 Kenneth James Chapin1†
Kenneth James Chapin1† Matthew R. Jones
Matthew R. Jones Daniel E. Winkler
Daniel E. Winkler Rob Massatti
Rob Massatti- 1US Geological Survey, Southwest Biological Science Center, Tucson, AZ, United States
- 2US Geological Survey, Southwest Biological Science Center, Flagstaff, AZ, United States
- 3Deaver Herbarium, Northern Arizona University, Flagstaff, AZ, United States
Uncovering the historical and contemporary processes shaping rare species with complex distributions is of growing importance due to threats such as habitat destruction and climate change. Species restricted to specialized, patchy habitat may persist by virtue of life history characteristics facilitating ongoing gene flow and dispersal, but they could also reflect the remnants of formerly widespread, suitable habitat that existed during past climate regimes. If formerly widespread species did not rely upon traits facilitating high dispersibility to persist, contemporary populations could be at high risk of extirpation or extinction. Fortunately, genomic investigations provide an opportunity to illuminate such alternative scenarios while simultaneously offering guidance for future management interventions. Herein, we test the role of these mechanisms in shaping patterns of genomic diversity and differentiation across a highly restricted and rare ecosystem: desert hanging gardens. We focus on Carex specuicola (Cyperaceae), a hanging garden obligate narrowly distributed in the Four Corners region of the southwestern United States that is listed as Threatened under the United States Endangered Species Act. Population structure and diversity analyses reveal that hanging garden populations are shaped by strong genetic drift, but that individuals in gardens are occasionally more closely related to individuals at other gardens than to individuals within the same garden. Similarly, gardens separated by long geographic distances may contain individuals that are more closely related compared to individuals in gardens separated by short geographic distances. Demographic modeling supports historical gene flow between some contemporary garden pairs, which is corroborated by low estimates of inbreeding coefficients and recent divergence times. As such, multiple lines of evidence support dispersal and gene flow across C. specuicola populations at both small and large spatial scales, indicating that even if C. specuicola was formerly more widespread, it may be well suited to persist in hanging gardens so long as suitable habitat remains available. Analyses like those demonstrated herein may be broadly applicable for understanding the short- and long-term evolutionary processes influencing rare species, and especially those having complex distributions across heterogeneous landscapes.
Introduction
Rare species with patchy distributions are susceptible to evolutionary processes that threaten long-term viability. For example, genetic drift due to small effective population sizes may result in the loss of genetic variation necessary to respond to selective pressures (Barrett and Schluter, 2008) and the fixation of deleterious mutations (Wright, 1931; Lynch et al., 1995; Allendorf et al., 2010). These circumstances are contemporaneously exacerbated by habitat degradation or loss and climate change, threats to global biodiversity that can further diminish or fragment species’ distributions (Brooks et al., 2002; Hanski, 2011; Tilman et al., 2017) and cause extirpations or extinctions. When a species’ declining abundance warrants management interventions, conservationists may seek to enhance connectivity and boost genetic diversity by restoring previously lost or degraded habitats or by promoting gene flow through efforts such as translocation (Hanski, 2011; Pavlova et al., 2017). However, the need to enhance genetic connectivity among locations should consider a species’ dispersal ability (Lowe and Allendorf, 2010; Reynolds et al., 2013; Kendrick et al., 2017). For example, some species, regardless of rarity, are able to maintain long-term viability by functioning as metapopulations, or multiple populations that are connected through dispersal (Hanski, 1998). As such, understanding the levels of connectivity and genetic diversity between populations distributed across heterogeneous landscapes is crucial to identifying and prioritizing conservation needs and informing appropriate management actions (e.g., Doerr et al., 2011).
Hanging garden ecosystems exemplify communities where organisms oftentimes have small populations scattered across a landscape. Hanging garden habitat occurs where water seeps from vertical rock faces (Welsh, 1989) and is often highly complex, for example, composed of small, isolated patches that are irregularly distributed across a topographically heterogeneous landscape (Malanson and Kay, 1980; Welsh, 1989). Some species occupying hanging gardens are obligates, resulting in distributions that mirror the complexity of their habitat (e.g., Palmquist et al., 2015). Concordantly, small, isolated, and scattered populations are prone to genetic stochasticity (i.e., inbreeding and drift) that can lead to extirpation or extinction (Nieminen et al., 2001; Keller and Waller, 2002; Reed, 2004). As a result, dispersal mechanisms promoting gene flow may be critical for the maintenance of populations and the overall long-term viability of a species. Challenges facing hanging garden species are further compounded by threats resulting from warmer and drier climatic conditions (Diffenbaugh et al., 2008; Redsteer et al., 2013; Elias et al., 2018; Gallo et al., 2020), invasive species, and changing land use regimes. Therefore, investigating the dispersal ability of hanging garden obligates may not only be broadly informative regarding rates of gene flow to maintain self-sustaining species, but it may also inform management interventions for species declining in abundance.
Exacerbating the evolutionary uncertainty resulting from the geospatial discontinuity of hanging garden habitat and species’ dispersal capabilities, the present distributions of obligate hanging garden species may result from alternative historical processes. For example, hanging gardens may represent paleorefugia resulting from vicariance events following the Last Glacial Maximum (Nekola, 1999). In this scenario, extirpation is expected to play a larger role in population-level processes compared to dispersal because more connected, continuous habitat (i.e., across more mesic valley bottoms) previously available to the species during cooler, wetter periods no longer exists (e.g., Avise, 2000). Similar vicariance scenarios have been supported in, for example, montane ecosystems, where once widespread species are now restricted to more limited distributions at higher elevations (Galbreath et al., 2010; Massatti and Knowles, 2016; Hodel et al., 2021). Alternatively, hanging garden specialists may be successful in their habitat due to life history strategies that allow them to persist despite the habitat’s complexity. In this scenario, a species’ history may be defined by its unique relationship to this specialized habitat, similar to species with limited distributions due to edaphic specialization (Corlett and Tomlinson, 2020). Accordingly, dispersal would be expected to play an important role in colonizing unoccupied habitat and maintaining genetic diversity within disparate populations.
What is evident by considering contemporary ecological dynamics, evolutionary processes, and phylogeographic hypotheses is that an integrative research approach is required to understand the conservation needs of rare, hanging garden obligates, especially considering continuing global change. Herein, we investigate population genomic patterns and evolutionary dynamics within Carex specuicola Howell (Cyperaceae; Navajo sedge), an obligate hanging garden sedge narrowly distributed across the Colorado Plateau Desert of the southwestern United States. We adopt the paleorefugium hypothesis as our null hypothesis given patterns resolved in other species (Palmquist et al., 2015) and prevailing assumptions implicit in C. specuicola’s management (detailed below; U.S. Fish and Wildlife Service, 2014). We use population genomic analyses to infer population structure, evidence of dispersal, and patterns of genomic diversity across the species’ range and demographic modeling to quantify divergence times and rates of historical gene flow between selected population pairs. Under the assumptions that the species’ current distribution results from vicariance and that extirpation is the predominant process influencing the species’ distribution, we predict that C. specuicola populations within hanging gardens will by highly differentiated and display large inbreeding coefficients (consistent with a lack of ongoing gene flow and small effective population sizes), population divergence times will be old, and that evidence for dispersal will be lacking. We include samples of C. specuicola’s putative sister taxon, C. utahensis Reznicek & Murray (Utah sedge), to ensure that inferred patterns of genomic structure, diversity, or population history are not misinterpreted signals of hybridization. Discerning the contemporary and historical processes influencing C. specuicola may help managers gain crucial insights into the species’ natural history and ability to persist naturally on heterogeneous landscapes and thereby inform the need for management interventions. More generally, the processes implicit in this system may illuminate how broadly conservation efforts may need to be applied to rare plants in specialized, remnant habitats facing global threats like climate change.
Materials and methods
Study system background
Carex specuicola is a perennial monocot endemic to the southern Colorado Plateau Desert in Arizona and Utah. The species is long-lived and occurs almost exclusively in hanging garden ecosystems (U.S. Fish and Wildlife Service, 2014), which house high numbers of endemic and rare plant species across this region (Welsh, 1989; Spence and Henderson, 1993; Fowler et al., 2007). In 1985, C. specuicola was only known from three localities (~700 individuals) and was listed as Threatened under the U.S. Endangered Species Act (ESA), with 600 square meters of critical habitat designated (U.S. Fish and Wildlife Service, 1987); since its listing, additional surveys have revealed 170 hanging garden sites occupied by C. specuicola (Rink, 2017). Occupied garden sites vary in size from 0.1–100 m2 and are isolated from one another by intervening dry habitats. Vegetative spread resulting from underground rhizomes, as opposed to sexual reproduction from seed, is hypothesized to account for within-garden spread (Hermann, 1970). Rates of gene flow among gardens are unknown, though low gene flow is an assumption implicit in the species’ management (U.S. Fish and Wildlife Service, 2014) based on a lack of seed traits promoting high dispersibility, presumed pollination by wind (Friedman and Barrett, 2009), and a short duration of pollen viability (Dafni and Firmage, 2000).
Carex specuicola may be impacted by gene flow with its partially sympatric putative sister taxon, C. utahensis. Carex utahensis is a regional endemic with a more northern distribution occupying seeps, wet slopes, wet meadows, and riparian bottomlands. The species was described based on morphological differences with C. specuicola (e.g., stiff versus flexuous culm, perigynium shape, and style number; Reznicek and Murray, 2013), but morphological overlap is not uncommon, which may suggest past or current hybridization or a recent divergence time between these taxa. Evidence of gene flow between the species may have critical implications for the long-term survival of C. specuicola and its federal protection (US Fish and Wildlife Service, 2014).
