for degradation (Shi et al., 2012). Besides, both P. gingivalis and A. actinomycetemcomitans-induced autophagy in THP-1 cells inhibited the production of intracellular IL-1β and ROS by promoting bacterial internalization via phagocytosis into the macrophages. This therefore restricted excessive inflammatory response (Park et al., 2017; Lee et al., 2020). Besides, recent studies found two compounds, lipoxin A4 and vitamin D could promote the autophagic activity upon P. gingivalis infection in macrophages (Niu et al., 2020; Zhao et al., 2021). Lipoxin A4 inhibited NLRP3 inflammasome and downstream caspase-1 and IL-1β in P. gingivalis LPS-infected RAW264.7 by promoting autophagy. This action was related to the phosphorylation of NF-κB (Zhao et al., 2021). Calcitriol could significantly enhance the colocalization of P. gingivalis with autophagosome and lysosome markers in U937-derived macrophages, which indicated that the autophagy process could degrade live P. gingivalis (Niu et al., 2020). In summary, the above studies elucidated the role of autophagy in suppressing P. gingivalis-induced inflammation in macrophages. On the one hand, autophagy inhibits the production and promotes the degradation of AIM2 and NLRP3 inflammasomes. On the other hand, autophagy promotes bacterial internalization through phagocytosis and limits excessive inflammatory responses.
The autophagic activity in DCs was different from macrophages upon infection. P. gingivalis recognizes different PRRs on the surface or in the cytoplasm of DCs, which can trigger different responses (Xie et al., 1997; den Dunnen et al., 2009). The PRRs included TLRs and C-type lectin receptors such as DC-SIGN. The activation of TLR2 by FimA fimbriae could inhibit Akt phosphorylation, whereas the activation of DC-SIGN by Mfa-1 fimbriae could promote Akt phosphorylation (El-Awady et al., 2015; Meghil et al., 2019). Phosphorylated Akt could phosphorylate and activate mTORC1, ultimately inhibiting its downstream effector ULK1 to decrease autophagic activity. The interaction between DC-SIGN and Mfa-1 P. gingivalis strains induced lower intracellular killing and higher intracellular content of P. gingivalis. Moreover, Mfa-1 P. gingivalis was majorly wrapped in single-membrane vesicles, where it survived intracellularly (El-Awady et al., 2015). The finding indicated that Mfa-1 P. gingivalis engaged with DC-SIGN promoted the evasion of xenophagy. However, the activation of TLR2 cannot escape the autophagic attack. In addition, transcription factor forkhead box O1 (FOXO1) was phosphorylated and inactivated by Akt in DCs infected with Mfa-1 positive strains Pg381, causing FOXO1 to relocate to the cytoplasm (Meghil et al., 2019). FOXO1 deletion could inhibit the migration of DCs to lymph nodes and reduce the capacity of DCs to promote the formation of plasma cells and decrease the production of bacteria-specific antibodies (Xiao et al., 2015). Thus, the inactivation of FOXO1 promotes the survival of intracellular P. gingivalis.
Autophagy Plays a Protective Role in Other Systematic Inflammatory Diseases
The invasion of oral P. gingivalis into blood circulation is related to several systemic inflammatory diseases. For example, P. gingivalis-induced periodontitis could increase the risk of atherosclerosis, and P. gingivalis could also be detected in human atherosclerotic plaques (Kozarov et al., 2005). Research found that P. gingivalis could induce human umbilical vein endothelial cells (HUVECs) ER stress-mediated apoptosis and autophagy. The autophagic response protects HUVEC from apoptosis and silencing of LC3 with siRNA significantly increased P. gingivalis-induced apoptosis (Hirasawa and Kurita-Ochiai, 2018).
Infection with P. gingivalis can also accelerate the development and progression of non-alcoholic fatty liver disease (NAFLD) (Yoneda et al., 2012), a disease characterized by intracellular accumulation of lipid droplets in hepatocytes. Zaitsu et al. (Zaitsu et al., 2016). found that internalized P. gingivalis was localized in autophagosomes or lysosomes rather than lipid droplets in HepG2 hepatocytes, indicating that P. gingivalis could not use the lipid droplets for nutritional purposes. However, lipid droplets could affect the formation of autolysosomes for degradation of P. gingivalis and increase the existence of P. gingivalis in the cells at an early phase of infection. Thus, delayed elimination of P. gingivalis prolonged the inflammation and cellular damage, which raise the risk for the development of NAFLD (Zaitsu et al., 2016).
P. gingivalis Inhibit or Escape Autophagy for Survival and may Promote Inflammation
The above researches preliminarily revealed that autophagy functioned as a defender to protect host cells against P. gingivalis in various periodontal tissue cells, osteoclasts, antigen-presenting cells, and other tissue cells. However, P. gingivalis can also interfere with the process of autophagy to escape from the host immune attack and then the inflammation shall persist.
P. gingivalis can Hide in Autophagosome for Survival
P. gingivalis can escape from autophagy to avoid degradation by hindering the combination of autophagosomes with lysosomes. In this situation, P. gingivalis could colonize and proliferate in a vacuole encapsulated by a monolayer membrane after its invasion. The autophagosome with a monolayer membrane is modified by P. gingivalis and cannot fuse with lysosomes to form autolysosomes, exerting its degradation effect. Instead of the lysosomal marker protein LAMP-1, only rough ER protein attaches to the surface around the vacuole (Dorn et al., 2001). Lee et al. (Lee et al., 2018). found P. gingivalis adhered to the surface of GECs and was trafficked into the ER-rich autophagosomes through lipid rafts. After initially localized in early endosomes in GECs, some P. gingival entered the late endosomes where they were then sorted to lysosomes for degradation, some were delivered to Rab11 and RalA positive recycling endosomes for exocytosis while others escaped from endosomes to autophagosomes (Takeuchi et al., 2011; Amano et al., 2014). Hiding in autophagosomes, P gingivalis could utilize the substances such as protein for its survival. Inhibition of autophagy by 3-MA or Atg5 siRNA significantly decreased the survival of internalized P. gingivalis in HGECs (Belanger et al., 2006; Lee et al., 2018). P. gingivalis could colonize and replicate in GECs by inhibiting pro-apoptotic Bad through Akt to facilitate its long-term survival (Yao et al., 2010).
P. gingivalis Inhibit Autophagy Through LAP
Epidemiological studies have also linked PD to age-related macular degeneration (AMD) (Winning and Linden, 2017). Recent researches reported that P. gingivalis invasion and autophagy evasion might contribute to the pathogenesis of retinal degenerative diseases (Arjunan et al., 2020). This study showed that P. gingivalis could invade the human-retinal pigment epithelial (ARPE) cells and was able to escape from the autophagic vesicles and traffic into single membrane structures, freely occupying the cytoplasm of ARPE cells (Arjunan et al., 2020). The underlying mechanism may be associated with MREG-dependent LC3-associated phagocytosis (LAP). Lysosome maturation required the binding of LC3 to melanoregulin (MREG), and the MREG-mediated LC3-dependent degradation pathway was an essential process for P. gingivalis clearance (Martinez et al., 2011; Frost et al., 2015). Frost et al. (Frost et al., 2015) observed the co‐localization of LC3 and MREG in GECs of patients with severe periodontitis, but not in mild periodontitis or healthy people. The result showed that the expression of LC3, Beclin-1, and MREG in periodontitis was less than that in healthy people. It is speculated that P. gingivalis may affect the expression of MREG and LC3, and inhibit the normal autophagic process in HGECs of periodontitis patients.
The Mechanism of Inhibiting Autophagy by P. gingivalis Varies in Different Strains
As mentioned above, P. gingivalis expressed two forms of fimbriae and could interact with different PRRs on the host cell, affecting the fate of engulfed bacteria. Both long and short fimbriae were critical for the initiation of inflammatory responses and alveolar bone loss in periodontitis (Umemoto and Hamada, 2003). However, the interaction between Mfa-1 positive strains Pg381 with DC-SIGN could promote Akt phosphorylation and decrease the expression of LC3-II, Rab5, and LAMP1, thus inducing lower intracellular killing of P. gingivalis in DCs (El-Awady et al., 2015). However, the activation of TLR2 by FimA positive strains cannot escape from autophagic attacks. A similar phenomenon was found in Pg381 strain-infected human coronary artery endothelial (HCAE) cells. The survival rate of intracellular P. gingivalis decreased when HCAE cells were pretreated with 3-MA or wortmannin (Dorn et al., 2001). However, the pathogenic mechanisms varied in different P. gingivalis strains (Rodrigues et al., 2012; Blasi et al., 2016). (Rodrigues et al., 2012). compared different P. gingivalis strains (PgW83, PgA7436, Pg381, and Pg33277) and their abilities of adherence, internalization, and survival rate in HCAE cells. Both PgW83 and Pg381 were trafficked through the autophagic pathway, but only Pg381 could survive, depending on the autophagic pathway. Furthermore, Pg381 strain infected-HACE cells resulted in the highest production of pro-inflammatory cytokines IL-6, IL-8, and TNF-a.
The Possible Mechanisms Involved in the Interaction Between Autophagy and P. gingivalis-Induced Inflammation
The above research summarized the different interactions between autophagy and P. gingivalis, which may affect inflammation. On the one hand, autophagy inhibits inflammation by targeting the intracellular pathogens for degradation; on the other hand, P. gingivalis can take advantage of autophagosomes to escape from the host attack and prolong the inflammation. Autophagy can also interact with inflammation directly. Therefore, the mechanisms of the specific interaction among autophagy, P. gingivalis, and inflammation are significant in exploring the pathogenesis and cure of periodontitis. Based on P. gingivalis and other similar pathogens infection, this chapter summarized the complex interaction through the NLRP3 inflammasome pathway and cGAMP-cGAS-STING signaling pathway. The possible mechanisms involved in the interaction between autophagy and P. gingivalis-induced inflammation are plotted in Figure 2.
