Mechanotransduction Mechanisms in Mitral Valve Physiology and Disease Pathogenesis
 Leah A. Pagnozzi
Leah A. Pagnozzi Jonathan T. Butcher
Jonathan T. Butcher- Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY, United States
The mitral valve exists in a mechanically demanding environment, with the stress of each cardiac cycle deforming and shearing the native fibroblasts and endothelial cells. Cells and their extracellular matrix exhibit a dynamic reciprocity in the growth and formation of tissue through mechanotransduction and continuously adapt to physical cues in their environment through gene, protein, and cytokine expression. Valve disease is the most common congenital heart defect with watchful waiting and valve replacement surgery the only treatment option. Mitral valve disease (MVD) has been linked to a variety of mechano-active genes ranging from extracellular components, mechanotransductive elements, and cytoplasmic and nuclear transcription factors. Specialized cell receptors, such as adherens junctions, cadherins, integrins, primary cilia, ion channels, caveolae, and the glycocalyx, convert mechanical cues into biochemical responses via a complex of mechanoresponsive elements, shared signaling modalities, and integrated frameworks. Understanding mechanosensing and transduction in mitral valve-specific cells may allow us to discover unique signal transduction pathways between cells and their environment, leading to cell or tissue specific mechanically targeted therapeutics for MVD.
Introduction
The mitral valve is a bicuspid valve that facilitates the flow of blood from the left atrium to the left ventricle. Mitral valve disease (MVD) affects 2.4% of the population and is a common congenital heart defect (1, 2). In adults, the most common disorders of the valve are mitral insufficiency (i.e., regurgitation), mitral stenosis, myxomatous degeneration, and mitral valve prolapse, with broad disease likely to include several of these effects.
The mitral valve leaflets consist of four layers that differ in extracellular matrix (ECM) composition and mechanical properties. The thickest layer of the valve, the fibrosa, is the main load bearing layer. It provides the majority of leaflet tensile strength through a thick layer of dense, aligned collagen fibers while a looser collagen network with increased glycosaminoglycan (GAG) and proteoglycan content provides compressive strength. The mitral valve is mechanically supported through the GAG-rich chordae tendinae which attach the mitral leaflets to the papillary muscles along the ventricular wall and maintain valve closure during systole. MVD results in altered mechanical and structural properties of the valve. Myxomatous mitral valves are characterized by leaflet enlargement, annular dilation, thickened and elongated chordae, GAG accumulation, loss of structure, increased compliance, and myxoid lesions. Disorganization and remodeling of the ECM and weakening of the chordae result in a loss of most of the valve’s mechanical properties and an overall thickened and enlarged leaflet. This in turn prevents the valve from fully closing causing symptoms of mitral regurgitation and prolapse.
The mitral valve is a dynamic structure which changes mechanically during the cardiac cycle; the constant flow of blood and opening and shutting of the valves exposes the tissue to a complex and demanding environment. The valve is subjected to bending, deformation, large area changes, shear stress, and heterogeneous strains in response to myocardial contraction, transvalvular pressure, and hemodynamic flow. The mitral valve exhibits a non-linear stress–strain relationship with complex viscoelastic and axial coupling behaviors (3, 4). These dynamic and adaptive interactions between the myocardial wall and valve leaflets ultimately impact the mechanical stress and strain experienced by cells through the ECM (5).
Cells and their ECM exhibit dynamic reciprocity, continuous, bidirectional interaction between cells and their ECM, in the growth and formation of tissue through mechanotransduction, the conversion of mechanical signals into biochemical responses (6). Cells and their ECM reorganize via a complex of mechanoresponsive elements (6) to physically regulate the spatiotemporal distribution of biochemical components maintaining homeostasis. MVD has been linked to a variety of mechano-active genes, such as extracellular components, mechanotransductive elements, and transcription factors. Mechanical stimulus in the microenvironment provides inductive signals of homeostasis and remodeling to the native cells—valve interstitial cells (VICs), and endothelial cells (VECs).
Valve endothelial cells reside on the exterior of the valve, maintain a non thrombogenic surface layer, and regulate immune and inflammatory reactions. The majority of valve cells are the VICs, a mesenchymal population that resides in all layers of the valve, distinct in their ability to differentiate into multiple phenotypes. There are five known phenotypes of VICs: embryonic progenitor endothelial/mesenchymal, quiescent, activated, progenitor, and osteoblastic VICs which may convert from one form to another. Most VICs in the healthy adult valve are quiescent with a small population of activated VICs to maintain base ECM remodeling. In pathological states, there is an increase in activated VICs which regulate repair and remodeling, which may lead to fibrosis and calcification. Inflammation, biochemical, and mechanical stimuli can induce activation of quiescent fibroblasts into myofibroblasts. VICs and VECs continuously remodel their environment by secreting and degrading ECM, and adapting their gene, protein, and cytokine expression to alter phenotype and function. These dynamic and adaptive interactions between the myocardial wall, flowing blood, and valve leaflets ultimately impact the mechanical stress and strain experienced by cells through the ECM. The movement, anisotropic deformation, and complex geometries of the mitral valve create a variety of ever changing mechanical cues between the cells and their matrix. ECM composition, fiber alignment, and compaction regulate cell deformation and thus mechanotransductive response. By focusing on broad classes of mechanosensing pathways as well as their integration in mechanotransduction, this review will explore the biomechanical mechanisms at play in the mitral valve microenvironment and mediators of mechanotransduction in this tissue.
Mechanobiology of Mitral Valvulogenesis
During valve development, the embryonic heart transforms from a myocardial tube into a complex, four chambered, mature structure. Valve cells differentiate from endocardial cells during gastrulation and by E9.5 valvulogenesis begins when the heart tube loops creating the primitive ventricle and atria. In these early embryos position sensing (7, 8) and force transduction instruct lineage allocation. Endothelial cells (ECs) of the endocardium form valve cushions in a GAG-rich cardiac jelly where, in response to growth factors, such as Transforming Growth Factor-β (TGF-β), they undergo endothelial to mesenchymal transition (EMT). ECs reorganize their actin architecture to permit migration, adhesion, and morphogenesis in the embryo. Knockout of cytoskeletal adaptors in ECs causes disorganized cytoskeletal organization, cell morphology, impaired focal adhesion development, and actin signaling, inhibiting EMT in embryonic mice (9). Atrioventricular endocardial cells adopt a cuboidal morphology prior to EMT which seems mediated by cardiac contraction- in mutants which lack heart contraction, endocardial cells fail to change shape and initiate EMT (10).
During EMT cell–cell contacts are downregulated and processes governing cell–matrix adhesions and cytoskeleton reorganization are upregulated (11). Cells acquire an invasive phenotype, allowing them to migrate into the cardiac jelly, degrade hyaluronan, and deposit collagen, versican, and proteoglycans to form mature leaflets. Cushion mesenchymal cells give rise to VICs post-EMT which organize their surrounding matrix into a fibrous, rigid tissue able to withstand the hemodynamic loading of the beating heart. Contractile VICs condense the ECM by pulling on it, creating cell–matrix alignment in response to mechanical cues (12, 13). During valvulogenesis, tension points are created which may promote the secretion and alignment of collagen fibrils from VICs in a manner similar to that seen during tendon development (14).
Mechanotrasnduction of hemodynamic shear and strain are crucial to valvulogenesis. In zebrafish embryos knockdown of oscillatory flow sensitive gene klf2a results in dysfunctional or absent leaflet formation despite no change in retrograde flow (15). klf2a is related to signaling through mechano-sensitive ion channels, which is discussed later in the Ion Channel section. Physical occlusion of the inflow or outflow tract in zebrafish embryos results in hearts with an abnormal third chamber, looping defects, and impaired valve formation. In the embryo, red blood cells themselves generate important shear fluctuations different than that of normal hemodynamic shear which may mechanically influence ECs (16). Zebrafish with transvalvular flow alterations fail to undergo atrioventricular valve maturation from two to four leaflets despite no alterations in contractility (17). Tissue strain from variations in pressure and cardiac contraction also mechanically drive valve formation in a similar fashion to cell–cell and cell–matrix contacts. Mutations that inhibit myocardial contractility in the embryo fail to form cushions with chemical inhibition of contraction inhibiting endocardial ring formation in a dose dependent fashion (18). Cytoskeletal adaptors in embryonic ECs mediate actin dynamics, and mutations in them disrupt EMT and valvulogenesis (19). The impact of strain alterations are time dependent as altered cardiac preload results in morphological defects in zebrafish embryos treated in earlier and later developmental stages without impacting groups treated at 30–36 h post fertilization (20). Alterations in cell–matrix homeostasis later in life may reactivate physical or chemical cues of valvulogenesis, particularly EMT, causing aberrant elongation, remodeling, and stiffening (21, 22).
Adherens Junctions and Cadherins
Adherens junctions are located at cell–cell contact points where they mediate cell adhesion, force, and signal transduction. Cells send out a finger-like lamellipodia to neighboring cells, which are stabilized by the acceptor lamellae with actin-myosin contractility. This actin finger determines the location and shape of the adherens junction and is co-localized with stress fibers in the neighboring cells. Adhesions are formed through integrin and cadherin interactions in both VECs and VICs at cell–cell and cell–integrin junctions, respectively. At adhesions, adhesion receptors interact with F-actin and adhesion proteins to regulate signaling, junction assembly, and maintenance. While traditionally recognized as distinct structures, adherens junctions and focal adhesions are intracellularly linked to the actin cytoskeleton, and activate the same signaling proteins and actin regulators (23).
Vinculin is a cytoplasmic actin binding protein, enriched at both cell–cell and cell–matrix adhesions, which regulates integrin dynamics and adhesion, stimulating polymerization, and remodeling through actin binding. Vinculin arranges itself in three domains: an integrin signaling layer, actin binding and force transducing layer, and actin regulatory layer. Vinculin is in an open active form in focal adhesions and a closed, inhibited form within the cytoplasm. In this inhibited form, the vinculin head domain interacts extensively with its tail in the integrin signaling layer and when these head–tail interactions are relieved (24), it migrates to the actin binding layer where it recruits proteins to regulate focal adhesion dynamics and cell migration (25).
Cadherins are calcium-dependent cell adhesion proteins composed of an extracellular region, a transmembrane domain, and cytoplasmic region. Cadherins connect the cortical actin cytoskeleton of neighboring cells and create zipper-like structures to maintain stable intercellular adhesion by regulating cortical tension and maintaining mechanical coupling between cells (26). In confluent monolayers, VICs with strong cell–cell contacts show weak expression of myofibroblastic marker α-smooth muscle actin (αSMA), suggesting cell contact inhibits myofibroblastic activation (27). In these conditions, cadherin protein complexes β-catenin and N-cadherin expression are decreased or absent (27). In aortic valve disease and development cell junction protein cadherin-11 (Cad-11) has been implicated in a variety of mechano-active defects and similar mechanisms may be at play in MVD. Cad-11, a known mediator of dystrophic calcification in calcific aortic valve disease, is strongly expressed in human calcified aortic leaflets with nodule formation dependent on strong cell–cell contacts (28) while cyclic strain upregulates Cad-11 and αSMA expression (29) in aortic VICs (AVICs). In canines with myxomatous valve disease, VE-cadherin was significantly decreased (30). Downregulation of VE-cadherin results in endothelial migration and EMT in zebrafish valvulogenesis (31) so similar expression in canines suggests a pathological proliferative and migratory endothelial phenotype (30).
Plakophilin-2 links cadherins to intermediate filaments in the cytoskeleton. In prolapsed mitral valves, increased Cad-11, N-cadherin, and aberrant presence of plakophilin-2 at the adherens junction, promotes latent TGF-β activation and pathological ECM remodeling (32). Cad-11 is expressed in chick mitral valves during development at the leaflet tips in endocardial cushion mesenchymal cells (31) and throughout the leaflets of remodeling valves in adults. In hyperlipidemic mice, Cad-11 expression was significantly increased in the aortic and mitral valves (33) inducing ECM remodeling and calcific nodule formation (34).