Sample collection, DNA isolation, and library preparation
We sampled leaf tissue from 190 Carex individuals across 20 sampling sites (1-12 putative independent genets per site) spanning the breadth of the C. specuicola (164 samples) distribution along with samples of C. utahensis (23 samples), C. curatorum (2 samples), and C. aurea (1 sample) (Figure 1 and Table 1); the latter two species often co-occur with C. specuicola and C. utahensis in hanging garden or adjacent mesic habitat and share phylogenetic affinities, making them appropriate outgroups in phylogenomic analyses. Four sampling sites were represented by close-proximity subsites separated by 60 m to 3.27 km (1.38 ± 1.16 km), which helped us understand relatedness at small spatial scales (Table 1). Proximate gardens were closely related (see Results) and were grouped for downstream analyses when indicated in the Methods. We extracted genomic DNA from leaf tissue using DNeasy 96 Plant Kits (QIAGEN, Germantown, MD, USA) following the manufacturer’s protocol. We then prepared ddRADseq libraries with individually barcoded samples using EcoRI and MspI restriction enzymes (Peterson et al., 2012). We pooled individual libraries and amplified them via polymerase chain reactions. We then isolated amplified genomic fragments ranging in length from 400 to 600 base pairs (bps) with a Pippin Prep (Sage Science, Beverly, MA, USA). We sequenced the final pooled library on a HiSeq 4000 (Illumina, San Diego, CA, USA) at the University of Oregon Genomics and Cell Characterization Core Facility to generate single-end, 100 bp reads.
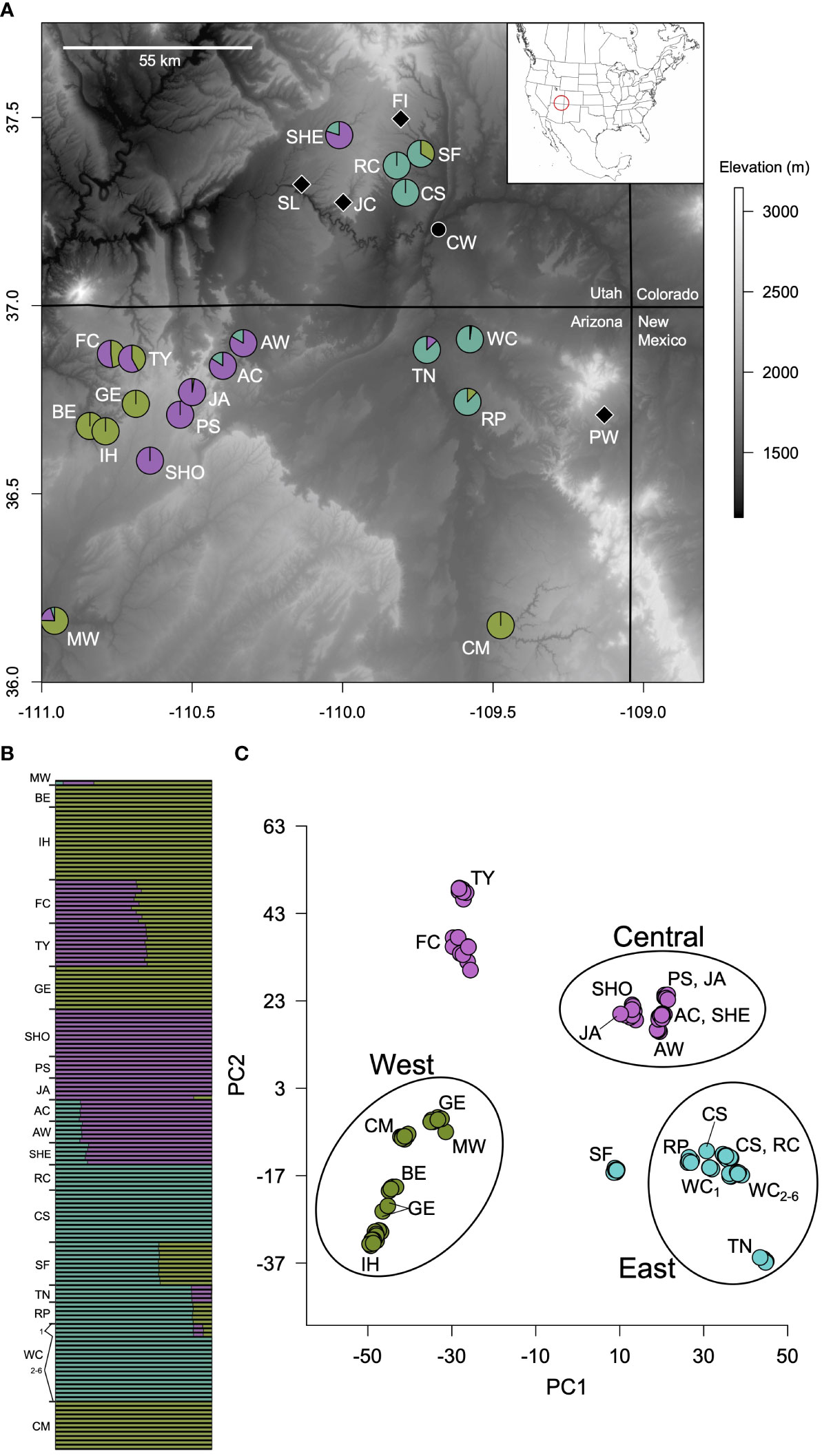
Figure 1 (A) Carex specuicola (circles) and C. utahensis (diamonds) sampling sites across Arizona and Utah. Colored pie charts for C. specuicola represent proportional ancestry coefficients averaged across individuals at a site; Chinle Wash (the black circle) was not included in Structure analyses due to putative admixture with C. utahensis. Subsites are grouped based on the uniformity of ancestry coefficients across individuals. (B) K = 3 Structure result showing ancestry coefficients across all individuals. (C) Principal component analysis of genomic variation within Carex specuicola. PC1 and PC2 account for 17.06% and 9.25% of variance, respectively. Individuals are colored according to their dominant ancestry coefficient. Sampling sites not contained within a circle are highly admixed according to Structure analyses. Individuals from sampling sites form tight clusters except for four individuals highlighted by black lines. Conservation implications for genetically defined populations (West, Central, East) are discussed in the text. See Table 1 for sites names associated with abbreviations.
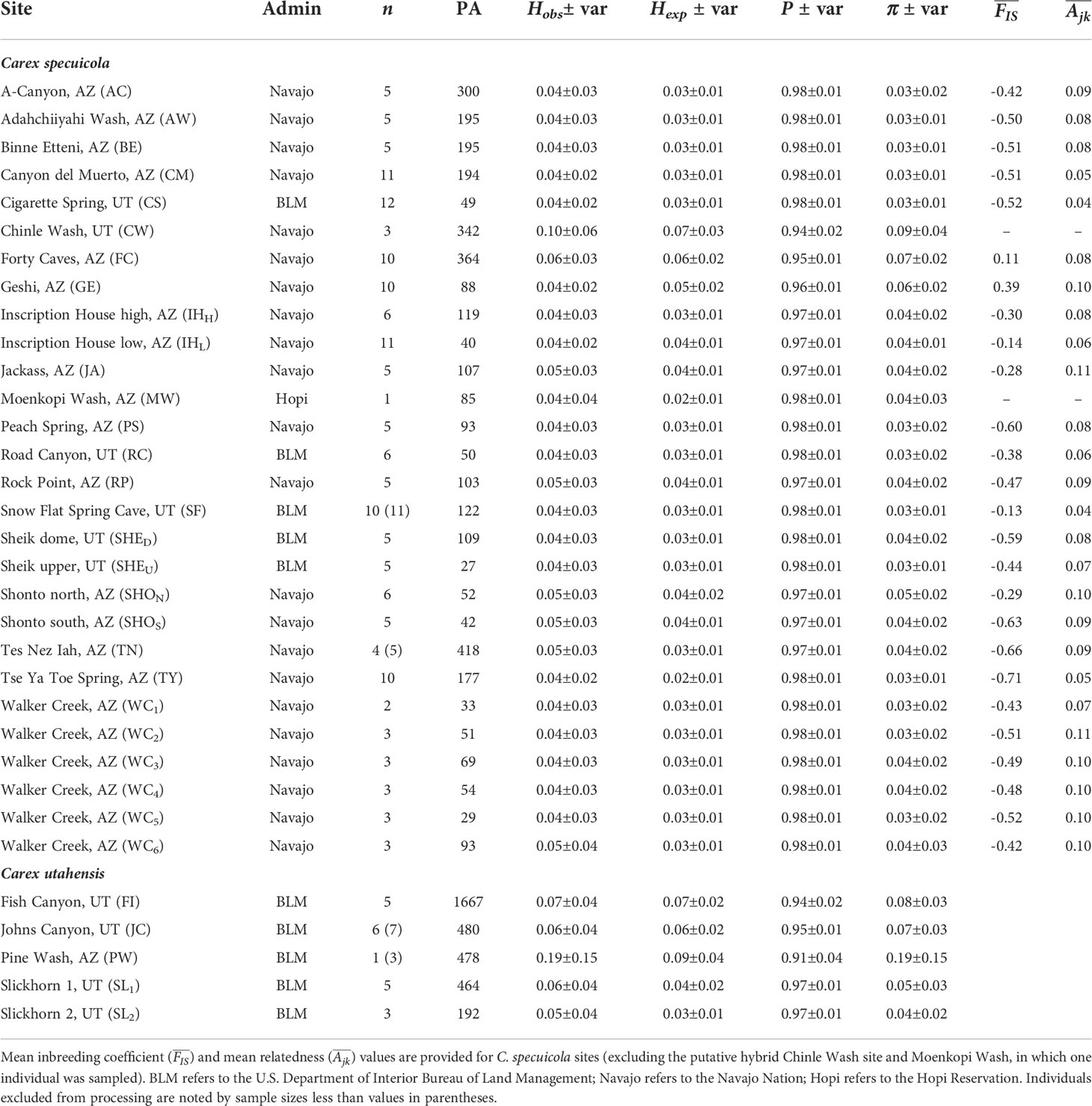
Table 1 Sampling site information for Carex specuicola and C. utahensis, including: Administrative unit (admin), sample size (n), private alleles (PA), observed and expected heterozygosity (Hobs, Hexp), major allele frequency (P), and nucleotide diversity (π).
