Invasive Streptococcus suis isolated in Spain contain a highly promiscuous and dynamic resistome
 Cristina Uruén
Cristina Uruén Jorge Gimeno
Jorge Gimeno Marina Sanz1,2
Marina Sanz1,2  Jesús Arenas
Jesús Arenas- 1Unit of Microbiology and Immunology, Faculty of Veterinary, University of Zaragoza, Zaragoza, Spain
- 2Institute Agrofood of Aragón-IA2, University of Zaragoza-CITA, Zaragoza, Spain
- 3Department of Animal Science, ETSEA, University of Lleida-Agrotecno, Lleida, Spain
- 4Department of Animal Production and Health, CITA, Zaragoza, Spain
Introduction: Streptococcus suis is a major pathogen for swine and human. Here we aimed to know the rates of antimicrobial resistance (AMR) in invasive S. suis isolates recovered along Spain between 2016 – 2021 and elucidate their genetic origin.
Methods: Antibiotic susceptibility testing was performed for 116 isolates of different genetic backgrounds and geographic origins against 18 antibiotics of 9 families. The association between AMR and genotypes and the origin of the isolates were statistically analyzed using Pearson´s chi-square test and the likelihood ratio. The antimicrobial resistant genes were identified by whole genome sequencing analysis and PCR screenings.
Results: High AMR rates (>80%) were detected for tetracyclines, spectinomycin, lincosamides, and marbofloxacin, medium (20-40%) for sulphonamides/trimethoprim, tiamulin, penicillin G, and enrofloxacin, and low (< 20%) for florfenicol, and four additional β-lactams. The occurrence of multidrug resistance was observed in 90% of isolates. For certain antibiotics (penicillin G, enrofloxacin, marbofloxacin, tilmicosin, and erythromycin), AMR was significantly associated with particular sequence types (STs), geographic regions, age of pigs, and time course. Whole genome sequencing comparisons and PCR screenings identified 23 AMR genes, of which 19 were previously reported in S. suis (aph(3’)-IIIa, sat4, aadE, spw, aac(6’)-Ie-aph(2’’)-Ia, fexA, optrA, erm(B), mef(A/E), mrs(D), mph(C), lnu(B), lsa(E), vga(F), tet(M), tet(O), tet(O/W/32/O), tet(W)), and 4 were novel (aph(2’’)-IIIa, apmA, erm(47), tet(T)). These AMR genes explained the AMR to spectinomycin, macrolides, lincosamides, tiamulin, and tetracyclines. Several genes were located on mobile genetic elements which showed a variable organization and composition. As AMR gene homologs were identified in many human and animal pathogens, the resistome of S. suis has a different phylogenetic origin. Moreover, AMR to penicillin G, fluoroquinolones, and trimethoprim related to mutations in genes coding for target enzymes (pbp1a, pbp2b, pbp2x, mraY, gyrA, parC, and dhfr). Bioinformatic analysis estimated traits of recombination on target genes, also indicative of gene transfer events.
Conclusions: Our work evidences that S. suis is a major contributor to AMR dissemination across veterinary and human pathogens. Therefore, control of AMR in S. suis should be considered from a One Health approach in regions with high pig production to properly tackle the issue of antimicrobial drug resistance.
1 Introduction
The Gram-positive coccus Streptococcus suis is a common inhabitant of the upper respiratory tract of pigs, but it is also the aetiological agent of streptococcal swine disease, a life-threatening infection characterized by the fast development of endocarditis, septicaemia, arthritis, and meningitis (Vötsch et al., 2018). It is a major cause of morbidity and mortality in sucking and transition pigs (Neila-Ibáñez et al., 2023), but can also cause infections in older pigs and sows. S. suis can be transmitted from pigs to humans by direct contact with animals, manipulation of pig meat, and consumption of raw meat. S. suis is also a zoonotic pathogen able to cause infections in humans as described in pigs (Goyette-Desjardins et al., 2014). Indeed, S. suis is one of the main meningitis-causing agents in several Asian countries.
Capsule is a major virulence factor for S. suis. The bacteria can express either one of the 29 different capsular polysaccharides (serotypes), but capsules 2, 3, 7, 8, 4, 1 1/2, and 9 are the most prevalent in clinical isolates from pigs (Goyette-Desjardins et al., 2014). Based on the antigenicity of the capsule, a serotyping scheme is used for routine diagnosis of strain virulence. Serotype distribution varies geographically, but serotype 2 predominates worldwide within clinical isolates (Goyette-Desjardins et al., 2014). In Spain, a recent study reported the most prevalent serotypes isolated from diseased pigs were 2 (21%), 1 (21%), 9 (19%), 3 (6%), and 7 (3%) (Petrocchi-Rilo et al., 2021). However, isolates of the same serotype show genetic heterogeneity, and thus genetic typing systems can better characterize this pathogen. Indeed, a Multi Locus Sequence Typing (MLST) scheme is available for S. suis and is globally used for epidemiological studies. Some sequence types (STs) are restricted to particular geographic regions (Goyette-Desjardins et al., 2014), for example, ST25 and ST28 in North America, while others are amply distributed, as ST1. Also, ST1 isolates were amply recovered from diseased people (Goyette-Desjardins et al., 2014).
Currently, the main immunization methods against S. suis in pig farming involve the use of commercial whole cells or bacterins prepared from clones recovered from diseased animals (Segura, 2015). However, the efficacy of bacterins is controversial (Corsaut et al., 2020), and there is a lack of tools to control safety, immunogenicity, or protective efficacy, leading to reliance on antibiotics as the primary defense against S. suis infections. This widespread antibiotic use contributes to high rates of antimicrobial resistance (AMR), particularly to tetracyclines, macrolides, β-lactams, aminoglycosides, sulfamethoxazole/trimethoprim (TMP), chloramphenicol, and fluoroquinolones, reported in clinical isolates recovered from diseased pigs and humans worldwide (Uruén et al., 2022). Many AMR genes were identified in S. suis (Dechêne-Tempier et al., 2021; Uruén et al., 2022). Some of them are placed on mobile genetic elements (MGE) such as integrative conjugative elements (ICEs), integrative mobile elements (IMEs), plasmids, transposons, genomic islands, or prophages. Thus AMR genes can easily migrate within strains (Dechêne-Tempier et al., 2021; Uruén et al., 2022). Notably, MGEs can be transferred from S. suis to different human streptococci pathogens such as Streptococcus agalactiae or Streptococcus pyogenes (Marini et al., 2015) or to pathogenic enterococci such as Enterococcus faecalis (Huang et al., 2016). This highlights S. suis as a relevant reservoir of AMR genes with potential implications for public health (Marini et al., 2015).
Spain is the highest pig producer in Europe, and it is hardly affected by S. suis infections. A recent study revealed that the percentage of units of sucking and nursery piglets clinically affected by S. suis is around 81% (Neila-Ibáñez et al., 2023). Antibiotics are highly used to treat and prevent S. suis infections in Spain (Neila-Ibáñez et al., 2023). Despite this microorganism is a zoonotic pathogen that can contribute to AMR dissemination, the rate of AMR for different antibiotics and their resistance mechanisms have been poorly studied. This knowledge is the basis for the design-built strategies to control S. suis disease in pigs and to minimize AMR spread. Therefore, we aimed here to know the rates of AMR for 18 different antibiotics in invasive isolates of S. suis recovered around Spain and to elucidate their genetic origin.
2 Material and methods
2.1 Bacterial isolates and growth conditions
A panel of 116 S. suis clinical isolates was selected in this study (Supplementary Table S1) from our S. suis collection described in our previous work (Uruén et al., 2024). The selective sampling criteria were i) only one isolate from the same farm, to avoid over-representation of endemic clones; ii) isolates should represent the most productive Autonomous Communities, iii) only isolates from internal organs or blood in which only one clone (serotype) was detected (to rule out the selection of opportunistic clones), iv) isolates recovered from the period 2014-2021 to have an indication of the current circulating Ss. 156 out of 843 isolates that matched the selective criteria were analyzed. All isolates were previously characterized in terms of serotyping, ST, and the presence of virulence-associated factors (Uruén et al., 2024).
Isolates were cultured onto BD™ Columbia Agar with 5% Sheep Blood (Columbia, Heidelberg, Germany) or Todd-Hewitt Agar (Oxoid Ltd., Hampshire, England) at 37°C in a candle jar for 18-24 hours. Todd-Hewitt liquid cultures were started from colonies grown on solid agar adjusted to an Optical Density of 600 nm (OD600) of 0.05. For antibiotic sensibility testing, bacteria were grown in Muëller-Hinton Broth (MHB) as described below.
2.2 Antibiotic susceptibility testing
Minimum Inhibitory Concentration (MIC) was determined in 18 antibiotics from 9 families using the broth microdilution method. Bacteria were thawed, propagated on blood agar, and incubated at 37°C in candle jars for 18-24 h. Three to five colonies were picked up and emulsified in Cation Adjusted MHB to obtain turbidity of 0.5 McFarland standard (Sensititre™ nephelometer V3011). Bacterial suspensions were further diluted in Cation Adjusted MHB with 2.5-5% Lysed Horse Blood to reach a final inoculum concentration of 5 x 105 colony-forming units in 1 ml. A panel of 11 antibiotics (penicillin G, ampicillin, amoxicillin, ceftiofur, cefquinome, sulfamethoxazole/Timetropim (TMP), tiamulin, florfenicol, enrofloxacin, marbofloxacin, and doxycycline) was tested in customized 96-well microtiter plates (Sensititre, Trek Diagnostic Systems Inc., East Grinstead, UK). Plates were reconstituted by adding 100 μl/well of the inoculum. Besides, a panel of 7 antibiotics (spectinomycin, erythromycin, tilmicosin, tylosin, clindamycin, lincomycin, and tetracycline) were tested in 96-well microtiter U bottom plates (Deltalab, Barcelona, Spain). Antibiotic dilutions were prepared following the recommendations of ISO 20776-1:2019, and 100 μl of each antibiotic was added to each well together with the bacterial inoculum as depicted above. The MIC value was established as the lowest drug concentration inhibiting visible growth. Streptococcus pneumoniae (ATCC 49619™) was included as internal quality control following CLSI recommendations (2019). The resistance/susceptibility to each antibiotic was determined based on breakpoints established in EUCAST (EUCAST, 2023) and CLSI (CLSI, 2019), and when not available, ECOFF 95% or literature was used.
2.3 Statistical analysis
Associations between AMR, the genotype of isolates, geographical distribution, age of animals, and year of isolation were analyzed using Pearson’s chi-square test, however, when comparing two categories and more than 20% of the cells had a frequency lower than 5 units, the likelihood ratio test was used. Associations were considered statistically significant when the p-value was lower than 0.05. In addition, adjusted standardized residues (ASR) were calculated and analyzed. When the ASR value is higher than 1.96, the frequency is significantly higher than expected and the relationship is considered positively significant; if the ASR value is lower than -1.96, the frequency is significantly lower than expected, and the association is considered negatively significant. When the ASR value is between -1.96 and 1.96 the association between variables is not statistically significant. All statistical analyses were performed with SPSS software version 26 (IBM Corporation, Armonk, NY, USA). In some analyses the risk ratio (RR) was calculated as in (Viera, 2008). Shortly, RR was estimated with the formula where a is the number of resistant isolates with the mutations, c is the number of resistant isolates with no mutations, b is the number of susceptible isolates with the mutations, and d is the number of susceptible isolates with no mutations.
2.4 Chromosomal DNA extraction
The chromosomal DNA was obtained using the Chromosomal DNA extraction kit Wizard® Genomic DNA Purification Kit (Promega, USA), according to manufacturer instructions, and used as template DNA for PCRs and whole genome sequencing. The DNA integrity was evaluated in a 0.7% agarose gel, and the quality and quantity of DNA were measured with a Nanodrop spectrophotometer (Implen, Madrid, Spain) and a Qubit 4 fluorimeter (Thermo Fisher Scientific, DE, USA).
2.6 Whole genome sequencing and bioinformatics analyses
Whole genome sequencing was performed in 11 S. suis isolates (Ss_02, Ss_69, Ss_70, Ss_80_ Ss_92, Ss_93, Ss_100, Ss_121, Ss_156, Ss_166, and Ss_167). The selective criteria were i) isolates that showed resistance to most antibiotics tested in this study, particularly isolates with different AMR patterns, and ii) represent all major genetic clusters and some genetically unrelated singletons. This was also considering 19 previously sequenced genomes (Ss_08, Ss_20, Ss_21, Ss_22, Ss_24, Ss_45, Ss_46, Ss_48, Ss_51, Ss_52, Ss_53, Ss_72, Ss_84, Ss_106, Ss_107, Ss_109, Ss_115, Ss_124, and Ss_134) (Uruén et al., 2024). For whole genome sequencing, the Illumina HiSeq 2500 system was used. The preparation of the DNA-Seq libraries was performed at STAB Vida Lda (Caparica, Portugal), followed by sequencing by HiSeq 2500. Quality control of the raw reads was conducted using FastQC v.0.11.7 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Trimmomatic v.0.38 was employed for trimming, aiming to eliminate low-quality reads and adaptors (Bolger et al., 2014). The bowtie2 tool was used to remove any potential contamination by human DNA (Homo sapiens (b38):hg38). Lastly, the SPAdes tool was used to assemble the sequences, matching the trimmed short-read Illumina sequences. The quality of the assembly was assessed using Quast 5.0.2 (Gurevich et al., 2013). For pangenome relatedness, firstly, sequences of the 30 genomes of our collection and 8 genomes of reference strains were analyzed with Prokka tool (Seemann, 2014). The obtained data were used to construct a maximum-likelihood phylogenetic tree using the Roary tool to obtain accessory binary genes in Newick format (Page et al., 2015).