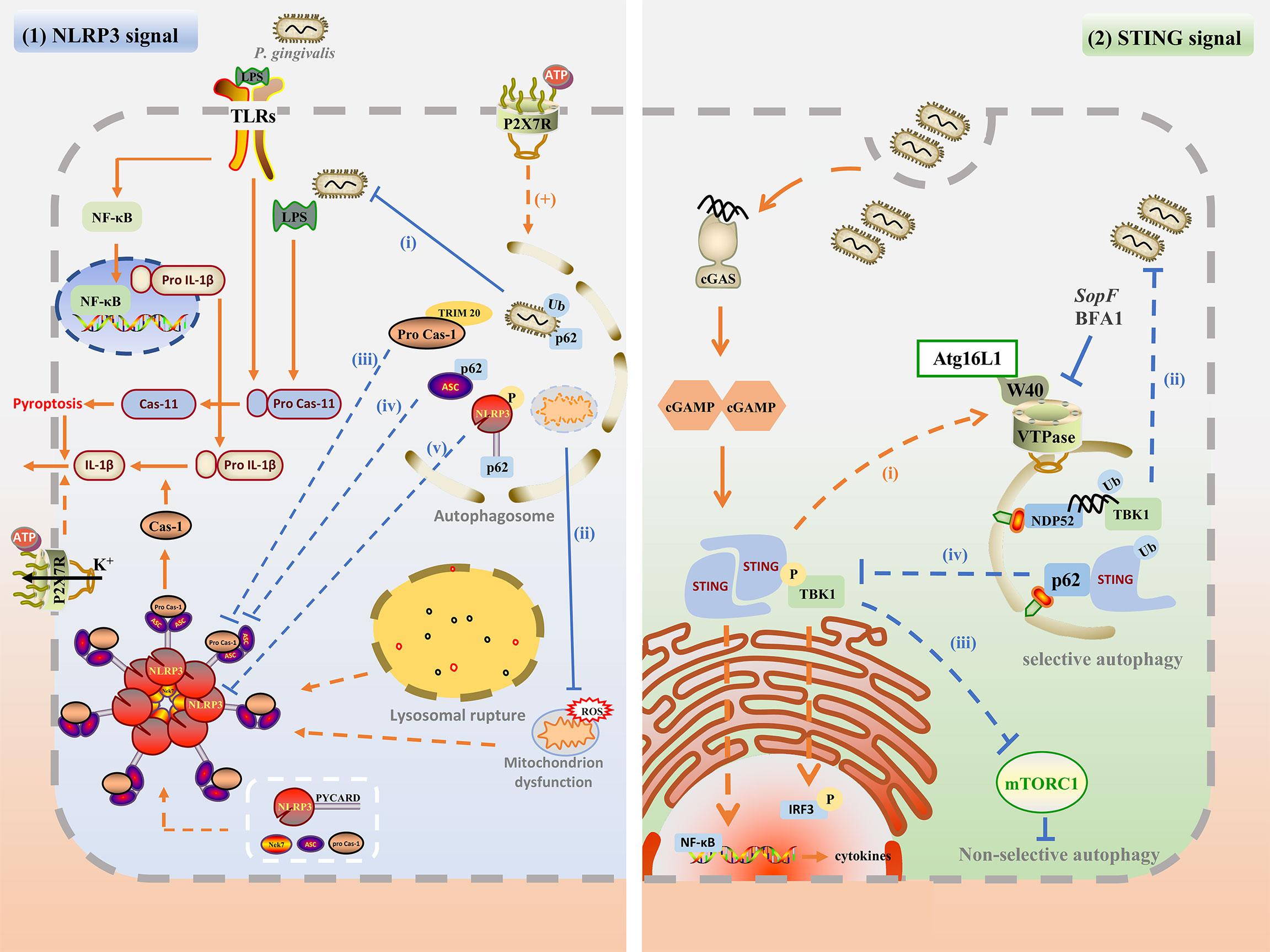
Figure 2 The possible mechanisms involved in the interaction between autophagy and P. gingivalis-induced inflammation. (1) Upon P. gingivalis infection, NLRP3 inflammasome can be activated by TLR/NF-κB signal, ROS generation, P2X7R and potassium efflux, and lysosomal rupture. The assembly of NLRP3 inflammasome promotes caspase-1 maturation as well as active IL-1β secretion, and pyroptosis. Autophagy may inhibit NLPR3 inflammasome by degrading intracellular P. gingivalis(i), dysfunctional mitochondrion (ii), phosphorylated NLRP3-PYCARD (iii), ubiquitinated ASC (iv), and pro-caspase-1 (v). Besides, P2RX7 activation may promote the autophagy flux, which is beneficial for bacterial degradation. (2) Autophagy may regulate the cGAS-cGAMP-STING pathway. cGAS may detect P. gingivalis infection by its dsDNA, and activate downstream cGAMP and STING. STING recruits and phosphorylates TBK and activated TBK1 kinase phosphorylates downstream IRF3. STING also activates NF-κB to induce IFN and other cytokines. Activated STING may recruit Atg16L1 and interact with VTPase via its WD40 domain to promote autophagy for degradation of P. gingivalis (i). Cytosolic dsDNA may be directly targeted for autophagosome via ubiquitin, TBK1, and NDP52-mediated xenophagy for degradation (ii). Besides, STING and TBK1 kinase may promote non-selective autophagy by inhibiting mTORC1 (iii). TBK1 kinase may also recruit and phosphorylates p62, enhancing the affinity of p62 with ubiquitinated STING and promoting selective autophagy.
Autophagy may Negatively Regulate Inflammation By Interacting With the NLRP3 Inflammasome Pathway
NLRP3 Inflammasome Pathway Introduction
NLRP3 inflammasome is a multiprotein complex that contains adapter protein apoptosis-associated speck-like protein (ASC) and procaspase-1. Its biochemical function is to activate caspase-1, which leads to the maturation of IL-1β and IL-18 (Shao et al., 2015; He et al., 2016a). Recent studies found a serine/threonine kinase named NEK7, which is bound to the leucine-rich repeat domain of NLRP3. NEK7 is also an essential mediator of NLRP3 activation (He et al., 2016b; Shi et al., 2016).
The NLRP3 inflammasome pathway activation involves two steps. In the initiating step, TLRs sense the invading P. gingivalis outside the cell and in intracellular endosomes and lysosomes (Medzhitov, 2001; Zhou et al., 2005; Zhang J. et al., 2018), leading to activation of NF-κB-mediated signaling, which in turn induces the transcription of NLRP3, pro-IL-1β (Bauernfeind et al., 2009; Hornung and Latz, 2010). The second step of inflammasome activation is the assembly of NLRP3, ASC, and pro-caspase-1 into a complex (Petrilli et al., 2007; Shao et al., 2015; Jo et al., 2016). P. gingivalis LPS also engages in the non-canonical inflammasome pathway (Ding et al., 2020; De Andrade et al., 2021), in which caspase-4 (known as caspase-11) is activated, promoting pyroptosis through cleavage of the pore-forming protein gasdermin D, resulting in maturation and release of IL-1β (Kayagaki et al., 2011; Hagar et al., 2013; Kayagaki et al., 2013). Different from the canonical NLRP3 inflammasome pathway, TLR4 senses the extracellular P. gingivalis LPS while caspase-4/11 senses the cytosolic LPS (Kayagaki et al., 2013; Downs et al., 2020; Xie et al., 2020)
The molecular mechanisms involved in NLRP3 inflammasome activation include mitochondrial ROS generation (Sorbara and Girardin, 2011; Heid et al., 2013), lysosomal rupture (Heid et al., 2013), pore formation, and potassium (K+) efflux (Petrilli et al., 2007). The P2X7 receptor is an ion channel activated by extracellular adenosine triphosphate (eATP) that induces IL-1β secretion through NLRP3 inflammasome activation (Park et al., 2014; Di Virgilio et al., 2017; Almeida-da-Silva et al., 2019).
Autophagy Plays a Protective Role in Inflammation by Interacting With the NLRP3 Inflammasome Pathway
Recent research reported that autophagy could suppress the activation of NLRP3 inflammasome. Enhancing autophagy with rapamycin reduced the expression of ASC as well as caspase-1 p20, and decreased the secretion of IL-1β, which in turn attenuated myocardial ischemia-reperfusion injury in diabetic rats (Zhang et al., 2020). Besides, Atg16L1-deficient macrophages produce high amounts of cleaved caspase-1 and IL-1β following stimulation of LPS (Saitoh et al., 2008). However, the mechanism of how autophagy suppressed the activation and assembly of inflammasomes upon P. gingivalis infection is intricate. It is supposed that this may be linked to clearing pathogens, degrading inflammasome components, and interrupting the molecular mechanism (Cao et al., 2019).
Xenophagy effectively limits intracellular bacterial survival and replication. Cytoplasmic bacteria, such as Salmonella. Typhimurium (S. Typhimurium), can be enveloped by ubiquitin proteins and recognized by autophagy receptors, such as p62, NDP52, OPTN, and TAX1BP1. With these receptors’ help, cytoplasmic bacteria are targeted to autophagosome (Thurston et al., 2009; Wild and Farhan, 2011; Tumbarello et al., 2015; Lee et al., 2022). In addition to ubiquitin, galectin-8, a cytosolic lectin, detects S. Typhimurium invasion by binding host glycans after being exposed to damaged Salmonella-containing vacuole (SCV). Galectin-8 can recruit NDP52 transiently and then NDP52 are recruited in a ubiquitin-dependent manner to activate xenophagy (Thurston et al., 2012). Although xenophagy machinery of targeting bacteria for degradation is well established, the ubiquitination mechanism and how autophagy receptor senses bacterial, especially P. gingivalis, are not fully clear. Only p62 and NDP52 receptors are demonstrated to bind to cytosolic P. gingivalis so far (Lee et al., 2018), and whether OPTN and TAXIBP1 or other ubiquitin adaptor proteins could target P. gingivalis to autophagosome is not clear. However, infecting P. gingivalis can be wrapped in ER-rich-double-membrane autophagosomal-vacuoles for survival. Inhibiting autophagy with 3-MA or ATG5 siRNA significantly reduced the viability of intracellular P. gingivalis. Besides, p62 and NDP52 receptors only bind to cytosolic free P. gingivalis while the vacuolar-P. gingivalis, which reveals a novel mechanism of P. gingivalis survival from xenophagy (Lee et al., 2018).