Integrins
Integrins regulate and respond to force by connecting the ECM to the cytoskeleton. Composed of an α and β subunit which combine to approximately 24 unique heterodimers (35, 36), integrins bind to different ECM proteins and interact with cell-surface ligands, transmembrane proteins, proteases, and growth factors (37). Integrins receive and transmit signals from both sides of the plasma membrane (38, 39). Cytoskeletal contractions pull on integrin links to the matrix, deforming binding proteins that connect actin to focal adhesion proteins and integrin to arginine–glycine–aspartate (RGD) containing proteins, altering gene and protein expression (40). RGD is the main integrin binding domain in ECM proteins common to the mitral valve: collagens, laminin, fibrillin, and fibronectin (41, 42).
Several adhesive peptides control integrin-mediated cell adhesion. VICs strongly express the α2 and β1 subunits and α5β1 integrin (43, 44) Collagen I mimetic DGEA binds integrin α2β1 and promotes adhesion and ECM deposition in VICs (45). The α2β1 integrin is necessary in coupling VICs to collagen I, propagating VIC contraction into leaflet force generation (46). In combination with RGD, peptide VAPG with affinity to laminin and elastin, along with DGEA downregulate myofibroblastic and osteogenic differentiation in VICs (45). Blocking integrin receptor 67LR, with affinities to laminin and elastin, resulted in formation of calcific nodules (47) suggesting an anticalcific effect in binding. Disruption of VIC binding via the α5β1 integrin or the 67-kDa laminin receptor had a dramatic calcification-stimulating effect. Binding via the α2β1 integrin did not alter calcification or VIC phenotype; blocking α5β1 resulted in calcification in AVICs (43) and is likely to have similar pathology in mitral valves.
Integrins bind to and activate TGF-β, which modulates cell growth, adhesion, migration, and ECM synthesis (48, 49). TGF-β secretion consists of three proteins: TGF-β, latency-associated protein (LAP), and latent TGF-β binding protein (LTBP), an ECM-binding protein. Several integrins activate latent TGF-β through binding to an RGD integrin binding site on LAP (50). Under high stress, TGF-β controls expression of αSMA, stress fiber formation, and differentiates quiescent fibroblasts into contractile myofibroblasts creating a positive feedback cycle (51). Mechanically conditioning ECM releases active TGFβ1 (52) demonstrating the role of force in fibroblast activation. VICs grown on stiff surfaces have strong cell-ECM adhesions, contractility, and myofibroblast differentiation (53). Shear flow induces TGFβ1 production and myofibroblast differentiation of fibroblasts in collagen gels (54). In both embryonic and adult VICs, a quiescent phenotype is maintained in unstressed collagen hydrogels; however, contractile expression, TGF-β, and matrix remodeling are upregulated in response to tension (55).
Latent TGF-β binding proteins interact with fibrillin, a large structural protein that polymerizes into extracellular microfibrils and contributes to the functional integrity of connective tissue (56). Mutations in fibrillin-1 cause Marfan Syndrome (MFS) and related disorders from dysregulated TGF-β activity. TGF-β cytokines act through various small GTPases such as RhoA and Rac1, which are implicated in valve disease and development (Figure 1). RhoA is a mechano-sensitive GTPase that acts complementary to Rac to control cell migration, differentiation, and proliferation. Filamin-A (FlnA) point mutations in mice, responsible for X-linked myxomatous valve disease (57), deregulate the balance between RhoA and Rac1 in favor of RhoA, altering downstream trafficking of β1 integrins (58) resulting in a myoxomatous phenotype by 2 months of age. For more information on GTPases, see section on Integrated Mechanotransduction at the end. FlnA mutations increase Erk signaling, a non canonical TGF-β driven kinase, which is present in mouse models of MFS (59). In murine aortic valves with an elastogenic defect, mice had latent hemodynamic AV disease from increased Erk1/2 activation, ECM disorganization, and inflammation (60). Both these mutant mice and aged mice display stiffened ECM, fibrosis, cell adhesion and fibronectin alterations, increased collagen expression, and decreased LTBP signaling (60) suggesting a similar mechanism may be driving integrin signaling in MVD.
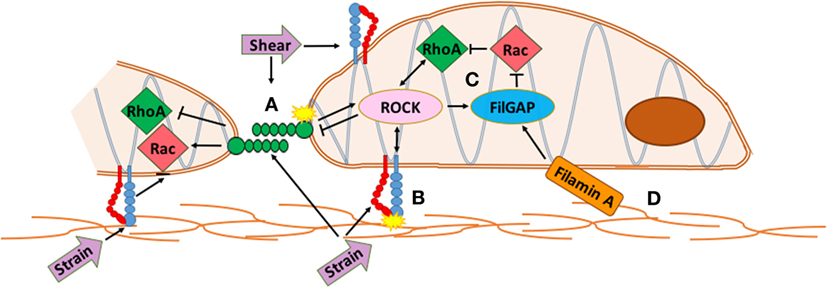
Figure 1. Integrated mechanotransduction of cadherins and integrins through the cytoskeleton and small GTPases. A. At cell–cell adhesion points coupled cadherins transduce strain and shear force into cytoskeletal remodeling and downstream signaling pathways. B. At cell–extracellular matrix (ECM) adhesions, strain is transduced through integrins into cytoskeletal remodeling and downstream signaling pathways. C. RhoA and Rac are mechano-sensitive small GTPases common to multiple methods of mechanotransduction in the mitral valve that act opposite and complementary to control cell migration, differentiation, and proliferation. RhoA regulates actin cytoskeleton and stress fiber formation while Rac1 regulates cell–cell adhesion, actin polymerization, lamellae protrustion, and cytoskeletal polarity. ROCK interacts with integrins and cadherins to mediate RhoA and Rac activity while FilGAP binds to filamin-A to control actin at cytoskeletal interfaces. D. The extracellular matrix interacts with the actin network directly through specific ECM components or through integrins and cadherins. See Section “Integrated Mechanotransduction” for more information.
Cilia
Primary cilia are solitary microtubule structures consisting of a basal body and projecting axoneme “antenna.” The axoneme senses the external environment and coordinates various signaling pathways, such as TGF-β (61) and calcium sinks (62), indicating a mechanosensory role (63). Primary cilia defects have been linked to various congenital cardiovascular diseases, such as heterotaxy and atrioventricular septal defects (64–66). Cilia are strongly expressed between stages E11.5 and E17.5 on the outflow tract cushions in aortic valvulogenesis, while they are lost in adult VICs (67).
Primary cilia restrain ECM expression during development and remodeling such that ablation of primary cilia during aortic valvulogenesis results in highly penetrant bicuspid valve phenotype (67). Primary cilia loss in arterial ECs sensitizes them toward BMP mediated osteogenic differentiation (68), inflammatory gene expression, and decreased eNOS activity (69). Exome sequencing of chemically mutagenized mice revealed mutations in 61 recessive congenital heart disease genes, 34 of them cilia related (66); cilia axoneme mutants caused outflow tract and atrioventricular septation (70). In polycystic kidney disease (PKD), a genetic disorder with TGF-β mediated abnormalities, there is a 10-fold increase of mitral valve prolapse tied to defective protein localization in the primary cilia (71–73). Mitral insufficiency has been seen in infantile nephronophthisis (74), structural defects in Ellis–van Creveld syndrome (75, 76), severe mitral regurgitation and structural defects in Kartagener’s syndrome (77, 78), and rheumatic valvular insufficiency in Bardet–Biedl syndrome (79).
Ion Channels
Mechano-sensitive channels (MCs) are a class of membrane ion channels that detect and respond to force, converting it into electrical or biochemical signals (80, 81). There is increasing evidence MCs play a key role in regulating endothelial response to shear flow (82–84). Cilia coupled with calcium channels (Figure 2) transduce shear stress during zebrafish valvulogenesis; endothelial cilia deflect with blood flow correlating to expression of calcium channel gene polycystin-2 (PKD2), increasing endothelial calcium levels, and altering vascular formation (85). Cilia response is mediated by transient receptor channels such as Trpv4 and Trpp2 which are expressed during valve development (86). mRNA expression of Piezo1, a mechanically activated cation channel, has been seen in murine hearts (87) while its loss in ECs causes stress fiber and cell orientation (88) deficits in response to shear stress, profound vascular defects, and embryonic lethality within days of the heart beating (89).
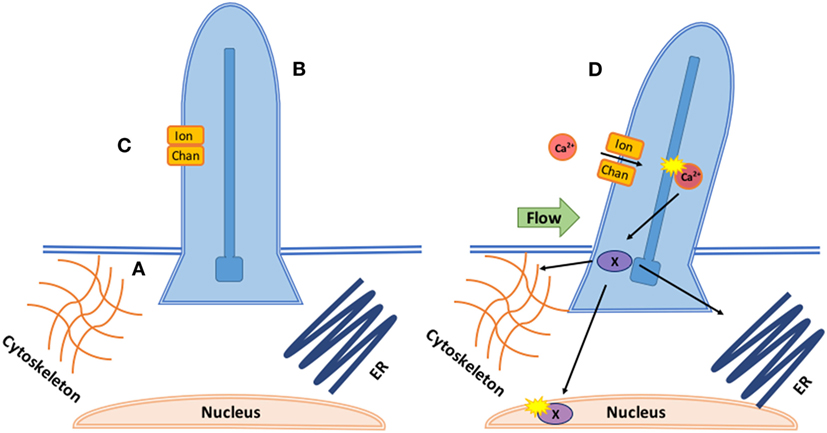
Figure 2. Coupled mechanotransduction of cilia and ion channels. A. The basal body is a modified centriole that sits at the bottom of the cilia and provides the origin point for new cilia. They provide a symmetric template for the axoneme structure and dictate the position and orientation of the cilia, ensuring correct cilia-driven fluid flows and response to flow. B. The axoneme is the most prominent component of cilia consisting of nine microtubules. Its major function is cell signaling as the axoneme senses and coordinates mechanical and chemical responses, bending the cilia and altering downstream signaling. Intraflagellar transport brings cargo into the cilia along the axoneme with kinesin and out with dynein. C. In response to axoneme bending, stretch or calcium responsive ion channels, such as the polycystins (polycystic kidney diseases), open, allowing for calcium- or ATP-dependent signaling to occur inside the ciliary body. D. In response to calcium influx through the ion channel, downstream transcription factors (X) are phosphorylated and translocated to interact with the cytoskeleton, nucleus, and endoplasmic reticulum.
Malfunction of MCs results in broad cardiovascular pathology such as arrthymias (90), hypertension (91), and PKD (92). PKD2 is localized to the cilia in vascular ECs. In mouse embryos it is required for right-left axis determination with knockouts displaying severe cardiac structural defects by E18 (93). PKD2 is mutated in PKD and murine mutants lose the ability to generate nitric oxide (NO) in response to shear flow which may promote high blood pressure (94). PKD2 defects may prolong channel activity by preventing calcium from leaving small compartments, such as cilia (95). Prolongation of the QT interval has been associated with myxomatous mitral valve related sudden cardiac death (96, 97). Mutations in sodium voltage-gated channel V account for 5–10% of long QT cases and have been comorbid with desmoplakin mutations, a protein responsible for mechanical coupling of cardiac myocytes with known overlap in channelopathies (97, 98). Oscillatory flow stimulates klf2a expression, a key transcription factor in valvulogenesis, and knockdown results in dysfunctional or absent leaflet formation (15). Oscillatory flow through Trpv4 and Trpp2 (99) modulate the endocardial calcium response and control klf2a expression in zebrafish, with absence of either resulting in severe valve defects. klf2a misexpression during angiogenesis occurs in the absence of flow, with downregulation of β1 integrin rescuing overgrowth and maintaining endothelial quiescence (100).