Genomic data preprocessing
After removing five samples that yielded low quality DNA (Table 1), we processed and aligned raw sequence data of 185 samples using Stacks 2.41 (Rochette et al., 2019). First, we cleaned raw data by removing reads with uncalled bases or an average phred-scaled quality score below 22 within a 15 bp sliding window. We also trimmed the last 5 bp because quality scores decline sharply towards the end of reads. We determined the appropriate de novo assembly and filtering parameters in Stacks following Rochette and Catchen (2017). We first processed a test dataset of 19 samples (16 C. specuicola, 3 C. utahensis) while varying the parameters M (the maximum nucleotide distance allowed between stacks) and n (the number of mismatches allowed between loci during catalog construction) from 1 to 9 and while keeping M = n and setting m = 3 (minimum depth of coverage to create a stack). Following this procedure, we processed the full dataset with M = 4 and n = 4 because the total number of loci retained in 80% of individuals (r80) and the distribution of the number of single nucleotide polymorphisms (SNPs) per locus appeared to stabilize at these values (Figure S1). We calculated genetic summary statistics across sampling sites of C. specuicola and C. utahensis in the populations program in Stacks using filtering parameters that removed loci with heterozygosity > 75% (–max-obs-et 0.75) and loci not present in at least 50% of individuals in a sampling site (-r 0.5).
Discerning the relationship between C. specuicola and C. utahensis
To ensure that downstream analyses were not influenced by admixture with C. utahensis, we inferred relationships among Carex species using a maximum-likelihood phylogenetic analysis in PhyML 3.0 (Guindon et al., 2010). Model selection was implemented using smart model selection (Lefort et al., 2017) and nodal support values were calculated from 100 bootstrap iterations. The phylogeny was rooted using C. aurea and C. curatorum as outgroups. Based on the placement of Chinle Wash between C. specuicola and C. utahensis (see Results), we performed f3-admixture tests with treemix v1.13 (Pickrell and Pritchard, 2012), which use information on the frequency of shared SNPs among three populations to infer whether the relationship between the three populations is best explained by admixture as opposed to a simple bifurcating tree (Peter, 2016). A negative f3 statistic indicates that a population results from admixture. We calculated f3 statistics for the Chinle Wash (C. specuicola) site relative to parental C. specuicola and C. utahensis sites using a black-jackknife approach with 160 bins of 100 random SNPs to estimate standard errors.
Resolving genomic patterns and processes within C. specuicola
To gain a better understanding of the processes influencing hanging gardens, we estimated mean pairwise Hudson’s FST with KRIS (sensu Bhatia et al., 2013) between hanging gardens with hierfstat (Goudet and Jombart, 2020) in R 4.1.2 (R Core Team, 2021). These data were leveraged to test for isolation-by-distance (IBD) patterns across the species’ range and for each genetically defined group identified by Structure analyses (see below). In all cases, we used Mantel tests (1x105 permutations) and linear regressions to test for a relationship between genetic and geographic distance matrices (Mantel, 1967; Bougeard and Dray, 2018). Next, we calculated mean inbreeding coefficients (FIS) in hierfstat and mean relatedness (unadjusted mean Ajk; Yang et al., 2010) between samples within sites using the -relatedness function of vcftools (Danecek et al., 2011).
To investigate intraspecific genomic structure, we employed two approaches including 1) Bayesian clustering implemented in Structure v2.3.4 (Pritchard et al., 2000) and 2) principal coordinates analysis (PCA) implemented in the adgenet package (Jombart, 2008) in R. Given that these methods complement one another due to their reliance on different assumptions (Jombart et al., 2008; Frantz et al., 2009), we assess their results in tandem to determine biologically relevant, genetically defined groups instead of using model selection methods that can be misleading (i.e., Janes et al., 2017). For Structure, we tested K values (representing the number of genetically defined populations) from 1 to 8, performing 5 iterations under each K value with 20,000 burn-in and 100,000 regular repetitions. We excluded Chinle Wash individuals because they showed evidence of C. utahensis genomic identity (see Results).
To further investigate the spatial distribution of genetic diversity, we used the program EEMS (Estimated Effective Migration Surfaces; Petkova et al., 2016), which models the relationship between genetic variation and geography using Markov chain Monte Carlo sampling to explore large parameter spaces (Petkova et al., 2016). This procedure results in a visualization illustrating the geographic areas where occupied sites are expected to have higher or lower effective diversities, on average, based on patterns across the entire empirical data. We ran EEMS assuming a stepping-stone migration model with either 50, 100, or 200 subpopulations (termed demes in EEMS) spaced in a grid across a triangular range encompassing all sampled localities. We excluded Moenkopi Wash because we sampled only one individual from this site and grouped proximate gardens (see Table 1). We performed 3 independent runs for each subpopulation value to ensure model convergence. We used default hyper-parameter values and tuned the proposal variances until proposal acceptance rates reached 20-40%. We ran EEMS for 2 million iterations with a burn-in of 1 million iterations and thinning iteration of 9999.
We performed demographic modeling to estimate divergence times (t), ancestral effective population sizes (Nanc), contemporary effective population sizes (Ne), and levels of historical gene flow between pairs of closely related C. specuicola hanging garden sites. Specifically, we explored the pairwise evolutionary history of the Forty Caves and Tse Ya Toe Spring sites; Geshi and Canyon del Muerto sites; and Cigarette Spring and Road Canyon sites. Each pair of hanging gardens was closely related according to PCA, Structure, and phylogenetic analyses, though we excluded two Geshi individuals that may have resulted from a recent dispersal event (see Results).
We used ∂a∂i (Gutenkunst et al., 2009) to infer demographic parameters using a folded site frequency spectrum (SFS; i.e., major/minor allele calls rather than derived/ancestral). We scaled parameters by θ, assuming the tomato neutral mutation rate (m = 1 × 10-8; Lin et al., 2014) and a 337:1 ratio of callable sites:SNPs, based on output from Stacks. For each pair of sites, we tested two demographic models: 1) no migration, 2) isolation-with-migration (IM). The ‘no migration’ model involved a split between sites at t generations in the past and allowed for instantaneous population size changes immediately following divergence. The ‘IM’ model was identical but included a symmetrical migration rate (m; migrants per generation) between sites immediately following divergence t generations ago that remained constant up to the present day.
For each model, we performed 20 independent runs starting with parameter values sampled randomly across the following uniform distributions: 0.01 < n < 100.00, 0 < 2Nanct < 20, 0 < 2Nancm < 5; where n is the relative change in population size compared to Nanc. We determined which of the two maximum likelihood demographic models produced the best overall fit to the data using a composite-likelihood ratio test with the Godambe Information Matrix (GIM; Coffman et al., 2016) and Akaike Information Criterion scores (AIC; Akaike, 1973). We estimated confidence intervals for each parameter with the Godambe Information Matrix with 100 bootstrap data sets comprising 50% of SNPs randomly selected from the full data set. We validated the final model by comparing the predicted SFS to the observed SFS for each population.
Results
We genotyped 168,387 sequence sites from 343 million reads (± SD = 1,507,587 ± 379,466; sequence coverage = 39.7 ± 8.5). We removed 124,524 of 168,387 loci that did not pass filtering criteria set in the populations program, which resulted in a vcf file containing 43,863 variant sites (n = 185) used for demographic modeling, to calculate summary statistics, and in EEMS. Variant sites were also summarized per sampling location to generate a phylip file used for phylogenetic analysis. Limiting the dataset to one SNP per locus and excluding outgroups and putative hybrid individuals resulted in a dataset with 6827 variant sites (n = 158) used in Structure and PCA. Data are available in Massatti (2022).
Interactions between C. specuicola and C. utahensis
The maximum likelihood phylogeny of the four Carex species demonstrates the close and highly supported relationships within and between C. specuicola and C. utahensis (Figure 2). Most nodes have support values >95%, except for a few instances within the C. specuicola and C. utahensis clades. All proximate garden sites (i.e., those located within 3.27 km) clustered together with high bootstrap support. The only exception to the pattern of close intraspecific relationships is represented by Chinle Wash, which is sister to a clade containing the remaining C. specuicola gardens. As this may be a sign of past or ongoing gene flow with C. utahensis, we calculated f3 statistics to identify a signal of hybridization. However, we found no C. specuicola × C. utahensis comparison that yielded a negative f3 value ( ± SEM = 0.0202 ± 0.0018).