To identify the AMR genes from the 30 genomes of the isolates, the ABRicate tool against ResFinder and NCBI bacterial AMR gene database was used. ICEs and IMEs were identified with ICEberg 2.0 database. When required, DNA sequences were aligned with MAFFT (https://www.ebi.ac.uk/Tools/msa/mafft/). To search for traits of recombination, a full exploratory recombination scan using all methods of the program was carried out on DNA fragments with the RDP4 program (Martin et al., 2015). Recombination points were considered when 3 or more algorithm tests were positive. The non-synonymous/synonymous substitution rate ratio (dN/dS) was estimated for each gene individually. To do that, the gene nucleotide sequence was aligned with MAFFT, and a maximum-likelihood phylogeny tree was elaborated with MEGA11 (Tamura et al., 2021). The tree and coding sequences of each gene were introduced in HyPhy program (Kosakovsky Pond et al., 2020), and the single-likelihood ancestor counting method (SLAC) was carried out with a 95% confidence interval.
2.5 Detection of AMR genes by PCR
To identify AMR genes in the entire population, PCR screenings were performed targeting those AMR genes detected in our genomes (described above) and those reported in the literature as highly prevalent in different studies. Together, a panel of 38 AMR genes (including antibiotic targeted genes) were examined, comprising aph(3’)-IIIa, ant1, aacA, aadE, ant(9’)-Ia, aac(6’)-Ie-aph(2’’), sat4, spw, apmA, aph(2’’)-IIIa, pbp2x, pbp2b, pbp1a, mraY, fexA, optrA, gyrA, parC, erm(B), mef(A/E), mrs(D), erm(47), mph(B), mph(C), cfr, lnu(B), lsa(E), vga(F), dhfr, tet(O), tet(M), tet(T), tet(L), tet(K), tet(W), tet(40), tet(44), tet(O/W/32/O). For PCR assays, 1 µl of chromosomic DNA, 0.25 µM of different primer combinations (Supplementary Table S2), and 400 µM dNTPs, were mixed with 0.5 U Taq DNA polymerase and PCR buffer (Biotools, Madrid, Spain). The mix was first incubated for 10 min at 94°C, followed by 30 cycles of 45 sec at 94°C, 30 sec at the annealing temperature described in Supplementary Table S2, 1 min per 1 kb of expected PCR product size at 72°C (Supplementary Table S2), and a final extension of 7 min at 72°C. The reactions were performed in a thermocycler (Biometra TRIO, Madrid, Spain). Amplicons were visualized in 1% agarose gels stained with Green®Nucleic Acid Stain (Sigma-Aldrich, Darmstadt, Germany) in an iBright Imaging System (Thermo Fisher Scientific, DE, USA). All PCR products obtained in this work were purified for sequencing using PCR Product Pre-Sequencing Kit (Thermo Fisher Scientific, DE, USA), and, when required, sequenced at STAB Vida Lda (Caparica, Portugal). The sequence of pbp2x, pbp2b, pbp1a, mraY, dhfr, gyrA, and gyrB was extracted from the whole genome sequencing analysis and analyzed independently. Partial sequencing was performed in pbps and mraY genes in isolate Ss_104, in dhfr gene in isolates Ss_82, Ss_85, Ss_104, and Ss_138, in gyrA and parC genes in isolates Ss_09, Ss_168, and Ss_170. When required, DNA sequences were translated with MEGA and used for sequence comparisons.
3 Results
3.1 Origin of the isolates
A total of 116 invasive isolates of S. suis from diseased pigs with symptoms of streptococcal swine disease were studied here (Supplementary Table S1). Isolates were recovered from pigs located in farms of 13 Spanish Autonomous Communities, comprising high swine producers (8-3x106 pigs/year): Aragón (n=43), Cataluña (n=24), Castilla y Leon (n=16); moderate swine producers (1–3x106 pigs/year): Andalucía (n=5), Castilla La Mancha (n=8), Murcia (n=8), Galicia (n=2), Valencia (n=4); and, low swine producers (< 1x106 pigs/year): Cantabria (n=1), Extremadura (n=1), Madrid (n=1), Navarra (n=2), País Vasco (n=1). The sampling period was from 2016 to 2021. Isolates were from suckling piglets (n=11), transition pigs (n=74), and fattening pigs (n=3), but the age of the host was non-defined for 28 isolates. All isolates were previously characterized by ST, serotype, and virulence factor profile (Supplementary Table S1) as published in our previous work (Uruén et al., 2024). The four most frequent STs were ST1 (n=28), ST123 (n=22), ST29 (n=10), and ST3 (n=6). Also, few isolates were of ST1552 (n=3), ST24 (n=2), ST1642 (n=2), and ST94 (n= 3), and 40 belonged each one to a single ST, including ST13, ST16, ST17, ST198, ST141, ST198, ST949, ST1621, ST1622, ST1623, ST1624, ST1625, ST1626, ST1627, ST1628, ST1629, ST1630, ST1631, ST1632, ST1633, ST1634, ST1635, ST1636, ST1637, ST1638, ST1639, ST1640, ST1641, ST1644, ST1645, ST1649, ST1650, ST1651, ST1652, ST1653, ST1654, ST1822, ST1824, ST1825, ST2275. These STs were grouped in six phylogenetic eBurst Groups (eBGs): eBG1 (n=42), eBG2 (n=3), eBG3 (n=4), eBG4 (n=12), eBG5 (n=33), otherwise singletons (n=22) [characterized in Uruén et al. (2024)]. eBG2, eBG3, and eBG6 isolates belong to clonal complex (CC) CC16, CC24, and CC1628/1633, respectively. All isolates of eBG1 belong to CC1, but one isolate is unrelated. All isolates of eBG4 belong to CC29 but one is of CC28. eBG5 contain isolates of CC94 (n=4), CC123 (n=28), and one unrelated. Moreover, isolates were distributed in 11 serotypes, where serotype 9 prevailed (n=33), followed by serotype 1 (n=21) and serotype 2 (n=13). 14 isolates were non-tippable. The core genome of the 30 isolates used in this study and 8 public genomes was analyzed phylogenetically (Supplementary Figure S1). Two main clusters were generated. A cluster was constituted by all isolates of eBG1 and public genomes A7, O1/7, S10 and 2016UMN29656, all of them of CC11 but of different STs (ST1, ST3 and ST1642) and serotypes (1, 2, 1/2, 9 and 14). The other cluster was sub-grouped into two clusters. One cluster was constituted of two isolates and the public genome of 861160. They were of eBG2 (CC16) and singletons (CC20) that belonged to different serotypes (2, 4, 9) and STs (ST29, ST16, ST949). The remaining cluster contained singletons, and isolates of eBG5, eBG4, and eBG5. Isolates of eBG5 were of CC123 and CC94 and of serotypes 9 and 4 and eBG4 (CC29, ST29, non-tippable) and clustered together with genome SH1510. These genomes were phylogenetically related to isolates of eBG3 (CC24, ST1637, and ST24) than singletons. Hence, the collection of isolates studied here is very diverse regarding isolation origin (geographic location, host, and isolation year) and genetic background.
3.2 AMR profiles
The susceptibility of S. suis isolates against 18 antibiotics of 9 families was measured by the MIC. Included were one aminoglycoside (spectinomycin), five β-lactams (amoxicillin, ampicillin, cefquinome, ceftiofur, and penicillin G), one amphenicol (florfenicol), two fluoroquinolones (enrofloxacin, marbofloxacin), two lincosamides (clindamycin and lincomycin), one pleuromutilin (tiamulin), three macrolides (tilmicosin, tylosin, and erythromycin), sulfamethoxazole/TMP, and two tetracyclines (doxycycline and tetracycline). Amoxicillin, cefquinome, ceftiofur, and penicillin G are widely used to treat S. suis infections, the remaining antibiotics are of extensive use in the pig production industry to treat a variety of bacterial infectious diseases. The MIC value and the resulting phenotype for each isolate are listed in Supplementary Table S1, and the distribution of the MICs within the population is summarized in Figure 1. Results evidence variations in the distribution of the population for the different antibiotics. Hence, the distribution of MIC values for macrolides, tetracyclines, florfenicol, sulfamethoxazole/TMP, and clindamycin was bimodal, suggesting clear differences between susceptible and resistant populations. In contrast, the MIC distribution for fluoroquinolones and spectinomycin was normal, and for β-lactams, tiamulin, and lincosamides was right-skewed. Considering established and/or calculated breakpoints, high AMR rates were detected for tetracyclines (93%), macrolides (84%, 85%, 86% for erythromycin, tilmicosin, and tylosin, respectively), lincosamides (88% and 97% for clindamycin and lincomycin, respectively), and the fluoroquinolone marbofloxacin (81%). Medium AMR rates were detected for sulfamethoxazole/TMP (29%), tiamulin (19%), β-lactam penicillin G (32%), and enrofloxacin (30%). For florfenicol (5%), and the remaining β-lactams (2%) (amoxicillin, ampicillin, cefquinome, and ceftiofur), AMR rates were low. The occurrence of multidrug resistance, i.e., simultaneous AMR to antibiotics of at least three different families, was identified in 90% of the population. Up to 32 multi-resistance patterns were detected (Supplementary Figure S2). The most prevalent pattern (27% of isolates) was multi-resistance to fluoroquinolones, lincosamides, macrolides, and tetracyclines, although some isolates of this group were susceptible to certain antibiotics of the cited families. Conversely, isolate Ss_166, which belonged to this group, exhibited resistance to 14 antibiotics. The remaining patterns contained around or less than 10% of the isolates.
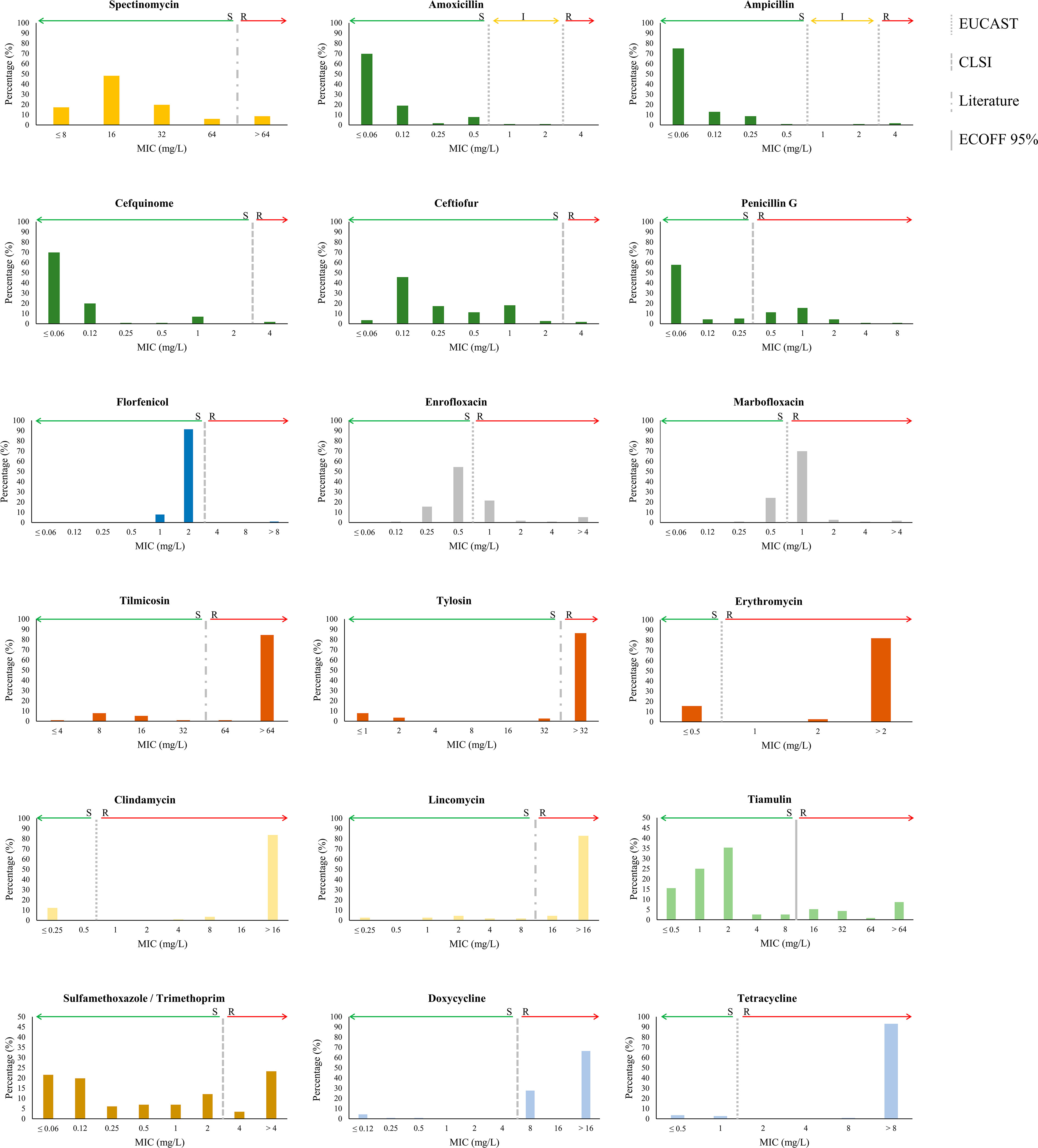
Figure 1 Distribution of MIC values for 18 antibiotics tested in 116 invasive S. suis isolates. Antibiotics of the same class are similarly coloured; the aminoglycosides (yellow), the β-lactams (dark green), the amphenicols (dark blue), the fluoroquinolones (grey), the lincosamides (light yellow), the macrolides (brown), the pleuromutilins (light green), the sulphonamides/TMP (ochre), and the tetracyclines (light blue). Breakpoints of resistance are indicated with vertical lines as determined by EUCAST, CLSI, literature reports, and ECOFF 95%.
