Stress Coping Strategies in the Heart: An Integrated View
 Marek Michalak
Marek Michalak Luis B. Agellon
Luis B. Agellon- 1Department of Biochemistry, University of Alberta, Edmonton, AB, Canada
- 2School of Human Nutrition, McGill University, Ste. Anne de Bellevue, QC, Canada
The heart is made up of an ordered amalgam of cardiac cell types that work together to coordinate four major processes, namely energy production, electrical conductance, mechanical work, and tissue remodeling. Over the last decade, a large body of information has been amassed regarding how different cardiac cell types respond to cellular stress that affect the functionality of their elaborate intracellular membrane networks, the cellular reticular network. In the context of the heart, the manifestations of stress coping strategies likely differ depending on the coping strategy outcomes of the different cardiac cell types, and thus may underlie the development of distinct cardiac disorders. It is not clear whether all cardiac cell types have similar sensitivity to cellular stress, how specific coping response strategies modify their unique roles, and how their metabolic status is communicated to other cells within the heart. Here we discuss our understanding of the roles of specialized cardiac cells that together make the heart function as an organ with the ability to pump blood continuously and follow a regular rhythm.
Introduction
Non-communicable diseases, in particular cardiovascular diseases and diabetes, now account for over half of all mortalities in the world (1). Acquired heart diseases have devastating consequences and are the most common cause of adult death. Environmental factors, lifestyle choices, and gene variations collectively contribute to the dysfunction of cardiac cells and development of a variety of heart diseases which when left untreated lead to diminished pump function, heart failure, and eventual death (2). There are many specific risk factors for heart diseases, and recent reports indicate that many diseases of the heart are triggered by and/or associated with stress and cellular stress coping response mechanisms (3). For example, the energy dense diets commonly consumed in industrialized societies, and those undergoing nutrition transition, are associated with cardiovascular diseases (1, 4–7). Myocarditis, a disease caused by viral infection, is characterized by inflammation and disruption of the electrical pathways that support normal sinus rhythm which triggers heart failure (8). The ability of cardiac cells to respond to cellular stress is important for the maintenance of cardiac health (3). To carry out its primary purpose as an organ, the heart takes advantage of the functions of specialized cell types to manage energetics, electrical conduction, mechanical work and tissue remodeling (Box 1, Figure 1). The aim of this review is to discuss the coordination of the various activities occurring simultaneously in the different cell types that make up the heart and enable it to continuously pump blood within the body.
Box 1. The Heart.
The heart is a complex biological structure that contracts rhythmically and continuously to distribute blood to the lungs and the rest of the body. It is comprised of unique cell types with dedicated functions that interact together forming a complex networked organ (Figure 1). The human heartbeats an average 80 times a minute to circulate up to 8,000 l of blood a day. Each cycle of the continuous cardiac sinus rhythm is comprised of a systole and a diastole and depends on the cardiac action potential transmitted as electrical impulses generated by the cardiac conductive system (SA, sinoatrial node and AV, atrioventricular node) that functions independently of the central nervous system. Cardiac cells also have endocrine functions releasing natriuretic peptides and cytokines to influence the blood pumping function of the heart. Of the many cell types found in the heart, atrial and ventricular cardiomyocytes form the heart myocardium. Cardiomyocytes occupy the majority of the volume of the mammalian heart (9). Of all nonmyocytes in the healthy adult heart, over 60% are endothelial cells, 5% are hematopoietic-derived cells and 20% are fibroblasts (10), with the remainder being made up of Purkinje and pacemaker cells, smooth muscle cells, and stem cells. Endothelial cells form the endocardium, the interior lining of cardiac valves, whereas smooth muscle cells contribute to coronary arteries and inflow and outflow vasculature. The pericardium is enriched in fibroblasts and connective tissue and allows the heart to function in a frictionless environment. Cardiac stem cells play a role in cardiac repair (11). Pacemaker cells and cells that make up Purkinje fibers in the electrical conduction system are specialized cardiomyocytes that produce and conduct electrical impulses. Cardiac macrophages are interspersed among other cardiac cell types (12–14).
Cardiomyocytes: Cardiomyocytes contain highly organized sarcomeres that facilitate contraction to fulfill the mechanical work of the heart (15). Neonatal and adult cardiomyocytes, HL-1 cardiomyocytes, ANF-T-antigen immortalized cardiomyocytes, and H2C9 myoblasts have been used in culture to study cardiomyocyte function (15). Pluripotent embryonic stem cells and induced pluripotent stem (iPS) cells have also been used to generate cardiomyocytes (16), and these cells contract and relax spontaneously in culture. Cardiac hypertrophy is primarily the result of cardiac myocyte enlargement and this feature can be experimentally recapitulated in tissue culture. Study of these models have yielded detailed knowledge about signaling pathways that control cardiomyocyte activity and have served as platforms for screening of pharmacological agents targeting cardiomyocyte-specific functions related to energetics, contractile and electrical functions. Atrial and ventricular cardiomyocytes do not have identical gene expression programs or Ca2+ handling (17–19). For example, atrial cardiomyocytes, unlike ventricular cardiomyocytes, possess endocrine function to influence blood pressure, renal function and ion balance (20, 21).
Pacemaker Cells: Cardiac pacemaker cells control the heart rate. These cells are concentrated in the SA node and are responsible for generating and propagating an action potential to the AV node to coordinate the mechanical action of cardiomyocytes. Pacemaker cells are sensitive to β-adrenergic stimulation and also might be controlled by micropeptides that affect their Ca2+ handling (22, 23). Dysfunction of pacemaker cells causes cardiac arrhythmias and heart block, which have been linked to heart failure.
Purkinje Cells: These cells form cardiac Purkinje fibers which specialize in rapid transmission of electric impulses in the heart. These cells predominantly express connexin40 and their action potential is longer than that of ventricular cardiomyocytes. Many ventricular arrhythmias, including long QT syndrome, are initiated in the Purkinje fiber conduction system (24).
Fibroblasts: Cardiac fibroblasts are highly responsive to cardiac injury and produce components of the extracellular matrix (ECM) such as periostin, fibronectin, collagen type I, III, V, VI (25, 26). Cardiac stem cells and endothelial cells may also contribute to ECM assembly (27). Cardiac fibroblasts also participate in matrix degradation, conduction system insulation, cardiomyocytes electrical coupling, vascular maintenance, and hemodynamic stress sensing (28, 29). These cells mediate the formation of fibrotic scars in response to cardiac injury (27).
Endothelial Cells: Cardiac endothelial cells are found in the aorta, coronary artery, pulmonary artery, and microvasculature. They play a role in regulating and maintaining the endocardium, and in the myocardial capillaries where endothelial cells directly interact with adjacent cardiomyocytes (30). Cardiac endothelial cells, like vascular endothelial cells, produce and release a variety of autocrine and paracrine agents, such as nitric oxide (NO), endothelin, prostaglandin I, and angiotensin II, which directly influence cardiac metabolism, growth, contractile performance, and rhythmicity (30). Endothelial dysfunction due to increased production of reactive oxygen species (ROS) and reduced synthesis of NO are key features of myocardial ischemia/reperfusion injury (31).
Pericytes: Pericytes control important physiological processes such as angiogenesis, blood flow and vascular permeability (32, 33). In the heart, these cells play a role in cardiac regeneration. Dysfunction of cardiac pericytes contributes to cardiovascular diseases including hypertension, myocardial edema, and post-ischemic coronary no-reflow.
Smooth Muscle Cells: Coronary artery smooth muscle cells are an integral part of cardiac vasculature and contribute to inflow and outflow vasculature. Coronary vasculature supplies the heart with nutrient-containing blood. Atherosclerosis or thrombosis can cause acute disruption of vascular function leading to myocardial infarction (34). As atherosclerosis worsens, the presence of LDL and atherogenic cytokines stimulate vascular smooth muscle cells to alter extracellular matrix composition and thus cause pathogenic vascular remodeling (35).
Macrophages: Macrophages are typically viewed as mobile and appear at sites of injury in response to stress and inflammation (36). However, certain populations of macrophages that are found in some organs, such as in the heart, are produced during organogenesis (36, 37). Resident cardiac macrophages are interspersed between cardiac cells (12, 14, 36) and have recently been implicated in the cardiac electric conduction system (37). Macrophages appear to be integral components of the AV node, connecting the atrial cardiomyocytes with ventricular cardiomyocytes via connexin43-containing gap junctions (37). Dysfunction of AV macrophages or inability of macrophages to communicate with cardiomyocytes induces AV block (37). Blood monocyte-derived and cardiac resident macrophages also contribute to inflammatory and remodeling stages of cardiac disease by phagocytosis of necrotic and apoptotic cells, release of proteases, pro-inflammatory cytokines/chemokines and ROS, and promotion of collagen production (38). Each of the different cell types that make up the heart fulfill unique roles to promote the function of the heart as an organ. Aberration of specific functions provided by each of the specialized cardiac cell types likely manifest in different ways and explains the heterogeneity of cardiac diseases. In acquired cardiac disorders, the insults that perturb cellular functions originate from the environment. The questions that arise relate to the relative sensitivity of the different cardiac cell types to specific stressors, the manner in which the different cell types respond to these stressors, and whether the status of affected cells is communicated to other cell types that make up the organ.