The activation of NLRP3 inflammasome and its components could be inhibited by autophagy. During the priming step of NLRP3 inflammasome activation, autophagy can inhibit NF-κB activation and the expression of pro-IL-1β (Wu et al., 2020), but the mechanism is not clear. During the process of inflammasomes’ assembly, the ubiquitination or phosphorylation modification of components can trigger selective autophagy. Spalinger et al. (Spalinger et al., 2017) found that only phosphorylated NLRP3 is found in autophagosomes and NLRP3 lacking phosphorylation sites cannot be recruited to phagophores upon inflammasome activation. Phosphorylated NLRP3 binds to p62 and is recruited into phagophores for degradation, which is dependent on NLRP3-PYCARD interaction (Spalinger et al., 2017). NLRP3 does not interact with SQSTM1in PYCARD-deficient cells, indicating that PYCARD may be the tag recognized by p62 for selective autophagy. The specific interaction and mechanism are worth further exploration. In addition to NLRP3, ASC under ubiquitination can recruit p62. With its assistance, ASC is delivered to autophagosomes for degradation in THP-1 treated with LPS and ATP (Shi et al., 2012). Besides to ubiquitination or phosphorylation modification-mediated selective autophagy, TRIMs family protein-mediated a highly exact process termed precision autophagy also plays an important role in the inactivation of NLRP3 inflammasome (Kimura et al., 2016). TRIM family proteins functioned as receptor-regulator proteins, both recognized targets and mediated autophagy. Different from selective autophagy, TRIMs could recognize targets through a direct protein-protein binding way without ubiquitin or galectin intermediates. And TRIMs function as platforms for the assembly of the core regulators of autophagy, such as the ULK1 complex or Atg16L1 complex (Kimura et al., 2016). For example, TRIM20 directly recognizes NLRP3 inflammasome and its component procaspase-1 and presents them for autophagic degradation (Kimura et al., 2015). The receptor and regulatory features enable TRIMs to mediate the inflammasome degradation via a highly exact process termed precision autophagy.
Autophagy also interacts with the molecular mechanisms involved in NLRP3 inflammasome activation, such as ROS clearance and P2X7 receptor. It has been demonstrated that P. gingivalis LPS-induced cytosolic ROS plays an important role in the pathogenesis of periodontitis (Chapple and Matthews, 2007; Golz et al., 2014). Mitophagy could preserve mitochondrial integrity and eliminate ROS through the degrading of damaged mitochondria, thereby reducing NLRP3 inflammasome activation (Lin et al., 2019). P2X7 receptor expression is enhanced in the P. gingivalis infection model, which is essential for NLRP3 activation promotes. The activation of the P2X7 receptor could promote P. gingivalis elimination in macrophages (Almeida-da-Silva et al., 2019). The possible mechanism may be the activation of P2RX7 can activate the AMPK/ULK1 pathway to promote autophagy flux for degradation of intracellular pathogens (Dong et al., 2021).
Autophagy may Regulate Inflammation by Interacting With the cGAS-cGAMP-STING Pathway
It is demonstrated that the cGAS-cGAMP-STING pathway-mediated interferon (IFN) activation plays an essential role in the resistance to DNA viral infection (Cai et al., 2014). However, emerging evidence has linked the cGAS-cGAMP-STING pathway with autophagy and both pro-inflammation and anti-bacteria effects upon infection have been reported (Gui et al., 2019).
As for pro-inflammatory activity, cyclic GMP-AMP synthase (cGAS) detects infections by binding to cytosolic DNA from bacteria or viruses (Sun et al., 2013; Collins et al., 2015). Upon recognition of double-stranded DNA, cGAS produces cyclic GMP-AMP (cGAMP), a second messenger for signal transduction (Kato et al., 2017). cGAMP binds to the adaptor protein STING, which promotes STING dimerization and translocation from the ER to perinuclear structures or Golgi apparatus (Ishikawa and Barber, 2008). STING recruits and activates TANK binding kinase 1 (TBK1), phosphorylating the IFN regulatory factor 3 (IRF3) (Abe and Barber, 2014; Liu et al., 2015). STING also activates NF-κB, which functions together with IRF3 to lead to the production of type 1 IFN and other inflammatory cytokines (Shu and Wang, 2014).
As for anti-bacteria activity, the activation of the cGAS-STING pathway and TBK1 kinase can lead to ubiquitin-mediated xenophagy for the degradation of cytosolic dsDNA and bacteria (Watson et al., 2012; Watson et al., 2015; Gui et al., 2019). In addition to activating transcriptional responses, dsDNA is also a potent inducer of autophagy. Waston et al. (Watson et al., 2012) transfected plasmid dsDNA into LC3-GFP BMDMs and found that ubiquitin, TBK1, and NDP52 all colocalized to DNA puncta after transfection, indicating that cytosolic dsDNA was directly targeted for autophagosome via ubiquitin-mediated xenophagy, in which STING played an essential role. On the other hand, Collins et al. (Collins et al., 2015) found that M. tuberculosis infection-induced cGAS -STING pathway activation in THP-1. The absence of cGAS or STING significantly reduced the colocalization of LC3-II with M. tuberculosis, which was more permissive for M. tuberculosis survival. Besides to selective autophagy, TBK1 also promotes non-selective autophagy by suppressing the mTORC1 activity (Hasan et al., 2017). However, bacteria have involved the mechanism of escaping STING-induced autophagy. During the infection of Salmonella, STING-induced autophagy is mediated by recruiting Atg16L1 to interact with the vacuolar ATPase (VTPase) via its WD40 domain. The process can be blocked by bafilomycin A1 (BFA1), an autophagy inhibitor, and SopF, a T3SS effector from Salmonella, for they can bind to and inhibit the V-ATPase (Xu et al., 2019; Fischer et al., 2020). Furthermore, WIPI2-deficient cells showed less LC3 lipidation after treatment with cGAMP, indicating that Atg16L1 is essential in STING-induced autophagy. However, ULK1 and Beclin-1 showed less influence on STING-induced autophagy (Gui et al., 2019). Although the cGAS-STING pathway plays an important protective role in the elimination of bacteria by autophagy, the specific mechanism of how the cGAS-STING pathway interacts with the autophagy pathway remains unclear.
Interestingly, the association between the cGAS-STING pathway and autophagy is like a negative feedback loop. Once the cGAS-STING pathway is activated to cGAS, TBK1 kinase recruits and phosphorylates p62, enhancing the affinity of p62 with ubiquitinated STING. As a consequence, STING is degraded through selective autophagy, attenuating the cGAS-cGAMP-STING pathway response (Prabakaran et al., 2018).
However, similar studies of the cGAS-cGAMP-STING pathway are hardly found in the P. gingivalis infection model. Combining the mechanism of how P. gingivalis-induced autophagy and escape from autophagy with the cGAS-cGAMP-STING pathway, we make a summary as follows. On the one hand, internalized P. gingivalis and its LPS may activate the NLRP3 inflammasome, promoting Caspase-1 and IL-1β maturation and secretion. P. gingivalis dsDNA may activate the cGAS-cGAMP-STING pathway (Wan et al., 2020).On the other hand, the non-selective autophagy and LAP are like a defender fighting against the internalized P. gingivalis. This process is similar to innate immunity, resulting in the majority of the bacteria being delivered into the autolysosome for degradation. However, some P. gingivalis can escape from autophagy degradation by inhibiting the initiation and development of non-selective autophagy, reducing the fusion of the autophagosome with lysosome and hiding into the autophagosomal-vacuoles. In this condition, STING-induced autophagy and ubiquitination-mediated xenophagy may target the remaining P. gingivalis for degradation. This process resembles adaptive immunity. Popularly speaking, STING pathway-mediated autophagy is like another branch of the autophagy signaling pathway, but they can all end with the formation of autolysosomes for the degradation of pathogens.
Summary
There is no doubt that the interaction between autophagy and P. gingivalis accounts for the balance of pro-inflammation and anti-inflammation to some extent, providing evidence for the possible mechanism of the pathogenesis of numerous inflammatory diseases, including periodontitis. In this review, we elaborate that autophagy majorly plays a protective role in defense against P. gingivalis infection and mediates P. gingivalis clearance to inhibit its inflammatory process. Besides, autophagy also interacts directly with the inflammatory processes, such as the NLPR3 inflammasome and cGAS-STING pathway, to restrict excessive inflammation (Figure 2). However, P. gingivalis and other bacteria have evolved the mechanism to escape or even hijack autophagy (Table 1), which may be unfavorable to the maintenance of host homeostasis. Therefore, it is of utmost importance to understand the molecular mechanisms of the interaction among autophagy, P. gingivalis, and inflammation. Such mechanisms ensure the normal anti-bacterial response that avoids excessive inflammatory damage and favors the maintenance of tissue homeostasis.
However, given the recent advanced researches on the interaction between autophagy, P. gingivalis, and inflammation, there is still a lack of critical evidence to elucidate the specific mechanism. 1) How P. gingivalis or other bacteria are modified and where are the accurate modification sites. 2) The specific mechanism of how P. gingivalis or other bacteria escape from autophagy is unspecific in most studies. The mechanism may be associated with the bacterial mutant virulence factors, host cell and receptors, and the specific interaction between the factors with the autophagy pathway. Precise interaction targets in the mechanism of bacteria capturing and escaping process are essential but still unclear. 3) The research on the anti-bacterial role of the cGAS-STING pathway and its interaction with autophagy and inflammation is still unclear and worth further exploration. 4) The evidence that autophagy plays a protective role in inflammatory disease is insufficient. Whether autophagy interacts directly with inflammation, such as the NLRP3 inflammasome pathway or other inflammatory factors is still unclear in P. gingivalis infection. On the other hand, there are few researches focused on the role of inflammation in autophagy. Excessive inflammation may interrupt the autophagy activity and in turn, forms a negative feedback effect on inflammation.