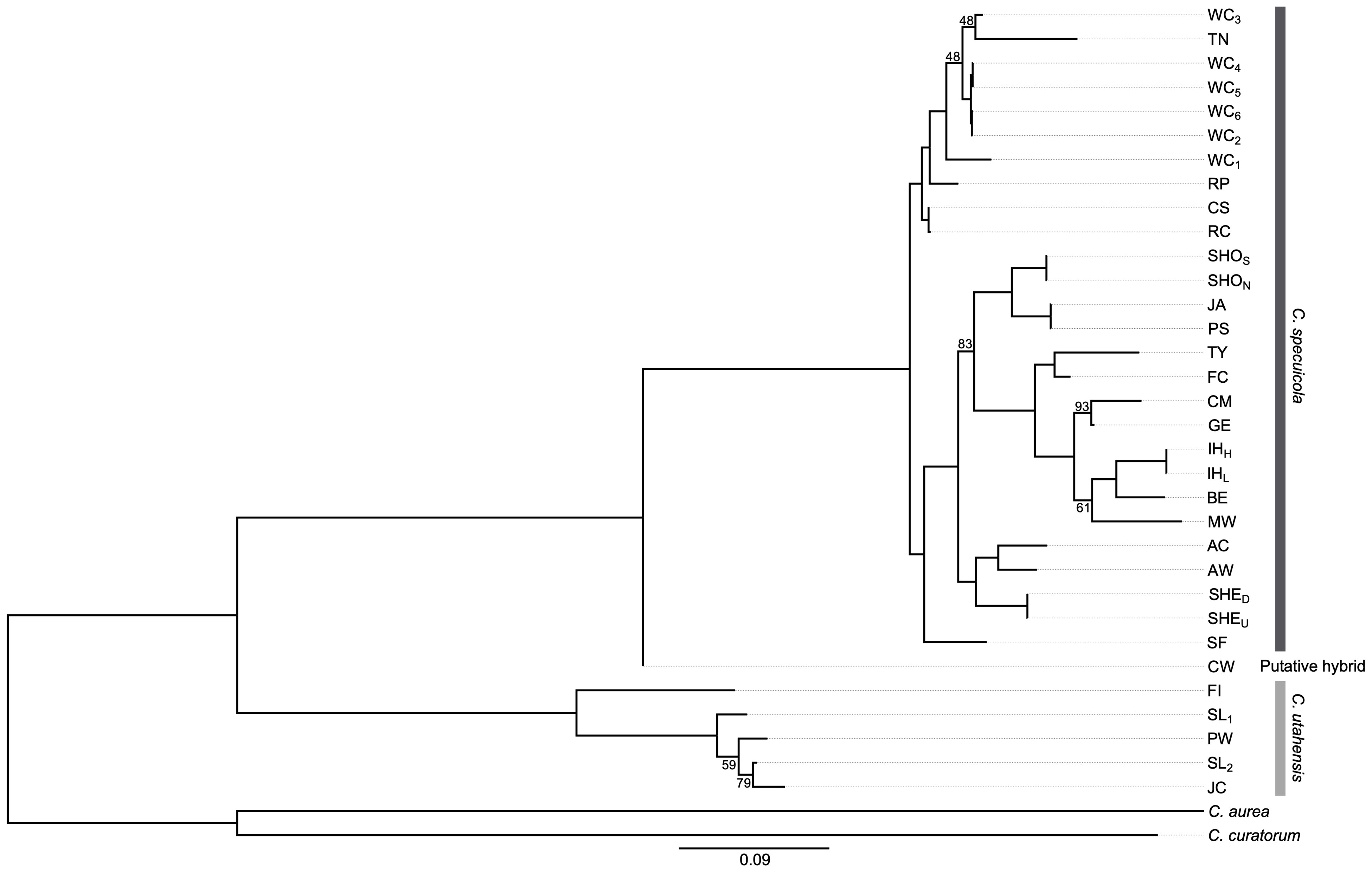
Figure 2 Maximum likelihood phylogeny for Carex samples inferred with PhyML (Guindon et al., 2010) using smart model selection (Lefort et al., 2017). Tip labels indicate sampling sites and are defined in Table 1. All nodes were highly supported (>95% bootstrap values) except where noted.
Population structure and genomic patterns within C. specuicola
Observed and expected heterozygosities within C. specuicola were generally low ( = 0.04 and 0.03, respectively), as were nucleotide diversities ( = 0.04; Table 1). Private alleles among sites ranged from 27-418 (Table 1). Genetic differentiation (i.e., FST) between pairs of hanging garden sites was high, with an overall ± SD = 0.49 ± 0.14 (Table S1), indicating substantial population structure even across small spatial scales. For instance, Adahchiiyahi Wash and A-Canyon are separated by only ca. 4 km but had a FST = 0.4. Across 325 pairwise FST values, the highest FST was between Tse Ya Toe Spring and Tes Nez Iah (FST= 0.68). Pairwise FST values between subsites ranged from -0.10–0.38 (Table S1), with both the highest and lowest FST values among Walker Creek subsites (Table S1). Geographic and genetic distance were significantly correlated in both Mantel tests and linear regressions across all hanging gardens and within Structure defined genetic clusters (Figure 3; Table S2).
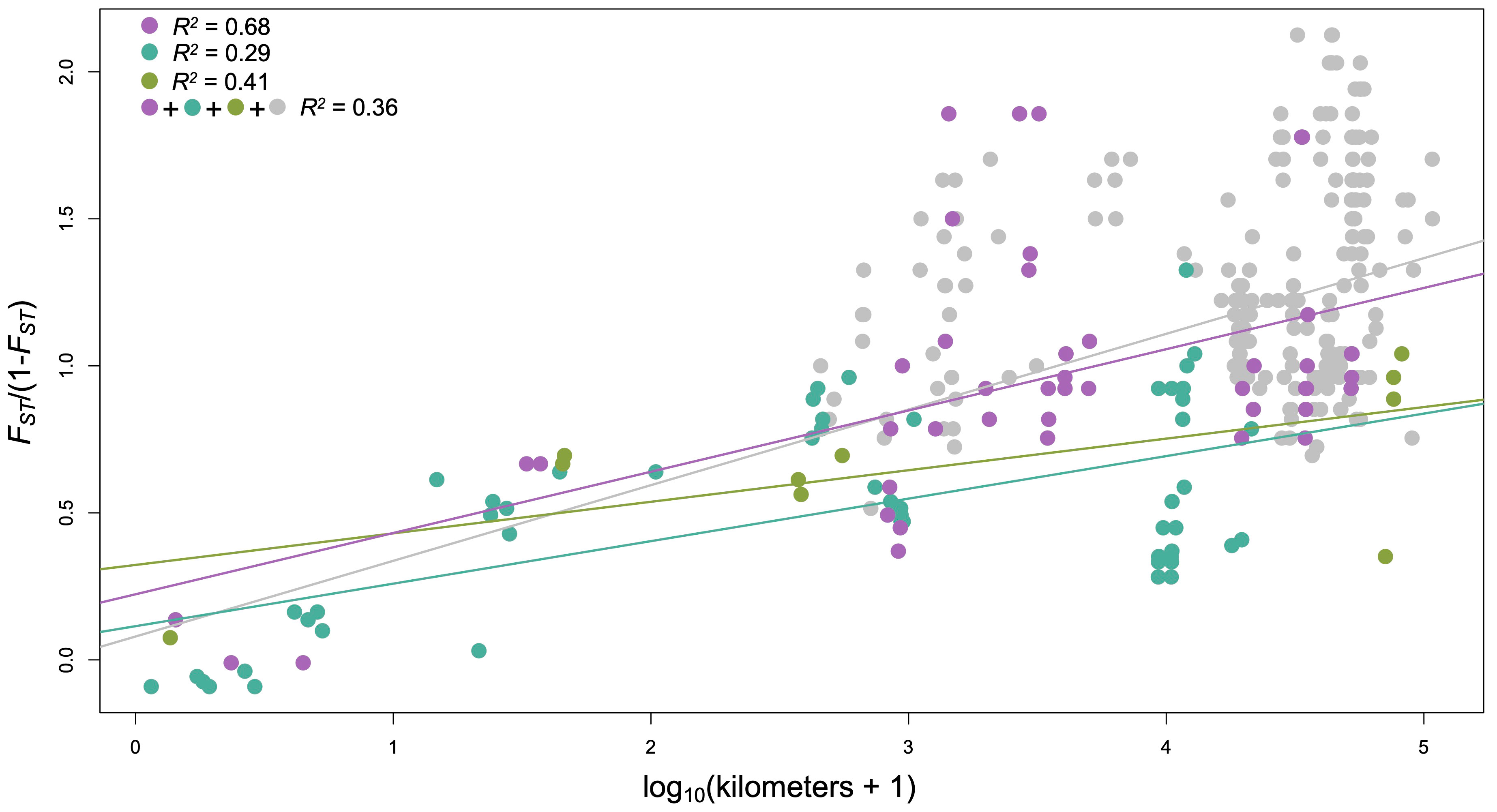
Figure 3 Correlations of geographic and genetic distance within Structure-defined groups (at K = 3; colors match those used in Figure 1) and for all comparisons across genetically defined populations (gray + colored points). See Table S2 for additional detail.
We estimated levels of inbreeding (FIS) and relatedness (Ajk) to investigate evidence of clonal reproduction within hanging gardens. FIS and Ajk were negative at most sites, suggesting that neither inbreeding nor clonal reproduction is widespread (Table 1). However, we found significantly positive FIS values across two sites (Forty Caves and Geshi), which may indicate recent colonization, inbreeding, or cryptic population structure. At Geshi, was no longer strongly elevated when calculated separately for individuals that group with Inscription House Spring (−0.14) or Canyon del Muerto (= −0.002), supporting cryptic population structure as the probable driver of the pattern.
Structure and PCA analyses resolve similar patterns of genetic structure across C. specuicola hanging gardens. At K = 2, hanging gardens are composed entirely of one genetic axis or the other (Figure S2) and only two sampling sites (Forty Caves and Moenkopi Wash) show evidence of admixture. At K = 3, one of the genetic axes splits into two, while the other genetic axis remains relatively consistent. The K = 3 Structure solution reiterates relationships discerned by the PCA, in which individuals group into three primary clusters (Figure 1B). All individuals from a garden cluster with one another in PC space except for four, including two from Geshi that are more closely related to Inscription House/Binne Etteni, one from Jackass that clusters with Shonto, and one from Cigarette Spring that approaches individuals from Rock Point (Figure 1C). In relation to geography, genetic ancestry of individuals from the Sheik and Canyon del Muerto sites was more closely aligned to individuals from distant sites (e.g., Adahchiiyahi Wash and Geshi, respectively) than to individuals from the nearest sites. We also observed mixed ancestry at the Snow Flat Spring Cave, Rock Point, and Moenkopi Wash in which the minor ancestry component was present in geographically distant gardens (Figure 1B). The K = 4 model highlights a close relationship between Geshi and Canyon del Muerto that is not evident in the PCA, as well as illustrates the mismatch of the two aforementioned Geshi individuals (Figure S2). Structure analyses at higher K values did not provide biologically relevant or interpretable patterns.
