Parallel paired electrolysis of green oxidizing agents by the combination of a gas diffusion cathode and boron-doped diamond anode
 Christin M. Hamm1
Christin M. Hamm1  Selina Schneider1 Stefanie Hild1 Rieke Neuber2 Thorsten Matthée2 Jens Krümberg3
Selina Schneider1 Stefanie Hild1 Rieke Neuber2 Thorsten Matthée2 Jens Krümberg3  Markus Stöckl1* Klaus-Michael Mangold1
Markus Stöckl1* Klaus-Michael Mangold1  Jürgen Kintrup4
Jürgen Kintrup4- 1DECHEMA-Forschungsinstitut, Frankfurt am Main, Germany
- 2CONDIAS GmbH, Itzehoe, Germany
- 3Eilenburger Elektrolyse und Umwelttechnik GmbH, Eilenburg, Germany
- 4Covestro Deutschland AG, Leverkusen, Germany
The generation of “green” oxidizing agents by electrochemical synthesis opens the field for sustainable, on-demand, and on-site production, which is often based on non-critical starting materials. In this study, electrosyntheses were carried out on different cathode and anode materials. In half-cell experiments, the cathodic synthesis of peracetic acid (PAA) was investigated on gas diffusion electrodes (GDEs), reaching 22.6 mmol L−1 of PAA with a current efficiency (CE) of 7.4%. Moreover, peroxodicarbonate (PODIC®) was produced anodically on boron-doped diamond (BDD) electrodes with concentrations as high as 42.7 mmol L−1 PODIC® and a CE of 30.3%. Both cathodic and anodic processes were individually examined and improved. Finally, the half-cell reactions were combined as a proof of concept in a parallel paired electrolysis cell for the first time to achieve an increased overall CE.
Introduction
In contrast to classical and well-established synthesis processes in the chemical industry, whose high energy demand is mainly covered by fossil fuels, electrochemical synthesis offers a sustainable and eco-friendly alternative (Biddinger and Modestino, 2020). The use of regenerative electricity not only avoids fossil energy sources but also prevents the application of hazardous oxidizing and reducing agents. Moreover, electrochemical synthesis routes are often energy-saving and cost-efficient as they can often be shortened in comparison to classical synthesis routes (Frontana-Uribe et al., 2010; Kreysa et al., 2014; Yan et al., 2017; Möhle et al., 2018; Blanco and Modestino, 2019; Janssens-Maenhout et al., 2019). It, thus, hardly comes as a surprise that electrochemical synthesis is increasingly becoming a focused topic with a wide application field in modern organic and inorganic synthesis (Wiebe et al., 2018; Pollok and Waldvogel, 2020; Zhu et al., 2021). Despite all its advantages, electrochemical synthesis still mainly focuses on the reaction at the working electrode, although the reaction at the counter electrode can have a great impact on the performance and selectivity of the electrochemical process at the working electrode (Klein and Waldvogel, 2022). As the counter electrode is often chosen just to reduce terminal voltage, the electrochemical reaction at the counter electrode remains unused in regard to the formation of valuable products. To exploit the whole potential of an electrolysis cell, the combination of two processes in a paired electrolysis cell and, thus, the formation of (valuable) products on both electrodes is an elegant way to increase process performance (Ibanez et al., 2017; Hilt, 2020; McKenzie et al., 2022). In the net reaction, 100% current efficiency (CE) per electrode—and, therefore, a combined CE of 200% in the whole cell—can be achieved. Hereby, paired electrolysis cells can operate in different modes. For example, in the linear paired electrolysis cell, the same product is obtained on both the anode and cathode (Figure 1A). In contrast, two different products are obtained in a parallel paired electrolysis cell (Figure 1B) (McKenzie et al., 2022). The synthesis of widely used chemicals such as “green” oxidizing agents in a paired electrolysis offers the universal implementation of an environmentally friendly process from the reactants over the process up to the products and was, therefore, set as the final scope of this study.
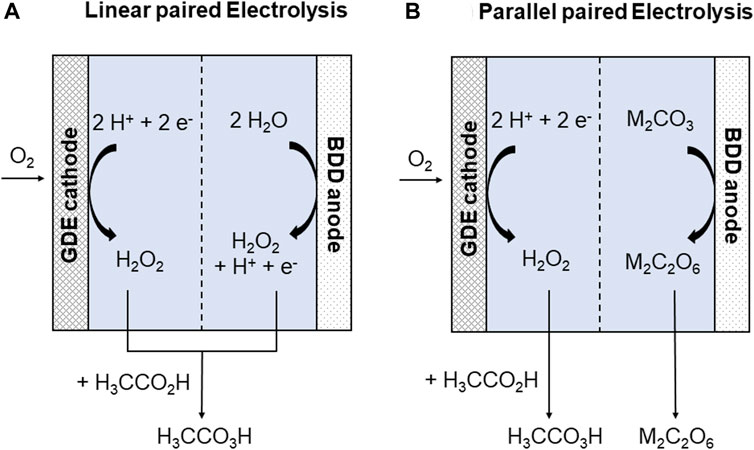
FIGURE 1. General electrochemical synthesis of hydrogen peroxide and peracetic acid on the GDE and of peroxodicarbonates on BDD electrodes in a linear (A) and parallel (B) paired electrolysis. M = H, alkali metal.