Caveolae
Caveolae are small plasma membrane invaginations made up of Caveolin (Cav) and Cavin proteins, glycosphingolipids, and cholesterol. Caveolae respond to mechanical stress by flattening into the membrane, increasing surface area to relieve tension, while confining receptors and signaling molecules (101, 102). Caveolae participate in a dynamic cycle of flattening and reassembly in response to mechanical stress independent the actin cytoskeleton. In vascular smooth muscle cells (103), cardiomyocytes (104), and aortic ECs, translocation of Cav1 to non-caveolar membrane domains during flattening is required for strain and flow induced Erk expression (105). Rho and Rac GTPases (104), Src (106) and MAP kinases (107), and calcium (108) expression are also modulated by caveolae mechanotransduction.
Genomic analysis in canine myxomatous valve disease identified caveolar mediated endocytosis as a canonical pathway relevant to MVD (12). This pathway controls EC growth and migration through endocytosis of cholesterol-enriched membrane microdomain (CEMM) internalization when integrins are uncoupled during cell detachment from the ECM. Integrins target Rac to CEMMs where it interacts with downstream effectors to induce signaling (109, 110). In caveolin-1 knockdowns, TGF-β, fibroblast activation, and collagen gene expression increases in human lung fibroblasts (111). In canines with chordal rupture induced mitral regurgitation, caveolar invagination decreased Erk signaling, regulating hypertrophic remodeling in response to volume overload (112). Positive caveolin staining and caveolae structures have been seen on aortic VECs (113) and may be conserved in mitral valves.
Glycocalyx
The glycocalyx (GC) are abundant proteoglycan complexes that cover the surface of ECs and maintain endothelial barrier integrity. They are composed of the syndecan, a transmembrane core protein, and membrane anchored GAGs (114). GC control NO production (115) in vascular ECs by transducing shear stress to the cytoskeleton (116, 117) resulting in intracellular signaling and NO production (118, 119). Breakdown of the GC results in dissolution of tight junctions (120) and production of NO is dependent on calcium intake from TRP channels (121).
Syndecans (Sdcs) are members of a proteoglycan family of adhesion transmembrane receptors (122, 123). There are four mammalian Sdcs that bind to ECM, cell adhesion molecules, and growth factors (43). While no Sdcs are expressed in healthy aortic or mitral VICs (43), Sdc1 is strongly expressed on the vascular EC surface (124) and GCs are broadly expressed on the mitral endothelium in hypercholesterolemic rabbits (125). GCs and Sdcs are implicated in inflammatory (126, 127) and vascular diseases in the context of heart failure (128–130), myocardial dysfunction (131), and myocardial infarct (132). Sdc1-null mice with myocardial infarction display enhanced endothelial adhesion, trans endothelial migration of inflammatory cells, matrix remodeling, and fibrosis (133) as well as attenuated angiotensin II-induced dysfunction (134). Oxidized LDL cholesterol degrades GCs and enhances adherence of leukocytes to the endothelial surface in mouse vascular models (135). Immune involvement provides a potential avenue to MVD given the autoimmune role in rheumatic valve disease.
Nuclear
Many mechanosensing modalities are physically coupled to the cytoskeleton filaments which in turn link to nuclear scaffolds, chromatin, and nuclear DNA (136–138). Forces applied to the cell surface cause structural changes to the nucleus (139, 140). As such, the nuclear aspect ratio (NAR) can be used as an index of cellular deformation due to the correlating deformation and directionality of the nucleus to the cell. In the mitral valve, NAR analysis determined VICs in the fibrosa and ventricularis layers deform more than the atrialis and spongiosa (141). MVICs also display cytoplasmic uncoupling from nuclear deformation under hyper-physiological strain levels (142) which may have phenotype and ECM remodeling consequences.
Lamins, nuclear intermediate filaments, are dense protein networks capable of forming stable structures within the nucleoplasm and have a crucial role in DNA/RNA synthesis and transcription (137). Dilated cardiomyopathy (143, 144) a laminopathy, causes volume overload and functional mitral regurgitation. Lamin A/C mutant mouse cells have impaired activation of mechano-sensitive transcription factor MRTF-A which causes cardiac myofibroblastic differentiation (145, 146) and activates vinculin and actin (147). Linker Nucleuskeleton and Cytoskeleton (LINC) proteins are key mechanotransductive structures between the cytoskeleton and nucleus. They include nesprin which connects LINC to the cytoskeleton and SUN which anchors LINC in the nucleus through lamin interactions and chromatin binding proteins (148). Nesprin is subject to actin-myosin mediated tension in adherent fibroblasts, which is reduced in fibroblasts from Hutchinson–Gilford progeria patients, a multisystem laminopathy (149). Nesprin also interacts with common intracellular signaling pathways such as Erk1/2 (67) and β catenin (150). Nesprin knockdown in ECs cripples nuclear deformation and cell orientation during cyclic strain, but increases focal adhesions (151). Nesprin knockout cells have altered morphology, polarization, and migration (152).
5-HT Serotonin
Multi-valve pathology (153) is seen after exposure to serotonergic drugs fenfluramine, dexfenfluramine, ergotamine, and methysergide (154) as well as ergot-derived dopamine agonists pergolide (155), cabergoline (156), and bromocriptine (157). Fenfluramine binds to serotonin or 5-hydroxytryptamine (5-HT) receptors 5-HT2A, 5-HT2B, and 5-HT2C with porcine aortic and mitral VICs expressing 5-HT2A and 5-HT2B receptor transcripts, suggesting valve fibrosis (158) after exposure to fenfluramine, ergot drugs, and 5-HT is a result of 5-HT2A and 5-HT2B stimulation. In ligand screening studies 5-HT2B is the commonly activated serotonin receptor of drugs associated with valvular heart disease (159) with myxomatous canine valves upregulating 5-HT2B receptor mRNA (160) and proteins (161). The 5-HT2B receptor is required for heart development (162) regulating differentiation and proliferation of cardiac tissue; 5-HT transporter deficient mice develop cardiac fibrosis, and valvulopathy (163).
5-HT2B increases MVIC proliferation and ECM production through common mechano-active signaling modalities. 5-HT2B receptor activation increases MAPK activity through Erk1/2 (164, 165) as well as Src family kinases (166), resulting in cell proliferation, while addition of 5-HT to canine MVIC cultures increases collagen and GAG synthesis through H-proline and H-glucosamine incorporation respectively (164). Cross-talk may occur between the TGF-β and 5-HT pathways under elevated mechanical stresses. During atrioventricular valve development in chick embryos, 5-HT induces pathological modeling effects through a TGF-β3-dependent mechanism causing tissue stiffening, contractile gene expression, and collagen expression (167). In myxomatous mitral valves, 5-HT2B receptor expression is co-localized with αSMA expression (168); neonatal rat cardiac fibroblasts treated with 5-HT upregulated αSMA expression marking fibroblast differentiation and TGF-β signaling (169). AVICs treated with 5-HT show increased TGF-β1 and 5-HT2A (170) expression while serotonin transporter (SERT) knockout embryonic mice increased expression of TGF-β1, αSMA, and 5-HT2A in the whole heart (171). At the tissue scale, treating an AVIC seeded construct with a 5-HT2B agonist acutely decreases tone generation of the cells, tissue alignment, and increases the tensile modulus along the primary fiber alignment axis (172). Similar mechanisms may be at play in 5-HT-related MVD.
While 5-HT alters the MV microenvironment and global valve mechanics, it may also be a direct mechanomodulator as proposed in Figure 3 below. In both aortic banded rats and neonatal rat cardiomyocytes, mechanical stress enhances 5-HT2B signaling in ventricular models of pressure induced cardiomyopathy (173). Serotonin induced a positive inotropic response in the papillary muscles and increased 5-HT2B receptor expression in hypertrophic rats with post infarction heart failure which correlated to degree of hypertrophy (174). Cyclic stretch upregulates 5-HT2A and 5-HT2B receptor expression in porcine aortic valve cusps causing AVIC proliferation and ECM remodeling (175). Cell proliferation, collagen synthesis, and tissue stiffness in response to cyclic stretch seem to be specifically modulated by the 5-HT2A receptor in the aortic valve (176) while unstrained in vitro experiments in MVs implicate the 5-HT2B receptor. Static and cyclic strain increase expression of myxomatous effector proteins, chondrogenic markers, and markers of the myofibroblastic phenotype compared to unstrained controls in myxomatous canine MVs (177). Interestingly, in both strain conditions, expression of serotoninsynthetic enzymes increased with higher serotonin levels in the media of cyclically strained valves suggesting mitral valves are capable of local serotonin synthesis and may be mechanically modulated (177). Myofibroblastic phenotype markers, matrix catabolic enzymes, cathepsins, matrix metalloproteases, and GAGS increased with increasing cyclic strain in cultured sheep MVs with serotonin present in the media of cyclically strained valves with concentration correlating to percent strain; inhibition of serotonin reduced these strain mediated protein expression patterns (178).
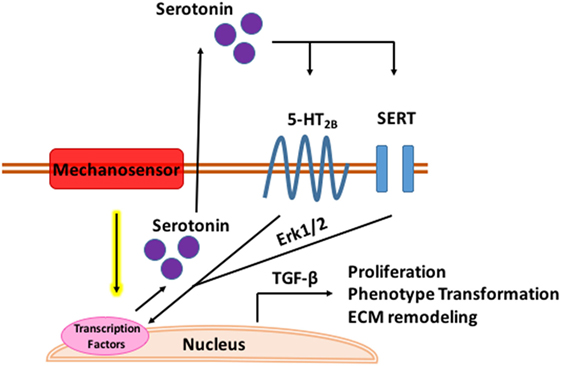
Figure 3. Mechanomodulation of mitral valve disease through serotonin. Tensile strain upregulates serotonin synthesis through a mechanosensory mechanism. Serotonin interacts with the serotonin type 2B receptor and serotonin transporter (SERT) in the mitral valve activating Erk1/2 through G-protein stimulation. Erk1/2 is phosphorylated in the nucleus where it induces TGF-β signaling and transcription of genes mediating myxomatous disease.
Integrated Mechanotransduction
It is likely individual methods of mechanotransduction work in concert through common signaling pathways. Multi faceted proteins such as small GTPases coupled with an integrated framework, such as the cytoskeleton, implicate a coordinated sensing and transduction network of shared, simultaneous components as illustrated in Figure 4.
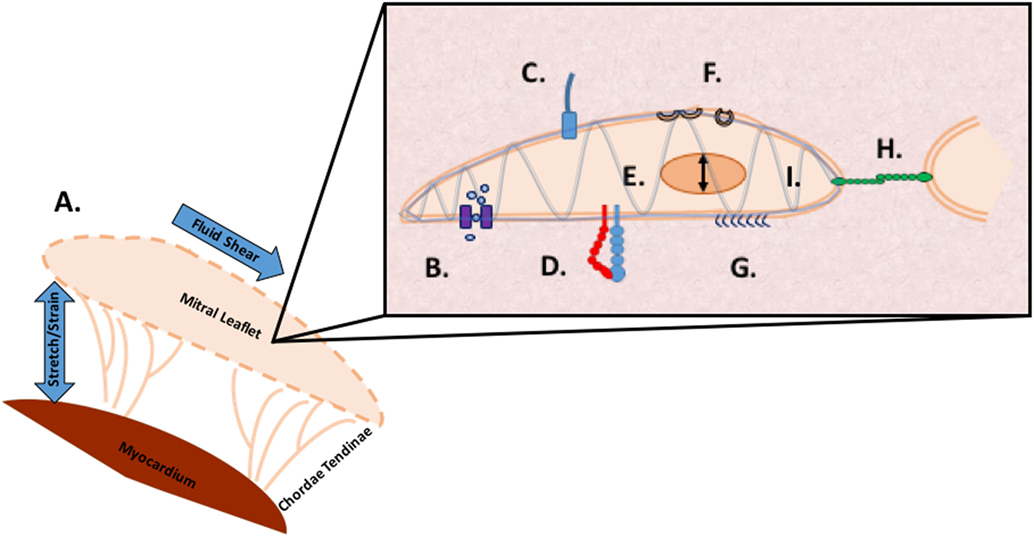
Figure 4. Methods of mechanosensing in the mitral valve. A. At a global level, the valve is subjected to flexure as the valve opens, shear as the blood flows through the valve, flexure as the valve closes, and tension as the valve seals shut to prevent regurgitation. At a microscopic level, mechanotransduction converts these extracellular forces into intracellular signaling through multiple cellular apparatuses. B. Mechano-sensitive ion channels convert mechanical force exerted on the cell membrane into electrical or biochemical signals. C. The axoneme of primary cilia convert extracellular cues into various signaling pathways as well as coupling transduction with voltage-gated channels D. Integrins are the main receptors connecting the cytoskeleton to the extracellular matrix (ECM) and transmit mechanical stress across the plasma membrane E. In nuclear deformation physical force is transmitted across the nuclear envelope to the nuclear interior where they modulate gene expression from physical deformation of genetic material F. Caveolae flatten into the plasma membrane when stimulated by cell-surface tension, relieving tension and physically sequestering proteins, growth hormones, and cytokines G. The glycocalyx transmits fluid shear stress to the cell through core proteins which connect to the actin cytoskeleton and cell membrane mediating cell signaling H. Cadherins are cell adhesion proteins that create zipper like structures at cell junctions to maintain stable intercellular adhesion and mechanical coupling between cells and the adherens junction to transform mechanical to chemical signals as well as interacting with integrins through actin filaments I. Directly or indirectly, the load bearing cytoskeleton is common to the various mechanosensing modalities. Often clustering at focal adhesions, the cytoskeleton rapidly transmits ECM stimulus into cellular response through actin filament reorganization.