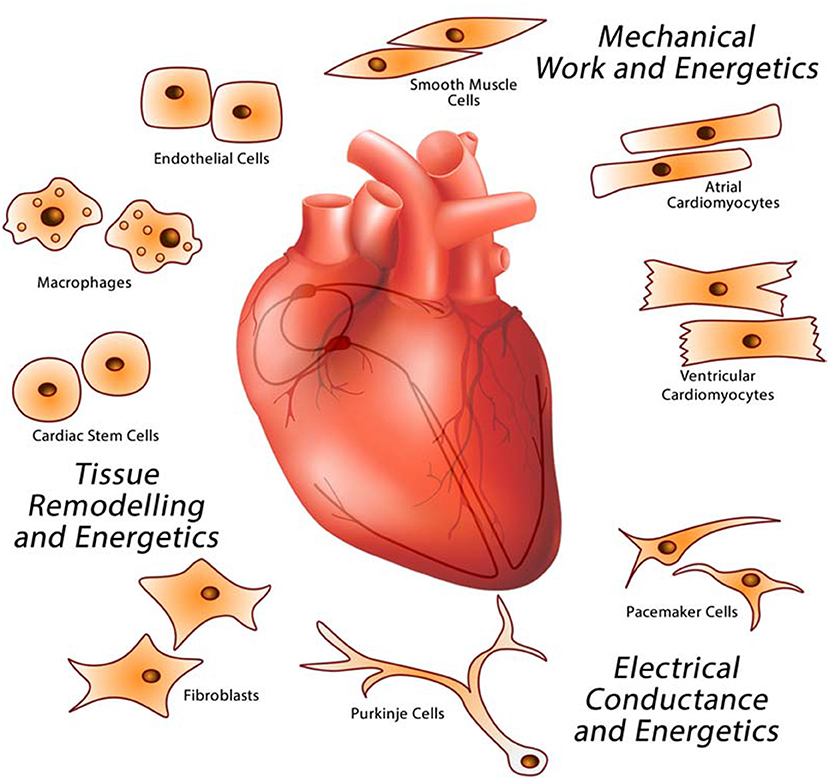
Figure 1. Major cardiac cell types that make up the heart. The heart is a multicellular organ comprised of different cell types responsible for providing specific functions related to electrical conductance (macrophages, pacemaker, and Purkinje cells), mechanical work (cardiomyocytes), and tissue remodeling (endothelial cells, fibroblasts, macrophages, and stem cells).
Cellular Stress Coping Strategies in the Heart
External factors are important risk factors for promoting the development of acquired cardiac disorders. For example, in viral infection, viruses hijack cellular processes and divert cellular resources toward viral replication. Xenobiotics that directly damage or disrupt protein function have detrimental impacts on the integrity of the cellular machinery. Similarly, deficiency of certain types of nutrients such as minerals and vitamins or excess nutrients that serve as fuel, negatively affect cellular metabolism since many of these compounds serve as enzyme co-factors as well as regulate gene expression (7). Lack of oxygen, as in ischemia, compromises the ability of cells to carry out oxidative phosphorylation. Long term perturbation of cellular energetics subsequently leads to loss of proteostasis (unfolded proteins, damaged proteins) and lipidostasis (damaged lipids, disordered membranes) within the cellular reticular network (CRN) (Box 2, Figure 2) (39). These situations have detrimental consequences to the heart as the organ requires a continuous supply of ATP to maintain its function.
Box 2. The Cellular Reticular Network (CRN).
All cardiac cells, as with other eukaryotic cells, consist of intracellular organelles namely the ER, Golgi apparatus, lysosomes, membranous components of the endocytic pathway, peroxisomes, and nuclear envelope (luminally connected to the ER) (Figure 2). We propose that these organelles are organized into the cellular reticular network (CRN) (3, 39), a linked intracellular network of specialized membrane systems, which work in concert to facilitate cell function. Organelles coordinate their activities to maintain homeostasis and fulfill their cellular roles under a variety of metabolic conditions. For example, the ER is responsible for many key cellular processes including the synthesis, folding, posttranslational modification, and transport of proteins; synthesis of lipids and sterols; assembly and trafficking of membranes; drug and xenobiotic detoxification; storage and release of intracellular Ca2+; regulation of gene expression, cellular energetics; and signaling to the nucleus, cytoplasm, mitochondria, and plasma membrane (3, 40, 41). Nearly all these processes utilize energy and thus rely on mitochondria for ATP production. Synthesis of membrane-bound and exported proteins involves the Golgi apparatus. Moreover, synthesis of cellular lipids involves the activities of enzymes resident in the ER, mitochondria, and peroxisomes (42). Coordination of these cellular processes is achieved in part through membrane contact sites (MCS), which are formed between closely apposed organellar membranes including the plasma membrane (Figure 2) (43–50).
The ER is the dominating component of the CRN. In some cells, the ER is further subspecialized into rough ER, such as in secretory cell types, and smooth ER, such as in cells that actively synthesize and metabolize lipids. In cardiomyocytes and smooth muscle cells, in addition to perinuclear rough and smooth ER, the ER is further subspecialized into sarcoplasmic reticulum (SR) which is instrumental in the regulation of excitation-contraction (E-C) coupling to facilitate muscle contraction and relaxation (51). There are two well defined structural and functional regions of the SR: the longitudinal SR, a membrane network around myofibrils extending into the junctional SR, a membrane network with multiple MCS with T-tubules, which are invaginations in the plasma membranes of muscle cells such as cardiomyocytes (Figure 2) (51, 52). The longitudinal SR is enriched with Ca2+-transport ATPase (SERCA), responsible for rapid removal of Ca2+ from the cytoplasm to initiate muscle relaxation (51, 52). The junctional SR contains the ryanodine receptor/Ca2+ channel (RyR), responsible for Ca2+ release to the cytoplasm to initiate muscle contraction, and calsequestrin, a major Ca2+ binding protein of muscle (51–53). The existence of the SR in cardiomyocytes and smooth muscle cells allows these cells to carry out cellular processes related to mechanical functions, notably E-C coupling, without compromising other important Ca2+ requiring cellular processes that are normally associated with rough ER and smooth ER (54).
Overall, the ER/SR remains the main driver of intracellular Ca2+ signaling and the maintainer of cellular Ca2+ homeostasis. Other components of the CRN contribute to Ca2+ homeostasis and signaling (55), and this likely is facilitated through MCS that link the various organelles. The release of Ca2+ from ER stores triggers Ca2+ influx from extracellular space via the store-operated Ca2+ entry (SOCE) mechanism (56–58). SOCE is the primary mechanism for replenishment of depleted ER Ca2+ stores in virtually all non-excitable cells (56–58). The main molecular components of the SOCE include the ER-associated luminal Ca2+ sensor STIM (stromal interacting molecule) and plasma membrane Ca2+ channel ORAI (56–58). In cardiomyocytes, SOCE operates in parallel with the E-C coupling mechanism to maintain basal Ca2+ homeostasis (59–63), but it is not yet clear if SOCE and cardiac E-C coupling mechanisms interact. In cardiomyocytes, increased Ca2+ influx due to SOCE prolongs the duration of the action potential, a contributing factor to the development of cardiac diseases (64).
The advantage of a well-developed CRN is the compartmentalization of different cellular processes such as E-C coupling, protein and lipid synthesis, ATP synthesis, and Ca2+ signaling, within optimal environments. For the CRN to work properly and have the ability to respond to cellular stress there must be a mechanism to allow efficient signal transmission between different compartments of the network. In the context of organs, such as the heart, intracellular communication within the CRN inevitably leads to intercellular communication that enables the function of the organ as the whole. Although not intensively studied in the heart, with the exception of junctional SR-T-tubules connection, MCS are important structures enabling the specialized roles of different cardiac cells. SR-T-tubules MCS play a role in initiation of the E-C coupling (51) while STIM-ORAI MCS support intracellular Ca2+ signaling in all cardiac cells (63). ER/SR-mitochondria MCS are necessary for energy supply of mechanical and electrical functions of the heart (65). Mitochondria-ER contacts are also the sites of mitochondrial fission (66) and are associated with mitochondrial DNA replication (67). Furthermore, MCS are also involved in the recruitment of the mitophagy apparatus for removal of defective mitochondria (68). Under ER/SR stress conditions there is a proliferation of MCS between ER/SR and mitochondria to increase the efficiency of Ca2+ transfer into mitochondria from the ER/SR as part of the adaptive response (69, 70).