Peracetic acid (PAA) is a strong oxidizing agent with remarkable oxidizing properties. The industrial production of PAA is based on the chemical reaction between hydrogen peroxide and acetic acid in the presence of sulfuric acid as a catalyst (Saha et al., 2003a; Zhao et al., 2007; Zhao et al., 2008; Jolhe et al., 2015). Its formation is an equilibrium reaction with hydrogen peroxide, and usually, concentrations up to 15% are used for the most applications (Van et al., 2014; Doll et al., 2015; Boyce, 2016; Johnston et al., 2016; Luukkonen and Pehkonen, 2017). In higher concentrations, PAA is chemically unstable and sensitive to external heating. These properties place high demands on transport, storage, and handling and make PAA expensive (Pettas and Karayannis, 2004; Zhao et al., 2007; Zhao et al., 2008; Ni et al., 2016; Wang, 2018; Zhang et al., 2018). In addition to the general advantages of electrochemical synthesis, the in situ formation of PAA reduces costs and increases safety when produced on site. As H2O2 is needed as the starting material for PAA synthesis, gas diffusion electrodes (GDEs) are a good choice of cathode material since they allow the direct reduction of oxygen to H2O2 and minimize the parasitic formation of hydrogen during electrolysis as a side reaction. Based on this, the (indirect) electrochemical synthesis of PAA via the electrosynthesis of H2O2 on GDE was reported by Saha et al. (2004), who obtained PAA concentrations of 6.3 mM (CE = 28%, Pt added) and 2.1 mM (CE = 9.5%, Pt free GDE) at a GDE surface of 19 cm2 and a current density of 10 mA cm−2 in 1 M CH3COOH.
Another substance group that can be used as “green” oxidizers in a broad range of applications and can be obtained by eco-friendly electrochemical synthesis is peroxo compounds such as peroxodicarbonates (PODIC®, M2C2O6, M = alkaline metal) (Velazquez-Peña et al., 2013; Groenen Serrano, 2021; Ziogas et al., 2022; Zirbes et al., 2023). For Na2C2O6, several groups showed its electrochemical synthesis on platinum anodes by electrolyzing an aqueous solution of sodium carbonate at current densities between 200 mA cm-2 and 1 A cm-2 and at low temperatures (Wiel et al., 1971a; Wiel et al., 1971b; Manoharan et al., 2000; Zhang and Oloman, 2005), and its synthesis specific to boron-doped diamond (BDD) was first described by Saha et al. (2003b). In contrast, investigations in the synthesis of potassium peroxodicarbonate—especially on BDD electrodes—are rare (Constam and von Hansen, 1896; Hansen and Elektrotech, 1897). A good overview of the synthesis of different peroxodicarbonates was reported by Seitz et al. (2022). In addition to basic experiments with aqueous K2CO3 solutions as the BDD electrode and by the use of different ternary electrolyte compositions in mixtures of M2CO3 (M = H, Na, K), they received the best CE results of 42% (A = 3 cm2, j = 2.88 A cm−2) and 24% at an enlarged BDD of 10.8 cm2 (j = 0.8 mA cm−2). Furthermore, they proved the use of the obtained peroxodicarbonate mixtures as green oxidizers for N- and S-oxidations such as for epoxidations (Seitz et al., 2022). In addition, further investigations into the improvement of the electrochemical synthesis of K2C2O6 on BDD, with special regard to the enlargement of the electrode, are still pending.
In this study, we present the electrochemical synthesis of PAA via H2O2 synthesis on the GDE and of PODIC® on BDD using particularly large electrodes with a geometrical surface area of 24 cm2. In general, the combination of the GDE and BDD in one setup is well known, e.g., for the synthesis of only hydrogen peroxide or PAA (Moraleda et al., 2016; Llanos et al., 2019; Tawabini et al., 2020; Muddemann et al., 2021). Herein, the proof-of-concept combination of the parallel paired synthesis of PAA and PODIC® is shown for the first time. After describing the optimization of the individual half-cell syntheses including material screening of different carbon-based GDEs, we combined the cathodic GDE process with the anodic BDD process in an enlarged cell setup with 35 cm2 electrode geometrical surface areas. The resulting parallel electrolysis setup was investigated in relation to its CEs and the improvement of the process.
Materials and methods
For the first material screening of the GDEs, we used an H-cell setup with a 5 cm2 geometrical electrode surface area. For further optimization of the reaction on the cathode and anode including the cathodic PAA synthesis, we designed a half-cell setup for both the GDE and the BDD reaction with an active geometrical area of 24 cm2 for each electrode. The anode and cathode chambers were separated with a Nafion® 115 membrane. For the combination of both processes, a flow cell (Figure 2) was used, which was designed by Eilenburger Elektrolyse-und Umwelttechnik GmbH (EUT) with 35 cm2 geometrical electrode surface area each. In the half-cell setup, both chambers were separated with a Nafion® 115 membrane. The GDEs were provided by Covestro Deutschland AG. We used graphite (1), carbon black (CB, 2), carbon nanotubes (CNTs, 3), carbon fiber (4), glassy carbon (5), and CNTs with additional silver (6) containing GDEs for synthesis. The BDD was a DIACHEM® electrode by CONDIAS GmbH (24 cm2 planar BDD electrode; 35 cm2 structured BDD electrode; see Supplementary Figure S10). Electrolyses were conducted in single experiments (n = 1), and if not stated otherwise, experiments were conducted without temperature control.