Both RhoA and Rac GTPases mediate endothelial–mesenchymal transition during valvulogenesis (179, 180), while in adult VICs RhoA regulates actin cytoskeleton and stress fiber formation as Rac regulates cell–cell adhesion, actin polymerization, lamellae protrustion, and cytoskeletal polarity (181). Altering the actin network geometry by overexpressing Rac1 GTPase so precursor actin bundles are suppressed at free borders, changes adherens junction shape, and increases lamellae protrusions (182, 183). RhoA signaling couples cadherin based adhesion with actimyosin contractility (184). Rac1 and RhoA interact in a spatiotemporal manner with adherens junction proteins to coordinate opening and closing of endothelial junctions (185). FilGAP, a Rac GTPase-activating Protein, binds FlnA to control actin remodeling (186) and is present at focal adhesions but more directly present at cytoskeletal interfaces where FlnA and the β integrin cytoplasmic tail interact to form a binding pocket for opposing β strands (187, 188). FlnA is an actin binding protein widely expressed during valvulogenesis, which anchors transmembrane proteins to the cytoskeleton and mediates remodeling events in response to stimulus.
In both embryonic and adult VICs, a quiescent phenotype is maintained when they are cultured in unstressed collagen hydrogels; however, contractile expression, TGF-β, and matrix remodeling are upregulated in response to mechanical tension (55, 189). During development, this quiescent phenotype transition is governed by decreasing αSMA following decreased RhoA-GTPase expression (11, 190). Cyclic stretch of embryonic valve progenitor cells activates RhoA in acute response to the mechanical stimulus and is later switched to chronic Rac1 activation through FilGAP (191). RhoA mediates myofibroblastic activation during this acute signaling while chronic cyclic strain deactivates RhoA, enabling Rac1 to compact the matrix. Mutations in FlnA are responsible for X-linked myxomatous valve disease (192) by weakening FilGAP binding (193) and disrupting GTPase regulation (58) which alters cytoskeletal remodeling ability. Rac-1 knockdown in embryonic kidney cells abrogated PKD1-mediated signaling suggesting a critical role for small GTPases in PKD, providing insight into ciliary and voltage-gated signaling (194). In Bardet–Biedl syndrome, RhoA levels are upregulated but treatment of mutant cells with RhoA inhibitors restores cilia length and number as well as actin cytoskeleton integrity (195). In vascular SMC, 5-HT induced mitogenesis relies on Rho-mediated translocation of Erk1/2 (196) and induces Smad activation in bovine and human pulmonary artery SMCs via RhoA (197). 5-HT potentiates TGF-β3 expression in cushions which then induces contractile gene expression through RhoA (167).
The cytoskeleton provides an integrated framework for communication by physically connecting distant parts of the cell (145), rapidly transmitting mechanical information and modulating signal transduction through posttranslational modification, remodeling, and reorganization. Mechanical activation of Src 50 µm from the point of force application in vascular smooth muscle cells takes less than 300 ms through actin stress fibers, orders of magnitude faster than reaction-diffusion signaling cascades (145, 198). Disrupting actin filaments (199) as well as relieving stress fiber prestress (200) impairs rapid long distance mechanotransduction. Association with cadherins and integrins produces a critical interface through which actin filaments are exposed to forces from the ECM. Integrins and cadherins share similar mechanotransductive mechanisms in their interactions with the actin cytoskeleton, recruitment of common adhesion components, and extensive cross-talk (200, 201). Both integrins and cadherins stimulate Rho and Rac GTPases resulting in cytoskeleton remodeling in response to adhesion (201, 202).
The actin cytoskeleton provides structural stability to GC in ECs under shear stress (203). Depolymerizing actin weakens the anchoring strength of core proteins that support the GC such that the GC layer is ablated under shear stress; this is potentially due to altered mechanotransduction (203). Caveolae associate and align with stress fibers (204, 205) through FlnA actin binding domains. Knock down of FlnA increases the lateral movement of Cav1 and reduces stress fiber alignment of the caveolae (206). Inhibiting actin polymerization increases the abundance of caveolar rosettes and increases Cav1 (207, 208) clustering while increasing stress fiber formation decreases caveolar rosettes (209). Caveolae, specifically Cav1 interactions (210, 211), regulate RhoA-mediated actomyosin contractility (209). Cav1 and RhoA are localized to the same membrane invaginations (212), physically interacting to induce cytoskeletal reorganization in response to force (104). Like FlnA mutations, alterations to the ECM change cytoskeletal structure and function which can result in pathological signaling and remodeling. Erk activity specifically localizes to regions of matrix metallopeptidase 2 expression (213), an ECM degrading enzyme, which is significantly increased in clinical patients with floppy mitral valves and mitral valve prolapse (214). A variety of collagen mutations result in mitral valve prolapse, aortic root dilation, and a host of structural defects (215, 216).
The mitral valve exists in a complex environment where global mechanical deformation alters cell phenotype and ECM remodeling (217) in the microenvironment in a synergistic and reciprocating fashion. It is increasingly apparent that multiple mechanobiological regulatory modalities exist and are interconnected through shared components. Much like our five senses, multiple methods of mechanosensing coexist in the same cell, interacting with each other and the environment. In cells with a disrupted sense, mechanical stimulus may seem preferentially potent in one sense compared to a wild-type cell, causing pathological signaling and remodeling. The interconnected pathways and frameworks of mechanotransduction can be thought of as a network in search of homeostasis; superior treatments may seek to rebalance the network instead of focusing on a solitary gene or protein defect. Increasing our understanding of how cells interact with their environment through mechanosensing and mechanotransduction provides potential therapeutic targets in valve disease by altering the environment, cellular perception of the environment, or communication with the environment in a profound and regenerative manner.
Author Contributions
JB suggested the subject of the review, recommended resources, direction of the review, suggested types of figures to include, and provided extensive editing. LP is a Ph.D. candidate in JB’s group and based on the recommendations of JB did an extensive literature review, drafted the article, created figures, and charted the direction and subject matter contained in the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by funding from the National Institutes of Health (HL110328 and HL128745) and the National Science Foundation (DGE-1650441).
References
1. Go A, Mozaffarian D, Roger V, Benjamin E, Berry J, Borden W, et al. Heart disease and stroke statistics – 2013 update: a report from the American Heart Association. Circulation (2012) 127(1):e6–245. doi:10.1161/CIR.0b013e31828124ad
2. Chehab G, El-Rassi I, Chokor I, Hammoud C, Saliba Z. Epidemiology of mitral valve disease in pediatrics: a Lebanese study. J Med Liban (2011) 59(4):197–201.
4. Sacks MS. Biaxial mechanical evaluation of planar biological materials. J Elast (2000) 61:199–246. doi:10.1023/A:1010917028671
5. Wang J, Thampatty B. An introductory review of cell mechanobiology. Biomech Model Mechanobiol (2006) 5(1):1–16. doi:10.1007/s10237-005-0012-z
6. Wang N, Butler J, Ingber D. Mechanotransduction across the cell surface and through the cytoskeleton. Science (1993) 260(5111):1124–7. doi:10.1126/science.7684161
7. Tarkowski A, Wroblewska J. Development of blastomeres of mouse eggs isolated at the 4- and 8-cell stage. J Embryol Exp Morphol (1968) 18(1):155–80.
8. Hillman N, Sherman M, Graham C. The effect of spatial arrangement on cell determination during mouse development. J Embryol Exp Morphol (1972) 28(2):263–78.