EEMS runs produced strong correlations between observed and expected genetic dissimilarity both within and between demes, indicating good model fit (Petkova et al., 2016). Effective diversity surfaces were similar across different deme models (Figures 4, S3); as a result, we report the surface developed using 200 subpopulations because it most closely reflects the number of known occupied hanging garden sites (170 sites; Rink, 2017). The effective diversity surfaces revealed relatively high genetic diversity (25-30% higher than average) in the far western portion of the range (i.e., Binne Etteni, Inscription House Spring; Geshi; Forty Caves; and Tse Ya Toe Spring; Figure 1) and lower than average diversity to the east. This is generally in agreement with the expected heterozygosity and nucleotide diversity of sites, which were both higher in the western portion of the range (Table 1).
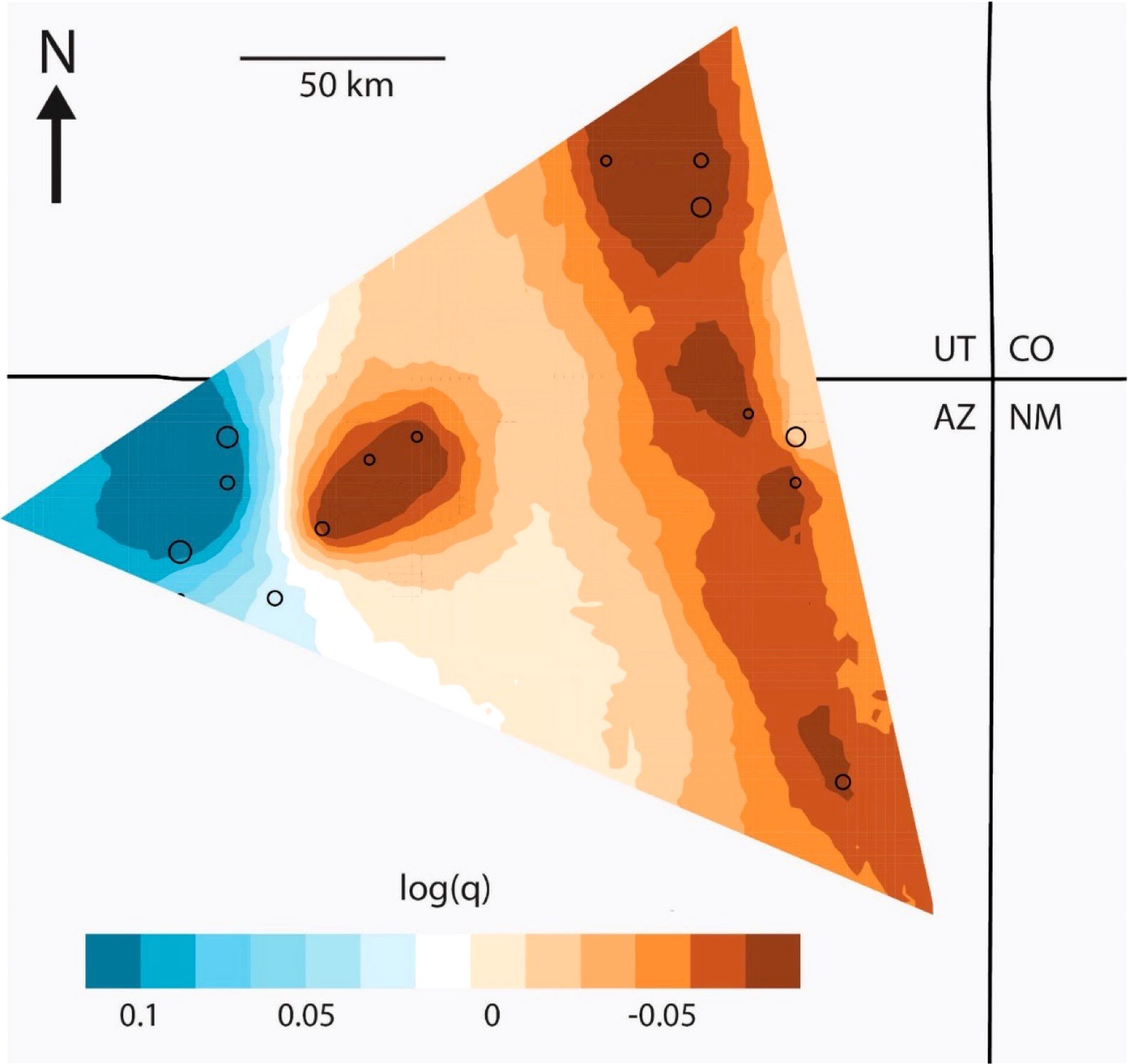
Figure 4 Effective diversity surface estimated with EEMS (200 demes) where blues indicate regions of relatively high effective diversity (q) and oranges indicate regions of relatively low effective diversity (here, log10(q) = 0.1 indicates ca. 26% higher diversity compared to the average across the range; Petkova et al., 2016). Circles indicate occupied deme localities with the size of the circle scaled to the number of samples.
Modeling of population history between pairs of closely related hanging garden sites generally revealed small effective population sizes and recent divergence times, although support for gene flow between sites was mixed. We found strong support for an IM model over a no migration model (GIM p< 0.001, ΔAIC = 40) between Forty Caves and Tse Ya Toe Spring (FST = 0.4, 4 km distance); however, confidence intervals for divergence time, migration rate, and population size were wide (Table 2). Our maximum likelihood estimates place the Forty Caves–Tse Ya Toe Spring divergence time at 2595 generations ago with ca. 2.5 migrants per generation following divergence. Forty Caves had the largest effective population size of any hanging garden sites (maximum likelihood Ne= 1089), while Tse Ya Toe Spring was characterized by a very small effective population size (maximum likelihood Ne= 48). The common ancestor of Forty Caves and Tse Ya Toe Spring also had a relatively large effective population size of an estimated 4340 but confidence intervals around these parameter estimates were large. Between Canyon del Muerto and Geshi (FST=0.26, 127 km), we found support for a no migration (ΔAIC = 7) model that included very low effective population sizes (Canyon del Muerto Ne= 8–17; Geshi Ne= 0–764) and recent divergence between sites (334–714 generations ago; Table 2). Finally, for Cigarette Spring and Road Canyon (FST= 0.03, 3 km distance), no migration and IM models explained variation in the data equally well (ΔAIC = 1.7). Effective population sizes were small (e.g., Cigarette Spring Ne= 12-261; Road Canyon Ne= 0-164) and divergence times were recent (0-302 generations ago; Table 2) regardless of whether gene flow was included in the model.
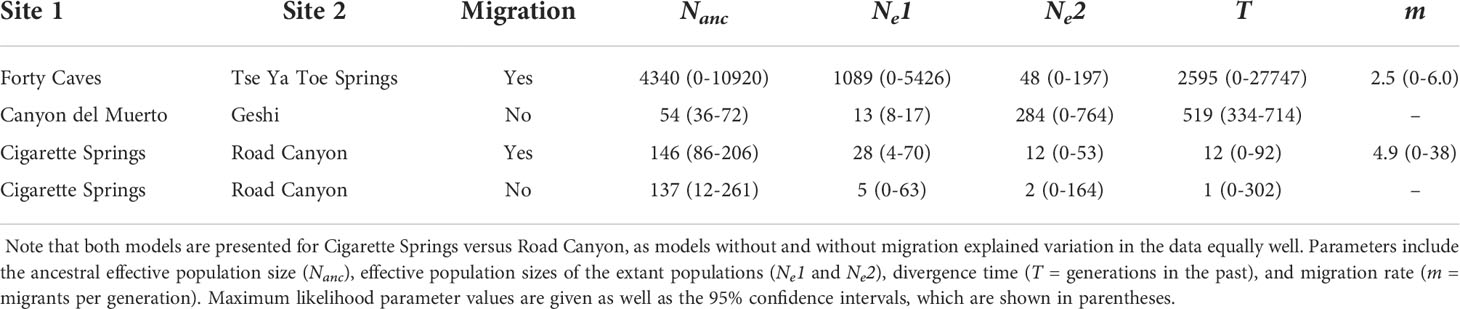
Table 2 Estimates of demographic parameters for pairs of hanging garden sites for the best supported model (i.e., Migration, which represents a model with migration [Yes] or without migration [No]).
Discussion
Uncovering the contemporary and historical processes shaping rare species with complex distributions is of growing importance due to threats of habitat destruction and climate change (Lowe and Allendorf et al., 2010; Hanski, 2011). Our investigations of Carex specuicola, a rare sedge with habitat requirements that restrict it to geographically separated, small sites, paint a compelling picture of dispersal capacity, strong genetic drift, and limited hybridization with its putative sister taxon. Our data provide insights into the natural history of this poorly understood species and may guide potential management actions.
The history of hybridization among Carex species
Carex specuicola and C. utahensis are closely related Colorado Plateau endemics within the “Carex parryana” complex of Carex sect. Racemosae (Reznicek and Murray, 2013; Massatti et al., 2016). Our analyses suggest that C. specuicola and C. utahensis form reciprocally monophyletic groups (Figure 2) and that hybridization appears to be rare. The conflicting results between the f3 tests and the phylogenetic hypothesis for Chinle Wash are interesting, though a non-negative f3 statistic does not mean that admixture did not occur; strong genetic drift, which appears common across C. specuicola hanging gardens, can quickly erode signatures of admixture in the f3 statistic (Patterson et al., 2012). Thus, the lack of an f3 admixture signature in Chinle Wash may be consistent with relatively old admixture. Investigating the broader genomic and evolutionary consequences of hybridization and dating the divergence between these species are intriguing areas of future research; such studies may also provide additional insight into the historical factors influencing diversification across this spatially complex and environmentally dynamic region (Kadereit and Abbott, 2021).