The emerging concept is that MCS provide structures and additional mechanisms for efficient sensing, signaling and exchange of molecules among the different components of the CRN. In the heart, this offers individual cardiac cells the capability to facilitate a rapid intracellular movement of molecules both under physiological conditions and in response to stress. Understanding the formation, function and maintenance of MCS in the CRN under physiological and pathological settings will provide insight into the importance of the CRN in the elaboration of adaptive responses that are stimulated by cellular stress coping response mechanisms.
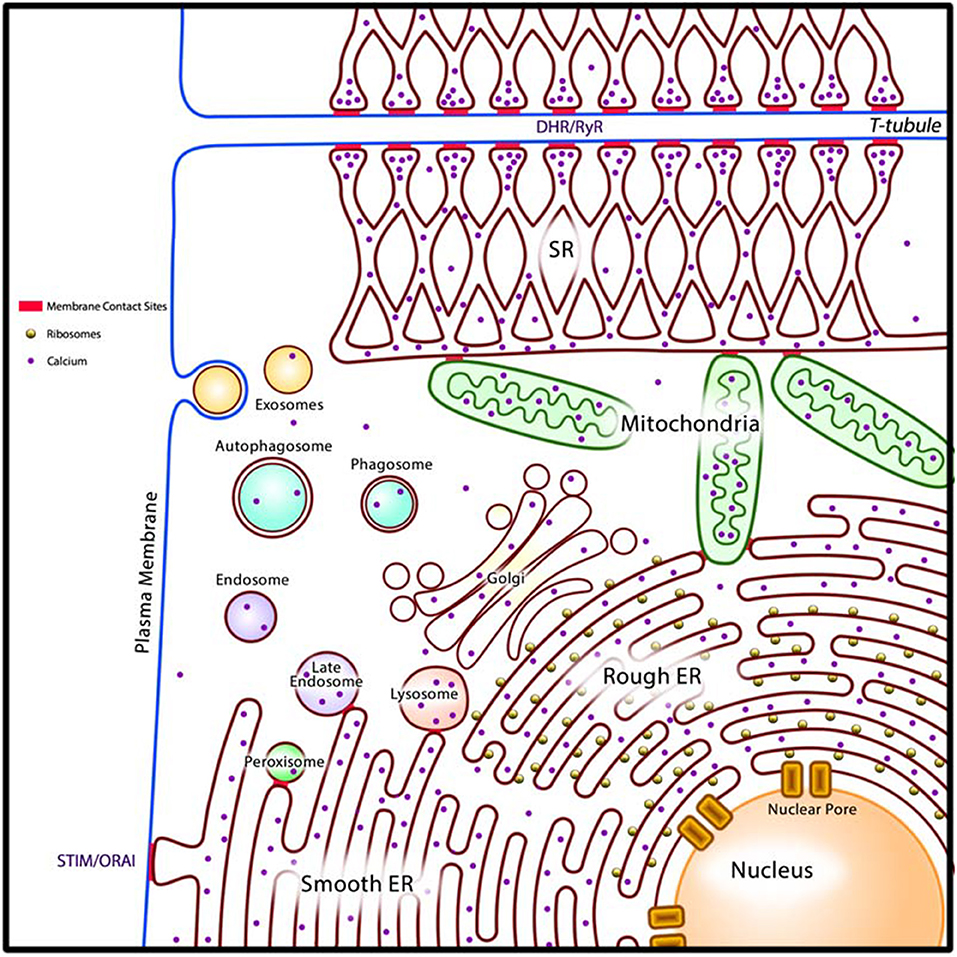
Figure 2. A generalized model of the cardiac cellular reticular network (CRN). Intracellular organelles (rough and smooth ER, SR in cardiomyocytes and smooth muscle cells, Golgi apparatus, lysosomes, membranous components of the endocytic pathway, peroxisomes, and nuclear envelope) are organized into the cellular reticular network (CRN). The CRN is linked by membrane contact sites (MCS, in red) allowing for rapid exchange of molecules between component organelles. In cardiomyocytes the MCS that join SR and T-tubules (DHR/RyR) allow communication between the plasma membrane and the SR to drive E-C coupling. Smooth ER form MCS with the plasma membrane (STIM/ORAI) to drive store-operated Ca2+ entry (SOCE). DHR, plasma membrane dihydropyridine Ca2+ channel receptor; RyR, SR ryanodine receptor/Ca2+ channel; STIM, ER Ca2+ sensor; ORAI, plasma membrane Ca2+ channel; ER, endoplasmic reticulum.
To mitigate cellular stress and re-establish homeostasis, cardiac cells must activate stress coping response mechanisms (3, 71, 72) (Figure 3). At the cellular level, failure to adapt to cellular stress leads to cell death. For survival, cells must successfully cope with the cellular stress, either by eliminating the source of the stress or by altering cellular metabolism to circumvent the negative effects of long term cellular stress. In the latter case, modification of cellular function to regain homeostasis without eliminating cellular stress permits cells to escape cell death but contributes detrimental effects to the organism since the adapted cells have modified function which is reflected in changes their gene expression program that can also be propagated to descendant cells through epigenetic modification of the genome (72). Indeed, epigenome remodeling of cardiac cells has been associated with aging, exposure to environmental factors and cardiac disease states (73–77).
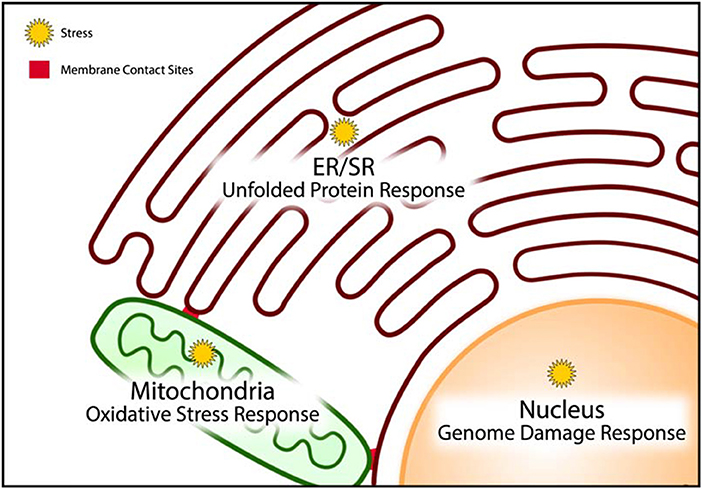
Figure 3. Stress coping response mechanisms. Cellular organelles have resident stress coping response mechanisms that are designed to mitigate the impact of stress and minimize damage caused by stress. The unfolded protein response, oxidative stress response, and genome damage response mechanisms operate predominantly in the endoplasmic/sarcoplasmic reticulum (ER/SR), mitochondria, and nucleus, respectively.
The unfolded protein response (UPR) pathway is a well described stress coping mechanism to regain proteostasis in the ER by activation of several signaling pathways associated with ER-associated protein degradation (ERAD), autophagy, apoptosis and inflammatory signaling (3, 78–83) (Figure 3). Proteostasis is essential to all the components of the CRN, but it is especially important for the ER to maintain proteostasis because this organelle is the site of membrane protein and lipid synthesis (Box 2, Figure 2). If uncorrected, the loss of proteostasis inevitably causes loss of lipidostasis since membrane bound proteins may not be able to function properly in an environment with aberrant lipid composition (84). For example, a recent study revealed that membrane lipid composition is closely tied to the ability of SERCA to fulfill its functions in Ca2+ transport (85) and therefore cardiac relaxation.
It is important to note that the UPR has a high demand for energy to relieve ER stress. ATP synthesis through oxidative phosphorylation in mitochondria creates ROS. These highly reactive species are normally quenched by an elaborate and efficient scavenging system, involving a variety of endogenous antioxidant proteins (such as glutathione, thioredoxin) and transcription factors (such as Nrf2, NF-kB) (86–88), thereby preventing chemical damage to cellular proteins and DNA. ROS are also formed by metal catalyzed oxidation, various oxidases, uncoupled NO synthase and myeloperoxidase from infiltrating monocytes and neutrophils (89). Although ROS are commonly described as damaging agents, recent studies suggest that ROS may also function as signaling molecules. Increased and uncontrolled oxidation reactions within the mitochondria induce mitochondrial membrane permeability transition (MMPT) causing the leakage of mitochondrial contents, such as cytochrome C, ROS and Ca2+, into the cytoplasm (Figure 3). This event is indicative of unrecoverable mitochondrial damage and signals the initiation of controlled cell death by apoptosis (89–91). In doxorubicin-induced cardiac toxicity, mitochondrial localization of Bnip3, a BH3-only protein Bcl-2-like interacting protein 3, increases ROS production, and induces the loss of mitochondrial membrane potential, driving MMPT pore opening and leading to both apoptotic and necrotic cell death (92).