9. Hove J, Köster R, Forouhar A, Acevedo-Bolton G, Fraser S, Gharib M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature (2003) 421(6919):172–7. doi:10.1038/nature01282
10. Beis D, Bartman T, Jin SW, Scott IC, D’Amico LA, Ober EA, et al. Genetic and cellular analyses of zebrafish atrioventricular cushion and valve development. Development (2005) 132(18):4193–204. doi:10.1242/dev.01970
11. Nakajima Y, Mironov V, Yamagishi T, Nakamura H, Markwald R. Expression of smooth muscle alpha-actin in mesenchymal cells during formation of avian endocardial cushion tissue: a role for transforming growth factor β3. Dev Dyn (1997) 209(3):296–309. doi:10.1002/(SICI)1097-0177(199707)209:3<296::AID-AJA5>3.0.CO;2-D
12. Butcher J, McQuinn T, Sedmera D, Turner D, Markwald R. Transitions in early embryonic atrioventricular valvular function correspond with changes in cushion biomechanics that are predictable by tissue composition. Circ Res (2007) 100(10):1503–11. doi:10.1161/CIRCRESAHA.107.148684
13. Butcher J, Norris R, Hoffman S, Mjaatvedt C, Markwald R. Periostin promotes atrioventricular mesenchyme matrix invasion and remodeling mediated by integrin signaling through Rho/PI 3-kinase. Dev Biol (2007) 302(1):256–66. doi:10.1016/j.ydbio.2006.09.048
14. Kapacee Z, Richardson S, Lu Y, Starborg T, Holmes D, Baar K, et al. Tension is required for fibripositor formation. Matrix Biol (2008) 27(4):371–5. doi:10.1016/j.matbio.2007.11.006
15. Vermot J, Forouhar A, Liebling M, Wu D, Plummer D, Gharib M, et al. Reversing blood flows act through klf2a to ensure normal valvulogenesis in the developing heart. PLoS Biol (2009) 7(11):e1000246. doi:10.1371/journal.pbio.1000246
16. Freund J, Vermot J. The wall-stress footprint of blood cells flowing in microvessels. Biophys J (2014) 106(3):752–62. doi:10.1016/j.bpj.2013.12.020
17. Kalogirou S, Malissovas N, Moro E, Argenton F, Stainier D, Beis D. Intracardiac flow dynamics regulate atrioventricular valve morphogenesis. Cardiovasc Res (2014) 104(1):49–60. doi:10.1093/cvr/cvu186
18. Bartman T, Walsh E, Wen K, McKane M, Ren J, Alexander J, et al. Early myocardial function affects endocardial cushion development in zebrafish. PLoS Biol (2004) 2(5):E129. doi:10.1371/journal.pbio.0020129
19. Clouthier D, Harris C, Harris R, Martin C, Puri M, Jones N. Requisite role for Nck adaptors in cardiovascular development, endothelial-to-mesenchymal transition, and directed cell migration. Mol Cell Biol (2015) 35(9):1573–87. doi:10.1128/MCB.00072-15
20. Johnson B, Bark D, Van Herck I, Garrity D, Dasi L. Altered mechanical state in the embryonic heart results in time-dependent decreases in cardiac function. Biomech Model Mechanobiol (2015) 14(6):1379–89. doi:10.1007/s10237-015-0681-1
21. Balachandran K, Alford P, Wylie-Sears J, Goss J, Grosberg A, Bischoff J, et al. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci U S A (2011) 108(50):19943–8. doi:10.1073/pnas.1106954108
22. Maleki S, Kjellqvist S, Paloschi V, Magné J, Branca R, Du L, et al. Mesenchymal state of intimal cells may explain higher propensity to ascending aortic aneurysm in bicuspid aortic valves. Sci Rep (2016) 6(1):35712. doi:10.1038/srep35712
23. Mui K, Chen C, Assoian R. The mechanical regulation of integrin-cadherin crosstalk organizes cells, signaling and forces. J Cell Sci (2016) 129(6):1093–100. doi:10.1242/jcs.183699
24. Case L, Baird M, Shtengel G, Campbell S, Hess H, Davidson M, et al. Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nat Cell Biol (2015) 17(7):880–92. doi:10.1038/ncb3180
25. Humphries J, Wang P, Streuli C, Geiger B, Humphries M, Ballestrem C. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J Cell Biol (2007) 179(5):1043–57. doi:10.1083/jcb.200703036
26. Sheetz M, Dai J. Modulation of membrane dynamics and cell motility by membrane tension. Trends Cell Biol (1996) 6(3):85–9. doi:10.1016/0962-8924(96)80993-7
27. Xu S, Liu A, Kim H, Gotlieb A. Cell density regulates in vitro activation of heart valve interstitial cells. Cardiovasc Pathol (2012) 21(2):65–73. doi:10.1016/j.carpath.2011.01.004
28. Hutcheson J, Chen J, Sewell-Loftin M, Ryzhova L, Fisher C, Su Y, et al. Cadherin-11 regulates cell-cell tension necessary for calcific nodule formation by valvular myofibroblasts. Arterioscler Thromb Vasc Biol (2012) 33(1):114–20. doi:10.1161/ATVBAHA.112.300278
29. Chen J, Ryzhova L, Sewell-Loftin M, Brown C, Huppert S, Baldwin H, et al. Notch1 mutation leads to valvular calcification through enhanced myofibroblast mechanotransduction. Arterioscler Thromb Vasc Biol (2015) 35(7):1597–605. doi:10.1161/ATVBAHA.114.305095
30. Lu C, Liu M, Culshaw G, Clinton M, Argyle D, Corcoran B. Gene network and canonical pathway analysis in canine myxomatous mitral valve disease: a microarray study. Vet J (2015) 204(1):23–31. doi:10.1016/j.tvjl.2015.02.021
31. Armstrong E, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res (2004) 95(5):459–70. doi:10.1161/01.RES.0000141146.95728.da
32. Rizzo S, Basso C, Lazzarini E, Celeghin R, Paolin A, Gerosa G, et al. TGF-beta1 pathway activation and adherens junction molecular pattern in nonsyndromic mitral valve prolapse. Cardiovasc Pathol (2015) 24(6):359–67. doi:10.1016/j.carpath.2015.07.009
33. Zhou J, Bowen C, Lu G, Knapp C III, Recknagel A, Norris R, et al. Cadherin-11 expression patterns in heart valves associate with key functions during embryonic cushion formation, valve maturation and calcification. Cells Tissues Organs (2013) 198(4):300–10. doi:10.1159/000356762
34. Sung D, Bowen C, Vaidya K, Zhou J, Chapurin N, Recknagel A, et al. Cadherin-11 overexpression induces extracellular matrix remodeling and calcification in mature aortic ValvesHighlights. Arterioscler Thromb Vasc Biol (2016) 36(8):1627–37. doi:10.1161/ATVBAHA.116.307812
36. Luo B, Carman C, Springer T. Structural basis of integrin regulation and signaling. Annu Rev Immunol (2007) 25(1):619–47. doi:10.1146/annurev.immunol.25.022106.141618
37. Van der Flier A, Sonnenberg A. Function and interactions of integrins. Cell Tissue Res (2001) 305(3):285–98. doi:10.1007/s004410100417
38. Calderwood DA, Yan B, de Pereda JM, Alvarez BG, Fujioka Y, Liddington RC, et al. The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem (2002) 277(24):21749–58. doi:10.1074/jbc.M111996200
39. Schwartz M. Integrin signaling revisited. Trends Cell Biol (2001) 11(12):466–70. doi:10.1016/S0962-8924(01)02152-3
40. Wipff P, Hinz B. Integrins and the activation of latent transforming growth factor β1 – an intimate relationship. Eur J Cell Biol (2008) 87(8–9):601–15. doi:10.1016/j.ejcb.2008.01.012
41. Plow E, Haas T, Zhang L, Loftus J, Smith J. Ligand binding to integrins. J Biol Chem (2000) 275(29):21785–8. doi:10.1074/jbc.R000003200
42. Midwood K, Schwarzbauer J. Elastic fibers: building bridges between cells and their matrix. Curr Biol (2002) 12(8):R279–81. doi:10.1016/S0960-9822(02)00800-X
43. Latif N, Sarathchandra P, Taylor P, Antoniw J, Yacoub M. Molecules mediating cell–ECM and cell–cell communication in human heart valves. Cell Biochem Biophys (2005) 43(2):275–88. doi:10.1385/CBB:43:2:275
44. Danen E, Sonnenberg A. Integrins in regulation of tissue development and function. J Pathol (2003) 200(4):471–80. doi:10.1002/path.1416
45. Wu Y, Jane Grande-Allen K, West J. Adhesive peptide sequences regulate valve interstitial cell adhesion, phenotype and extracellular matrix deposition. Cell Mol Bioeng (2016) 9(4):479–95. doi:10.1007/s12195-016-0451-x
46. Stephens E, Durst C, Swanson J, Grande-Allen K, Ingels N, Miller D. Functional coupling of valvular interstitial cells and collagen via α2β1 integrins in the mitral leaflet. Cell Mol Bioeng (2010) 3(4):428–37. doi:10.1007/s12195-010-0139-6
47. Gu X, Masters K. Regulation of valvular interstitial cell calcification by adhesive peptide sequences. J Biomed Mater Res A (2010) 93(4):1620–30. doi:10.1002/jbm.a.32660
48. McCartney-Francis N, Frazier-Jessen M, Wahl S. TGF-β: a balancing act. Int Rev Immunol (1998) 16(5–6):553–80. doi:10.3109/08830189809043009
49. Massagué J. TGF-β signal transduction. Annu Rev Biochem (1998) 67(1):753–91. doi:10.1146/annurev.biochem.67.1.753
50. Munger J, Sheppard D. Cross talk among TGF-signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol (2011) 3(11):a005017–005017. doi:10.1101/cshperspect.a005017
51. Hinz B, Celetta G, Tomasek J, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell (2001) 12(9):2730–41. doi:10.1091/mbc.12.9.2730
52. Klingberg F, Chow M, Koehler A, Boo S, Buscemi L, Quinn T, et al. Prestress in the extracellular matrix sensitizes latent TGF-β1 for activation. J Cell Biol (2014) 207(2):283–97. doi:10.1083/jcb.201402006
53. Tomasek J, Gabbiani G, Hinz B, Chaponnier C, Brown R. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol (2002) 3(5):349–63. doi:10.1038/nrm809
54. Ng C, Hinz B, Swartz MA. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J Cell Sci (2005) 118(20):4731–9. doi:10.1242/jcs.02605
55. Gould R, Chin K, Santisakultarm T, Dropkin A, Richards J, Schaffer C, et al. Cyclic strain anisotropy regulates valvular interstitial cell phenotype and tissue remodeling in three-dimensional culture. Acta Biomater (2012) 8(5):1710–9. doi:10.1016/j.actbio.2012.01.006
56. Sakai L, Keene D, Renard M, De Backer J. FBN1: the disease-causing gene for Marfan syndrome and other genetic disorders. Gene (2016) 591(1):279–91. doi:10.1016/j.gene.2016.07.033
57. Alberts J, van Tintelen J, Oomen T, Bergman J, Halley D, Jongbloed J, et al. Screening of TGFBR1, TGFBR2, and FLNA in familial mitral valve prolapse. Am J Med Gen A (2013) 164(1):113–9. doi:10.1002/ajmg.a.36211
58. Duval D, Lardeux A, Le Tourneau T, Norris R, Markwald R, Sauzeau V, et al. Valvular dystrophy associated filamin A mutations reveal a new role of its first repeats in small-GTPase regulation. Biochim Biophys Acta (2014) 1843(2):234–44. doi:10.1016/j.bbamcr.2013.10.022
59. Holm T, Habashi J, Doyle J, Bedja D, Chen Y, van Erp C, et al. Noncanonical TGF signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science (2011) 332(6027):358–61. doi:10.1126/science.1192149
60. Angel P, Narmoneva D, Sewell-Loftin M, Munjal C, Dupuis L, Landis B, et al. Proteomic alterations associated with biomechanical dysfunction are early processes in the Emilin1 deficient mouse model of aortic valve disease. Ann Biomed Eng (2017) 45(11):2548–62. doi:10.1007/s10439-017-1899-0
61. Clement C, Ajbro K, Koefoed K, Vestergaard M, Veland I, Henriques de Jesus M, et al. TGF-β signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep (2013) 3(6):1806–14. doi:10.1016/j.celrep.2013.05.020
62. Delling M, DeCaen P, Doerner J, Febvay S, Clapham D. Primary cilia are specialized calcium signalling organelles. Nature (2013) 504(7479):311–4. doi:10.1038/nature12833
63. Jin X, Muntean B, Aal-Aaboda M, Duan Q, Zhou J, Nauli S. L-type calcium channel modulates cystic kidney phenotype. Biochim Biophys Acta (2014) 1842(9):1518–26. doi:10.1016/j.bbadis.2014.06.001
64. Hoffmann A, Peterson M, Friedland-Little J, Anderson S, Moskowitz I. Sonic hedgehog is required in pulmonary endoderm for atrial septation. Development (2009) 136(10):1761–70. doi:10.1242/dev.034157
65. Hoffmann A, Yang X, Burnicka-Turek O, Bosman J, Ren X, Steimle J, et al. Foxf genes integrate Tbx5 and Hedgehog pathways in the second heart field for cardiac septation. PLoS Genet (2014) 10(10):e1004604. doi:10.1371/journal.pgen.1004604
66. Li Y, Klena N, Gabriel G, Liu X, Kim A, Lemke K, et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature (2015) 521(7553):520–4. doi:10.1038/nature14269
67. Toomer K, Fulmer D, Guo L, Drohan A, Peterson N, Swanson P, et al. A role for primary cilia in aortic valve development and disease. Dev Dyn (2017) 246(8):625–34. doi:10.1002/dvdy.24524
68. Sánchez-Duffhues G, de Vinuesa A, Lindeman J, Mulder-Stapel A, DeRuiter M, Van Munsteren C, et al. SLUG is expressed in endothelial cells lacking primary cilia to promote cellular calcification. Arterioscler Thromb Vasc Biol (2015) 35(3):616–27. doi:10.1161/ATVBAHA.115.305268
69. Dinsmore C, Reiter J. Endothelial primary cilia inhibit atherosclerosis. EMBO Rep (2016) 17(2):156–66. doi:10.15252/embr.201541019
70. Willaredt M, Gorgas K, Gardner H, Tucker K. Multiple essential roles for primary cilia in heart development. Cilia (2012) 1(1):23. doi:10.1186/2046-2530-1-23
71. Kathem SH, Mohieldin AM, Nauli SM. The roles of primary cilia in polycystic kidney disease. AIMS Mol Sci (2013) 1(1):27–46. doi:10.3934/molsci.2013.1.27
72. Sessa A, Righetti M, Battini G. Autosomal recessive and dominant polycystic kidney diseases. Minerva Urol Nefrol (2004) 56(4):329–38.