Author Contributions
Writing-original draft preparation, SK; writing - review and editing, SK, P-HD, AD; supervision, P-HD, HW; project administration and funding acquisition, P-HD. All authors critically reviewed the manuscript before submission. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China under grants No. 81870765 and 82170953.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abe, T., Barber, G. N. (2014). Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-kappaB Activation Through TBK1. J. Virol. 88 (10), 5328–5341. doi: 10.1128/JVI.00037-14
Almeida-da-Silva, C. L. C., Ramos-Junior, E. S., Morandini, A. C., Rocha, G. D. C., Marinho, Y., Tamura, A. S., et al. (2019). P2X7 Receptor-Mediated Leukocyte Recruitment and Porphyromonas Gingivalis Clearance Requires IL-1beta Production and Autocrine IL-1 Receptor Activation. Immunobiology 224 (1), 50–59. doi: 10.1016/j.imbio.2018.10.008
Amano, A., Chen, C., Honma, K., Li, C., Settem, R. P., Sharma, A. (2014). Genetic Characteristics and Pathogenic Mechanisms of Periodontal Pathogens. Adv. Dent. Res. 26 (1), 15–22. doi: 10.1177/0022034514526237
An, Y., Liu, W., Xue, P., Zhang, Y., Wang, Q., Jin, Y. (2016). Increased Autophagy is Required to Protect Periodontal Ligament Stem Cells From Apoptosis in Inflammatory Microenvironment. J. Clin. Periodontol. 43 (7), 618–625. doi: 10.1111/jcpe.12549
Arasaki, K., Mikami, Y., Shames, S. R., Inoue, H., Wakana, Y., Tagaya, M. (2017). Legionella Effector Lpg1137 Shuts Down ER-Mitochondria Communication Through Cleavage of Syntaxin 17. Nat. Commun. 8, 15406. doi: 10.1038/ncomms15406
Arjunan, P., Swaminathan, R., Yuan, J., Al-Shabrawey, M., Espinosa-Heidmann, D. G., Nussbaum, J., et al. (2020). Invasion of Human Retinal Pigment Epithelial Cells by Porphyromonas Gingivalis Leading to Vacuolar/Cytosolic Localization and Autophagy Dysfunction In-Vitro. Sci. Rep. 10 (1), 7468. doi: 10.1038/s41598-020-64449-8
Armitage, G. C. (2004). Periodontal Diagnoses and Classification of Periodontal Diseases. Periodontol. 2000 34, 9–21. doi: 10.1046/j.0906-6713.2002.003421.x
Bah, A., Vergne, I. (2017). Macrophage Autophagy and Bacterial Infections. Front. Immunol. 8. doi: 10.3389/fimmu.2017.01483
Barnett, T. C., Liebl, D., Seymour, L. M., Gillen, C. M., Lim, J. Y., Larock, C. N., et al. (2013). The Globally Disseminated M1T1 Clone of Group A Streptococcus Evades Autophagy for Intracellular Replication. Cell Host Microbe 14 (6), 675–682. doi: 10.1016/j.chom.2013.11.003
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., MacDonald, K., Speert, D., et al. (2009). Cutting Edge: NF-kappaB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 183 (2), 787–791. doi: 10.4049/jimmunol.0901363
Belanger, M., Rodrigues, P. H., Dunn, W. A., Jr., Progulske-Fox, A. (2006). Autophagy: A Highway for Porphyromonas Gingivalis in Endothelial Cells. Autophagy 2 (3), 165–170. doi: 10.4161/auto.2828
Bernales, S., McDonald, K. L., Walter, P. (2006). Autophagy Counterbalances Endoplasmic Reticulum Expansion During the Unfolded Protein Response. PloS Biol. 4 (12), e423. doi: 10.1371/journal.pbio.0040423
Blasi, I., Korostoff, J., Dhingra, A., Reyes-Reveles, J., Shenker, B. J., Shahabuddin, N., et al. (2016). Variants Ofporphyromonas Gingivalislipopolysaccharide Alter Lipidation of Autophagic Protein, Microtubule-Associated Protein 1 Light Chain 3, LC3. Mol. Oral. Microbiol. 31 (6), 486–500. doi: 10.1111/omi.12141
Bostanci, N., Belibasakis, G. N. (2012). Porphyromonas Gingivalis: An Invasive and Evasive Opportunistic Oral Pathogen. FEMS Microbiol. Lett. 333 (1), 1–9. doi: 10.1111/j.1574-6968.2012.02579.x
Bullon, P., Cordero, M. D., Quiles, J. L., Ramirez-Tortosa, M. D., Gonzalez-Alonso, A., Alfonsi, S., et al. (2012). Autophagy in Periodontitis Patients and Gingival Fibroblasts: Unraveling the Link Between Chronic Diseases and Inflammation. BMC Med. 10, 122–133. doi: 10.1186/1741-7015-10-122
Cai, X., Chiu, Y. H., Chen, Z. J. (2014). The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell 54 (2), 289–296. doi: 10.1016/j.molcel.2014.03.040
Cao, Z., Wang, Y., Long, Z., He, G. (2019). Interaction Between Autophagy and the NLRP3 Inflammasome. Acta Biochim. Biophys. Sin. 51 (11), 1087–1095. doi: 10.1093/abbs/gmz098
Casanova, J. E. (2017). Bacterial Autophagy: Offense and Defense at the Host-Pathogen Interface. Cell Mol. Gastroenterol. Hepatol. 4 (2), 237–243. doi: 10.1016/j.jcmgh.2017.05.002
Castrejon-Jimenez, N. S., Leyva-Paredes, K., Hernandez-Gonzalez, J. C., Luna-Herrera, J., Garcia-Perez, B. E. (2015). The Role of Autophagy in Bacterial Infections. Biosci. Trends 9 (3), 149–159. doi: 10.5582/bst.2015.01035
Chang, M. C., Tsai, Y. L., Chen, Y. W., Chan, C. P., Huang, C. F., Lan, W. C., et al. (2013). Butyrate Induces Reactive Oxygen Species Production and Affects Cell Cycle Progression in Human Gingival Fibroblasts. J. Periodontal Res. 48 (1), 66–73. doi: 10.1111/j.1600-0765.2012.01504.x
Chapple, I. L., Matthews, J. B. (2007). The Role of Reactive Oxygen and Antioxidant Species in Periodontal Tissue Destruction. Periodontol. 2000 43, 160–232. doi: 10.1111/j.1600-0757.2006.00178.x
Choy, A., Dancourt, J., Mugo, B., O'Connor, T. J., Isberg, R. R., Melia, T. J., et al. (2012). The Legionella Effector RavZ Inhibits Host Autophagy Through Irreversible Atg8 Deconjugation. Science 338 (6110), 1072–1076. doi: 10.1126/science.1227026
Collins, A. C., Cai, H., Li, T., Franco, L. H., Li, X. D., Nair, V. R., et al. (2015). Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium Tuberculosis. Cell Host Microbe 17 (6), 820–828. doi: 10.1016/j.chom.2015.05.005
Cosin-Roger, J., Simmen, S., Melhem, H., Atrott, K., Frey-Wagner, I., Hausmann, M., et al. (2017). Hypoxia Ameliorates Intestinal Inflammation Through NLRP3/mTOR Downregulation and Autophagy Activation. Nat. Commun. 8 (1), 98. doi: 10.1038/s41467-017-00213-3
De Andrade, K. Q., Almeida-da-Silva, C. L. C., Ojcius, D. M., Coutinho-Silva, R. (2021). Differential Involvement of the Canonical and Noncanonical Inflammasomes in the Immune Response Against Infection by the Periodontal Bacteria Porphyromonas Gingivalis and Fusobacterium Nucleatum. Curr. Res. Microb. Sci. 2, 100023. doi: 10.1016/j.crmicr.2021.100023
den Dunnen, J., Gringhuis, S. I., Geijtenbeek, T. B. (2009). Innate Signaling by the C-Type Lectin DC-SIGN Dictates Immune Responses. Cancer Immunol. Immunother. 58 (7), 1149–1157. doi: 10.1007/s00262-008-0615-1
DeSelm, C. J., Miller, B. C., Zou, W., Beatty, W. L., van Meel, E., Takahata, Y., et al. (2011). Autophagy Proteins Regulate the Secretory Component of Osteoclastic Bone Resorption. Dev. Cell 21 (5), 966–974. doi: 10.1016/j.devcel.2011.08.016
Ding, P. H., Yang, M. X., Wang, N. N., Jin, L. J., Dong, Y., Cai, X., et al. (2020). Porphyromonas Gingivalis-Induced NLRP3 Inflammasome Activation and Its Downstream Interleukin-1beta Release Depend on Caspase-4. Front. Microbiol. 11, 1881. doi: 10.3389/fmicb.2020.01881
Di Virgilio, F., Dal Ben, D., Sarti, A. C., Giuliani, A. L., Falzoni, S. (2017). The P2X7 Receptor in Infection and Inflammation. Immunity 47 (1), 15–31. doi: 10.1016/j.immuni.2017.06.020
Dong, Z., Wei, Y., Tao, M., Zhang, L. (2021). Activation of the Purinergic Receptor P2X7 Improves Hepatosteatosis by Promoting Lipophagy. FEBS Lett. 595 (22), 2768–2780. doi: 10.1002/1873-3468.14207
Dong, N., Zhu, Y., Lu, Q., Hu, L., Zheng, Y., Shao, F. (2012). Structurally Distinct Bacterial TBC-Like GAPs Link Arf GTPase to Rab1 Inactivation to Counteract Host Defenses. Cell 150 (5), 1029–1041. doi: 10.1016/j.cell.2012.06.050