Population structure, gene flow, and metapopulation dynamics
We explored patterns of population structure and demographic history to infer the factors influencing the formation of and interaction among C. specuicola hanging gardens. We uncovered a broad spatial pattern of genetic differentiation consistent with IBD (Figure 3) and patterns of ancestry wherein geographically adjacent sites tended to be closely related (Figure 1). At small spatial scales, as represented by our sampling of proximate subsites, individuals were almost always found to be highly related (Table S1). Moreover, demographic inference supported a model of continued gene flow since divergence between Forty Caves and Tse Ya Toe Springs (separated by 3.8 km; Table 2), and we found that four individuals did not cluster with individuals from the same garden, but rather with individuals at nearby gardens, potentially indicating recent dispersal events (Figure 1C). Analyses supporting gene flow across the species’ range may explain why inbreeding coefficients and relatedness values are low despite FST values that may indicate strong genetic drift.
In tandem with evidence indicating that gene flow among adjacent sites influences patterns of genetic structure, we uncovered genetic patterns indicating C. specuicola is capable of long-distance dispersal events. For example, although the Geshi and Canyon del Muerto sites are 127 km apart, they were found to be closely related based on PCA, Structure, and phylogenetic results. Moreover, their estimated divergence time (519 generations) is more recent than is the divergence time of the spatially proximate Forty Caves and Tse Ya Toe Springs sites (2595 generations; Table 2). Furthermore, we found that Sheik individuals are more closely related to individuals from Adahchiiyahi Wash and A-Canyon (72 and 76 km, respectively) than to nearby sites like Road Canyon (21 km; Figure 2). Long-distance dispersal requires a mechanism, which is not evident in the intrinsic seed traits of Carex species; for example, seeds lack structures that facilitate dispersal by wind (e.g., a pappus as in Asteraceae species) or by hitchhiking (e.g., barbed seeds as in some Boraginaceae species). However, Wilson et al. (2008) found that Carex seeds can pass intact and viable through the digestive systems of waterfowl and gallinaceous birds, potentially implicating zoochory. Within desert ecosystems, mesic plant communities like hanging gardens may be important sources of food, water, and shelter for birds (Malanson and Kay, 1980; Welsh, 1989), thereby providing a direct avenue of propagule dispersal among the gardens distributed across otherwise heterogeneous, complex, environmentally unsuitable terrain.
Population genetic patterns and inferred demographic processes suggest that C. specuicola may have metapopulation-like dynamics. Metapopulations are governed by recurrent extinction and colonization processes (Pannell and Charlesworth, 2000), leading to temporary occupancy of habitat patches (Thomas, 1994). In this system, the recent estimated divergence times between pairs of hanging garden sites (Table 2) are consistent with rapid population turnover of occupied/unoccupied sites; furthermore, the range of C. specuicola’s hanging garden FST values reflects biological systems in which colonization occurs regularly (Knowles and Alvarado-Serrano, 2010). Population census data support these processes; for instance, Hevron (1990) recorded negative survey results (i.e., no C. specuicola found) at 10 sites in the Forty Caves area that were subsequently resurveyed by Rink (2017), who located C. specuicola at 6 of the 10 sites. Likewise, extirpation at one site has been documented (Rink and Hazelton, 2014). Extinction and colonization within metapopulations is expected to strongly reduce population genetic diversity (i.e., effective population size; Harrison and Hastings, 1996; Pannell and Charlesworth, 2000). Consistent with this, we find low relative genetic diversity and small effective populations across most of the range (Figure 4, Table 1). However, the western portion of C. specuicola’s range has relatively high genetic diversity (Figure 4), which may reflect larger and more stable populations and thus a source of the metapopulation; this hypothesis requires further testing with additional census and genetic data. In light of geographic patterns of genetic diversity, hanging garden ecosystems may be biogeographically similar to oceanic and sky island systems, where species composition is determined by dispersal limitations, extirpation, and habitat size (MacArthur and Wilson, 1963; Malanson and Kay, 1980; Atwood et al., 2011; Vásquez et al., 2016; He et al., 2019).
We presume that similar intraspecific dynamics occurred throughout the phylogeographic history of C. specuicola, specifically regarding gene flow and dispersal. Considering the paleorefugium hypothesis (our null hypothesis), our results do not support that this system is dominated by persistent extirpations (Nekola, 1999) to the point that the species’ long-term survival is at risk (assuming the maintenance of suitable hanging garden habitat). In a scenario where C. specuicola was widespread across suitable habitat during cooler and wetter periods (Spence and Henderson, 1993; Coats et al., 2008) and maintained similar levels of dispersal, population genetic structure was presumably more homogenous across the species’ distribution. Subsequent environmental warming and drying restricting C. specuicola to paleorefugia (i.e., hanging gardens) would have restricted gene flow, but it is unclear whether range fragmentation in the face of ongoing gene flow would be able to produce the patterns of small effective population sizes and large FST values inferred herein.
The alternative hypothesis to a system dominated by paleorefugia – that C. specuicola has always been a hanging garden specialist – seems more consistent with our data. Namely, groups of individuals within gardens do not display large inbreeding coefficients or a lack of dispersal. While support for the paleorefugium hypothesis in a co-distributed hanging garden species (Anticlea elegans subsp. vaginata – a monocot in the family Melanthiaceae) was in part garnered from the distribution of the species’ sister taxon in montane habitat of the San Juan Mountains to the northeast of this study region (Palmquist et al., 2015), we note that C. specuicola is within a clade of species that is not restricted to higher elevations (Massatti et al., 2016). In other words, while vicariance presumably influenced diversification of Anticlea elegans as the climate warmed after the Last Glacial Maximum and the parent taxon retreated to (or only persisted in) higher elevations, a similar diversification event involving a montane sister taxon is not supported for C. specuicola. However, this hypothesis does not preclude C. utahensis (or an ancestor of the two species) having a more widespread distribution during cooler and wetter times, and perhaps a drying and warming climate may have reduced gene flow between these species to the point where they are now largely reproductively isolated. Similar diversification processes are likely responsible for diversification within Carex (Martín-Bravo et al., 2019, Hodel et al., 2021), as well as within other clades distributed across topographically and environmentally heterogeneous landscapes (Jones et al., 2021; Kadereit and Abbott, 2021). A spatially and temporally explicit modeling framework may be the best way to test alternative hypotheses (e.g., Bemmels et al., 2016; Massatti and Knowles, 2016; Ortego and Knowles, 2020). Regardless of C. specuicola’s true history, our data indicate that the species maintains gene flow and can colonize hanging gardens through dispersal processes.
Conservation and management implications
Understanding dispersal, colonization, gene flow, and interspecific dynamics in rare species distributed across isolated habitat is essential for developing effective conservation strategies. Our data indicate that hybridization with C. utahensis is rare and geographically restricted, which alleviates admixture concerns regarding the persistence of C. specuicola. Furthermore, gene flow and dispersal processes influence population structure and genetic diversity. In light of these results, we suggest that future habitat loss due to climate change, drought, or other factors represent the major threat to this species, given that fewer hanging gardens translates to less habitat for C. specuicola to potentially inhabit. As hanging garden habitat is hard to survey given accessibility issues, a management plan involving recurrent genetic surveys may indicate whether declining habitat quality is influencing genetic diversity and structure (Paz-Vinas et al., 2018). To this end, we note that our investigation provides baseline data that can inform future surveys. The preservation of multiple, genetically distinct populations is crucial to maintain existing genetic diversity and to support colonization of new sites if hanging garden populations stochastically extirpate. Further, the maintenance of redundant sites with similar genetic profiles may prevent stochastic events from reducing genetic richness. Given the congruence of genetic patterns between PCA and the Structure K = 3 analysis, we identify three primary populations that may have utility in management applications, including the East population (represented herein by Road Canyon, Cigarette Springs, Walker Creek sites, Rock Point, and Tes Nez Iah); the Central population (represented herein by Sheik, Adahchiiyahi Wash, A-Canyon, Jackass, Peach Spring, and Shonto); and the West population (represented herein by Geshi, Binne Etteni, Inscription House, Moenkopi Wash, and Canyon del Muerto). However, due to the dispersal biology of the species, we emphasize that it may be difficult to predict the genetic ancestry of a site with high confidence (e.g., Massatti and Winkler, 2022), especially as distance increases from a site with known ancestry.
Although our work provides important insights into the biology of this species and potential conservation strategies, future research is essential to better understand whether it can persist under current conditions and how it may respond to future challenges. In particular, characterizing dispersal distance and the spatial structure of suitable habitat are crucial for understanding capacity for long-term persistence of metapopulations (Grilli et al., 2015; Hanski et al., 2017). In addition, investigating potential adaptive loci could provide insights into genetic or environmental factors that influence metapopulation size and persistence (e.g., PGI in butterflies, Hanski et al., 2017). For instance, future work could explore spatial patterns and potential genetic underpinnings of drought tolerance, which could be integrated into conservation strategies to facilitate adaptation under climate change. Ultimately, strategies for managing rare species depend upon understanding how historical and contemporary processes affect intraspecific patterns and processes with an eye on what the future may hold; genetic investigations are a prime mechanism to meet these needs.