73. Lumiaho A, Ikäheimo R, Miettinen R, Niemitukia L, Laitinen T, Rantala A, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis (2001) 38(6):1208–16. doi:10.1053/ajkd.2001.29216
74. Tory K, Rousset-Rouvière C, Gubler M, Morinière V, Pawtowski A, Becker C, et al. Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int (2009) 75(8):839–47. doi:10.1038/ki.2008.662
75. Hills C, Kochilas L, Schimmenti L, Moller J. Ellis–van Creveld syndrome and congenital heart defects: presentation of an additional 32 cases. Pediatr Cardiol (2011) 32(7):977–82. doi:10.1007/s00246-011-0006-9
76. O’Connor M, Rider N, Thomas Collins R, Hanna B, Holmes Morton D, Strauss K. Contemporary management of congenital malformations of the heart in infants with Ellis–van Creveld syndrome: a report of nine cases. Cardiol Young (2010) 21(02):145–52. doi:10.1017/S1047951110001587
77. Thiam M, Gning S, Faye M, Fall P, Mbaye A, Charpentier P. Kartagener’s syndrome: a case report. Dakar Med (2002) 47(1):100–2.
78. Kim S, Sir J, Lee B, Kwak C, Kim S, Cho W, et al. Severe mitral regurgitation in a young female with pansinusitis and bronchiectasis. Respir Med CME (2008) 1(2):185–7. doi:10.1016/j.rmedc.2008.04.007
79. Purkait R, Roy B, Samanta T, Mallick A, Sinhamahapatra T. Rheumatic valvular insufficiency in Bardet-Biedl syndrome: a case report. J Indian Med Assoc (2012) 110(9):651–2.
80. Baumgarten CM. Origin of mechanotransduction: stretch-activated ion channels. Madame Curie Bioscience Database. Austin, TX: Landes Bioscience (2000).
81. Kung C. A possible unifying principle for mechanosensation. Nature (2005) 436(7051):647–54. doi:10.1038/nature03896
82. Barakat A, Lieu D, Gojova A. Secrets of the code: do vascular endothelial cells use ion channels to decipher complex flow signals? Biomaterials (2006) 27(5):671–8. doi:10.1016/j.biomaterials.2005.07.036
83. Ohno M, Cooke J, Dzau V, Gibbons G. Fluid shear stress induces endothelial transforming growth factor beta-1 transcription and production. Modulation by potassium channel blockade. J Clin Invest (1995) 95(3):1363–9. doi:10.1172/JCI117787
84. Malek A, Izumo S. Molecular aspects of signal transduction of shear stress in the endothelial cell. J Hypertens (1994) 12(9):989–1000. doi:10.1097/00004872-199409000-00001
85. Goetz J, Steed E, Ferreira R, Roth S, Ramspacher C, Boselli F, et al. Endothelial cilia mediate low flow sensing during zebrafish vascular development. Cell Rep (2014) 6(5):799–808. doi:10.1016/j.celrep.2014.01.032
86. Warren D, Tajsic T, Porter L, Minaisah R, Cobb A, Jacob A, et al. Nesprin-2-dependent ERK1/2 compartmentalisation regulates the DNA damage response in vascular smooth muscle cell ageing. Cell Death Differ (2015) 22(9):1540–50. doi:10.1038/cdd.2015.12
87. Coste B, Mathur J, Schmidt M, Earley T, Ranade S, Petrus M, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science (2010) 330(6000):55–60. doi:10.1126/science.1193270
88. Ranade S, Qiu Z, Woo S, Hur S, Murthy S, Cahalan S, et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci U S A (2014) 111(28):10347–52. doi:10.1073/pnas.1409233111
89. Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow M, et al. Piezo1 integration of vascular architecture with physiological force. Nature (2014) 515(7526):279–82. doi:10.1038/nature13701
90. Peyronnet R, Nerbonne J, Kohl P. Cardiac mechano-gated ion channels and arrhythmias. Circ Res (2016) 118(2):311–29. doi:10.1161/CIRCRESAHA.115.305043
91. Köhler R, Distler A, Hoyer J. Increased mechanosensitive currents in aortic endothelial cells from genetically hypertensive rats. J Hypertens (1999) 17(3):365–71. doi:10.1097/00004872-199917030-00009
92. Grieben M, Pike A, Shintre C, Venturi E, El-Ajouz S, Tessitore A, et al. Structure of the polycystic kidney disease TRP channel polycystin-2 (PC2). Nat Struct Mol Biol (2016) 24(2):114–22. doi:10.1038/nsmb.3343
93. Pennekamp P, Karcher C, Fischer A, Schweickert A, Skryabin B, Horst J, et al. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr Biol (2002) 12(11):938–43. doi:10.1016/S0960-9822(02)00869-2
94. AbouAlaiwi W, Takahashi M, Mell B, Jones T, Ratnam S, Kolb R, et al. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res (2009) 104(7):860–9. doi:10.1161/CIRCRESAHA.108.192765
95. DeCaen P, Liu X, Abiria S, Clapham D. Atypical calcium regulation of the PKD2-L1 polycystin ion channel. Elife (2016) 5:e13413. doi:10.7554/eLife.13413
96. Priori S. Task force on sudden cardiac death of the European Society of Cardiology. Eur Heart J (2001) 22(16):1374–450. doi:10.1053/euhj.2001.2824
97. Missov E, Cogswell R. Sudden cardiac death, mitral valve prolapse, and long QT syndrome. Am J Med (2015) 128:e37–8. doi:10.1016/j.amjmed.2015.05.030
98. Haas J, Frese K, Peil B, Kloos W, Keller A, Nietsch R, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J (2014) 36(18):1123–35. doi:10.1093/eurheartj/ehu301
99. Heckel E, Boselli F, Roth S, Krudewig A, Belting HG, Charvin G, et al. Oscillatory flow modulates mechanosensitive klf2a expression through trpv4 and trpp2 during heart valve development. Curr Biol (2015) 25:1354–61. doi:10.1016/j.cub.2015.03.038
100. Renz M, Otten C, Faurobert E, Rudolph F, Zhu Y, Boulday G, et al. Regulation of β1 integrin-Klf2-mediated angiogenesis by CCM proteins. Dev Cell (2015) 32(2):181–90. doi:10.1016/j.devcel.2014.12.016
101. Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol (2000) 1:31–9. doi:10.1038/35036052
102. Patel H, Murray F, Insel P. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu Rev Pharmacol Toxicol (2008) 48(1):359–91. doi:10.1146/annurev.pharmtox.48.121506.124841
103. Boyd N, Park H, Yi H, Boo Y, Sorescu G, Sykes M, et al. Chronic shear induces caveolae formation and alters ERK and Akt responses in endothelial cells. Am J Physiol Heart Circ Physiol (2003) 285(3):H1113–22. doi:10.1152/ajpheart.00302.2003
104. Kawamura S, Miyamoto S, Brown J. Initiation and transduction of stretch-induced RhoA and Rac1 activation through caveolae. J Biol Chem (2003) 278(33):31111–7. doi:10.1074/jbc.M300725200
105. Kawabe J, Okumura S, Lee MC, Sadoshima J, Ishikawa Y. Translocation of caveolin regulates stretch-induced ERK activity in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol (2004) 286(5):H1845–52. doi:10.1152/ajpheart.00593.2003
106. Sawada Y, Tamada M, Dubin-Thaler B, Cherniavskaya O, Sakai R, Tanaka S, et al. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell (2006) 127(5):1015–26. doi:10.1016/j.cell.2006.09.044
107. Furuchi T, Anderson R. Cholesterol depletion of caveolae causes hyperactivation of extracellular signal-related kinase (ERK). J Biol Chem (1998) 273(33):21099–104. doi:10.1074/jbc.273.33.21099
108. Isshiki M, Anderson R. Function of caveolae in Ca2+ entry and Ca2+-dependent signal transduction. Traffic (2003) 4(11):717–23. doi:10.1034/j.1600-0854.2003.00130.x
109. Del Pozo M, Kiosses W, Alderson N, Meller N, Hahn K, Schwartz M. Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat Cell Biol (2002) 4(3):232–9. doi:10.1038/ncb759
110. Del Pozo M, Price LS, Alderson NB, Ren XD, Schwartz MA. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J (2000) 19(9):2008–14. doi:10.1093/emboj/19.9.2008
111. Del Galdo F, Lisanti M, Jimenez S. Caveolin-1, transforming growth factor-β receptor internalization, and the pathogenesis of systemic sclerosis. Curr Opin Rheumatol (2008) 20(6):713–9. doi:10.1097/BOR.0b013e3283103d27
112. Liu Y, Dillon A, Tillson M, Makarewich C, Nguyen V, Dell’Italia L, et al. Volume overload induces differential spatiotemporal regulation of myocardial soluble guanylyl cyclase in eccentric hypertrophy and heart failure. J Mol Cell Cardiol (2013) 60:72–83. doi:10.1016/j.yjmcc.2013.03.019
113. Rajamannan N, Springett M, Pederson L, Carmichael S. Localization of caveolin 1 in aortic valve endothelial cells using antigen retrieval. J Histochem Cytochem (2002) 50(5):617–27. doi:10.1177/002215540205000503
114. Kim Y, Nijst P, Kiefer K, Tang W. Endothelial glycocalyx as biomarker for cardiovascular diseases: mechanistic and clinical implications. Curr Heart Fail Rep (2017) 14(2):117–26. doi:10.1007/s11897-017-0320-5
115. Bartosch A, Mathews R, Tarbell J. Endothelial glycocalyx-mediated nitric oxide production in response to selective AFM pulling. Biophys J (2017) 113(1):101–8. doi:10.1016/j.bpj.2017.05.033
116. Siegel G, Malmsten M, Ermilov E. Anionic biopolyelectrolytes of the syndecan/perlecan superfamily: physicochemical properties and medical significance. Adv Colloid Interface Sci (2014) 205:275–318. doi:10.1016/j.cis.2014.01.009
117. Korte S, Wiesinger A, Straeter A, Peters W, Oberleithner H, Kusche-Vihrog K. Firewall function of the endothelial glycocalyx in the regulation of sodium homeostasis. Pflugers Arch (2011) 463(2):269–78. doi:10.1007/s00424-011-1038-y
118. Weinbaum S, Zhang X, Han Y, Vink H, Cowin S. Mechanotransduction and flow across the endothelial glycocalyx. Proc Natl Acad Sci U S A (2003) 100(13):7988–95. doi:10.1073/pnas.1332808100
119. Florian J, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res (2003) 93(10):e136–42. doi:10.1161/01.RES.0000101744.47866.D5
120. Radeva MY, Waschke J. Mind the gap: mechanisms regulating the endothelial barrier. Acta Physiol (2017). doi:10.1111/apha.12860
121. Dragovich M, Chester D, Zhang X. Mechanotransduction of the endothelial glycocalyx mediates nitric oxide production through activation of TRP channels. Biophys J (2016) 110(3):23a. doi:10.1152/ajpcell.00288.2015
122. Carey D. Syndecans: multifunctional cell-surface co-receptors. Biochem J (1997) 327(1):1–16. doi:10.1042/bj3270001
123. Tumova S, Woods A, Couchman J. Heparan sulfate proteoglycans on the cell surface: versatile coordinators of cellular functions. Int J Biochem Cell Biol (2000) 32(3):269–88. doi:10.1016/S1357-2725(99)00116-8
124. Becker B, Jacob M, Leipert S, Salmon A, Chappell D. Degradation of the endothelial glycocalyx in clinical settings: searching for the sheddases. Br J Clin Pharmacol (2015) 80(3):389–402. doi:10.1111/bcp.12629
125. Sarphie T. A cytochemical study of the surface properties of aortic and mitral valve endothelium from hypercholesterolemic rabbits. Exp Mol Pathol (1986) 44(3):281–96. doi:10.1016/0014-4800(86)90042-0