Under optimal conditions, mitochondria form a tight network that efficiently synthesize ATP without the accumulation of damaging ROS (93, 94) and this might be associated with longevity. The mitochondrial network has been shown to break down functionally and morphologically under conditions of fuel excess, and is accompanied by increased production and accumulation of ROS (95–97). At times of fuel insufficiency, autophagy may serve as a source of substrates in addition to the removal of defective cellular components. Thus, effective communication among the components of the CRN (Figure 2) and coordination of stress coping response mechanisms likely enables the cell to minimize damage to itself as well as to prevent the spread of damaging molecules to neighboring cells. The coordination of these strategies is perhaps best illustrated by the integrated induction of MMPT from the UPR to signal cell death in the case of unrecoverable damage, and by autophagy to remove damaged cellular components that can be used as temporary sources of building materials or fuel substrates (Figure 3) (98).
At present, it is not known if all cardiac cell types have similar sensitivity to inducers of cellular stress, whether the specific stress sensors and coping strategies are configured in the same way in all cardiac cell types, how specific coping response strategies modify their unique roles, and how their metabolic status is communicated to other cells within the heart.
Cardiac Energetics and Stress Coping Responses
Cellular energetics is a basic function of all cells. Like other cells in the body, cardiac cells can use a variety of substrates to produce the energy to maintain cellular processes associated with growth and proliferation, cell signaling and metabolic homeostasis. Cardiomyocytes of the fetal heart generate ATP mainly by glycolysis whereas cardiomyocytes of the adult heart prefer fatty acids as fuel to meet the high energy demand of mechanical work (99). The switch of fuel preference in fetal and adult cardiomyocytes is likely related to greater availability of oxygen in the adult heart, which is necessary for efficient burning of fatty acids. Reliance on anaerobic glycolysis, such as during extended periods of work, produces a condition of ATP deficit and excess lactic acid, which exacerbates cellular stress (100). Coincidentally, the fuel preference of cardiomyocytes in the failing adult heart shifts from fatty acids to glucose, reflecting reduced oxygen availability and oxidative capacity (99, 101). Amino acids generated by autophagy may be an important source of energy for cardiomyocytes during severe cellular stress conditions. In general, fuel selection in cardiomyocytes is influenced by nutrient availability, namely excess or deficiency of fatty acids, glucose, oxygen, and calcium. Long term consumption of the Western-style diet, a condition of excess energy input, is typically associated with the development of cardiac ischemia which often end in heart failure (1, 7, 102, 103), although the morbidity of acquired cardiac disorders is influenced by gene variations (104).
Cardiac energetics has been the topic of intensive studies for decades. Early studies employed intact hearts taken from animals to measure energy requirement and utilization (105). More recent studies using transgenic and targeted gene disruption technologies in mice permitted the direct evaluation of many genes on cardiac metabolic efficiency and disease susceptibility (106). Animal studies, in conjunction with studies using isolated cardiomyocytes, have revealed that cellular stress is a key feature that initiates a pathogenesis of acquired cardiac disorders (107). Even though cardiomyocytes exhibit a preference for fatty acids as a fuel substrate, exposure to excessive amounts of fatty acids induce lipotoxicity due to elevation in the synthesis of lipid signaling molecules such as diacylglycerol and ceramide, accumulation of ROS, and increased fat synthesis and accretion (108, 109). Moreover, acyl-CoAs have been shown to act on histone deacetylases, affecting the epigenome and thus the gene expression program (110, 111).
In recent years, there has been substantial progress toward our understanding of how cardiomyocytes respond to cellular stress (3, 72, 79, 112). However, other cardiac cell types also contribute key energy-requiring roles to deliver heart function, such as ion transport to coordinate electrical conductance and cell proliferation during cardiac remodeling to maximize mechanical capacity while minimizing the negative impacts of changes in cardiomyocyte morphology. At very high levels, ROS induce an inflammatory response involving macrophages and cardiac fibroblasts (113, 114). Increased lactic acid synthesis has been shown to stimulate vascular smooth muscle cells (115). The mechanisms involved in orchestrating the coordinated adaptations of different cardiac cell types is not well understood.
Cardiac Electrical Conductance and Stress Coping Responses
Electrical impulses that drive the cardiac rhythm originate from SA node and then spreads rapidly to the pacemaker cells in the AV node. The pacemaker cells produce action potential that propagates to cardiomyocytes to initiate the influx of extracellular Ca2+ into the cardiomyocytes to drive the E-C coupling mechanism. The E-C coupling links the electrical activation of the surface membrane to mechanical force, i.e., the contraction. This process requires coordinated movement of Ca2+ at the level of cardiomyocyte CRN (Figure 2) (51). Gap junctions between cardiomyocytes are important for mediating electrical cell-cell communication by ion signal spread (Figure 4). These structures are not equally distributed across the myocardium but high numbers are found localized at the ends of cardiomyocytes. This arrangement can affect electrical and metabolic coupling of cardiac cells, and influence both the propagation of the conduction velocity and the direction of the signal spread along adjacent cardiomyocytes. Importantly, the function of gap junctions and expression of gap junction components are sensitive to cell stress (116, 117).
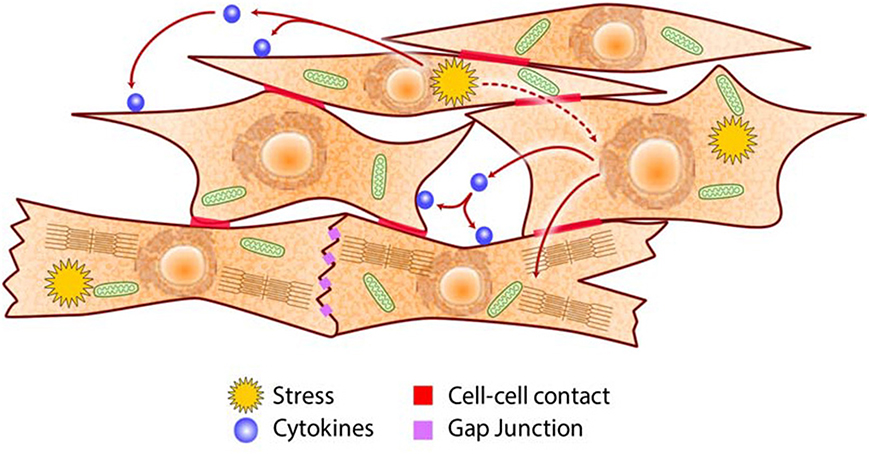
Figure 4. Schematic of intercellular communications in the heart. Cardiac cells communicate via cell-cell contacts (e.g., adhesion molecules, gap junctions), paracrine/autocrine signaling (e.g., cytokines, growth factors), and metabolites (e.g., ROS, adenosine, NO) to coordinate their specific roles in generating the heartbeat and in adapting to metabolic challenges.
A growing list of potentially lethal cardiac arrhythmias including catecholaminergic polymorphic ventricular tachycardia, atrial fibrillation, long QT syndrome and hypertrophic cardiomyopathy, are linked to activation of cellular stress coping mechanisms triggered by environmental factors. Atrial fibrillation is one of the most common cardiac arrhythmias associated with stress and inflammation of cardiac tissue (118, 119). At the molecular level, dysregulation of Ca2+ handling in the SR is the major factor contributing to the development of atrial fibrillation (119, 120). Modification of Ca2+ handling proteins that lead to increased Ca2+ release from the SR result in the large rise in cytoplasmic and mitochondrial Ca2+ concentration (121–123) causing mitochondrial dysfunction, reduced ATP synthesis, increased ROS production (124–126), activation of UPR, attenuation of Ca2+-independent transient outward K+ channel and apoptosis (Figure 3). All these features have been associated with cardiac arrhythmias in failing hearts (122, 127, 128). Activation of UPR also drives acquired arrhythmias (122). For example, PERK activation downregulates the Na+ channels and decreases Na+ current density (127, 128), reduces cardiac Kv4.3 channel conductance (128) and consequently cardiac conduction velocity. Inhibition of PERK prevents downregulation of these ion channels (128). Impaired glycosylation of the Nav1.5 contributes to stimulation of UPR due to misfolding and impaired trafficking of the ion channel also leading to arrhythmias (129).