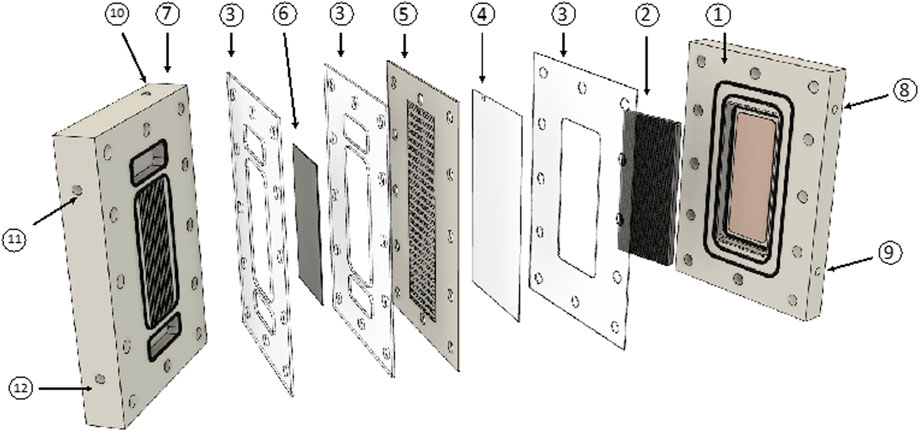
FIGURE 2. Exploded assembly of the electrochemical flow cell with the GDE and BDD with 35 cm2 geometrical electrode surface area each. 1 = cell body for the anode with o-rings and copper-contact; 2 = BDD; 3 = seal; 4 = Nafion® membrane; 5 = spacer with flow field; 6 = GDE; 7 = cell body for the cathode with a gas chamber; 8 = anolyte outlet; 9 = anolyte inlet; 10 = gas inlet (gas outlet is not visible); 11 = catholyte outlet; and 12 = catholyte inlet. Some parts are twisted for better visibility.
Further information on materials and methods, such as the size of the electrolysis cells and product quantification, is provided in Supplementary Material.
Results and discussion
Material screening of the gas diffusion electrode for H2O2 synthesis
For the cathodic synthesis of hydrogen peroxide as an educt for PAA synthesis, different carbon materials are tested (Supplementary Table S1). As carbon itself is the catalyst used to produce hydrogen peroxide, no further catalysts are needed for its production with the GDE. We, therefore, focused on five different carbon materials to evaluate the effect of their different surface structures on hydrogen peroxide synthesis. Even though we used two different CNT cathodes, one CNT-GDE included additional silver, since silver might not catalyze hydrogen peroxide production, but it could have a positive effect on PAA formation as it is known for platinum (Saha et al., 2004). The GDEs were screened in H-cell experiments of 30 min in a 0.5 M Na2SO4 aqueous electrolyte solution (V = 100 mL) with a geometrical GDE surface area of 5 cm2 under variation of the current density (20 mA cm−2, 60 mA cm−2, and 100 mA cm−2). The quantification of hydrogen peroxide was performed via iodine titration (n = 2). Using graphite and glassy carbon, no H2O2 was detected; therefore, those materials were not chosen for further experiments. As the four residual materials produced hydrogen peroxide at current densities between 20 mA cm−2 and 100 mA cm−2, we chose CB and CNT-based GDEs for our further experiments, since they showed the best CEs even at higher current densities. The CE for the CB GDE was the highest at 84% at j = 20 mA cm−2 and 57% at j = 100 mA cm−2. The latter is slightly better than the data reported by Cordeiro-Junior et al. (2022) for GDEs with 40% PTFE load (∼40%) but worse in comparison with a GDE with 20% PTFE load (∼60%); the CE reported in the literature increases with an increase in current density while we observed a decrease in the CE. Compared with this, the CE of the CNT-based GDE was lower at j = 20 mA cm−2 (58%), but it remained relatively stable even at higher current densities, making it adequate for further experiments. A detailed overview of materials and reached CEs for the respective current densities is provided in Supplementary Table S1.
Half-cell experiment 1: investigation of the PAA formation
In the next step, we probed the influence of different parameters on the electrochemical synthesis of PAA via H2O2 on CB- and CNT-based GDEs, and we started with the variation in the current density (20 mA cm−2, 60 mA cm−2, and 100 mA cm−2). Therefore, the setup was changed to a flow reactor with a geometrical GDE surface area of 24 cm2 and a flow rate of 50 mL min-1. The quantification of PAA next to hydrogen peroxide was performed via a two-step titration with Ce(SO4)2, followed by iodometry (see Supplementary Material for additional details). The catholyte (V = 100 mL) was changed from Na2SO4 to acetic acid as it is needed as an educt for further reaction with hydrogen peroxide. Initially, the current density was varied in a 2.5 M aqueous CH3COOH/CH3COONa solution for 30 min electrolysis experiments.
For both tested GDEs, PAA concentration increased with an increase in current density (Supplementary Figure S5A) to maximum values of 3.6 mmol L−1 and 5.6 mmol L−1 for CB and CNT GDEs at j = 100 mA cm−2, respectively. At the same time, the CE for PAA decreased from 3.9% (CB) and 3.1% (CNT) at a current density of 20 mA cm−2 to 1.6% (CB) and 2.5% (CNT) at a current density of 100 mA cm−2 (Supplementary Figure S5B), which is significantly less than that in the study by Saha et al. (2004). Nevertheless, at this point, the GDE surface was 1.3 times larger, and further improvement in the herein-reported synthesis is still pending. As it is assumed that PAA is not synthesized directly on the GDE but by the reaction with H2O2, the applied GDE should not have a direct impact on the formation of PAA. Nevertheless, slightly higher PAA concentration could be reached on the CNT-based GDE in comparison to the CB-based GDEs even though our former experiments showed a higher concentration of H2O2 formed on CB. This small difference can be an indication that the formation of PAA is, to some extent, based on the reaction of the electrolyte with active oxygen formed on the GDE instead of the assumed reaction with the H2O2 formed on the GDE, as described by Saha et al. (2004). All in all, the impact of the GDE on the formation of PAA is negligible, and we used the CB-based GDE in the next step as it shows better results for the production of H2O2.