3.3 Distribution of AMR
The association between AMR, MLST-based genotypes, and the origin of the isolates was statistically analyzed. The high AMR rates for tetracyclines, macrolides, and lincosamides prevented significant associations with these antibiotics. But statistically significant associations were found with other antibiotics; for instance, the AMR rate for penicillin G was significantly high within isolates of ST123 (91%; p<0.001, ASR=6.7), all of them recovered from very distant geographic locations (Supplementary Table S1). ST123 belongs to the phylogenetic group eBG5 and is constituted of several other STs (i.e., ST94, ST1621, ST1622, ST1632, ST1640, ST1641, ST1650 and ST1652), 60% of which were susceptible to penicillin G. Thus, apparently, AMR for penicillin G is higher in ST123 isolates than the rest isolates. In contrast, AMR for penicillin G was significantly low in ST1 and ST29 isolates, both of them of eBG1. Also, AMR rates for enrofloxacin (48.1%; p=0.001, ASR=3.3), and marbofloxacin (100%; p=0.001, ASR=3.4) were significantly high in ST1 isolates. Finally, the AMR rates for clindamycin and tiamulin were also high in isolates of eBG5 (97%; p=0.035, ASR=1.9), eBG6 (100%; p<0.001, ASR=2.9), and singletons (65%; p<0.001, ASR=5.8). Notably, the AMR rates for tilmicosin and erythromycin varied within geographic regions; it was significantly high in isolates recovered from Aragón (93%; ASR=1.8), Cataluña (91%; ASR=1), Castilla La Mancha (87%; ASR=0.2), and Murcia (87%; ASR=0.3). Also, high AMR rates for erythromycin occurred in isolates recovered from Aragón (93%; ASR=1.9) and Cataluña (91%; ASR=1.1). Yet, certain Autonomous Communities exhibited high AMR rates for some antibiotics, but these associations had low statistical power, including AMR rates for spectinomycin in the Valencian Community (50%; p>0.05, ASR=2.8) or for penicillin G in Aragón (44.2%; p>0.05; ASR=2). Interestingly, the AMR rate for tiamulin was significantly high in fattening (66.7%; p=0.032; ASR=2.1) and suckling pigs (45.5%; p=0.032; ASR=2.4) as compared to transition pigs. When the AMR rates were compared within isolation years, the resistance to most β-lactams (amoxicillin, ampicillin, ceftiofur, and cefquinome) increased significantly over time (p=0.015 - 0.027; ASR=3.7). Together, our statistical analysis shows that the AMR rates for certain antibiotics varied according to genetic background (penicillin, enrofloxacin, marbofloxacin, clindamycin, and tiamulin), place of isolation (tilmicosin, erythromycin), growth stage of the host (tiamulin), and isolation year (penicillin G) of the isolates.
3.4 Identification of AMR determinants
To identify AMR genes in our population, we analyzed the genome of a subset of 30 S. suis isolates using bioinformatics tools. We make use of the genome of 19 isolates that were recently sequenced by us. Additionally, we sequenced 11 novel isolates (see details in material and methods). Isolates were resistant to multiple antibiotics, including aminoglycosides (n=4), β-lactams (n=10), fluoroquinolones (n=16), lincosamides (n=28), macrolides (n=25), pleuromutilins (n=9), sulphonamides/TMP (n=8), tetracyclines (n=29) (Supplementary Figure S1). A total of 19 AMR genes were identified, comprising aph(3’)-IIIa, sat4, aadE, spw, aph(2’’)-IIIa, apmA, fexA, optrA, erm(B), erm(47), mef(A/E), lnu(B), lsa(E), tet(M), tet(O), tet(W), tet(O/W/32/O), tet(T), and vga(F). All those genes were previously reported in drug-resistant S. suis (Dechêne-Tempier et al., 2021; Uruén et al., 2022), except for apmA, aph(2’’)-IIIa, erm(47), tet(T)) that were detected in other species (Table 1). Then, specific multi-PCR assays targeting the aforementioned genes were performed in the entire population. In addition, we analyzed 12 genes reported as prevalent in AMR S. suis, comprising ant1, aacA, ant(9’)-Ia, aac(6’)-Ie-aph(2’’), mrs(D), mph(C), cfr, tet(L), tet(K), tet(40), tet(44).
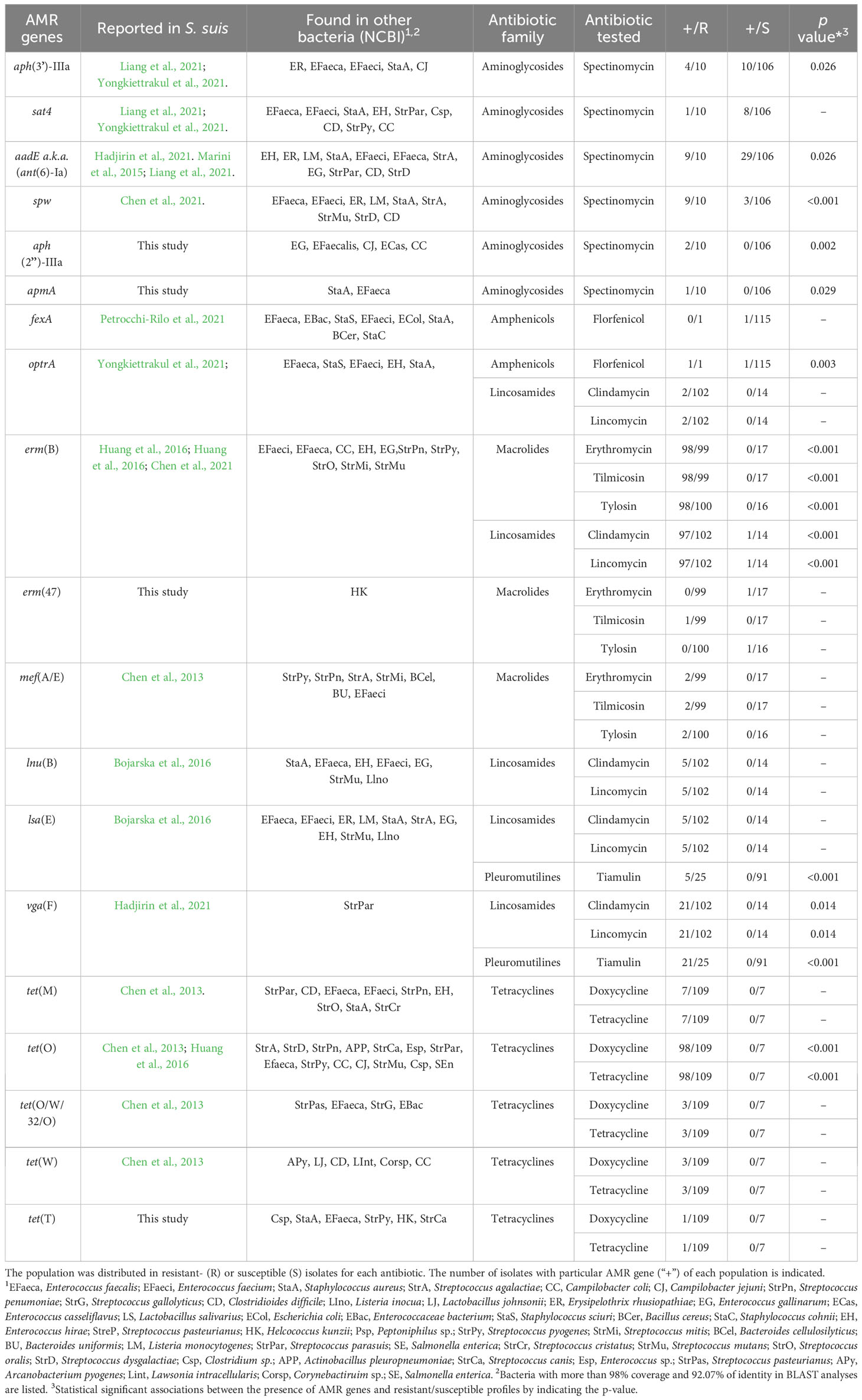
Table 1 AMR genes detected in 116 invasive isolates of S. suis recovered in Spain and their association with antibiotic resistance.
3.4.1 Aminoglycosides
Aminoglycoside resistance in S. suis is produced by enzymatic modification of aminoglycosides, target site modification, and efflux pumps. Aminoglycoside modifying enzymes include Streptothricin N-acetyltransferase (sat4), diverse aminoglycoside O-nucleotidyltransferases (ant, spw), aminoglycoside O-phosphotransferases (aph) or aminoglycoside N-acetyltransferase–O-phosphotransferases (ant-aph). aph(3’)-IIIa, sat4, spw, aph(2’’)-IIIa, apmA, and aadE genes were detected in our population, and their distribution is indicated in Table 1. Interestingly, the spw gene was present in 9 out of 10 (90%) streptomycin-resistant isolates as compared to 3 out of 106 (3%) streptomycin-susceptible isolates. Sequencing of the spw gene in three spectinomycin-susceptible and three spectinomycin-resistant isolates showed 100% amino acid identity among isolates. Also, the promoter region had no mutations, except for a spectinomycin-susceptible isolate (Ss_125) that had few nucleotide alterations (Supplementary Figure S3A). These mutations may cause a lack or reduced gene expression for this isolate but did not clarify the lack of phenotype for the remaining two susceptible isolates. The aph(3´)-IIIa gene was present in 36% of the spectinomycin-resistant isolates and 9% of spectinomycin-susceptible isolates. In contrast, aadE gene was present in 90% of spectinomycin-resistant isolates and 28% of spectinomycin-susceptible isolates. The sat4 gene was present in 9% and 7% of susceptible and resistant isolates, respectively. apmA and aph(2´)-IIIa were uniquely detected in one and two spectinomycin-resistant isolates, respectively. Statistical analysis revealed that aph(3´)-IIIa, aadE, spw, aph(2”)-IIIa and apmA were significantly associated with spectinomycin-resistant isolates (p<0.05, Table 1). Notably, spw showed the strongest association (p<0.001). Together, these data indicate that spectinomycin resistance is mainly caused by spw, aadE, and aph(3´)-IIIa. Interestingly, high MIC values were correlated with the co-existence of more than two AMR genes (p<0.001), indicative that their accumulation increases spectinomycin resistance.
The aph(2”)-IIIa and apmA genes were not reported in S. suis yet. To explore their origin, both genes, and their genetic context, were examined in isolates Ss_48 and Ss_107 that carry aph(2´)-IIIa and apmA, respectively, and compared with public genomes (Figure 2). The plasmid pEW1611 of Enterococcus casseliflavus strain EW1611 contains aph(2”)-IIIa, but their flanking regions have very little homology with those in S. suis isolate Ss_48. In contrast, the genome of S. suis strain Ssu_584 which lacked aph(2”)-IIIa, contained 22,324 bp with 97% of homology with several flanking regions of aph(2´)-IIIa in Ss_48 (Figure 2). This region corresponded to an ICE that carried multiple AMR genes including tet(O), aadE, spw, lnu(B), lsa(E), erm(B). Besides that, the gene apmA is located in the plasmid pKKKS49 of Staphylococcus aureus strain ST398, but their flanking regions lack any homology to the genome sequences of isolate Ss_107. Interestingly, the genome of S. suis YS165 contains an ICE (ICESsuYS165) that lacks apmA but harbors a sequence of 13,091 bp with 97% of nucleotide homology with several flanking regions of apmA of isolate Ss_107 (Figure 2). Together, these analyses suggest that aph(2´)-IIIa and apmA were transferred from another bacteria to S. suis through MGEs.
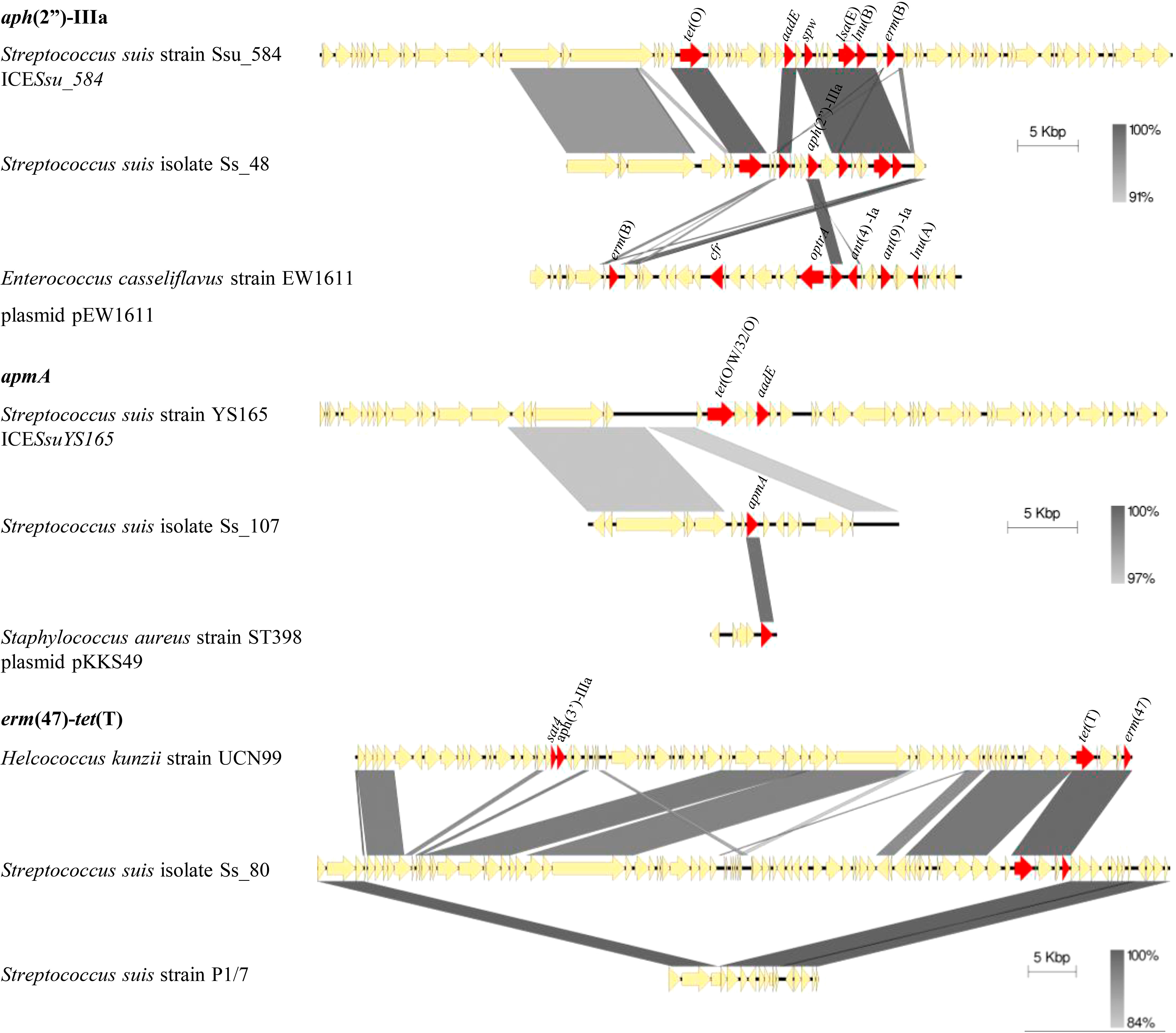
Figure 2 Genetic organization of aph(2’’)-IIIa, ampA, and erm(47) genes in S. suis isolates and other public genomes as indicated. AMR genes are red coloured and gene names are indicated.