Endothelial cells control vascular tone by releasing vasodilators including NO, prostacyclin and endothelin-1, angiotensin II which affect E-C coupling (130). Endothelial cells lack gap junctions and their communication with neighboring cells, in particular cardiomyocytes, must occur by gap junctions-independent mechanisms. Endothelin-1 secreted by endothelial cells mediates electrical remodeling during heart failure by downregulating the expression and phosphorylation of gap junction proteins connexin40 and connexin43 in cardiomyocytes, as well as by reducing Nav1.5 protein abundance and Na+ channel conductance (131). In contrast, cardiac fibroblasts are directly coupled to cardiomyocytes by gap junctions (132) and also express voltage sensitive and mechanosensitive ion channels (133, 134) that likely modulate cardiac electric activity. Finally, resident cardiac macrophages were recently found in the distal AV node. These cells are permanently attached to AV node pacemaker cells via connexin43-containing gap junctions and are essential for both normal and aberrant cardiac electrical conduction (37).
Even though a large body of information on the biology of cardiac cells has been accumulated, the nature of cardiac cell type-specific stress sensors and the outcomes of their stress coping response mechanisms that deal with electrical properties of the heart are not well studied.
Cardiac Mechanical Work and Stress Coping Responses
The main role of cardiomyocytes is to carry out mechanical function, but these cells also contribute to electrical conductance. Mechanical coupling between cardiomyocytes ensures that the outcome of action potential pacing synchronizes cardiomyocyte contraction to produce a coherent heartbeat (135). Considering an average of 80 heartbeats per minute, cardiomyocytes are well equipped to deal with repeated stretch-induced mechanical stresses. Prolonged mechanical force overload activates UPR, induces inflammatory mediators and causes ventricular hypertrophy (136). Long-term mechanical stretch-induced ER/SR stress may also affect other cardiac cell types, although their sensitivity to this stressor is not known. Disruptions in E-C coupling cause ER/SR stress and congestive heart failure (137). Increased abundance of calreticulin, a Ca2+ binding ER/SR resident chaperone, is associated with the failing human heart (127, 138). In mice, forced overexpression of calreticulin in cardiomyocytes increases cardiomyocytes ER/SR Ca2+ capacity and mechanical work potential, but also activates the IRE1α branch of UPR and eventually leads to cardiomyopathy (116, 139). Calreticulin overexpression also causes the reduction of gap junction protein abundance in the heart indicating a defect in cell-cell communication (116).
Heart failure is a typical result of extended periods of extreme mechanical activity (140). Remarkably, mechanical unloading with left ventricular assist devices improves Ca2+ handling and reduces UPR status in myocardial tissue of patients with advanced heart failure (141). The activation of the PERK branch of UPR in cardiomyocytes is associated with pressure overload-induced congestive heart failure (142) and affects multiple cardiac genes including increased expression and stability of Na+ voltage-gated channel α5 and Kv4.3 channel (127). Other molecular components of the CRN are also involved. For example, tribbles homolog 3 (TRB3), a neuronal cell death-inducible putative protein kinase, is increased by ER/SR stress in cultured cardiomyocytes where it plays a role in stretch-induced cardiomyocyte apoptosis (143). E3 ubiquitin-protein ligase, HMG-CoA reductase degradation 1 (Hrd1) and ERAD are essential components of UPR in cardiomyocytes. Hrd1 is involved in preserving heart structure and function in a mouse model of cardiac hypertrophy (144). Nogo-C, a ubiquitously expressed ER protein is necessary for ischemia-induced cardiomyocyte apoptosis and cardiac dysfunction (145). Eva-1 homolog A (EVA1A), an ER-associated protein, improves cardiac function and inhibits cardiac hypertrophy and fibrosis by increasing autophagy (146). Stress coping response mechanisms induce changes within the CRN of cardiac cells and promotion of molecular constituents designed to preserve heart function (Figure 3).
Cardiomyocytes are the workhorse of the cardiac mechanical function and considerable information about cellular stress coping response mechanisms in these cells have been gained. However, the link between UPR and mechanical function of the heart and how the cardiomyocyte stress status is communicated to other cardiac cell types under unfavorable metabolic situations remains to be established.
Cardiac Tissue Remodeling and Stress Coping Responses
Cardiac remodeling is initially an adaptive response to hemodynamic stress and cardiac injury. In association with neurohormonal activation, cardiac remodeling can lead to cardiac diseases as a result of changes in gene expression program (e.g., re-expression of fetal genes) and cell morphology (volume, mass, shape). Cardiac tissue remodeling is stimulated by a variety of stressful conditions such as myocardial infarction, hypertension, myocarditis, idiopathic dilated cardiomyopathy or volume overload, and is associated with increased collagen synthesis induced by TGFβ, endothelin-1, angiotensin II and connective tissue growth factors culminating in cardiac fibrosis (147, 148). Communication among cardiomyocytes, fibroblasts and endothelial cells influences the biology of the cardiac ECM and is a key aspect of cardiac remodeling (147, 149–151). Endogenous pluripotent cardiac stem cells play an important part in cardiac remodeling and repair as they are capable of differentiating into cardiomyocytes, smooth muscle cells and endothelial cells (152–155).
Cardiac fibroblasts are central to the development of cardiac fibrosis but cardiomyocytes, endothelial cells and macrophages also contribute to the process. Increased abundance of TGFβ promote fibroblast proliferation and differentiation to myofibroblasts, activation of metalloproteinases in response to activation of β-adrenergic, angiotensin II and endothelin I receptors (147, 156, 157). Cardiac remodeling is also characterized by changes in cardiomyocytes contractile proteins and alterations in the handling of Ca2+ within the cardiomyocyte CRN (65, 147). For example, while increasing calreticulin abundance in the adult cardiomyocytes improves ER/SR Ca2+ capacity and delays SOCE, it also stimulates UPR (Box 2, Figure 3) which promotes the increase of cardiac TGFβ abundance that in turn induces increased collagen deposition and severe cardiac fibrosis (116, 139). Remarkably, pharmacological inhibition of UPR activation with tauroursodeoxycholic acid (TUDCA), a proteostasis promoter (158), inhibits the activation of IRE1α branch of UPR and averts the development of cardiac fibrosis (139, 159). Resveratrol, another proteostasis promoter (158), similarly reduces cardiac fibrosis in transverse aortic constriction induced heart failure (160, 161).
Endothelial cells, in pressure overload stressed myocardium, contribute to cardiac remodeling by regulating cardiomyocytes growth and modulating mechanical/electrical functions in a paracrine fashion (30, 130, 162, 163). Stress-induced endothelial cells impact on cardiomyocytes, driving cardiac hypertrophy (164–166). The inhibition of UPR with proteostasis promoter 4-phenylbutyrate prevents endothelial cell apoptosis, inflammation in the aorta, and development of the thoracic aortic aneurysm associated with degeneration of vascular smooth muscle cells (167). Endothelial-to-mesenchymal transition gives rise to cells that have fibroblast-like characteristics but still express endothelial markers, further enhancing cardiac remodeling and fibrotic events (168). Macrophages contribute to myocardial infarction-induced cardiac remodeling after recruitment to the ischemic myocardium to promote infarct repair and healing (169).
Stress-induced cardiac remodeling involves many cardiac cell types and is coordinated via gap junction-dependent cell-cell communication as well as both paracrine and autocrine signaling. However, each of the different cardiac cell types likely elaborate unique adaptive outcomes that have consequences not only on their own metabolic status but also that of their neighboring cells.
Coordination of Cardiac Cell Roles in the Heart
The heart is a remarkably elegant and adaptable machine that can maintain its blood pumping action under a wide variety of metabolic conditions. To deliver a single heartbeat, an action potential generated by AV node pacemaker cells that is triggered by influx of Na+, causes membrane depolarization that opens L-type Ca2+ channels on the surface membrane and T-tubules of cardiomyocytes. The resulting entry of Ca2+ induces the opening of RyR Ca2+ channel to release Ca2+ into the cytoplasm, initiating muscle contraction. To relax, the Ca2+ is removed from cytoplasm by SERCA and Na+/Ca2+ exchanger. The Ca2+ signal is propagated as a wave via gap junctions along adjacent cardiomyocytes to drive contraction and relaxation cycles of the whole organ. For the heart to function properly, Ca2+ handling by cardiomyocytes must be tightly regulated, and Ca2+ concentration must be appropriately high in systole and low in diastole. Thus, the heartbeat is a result of cooperative roles carried out by several cell types that communicate with each other using a variety of methods (Figure 4). It is crucial to ensure a sufficient supply of ATP in the heart to meet changing metabolic demands, since a deficit in energy will cause the breakdown in the coordination of cardiac cell roles which eventually culminates in the loss of heart function and death of the organism.