In a second set of experiments, we varied the electrolyte solution consisting of different aqueous alkaline acetate salt solutions and mixtures in 3 h electrolysis. As it showed the best results for the CE of both H2O2 and PAA, a current density of 20 mA cm−2 was applied. The obtained CEs for the PAA formation in different electrolytes over time in Figure 3B (see Supplementary Figure S6 for concentration and the data for H2O2). The best results to produce H2O2 were obtained in 5 M CH3COOK and in a mixture of 2.5 M CH3COOH/CH3COOK (1:1) with CEs over 80%. For the formation of PAA, the 2.5 M CH3COOH/CH3COOK (1:1) electrolyte also showed significant production of acid as we obtained concentrations of approximately 22 mmol L−1 at a CE of ca. 10% after 3 h. Any other tested electrolytes resulted in the formation of only 11 mmol L−1 PAA or less. As for the indirect electrochemical PAA synthesis, the catholyte had to change from an aqueous Na2SO4 solution (pHstart = 6.9; pHend = 11–12) to an acetic acid- or acetate-containing catholyte, pH values of the catholyte changed too. The pH values at the end of PAA electrolysis were acidic, ranging from 2.1 (CH3COOH) to 5.2 (both buffered systems), or alkaline, between 10.8 and 14.5 (acetate salts). Even though a more acidic pH should have a positive impact on PAA formation, the CH3COOH/CH3COOK buffer showed significantly better PAA yields than the CH3COOH or CH3COOH/CH3COONa catholytes. Therefore, it was suggested that not only the pH value but also the used salt might have an influence on the PAA synthesis. Compared with the literature, the obtained CEs for the formation of both H2O2 and PAA are in good agreement. As for the first described indirect synthesis of PAA outgoing from H2O2 that was produced on GDEs, Saha et al. (2003c) reached a maximum CE of 12% (H2O2) and ca. 4% (PAA) at a current density of 10 mA cm-2 and a 1 M CH3COOH electrolyte. In a later study, they were able to increase the CE up to 23% (H2O2) and 9.5% (PAA) by the addition of Nafion-H to the electrolyte (Saha et al., 2004). Furthermore, Merk et al. (2001) showed in a patent that the use of acetylsalicylic acid as an acetate transfer reagent, in combination with NaOH and acetic acid, can shorten the reaction time, resulting in concentrations of even 31 mmol L−1 PAA after 20 min at current densities of j = 100 mA cm2.
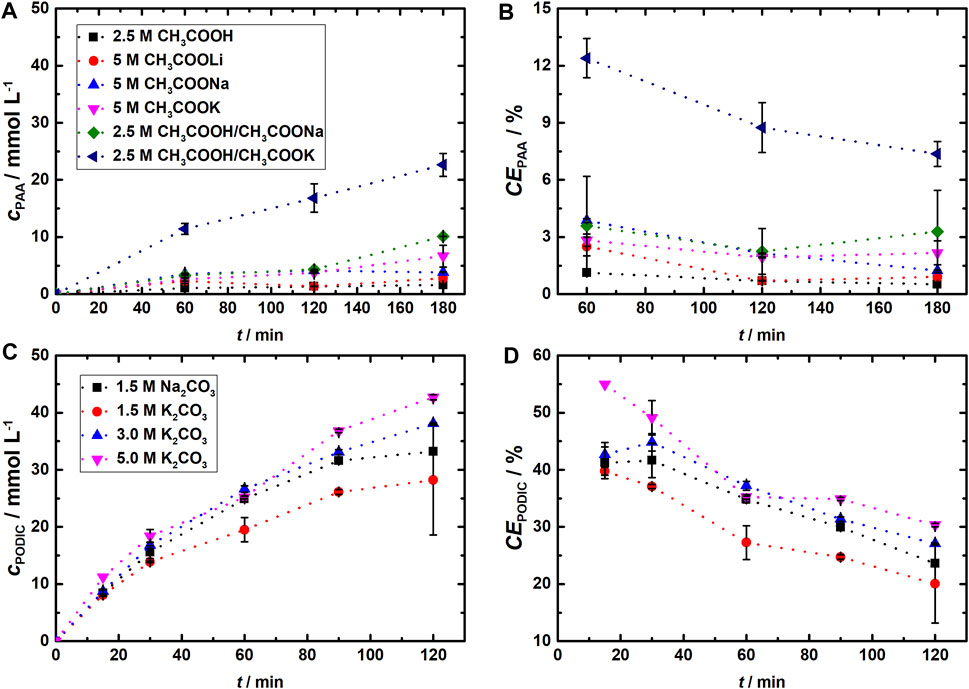
FIGURE 3. Variation in the electrolytes in the different half-cell experiments and resulting concentrations and CEs for the synthesis of PAA on the CB GDE (A, B) in 100 mL catholyte and of PODIC® on BDD (C, D) in 150 mL anolyte. j = 20 mA cm−2. The geometrical electrode surface area of each electrode was 24 cm2, and the flow rate was maintained at 50 mL min−1.
As our half-cell experiments showed the best results (the second-highest H2O2 and by far the highest PAA concentrations and CEs) at a current density of 20 mA cm−2 with a 2.5 M potassium acetate buffer solution, we applied this setup in the combined cell.