3.4.2 β-lactams
In Gram-positive bacteria, the resistance to β-lactams is mostly caused by mutations in the sequences that code for transpeptidase domains of Penicillin Binding Proteins (PBPs), which are targets for β-lactams, and mraY that code for an enzyme involved in the synthesis of a peptidoglycan precursor. There are up to six pbp genes (pbp1a, pbp1b, pbp2a, pbp2b, pbp2x, pbp3) in S. suis, but mutations in pbp1a, pbp2b, pbp2x and mraY were mainly related to β-lactams resistance (Bamphensin et al., 2021; Hadjirin et al., 2021). Hence, to investigate the origin of penicillin G resistance in our isolates, the sequence of pbps and mraY were analyzed in 31 S. suis isolates, including 20 penicillin G-susceptible and 11 penicillin G-resistant isolates. Compared with the S. suis genome of P1/7, which is susceptible to β-lactams, a total of 399 mutation sites were identified in the 31 isolates analyzed, comprising 37, 128, 185, and 49 sites in PBP1A, PBP2B, PBP2X, and MraY, respectively. Importantly, a significant abundance of amino acid substitutions occurred in PBP1A, PBP2B, PBP2X, and MraY (1.3%, 5.3%, 7.4% and 2.8%, respectively) of penicillin G-resistant isolates compared to penicillin G-susceptible isolates (0.4%, 0.7%, 0.6% y 0.1%, respectively) (p<0.05) (Figure 3A); many of these mutations occurred in the transpeptidase domain (Figure 3A). Notably, 301 substitutions were exclusively present in penicillin G-resistant isolates (9 in PBP1A, 90 in PBP2B, 157 in PBP2X, and 45 in MraY), and the most prevalent were G727D (36.4%) in PBP1A; A562D/P (63.6%) and Q668E/L, Q678T/P/N, Q685N/H, and H688Y (54.5%) in PBP2B; N631K/T/S, Q655E/N/Y, I680A/P/L/S, and I701L/V (90.9%) in PBP2X; A630T/G and Q636K/T/R (81.8%), N616S and A666S (72.7%), and T70A/V, F72L, L74M, and V91L (45.5%) in MraY. A total of 30 and 4 mutations in PBPs and MraY, respectively, were present in at least 70% and 40% of penicillin G-resistant isolates, respectively. Indeed, these mutations were statistically associated with penicillin G resistance (p<0.05). Then, the RR was calculated. An RR value higher than 1 is interpreted as an increased probability of the association between the genotypic event and the phenotype, and an RR lower than 1 as a negative association. A high RR was detected in 1, 4 and 5 mutations in PBP1A, PBP2B, and PBP2X, respectively (Figure 3B). Notably, no RR could be calculated for 6 mutations in PBP2B and 3 in PBP2X due to their high prevalence in resistant isolates and low in susceptible isolates (indicated as ND in Figure 3B). Most of these substitutions are located in the sequences that encode for the transpeptidase domain of the cited enzymes (Figure 3B).
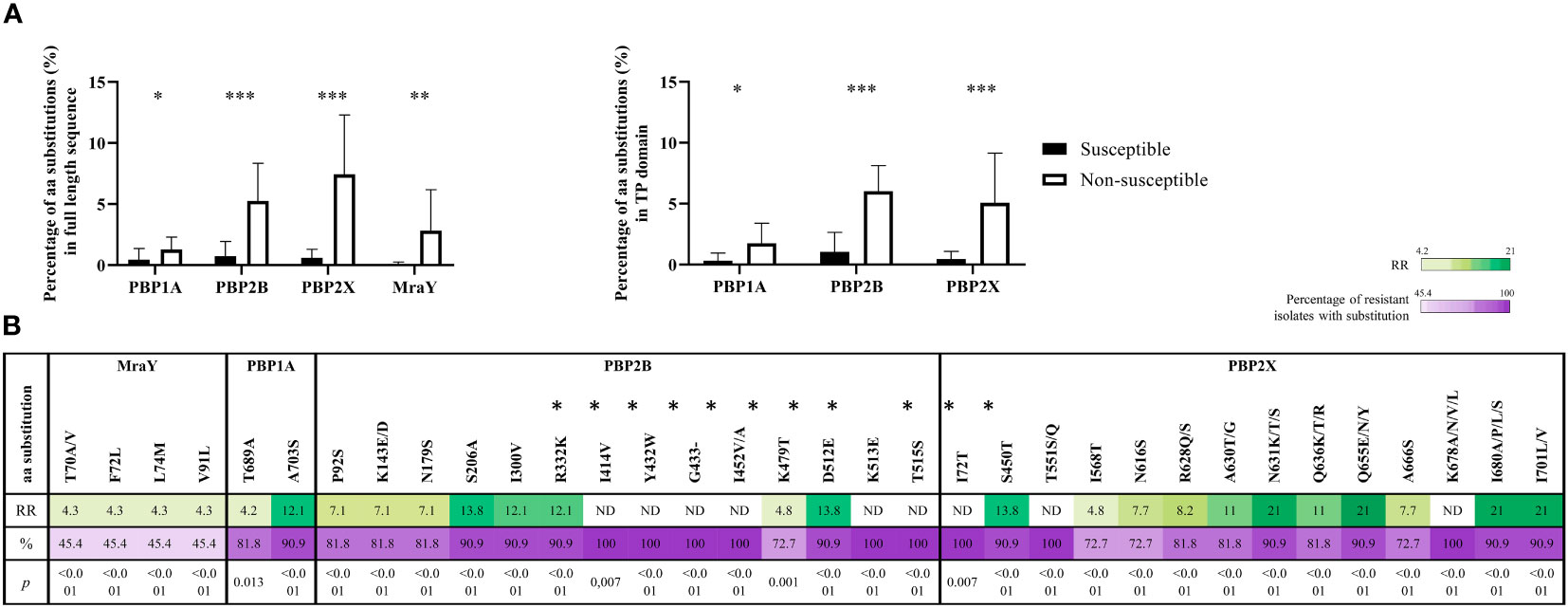
Figure 3 Genetic differences of Penicillin G resistant and susceptible isolates. (A) Amino acidic variability of penicillin-binding proteins (PBPs) and MraY proteins in resistant and susceptible S. suis isolates. The variability is shown for the full-length proteins (left graft) and transpeptidase domain (right graft). (B) Risk ratio (RR) for amino acid alterations in the sequences of MraY, PBP1A, PBP2B, and PBP2X. A total of 32 alterations were significantly associated and were present in 70% of penicillin G-resistant isolates (p< 0.05). Values non-defined (ND) correspond to alterations present in all resistant isolates, and therefore RR could not be calculated. The colour scale is proportional to the RR value. Mutations located in transpeptidase domain are indicated with an asterisk.
Because of the large abundance of mutations found in PBPs, the level of genetic conservation of pbps and mraY genes was analyzed by calculating the dN/dS substitutions. dN/dS is a metric parameter commonly interpreted in terms of the amount of amino acid diversity generated by codon substitutions (Del Amparo et al., 2021). It is widely used to evaluate selection in protein coding sequences in a large variety of microorganisms, including viruses (Meyer et al., 2015) and bacteria (Rocha, 2006; Arenas et al., 2008). Positive selection was considered when the dN/dS is higher than 1, and negative selection when dN/dS is lower than 1. Positively or negatively coding sites were considered when the significance level was less than 0.05. The dN/dS of pbp1a, pbp2x, pbp2b, and mraY ranged from 0.13 to 0.05 (Table 2) evidencing negative selection for amino acid replacement. Actually, several negative selection sites were identified in all genes. Yet, pbp2x contained a positive selection site (Q636K/T/R) with a high prevalence in resistant isolates (Figure 3B). Then, to understand the origin of the mutations, the presence of putative recombination events was analyzed. Many putative recombination breakpoints (Supplementary Table S3) were estimated and located within the pbp1a, pbp2b, and pbp2x-mraY and their flanking regions (Supplementary Figure S4). Together, our analysis supports that penicillin G resistance in our isolates could be caused by polymutations in pbp and mraY genes, some of which could be likely acquired by horizontal genetic transfer.
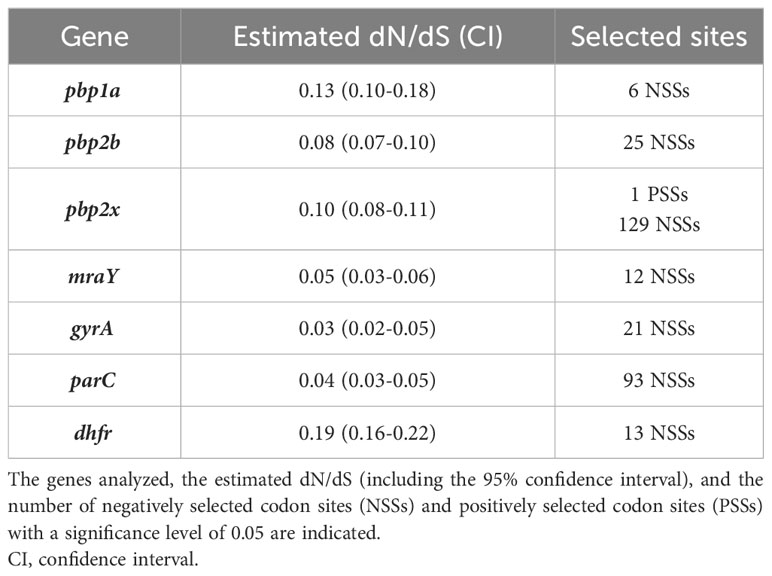
Table 2 Estimated dN/dS for AMR determinants for β-lactams, fluoroquinolones, and TMP in 30 S. suis isolates.
3.4.3 Amphenicols
Amphenicol resistance can be caused by rRNA 23S modification (cfr, optrA), enzymatic inactivation of amphenicols (catA), and active efflux (fexA). The cfr gene was not detected in our population. The optrA gene was detected in the florfenicol-resistant isolate and in a florfenicol-susceptible isolate out of 115. The fexA gene was detected in one florfenicol-susceptible isolate (Ss_49), which also carried the optrA gene. PCR analysis in Ss_49 evidenced that both genes were located in tandem on the chromosome, and sequencing analysis revealed an intact optrA gene. Thus, apparently, the resistance to florfenicol could be determined by optrA (p=0.003), but for unknown reasons, its function is lacking in isolate Ss_49.
3.4.4 Fluoroquinolones
Fluoroquinolone resistance is determined by mutations in genes involved in DNA replication, including gyrA, gyrB, parC, and parE (Dechêne-Tempier et al., 2021; Uruén et al., 2022). Particularly in S. suis, fluoroquinolone resistance is caused by specific mutations in GyrA (S81 and E85) and ParC (S79) (Hadjirin et al., 2021). Hence, we searched for these mutations in 33 S. suis isolates with a different AMR profile to fluoroquinolones. Pre-established resistance mutations were detected in isolates Ss_09, Ss_166, Ss_168, and Ss_170 which were resistant to enrofloxacin and marbofloxacin (Supplementary Figures S3B, C) but lacked fluoroquinolone-susceptible isolates. Yet, isolate Ss_80, which manifested resistance to marbofloxacin and enrofloxacin, and isolates Ss_02, Ss_21, Ss_22, Ss_24, Ss_45, Ss_48, Ss_70, Ss_72, Ss_84, Ss_92, Ss_100, Ss_106, Ss_115, and Ss_121, which were only resistant to marbofloxacin, lacked the cited mutations. In addition, the amino acid sequences of GyrB and ParE were analyzed in 10 isolates (3 susceptible to both antibiotics, 5 resistant to marbofloxacin, and 2 resistant to both antibiotics). A total of 4 amino acid substitutions were identified in GyrB in all isolates (E219D, V221I, L409F, and A427T), but two of them (E219D and V221I) were only in isolate Ss_166 that manifested resistance to both antibiotics. Besides, a total of 13 amino acid substitutions were detected in ParE, but no one was associated with fluoroquinolone resistance. The dN/dS values for gyrA and parC were 0.03 and 0.04 (Table 2), respectively, indicating they are highly conserved genes. Surprisingly, estimates of recombination revealed multiple recombination events (Supplementary Table S3) along their flanking regions (Supplementary Figure S4), also suggesting that the acquisition of such specific mutations can be also through horizontal gene transfer. To summarize, resistance to enrofloxacin is mostly attributed to pre-established mutations in GyrA and ParC, but resistance to marbofloxacin was not fully elucidated.