The heart is dependent on key nutrients (minerals such as calcium, potassium, and magnesium for conduction of electrical impulse; simple sugars, fatty acids and amino acids as sources of energy to fuel muscle contraction and relaxation; iron and oxygen for efficient synthesis of ATP) to accomplish its function. Severe deficiency or large excess of nutrients, which originate from external sources, cause loss of nutrient/energy homeostasis that in turn cause the disruption of proteostasis and lipidostasis in the CRN (Figures 2, 3) (7, 170–172). How does the heart perform its function continuously when the cells that constitute the organ are faced with cellular stress? Cardiac cells experiencing stress not only act to restore their own cellular homeostasis but also emit signals that induce pathways in neighboring cells (Figure 4) designed to preserve the function and integrity of the organ as a whole. Autocrine/paracrine signaling involves cytokines such as angiotensin II, endothelin-1, TGFβ, VEGF, FGF, PDGF, MCP-1, and leptin that interact with cell surface receptors. Endothelial cells control cardiomyocytes E-C coupling by releasing vasodilators including NO, prostacyclin and endothelin-1, angiotensin II, natriuretic peptides, ROS, kinins, adenosine, and others. Cytokine activated endothelial cells express adhesion molecules (intracellular adhesion molecule-1, ICAM-1; vascular cell adhesion molecule-1, VCAM-1) that recruit and promote infiltration of immune cells into myocardium in response to stress stimuli (173). E-C coupling is sensitive to metabolic stress that induces signaling cascades that modify ion channels abundance and behavior, causing electrophysiological and structural remodeling in the heart that promote arrhythmogenesis (174, 175). Communication between cardiomyocytes, fibroblasts and endothelial cells, is a key aspect of cardiac remodeling and influences biology of the ECM components in the heart (Figure 4). Under stress conditions there is increased release of TGFβ from macrophages and fibroblasts that promotes fibroblasts activation, differentiation into myofibroblasts, production of ECM and cardiac remodeling. Upon cardiac injury there is an increased abundance of ECM promoting cardiac remodeling (e.g., fibrosis, angiogenesis) or ECM break down releasing cytokines bound to the ECM. Increases in NO release from endothelial cells activate guanylyl cyclase causing increased formation of cGMP and vasodilation increasing coronary blood flow to relieve ischemia. This ultimately results in promotion of protein synthesis including sarcomere proteins, hypertrophy, activation of the fetal cardiac gene expression program, fibrillogenesis and angiogenesis. Metabolites such as adenosine, NO, and ROS released by one stressed cardiac cell interact with transporters and receptors that activate signaling cascades and modify metabolic pathways of another cardiac cell. This unique arrangement of cardiac cells and specific signaling system (Figure 4) developed for cell-cell communication via direct contacts or via autocrine/paracrine signaling, are fundamental for the function of the heart as an organ and play important role under stress conditions in helping to maintain heart function.
It is likely that altered cellular function induced by stress coping response strategies (Figure 3) are meant to operate only during stress conditions. In the case of nutrient excess as in diet-induced cardiac disorders, cardiac cells are subject to prolonged exposure of nutrient overload causing persistence of altered metabolic functions that in the end drives the pathogenesis of cardiac disease (3, 7). In animal models of heart failure, administration of proteostasis promoters, such as TUDCA and resveratrol (158), have demonstrated efficacy in preventing the progression of cardiac fibrosis indicating that it is possible to delay terminal heart failure through therapeutic manipulation of stress coping response mechanisms such as UPR (139, 159–161). At present, it is not clear how the roles of the different cardiac cell types are modified by long-term cellular stress, or how these cells effectively communicate to coordinate their activities under unfavorable conditions.
Challenges
Many animal models (mouse, rat, hamster, rabbit, dog, and pig) are commonly used in cardiovascular research (176, 177). Transgenic and targeted gene disrupted mice have permitted the evaluation of the function of virtually any gene in vivo, and together with primary and established cell cultures (cardiomyocytes, cardiac fibroblasts, vascular cells, and more recently iPS-derived cardiomyocytes) (15), these experimental models have been instrumental in understanding the pathophysiology of cardiac diseases. Nevertheless, despite the massive amount of information that has been gathered so far about the cardiovascular system, gaps in knowledge regarding cell-cell communication and how cardiac cell roles are coordinated still exist. One challenge that is particularly difficult to overcome relates to the inherent limitation of animal models, even humanized animal models, to fully recapitulate human biology. The impact of biological sex as an important modifier is another shortcoming of the existing knowledge base. Heart disease develops and presents differently in males and females (178–181). Yet little is known about the precise mechanisms that underlie sex differences and how these impact on prognosis, morbidity or therapeutic choices. There is evidence showing that male and female cardiac cells have distinct gene expression programs (182), and therefore differ in metabolic capacity. Fortunately, there is now a movement to include both sexes, not only in in vivo studies but also in in vitro studies, as well as to analyze their responses separately. The influence of extrinsic factors should also be taken into careful consideration. For example, palmitic acid has been the choice energy substrate used in numerous metabolic studies employing the isolated perfused heart model (183), yet palmitic acid is a potent inducer of UPR, autophagy, Ca2+ depletion, apoptosis and mitochondrial stress (184–190). Moreover, the importance of the extracellular matrix and intercellular communication among cardiac cells is not adequately addressed in many experimental designs. Cardiac cells grown in tissue culture lack important contextual information, such cell-cell contact between different cell types (Box 1, Figure 4) and other cues that indicate the metabolic status of neighboring cardiac cells as well as the heart itself. Analysis of abundance or phosphorylation status of specific proteins in cardiac tissue homogenates does not provide information about the status of specific mechanisms, such as stress coping responses (Figure 3), operating in each of the different cardiac cell types. These considerations are vital to the understanding of how the different cell activities are integrated to sustain a continuous rhythm.
Summary
The heart is able to perform its blood pumping function due to the collaboration of different cell types that make up its structure. Specialized cell types are responsible for the principal activities of cardiac tissues, namely generation and propagation of the electrical impulse, facilitation of the rhythmic contraction and relaxation cycles, and the orchestration of the cardiac remodeling, which all operate coordinately to maintain a regular heartbeat throughout the lifetime of the organism. In response to stress inducers, especially to nutrient overload which disrupts cellular energy homeostasis, all cells employ the same basic cellular stress coping response mechanisms that operate in various organelles to sense and rectify metabolic disturbances in the cellular reticular network. In the context of the heart, the manifestations of stress coping strategies likely differ depending on the coping strategy outcomes of the different cardiac cell types, and thus may underlie the development of distinct cardiac disorders. Adaptive outcomes normally cease to operate when cellular stress is resolved, whereas long term exposure of cardiac cells to stress induce persistent changes in cell activity that eventually promote heart disease. In the case of nutrient excess, reduction in the intake of energy dense nutrients may be the first and crucial step toward the recovery of normal cardiac cell function. In addition, it may also be possible to delay complete heart failure in advanced cases of acquired cardiac diseases by simultaneous therapeutic targeting of cellular stress coping mechanisms operating in various organelles in order to accelerate the recovery of proteostasis and lipidostasis.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Work in our laboratories is supported by grants from the Canadian Institutes of Health Research (CIHR) grants MOP-15291, MOP-15415, and PS-153325 to MM; a generous donation from the Kenneth and Sheelagh McCourt family; CIHR grant MOP-15291 and PS-153325 to LA; Natural Sciences and Engineering Research Council of Canada grant to LA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jonathan Parrish for help with the figures. Research in our laboratories is supported by grants from the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada.
Abbreviations
ANF, Atrial natriuretic factor; AV, atrioventricular node; CHOP, C/EBP-homologous protein; CRN, cellular reticular network; ECM, extracellular matrix; E-C, excitation-contraction; ER, endoplasmic reticulum; ERAD, ER associated protein degradation; iPS, inducible pluripotent stem cells; IRE1α, inositol-requiring kinase 1α; MCS, membrane contact sites; MMPT, mitochondrial membrane permeability transition; ORAI, Ca2+ release-activated Ca2+ channel protein; PERK, ER kinase dsRNA-activated protein kinase-like ER kinase; ROS, reactive oxygen species; RyR, ryanodine receptor; SA, sinoatrial node; SR, sarcoplasmic reticulum; SERCA, ER/SR Ca2+-transport ATPase; SOCE, store-operated Ca2+ entry; STIM, stromal interaction molecule; TGFβ, transforming growth factor β; UPR, unfolded protein response.