Half-cell experiment 2: investigation of the anodic PODIC® formation on BDD
In addition to the synthesis of PAA on GDEs, we investigated the anodic synthesis of PODIC® on a planar BDD in a reactor setup with a geometrical BDD surface area of 24 cm2 and an electrolyte volume of 150 mL. Based on the better solubility of K2CO3 in water in comparison to its sodium analogous (30.7 g Na2CO3/100 g H2O or 2.9 mol L−1 and 111 g/0.8 mol K2CO3/100 g H2O or 8.0 mol L−1 at 25°C) (Haynes et al., 2016), its use permits a great variety of the electrolyte by adjusting the salt concentration. Therefore, we investigated the dependence of the PODIC® formation on BDD under variation in the concentration of the carbonate-based electrolytes up to highly concentrated solutions of 5 mol L−1 K2CO3. To characterize and compare the results of our flow cell with early reports, an electrolysis with 1.5 M Na2CO3 solution was carried out in the beginning at current densities of 20 mA cm−2, 50 mA cm−2, and 100 mA cm−2. The amount of percarbonates was determined via iodine titration.
The obtained concentration of Na2C2O6 in our initial experiments is in the same range as can be found in the literature; however, the CEs (Figure 3D) are lower than those in the studies of Chardon et al. (2017). On the basis of the Na2C2O6 synthesis in our flow cell, we repeated it starting from a 1.5 M K2CO3 and with higher concentrated K2CO3 electrolytes up to 5 M aqueous solutions. In all experiments, a saturation level is reached after 60–90 min at current densities less than 100 mA cm-2. At a current density of 100 mA cm−2, the concentration of K2C2O6 slowly decreased after 60 min except for the 5 M K2CO3 solution. Moreover, the highest starting concentration of the K2CO3 solutions of 5 M resulted in a 35% higher concentrated peroxodicarbonate solution of 135 mM at 100 mA cm−2 compared to the 1.5 M Na2CO3 electrolyte solution. Notably, in this context, the pH values are still relatively high for the different experiments outgoing from 5 M K2CO3. They lie within the range of 10.95–11.55, which is considered favorable for the formation of peroxodicarbonates according to Chardon et al. (2017). Due to the probably necessary formation of bicarbonates at pH values between 11.4 and 9, the resulting ratio of carbonates and bicarbonates affects the peroxodicarbonate synthesis directly and prevents it in reverse at higher (only carbonates) or lower (only bicarbonates) pH values (pKa1 = 6.35 and pKa2 = 10.33 of H2CO3) (Haynes et al., 2016; Chardon et al., 2017). Moreover, the carbonates react as a base, and therefore, the resulting alkaline pH stabilizes the peroxodicarbonates.
In comparison to the results of the study by Seitz et al. (2022), the obtained concentrations of K2C2O6 are lower (258 mM starting from 1.5 mM K2CO3 and 275 mM starting from 2.25 M K2CO3 and 800 mA cm−2). Although their applied current density was eight times higher than that in our experiments, their obtained CE was lower, with a maximum of 23% in comparison to the 56% we report in this study.
Synthesis of peracetic acid and potassium peroxodicarbonate in a parallel paired electrolysis cell
Based on the results of the half-cell experiments, we used a 5 M K2CO3 solution as the anolyte and a mixture of 2.5 M CH3COOH and 2.5 M CH3COOK (1:1 ratio) as the catholyte solution for the synthesis of PAA and PODIC® in a parallel paired electrolysis setup. The geometrical electrode surface areas were 35 cm2 each, and the electrolyte volumes were 150 mL. The experiments were performed with CB- and CNT-based GDEs as cathodes for a duration of 2 h. Due to the cell design of the up-scaled 35 cm2 electrolysis cell by EUT, a structured BDD electrode (BDD on the silicon substrate, see Supplementary Figure S10) was employed as an anode. The structured flow channel regime of the electrode was employed to allow the flow of the anolyte, to mechanically stabilize the ion exchange membrane, and also, to allow the application of electrolytes with decreased conductivities, even though the design might lead to a decrease in electrolysis performance due to inhomogeneous distribution of local current densities. The current density was set at 50 mA cm-2 (referring to the geometrical surface areas) to achieve a balance between high productivity and high selectivity of the reactions; the corresponding results are plotted in Supplementary Figure S9. The anodic formation of K2C2O4 was independent of the gas diffusion counter electrode. A maximum concentration of 75 mmol L−1 with a corresponding CE of 14% was obtained at the end of the electrolysis. The CE decreased relatively quickly from initially 38% after 0.5 h (Supplementary Figure S9B) as the concentration of PODIC® reached a saturation level after 2 h, as and it was observed for the synthesis in the half-cell setup. Nevertheless, the CE of the PODIC® synthesis in the half-cell experiments was four times higher than that in the combined cell. At the cathodic side employing a CNT-based GDE, a maximum concentration of 11.3 mmol L−1 PAA with a CE of 2.0% (Supplementary Figures S9C, D, respectively) was obtained, which is less than a quarter of the CE that can be reached after 2 h under the same conditions in the half-cell setup with a CB-based cathode. The use of CB in the parallel paired electrolysis ended in an even smaller PAA concentration and CE of 8.5 mmol L−1 and 1.5% after 2 h.
To the best of our knowledge, the first synthesis of PAA and PODIC® in a parallel paired electrolysis cell was demonstrated with this study. The presented proof of concept indicates that the combination of the individually described half-cell reactions is possible in general. However, a high variation in the efficiencies between the half-cell experiments and the parallel paired electrolysis setup was observed. The combination of the BDD and GDE and the related changes in cell and electrode design to operate both electrodes in one setup are the main changes in the setup of the parallel paired electrolysis cell and might decrease electrolysis performance. Moreover, the different structure of the BDD (planar in the half-cell experiments vs. structured in the paired setup) can cause a major change in the local current densities under BDD and hydrodynamic conditions and, therefore, affect the local PODIC® synthesis. Furthermore, differences in the pH values must be pointed out. In the paired electrolysis, the pH values of both the catholyte and anolyte were in the alkaline range with values between 10 and 11 after 2 h of synthesis. In contrast to this, the pH value of the catholyte in the half-cell experiments was at 5.2 after 3 h at j = 20 mA cm−2. As an acidic pH value improves the PAA formation, this might have had a major influence on the worsening of the process. Nevertheless, the presented proof of principle showed the combination of the individual half-cell reactions, leaving room for improvement in future studies.