3.4.5 Macrolides, lincosamides and pleuromutilins
Resistance to macrolides and lincosamides is caused by ribosome methylation by sRNA methyltransferases (encoded by erm and cfr genes), antibiotic modifying enzymes such as phosphotransferases and esterases (encoded by mph and lin genes), and specific flux pumps (encoded by mef or mrs(D) genes) (Dechêne-Tempier et al., 2021; Uruén et al., 2022). Only erm(B) and mef(A/E) were detected in our population. Most of the macrolide-resistant isolates (99%) harbored erm(B), and this gene was not present in susceptible isolates. Isolate Ss_80, which manifested tilmicosin-resistance, but was susceptible to tylosin and erythromycin, did not carry erm(B) nor mef(A/E). Notably, isolates that carried both genes had enhanced MIC values for erythromycin (>2 mg/L), tilmicosin (>64 mg/L), and tylosin (32 mg/L) (p<0.001). Interestingly, the genome of the isolate Ss_80 contained an erm(47) gene that is linked to erythromycin resistance in other bacteria (Table 1), but not reported in S. suis. Inspection of the upstream flanking region of the erm(47) gene exhibited a large sequence of 67,715 nucleotides that was not present in the public genomes of S. suis (Figure 2). Blast searchers using this sequence as a query revealed that 55.9% of the nucleotide sequence had >90% of homology with sequences of the genome of Helcococcus kunzii strain UCN99 that contains an erm(47)-containing element and other AMR genes such as tet(T), sat4, and aph(3’)-IIIa (Figure 2). In addition, these regions contain sequences of different sizes that share homology (> 95%) with different bacteria, such as the plasmid pAPRE01 of Anaerococcus prevotii DSM 20548, and the genome of Peptoniphilus sp. CBA3646, Amylolactobacillus amylophilus DSM 20533, Streptococcus parasuis SUT-286, and E. faecalis isolate 27688. Notably, the upstream and downstream flanking regions of the erm(47)-containing element in isolate Ss_80 corresponded to a type I restriction-modification system gene of P1/7 (Figure 2). Moreover, the GC content of erm(47) and tet(T) was 25.9% and 32%, respectively, which deviates from the rest of the genome (42.5%) (Zhang et al., 2011). Together, these data suggest that erm(47) and tet(T) genes have traveled from another bacterial species to S. suis through MGEs.
AMR determinants of lincosamide resistance in S. suis are lnu(B), lsa(E), oprtA, vga(F) (Dechêne-Tempier et al., 2021; Uruén et al., 2022). erm(B) (95.1%), vga(F) (20.6%), lsa(E) (4.9%), and lnu(B) (4.9%) were only present in clindamycin- and lincomycin-resistant isolates in our population (Table 1), except for isolate Ss_31 that was susceptible to clindamycin and lincomycin but carried erm(B) gene. Sequencing of the full-length erm(B) gene in Ss_31 and the adjacent promoter region showed no differences with the homologs in three lincosamide-resistant isolates (data not shown). Statistical analysis revealed that erm(B) (p<0.001) and vga(F) (p=0.014) were significantly associated with lincosamide and clindamycin-resistant isolates, but the low prevalence of lsa(E) and lnu(B) prevented statistical power. Interestingly, lincosamide-resistant isolates with more than 2 AMR genes manifested a higher MIC than 16 mg/L to lincomycin and clindamycin (p<0.001). Apparently, accumulation of the cited AMR genes enhances resistance to lincomycin and clindamycin.
Pleuromutilin resistance has been associated with ribosome protection proteins (encoded by lsa(E) and vga(F)) in S. suis. vga(F) and lsa(E) were present in 21 and 5 out of 25 tiamulin-resistant isolates, respectively, and they were missing in tiamulin-susceptible isolates, except for isolate Ss_52 which contained vga(F). Ss_52 manifested a MIC value of 8 mg/L, just below the established breakpoint (Figure 1). Statistical analysis showed that both genes, vga(F) and lsa(E), were significantly related to tiamulin resistance (p<0.001). Notably, 2 isolates (Ss_48 and Ss_147) that manifest the highest MIC value (>64 mg/L) carried both genes.
3.4.6 Sulfonamides/TMP
It has been reported that TMP resistance is caused by enzymatic modification (encoded by dfrF, dfrK) of the dihydropteroate synthase (encoded by dhfr), and a particular mutation in dhfr gene (I102L) or its promoter (A5G) (Hadjirin et al., 2021). dfrF and dfrK genes were not detected in our isolates. Then, the sequence of dhfr was analyzed in 35 isolates, including 12 sulfamethoxazole/TMP-resistant (Ss_45, Ss_52, Ss_53, Ss_70, Ss_82, Ss_85, Ss_104, Ss_106, Ss_107, Ss_109, Ss_134, and Ss_138) and 23 sulfamethoxazole/TMP-susceptible isolates (Ss_02, Ss_08, Ss_20, Ss_21, Ss_22, Ss_24, Ss_46, Ss_48, Ss_51, Ss_69, Ss_72, Ss_80, Ss_84, Ss_92, Ss_93, Ss_100, Ss_115, Ss_121, Ss_124, Ss_156, Ss_166, Ss_167, and Ss_168). Amino acidic alteration I102L in dhfr gene was identified in all sulfamethoxazole/TMP-resistant isolates and three susceptible isolates (Ss_08, Ss_100, and Ss_166) (Supplementary Figure S3D). Several additional mutations were found in sulfamethoxazole/TMP-susceptible and resistant isolates, but none were attributed to resistant isolates. Curiously, isolates Ss_08, Ss_100, and Ss_166 manifested MIC values of 2 mg/L (Ss_08 and Ss_100) or 1 mg/L (Ss_166), just below the pre-established breakpoint (Figure 1). The dN/dS value for dhfr gene was 0.18 indicating that the gene is indeed conserved (Table 2), but several points of recombination (Supplementary Table S3) were estimated in the intergenic region located upstream of dhfr gene and in a few downstream genes (Supplementary Figure S4).
3.4.7 Tetracyclines
Tetracycline resistance in S. suis has been related to the presence of efflux pumps (encoded by tet(B, K, L, 40)) and ribosome protection proteins (encoded by tet(M, O, S, W, 44)). We only detected tet(O), tet(M), tet(W), and tet(O/W/32/O) in our population. All genes were detected only in tetracycline- and doxycycline-resistant isolates. tet(O) prevailed (89% of resistant isolates), followed by tet(M) (6.4%), tet(W) (2.7%), and tet(O/W/32/O) (2.7%) (Table 1). Statistical analysis revealed that tet(O) was significantly associated with tetracycline and doxycycline resistance (p<0.001), but the low prevalence of the remaining genes and the reduced number of susceptible isolates prevented statistical associations for the remaining AMR genes. However, the presence of 2 AMR genes was significantly related to higher MIC values for doxycycline and tetracycline (p<0.001). Isolate Ss_80 was resistant to tetracycline but did not carry any of the cited AMR genes. Whole genome sequence analysis of Ss_80 evidenced the presence of tet(T) gene located on the chromosome close to erm(47) (Figure 2). tet(T) gene was not previously reported in S. suis but was associated with tetracycline resistance in S. pyogenes (Clermont et al., 1997).
3.5 Location of AMR genes in MGEs
The 30 genomes of our isolate collection were used for detecting MGEs, analyzing their structure, and if they transfer AMR genes. A total of 35 ICEs, 20 IMEs, and 2 conjugative elements were identified in the 30 genomes of the invasive isolates analyzed here (Supplementary Table S4). 18 out of 60 of these MGEs carried at least one or more AMR genes, including aph(3´)-IIIa, sat4, aadE, erm(B), tet(O), and tet(M). Notably, all of them carried at least erm(B) and/or tet(O), except Ss_51, which only carried tet(M). Ss_08 carried an ICE harboring 5 AMR genes (aph(3’)-IIIa, sat4, aadE, erm(B), and tet(O)). MGEs had different gene content and length, but shared regions of different size (Figure 4), mainly the upstream region that harbors genes coding for Type IV Secretion System. However, downstream regions were shared between MGEs. This evidence the occurrence of many recombination events between MGEs resulting in the gain or loss of genes, and thus explains the large repertoire of MGEs and the variable combinations of AMR genes (Figure 2). Moreover, identical IMEs were identified in isolates of the same genetic background. Examples included isolates Ss_100, Ss_106, and Ss_156 of ST123 that contained the same IME of 5,994 bp. These isolates were recovered in neighbor Autonomous Communities (i.e., Aragón and Cataluña). But, curiously, isolates Ss_20 and Ss_92 of ST29 contained a similar 5,987 bp IME but were recovered from geographically distant Autonomous Communities (i.e., Aragón and Extremadura). Anyhow, these data demonstrate that some AMR genes are located in MGEs which may serve for their transference between S. suis isolates. The presence of multiple AMR genes in MGEs may explain the rapid occurrence of multi-drug resistance.
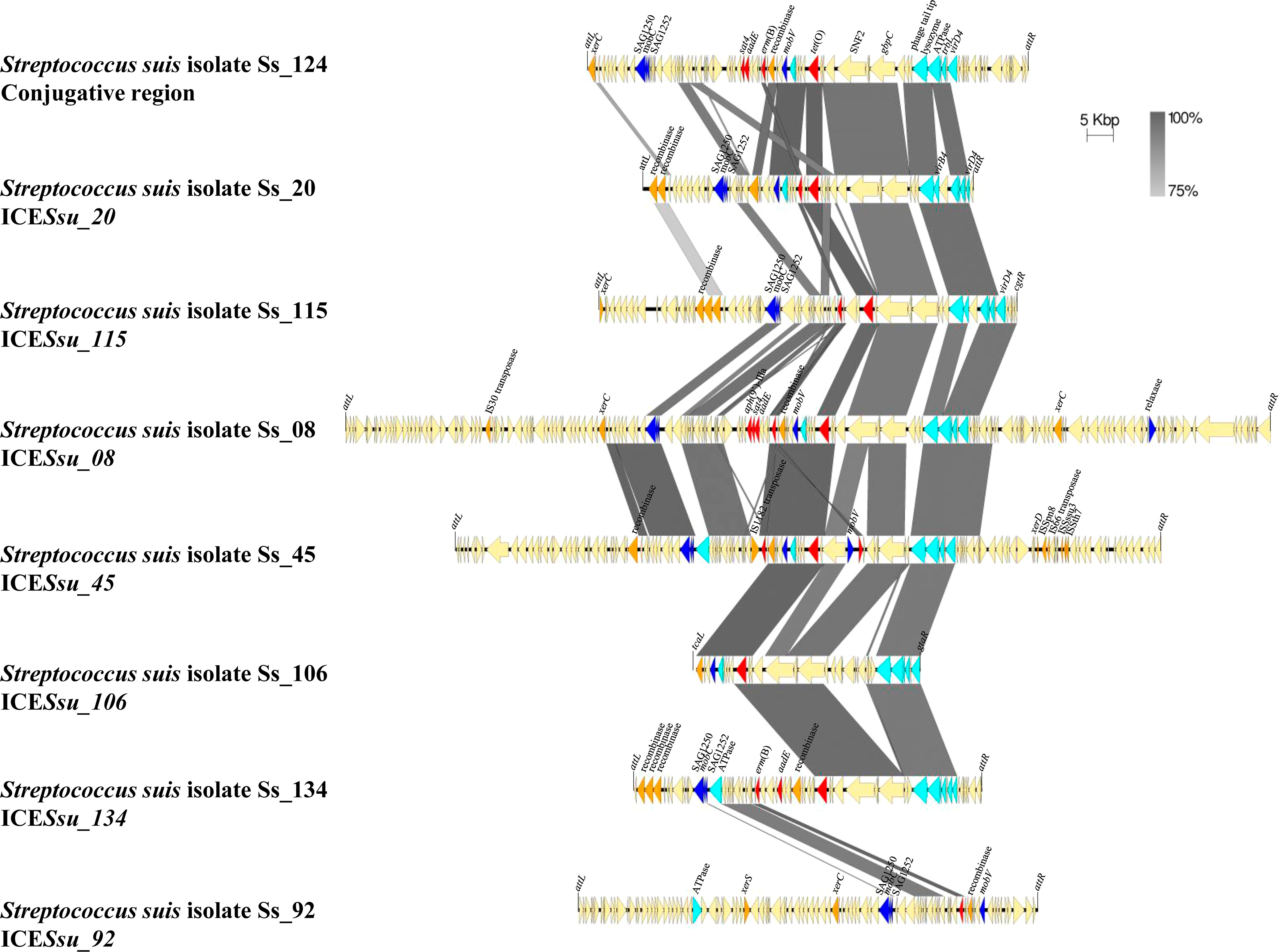
Figure 4 Comparison of the genetic organization of eight MGEs detected in S. suis isolates. Nucleotide sequences of 1 conjugative region and 7 ICEs carrying AMR genes are compared. Light blue arrows indicate genes coding for Type IV Secretion System, dark blue arrows indicate gene coding for relaxases, orange arrows indicate genes coding for integrases, and red arrows indicate AMR genes. Integration sites are shown.
3.6 Dissemination of AMR genes of S. suis among pathogenic bacteria
The GC content of several AMR genes present in our S. suis genomes, including erm(47) (25.9%), lnu(B) (31.1%), tet(T) (32%), lsa(E) (32.4%), aadE (32.9%) and erm(B) (33.1%), apmA (44.6%), and aph(3’)-IIIa (44.9%), deviates from the rest of genome (41.2%) (Zhang et al., 2011). This suggests that many of them were acquired from another bacterium. We performed nucleotide BLAST searches to investigate their dispersion in public genomes. A summary of major hits is listed in Table 1. For example, when using spw gene of the isolate Ss_48 as a query in BLAST, we identified homologs in many genomes of different pathogenic and non-pathogenic bacteria, including 33 Enterococcus faecium, 24 E. faecalis, 9 S. aureus, 7 Erysipelothrix rhusiopathiae, 4 Clostridioides difficile, diverse streptococci species (3 Streptococcus agalactiae, 1 Stretptococcus dysgalactiae, 2 Streptococcus mutans, and 1 Streptococcus parasuis), and 1 Listeria monocytogenes. In the genome of L. monocytogeneses UA159, the spw homolog is located in a genomic island together with other AMR genes (aph(3’)-IIIa, aadE, spw, lnu(B), and lsa(E)) flanked by genes coding for two transposases (Figure 5A). Notably, aadE, spw, lnu(B) and lsA(E) of L. monocytogenesis UA159 are homologs (100% identity) to those of S. suis isolate Ss_48 although the genetic organization varied. But, also homologs (100% identify) of the S. suis aadE gene were identified in 25 genomes of E. faecium, 21 of E. faecalis, 3 of E. rhusiopathiae, 20 of S. aureus, 4 of C. difficile, 1 of L. monocytogenes, and various streptococci species (3 S. agalactiae, 1 S. dysgalactiae, and 2 S. parasuis) (Table 1). However, their genomic organization varied among genomes. For example, in the genome of S. aureus strain N08CSA36, the aadE gene is located together with 9 additional AMR genes (blaI, blaR1, blaZ, aph(2’’)-Ia, erm(C), tet(L), spw, lsa(E), and lnu(B)), and genes coding for diverse transposases, relaxases and a mobV recombinase, the latter is also in S. suis Ss_48 (Figure 5A). Also, a homolog (100% identity) of the lsa(E) gene of isolate Ss_48 was detected in S. mutans strain NH1-T1-2, which was located together with aadE, spw, and lnu(B) genes and a recombinase (Figure 5A). The presence of shared regions between genomes, genes coding for recombinases and transposases and several insertion sequences may contribute to the allelic exchange and the gain or loss of AMR genes.