Dooley, H. C., Razi, M., Polson, H. J., Girardin, S. E., Tooze, S. A. (2014). WIPI2 Links LC3 Conjugation With PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12-5-16l1. Mol. Cell 55 (2), 238–252. doi: 10.1016/j.molcel.2014.05.021
Dorn, B. R., Dunn, W. A., Jr., Progulske-Fox, A. (2001). Porphyromonas Gingivalis Traffics to Autophagosomes in Human Coronary Artery Endothelial Cells. Infect. Immun. 69 (9), 5698–5708. doi: 10.1128/iai.69.9.5698-5708.2001
Downs, K. P., Nguyen, H., Dorfleutner, A., Stehlik, C. (2020). An Overview of the non-Canonical Inflammasome. Mol. Aspects Med. 76, 100924. doi: 10.1016/j.mam.2020.100924
Ebersole, J. L., Al-Sabbagh, M., Dawson, D. R., 3rd (2020). Heterogeneity of Human Serum Antibody Responses to P. Gingivalis in Periodontitis: Effects of Age, Race/Ethnicity, and Sex. Immunol. Lett. 218, 11–21. doi: 10.1016/j.imlet.2019.12.004
El-Awady, A. R., Miles, B., Scisci, E., Kurago, Z. B., Palani, C. D., Arce, R. M., et al. (2015). Porphyromonas Gingivalis Evasion of Autophagy and Intracellular Killing by Human Myeloid Dendritic Cells Involves DC-SIGN-TLR2 Crosstalk. PloS Pathog. 10 (2), e1004647. doi: 10.1371/journal.ppat.1004647
Fan, X. Y., Tian, C., Wang, H., Xu, Y., Ren, K., Zhang, B. Y., et al. (2015). Activation of the AMPK-ULK1 Pathway Plays an Important Role in Autophagy During Prion Infection. Sci. Rep. 5, 14728. doi: 10.1038/srep14728
Feng, Y., He, D., Yao, Z., Klionsky, D. J. (2014). The Machinery of Macroautophagy. Cell Res. 24 (1), 24–41. doi: 10.1038/cr.2013.168
Fischer, T. D., Wang, C., Padman, B. S., Lazarou, M., Youle, R. J. (2020). STING Induces LC3B Lipidation Onto Single-Membrane Vesicles via the V-ATPase and ATG16L1-WD40 Domain. J. Cell Biol. 219 (12), 17–29. doi: 10.1083/jcb.202009128
Fortier, A., de Chastellier, C., Balor, S., Gros, P. (2007). Birc1e/Naip5 Rapidly Antagonizes Modulation of Phagosome Maturation by Legionella Pneumophila. Cell Microbiol. 9 (4), 910–923. doi: 10.1111/j.1462-5822.2006.00839.x
Frost, L. S., Lopes, V. S., Bragin, A., Reyes-Reveles, J., Brancato, J., Cohen, A., et al. (2015). The Contribution of Melanoregulin to Microtubule-Associated Protein 1 Light Chain 3 (LC3) Associated Phagocytosis in Retinal Pigment Epithelium. Mol. Neurobiol. 52 (3), 1135–1151. doi: 10.1007/s12035-014-8920-5
Ganley, I. G., Lam du, H., Wang, J., Ding, X., Chen, S., Jiang, X. (2009). ULK1.ATG13.FIP200 Complex Mediates mTOR Signaling and is Essential for Autophagy. J. Biol. Chem. 284 (18), 12297–12305. doi: 10.1074/jbc.M900573200
Glick, D., Barth, S., Macleod, K. F. (2010). Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 221 (1), 3–12. doi: 10.1002/path.2697
Golz, L., Memmert, S., Rath-Deschner, B., Jager, A., Appel, T., Baumgarten, G., et al. (2014). LPS From P. Gingivalis and Hypoxia Increases Oxidative Stress in Periodontal Ligament Fibroblasts and Contributes to Periodontitis. Mediators Inflammation 2014, 986264. doi: 10.1155/2014/986264
Gui, X., Yang, H., Li, T., Tan, X., Shi, P., Li, M., et al. (2019). Autophagy Induction via STING Trafficking is a Primordial Function of the cGAS Pathway. Nature 567 (7747), 262–266. doi: 10.1038/s41586-019-1006-9
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K., Miao, E. A. (2013). Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 341 (6151), 1250–1253. doi: 10.1126/science.1240988
Hagio-Izaki, K., Yasunaga, M., Yamaguchi, M., Kajiya, H., Morita, H., Yoneda, M., et al. (2018). Lipopolysaccharide Induces Bacterial Autophagy in Epithelial Keratinocytes of the Gingival Sulcus. BMC Cell Biol. 19 (1), 18. doi: 10.1186/s12860-018-0168-x
Hajishengallis, G., Lamont, R. J. (2014). Breaking Bad: Manipulation of the Host Response by Porphyromonas Gingivalis. Eur. J. Immunol. 44 (2), 328–338. doi: 10.1002/eji.201344202
Harris, J., Hartman, M., Roche, C., Zeng, S. G., O'Shea, A., Sharp, F. A., et al. (2011). Autophagy Controls IL-1beta Secretion by Targeting Pro-IL-1beta for Degradation. J. Biol. Chem. 286 (11), 9587–9597. doi: 10.1074/jbc.M110.202911
Hasan, M., Gonugunta, V. K., Dobbs, N., Ali, A., Palchik, G., Calvaruso, M. A., et al. (2017). Chronic Innate Immune Activation of TBK1 Suppresses Mtorc1 Activity and Dysregulates Cellular Metabolism. Proc. Natl. Acad. Sci. U.S.A. 114 (4), 746–751. doi: 10.1073/pnas.1611113114
He, Y., Hara, H., Nunez, G. (2016a). Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 41 (12), 1012–1021. doi: 10.1016/j.tibs.2016.09.002
Heid, M. E., Keyel, P. A., Kamga, C., Shiva, S., Watkins, S. C., Salter, R. D. (2013). Mitochondrial Reactive Oxygen Species Induces NLRP3-Dependent Lysosomal Damage and Inflammasome Activation. J. Immunol. 191 (10), 5230–5238. doi: 10.4049/jimmunol.1301490
He, Y., Zeng, M. Y., Yang, D., Motro, B., Nunez, G. (2016b). NEK7 is an Essential Mediator of NLRP3 Activation Downstream of Potassium Efflux. Nature 530 (7590), 354–357. doi: 10.1038/nature16959
Hirasawa, M., Kurita-Ochiai, T. (2018). Porphyromonas Gingivalis Induces Apoptosis and Autophagy via ER Stress in Human Umbilical Vein Endothelial Cells. Mediators Inflammation 2018, 1967506. doi: 10.1155/2018/1967506
Hornung, V., Latz, E. (2010). Critical Functions of Priming and Lysosomal Damage for NLRP3 Activation. Eur. J. Immunol. 40 (3), 620–623. doi: 10.1002/eji.200940185
How, K. Y., Song, K. P., Chan, K. G. (2016). Porphyromonas Gingivalis: An Overview of Periodontopathic Pathogen Below the Gum Line. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00053
Huang, J., Brumell, J. H. (2014). Bacteria-Autophagy Interplay: A Battle for Survival. Nat. Rev. Microbiol. 12 (2), 101–114. doi: 10.1038/nrmicro3160
Huang, X., Kuang, S., Shen, Z., Liang, M., Lin, Z. (2020). High Glucose Disrupts Autophagy Lysosomal Pathway in Gingival Epithelial Cells via ATP6V0C. J. Periodontol. 91 (5), 705–714. doi: 10.1002/JPER.19-0262
Hu, W., Chan, H., Lu, L., Wong, K. T., Wong, S. H., Li, M. X., et al. (2020). Autophagy in Intracellular Bacterial Infection. Semin. Cell Dev. Biol. 101, 41–50. doi: 10.1016/j.semcdb.2019.07.014
Huck, O., Mulhall, H., Rubin, G., Kizelnik, Z., Iyer, R., Perpich, J. D., et al. (2020). Akkermansia Muciniphila Reduces Porphyromonas Gingivalis-Induced Inflammation and Periodontal Bone Destruction. J. Clin. Periodontol. 47 (2), 202–212. doi: 10.1111/jcpe.13214
Humphrey, L. L., Fu, R., Buckley, D. I., Freeman, M., Helfand, M. (2008). Periodontal Disease and Coronary Heart Disease Incidence: A Systematic Review and Meta-Analysis. J. Gen. Intern. Med. 23 (12), 2079–2086. doi: 10.1007/s11606-008-0787-6
Ishikawa, H., Barber, G. N. (2008). STING is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 455 (7213), 674–678. doi: 10.1038/nature07317
Itakura, E., Kishi-Itakura, C., Mizushima, N. (2012). The Hairpin-Type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion With Endosomes/Lysosomes. Cell 151 (6), 1256–1269. doi: 10.1016/j.cell.2012.11.001
Jager, S., Bucci, C., Tanida, I., Ueno, T., Kominami, E., Saftig, P., et al. (2004). Role for Rab7 in Maturation of Late Autophagic Vacuoles. J. Cell Sci. 117 (Pt 20), 4837–4848. doi: 10.1242/jcs.01370
Jia, L., Wu, R., Han, N., Fu, J., Luo, Z., Guo, L., et al. (2020). Porphyromonas Gingivalis and Lactobacillus Rhamnosus GG Regulate the Th17/Treg Balance in Colitis via TLR4 and TLR2. Clin. Transl. Immunol. 9 (11), e1213. doi: 10.1002/cti2.1213
Jo, E. K., Kim, J. K., Shin, D. M., Sasakawa, C. (2016). Molecular Mechanisms Regulating NLRP3 Inflammasome Activation. Cell Mol. Immunol. 13 (2), 148–159. doi: 10.1038/cmi.2015.95
Kassebaum, N. J., Smith, A. G. C., Bernabe, E., Fleming, T. D., Reynolds, A. E., Vos, T., et al. (2017). Global, Regional, and National Prevalence, Incidence, and Disability-Adjusted Life Years for Oral Conditions for 195 Countries 1990-2015: A Systematic Analysis for the Global Burden of Diseases, Injuries, and Risk Factors. J. Dent. Res. 96 (4), 380–387. doi: 10.1177/0022034517693566