References
1. Collaborators GBDRF. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet (2017) 390:1345–422. doi: 10.1016/S0140-6736(17)32366-8
2. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. (2008) 358:1370–80. doi: 10.1056/NEJMra072139
3. Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu Rev Physiol. (2013) 75:49–67. doi: 10.1146/annurev-physiol-030212-183707
4. Lao XQ, Ma WJ, Sobko T, Zhang YH, Xu YJ, Xu XJ, et al. Dramatic escalation in metabolic syndrome and cardiovascular risk in a Chinese population experiencing rapid economic development. BMC Public Health (2014) 14:983. doi: 10.1186/1471-2458-14-983
5. Bosu WK. An overview of the nutrition transition in West Africa implications for non-communicable diseases. Proc Nutr Soc. (2015) 74:466–77. doi: 10.1017/S0029665114001669
6. DiNicolantonio JJ, Lucan SC, O'Keefe JH. The evidence for saturated fat and for sugar related to coronary heart disease. Prog Cardiovasc Dis. (2016) 58:464–72. doi: 10.1016/j.pcad.2015.11.006
7. Chen Y, Michalak M, Agellon LB. Importance of nutrients and nutrient metabolism in human health. Yale J Biol Med. (2018) 91:95–103.
8. Fung G, Luo H, Qiu Y, Yang D, McManus B. Myocarditis. Circ Res. (2016) 118:496–514. doi: 10.1161/CIRCRESAHA.115.306573
9. Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, et al. Dynamics of cell generation and turnover in the human heart. Cell (2015) 161:1566–75. doi: 10.1016/j.cell.2015.05.026
10. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D'Antoni ML, Debuque R, et al. Revisiting cardiac cellular composition. Circ Res. (2016) 118:400–9. doi: 10.1161/CIRCRESAHA.115.307778
11. Hou J, Wang L, Jiang J, Zhou C, Guo T, Zheng S, et al. Cardiac stem cells and their roles in myocardial infarction. Stem Cell Rev. (2013) 9:326–38. doi: 10.1007/s12015-012-9421-4
12. Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS ONE (2012) 7:e36814. doi: 10.1371/journal.pone.0036814
13. Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci USA. (2014) 111:16029–34. doi: 10.1073/pnas.1406508111
14. Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res. (2014) 115:284–95. doi: 10.1161/CIRCRESAHA.115.303567
15. Peter AK, Bjerke MA, Leinwand LA. Biology of the cardiac myocyte in heart disease. Mol Biol Cell. (2016) 27:2149–60. doi: 10.1091/mbc.e16-01-0038
16. Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. (2015) 117:80–8. doi: 10.1161/CIRCRESAHA.117.305365
17. Kemeny-Suss N, Kasneci A, Rivas D, Afilalo J, Komarova SV, Chalifour LE, et al. Alendronate affects calcium dynamics in cardiomyocytes in vitro. Vascul Pharmacol. (2009) 51:350–8. doi: 10.1016/j.vph.2009.09.002
18. Maass AH, De Jong AM, Smit MD, Gouweleeuw L, de Boer RA, Van Gilst WH, et al. Cardiac gene expression profiling - the quest for an atrium-specific biomarker. Neth Heart J. (2010) 18:610–4. doi: 10.1007/s12471-010-0844-8
19. Bootman MD, Smyrnias I, Thul R, Coombes S, Roderick HL. Atrial cardiomyocyte calcium signalling. Biochim Biophys Acta (2011) 1813:922–34. doi: 10.1016/j.bbamcr.2011.01.030
20. Ogawa T, de Bold AJ. The heart as an endocrine organ. Endocr Connect. (2014) 3:R31–44. doi: 10.1530/EC-14-0012
21. Clerico A, Passino C, Franzini M, Emdin M. Natriuretic peptides as biomarkers of cardiac endocrine function in heart failure: new challenges and perspectives. Future Cardiol. (2016) 12:573–84. doi: 10.2217/fca-2016-0013
22. Anderson DM, Makarewich CA, Anderson KM, Shelton JM, Bezprozvannaya S, Bassel-Duby R, et al. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Sci Signal. (2016) 9:ra119. doi: 10.1126/scisignal.aaj1460
23. Makarewich CA, Olson EN. Mining for micropeptides. Trends Cell Biol. (2017) 27:685–96. doi: 10.1016/j.tcb.2017.04.006
24. Boyden PA, Hirose M, Dun W. Cardiac Purkinje cells. Heart Rhythm (2010) 7:127–35. doi: 10.1016/j.hrthm.2009.09.017
25. Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, et al. Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation (2017) 136:549–61. doi: 10.1161/CIRCULATIONAHA.116.026238
26. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. (2017) 127:3770–83. doi: 10.1172/JCI94753
27. Ivey MJ, Tallquist MD. Defining the cardiac fibroblast. Circ J. (2016) 80:2269–76. doi: 10.1253/circj.CJ-16-1003
28. Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ. Origin of cardiac fibroblasts and the role of periostin. Circ Res. (2009) 105:934–47. doi: 10.1161/CIRCRESAHA.109.201400
29. Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. (2009) 105:1164–76. doi: 10.1161/CIRCRESAHA.109.209809
30. Brutsaert DL. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev. (2003) 83:59–115. doi: 10.1152/physrev.00017.2002
31. Yang Q, He GW, Underwood MJ, Yu CM. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: perspectives and implications for postischemic myocardial protection. Am J Transl Res. (2016) 8:765–77.
32. Volz KS, Jacobs AH, Chen HI, Poduri A, McKay AS, Riordan DP, et al. Pericytes are progenitors for coronary artery smooth muscle. Elife (2015) 4:e10036. doi: 10.7554/eLife.10036
33. Avolio E, Madeddu P. Discovering cardiac pericyte biology: from physiopathological mechanisms to potential therapeutic applications in ischemic heart disease. Vascul Pharmacol. (2016) 86:53–63. doi: 10.1016/j.vph.2016.05.009
34. Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature (2012) 487:325–9. doi: 10.1038/nature11260
35. Badimon L, Padro T, Vilahur G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur Heart J Acute Cardiovasc Care (2012) 1:60–74. doi: 10.1177/2048872612441582
36. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity (2014) 40:91–104. doi: 10.1016/j.immuni.2013.11.019
37. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell (2017) 169:510–522.e20. doi: 10.1016/j.cell.2017.03.050
38. Sager HB, Kessler T, Schunkert H. Monocytes and macrophages in cardiac injury and repair. J Thorac Dis. (2017) 9:S30–5. doi: 10.21037/jtd.2016.11.17
39. Agellon LB, Michalak M. The endoplasmic reticulum and the cellular reticular network. Adv Exp Med Biol. (2018) 981:61–76. doi: 10.1007/978-3-319-55858-5_4
40. Baumann O, Walz B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int Rev Cytol. (2001) 205:149–214. doi: 10.1016/S0074-7696(01)05004-5
41. Krebs J, Agellon LB, Michalak M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: an integrated view of calcium signaling. Biochem Biophys Res Commun. (2015) 460:114–21. doi: 10.1016/j.bbrc.2015.02.004
42. van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. (2008) 9:112–24. doi: 10.1038/nrm2330
43. Prinz WA. Bridging the gap: membrane contact sites in signaling, metabolism, and organelle dynamics. J Cell Biol. (2014) 205:759–69. doi: 10.1083/jcb.201401126
44. Filadi R, Pozzan T. Generation and functions of second messengers microdomains. Cell Calcium (2015) 58:405–14. doi: 10.1016/j.ceca.2015.03.007
45. Penny CJ, Kilpatrick BS, Eden ER, Patel S. Coupling acidic organelles with the ER through Ca2+ microdomains at membrane contact sites. Cell Calcium (2015) 58:387–96. doi: 10.1016/j.ceca.2015.03.006
46. Phillips MJ, Voeltz GK. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol. (2016) 17:69–82. doi: 10.1038/nrm.2015.8
47. Joshi AS, Zhang H, Prinz WA. Organelle biogenesis in the endoplasmic reticulum. Nat Cell Biol. (2017) 19:876–82. doi: 10.1038/ncb3579
48. Barneda D, Christian M. Lipid droplet growth: regulation of a dynamic organelle. Curr Opin Cell Biol. (2017) 47:9–15. doi: 10.1016/j.ceb.2017.02.002
49. Prudent J, McBride HM. The mitochondria-endoplasmic reticulum contact sites: a signalling platform for cell death. Curr Opin Cell Biol. (2017) 47:52–63. doi: 10.1016/j.ceb.2017.03.007