Conclusion
In this study, the cathodic synthesis of PAA via H2O2 synthesis on the GDE and the anodic synthesis of PODIC® on BDD in single half-cell setups were evaluated. With active geometrical electrode surface areas of 24 cm2, 22.6 mmol L−1 of PAA (CE = 7.4%, j = 20 mA cm−2, and t = 3 h) in 2.5 M CH3COOH/CH3COOK and 42.7 mmol L−1 of PODIC® (CE = 30.3%, j = 20 mA cm−2, and t = 2 h) in 5.0 M K2CO3 were obtained. The combination of the cathodic GDE and anodic BDD reaction was realized in a parallel paired electrolysis setup with enlarged geometrical electrode surface areas of 35 cm2 each. The combination in a parallel paired electrolysis resulted in the worsening of the single processes and does not lead to a simple addition of the CEs of the individual half-cell reactions. The cell setup and changes in the pH values of the catholyte to unfavorable alkaline conditions are hypothesized as the most significant parameters. Nevertheless, the experiments in the parallel paired test setup demonstrate the simultaneous synthesis of oxidizing agents in both electrochemical half-cells and the first proof of principle of a parallel paired electrolysis for the anodic PODIC® and cathodic PAA synthesis. Future work for the adjustment of parameters and of the combined cell design, such as the experimental setup in general including a stable acidic catholyte, will be necessary to improve this promising approach to reach at least similar results compared to the half-cell experiments.
Data availability statement
The data that support the findings of this study are available in the Supplementary Material of this article.
Author contributions
CH: conceptualization, data curation, investigation, methodology, and writing–review and editing. SS: data curation and writing–original draft. SH: data curation, investigation, and writing–review and editing. RN: funding acquisition, resources, and writing–review and editing. TM: funding acquisition and writing–review and editing. JKr: funding acquisition, resources, and writing–review and editing. MS: writing–review and editing. K-MM: conceptualization, funding acquisition, resources, supervision, and writing–review and editing. JKi: writing–review and editing.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by the German Federal Ministry of Education and Research under the project ZellCoDia (FKZ 13XP5022A, FKZ 13XP5022B, FKZ 13XP5022C, and FKZ 13XP5022D). Open Access publication was supported by Deutsche Forschungsgemeinschaft (DFG—German Research Foundation), 512514405.
Acknowledgments
The authors thank their project partners. Furthermore, they thank the DFI workshop for the construction of the flow cell for the half-cell experiments and Haithem Ajlani for his support.
Conflict of interest
Authors RN and TM were employed by CONDIAS GmbH. JKr was employed by Eilenburger Elektrolyse und Umwelttechnik GmbH. JKi was employed by Covestro Deutschland AG.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fctls.2024.1323322/full#supplementary-material
References
Biddinger, E. J., and Modestino, M. A. (2020). Electro-organic syntheses for green chemical manufacturing. Electrochem. Soc. Interface 29, 43–47. doi:10.1149/2.f06203if
Blanco, D. E., and Modestino, M. A. (2019). Organic electrosynthesis for sustainable chemical manufacturing. Trends Chem. 1, 8–10. doi:10.1016/j.trechm.2019.01.001
Boyce, J. M. (2016). Modern technologies for improving cleaning and disinfection of environmental surfaces in hospitals. Antimicrob. Resist. Infect. Control. 5, 10. doi:10.1186/s13756-016-0111-x
Chardon, C. P., Matthée, T., Neuber, R., Fryda, M., and Comninellis, C. (2017). Efficient electrochemical production of peroxodicarbonate applying DIACHEM® diamond electrodes. ChemistrySelect 2, 1037–1040. doi:10.1002/slct.201601583
Constam, E. J., and von Hansen, A. (1896). ELEKTROLYTISCHE DARSTELLUNG EINER NEUEN KLASSE OXYDIERENDER SUBSTANZEN. Z. Elektrochem. 3, 137–144. doi:10.1002/bbpc.189600032
Cordeiro-Junior, P. J. M., Lobato Bajo, J., Lanza, M. R. D. V., and Rodrigo Rodrigo, M. A. (2022). Highly efficient electrochemical production of hydrogen peroxide using the GDE technology. Ind. Eng. Chem. Res. 61, 10660–10669. doi:10.1021/acs.iecr.2c01669
Doll, M., Morgan, D. J., Anderson, D., and Bearman, G. (2015). Touchless technologies for decontamination in the hospital: a review of hydrogen peroxide and UV devices. Curr. Infect. Dis. Rep. 17, 44. doi:10.1007/s11908-015-0498-1
Frontana-Uribe, B. A., Little, R. D., Ibanez, J. G., Palma, A., and Vasquez-Medrano, R. (2010). Organic electrosynthesis: a promising green methodology in organic chemistry. Green Chem. 12, 2099. doi:10.1039/c0gc00382d
Groenen Serrano, K. (2021). A critical review on the electrochemical production and use of peroxo-compounds. Curr. Opin. Electrochem. 27, 100679. doi:10.1016/j.coelec.2020.100679
Hansen, A. V., and Elektrotech, Z. (1897). Electrochemical synthesis of inorganic compounds. Elektrochem. 3, 445–448.