Kato, K., Omura, H., Ishitani, R., Nureki, O. (2017). Cyclic GMP-AMP as an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Annu. Rev. Biochem. 86, 541–566. doi: 10.1146/annurev-biochem-061516-044813
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-Canonical Inflammasome Activation Targets Caspase-11. Nature 479 (7371), 117–121. doi: 10.1038/nature10558
Kayagaki, N., Wong, M. T., Stowe, I. B., Ramani, S. R., Gonzalez, L. C., Akashi-Takamura, S., et al. (2013). Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 341 (6151), 1246–1249. doi: 10.1126/science.1240248
Kimura, T., Jain, A., Choi, S. W., Mandell, M. A., Schroder, K., Johansen, T., et al. (2015). TRIM-Mediated Precision Autophagy Targets Cytoplasmic Regulators of Innate Immunity. J. Cell Biol. 210 (6), 973–989. doi: 10.1083/jcb.201503023
Kimura, T., Mandell, M., Deretic, V. (2016). Precision Autophagy Directed by Receptor Regulators - Emerging Examples Within the TRIM Family. J. Cell Sci. 129 (5), 881–891. doi: 10.1242/jcs.163758
Kinane, D. F., Stathopoulou, P. G., Papapanou, P. N. (2017). Periodontal Diseases. Nat. Rev. Dis. Primers 3, 17038. doi: 10.1038/nrdp.2017.38
Kissova, I., Salin, B., Schaeffer, J., Bhatia, S., Manon, S., Camougrand, N. (2007). Selective and non-Selective Autophagic Degradation of Mitochondria in Yeast. Autophagy 3 (4), 329–336. doi: 10.4161/auto.4034
Kozarov, E. V., Dorn, B. R., Shelburne, C. E., Dunn, W. A., Jr., Progulske-Fox, A. (2005). Human Atherosclerotic Plaque Contains Viable Invasive Actinobacillus Actinomycetemcomitans and Porphyromonas Gingivalis. Arterioscler. Thromb. Vasc. Biol. 25 (3), e17–e18. doi: 10.1161/01.ATV.0000155018.67835.1a
Krause, K., Caution, K., Badr, A., Hamilton, K., Saleh, A., Patel, K., et al. (2018). CASP4/caspase-11 Promotes Autophagosome Formation in Response to Bacterial Infection. Autophagy 14 (11), 1928–1942. doi: 10.1080/15548627.2018.1491494
Lee, Y. J., Kim, J. K., Jung, C. H., Kim, Y. J., Jung, E. J., Lee, S. H., et al. (2022). Chemical Modulation of SQSTM1/p62-Mediated Xenophagy That Targets a Broad Range of Pathogenic Bacteria. Autophagy, 1–20. doi: 10.1080/15548627.2022.2054240
Lee, H. A., Park, M. H., Song, Y., Na, H. S., Chung, J. (2020). Role of Aggregatibacter Actinomycetemcomitans-Induced Autophagy in Inflammatory Response. J. Periodontol 91 (12), 1682–1693. doi: 10.1002/JPER.19-0639
Lee, K., Roberts, J. S., Choi, C. H., Atanasova, K. R., Yilmaz, O. (2018). Porphyromonas Gingivalis Traffics Into Endoplasmic Reticulum-Rich-Autophagosomes for Successful Survival in Human Gingival Epithelial Cells. Virulence 9 (1), 845–859. doi: 10.1080/21505594.2018.1454171
Li, Y., Jacox, L. A., Coats, S., Kwon, J., Xue, P., Tang, N., et al. (2021). Roles of Autophagy in Orthodontic Tooth Movement. Am. J. Orthod Dentofacial Orthop. 159 (5), 582–593. doi: 10.1016/j.ajodo.2020.01.027
Lin, Q., Li, S., Jiang, N., Shao, X., Zhang, M., Jin, H., et al. (2019). PINK1-Parkin Pathway of Mitophagy Protects Against Contrast-Induced Acute Kidney Injury via Decreasing Mitochondrial ROS and NLRP3 Inflammasome Activation. Redox Biol. 26, 101254. doi: 10.1016/j.redox.2019.101254
Li, S. J., Sun, S. J., Gao, J., Sun, F. B. (2016). Wogonin Induces Beclin-1/PI3K and Reactive Oxygen Species-Mediated Autophagy in Human Pancreatic Cancer Cells. Oncol. Lett. 12 (6), 5059–5067. doi: 10.3892/ol.2016.5367
Li, J., Sun, Z., Lin, Y., Yan, Y., Yan, H., Jing, B., et al. (2021). Syndecan 4 Contributes to Osteoclast Differentiation Induced by RANKL Through Enhancing Autophagy. Int. Immunopharmacol. 91, 107275. doi: 10.1016/j.intimp.2020.107275
Liu, S., Cai, X., Wu, J., Cong, Q., Chen, X., Li, T., et al. (2015). Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 347 (6227), aaa2630. doi: 10.1126/science.aaa2630
Liu, J., Wang, X., Zheng, M., Luan, Q. (2018). Lipopolysaccharide From Porphyromonas Gingivalis Promotes Autophagy of Human Gingival Fibroblasts Through the PI3K/Akt/mTOR Signaling Pathway. Life Sci. 211, 133–139. doi: 10.1016/j.lfs.2018.09.023
Li, S., Wandel, M. P., Li, F., Liu, Z., He, C., Wu, J., et al. (2013). Sterical Hindrance Promotes Selectivity of the Autophagy Cargo Receptor NDP52 for the Danger Receptor Galectin-8 in Antibacterial Autophagy. Sci. Signal 6 (261), ra9. doi: 10.1126/scisignal.2003730
Losier, T. T., Akuma, M., McKee-Muir, O. C., LeBlond, N. D., Suk, Y., Alsaadi, R. M., et al. (2019). AMPK Promotes Xenophagy Through Priming of Autophagic Kinases Upon Detection of Bacterial Outer Membrane Vesicles. Cell Rep. 26 (8), 2150–2165.e2155. doi: 10.1016/j.celrep.2019.01.062
Maho, H., Nobumichi, F., Atsushi, M., Akiko, N., Akitsugu, Y., Naonobu, F., et al. (2013). Autophagosomes Form at ER-Mitochondria Contact Sites. Nature 495 (7441), 389–393. doi: 10.1038/nature11910
Martinez, J., Almendinger, J., Oberst, A., Ness, R., Dillon, C. P., Fitzgerald, P., et al. (2011). Microtubule-Associated Protein 1 Light Chain 3 Alpha (LC3)-Associated Phagocytosis is Required for the Efficient Clearance of Dead Cells. Proc. Natl. Acad. Sci. U.S.A. 108 (42), 17396–17401. doi: 10.1073/pnas.1113421108
Medzhitov, R. (2001). Toll-Like Receptors and Innate Immunity. Nat. Rev. Immunol. 1 (2), 135–145. doi: 10.1038/35100529
Meghil, M. M., Tawfik, O. K., Elashiry, M., Rajendran, M., Arce, R. M., Fulton, D. J., et al. (2019). Disruption of Immune Homeostasis in Human Dendritic Cells via Regulation of Autophagy and Apoptosis by Porphyromonas Gingivalis. Front. Immunol. 10. doi: 10.3389/fimmu.2019.02286
Mizushima, N., Komatsu, M. (2011). Autophagy: Renovation of Cells and Tissues. Cell 147 (4), 728–741. doi: 10.1016/j.cell.2011.10.026
Mizushima, N., Levine, B. (2020). Autophagy in Human Diseases. N. Engl. J. Med. 383 (16), 1564–1576. doi: 10.1056/NEJMra2022774
Mizushima, N., Levine, B., Cuervo, A. M., Klionsky, D. J. (2008). Autophagy Fights Disease Through Cellular Self-Digestion. Nature 451 (7182), 1069–1075. doi: 10.1038/nature06639
Mizushima, N., Noda, T., Yoshimori, T., Tanaka, Y., Ohsumi, Y. A. (1998). A Protein Conjugation System Essential for Autophagy. Nature 395 (6700), 395–398. doi: 10.1038/26506
Niu, L., Chen, S., Yang, X., Ma, C., Pan, C., Wang, H., et al. (2020). Vitamin D Decreases Porphyromonas Gingivalis Internalized Into Macrophages by Promoting Autophagy. Oral. Dis. 27 (7), 1775–1788. doi: 10.1111/odi.13696
Niu, H., Xiong, Q., Yamamoto, A., Hayashi-Nishino, M., Rikihisa, Y. (2012). Autophagosomes Induced by a Bacterial Beclin 1 Binding Protein Facilitate Obligatory Intracellular Infection. Proc. Natl. Acad. Sci. U.S.A. 109 (51), 20800–20807. doi: 10.1073/pnas.1218674109
Ogawa, T., Yagi, T. (2010). Bioactive Mechanism of Porphyromonas Gingivalis Lipid A. Periodontol 54 (1), 71–77. doi: 10.1111/j.1600-0757.2009.00343.x
Ogawa, M., Yoshimori, T., Suzuki, T., Sagara, H., Mizushima, N., Sasakawa, C. (2005). Escape of Intracellular Shigella From Autophagy. Science 307 (5710), 727–731. doi: 10.1126/science.1106036
Park, M. H., Jeong, S. Y., Na, H. S., Chung, J. (2017). Porphyromonas Gingivalisinduces Autophagy in THP-1-Derived Macrophages. Mol. Oral. Microbiol. 32 (1), 48–59. doi: 10.1111/omi.12153
Park, E., Na, H. S., Song, Y. R., Shin, S. Y., Kim, Y. M., Chung, J. (2014). Activation of NLRP3 and AIM2 Inflammasomes by Porphyromonas Gingivalis Infection. Infect. Immun. 82 (1), 112–123. doi: 10.1128/IAI.00862-13
Periyasamy, P., Guo, M. L., Buch, S. (2016). Cocaine Induces Astrocytosis Through ER Stress-Mediated Activation of Autophagy. Autophagy 12 (8), 1310–1329. doi: 10.1080/15548627.2016.1183844