126. Chignalia A, Yetimakman F, Christiaans S, Unal S, Bayrakci B, Wagener B, et al. The glycocalyx and trauma. Shock (2016) 45(4):338–48. doi:10.1097/SHK.0000000000000513
127. Johansson P, Stensballe J, Rasmussen L, Ostrowski S. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann Surg (2011) 254(2):194–200. doi:10.1097/SLA.0b013e318226113d
128. Bielecka-Dabrowa A, Gluba-Brzózka A, Michalska-Kasiczak M, Misztal M, Rysz J, Banach M. The multi-biomarker approach for heart failure in patients with hypertension. Int J Mol Sci (2015) 16(5):10715–33. doi:10.3390/ijms160510715
129. Tromp J, van der Pol A, Klip I, de Boer R, Jaarsma T, van Gilst W, et al. Fibrosis marker syndecan-1 and outcome in patients with heart failure with reduced and preserved ejection fraction. Circ Heart Fail (2014) 7(3):457–62. doi:10.1161/CIRCHEARTFAILURE.113.000846
130. Meyer S, van der Meer P, van Deursen V, Jaarsma T, van Veldhuisen D, van der Wal M, et al. Neurohormonal and clinical sex differences in heart failure. Eur Heart J (2013) 34(32):2538–47. doi:10.1093/eurheartj/eht152
131. Bielecka-Dabrowa A, Michalska-Kasiczak M, Gluba A, Ahmed A, Gerdts E, von Haehling S, et al. Biomarkers and echocardiographic predictors of myocardial dysfunction in patients with hypertension. Sci Rep (2015) 5(1):8916. doi:10.1038/srep08916
132. Ostrowski S, Pedersen S, Jensen J, Mogelvang R, Johansson P. Acute myocardial infarction is associated with endothelial glycocalyx and cell damage and a parallel increase in circulating catecholamines. Crit Care (2013) 17(1):R32. doi:10.1186/cc12532
133. Vanhoutte D, Schellings M, Gotte M, Swinnen M, Herias V, Wild M, et al. Increased expression of syndecan-1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation (2007) 115(4):475–82. doi:10.1161/CIRCULATIONAHA.106.644609
134. Schellings M, Vanhoutte D, van Almen G, Swinnen M, Leenders J, Kubben N, et al. Syndecan-1 amplifies angiotensin II-induced cardiac fibrosis. Hypertension (2010) 55(2):249–56. doi:10.1161/HYPERTENSIONAHA.109.137885
135. Constantinescu A, Vink H, Spaan JA. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler Thromb Vasc Biol (2003) 23(9):1541–7. doi:10.1161/01.ATV.0000085630.24353.3D
136. Ingber D. The riddle of morphogenesis: a question of solution chemistry or molecular cell engineering? Cell (1993) 75(7):1249–52. doi:10.1016/0092-8674(93)90612-T
137. Mattout-Drubezki A, Gruenbaum Y. Dynamic interactions of nuclear lamina proteins with chromatin and transcriptional machinery. Cell Mol Life Sci (2003) 60(10):2053–63. doi:10.1007/s00018-003-3038-3
138. Booth-Gauthier E, Alcoser T, Yang G, Dahl K. Force-induced changes in subnuclear movement and rheology. Biophys J (2012) 103(12):2423–31. doi:10.1016/j.bpj.2012.10.039
139. Fey E, Wan KM, Penman S. Epithelial cytoskeletal framework and nuclear matrix-intermediate filament scaffold: three-dimensional organization and protein composition. J Cell Biol (1984) 98(6):1973–84. doi:10.1083/jcb.98.6.1973
140. Maniotis A, Chen C, Ingber D. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci U S A (1997) 94(3):849–54. doi:10.1073/pnas.94.3.849
141. Lee C, Carruthers C, Ayoub S, Gorman R, Gorman J, Sacks M. Quantification and simulation of layer-specific mitral valve interstitial cells deformation under physiological loading. J Theor Biol (2015) 373:26–39. doi:10.1016/j.jtbi.2015.03.004
142. Lee C, Amini R, Gorman R, Gorman J, Sacks M. An inverse modeling approach for stress estimation in mitral valve anterior leaflet valvuloplasty for in-vivo valvular biomaterial assessment. J Biomech (2014) 47(9):2055–63. doi:10.1016/j.jbiomech.2013.10.058
143. Muchir A, Wu W, Choi J, Iwata S, Morrow J, Homma S, et al. Abnormal p38 mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum Mol Genet (2012) 21(19):4325–33. doi:10.1093/hmg/dds265
144. Emerson L, Holt M, Wheeler M, Wehnert M, Parsons M, Ellis J. Defects in cell spreading and ERK1/2 activation in fibroblasts with lamin A/C mutations. Biochim Biophys Acta (2009) 1792(8):810–21. doi:10.1016/j.bbadis.2009.05.007
145. Wang N, Tytell J, Ingber D. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol (2009) 10(1):75–82. doi:10.1038/nrm2594
146. Small E, Thatcher J, Sutherland L, Kinoshita H, Gerard R, Richardson J, et al. Myocardin-related transcription factor-A controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res (2010) 107(2):294–304. doi:10.1161/CIRCRESAHA.110.223172
147. Olson E, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol (2010) 11(5):353–65. doi:10.1038/nrm2890
148. Chang W, Worman H, Gundersen G. Accessorizing and anchoring the LINC complex for multifunctionality. J Cell Biol (2015) 208(1):11–22. doi:10.1083/jcb.201409047
149. Arsenovic P, Ramachandran I, Bathula K, Zhu R, Narang J, Noll N, et al. Nesprin-2G, a component of the nuclear LINC complex, is subject to myosin-dependent tension. Biophys J (2016) 110(1):34–43. doi:10.1016/j.bpj.2015.11.014
150. Zhang Q, Minaisah R, Ferraro E, Li C, Porter L, Zhou C, et al. N-terminal nesprin-2 variants regulate β-catenin signalling. Exp Cell Res (2016) 345(2):168–79. doi:10.1016/j.yexcr.2016.06.008
151. Chancellor T, Lee J, Thodeti C, Lele T. Actomyosin tension exerted on the nucleus through nesprin-1 connections influences endothelial cell adhesion, migration, and cyclic strain-induced reorientation. Biophys J (2010) 99(1):115–23. doi:10.1016/j.bpj.2010.04.011
152. Isermann P, Lammerding J. Nuclear mechanics and mechanotransduction in health and disease. Curr Biol (2013) 23(24):R1113–21. doi:10.1016/j.cub.2013.11.009
153. Connolly H, Crary J, McGoon M, Hensrud D, Edwards B, Edwards W, et al. Valvular heart disease associated with fenfluramine–phentermine. N Eng J Med (1997) 337(9):581–8. doi:10.1056/NEJM199708283370901
154. Redfield M. Valve disease associated with ergot alkaloid use: echocardiographic and pathologic correlations. Ann Intern Med (1992) 117(1):50. doi:10.7326/0003-4819-117-1-50
155. Zanettini R, Antonini A, Gatto G, Gentile R, Tesei S, Pezzoli G. Valvular heart disease and the use of dopamine agonists for Parkinson’s disease. N Engl J Med (2007) 356:39–46. doi:10.1056/NEJMoa054830
156. Pinero A, Marcos-Alberca P, Fortes J. Cabergoline-related severe restrictive mitral regurgitation. N Engl J Med (2005) 353(18):1976–7. doi:10.1056/NEJM200511033531822
157. Serratrice J, Disdier P, Habib G, Viallet F, Weiller P. Fibrotic valvular heart disease subsequent to bromocriptine treatment. Cardiol Rev (2002) 10(6):334–6. doi:10.1097/00045415-200211000-00005
158. Fitzgerald L, Burn T, Brown B, Patterson J, Corjay M, Valentine P, et al. Possible role of valvular serotonin 5-HT2B receptors in the cardiopathy associated with fenfluramine. Mol Pharmacol (2000) 57(1):75–81.
159. Rothman R, Baumann M, Savage J, Rauser L, McBride A, Hufeisen S, et al. Evidence for possible involvement of 5-HT2B receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation (2000) 102(23):2836–41. doi:10.1161/01.CIR.102.23.2836
160. Oyama M, Chittur S. Genomic expression patterns of mitral valve tissues from dogs with degenerative mitral valve disease. Am J Vet Res (2006) 67(8):1307–18. doi:10.2460/ajvr.67.8.1307
161. Disatian S, Orton EC. Autocrine serotonin and transforming growth factor beta 1 signaling mediates spontaneous myxomatous mitral valve disease. J Heart Valve Dis (2009) 19(1):71–8.
162. Nebigil C, Choi D, Dierich A, Hickel P, Le Meur M, Messaddeq N, et al. Serotonin 2B receptor is required for heart development. Proc Natl Acad Sci U S A (2000) 97(17):9508–13. doi:10.1073/pnas.97.17.9508
163. Mekontso-Dessap A, Brouri F, Pascal O, Lechat P, Hanoun N, Lanfumey L, et al. Deficiency of the 5-hydroxytryptamine transporter gene leads to cardiac fibrosis and valvulopathy in mice. Circulation (2005) 113(1):81–9. doi:10.1161/CIRCULATIONAHA.105.554667
164. Connolly JM, Bakay MA, Fulmer JT, Gorman RC, Gorman JH III, Oyama MA, et al. Fenfluramine disrupts the mitral valve interstitial cell response to serotonin. Am J Pathol (2017) 175:988–97. doi:10.2353/ajpath.2009.081101
165. Launay J, Birraux G, Bondoux D, Callebert J, Choi D, Loric S, et al. Ras involvement in signal transduction by the serotonin 5-HT2B receptor. J Biol Chem (1996) 271(6):3141–7. doi:10.1074/jbc.271.6.3141
166. Nebigil C, Launay J, Hickel P, Tournois C, Maroteaux L. 5-Hydroxytryptamine 2B receptor regulates cell-cycle progression: cross-talk with tyrosine kinase pathways. Proc Natl Acad Sci U S A (2000) 97(6):2591–6. doi:10.1073/pnas.050282397
167. Buskohl P, Sun M, Thompson R, Butcher J. Serotonin potentiates transforming growth factor-beta3 induced biomechanical remodeling in avian embryonic atrioventricular valves. PLoS One (2012) 7(8):e42527. doi:10.1371/journal.pone.0042527
168. Cremer S, Moesgaard S, Rasmussen C, Zois N, Falk T, Reimann M, et al. Alpha-smooth muscle actin and serotonin receptors 2A and 2B in dogs with myxomatous mitral valve disease. Res Vet Sci (2015) 100:197–206. doi:10.1016/j.rvsc.2015.03.020
169. Yabanoglu S, Akkiki M, Seguelas M, Mialet-Perez J, Parini A, Pizzinat N. Platelet derived serotonin drives the activation of rat cardiac fibroblasts by 5-HT2A receptors. J Mol Cell Cardiol (2009) 46(4):518–25. doi:10.1016/j.yjmcc.2008.12.019
170. Jian B, Xu J, Connolly J, Savani R, Narula N, Liang B, et al. Serotonin mechanisms in heart valve disease I: serotonin-induced up-regulation of transforming growth factor-β1 via G-protein signal transduction in aortic valve interstitial cells. Am J Pathol (2002) 161(6):2111–21. doi:10.1016/S0002-9440(10)64489-6
171. Pavone L, Spina A, Rea S, Santoro D, Mastellone V, Lombardi P, et al. Serotonin transporter gene deficiency is associated with sudden death of newborn mice through activation of TGF-β1 signalling. J Mol Cell Cardiol (2009) 47(5):691–7. doi:10.1016/j.yjmcc.2009.07.021
172. Capulli A, MacQueen L, O’Connor B, Dauth S, Parker K. Acute pergolide exposure stiffens engineered valve interstitial cell tissues and reduces contractility in vitro. Cardiovasc Pathol (2016) 25(4):316–24. doi:10.1016/j.carpath.2016.04.004
173. Liang Y, Lai L, Wang B, Juang S, Chang C, Leu J, et al. Mechanical stress enhances serotonin 2B receptor modulating brain natriuretic peptide through nuclear factor-κB in cardiomyocytes. Cardiovasc Res (2006) 72(2):303–12. doi:10.1016/j.cardiores.2006.08.003