Haynes, W. M., Lide, D. R., and Bruno, T. J. (2016). CRC handbook of chemistry and physicy. Boca Raton FL: CRC Press/Taylor And Francis.
Hilt, G. (2020). Basic strategies and types of applications in organic electrochemistry. ChemElectroChem 7, 395–405. doi:10.1002/celc.201901799
Ibanez, J. G., Frontana-Uribe, B. A., and Vasquez-Medrano, R. (2017). Paired electrochemical processes: overview, systematization, selection criteria, design strategies, and projection. Jo. Mex. Chemm. Soc. 60, 247–260.
Janssens-Maenhout, G., Crippa, M., Guizzardi, D., Muntean, M., Schaaf, E., Dentener, F., et al. (2019). EDGAR v4.3.2 Global Atlas of the three major greenhouse gas emissions for the period 1970–2012. Earth Syst. Sci. Data 11, 959–1002. doi:10.5194/essd-11-959-2019
Johnston, C., Callum, J., Mohr, J., Duong, A., Garibaldi, A., Simunovic, N., et al. (2016). Disinfection of human skin allografts in tissue banking: a systematic review report. Cell Tissue Bank. 17, 585–592. doi:10.1007/s10561-016-9569-2
Jolhe, P. D., Bhanvase, B. A., Patil, V. S., and Sonawane, S. H. (2015). Sonochemical synthesis of peracetic acid in a continuous flow micro-structured reactor. Chem. Eng. J. 276, 91–96. doi:10.1016/j.cej.2015.04.054
Klein, M., and Waldvogel, S. R. (2022). Counter electrode reactions—important stumbling blocks on the way to a working electro-organic synthesis. Angew. Chem. Int. Ed. 61, e202204140. doi:10.1002/anie.202204140
Kreysa, G., Ota, K., and Savinell, R. F. (2014). Encyclopedia of applied electrochemistry. New York, NY: Springer.
Llanos, J., Moraleda, I., Sáez, C., Rodrigo, M. A., and Cañizares, P. (2019). Reactor design as a critical input in the electrochemical production of peroxoacetic acid. J. Chem. Technol. Biotechnol. 94, 2955–2960. doi:10.1002/jctb.6101
Luukkonen, T., and Pehkonen, S. O. (2017). Peracids in water treatment: a critical review. Crit. Rev. Environ. Sci.Technol. 47, 1–39. doi:10.1080/10643389.2016.1272343
Manoharan, G., Muthu Mohamed, M., Raghavendran, N. S., and Narasimham, K. C. (2000). Electrochemical epoxidation catalyzed by manganese salen complex and carbonate with boron-doped diamond electrode. Trans. SAEST 35, 69–72.
McKenzie, E. C. R., Hosseini, S., Petro, A. G. C., Rudman, K. K., Gerroll, B. H. R., Mubarak, M. S., et al. (2022). Versatile tools for understanding electrosynthetic mechanisms. Chem. Rev. 122, 3292–3335. doi:10.1021/acs.chemrev.1c00471
Merk, T. L., Malchesky, P. S., and Liu, C.-C. (2001). Electrolytic synthesis of peracetic acid. US6387238B1.
Möhle, S., Zirbes, M., Rodrigo, E., Gieshoff, T., Wiebe, A., and Waldvogel, S. R. (2018). Modern electrochemical aspects for the synthesis of value-added organic products. Angew. Chem. Int. Ed. 57, 6018–6041. doi:10.1002/anie.201712732
Moraleda, I., Llanos, J., Sáez, C., Rodrigo, M. A., and Cañizares, P. (2016). Integration of anodic and cathodic processes for the synergistic electrochemical production of peracetic acid. Electrochem Commun. 73, 1–4. doi:10.1016/j.elecom.2016.10.010
Muddemann, T., Neuber, R., Haupt, D., Graßl, T., Issa, M., Bienen, F., et al. (2021). Improving the treatment efficiency and lowering the operating costs of electrochemical advanced oxidation processes. Processes 9, 1482. doi:10.3390/pr9091482
Ni, L., Mebarki, A., Jiang, J., Zhang, M., Pensee, V., and Dou, Z. (2016). Thermal risk in batch reactors: theoretical framework for runaway and accident. J. Loss Prev. Process Ind. 43, 75–82. doi:10.1016/j.jlp.2016.04.004
Pettas, I. A., and Karayannis, M. I. (2004). Simultaneous spectra-kinetic determination of peracetic acid and hydrogen peroxide in a brewery cleaning-in-place disinfection process. Anal. Chim. Acta 522, 275–280. doi:10.1016/j.aca.2004.07.010
Pollok, D., and Waldvogel, S. R. (2020). Electro-organic synthesis – a 21stcentury technique. Chem. Sci. 11, 12386–12400. doi:10.1039/d0sc01848a
Saha, M. S., Denggerile, A., Nishiki, Y., Furuta, T., and Ohsaka, T. (2003c). Synthesis of peroxyacetic acid using in situ electrogenerated hydrogen peroxide on gas diffusion electrode. Electrochem. Commun. 5, 445–448. doi:10.1016/s1388-2481(03)00097-3
Saha, M. S., Furuta, T., and Nishiki, Y. (2003b). Electrochemical synthesis of sodium peroxycarbonate at boron-doped diamond electrodes. Electrochem. Solid-State Lette. 6, D5. doi:10.1149/1.1576050
Saha, M. S., Nishiki, Y., Furuta, T., Denggerile, A., and Ohsaka, T. (2003a). A new method for the preparation of peroxyacetic acid using solid superacid catalysts. Tetrahedron Lett. 44, 5535–5537. doi:10.1016/s0040-4039(03)01276-0