Petrilli, V., Papin, S., Dostert, C., Mayor, A., Martinon, F., Tschopp, J. (2007). Activation of the NALP3 Inflammasome is Triggered by Low Intracellular Potassium Concentration. Cell Death Differ. 14 (9), 1583–1589. doi: 10.1038/sj.cdd.4402195
Polson, H. E., de Lartigue, J., Rigden, D. J., Reedijk, M., Urbe, S., Clague, M. J., et al. (2010). Mammalian Atg18 (WIPI2) Localizes to Omegasome-Anchored Phagophores and Positively Regulates LC3 Lipidation. Autophagy 6 (4), 506–522. doi: 10.4161/auto.6.4.11863
Prabakaran, T., Bodda, C., Krapp, C., Zhang, B. C., Christensen, M. H., Sun, C., et al. (2018). Attenuation of cGAS-STING Signaling is Mediated by a P62/SQSTM1-Dependent Autophagy Pathway Activated by TBK1. EMBO J. 37 (8), 17–33. doi: 10.15252/embj.201797858
Py, B. F., Lipinski, M. M., Yuan, J. (2007). Autophagy Limits Listeria Monocytogenes Intracellular Growth in the Early Phase of Primary Infection. Autophagy 3 (2), 117–125. doi: 10.4161/auto.3618
Randow, F. (2011). How Cells Deploy Ubiquitin and Autophagy to Defend Their Cytosol From Bacterial Invasion. Autophagy 7 (3), 304–309. doi: 10.4161/auto.7.3.14539
Randow, F., Munz, C. (2012). Autophagy in the Regulation of Pathogen Replication and Adaptive Immunity. Trends Immunol. 33 (10), 475–487. doi: 10.1016/j.it.2012.06.003
Ravenhill, B. J., Boyle, K. B., von Muhlinen, N., Ellison, C. J., Masson, G. R., Otten, E. G., et al. (2019). The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell 74(2), 320–329.e326. doi: 10.1016/j.molcel.2019.01.041
Rodrigues, P. H., Reyes, L., Chadda, A. S., Belanger, M., Wallet, S. M., Akin, D., et al. (2012). Porphyromonas Gingivalis Strain Specific Interactions With Human Coronary Artery Endothelial Cells: A Comparative Study. PloS One 7 (12), e52606. doi: 10.1371/journal.pone.0052606
Romagnoli, A., Etna, M. P., Giacomini, E., Pardini, M., Remoli, M. E., Corazzari, M., et al. (2012). ESX-1 Dependent Impairment of Autophagic Flux by Mycobacterium Tuberculosis in Human Dendritic Cells. Autophagy 8 (9), 1357–1370. doi: 10.4161/auto.20881
Rutger Persson, G. (2012). Rheumatoid Arthritis and Periodontitis - Inflammatory and Infectious Connections. Review of the Literature. J. Oral. Microbiol. 4, 11829–11944. doi: 10.3402/jom.v4i0.11829
Saitoh, T., Fujita, N., Jang, M. H., Uematsu, S., Yang, B. G., Satoh, T., et al. (2008). Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1beta Production. Nature 456 (7219), 264–268. doi: 10.1038/nature07383
Shaid, S., Brandts, C. H., Serve, H., Dikic, I. (2013). Ubiquitination and Selective Autophagy. Cell Death Differ. 20 (1), 21–30. doi: 10.1038/cdd.2012.72
Shao, B. Z., Xu, Z. Q., Han, B. Z., Su, D. F., Liu, C. (2015). NLRP3 Inflammasome and its Inhibitors: A Review. Front. Pharmacol. 6. doi: 10.3389/fphar.2015.00262
Sharma, V., Verma, S., Seranova, E., Sarkar, S., Kumar, D. (2018). Selective Autophagy and Xenophagy in Infection and Disease. Front. Cell Dev. Biol. 6. doi: 10.3389/fcell.2018.00147
Shi, C. S., Shenderov, K., Huang, N. N., Kabat, J., Abu-Asab, M., Fitzgerald, K. A., et al. (2012). Activation of Autophagy by Inflammatory Signals Limits IL-1beta Production by Targeting Ubiquitinated Inflammasomes for Destruction. Nat. Immunol. 13 (3), 255–263. doi: 10.1038/ni.2215
Shi, H., Wang, Y., Li, X., Zhan, X., Tang, M., Fina, M., et al. (2016). NLRP3 Activation and Mitosis are Mutually Exclusive Events Coordinated by NEK7, a New Inflammasome Component. Nat. Immunol. 17 (3), 250–258. doi: 10.1038/ni.3333
Shu, H. B., Wang, Y. Y. (2014). Adding to the STING. Immunity 41 (6), 871–873. doi: 10.1016/j.immuni.2014.12.002
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009). Autophagy Regulates Lipid Metabolism. Nature 458 (7242), 1131–1135. doi: 10.1038/nature07976
Singhrao, S. K., Harding, A., Poole, S., Kesavalu, L., Crean, S. (2015). Porphyromonas Gingivalis Periodontal Infection and Its Putative Links With Alzheimer's Disease. Mediators Inflammation 2015, 137357. doi: 10.1155/2015/137357
Socransky, S. S., Haffajee, A. D., Cugini, M. A., Smith, C., Kent, R. L., Jr. (1998). Microbial Complexes in Subgingival Plaque. J. Clin. Periodontol. 25 (2), 134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x
Song, L., Tan, J., Wang, Z., Ding, P., Tang, Q., Xia, M., et al. (2019). Interleukin17A Facilitates Osteoclast Differentiation and Bone Resorption via Activation of Autophagy in Mouse Bone Marrow Macrophages. Mol. Med. Rep. 19 (6), 4743–4752. doi: 10.3892/mmr.2019.10155
Sorbara, M. T., Girardin, S. E. (2011). Mitochondrial ROS Fuel the Inflammasome. Cell Res. 21 (4), 558–560. doi: 10.1038/cr.2011.20
Spalinger, M. R., Lang, S., Gottier, C., Dai, X., Rawlings, D. J., Chan, A. C., et al. (2017). PTPN22 Regulates NLRP3-Mediated IL1B Secretion in an Autophagy-Dependent Manner. Autophagy 13 (9), 1590–1601. doi: 10.1080/15548627.2017.1341453
Steele, S., Brunton, J., Ziehr, B., Taft-Benz, S., Moorman, N., Kawula, T. (2013). Francisella Tularensis Harvests Nutrients Derived via ATG5-Independent Autophagy to Support Intracellular Growth. PloS Pathog. 9 (8), e1003562. doi: 10.1371/journal.ppat.1003562
Sturgill-Koszycki, S., Schlesinger, P. H. (1994). Lack of Acidification in Mycobacterium Phagosomes Produced by Exclusion of the Vesicular. Science 263 (5147), 678–681. doi: 10.1126/science.8303277
Sun, L., Wu, J., Du, F., Chen, X., Chen, Z. J. (2013). Cyclic GMP-AMP Synthase is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 339 (6121), 786–791. doi: 10.1126/science.1232458
Takahama, M., Akira, S., Saitoh, T. (2018). Autophagy Limits Activation of the Inflammasomes. Immunol. Rev. 281 (1), 62–73. doi: 10.1111/imr.12613
Takeuchi, H., Furuta, N., Morisaki, I., Amano, A. (2011). Exit of Intracellular Porphyromonas Gingivalis From Gingival Epithelial Cells is Mediated by Endocytic Recycling Pathway. Cell Microbiol. 13 (5), 677–691. doi: 10.1111/j.1462-5822.2010.01564.x
Tattoli, I., Sorbara, M. T., Vuckovic, D., Ling, A., Soares, F., Carneiro, L. A., et al. (2012). Amino Acid Starvation Induced by Invasive Bacterial Pathogens Triggers an Innate Host Defense Program. Cell Host Microbe 11 (6), 563–575. doi: 10.1016/j.chom.2012.04.012
Thurston, T. L., Boyle, K. B., Allen, M., Ravenhill, B. J., Karpiyevich, M., Bloor, S., et al. (2016). Recruitment of TBK1 to Cytosol-Invading Salmonella Induces WIPI2-Dependent Antibacterial Autophagy. EMBO J. 35 (16), 1779–1792. doi: 10.15252/embj.201694491
Thurston, T. L., Ryzhakov, G., Bloor, S., von Muhlinen, N., Randow, F. (2009). The TBK1 Adaptor and Autophagy Receptor NDP52 Restricts the Proliferation of Ubiquitin-Coated Bacteria. Nat. Immunol. 10 (11), 1215–1221. doi: 10.1038/ni.1800
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A., Randow, F. (2012). Galectin 8 Targets Damaged Vesicles for Autophagy to Defend Cells Against Bacterial Invasion. Nature 482 (7385), 414–418. doi: 10.1038/nature10744
Tokutomi, F., Wada-Takahashi, S., Sugiyama, S., Toyama, T., Sato, T., Hamada, N., et al. (2015). Porphyromonas Gingivalis-Induced Alveolar Bone Loss is Accelerated in the Stroke-Prone Spontaneously Hypertensive Rat. Arch. Oral. Biol. 60 (6), 911–918. doi: 10.1016/j.archoralbio.2015.02.012
Tong, X., Gu, J., Song, R., Wang, D., Sun, Z., Sui, C., et al. (2018). Osteoprotegerin Inhibit Osteoclast Differentiation and Bone Resorption by Enhancing Autophagy via AMPK/mTOR/p70S6K Signaling Pathway In Vitro. J. Cell Biochem. 120 (2), 1630–1642. doi: 10.1002/jcb.27468
Tumbarello, D. A., Manna, P. T., Allen, M., Bycroft, M., Arden, S. D., Kendrick-Jones, J., et al. (2015). The Autophagy Receptor TAX1BP1 and the Molecular Motor Myosin VI Are Required for Clearance of Salmonella Typhimurium by Autophagy. PloS Pathog. 11 (10), e1005174. doi: 10.1371/journal.ppat.1005174