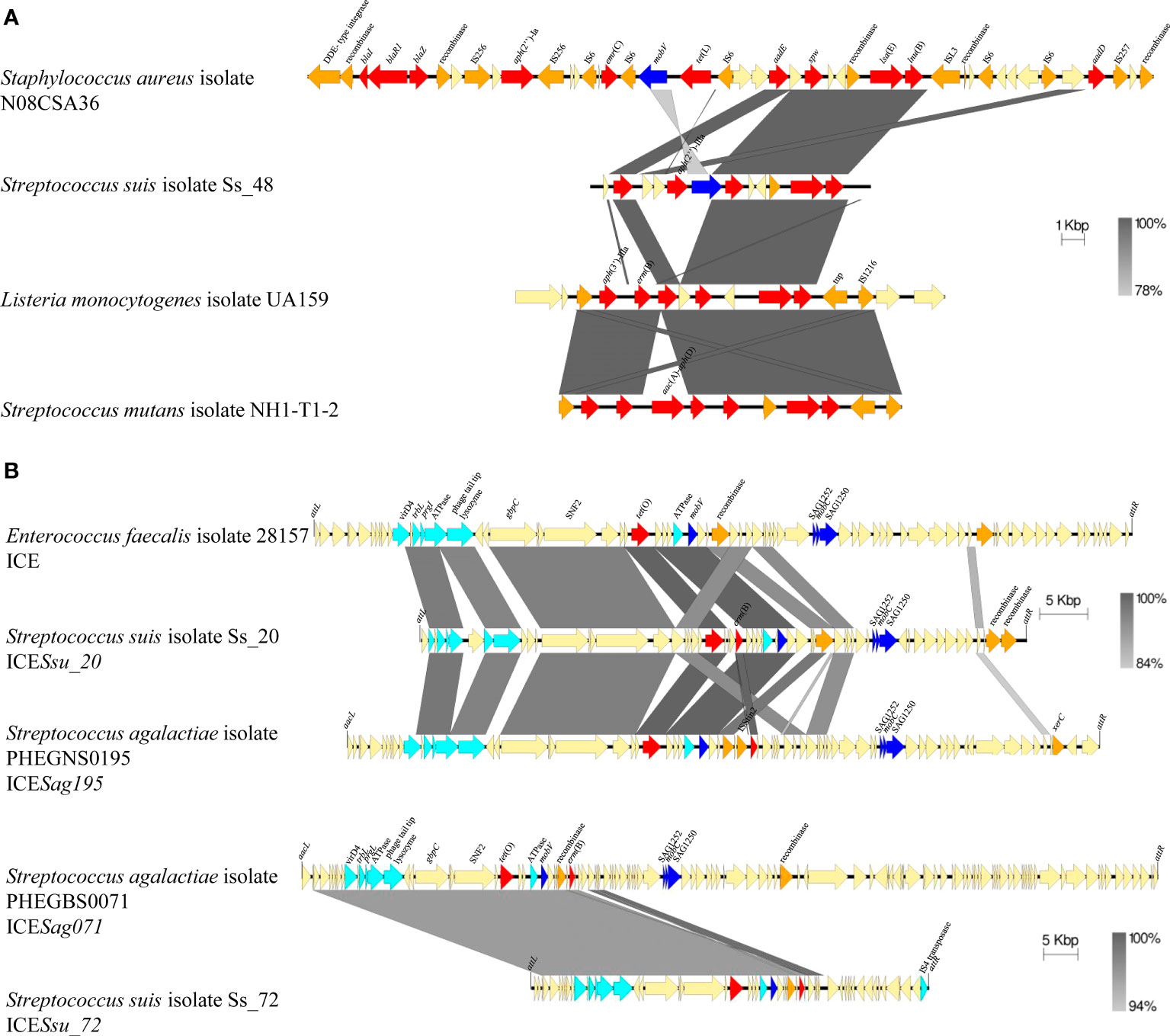
Figure 5 Comparison of the genetic organization of S. suis isolates with genomes of diverse pathogenic bacteria. (A) Comparison of genomic islands carrying AMR genes. (B) Comparison of ICEs carrying AMR genes. Light blue arrows indicate genes coding for Type IV Secretion System, dark blue arrows indicate gene coding for relaxases, orange arrows indicate genes coding for integrases, and red arrows indicate AMR genes. Integration site for ICEs is indicated.
Homologs of the erm(B) gene of isolate Ss_08 were also identified in 67 of E. faecium, 8 of E. faecalis, 1 of Campylobacter coli, 1 of S. agalactiae, and 1 of Streptococcus mitis. Also, homologs of tet(O) of S. suis isolate Ss_08 were detected in 25 genomes of S. agalactiae, 2 of S. pneumoniae, 1 of S. dysgalactiae, and 1 of E. faecalis with 100% identity, and tet(O) variants (>99% identity) in 3 genomes of Campylobacter jejuni, 2 of S. agalactiae, 2 of S. mutants, 1 of Staphylococcus coagulans, 1 of S. dysgalactiae, 1 of Salmonella enterica and 1 of Clostridium sp. (Table 1). In S. suis Ss_08 tet(O) is placed together with erm(B) in an ICE. A sequence of 17,306 bp shares a 50-98% coverage with more than 94% of identity with genomes of 42 bacterial species, such as S. agalactiae S5 (86% coverage, 94.45% identity), S. pyogenes strain TSPY453 (59% coverage, 94.6% identity), S. dysgalactiae strain DY107 (91% coverage, 95.5% identity), E. faecalis isolate 28157_4#173 (72% coverage, 99.9% identity), and several species of Clostridium including C. scindens strain BL389WT3D (75% coverage, 94.6% identity). Also, in S. suis isolate Ss_20, tet(O) and erm(B) genes are located in an ICE, which shares 37,584 bp and 37,979 bp (>90% identity) with ICEs placed in E. faecalis isolate 28157 and S. agalactiae isolate PHEGNS0195 (Figure 5B). Also, S. suis isolate Ss_72 contains an ICE with tet(O) and erm(B) genes in a different genomic organization but that share 41,726 bp (> 96% homology) with an ICE of S. agalactiae isolate PHEGBS0071 (Figure 5B) and 6807 bp (44-100% coverage, 84-100% identity) with the plasmid pELF_mdr in strain NUITM-VRE1 of E. faecium (52% of coverage, 99.6% identity), the plasmid pRE25 in E. faecalis strain RE25 (54% of coverage, 99.9% identity), genomes of 25 species, including S. agalactiae strain FDAARGOS_670 (100% coverage, 99.9% identity), S. mutans strain NH1-T1-1 (52% coverage, 99.8% identity), S. aureus strain AOUC-0915 (54% coverage, 96.4% identity), S. coagulans strain 1031373 (68% coverage, 95.8% identity), C. difficile strain CD161 (48% coverage, 95.6% identity), among others. In contrast to the cited AMR genes, homologs of vga(F), which GC content was similar to S. suis genome, were only identified in streptococci species (Table 1). In conclusion, several AMR genes identified in Spanish S. suis have a different phylogenetic origin and seem to be mobilized through different genetic mechanisms.
4 Discussion
High AMR rates for lincosamides, macrolides, and tetracyclines in S. suis have been reported globally (Uruén et al., 2020). A recent study testing 103 S. suis isolates from Spain revealed AMR rates for clindamycin and tylosin up to 87% (Petrocchi-Rilo et al., 2021), which confirms our results. Both antibiotics were used in pig production to treat a diversity of bacterial infectious diseases in Spain. Because S. suis is a commensal, the high AMR rates found here could be caused by its exposition to the cited antibiotics. Lincosamides and macrolides often show cross-resistance caused by mutual resistant mechanisms. The AMR for erythromycin and lincosamides found in this work were attributed to erm(B) which exhibited a high prevalence (>98%) in resistant isolates. erm(B) encodes for a methylase that modifies ribosomal 23S rRNA, often located in MGEs (Huang et al., 2016; Chen et al., 2021). Notably, we identified erm(B) located together with tet(O). This genetic linkage between both genes was early reported (Huang et al., 2016). The co-localization of both genes in MGEs could contribute to the high co-occurrence of erythromycin and tetracycline resistance.
The AMR rate for enrofloxacin (30%) was similar to that reported recently in Spain (Petrocchi-Rilo et al., 2021), but much higher than an earlier work (2005) (Vela et al., 2005), suggestive of an enhancement of AMR in the last decade. This rate is considerably higher than other European countries such as France (18%) (Vachee et al., 2008), Belgium (0.3%) (Callens et al., 2013), The Netherlands (0.6%) (van Hout et al., 2016), or Sweden (5.3%) (Werinder et al., 2020). Enrofloxacin-resistance was mainly caused by particular mutations in pre-established quinolone-resistance determining region of gyrA and parC genes as reported (Escudero et al., 2007), however, they do not fully explain marbofloxacin-resistance. Five marbofloxacin-resistant but enrofloxacin-susceptible isolates lacked pre-established genotypes. Also, mutations in gyrB and parE can contribute to enrofloxacin resistance in streptococci (González et al., 1998; Jorgensen et al., 1999). Sequencing of gyrB and parE in our isolates did not show differences between susceptible and resistant isolates. Alternatively, efflux pumps such as PmrA in S. pneumoniae (Gill et al., 1999) cause fluoroquinolone resistance, but no homologous were found in our genomes.
Tiamulin is broadly used to treat S. suis infections. The AMR rate for tiamulin (19%) matches that recently reported (12%) in Spain (Petrocchi-Rilo et al., 2021) and in some European countries such as Denmark (2020) (20%) (DANMAP, 2021) or England (2014) (23%) (Hernandez-Garcia et al., 2017). Notably, Thailand reported rates up to 80% (2019) (Yongkiettrakul et al., 2019). Tiamulin resistance was caused by vga(F) and lsa(E) genes, which code for ABC-F proteins with a function as ribosome-protection proteins. Hence, they can confer resistance to different ribosome-targeting antibiotics such as lincosamides and pleuromutilins (Uruén et al., 2020), as we find here.
The AMR rate for florfenicol (5%) was considerably lower than that recently reported (Petrocchi-Rilo et al., 2021) (14%), but in line with many European countries such as Denmark (2020) (0%) (Arndt et al., 2019) or The Netherlands (van Hout et al., 2016) (0.1%). Also, low resistance rates were reported in American countries such as Canada (2019) (0.5%) (Arndt et al., 2019) or USA (2016) (1%) (Hayer et al., 2020). The only florfenicol-resistant isolate contained an oprtA gene that encodes an ABC-F family protein producing ribosome protection. oprtA gen is often reported inside of MGEs (Huang et al., 2017; Shang et al., 2019; Zhang et al., 2020), sometimes together with other AMR genes. Actually, it was reported in S. suis isolates in China with a prevalence ranging from 11%-38% (Huang et al., 2019; Shang et al., 2019; Zhang et al., 2020). Also, we detected the fexA gene that codes for an efflux pump that exports amphenicols, but it could not be related to florfenicol resistance. Surprisingly, previous studies demonstrated a high prevalence of fexA (26%) in Spanish S. suis isolates by PCR screening (Petrocchi-Rilo et al., 2021), but its presence could not be related to chloramphenicol resistance. When we used primers described by the authors to detect fexA, an amplicon was produced in several isolates but sequencing revealed sequences of a putative transketolase-subunit. New primers were designed here, and all resulting amplicons were sequenced. We hypothesized that the prevalence of fexA previously reported in Spain is overestimated.
β-lactams are the gold-standard treatments against S. suis disease. AMR rates for β-lactams varied considerably among antibiotics being high for penicillin (32%). Low resistance rates of β-lactams (<10%) were reported in other European countries such as The Netherlands (van Hout et al., 2016) or The Czech Republic (Matiašovic et al., 2021). This huge difference could be caused by their extensive use in Spain, sometimes as metaphylactic therapies. Importantly, our statistical analysis revealed an enhancement of penicillin G-resistance in a 6-year period. Earlier work in Spain (2005) reported low AMR rates for penicillin (4%) and amoxicillin (0.7%) in 151 clinical isolates (Vela et al., 2005). Recent work (Petrocchi-Rilo et al., 2021) reported a low resistance to ampicillin (3%), and higher to ceftiofur (17%) and penicillin (26%). Altogether, it seems penicillin resistance is rapidly increasing in Spain. Notably, the low AMR rates in ST1 isolates and high AMR rates in ST123 isolates are in agreement with studies in other countries (Cucco et al., 2022).