Conclusion
Understanding the evolutionary history of a species restricted to specialized habitat has important ramifications when it is unclear if management interventions are required to promote the species’ persistence. Fortunately, analyses utilizing genomic data can illuminate historical and contemporary processes like migration, population divergence, dispersal ability, and population differentiation, which together may provide support for the factors influencing a species’ distribution. Herein, we investigated a range-restricted species obligate to a desert hanging garden ecosystem that either has life history characteristics allowing it to survive in this specialized habitat or that is composed of relict populations resulting from paleorefugia and therefore at high risk for extirpation. Despite the specialized nature of this system, similar circumstances apply to other narrowly endemic species in terrestrial and aquatic systems. Contrary to assumptions implicit in current guidance, genomic analyses suggested ongoing dispersal and genetically healthy populations and illustrate a dynamic system that likely depends on colonization to balance extirpation. This case study highlights the importance of applying genomic data to inform management to understand species needs.
Data availability statement
Data supporting the original contributions presented in the study are publicly available from the USGS ScienceBase Catalog: https://doi.org/10.5066/P9LLZ1XD (Massatti, 2022).
Author contributions
MJ, RM, and DW designed the conceptual and analytical research framework. GR coordinated and carried out field sampling. MJ, RM, and KC performed analyses. MJ wrote the first draft of the manuscript, which was completed by RM and KC. All authors edited the final version of the manuscript.
Funding
This research was supported by the U.S. Fish and Wildlife Service Cooperative Endangered Species Conservation Fund Traditional Section Six Grant Segment 23 through the Arizona Department of Agriculture. Additional support came from the BLM Plant Conservation Program and USGS Ecosystems Mission Area.
Acknowledgments
We thank Nora Talkington (Navajo Nation Herbarium), Tina Ayers (Deaver Herbarium), and Kirstin Phillips and Janet Gillette (Museum of Northern Arizona). Nora Talkington made many of the collections for this study. Max Taylor facilitated the collection of material in the southwest portion of the species’ range. Bill Hatcher and Dawn Kish helped with specimen collection in the San Juan River drainage. Eva Christ helped with sampling at Forty Caves. Collections were made under USFWS permit numbers TE10057B-0 and ESPER0003958 and with permission from the Hopi and Navajo tribes. The Museum of Northern Arizona permitted us to remove a small amount of leaf material from one specimen. Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcosc.2022.941002/full#supplementary-material
References
Akaike H. (1973). Information theory and an extension of the maximum likelihood principle. 2nd international symposium on information theory. Eds. Petrov B. N., Csaki F. (Budapest: Akademiai Kiado), 267–281.
Allendorf F. W., Hohenlone P. A., Luikart G. (2010). Genomics and the future of conservation genetics. Nat. Rev. Genet. 11, 697–709. doi: 10.1038/nrg2844
Atwood T. C., Young J. K., Beckmann J. P., Breck S. W., Fike J., Rhodes O. E., et al. (2011). Modeling connectivity of black bears in a desert sky island archipelago. Biol. Conserv. 144, 2851–2862. doi: 10.1016/j.biocon.2011.08.002
Avise J. C. (2000). Phylogeography: the history and formation of species Cambridge, MA: (Harvard University Press).
Barrett R. D., Schluter D. (2008). Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44. doi: 10.1016/j.tree.2007.09.008
Bemmels J. B., Title P. O., Ortego J., Knowles L. L. (2016). Tests of species-specific models reveal the importance of drought in postglacial range shifts of a Mediterranean-climate tree: insights from integrative distributional, demographic and coalescent modelling and ABC model selection. Mol. Ecol. 25, 4889–4906. doi: 10.1111/mec.13804
Bhatia G., Patterson N., Sankararaman S., Price A. L. (2013). Estimating and interpreting FST: the impact of rare variants. Genome Res. 23, 1514–1521. doi: 10.1101/gr.154831.113
Bougeard S., Dray S. (2018). Supervised multiblock analysis in r with the ade4 package. J. Stat. Software 86, 1–17. doi: 10.18637/jss.v086.i01
Brooks T. M., Mittermeier R. A., Mittermeier C. G., Da Fonseca G. A., Rylands A. B., Konstant W. R., et al. (2002). Habitat loss and extinction in the hotspots of biodiversity. Conserv. Biol. 16, 909–923. doi: 10.1046/j.1523-1739.2002.00530.x
Coats L. L., Cole K. L., Mead J. I. (2008). 50,000 years of vegetation and climate history on the Colorado plateau, Utah and Arizona, USA. Quaternary Res. 70, 322–338. doi: 10.1016/j.yqres.2008.04.006
Coffman A. J., Hsieh P. H., Gravel S., Gutenkunst R. N. (2016). Computationally efficient composite likelihood statistics for demographic inference. Mol. Biol. Evol. 33, 591–593. doi: 10.1093/molbev/msv255
Corlett R. T., Tomlinson K. W. (2020). Climate change and edaphic specialists: Irresistible force meets immovable object? Trends Ecol. Evol. 35, 367–376. doi: 10.1016/j.tree.2019.12.007
Dafni A., Firmage D. (2000). Pollen viability and longevity: practical, ecological and evolutionary implications. Plant Syst. Evol. 222, 113–132. doi: 10.1007/BF00984098
Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Diffenbaugh N. S., Giorgi F., Pal J. S. (2008). Climate change hotspots in the united states. Geophys. Res. Let. 35:1–5 doi: 10.1029/2008GL035075
Doerr V. A., Barrett T., Doerr E. D. (2011). Connectivity, dispersal behaviour and conservation under climate change: A response to hodgson. J. Appl. Ecol. 48, 143–147. doi: 10.1111/j.1365-2664.2010.01899.x
Elias E., Reyes J., Steele C., Rango A. (2018). Diverse landscapes, diverse risks: synthesis of the special issue on climate change and adaptive capacity in a hotter, drier southwestern united states. Climatic Change 148, 339–353. doi: 10.1007/s10584-018-2219-x
Fowler J. F., Stanton N. L., Hartman R L. (2007). Distribution of hanging garden vegetation associations on the Colorado plateau, USA. J. Bot. Res. Inst. Texas 1, 585–607.
Frantz A. C., Cellina S., Krier A., Schley L., Burke T. (2009). Using spatial Bayesian methods to determine the genetic structure of a continuously distributed population: clusters or isolation by distance? J. Appl. Ecol. 46, 493–505. doi: 10.1111/j.1365-2664.2008.01606.x
Friedman J., Barrett S. C. (2009). The consequences of monoecy and protogyny for mating in wind-pollinated Carex. New Phytol. 181, 489–497. doi: 10.1111/j.1469-8137.2008.02664.x
Galbreath K. E., Hafner D. J., Zamudio K. R., Agnew K. (2010). Isolation and introgression in the intermountain West: Contrasting gene genealogies reveal the complex biogeographic history of the American pika (Ochotona princeps). J. Biogeogr. 37, 344–362. doi: 10.1111/j.1365-2699.2009.02201.x
Gallo E. L., Meixner T., Lohse K. A., Yonce H. N. (2020). Estimating surface water presence and infiltration in ephemeral to intermittent streams in the southwestern US. Front. Water 2, 2. doi: 10.3389/frwa.2020.572950
Goudet J., Jombart T. (2020) Hierfstat: estimation and tests of hierarchical f-statistics. r package version 0.5-7. Available at: https://CRAN.R-project.org/package=hierfstat.
Grilli J., Barabas G., Allesina S. (2015). Metapopulation persistence in random fragmented landscapes. PloS Comput. Biol. 11, e1004251. doi: 10.1371/journal.pcbi.1004251
Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. System Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Gutenkunst R. N., Hernandez R. D., Williamson S. H., Bustamante C. D. (2009). Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PloS Genet. 5, e1000695. doi: 10.1371/journal.pgen.1000695
Hanski I. (2011). Habitat loss, the dynamics of biodiversity, and a perspective on conservation. AMBIO 40, 248–255. doi: 10.1007/s13280-011-0147-3
Hanski I., Schulz T., Wong S. C., Ahola V., Ruokolainen A., Ojanen S. P. (2017). Ecological and genetic basis of metapopulation persistence of the glanville fritillary butterfly in fragmented landscapes. Nat. Commun. 8, 14504. doi: 10.1038/ncomms14504
Harrison S., Hastings A. (1996). Genetic and evolutionary consequences of metapopulation structure. Trends Ecol. Evol. 11, 180–183. doi: 10.1016/0169-5347(96)20008-4
He K., Gutiérrez E. E., Heming N. M., Koepfli K. P., Wan T., He S., et al. (2019). Cryptic phylogeographic history sheds light on the generation of species diversity in sky-island mountains. J. Biogeog 46, 2232–2247. doi: 10.1111/jbi.13664
Hermann F. J. (1970). Manual of the carices of the rocky mountains and Colorado basin (No. 374) (Washington DC:US Department of Agriculture).
Hevron W. (1990). March 12 letter to the regional director, USFWS, found within the files of the Navajo natural heritage program.