Umemoto, T., Hamada, N. (2003). Characterization of Biologically Active Cell Surface Components of a Periodontal Pathogen. The Roles of Major and Minor Fimbriae of Porphyromonas Gingivalis. J. Periodontol. 74 (1), 119–122. doi: 10.1902/jop.2003.74.1.119
Waddington, R. J., Moseley, R., Embery, G. (2000). Reactive Oxygen Species: A Potential Role in the Pathogenesis of Periodontal Diseases. Oral. Dis. 6 (3), 138–151. doi: 10.1111/j.1601-0825.2000.tb00325.x
Wang, Z., Li, C. (2020). Xenophagy in Innate Immunity: A Battle Between Host and Pathogen. Dev. Comp. Immunol. 109, 103693. doi: 10.1016/j.dci.2020.103693
Wang, Y., Li, L., Hou, C., Lai, Y., Long, J., Liu, J., et al. (2016). SNARE-Mediated Membrane Fusion in Autophagy. Semin. Cell Dev. Biol. 60, 97–104. doi: 10.1016/j.semcdb.2016.07.009
Wan, D., Jiang, W., Hao, J. (2020). Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front. Immunol. 11. doi: 10.3389/fimmu.2020.00615
Watanabe, N., Yokoe, S., Ogata, Y., Sato, S., Imai, K. (2020). Exposure to Porphyromonas Gingivalis Induces Production of Proinflammatory Cytokine via TLR2 From Human Respiratory Epithelial Cells. J. Clin. Med. 9 (11), 3433–3445. doi: 10.3390/jcm9113433
Watson, R. O., Bell, S. L., MacDuff, D. A., Kimmey, J. M., Diner, E. J., Olivas, J., et al. (2015). The Cytosolic Sensor cGAS Detects Mycobacterium Tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 17 (6), 811–819. doi: 10.1016/j.chom.2015.05.004
Watson, R. O., Manzanillo, P. S., Cox, J. S. (2012). Extracellular M. Tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell 150 (4), 803–815. doi: 10.1016/j.cell.2012.06.040
Wei, W., An, Y., An, Y., Fei, D., Wang, Q. (2018). Activation of Autophagy in Periodontal Ligament Mesenchymal Stem Cells Promotes Angiogenesis in Periodontitis. J. Periodontol. 89 (6), 718–727. doi: 10.1002/jper.17-0341
Whitney, C., Ant, J., Moncla, B., Johnson, B., Page, R. C., Engel, D. (1992). Serum Immunoglobulin G Antibody to Porphyromonas Gingivalis in Rapidly Progressive Periodontitis: Titer, Avidity, and Subclass Distribution. Infect. Immun. 60 (6), 2194–2200. doi: 10.1128/IAI.60.6.2194-2200.1992
Wild, P., Farhan, H. (2011). Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science. Science 333, 228–233. doi: 10.1126/science.1205405
Wild, P., Farhan, H., McEwan, D. G., Wagner, S., Rogov, V. V., Brady, N. R., et al. (2011). Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science 333 (6039), 228–233. doi: 10.1126/science.1205405
Winning, L., Linden, G. J. (2017). Periodontitis and Systemic Disease: Association or Causality? Curr. Oral. Health Rep. 4 (1), 1–7. doi: 10.1007/s40496-017-0121-7
Wu, C. H., Gan, C. H., Li, L. H., Chang, J. C., Chen, S. T., Menon, M. P., et al. (2020). A Synthetic Small Molecule F240B Decreases NLRP3 Inflammasome Activation by Autophagy Induction. Front. Immunol. 11. doi: 10.3389/fimmu.2020.607564
Wu, Y. W., Li, F. (2019). Bacterial Interaction With Host Autophagy. Virulence 10 (1), 352–362. doi: 10.1080/21505594.2019.1602020
Xiao, W., Dong, G., Pacios, S., Alnammary, M., Barger, L. A., Wang, Y., et al. (2015). FOXO1 Deletion Reduces Dendritic Cell Function and Enhances Susceptibility to Periodontitis. Am. J. Pathol. 185 (4), 1085–1093. doi: 10.1016/j.ajpath.2014.12.006
Xie, H., Cai, S., Lamont, R. J. (1997). Environmental Regulation of Fimbrial Gene Expression in Porphyromonas Gingivalis. Infect. Immun. 65 (6), 2265–2271. doi: 10.1128/IAI.65.6.2265-2271.1997
Xie, M., Tang, Q., Yu, S., Sun, J., Mei, F., Zhao, J., et al. (2020). Porphyromonas Gingivalis Disrupts Vascular Endothelial Homeostasis in a TLR-NF-kappaB Axis Dependent Manner. Int. J. Oral. Sci. 12 (1), 28. doi: 10.1038/s41368-020-00096-z
Xu, Y., Zhou, P., Cheng, S., Lu, Q., Nowak, K., Hopp, A. K., et al. (2019). A Bacterial Effector Reveals the V-ATPase-ATG16L1 Axis That Initiates Xenophagy. Cell 178(3), 552–566.e520. doi: 10.1016/j.cell.2019.06.007
Yang, Z., Klionsky, D. J. (2010). Mammalian Autophagy: Core Molecular Machinery and Signaling Regulation. Curr. Opin. Cell Biol. 22 (2), 124–131. doi: 10.1016/j.ceb.2009.11.014
Yang, X., Niu, L., Pan, Y., Feng, X., Liu, J., Guo, Y., et al. (2020). LL-37-Induced Autophagy Contributed to the Elimination of Live Porphyromonas Gingivalis Internalized in Keratinocytes. Front. Cell Infect. Microbiol. 10. doi: 10.3389/fcimb.2020.561761
Yao, L., Jermanus, C., Barbetta, B., Choi, C., Verbeke, P., Ojcius, D. M., et al. (2010). Porphyromonas Gingivalis Infection Sequesters Pro-Apoptotic Bad Through Akt in Primary Gingival Epithelial Cells. Mol. Oral. Microbiol. 25 (2), 89–101. doi: 10.1111/j.2041-1014.2010.00569.x
Yim, W. W., Mizushima, N. (2020). Lysosome Biology in Autophagy. Cell Discovery 6, 6. doi: 10.1038/s41421-020-0141-7
Yoneda, M., Naka, S., Nakano, K., Wada, K., Endo, H., Mawatari, H., et al. (2012). Involvement of a Periodontal Pathogen, Porphyromonas Gingivalis on the Pathogenesis of non-Alcoholic Fatty Liver Disease. BMC Gastroenterol. 12, 16. doi: 10.1186/1471-230X-12-16
Yoshikawa, Y., Ogawa, M., Hain, T., Yoshida, M., Fukumatsu, M., Kim, M., et al. (2009). Listeria Monocytogenes ActA-Mediated Escape From Autophagic Recognition. Nat. Cell Biol. 11 (10), 1233–1240. doi: 10.1038/ncb1967
Zaitsu, Y., Iwatake, M., Sato, K., Tsukuba, T. (2016). Lipid Droplets Affect Elimination of Porphyromonas Gingivalis in HepG2 Cells by Altering the Autophagy-Lysosome System. Microbes Infect. 18 (9), 565–571. doi: 10.1016/j.micinf.2016.05.004
Zhang, D., He, Y., Ye, X., Cai, Y., Xu, J., Zhang, L., et al. (2020). Activation of Autophagy Inhibits Nucleotide-Binding Oligomerization Domain-Like Receptor Protein 3 Inflammasome Activation and Attenuates Myocardial Ischemia-Reperfusion Injury in Diabetic Rats. J. Diabetes Investig 11 (5), 1126–1136. doi: 10.1111/jdi.13235
Zhang, L., Hu, W., Cho, C. H., Chan, F. K., Yu, J., Fitzgerald, J. R., et al. (2018). Reduced Lysosomal Clearance of Autophagosomes Promotes Survival and Colonization of Helicobacter Pylori. J. Pathol. 244 (4), 432–444. doi: 10.1002/path.5033
Zhang, J., Yu, C., Zhang, X., Chen, H., Dong, J., Lu, W., et al. (2018). Porphyromonas Gingivalis Lipopolysaccharide Induces Cognitive Dysfunction, Mediated by Neuronal Inflammation via Activation of the TLR4 Signaling Pathway in C57BL/6 Mice. J. Neuroinflamm. 15 (1), 37. doi: 10.1186/s12974-017-1052-x
Zhao, J., Geng, W., Wan, K., Guo, K., Xi, F., Xu, X., et al. (2021). Lipoxin A4 Promotes Autophagy and Inhibits Overactivation of Macrophage Inflammasome Activity Induced by Pg LPS. J. Int. Med. Res. 49 (2), 300060520981259. doi: 10.1177/0300060520981259
Zheng, Y. T., Shahnazari, S., Brech, A., Lamark, T., Johansen, T., Brumell, J. H. (2009). The Adaptor Protein P62/SQSTM1 Targets Invading Bacteria to the Autophagy Pathway. J. Immunol. 183 (9), 5909–5916. doi: 10.4049/jimmunol.0900441
Zhong, Z., Umemura, A., Sanchez-Lopez, E., Liang, S., Shalapour, S., Wong, J., et al. (2016). NF-κb Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 164 (5), 896–910. doi: 10.1016/j.cell.2015.12.057
Keywords: autophagy, Porphyromonas gingivalis, inflammation, xenophagy, macroautophagy
Citation: Kang S, Dai A, Wang H and Ding P-H (2022) Interaction Between Autophagy and Porphyromonas gingivalis-Induced Inflammation. Front. Cell. Infect. Microbiol. 12:892610. doi: 10.3389/fcimb.2022.892610
Received: 09 March 2022; Accepted: 06 June 2022;
Published: 01 July 2022.
Edited by:
Jianzhu Liu, Shandong Agricultural University, ChinaCopyright © 2022 Kang, Dai, Wang and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pei-Hui Ding, phding@zju.edu.cn; Huiming Wang, whmwhm@zju.edu.cn