We detected a high amino acid diversity in PBPs in penicillin-resistant compared to susceptible isolates in order PBP2X > PBP2B > PBP1A. Previous reports showed that mutations in PBP2B and PBP2X confer moderate AMR, while in combination with mutations in PBP1A confer high AMR (Hadjirin et al., 2021). It has been postulated in other streptococci species that such mutations are acquired by interspecies gene transfer (Hakenbeck et al., 2012). Our bioinformatics analysis is in line with this hypothesis, as we found significant shreds of evidence of recombination within pbp genes. Furthermore, we detected particular mutations, only present or, highly associated with penicillin-resistant isolates; some were previously detected by other authors, for example, K479T, D512E in PBP2B or T551S in PBP2X (Hadjirin et al., 2021), but many others were not reported before (Bamphensin et al., 2021; Hadjirin et al., 2021; Lunha et al., 2023). Hence, penicillin resistance can be acquired by one or a combination of mutations that probably affect the binding of the enzyme to penicillin. However, the estimates of dN/dS for these genes indicated a strong negative selection for amino acid replacement. This matches their relevant role in bacterial growth. Therefore, genetic transference enables penicillin resistance, but the accumulation of mutations probably alters the properties and function of the enzymes compromising bacterial survival. Thus, only mutations that balance bacteria survival and antibiotic resistance are selected. Moreover, mutations in MraY have been related to β-lactam resistance in S. suis (Hadjirin et al., 2021). MraY transfers N-acetylmuramyl-pentapeptide-1phosphate to undecaprenyl phosphate to generate the peptidoglycan precursor. MraY is not a target for β-lactams, probably these mutations are compensatory for those in PBPs, and thus they improve bacterial fitness. Also, during the recombination process, adjacent genes to pbps may be affected, including ddlA, that form part of the biosynthesis of the peptidoglycan precursor, or omsC that encodes for a putative osmotically inducible protein, among others (Supplementary Figure S4). Their contribution to AMR or bacterial fitness should be elucidated in future studies.
To the best of our knowledge, we identified four novel AMR genes in S. suis. tet(T) gene encodes for a ribosomal-protection protein. They were identified in other streptococci (Clermont et al., 1997; Stefańska et al., 2022), or enterococci (Nishimoto et al., 2005). ampA gene encodes for an N-acetyltransferase to N2´of apramycin (Bordeleau et al., 2021). Apramycin is an antibiotic broadly used in veterinary medicine for decades. ampA gene was identified in diverse pathogens including S. aureus (Feßler et al., 2011) and Campylobacter (Fabre et al., 2018). It is often located on plasmids together with other AMR genes (Feßler et al., 2018) that favor its dissemination. Finally, erm(47) encodes for a protein sharing 44%-48% amino acid identified with Erythromycin methylases in Helcococcus kunzii (Guérin et al., 2016). H. kunzii is part of the skin microbiota of pigs and can become an opportunistic pathogen (Grattarola et al., 2010). Besides, aph(2’’)-IIIa is present in a variety of bacterial species including Enterococcus, Staphylococcus, or Campylobacter, among others. Also, blast searchers with several AMR genes identified homologs in genomes of many other bacteria, including human and animal pathogens. Some genes are located in MGEs which can facilitate horizontal gene transfer through conjugation. Our analysis evidenced that MGEs can contain a large repertoire of AMR genes that favor the rapid acquisition of multi-drug resistance and explain the different multi-drug resistance profiles. Together, our data support that Spanish S. suis has acquired AMR genes from other species through multiple and independent events. Considering experimental evidence that S. suis can transfer AMR genes to different pathogenic bacteria (Marini et al., 2015; Huang et al., 2016), these data point out that S. suis is a relevant contributor to the spread of AMR genes across human and animal pathogens.
5 Concluding remarks
To summarize, here we reported the AMR rates for different antibiotics in circulating clinical isolates of S. suis in Spain. High rates of resistance were found to lincosamides, tetracyclines, and erythromycin following a global trend. However, our study also reflects the emergence of resistance to enrofloxacin, sulphonamides, and penicillin G in Spain showing higher AMR rates than in studies reported in other European countries. Importantly, multidrug resistance was observed in 90% of the isolates. Indeed, particular actions must be taken to control AMR of S. suis in Spain. Additionally, our work also revealed the genetic mechanisms contributing to AMR for most antibiotics, involving genes coding for target-protecting proteins (optrA, erm(B), lsa(E), vga(F), tet(M), tet(O), tet(O/W/32/O), tet(W), tet(T), aph(2’’)-IIIa) antibiotic-modifying enzymes (aph(3’)-IIIa, sat4, aadE, spw, aac(6’)-Ie-aph(2’’)-Ia, mrs(D), mph(C), lnu(B), erm(47)), active efflux pumps (fexA, mef(A/E)), and mutations in chromosomal genes (pbp1a, pbp2b, pbp2x, mraY, gyrA, parC, and dhfr). Our genetic analysis and comparisons showed evidence for the genetic transference of AMR genes between S. suis isolates and strains of other species, which explains the broad AMR dissemination and multidrug resistance of S. suis.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Bioproject, PRJNA1037519, Ss_02 (ASM3380320v1), Ss_69 (ASM3380323v1), Ss_70 (ASM3380322v1), Ss_80 (ASM3380318v1), Ss_92 (ASM3380316v1), Ss_93 (ASM3380314v1), Ss_100 (ASM3380312v1), Ss_121 (ASM3380306v1), Ss_156 (ASM3380310v1), Ss_166 (ASM3380308v1), and Ss_167 (ASM3380302v1).
Author contributions
CU: Data curation, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. JG: Investigation, Writing – review & editing. MS: Investigation, Writing – review & editing. LF: Investigation, Methodology, Supervision, Writing – review & editing. CM: Investigation, Funding acquisition, Writing – review & editing. JA: Funding acquisition, Conceptualization, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work received funding from Gobierno de Aragón (Department of I+D+I projects in priority lines, Grant agreement LMP58_21), and Ministerio de Ciencia e Innovación/Agencia Española de Investigación MCIN/AEI/10.13039/501100011033, as appropriate, by ERDF A way of making Europe by the European Union or by the European Union NextGenerationEU/PRTR (Gran agreement PID2020-114617RB-100). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank Mateo del Pozo (Labopat, Spain), Maria Casas Amorieta (Ovislab, Spain) and Ana Fernandez and Jose L Arnal (Exopol, Spain) for kindly provide the S. suis isolates used in this study for research. We would like thanks Jorge Hugo (CITA, Spain) for support in bioinformatic analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1329632/full#supplementary-material
Supplementary Table 1 | S. suis isolates used in this study and the MIC values for 18 antibiotics. The geographic origin, ST, serotype, and host is indicated.
Supplementary Table 2 | Primers used in this study.
Supplementary Table 3 | Events of genomic recombination detected in 19 genomes of S. suis.
Supplementary Table 4 | Mobile genetic elements found in isolates of S. suis by whole genome sequencing.
Supplementary Figure 1 | Maximum-likelihood phylogenetic tree constructed with the nucleotide sequences of the 30 genomes of our S. suis collection (colored in black) and 8 reference genomes (colored in red). The genetic characteristics of serotype, ST, eBG and CC are given in the first 4 columns (color code as indicated in the figure legend). From column 5 onwards, the resistance profile to the 17 antimicrobials tested is shown in green when the isolate is susceptible, red when is resistant, and yellow when it has intermedia resistance. ST, Sequence Type; eBG, eBurst Group; CC, Clonal Complex; ser, Serotype; SPE, Spectinomycin; AMOX, Amoxicillin; AMP, Ampicillin; CEFQ, Cefquinome; CEFT, Ceftiofur; PG, Penicillin G; FLO, Florfenicol; ENR, Enrofloxacin; MAR, Marbofloxacin; CLI, Clindamycin; LIN, Lincomycin; ERY, Erythromycin; TIL, Tilmicosin; TYL, Tylosin; TIA, Tiamulin; S/TMP, Sulfamethoxazole/Trimethoprim; DOX, Doxycycline; TET, Tetracycline.
Supplementary Figure 2 | Distribution of multi-resistance patterns identified in 116 invasive S. suis isolates. The number of antibiotic families and the percentage of isolates in each pattern is indicated.
Supplementary Figure 3 | Alignment of sequences of AMR determinants in susceptible and resistant isolates. Alignments comprise (A) spw gene and the upstream sequence of one spectinomycin resistant and three spectinomycin-susceptible isolates, (B) GyrA sequences of fluoroquinolone resistant and susceptible isolates, (C) ParC of fluoroquinolone resistant and susceptible isolates, and (D) DHFR of sulfamethoxazole/TMP resistant and susceptible isolates. In panels b-d, the site position previously associated with antibiotic resistance are indicated, including positions 81 and 85 for GyrA, position 79 and 83 for ParC, and 102 position for DHFR. The sequence of the reference genome P1/7, which is susceptible to all antibiotics, is included. Susceptible isolates are green coloured, resistant are red coloured, and, in the case of fluoroquinolones, marbofloxacin resistant isolates are orange coloured otherwise indicated.
Supplementary Figure 4 | Genetic organization of different genes involved in AMR. The shadow indicates sequences where recombination breakpoints were estimated. HP, Hypothetical protein; ABC-t, ABC transporter ATP-binding protein.
References
Arenas, J., Abel, A., Sánchez, S., Marzoa, J., Berrón, S., van der Ley, P., et al. (2008). A cross-reactive neisserial antigen encoded by the NMB0035 locus shows high sequence conservation but variable surface accessibility. J. Med. Microbiol. 57 (Pt 1), 80–87. doi: 10.1099/jmm.0.47172-0
Arndt, E. R., Farzan, A., MacInnes, J. I., Friendship, R. M. (2019). Antimicrobial resistance of Streptococcus suis isolates recovered from clinically ill nursery pigs and from healthy pigs at different stages of production. Can. Vet. J. 60 (5), 519–522.
Bamphensin, N., Chopjitt, P., Hatrongjit, R., Boueroy, P., Fittipaldi, N., Gottschalk, M., et al. (2021). Non-penicillin-susceptible Streptococcus suis isolated from humans. Pathogens 10 (9), 1178. doi: 10.3390/pathogens10091178
Bojarska, A., Molska, E., Janas, K., Skoczyńska, A., Stefaniuk, E., Hryniewicz, W., et al. (2016). Streptococcus suis in invasive human infections in Poland: clonality and determinants of virulence and antimicrobial resistance. Eur. J. Clin. Microbiol. Infect. Dis. 35 (6), 917–925. doi: 10.1007/s10096-016-2616-x
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Bordeleau, E., Stogios, P. J., Evdokimova, E., Koteva, K., Savchenko, A., Wright, G. D. (2021). ApmA is a unique aminoglycoside antibiotic acetyltransferase that inactivates apramycin. mBio 12 (1), e02705–e02720. doi: 10.1128/mBio.02705-20
Callens, B. F., Haesebrouck, F., Maes, D., Butaye, P., Dewulf, J., Boyen, F. (2013). Clinical resistance and decreased susceptibility in Streptococcus suis isolates from clinically healthy fattening pigs. Microb. Drug Resist. 19 (2), 146–151. doi: 10.1089/mdr.2012.0131
Chen, L., Huang, J., Huang, X., He, Y., Sun, J., Dai, X., et al. (2021). Horizontal transfer of different erm(B)-carrying mobile elements among Streptococcus suis strains with different serotypes. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.628740
Chen, L., Song, Y., Wei, Z., He, H., Zhang, A., Jin, M. (2013). Antimicrobial susceptibility, tetracycline and erythromycin resistance genes, and multilocus sequence typing of Streptococcus suis isolates from diseased pigs in China. J. Vet. Med. Sci. 75 (5), 583–587. doi: 10.1292/jvms.12-0279
Clermont, D., Chesneau, O., De Cespédès, G., Horaud, T. (1997). New tetracycline resistance determinants coding for ribosomal protection in streptococci and nucleotide sequence of tet(T) isolated from Streptococcus pyogenes A498. Antimicrob. Agents Chemother. 41, 112–116. doi: 10.1128/AAC.41.1.11200ED29
CLSI (2019) CLSI M1. Available at: http://em100.edaptivedocs.net/GetDoc.aspx?doc=CLSI%20M100%20ED29:2019&sbssok=CLSI%20M100%20ED29:2019&format=HTML&hl=CLSI%20M100%202019 2019 (Accessed February 2022).
Corsaut, L., Misener, M., Canning, P., Beauchamp, G., Gottschalk, M., Segura, M. (2020). Field study on the immunological response and protective effect of a licensed autogenous vaccine to control Streptococcus suis infections in post-weaned piglets. Vaccines 8 (3), 384. doi: 10.3390/vaccines8030384
Cucco, L., Paniccià, M., Massacci, F. R., Morelli, A., Ancora, M., Mangone, I., et al. (2022). New sequence types and antimicrobial drug–resistant strains of Streptococcus suis in diseased pigs, Italy 2017–2019. Emerg. Infect. Dis. 28 (1), 139–147. doi: 10.3201/eid2801.210816
DANMAP 2020 (2021). Use of antimicrobial agents and occurrence of antimicrobial resistance in bacteria from food animals, food and humans in Denmark. Available at: https://www.ssi.dk/-/media/arkiv/subsites/antibiotikaresistens/danmap_2020_07102021_version-2_low.pdf
Dechêne-Tempier, M., Marois-Créhan, C., Libante, V., Jouy, E., Leblond-Bourget, N., Payot, S. (2021). Update on the mechanisms of antibiotic resistance and the mobile resistome in the emerging zoonotic pathogen Streptococcus suis. Microorganisms 9, 1765. doi: 10.3390/microorganisms9081765
Del Amparo, R., Branco, C., Arenas, J., Vicens, A., Arenas, M. (2021). Analysis of selection in protein-coding sequences accounting for common biases. Brief. Bioinform. 22 (5), bbaa431. doi: 10.1093/bib/bbaa431
Escudero, J. A., San Millan, A., Catalan, A., de la Campa, A. G., Rivero, E., Lopez, G., et al. (2007). First characterization of fluoroquinolone resistance in Streptococcus suis. Antimicrob. Agents Chemother. 51 (2), 777–782. doi: 10.1128/AAC.00972-06
EUCAST (2023) Breakpoint tables for interpretation of MICs and zone diameters. Version 13.1, 2023. Available at: https://www.eucast.org/clinical_breakpoints (Accessed November 2023).