Hodel R. G., Massatti R., Bishop S. G., Knowles L. L. (2021). Testing which axes of species differentiation underlie covariance of phylogeographic similarity among montane sedge species. Evol. 75, 349–364. doi: 10.1111/evo.14159
Janes J. K., Miller J. M., Dupuis J. R., Malenfant R. M., Gorrell J. C., Cullingham C. I., et al. (2017). The K= 2 conundrum. Molec. Ecol. 26, 3594–3602. doi: 10.1111/mec.14187
Jombart T. (2008). adegenet: an r package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Jombart T., Devillard S., Dufour A. B., Pontier D. (2008). Revealing cryptic spatial patterns in genetic variability by a new multivariate method. Heredity 101, 92–103. doi: 10.1038/hdy.2008.34
Jones M. R., Winkler D. E., Massatti R. (2021). The demographic and ecological factors shaping diversification among rare Astragalus species. Divers. Distrib. 27, 1407–1421. doi: 10.1111/ddi.13288
Kadereit J. W., Abbott R. J. (2021). Plant speciation in the quaternary. Plant Ecol. Divers. 14, 105–142. doi: 10.1080/17550874.2021.2012849
Keller L. F., Waller D. M. (2002). Inbreeding effects in wild populations. Trends Ecol. Evol. 5, 230–241. doi: 10.1016/S0169-5347(02)02489-8
Kendrick G. A., Orth R. J., Statton J., Hovey R., Ruiz Montoya L., Lowe R. J., et al. (2017). Demographic and genetic connectivity: the role and consequences of reproduction, dispersal and recruitment in seagrasses. Biol. Rev. 92, 921–938. doi: 10.1111/brv.12261
Knowles L., Alvarado-Serrano D. F. (2010). Exploring the population genetic consequences of the colonization process with spatio-temporally explicit models: insights from coupled ecological, demographic and genetic models in montane grasshoppers. Molec. Ecol. 19, 3727–3745. doi: 10.1111/j.1365-294X.2010.04702.x
Lefort V., Longueville J. E., Gascuel O. (2017). SMS: smart model selection in PhyML. Molec. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149
Lin T., Zhu G., Zhang J., Xu X., Yu Q., Zheng Z., et al. (2014). Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–1226. doi: 10.1038/ng.3117
Lowe W. H., Allendorf F. W. (2010). What can genetics tell us about population connectivity? Molec. Ecol. 19, 3038–3051. doi: 10.1111/j.1365-294X.2010.04688.x
Lynch M., Conery J., Burger R. (1995). Mutation accumulation and the extinction of small populations. Am. Nat. 146, 489–518. doi: 10.1086/285812
MacArthur R. H., Wilson E. O. (1963). An equilibrium theory of insular zoogeography. Evolution 17, 393–387. doi: 10.1111/j.1558-5646.1963.tb03295.x
Malanson G. P., Kay J. (1980). Flood frequency and the assemblage of dispersal types in hanging gardens of the narrows, Zion national park, Utah. Great Basin Nat. 40, 365–371.
Mantel N. (1967). The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220.
Martín-Bravo S., Jiménez-Mejías P., Villaverde T., Escudero M., Hahn M., Spalink D., et al. (2019). A tale of worldwide success: Behind the scenes of Carex (Cyperaceae) biogeography and diversification. J. Systematics Evol. 57, 695–718. doi: 10.1111/jse.12549
Massatti R. (2022). Carex specuicola genomic data for the southern Colorado plateau desert: U.S (Geological Survey data release). doi: 10.5066/P9LLZ1XD
Massatti R., Knowles L. L. (2016). Contrasting support for alternative models of genomic variation based on microhabitat preference: Species-specific effects of climate change in alpine sedges. Mol. Ecol. 25, 3974–3986. doi: 10.1111/mec.13735
Massatti R., Reznicek A. A., Knowles L. L. (2016). Utilizing RADseq data for phylogenetic analysis of challenging taxonomic groups: A case study in Carex sect. Racemosae. Am. J. Bot. 00: 1–14 103, 337–347. doi: 10.3732/ajb.1500315
Massatti R., Winkler D. E. (2022). Spatially explicit management of genetic diversity using ancestry probability surfaces. Methods Ecol. Evol 00: 1–14. doi: 10.1111/2041-210X.13902
Nekola J. C. (1999). Paleorefugia and neorefugia: the influence of colonization history on community pattern and process. Ecology 80, 2459–2473. doi: 10.1890/0012-9658(1999)080[2459:PANTIO]2.0.CO;2
Nieminen M., Singer M. C., Fortellius W., Schöps K., Hanski I. (2001). Experimental confirmation that inbreeding depression increases extinction risk in butterfly populations. Am. Nat. 157, 237–244. doi: 10.1086/318630
Ortego J., Knowles L. L. (2020). Incorporating interspecific interactions into phylogeographic models: A case study with Californian oaks. Mol. Ecol. 29, 4510–4524. doi: 10.1111/mec.15548
Palmquist E., Ayers T., Allan G. (2015). Genetic and morphometric assessment of the origin, population structure, and taxonomic status of Anticlea vaginata (Melanthiaceae). System Bot. 40, 56–68. doi: 10.1600/036364415X686332
Pannell J. R., Charlesworth B. (2000). Effects of metapopulation processes on measures of genetic diversity. Philos. Trans. R. Soc London - Ser. B Biol. Sci. 355, 1851–1864. doi: 10.1098/rstb.2000.0740
Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., Zhan Y., et al. (2012). Ancient admixture in human history. Genetics 192, 1065–1093. doi: 10.1534/genetics.112.145037
Pavlova A., Beheregaray B., Coleman R., Gilligan D., Harrisson K. A., Ingram B. A., et al. (2017). Severe consequences of habitat fragmentation on genetic diversity of an endangered Australian freshwater fish: a call for assisted gene flow. Evol. Applic. 10, 531–550. doi: 10.1111/eva.12484
Paz-Vinas I., Loot G., Hermoso V., Veyssiere C., Poulet N., Grenouillet G., et al. (2018). Systematic conservation planning for intraspecific genetic diversity. Proc. R. Soc B: Biol. Sci. 285, 20172746. doi: 10.1098/rspb.2017.2746
Peter B. M. (2016). Admixture, population structure, and f-statistics. Genetics 202, 1485–1501. doi: 10.1534/genetics.115.183913
Peterson B. K., Weber J. N., Kay E. H., Fisher H. S., Hoekstra H. E. (2012). Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PloS One 7, e37135. doi: 10.1371/journal.pone.0037135
Petkova D., Novembre J., Stephens M. (2016). Visualizing spatial population structure with estimated effective migration surfaces. Nat. Genet. 48, 94–100. doi: 10.1038/ng.3464
Pickrell J. K., Pritchard J. K. (2012). Inference of population splits and mixtures from genome-wide allele frequency data. PloS Genet. 8, e1002967. doi: 10.1371/journal.pgen.1002967
Pritchard J. K., Stephens M., Donnelly P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
R Core Team (2021). R: A language and environment for statistical computing (Vienna: R Foundation for Statistical Computing).
Redsteer M. H., Kelley K. B., Francis H., Block D. (2013). Increasing vulnerability of the Navajo people to drought and climate change in the southwestern united states: Accounts from tribal elders (Special report on indigenous people, marginalized populations and climate change. Cambridge: Cambridge University Press).
Reed D. H. (2004). Extinction risk in fragmented habitats. Anim. Conserv. Forum 7, 181–181. doi: 10.1017/S1367943004001313
Reynolds L. K., Waycott M., McGlathery K. J. (2013). Restoration recovers population structure and landscape genetic connectivity in a dispersal-limited ecosystem. J. Ecol. 101, 1288–1297. doi: 10.1111/1365-2745.12116
Reznicek A. A., Murray D. F. (2013). A re-evaluation of carex specuicola and the carex parryana complex (Cyperaceae). J. Bot. Res. Inst. Texas 7, 37–51.
Rink G. R. (2017). Survey for Carex specuicola Navajo sedge in support of the draft recovery plan, section 6 final report reference # 2015-2017-07.
Rink G. R., Hazelton A. (2014). Demography studies for Carex specuicola (Navajo sedge, Cyperaceae), section 6 final report reference # 2013-2014–07.
Rochette N. C., Catchen J. M. (2017). Deriving genotypes from RAD-seq short-read data using stacks. Nat. Protoc. 12, 2640–2659. doi: 10.1038/nprot.2017.123
Rochette N. C., Rivera-Colón A. G., Catchen J. M. (2019). Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 28, 4737–4754. doi: 10.1111/mec.15253
Spence J. R., Henderson N. R. (1993). Tinaja and hanging garden vegetation of capitol reef national park, southern Utah, U.S.A. J. Arid Environ. 24, 21–36. doi: 10.1006/jare.1993.1002
Thomas C. D. (1994). Extinction, colonization, and metapopulations: environmental tracking by rare species. Conserv. Biol. 8, 373–378. doi: 10.1046/j.1523-1739.1994.08020373.x
Tilman D., Clark M., Williams D. R., Kimmel K., Polasky S., Packer C. (2017). Future threats to biodiversity and pathways to their prevention. Nature 546, 73–81. doi: 10.1038/nature22900
U.S. Fish and Wildlife Service (1987). Navajo Sedge (Carex specuicola) recovery plan (Albuquerque, NM: U.S. Fish and Wildlife Service).
U.S. Fish and Wildlife Service (2014). Navajo Sedge (Carex specuicola) 5 year review: summary and evaluation (Phoenix, AZ: U.S. Fish and Wildlife Service).
Vásquez D. L., Balslev H., Hansen M. M., Sklenář P., Romoleroux K. (2016). Low genetic variation and high differentiation across sky island populations of Lupinus alopecuroides (Fabaceae) in the northern Andes. Alpine Bot. 126, 135–142. doi: 10.1007/s00035-016-0165-7
Wilson B. L., Brainerd R., Lytjen D., Newhouse B., Otting N. (2008). Field guide to the sedges of the pacific Northwest (Corvallis, OR: Oregon State University Press).
Wright S. (1931). Evolution in mendelian populations. Genetics 16, 97. doi: 10.1093/genetics/16.2.97
Keywords: demographic modeling, metapopulation, paleorefugia, hanging garden, phylogeography, population genetics, Cyperaceae, Colorado Plateau
Citation: Chapin KJ, Jones MR, Winkler DE, Rink G and Massatti R (2022) Evolutionary dynamics inform management interventions of a hanging garden obligate, Carex specuicola. Front. Conserv. Sci. 3:941002. doi: 10.3389/fcosc.2022.941002
Received: 10 May 2022; Accepted: 14 September 2022;
Published: 05 October 2022.
Edited by:
Hua Wu, Central China Normal University, ChinaReviewed by:
Maria Cristina Duarte, University of Lisbon, PortugalAndrés J. Cortés, Colombian Corporation for Agricultural Research (AGROSAVIA), Colombia
Copyright © 2022 Chapin, Jones, Winkler, Rink and Massatti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rob Massatti, cm1hc3NhdHRpQHVzZ3MuZ292
†These authors share first authorship