Saha, M. S., Nishiki, Y., Furuta, T., and Ohsaka, T. (2004). Electrolytic synthesis of peroxyacetic acid using in situ generated hydrogen peroxide on gas diffusion electrodes. J. Electrochem. Soc. 151, D93. doi:10.1149/1.1782971
Seitz, A., Kohlpaintner, P. J., van Lingen, T., Dyga, M., Sprang, F., Zirbes, M., et al. (2022). Concentrated aqueous peroxodicarbonate: efficient electrosyn- thesis and use as oxidizer in epoxidations, S-, and N-oxidations. Angew. Chem. Int. Ed. 61, e202117563. doi:10.1002/anie.202117563
Tawabini, B. S., Plakas, K. V., Fraim, M., Safi, E., Oyehan, T., and Karabelas, A. J. (2020). Assessing the efficiency of a pilot-scale GDE/BDD electrochemical system in removing phenol from high salinity waters Author links open overlay panel. Chemosphere 239, 124714. doi:10.1016/j.chemosphere.2019.124714
Van de Velde, F., Güemes, D. R., and Pirovani, M. E. (2014). Optimisation of the peracetic acid washing disinfection of fresh-cut strawberries based on microbial load reduction and bioactive compounds retention. Int. Jo. Food Scie. Technol. 49, 634–640. doi:10.1111/ijfs.12346
Velazquez-Peña, S., Sáez, C., Cañizares, P., Linares-Hernández, I., Martínez-Miranda, V., Barrera-Díaz, C., et al. (2013). Production of oxidants via electrolysis of carbonate solutions with conductive-diamond anodes. Chem. Eng. J. 230, 272–278. doi:10.1016/j.cej.2013.06.078
Wang, Y. W. (2018). Evaluation of self-heating models for peracetic acid using calorimetry. Process Saf. Environ. Prot. 113, 122–131. doi:10.1016/j.psep.2017.10.001
Wiebe, A., Gieshoff, T., Möhle, S., Rodrigo, E., Zirbes, M., and Waldvogel, S. R. (2018). Electrifying organic synthesis. Angew. Chem. Int. Ed. 57, 5594–5619. doi:10.1002/anie.201711060
Wiel, P. M. V. D., Janssen, L. J. J., and Hoogland, J. G. (1971a). The electrolysis of a carbonate-borate solution with a platinum anode—II. Relation between current efficiency and perborate concentration. Electrochim. Acta 16, 1227–1234. doi:10.1016/0013-4686(71)85111-3
Wiel, P. M. V. D., Janssen, L. J. J., and Hoogland, J. G. (1971b). Electrolysis of a carbonate-borate solution with a platinum anode—I. Current efficiency at perborate concentration of zero. Electrochim. Acta 16, 1217–1226. doi:10.1016/0013-4686(71)85110-1
Yan, M., Kawamata, Y., and Baran, P. S. (2017). Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Zhang, C., Brown, P. J. B., and Hu, Z. (2018). Thermodynamic properties of an emerging chemical disinfectant, peracetic acid. Sci. Total Environ. 621, 948–959. doi:10.1016/j.scitotenv.2017.10.195
Zhang, J., and Oloman, C. W. (2005). Electro-oxidation of carbonate in aqueous solution on a platinum rotating ring disk electrode. J. Appl. Electrochem. 35, 945–953. doi:10.1007/s10800-005-7078-2
Zhao, X., Cheng, K., Hao, J., and Liu, D. (2008). Preparation of peracetic acid from hydrogen peroxide, part II: kinetics for spontaneous decomposition of peracetic acid in the liquid phase. J. Mol. Catal. A Chem. 284, 58–68. doi:10.1016/j.molcata.2008.01.003
Zhao, X., Zhang, T., Zhou, Y., and Liu, D. (2007). Preparation of peracetic acid from hydrogen peroxide. J. Mol. Catal. A Chem. 271, 246–252. doi:10.1016/j.molcata.2007.03.012
Zhu, C., Ang, N. W. J., Meyer, T. H., Qiu, Y., and Ackermann, L. (2021). Organic electrochemistry: molecular syntheses with potential. ACS Cent. Sci. 7, 415–431. doi:10.1021/acscentsci.0c01532
Ziogas, A., Belda, J., Kost, H.-J., Magomajew, J., Sperling, R. A., and Wernig, P. (2022). Peroxodicarbonate: electrosynthesis and first directions to green industrial applications. Curr. Res. Green Sustain. Chem. 5, 100341. doi:10.1016/j.crgsc.2022.100341
Keywords: boron-doped diamond electrodes, gas diffusion electrode, paired electrolysis, peracetic acid, peroxodicarbonate
Citation: Hamm CM, Schneider S, Hild S, Neuber R, Matthée T, Krümberg J, Stöckl M, Mangold K-M and Kintrup J (2024) Parallel paired electrolysis of green oxidizing agents by the combination of a gas diffusion cathode and boron-doped diamond anode. Front. Catal. 4:1323322. doi: 10.3389/fctls.2024.1323322
Received: 17 October 2023; Accepted: 22 January 2024;
Published: 08 February 2024.
Edited by:
Mingxuan Sun, Shanghai University of Engineering Sciences, ChinaReviewed by:
Nils Kurig, RWTH Aachen University, GermanyLuciana Vieira, Fraunhofer Institute for Interfacial Engineering and Biotechnology, Germany
Copyright © 2024 Hamm, Schneider, Hild, Neuber, Matthée, Krümberg, Stöckl, Mangold and Kintrup. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Stöckl, markus.stoeckl@dechema.de