Fabre, A., Oleastro, M., Nunes, A., Santos, A., Sifré, E., Ducournau, A., et al. (2018). Whole-genome sequence analysis of multidrug-resistant Campylobacter isolates: a focus on aminoglycoside resistance determinants. J. Clin. Microbiol. 56 (9), e00390–e00318. doi: 10.1128/JCM.00390-18
Feßler, A. T., Kadlec, K., Schwarz, S. (2011). Novel apramycin resistance gene apmA in bovine and porcine methicillin-resistant Staphylococcus aureus ST398 isolates. Antimicrob. Agents Chemother. 55 (1), 373–375. doi: 10.1128/AAC.01124-10
Feßler, A. T., Wang, Y., Wu, C., Schwarz, S.. (2018). Mobile lincosamide resistance genes in staphylococci. Plasmid 99, 22–31. doi: 10.1016/j.plasmid.2018.06.002
Gill, M. J., Brenwald, N. P., Wise, R. (1999). Identification of an efflux pump gene, pmrA, associated with fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 43 (1), 187–189. doi: 10.1128/AAC.43.1.187
González, I., Georgiou, M., Alcaide, F., Balas, D., de la Campa, A. G. (1998). Fluoroquinolone resistance mutations in the parC, parE, and gyrA genes of clinical isolates of viridans group streptococci. Antimicrob. Agents Chemother. 42 (11), 2792–2798. doi: 10.1128/AAC.42.11.2792
Goyette-Desjardins, G., Auger, J. P., Xu, J., Segura, M., Gottschalk, M. (2014). Streptococcus suis, an important pig pathogen and emerging zoonotic agent—an update on the worldwide distribution based on serotyping and sequence typing. Emerg. Microbes Infect. 3 (6), e45. doi: 10.1038/emi.2014.45
Grattarola, C., Bellino, C., Tursi, M., Maggi, E., D'Angelo, A., Gianella, P., et al. (2010). Helcococcus kunzii isolated from a sow with purulent urocystitis. J. Clin. Microbiol. 48 (8), 3019–3020. doi: 10.1128/JCM.00554-10
Guérin, F., Isnard, C., Bucquet, F., Fines-Guyon, M., Giard, J. C., Burrus, V. (2016). Novel chromosome-encoded erm(47) determinant responsible for constitutive MLSB resistance in Helcococcus kunzii. J. Antimicrob. Chemother. 71 (11), 3046–3049. doi: 10.1093/jac/dkw290
Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29 (8), 1072–1075. doi: 10.1093/bioinformatics/btt086
Hadjirin, N. F., Miller, E. L., Murray, G. G. R., Yen, P. L. K., Phuc, H. D., Wileman, T. M., et al. (2021). Large-scale genomic analysis of antimicrobial resistance in the zoonotic pathogen Streptococcus suis. BMC Biol. 19 (1), 191. doi: 10.1186/s12915-021-01094-1
Hakenbeck, R., Brückner, R., Denapaite, D., Maurer, P. (2012). Molecular mechanisms of β-lactam resistance in Streptococcus pneumoniae. Future Microbiol. 7 (3), 395–410. doi: 10.2217/fmb.12.2
Hayer, S. S., Rovira, A., Olsen, K., Johnson, T. J., Vannucci, F., Rendahl, A., et al. (2020). Prevalence and time trend analysis of antimicrobial resistance in respiratory bacterial pathogens collected from diseased pigs in USA between 2006–2016. Res. Vet. Sci. 128, 135–144. doi: 10.1016/j.rvsc.2019.11.010
Hernandez-Garcia, J., Wang, J., Restif, O., Holmes, M. A., Mather, A. E., Weinert, L. A., et al. (2017). Patterns of antimicrobial resistance in Streptococcus suis isolates from pigs with or without streptococcal disease in England between 2009 and 2014. Vet. Microbiol. 207, 117–124. doi: 10.1016/j.vetmic.2017.06.002
Huang, J., Chen, L., Wu, Z., Wang, L. (2017). Retrospective analysis of genome sequences revealed the wide dissemination of optrA in Gram-positive bacteria. J. Antimicrob. Chemother. 72 (2), 614–616. doi: 10.1093/jac/dkw488
Huang, K., Song, Y., Zhang, Q., Zhang, A., Jin, M. (2016). Characterisation of a novel integrative and conjugative element ICESsD9 carrying erm(B) and tet(O) resistance determinants in Streptococcus suis, and the distribution of ICESsD9-like elements in clinical isolates. J. Glob. Antimicrob. Resist. 7, 13–18. doi: 10.1016/j.jgar.2016.05.008
Huang, J., Sun, J., Wu, Y., Chen, L., Duan, D., Lv, X., et al. (2019). Identification and pathogenicity of an XDR Streptococcus suis isolate that harbours the phenicol-oxazolidinone resistance genes optrA and cfr, and the bacitracin resistance locus bcrABDR. Int. J. Antimicrob. Agents. 54 (1), 43–48. doi: 10.1016/j.ijantimicag.2019.04.003
Jorgensen, J. H., Weigel, L. M., Ferraro, M. J., Swenson, J. M., Tenover, F. C. (1999). Activities of newer fluoroquinolones against Streptococcus pneumoniae clinical isolates including those with mutations in the gyrA, parC, and parE loci. Antimicrob. Agents Chemother. 43 (2), 329–334. doi: 10.1128/AAC.43.2.329
Kosakovsky Pond, S. L., Poon, A. F. Y., Velazquez, R., Weaver, S., Hepler, N. L., et al. (2020). HyPhy 2.5 - a customizable platform for evolutionary hypothesis testing using phylogenies. Mol. Biol. Evol. 37 (1), 295–299. doi: 10.1093/molbev/msz197
Liang, P., Wang, M., Gottschalk, M., Vela, A. I., Estrada, A. A., Wang, J., et al. (2021). Genomic and pathogenic investigations of Streptococcus suis serotype 7 population derived from a human patient and pigs. Emerg. Microbes Infect. 10 (1), 1960–1974. doi: 10.1080/22221751.2021.1988725
Lunha, K., Chumpol, W., Jiemsup, S., Samngamnim, S., Assavacheep, P., Yongkiettrakul, S. (2023). Relationship between penicillin-binding proteins alterations and β-lactams non-susceptibility of diseased pig-isolated Streptococcus suis. Antibiotics 12 (1), 158. doi: 10.3390/antibiotics12010158
Marini, E., Palmieri, C., Magi, G., Facinelly, B. (2015). Recombination between Streptococcus suis ICESsu32457 and Streptococcus agalactiae ICESa2603 yields a hybrid ICE transferable to Streptococcus pyogenes. Vet. Microbiol. 178 (1-2), 99–104. doi: 10.1016/j.vetmic.2015.04.013
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 1 (1), vev003. doi: 10.1093/ve/vev003
Matiašovic, J., Nedbalcová, K., Žižlavský, M., Fleischer, P., Pokludová, L., Kellnerová, D., et al. (2021). Streptococcus suis isolates—serotypes and susceptibility to antimicrobials in terms of their use on selected repopulated Czech pig farms. Pathogens 10 (10), 1314. doi: 10.3390/pathogens10101314
Meyer, A., Dierks, K., Hussein, R., Brillet, K., Brognaro, H., Betzel, C. (2015). Systematic analysis of protein-detergent complexes applying dynamic light scattering to optimize solutions for crystallization trials. Acta Crystallogr. F Struct. Biol. Commun. 71 (Pt 1), 75–81. doi: 10.1107/S2053230X14027149
Neila-Ibáñez, C., Napp, S., Pailler-García, L., Franco-Martínez, L., Cerón, J. J., Aragon, V., et al. (2023). Risk factors associated with Streptococcus suis cases on pig farms in Spain. Vet. Rec. 193 (5), e3056. doi: 10.1002/vetr.3056
Nishimoto, Y., Kobayashi, N., Alam, M. M., Ishino, M., Uehara, N., Watanabe, N. (2005). Analysis of the prevalence of tetracycline resistance genes in clinical isolates of Enterococcus faecalis and Enterococcus faecium in a Japanese hospital. Microb. Drug Resist. 11 (2), 146–153. doi: 10.1089/mdr.2005.11.146
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31 (22), 3691–3693. doi: 10.1093/bioinformatics/btv421
Petrocchi-Rilo, M., Martínez-Martínez, S., Aguarón-Turrientes, Á., Roca-Martínez, E., García-Iglesias, M. J., Pérez-Fernández, E., et al. (2021). Anatomical site, typing, virulence gene profiling, antimicrobial susceptibility and resistance genes of Streptococcus suis isolates recovered from pigs in Spain. Antibiotics 10 (6), 707. doi: 10.3390/antibiotics10060707
Rocha, E. P. (2006). The quest for the universals of protein evolution. Trends Genet. 22 (8), 412–416. doi: 10.1016/j.tig.2006.06.004
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30 (14), 2068–2069. doi: 10.1093/bioinformatics/btu153
Segura, M. (2015). Streptococcus suis vaccines: candidate antigens and progress. Expert Rev. Vaccines 14 (12), 1587–1608. doi: 10.1586/14760584.2015.1101349
Shang, Y., Li, D., Hao, W., Schwarz, S., Shan, X., Liu, B., et al. (2019). A prophage and two ICESa2603-family integrative and conjugative elements (ICEs) carrying optrA in Streptococcus suis. J. Antimicrob. Chemother. 74 (10), 2876–2879. doi: 10.1093/jac/dkz309
Stefańska, I., Kwiecień, E., Kizerwetter-Świda, M., Chrobak-Chmiel, D., Rzewuska, M. (2022). Tetracycline, macrolide and lincosamide resistance in Streptococcus canis strains from companion animals and its genetic determinants. Antibiotics 11 (8), 1034. doi: 10.3390/antibiotics11081034
Tamura, K., Stecher, G., Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38 (7), 3022–3027. doi: 10.1093/molbev/msab120
Uruén, C., Fernandez, A., Arnal, J. L., del Pozo, M., Casas, M., de Blas, I., et al. (2024). Genomic and phenotypic analysis of invasive Streptococcus suis isolated in Spain reveals genetic diversification and associated virulence traits. Vet. Res. In press. doi: 10.21203/rs.3.rs-3414110/v1
Uruén, C., Chopo-Escuin, G., Tommassen, J., Mainar-Jaime, R. C., Arenas, J.. (2020). Biofilms as promoters of bacterial antibiotic resistance and tolerance. Antibiotics 10 (1), 3. doi: 10.3390/antibiotics10010003
Uruén, C., García, C., Fraile, L., Tommassen, J., Arenas, J. (2022). How Streptococcus suis escapes antibiotic treatments. Vet. Res. 53 (1), 91. doi: 10.1186/s13567-022-01111-3
Vachee, A., Varon, E., Jouy, E., Meunier, D. (2008). Antibiotics susceptibility of Streptococcus and Enterococcus: data of Onerba network. Pathol. Biol. (Paris). 57 (3), 240–244. doi: 10.1016/j.patbio.2007.12.009
van Hout, J., Heuvelink, A., Gonggrijp, M. (2016). Monitoring of antimicrobial susceptibility of Streptococcus suis in the Netherlands 2013–2015. Vet. Microbiol. 194, 5–10. doi: 10.1016/j.vetmic.2016.03.014
Vela, A. I., Moreno, M. A., Cebolla, J. A., González, S., Latre, M. V., Domínguez, L., et al. (2005). Antimicrobial susceptibility of clinical strains of Streptococcus suis isolated from pigs in Spain. Vet. Microbiol. 105 (2), 143–147. doi: 10.1016/j.vetmic.2004.10.009
Viera, A. J. (2008). Odds ratios and risk ratios: what's the difference and why does it matter? South. Med. J. 101 (7), 730–734. doi: 10.1097/SMJ.0b013e31817a7ee4
Vötsch, D., Willenborg, M., Weldearegay, Y. B., Valentin-Weigand, P. (2018). Streptococcus suis–the “two faces” of a pathobiont in the porcine respiratory tract. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00480
Werinder, A., Aspán, A., Backhans, A., Sjölund, M., Guss, B., Jacobson, M. (2020). Streptococcus suis in Swedish grower pigs: occurrence, serotypes, and antimicrobial susceptibility. Acta Vet. Scand. 62 (1), 36. doi: 10.1186/s13028-020-00533-3
Yongkiettrakul, S., Maneerat, K., Arechanajan, B., Malila, Y., Srimanote, P., Gottschalk, M., et al. (2019). Antimicrobial susceptibility of Streptococcus suis isolated from diseased pigs, asymptomatic pigs, and human patients in Thailand. BMC Vet. Res. 15 (1), 5. doi: 10.1186/s12917-018-1732-5
Yongkiettrakul, S., Wongsurawat, T., Jenjaroenpun, P., Acheampong, D. A., Srimanote, P., Maneerat, K., et al. (2021). Genome sequences of antibiotic-resistant Streptococcus suis strains isolated from human patients and diseased and asymptomatic pigs in Thailand. Infect. Genet. Evol. 87, 104674. doi: 10.1016/j.meegid.2020.104674
Zhang, A., Yang, M., Hu, P., Wu, J., Chen, B., Hua, Y., et al. (2011). Comparative genomic analysis of Streptococcus suis reveals significant genomic diversity among different serotypes. BMC Genomics 12, 523. doi: 10.1186/1471-2164-12-523
Zhang, C., Zhang, P., Wang, Y., Fu, L., Liu, L., Xu, D., et al. (2020). Capsular serotypes, antimicrobial susceptibility, and the presence of transferable oxazolidinone resistance genes in Streptococcus suis isolated from healthy pigs in China. Vet. Microbiol. 247, 108750. doi: 10.1016/j.vetmic.2020.108750
Keywords: Streptococcus suis, antimicrobial resistance (AMR), antimicrobial resistant genes, mobile genetic elements, ICEs, IMEs, Streptococcus sp.
Citation: Uruén C, Gimeno J, Sanz M, Fraile L, Marín CM and Arenas J (2024) Invasive Streptococcus suis isolated in Spain contain a highly promiscuous and dynamic resistome. Front. Cell. Infect. Microbiol. 13:1329632. doi: 10.3389/fcimb.2023.1329632
Received: 29 October 2023; Accepted: 26 December 2023;
Published: 22 January 2024.
Edited by:
Mohammad Shahid, Arabian Gulf University, BahrainReviewed by:
Ben Pascoe, University of Oxford, United KingdomAnusak Kerdsin, Kasetsart University, Thailand
Copyright © 2024 Uruén, Gimeno, Sanz, Fraile, Marín and Arenas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesús Arenas, jaarenas@unizar.es