174. Brattelid T, Qvigstad E, Birkeland J, Swift F, Bekkevold S, Krobert K, et al. Serotonin responsiveness through 5-HT2A and 5-HT4 receptors is differentially regulated in hypertrophic and failing rat cardiac ventricle. J Mol Cell Cardiol (2006) 40(6):944–5. doi:10.1016/j.yjmcc.2006.03.081
175. Balachandran K, Bakay M, Connolly J, Zhang X, Yoganathan A, Levy R. Aortic valve cyclic stretch causes increased remodeling activity andenhanced serotonin receptor responsiveness. Ann Thorac Surg (2011) 92(1):147–53. doi:10.1016/j.athoracsur.2011.03.084
176. Balachandran K, Hussain S, Yap C, Padala M, Chester A, Yoganathan A. Elevated cyclic stretch and serotonin result in altered aortic valve remodeling via a mechanosensitive 5-HT2A receptor-dependent pathway. Cardiovasc Pathol (2012) 21(3):206–13. doi:10.1016/j.carpath.2011.07.005
177. Lacerda C, MacLea H, Kisiday J, Orton E. Static and cyclic tensile strain induce myxomatous effector proteins and serotonin in canine mitral valves. J Vet Cardiol (2012) 14(1):223–30. doi:10.1016/j.jvc.2011.12.002
178. Lacerda C, Kisiday J, Johnson B, Orton E. Local serotonin mediates cyclic strain-induced phenotype transformation, matrix degradation, and glycosaminoglycan synthesis in cultured sheep mitral valves. Am J Physiol Heart Circ Physiol (2012) 302(10):H1983–90. doi:10.1152/ajpheart.00987.2011
179. Knipe R, Tager A, Liao J. The rho kinases: critical mediators of multiple profibrotic processes and rational targets for new therapies for pulmonary fibrosis. Pharmacol Rev (2014) 67(1):103–17. doi:10.1124/pr.114.009381
180. Hartmann S, Ridley A, Lutz S. The function of Rho-associated kinases ROCK1 and ROCK2 in the pathogenesis of cardiovascular disease. Front Pharmacol (2015) 6:276. doi:10.3389/fphar.2015.00276
181. Price S, Norwood C, Williams J, McElderry H, Merryman W. Radiofrequency ablation directionally alters geometry and biomechanical compliance of mitral valve leaflets: refinement of a novel percutaneous treatment strategy. Cardiovasc Eng Technol (2010) 1(3):194–201. doi:10.1007/s13239-010-0018-2
182. Waterman-Storer C, Worthylake R, Liu B, Burridge K, Salmon E. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol (1999) 1(1):45–50. doi:10.1038/9018
183. Brevier J, Montero D, Svitkina T, Riveline D. The asymmetric self-assembly mechanism of adherens junctions: a cellular push–pull unit. Phys Biol (2008) 5(1):016005. doi:10.1088/1478-3975/5/1/016005
184. Priya R, Gomez G, Budnar S, Acharya B, Czirok A, Yap A, et al. Bistable front dynamics in a contractile medium: travelling wave fronts and cortical advection define stable zones of RhoA signaling at epithelial adherens junctions. PLoS Comput Biol (2017) 13(3):e1005411. doi:10.1371/journal.pcbi.1005411
185. Braga V, Del Maschio A, Machesky L, Dejana E. Regulation of cadherin function by Rho and Rac: modulation by junction maturation and cellular context. Mol Biol Cell (1999) 10(1):9–22. doi:10.1091/mbc.10.1.9
186. Ohta Y, Hartwig J, Stossel T. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat Cell Biol (2006) 8(8):803–14. doi:10.1038/ncb1437
187. Nakamura F, Pudas R, Heikkinen O, Permi P, Kilpeläinen I, Munday AD, et al. The structure of the GPIb-filamin A complex. Blood (2006) 107(5):1925–32. doi:10.1182/blood-2005-10-3964
188. Kiema T, Lad Y, Jiang P, Oxley C, Baldassarre M, Wegener K, et al. The molecular basis of filamin binding to integrins and competition with Talin. Mol Cell (2006) 21(3):337–47. doi:10.1016/j.molcel.2006.01.011
189. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J Cell Biol (2007) 179(6):1311–23. doi:10.1083/jcb.200704042
190. Townsend T, Wrana J, Davis G, Barnett J. Transforming growth factor-β-stimulated endocardial cell transformation is dependent on Par6c regulation of RhoA. J Biol Chem (2008) 283(20):13834–41. doi:10.1074/jbc.M710607200
191. Gould R, Yalcin H, MacKay J, Sauls K, Norris R, Kumar S, et al. Cyclic mechanical loading is essential for Rac1-mediated elongation and remodeling of the embryonic mitral valve. Curr Biol (2016) 26(1):27–37. doi:10.1016/j.cub.2015.11.033
192. Kyndt F, Gueffet J, Probst V, Jaafar P, Legendre A, Le Bouffant F, et al. Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation (2006) 115(1):40–9. doi:10.1161/CIRCULATIONAHA.106.622621
193. Nakamura F, Heikkinen O, Pentikäinen O, Osborn T, Kasza K, Weitz D, et al. Molecular basis of filamin A-FilGAP interaction and its impairment in congenital disorders associated with filamin A mutations. PLoS One (2009) 4(3):4928. doi:10.1371/journal.pone.0004928
194. Arnould T, Kim E, Tsiokas L, Jochimsen F, Grüning W, Chang J, et al. The polycystic kidney disease 1 gene product mediates protein kinase C α-dependent and c-Jun N-terminal kinase-dependent activation of the transcription factor AP-1. J Biol Chem (1998) 273(11):6013–8. doi:10.1074/jbc.273.11.6013
195. Hernandez V, Pravincumar P, Diaz-Font A, May-Simera H, Jenkins D, Knight M, et al. Bardet-Biedl syndrome proteins control cilia length through regulation of actin polymerisation. Cilia (2012) 1(Suppl 1):88. doi:10.1186/2046-2530-1-S1-P88
196. Liu Y, Suzuki YJ, Day RM, Fanburg BL. Rho kinase-induced nuclear translocation of Erk1/Erk2 in smooth muscle cell mitogenesis caused by serotonin. Circ Res (2004) 95(6):579–86. doi:10.1161/01.RES.0000141428.53262.a4
197. Liu Y, Ren W, Warburton R, Toksoz D, Fanburg B. Serotonin induces Rho/ROCK-dependent activation of Smads 1/5/8 in pulmonary artery smooth muscle cells. FASEB J (2009) 23(7):2299–306. doi:10.1096/fj.08-127910
198. Na S, Collin O, Chowdhury F, Tay B, Ouyang M, Wang Y, et al. Rapid signal transduction in living cells is a unique feature of mechanotransduction. Proc Natl Acad Sci U S A (2008) 105(18):6626–31. doi:10.1073/pnas.0711704105
199. Watanabe T, Sato K, Kaibuchi K. Cadherin-mediated intercellular adhesion and signaling cascades involving small GTPases. Cold Spring Harb Perspect Biol (2009) 1(3):a003020. doi:10.1101/cshperspect.a003020
200. Burridge K. Focal adhesions: transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu Rev Cell Dev Biol (1988) 4(1):487–525. doi:10.1146/annurev.cb.04.110188.002415
201. Parsons J, Horwitz A, Schwartz M. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol (2010) 11(9):633–43. doi:10.1038/nrm2957
202. Hu S, Wang N. Control of stress propagation in the cytoplasm by prestress and loading frequency. Mol Cell Biomech (2006) 3:49–60.
203. Li W, Wang W. Structural alteration of the endothelial glycocalyx: contribution of the actin cytoskeleton. Biomech Model Mechanobiol (2017). doi:10.1007/s10237-017-0950-2
204. Röhlich P, Allison A. Oriented pattern of membrane-associated vesicles in fibroblasts. J Ultrastruct Res (1976) 57(1):94–103. doi:10.1016/S0022-5320(76)80059-7
205. Rothberg K, Heuser J, Donzell W, Ying Y, Glenney J, Anderson R. Caveolin, a protein component of caveolae membrane coats. Cell (1992) 68(4):673–82. doi:10.1016/0092-8674(92)90143-Z
206. Muriel O, Echarri A, Hellriegel C, Pavon D, Beccari L, Del Pozo M. Phosphorylated filamin A regulates actin-linked caveolae dynamics. J Cell Sci (2011) 124(16):2763–76. doi:10.1242/jcs.080804
207. Mundy D, Machleidt T, Ying YS, Anderson RG, Bloom GS. Dual control of caveolar membrane traffic by microtubules and the actin cytoskeleton. J Cell Sci (2002) 115(22):4327–39. doi:10.1242/jcs.00117
208. Fujimoto T, Nakade S, Miyawaki A, Mikoshiba K, Ogawa K. Localization of inositol 1,4,5-trisphosphate receptor-like protein in plasmalemmal caveolae. J Cell Biol (1992) 119(6):1507–13. doi:10.1083/jcb.119.6.1507
209. Echarri A, Muriel O, Pavon D, Azegrouz H, Escolar F, Terron M, et al. Caveolar domain organization and trafficking is regulated by Abl kinases and mDia1. J Cell Sci (2012) 125(18):4413–4413. doi:10.1242/jcs.090134
210. Grande-García A, Echarri A, de Rooij J, Alderson N, Waterman-Storer C, Valdivielso J, et al. Caveolin-1 regulates cell polarization and directional migration through Src kinase and Rho GTPases. J Cell Biol (2007) 177(4):683–94. doi:10.1083/jcb.200701006
211. Ogata T, Ueyama T, Isodono K, Tagawa M, Takehara N, Kawashima T, et al. MURC, a muscle-restricted coiled-coil protein that modulates the Rho/ROCK pathway, induces cardiac dysfunction and conduction disturbance. Mol Cell Biol (2008) 28(10):3424–36. doi:10.1128/MCB.02186-07
212. Peng F, Wu D, Ingram A, Zhang B, Gao B, Krepinsky J. RhoA activation in mesangial cells by mechanical strain depends on caveolae and caveolin-1 interaction. J Am Soc Nephrol (2006) 18(1):189–98. doi:10.1681/ASN.2006050498
213. Sauls K, Toomer K, Williams K, Johnson A, Markwald R, Hajdu Z, et al. Increased infiltration of extra-cardiac cells in myxomatous valve disease. J Cardiovasc Dev Dis (2015) 2(3):200–13. doi:10.3390/jcdd2030200
214. Lima SM, Pitsis AA, Kelpis TG, Shahin MH, Langaee TY, Cavallari LH, et al. Matrix metalloproteinase polymorphisms in patients with floppy mitral valve/mitral valve prolapse (FMV/MVP) and FMV/MVP syndrome. Cardiology (2017) 138:179–85. doi:10.1159/000477656
215. Ahmad N, Richards A, Murfett H, Shapiro L, Scott J, Yates J, et al. Prevalence of mitral valve prolapse in Stickler syndrome. Am J Med Genet (2002) 116A(3):234–7. doi:10.1002/ajmg.a.10619
216. Malfait F, Wenstrup R, De Paepe A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genet Med (2010) 12(10):597–605. doi:10.1097/GIM.0b013e3181eed412
Keywords: mitral valve, valve disease, mechanotransduction, pathogenesis, biomechanics
Citation: Pagnozzi LA and Butcher JT (2017) Mechanotransduction Mechanisms in Mitral Valve Physiology and Disease Pathogenesis. Front. Cardiovasc. Med. 4:83. doi: 10.3389/fcvm.2017.00083
Received: 15 October 2017; Accepted: 07 December 2017;
Published: 22 December 2017
Edited by:
Joshua D. Hutcheson, Florida International University, United StatesReviewed by:
Mary Kathryn Sewell-Loftin, Washington University Medical Center, United StatesMark C. Blaser, Brigham and Women’s Hospital, United States
Copyright: © 2017 Pagnozzi and Butcher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan T. Butcher, jtb47@cornell.ed