50. Nunes-Hasler P, Demaurex N. The ER phagosome connection in the era of membrane contact sites. Biochim Biophys Acta (2017) 1864:1513–24. doi: 10.1016/j.bbamcr.2017.04.007
51. Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol. (2014) 76:107–27. doi: 10.1146/annurev-physiol-020911-153308
52. Wray S, Burdyga T. Sarcoplasmic reticulum function in smooth muscle. Physiol Rev. (2010) 90:113–78. doi: 10.1152/physrev.00018.2008
53. Rossi AE, Dirksen RT. Sarcoplasmic reticulum: the dynamic calcium governor of muscle. Muscle Nerve (2006) 33:715–31. doi: 10.1002/mus.20512
54. Michalak M, Opas M. Endoplasmic and sarcoplasmic reticulum in the heart. Trends Cell Biol. (2009) 19:253–9. doi: 10.1016/j.tcb.2009.03.006
55. Prins D, Michalak M. Organellar calcium buffers. Cold Spring Harb Perspect Biol. (2011) 3:197–212. doi: 10.1101/cshperspect.a004069
56. Smyth JT, Hwang SY, Tomita T, DeHaven WI, Mercer JC, Putney JW. Activation and regulation of store-operated calcium entry. J Cell Mol Med. (2010) 14:2337–49. doi: 10.1111/j.1582-4934.2010.01168.x
57. Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. (2012) 13:549–65. doi: 10.1038/nrm3414
58. Zhou Y, Cai X, Nwokonko RM, Loktionova NA, Wang Y, Gill DL. The STIM-Orai coupling interface and gating of the Orai1 channel. Cell Calcium (2017) 63:8–13. doi: 10.1016/j.ceca.2017.01.001
59. Ohba T, Watanabe H, Murakami M, Sato T, Ono K, Ito H. Essential role of STIM1 in the development of cardiomyocyte hypertrophy. Biochem Biophys Res Commun. (2009) 389:172–6. doi: 10.1016/j.bbrc.2009.08.117
60. Voelkers M, Salz M, Herzog N, Frank D, Dolatabadi N, Frey N, et al. Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes. J Mol Cell Cardiol. (2010) 48:1329–34. doi: 10.1016/j.yjmcc.2010.01.020
61. Hulot JS, Fauconnier J, Ramanujam D, Chaanine A, Aubart F, Sassi Y, et al. Critical role for stromal interaction molecule 1 in cardiac hypertrophy. Circulation (2011) 124:796–805. doi: 10.1161/CIRCULATIONAHA.111.031229
62. Zhu-Mauldin X, Marsh SA, Zou L, Marchase RB, Chatham JC. Modification of STIM1 by O-linked N-acetylglucosamine (O-GlcNAc) attenuates store-operated calcium entry in neonatal cardiomyocytes. J Biol Chem. (2012) 287:39094–106. doi: 10.1074/jbc.M112.383778
63. Collins HE, Zhu-Mauldin X, Marchase RB, Chatham JC. STIM1/Orai1-mediated SOCE: current perspectives and potential roles in cardiac function and pathology. Am J Physiol Heart Circ Physiol. (2013) 305:H446–58. doi: 10.1152/ajpheart.00104.2013
64. Troupes CD, Wallner M, Borghetti G, Zhang C, Mohsin S, von Lewinski D, et al. Role of STIM1 (Stromal Interaction Molecule 1) in hypertrophy-related contractile dysfunction. Circ Res. (2017) 121:125–36. doi: 10.1161/CIRCRESAHA.117.311094
65. Lopez-Crisosto C, Pennanen C, Vasquez-Trincado C, Morales PE, Bravo-Sagua R, Quest AFG, et al. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat Rev Cardiol. (2017) 14:342–60. doi: 10.1038/nrcardio.2017.23
66. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science (2011) 334:358–62. doi: 10.1126/science.1207385
67. Lewis SC, Uchiyama LF, Nunnari J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science (2016) 353:aaf5549. doi: 10.1126/science.aaf5549
68. Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. (2016) 35:1368–84. doi: 10.15252/embj.201593102
69. Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. (2011) 124(Pt 13):2143–52. doi: 10.1242/jcs.080762
70. De Stefani D, Rizzuto R, Pozzan T. Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem. (2016) 85:161–92. doi: 10.1146/annurev-biochem-060614-034216
71. Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. (2014) 21:396–413. doi: 10.1089/ars.2014.5851
72. Dicks N, Gutierrez K, Michalak M, Bordignon V, Agellon LB. Endoplasmic reticulum stress, genome damage, and cancer. Front Oncol. (2015) 5:11. doi: 10.3389/fonc.2015.00011
73. Yu XY, Geng YJ, Liang JL, Zhang S, Lei HP, Zhong SL, et al. High levels of glucose induce “metabolic memory” in cardiomyocyte via epigenetic histone H3 lysine 9 methylation. Mol Biol Rep. (2012) 39:8891–8. doi: 10.1007/s11033-012-1756-z
74. Gilsbach R, Preissl S, Gruning BA, Schnick T, Burger L, Benes V, et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun. (2014) 5:5288. doi: 10.1038/ncomms6288
75. Greco CM, Kunderfranco P, Rubino M, Larcher V, Carullo P, Anselmo A, et al. DNA hydroxymethylation controls cardiomyocyte gene expression in development and hypertrophy. Nat Commun. (2016) 7:12418. doi: 10.1038/ncomms12418
76. Rau CD, Vondriska TM. DNA methylation and human heart failure: mechanisms or prognostics. Circulation (2017) 136:1545–7. doi: 10.1161/CIRCULATIONAHA.117.029840
77. Zhang D, Li Y, Heims-Waldron DA, Bezzerides VJ, Guatimosim S, Guo Y, et al. Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ Res. (2018) 122:74–87. doi: 10.1161/CIRCRESAHA.117.311349
78. Groenendyk J, Sreenivasaiah PK, Kim do H, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res. (2010) 107:1185–97. doi: 10.1161/CIRCRESAHA.110.227033
79. Glembotski CC. Roles for ATF6 and the sarco/endoplasmic reticulum protein quality control system in the heart. J Mol Cell Cardiol. (2014) 71:11–5. doi: 10.1016/j.yjmcc.2013.09.018
80. Zhang C, Syed TW, Liu R, Yu J. Role of endoplasmic reticulum stress, autophagy, and inflammation in cardiovascular disease. Front Cardiovasc Med. (2017) 4:29. doi: 10.3389/fcvm.2017.00029
81. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature (2016) 529:326–35. doi: 10.1038/nature17041
82. Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. (2017) 13:477–91. doi: 10.1038/nrneurol.2017.99
83. Arrieta A, Blackwood EA, Glembotski CC. ER protein quality control and the unfolded protein response in the heart. Curr Top Microbiol Immunol. (2018) 414:193–213. doi: 10.1007/82_2017_54
84. Jung J, Michalak M, Agellon LB. Endoplasmic reticulum malfunction in the nervous system. Front Neurosci. (2017) 11:220. doi: 10.3389/fnins.2017.00220
85. Norimatsu Y, Hasegawa K, Shimizu N, Toyoshima C. Protein-phospholipid interplay revealed with crystals of a calcium pump. Nature (2017) 545:193–8. doi: 10.1038/nature22357
86. Giudice A, Arra C, Turco MC. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol Biol. (2010) 647:37–74. doi: 10.1007/978-1-60761-738-9_3
87. Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res. (2018) 1865:721–33. doi: 10.1016/j.bbamcr.2018.02.010
88. Miller CG, Holmgren A, Arner ESJ, Schmidt EE. NADPH-dependent and -independent disulfide reductase systems. Free Radic Biol Med. (2018) 127:248–61. doi: 10.1016/j.freeradbiomed.2018.03.051
89. Kornfeld OS, Hwang S, Disatnik MH, Chen CH, Qvit N, Mochly-Rosen D. Mitochondrial reactive oxygen species at the heart of the matter: new therapeutic approaches for cardiovascular diseases. Circ Res. (2015) 116:1783–99. doi: 10.1161/CIRCRESAHA.116.305432
90. Topf U, Wrobel L, Chacinska A. Chatty mitochondria keeping balance in cellular protein homeostasis. Trends Cell Biol. (2016) 26:577–86. doi: 10.1016/j.tcb.2016.03.002
91. Suomalainen A, Battersby BJ. Mitochondrial diseases the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol. (2018) 19:77–92. doi: 10.1038/nrm.2017.66
92. Dhingra R, Margulets V, Chowdhury SR, Thliveris J, Jassal P, Fernyhough GW, et al. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci USA. (2014) 111:E5537–44. doi: 10.1073/pnas.1414665111
93. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell (2015) 163:560–9. doi: 10.1016/j.cell.2015.10.001
94. Cortassa S, O'Rourke B, Aon MA. Redox-optimized ROS balance and the relationship between mitochondrial respiration and ROS. Biochim Biophys Acta (2014) 1837:287–95. doi: 10.1016/j.bbabio.2013.11.007
95. Gao AW, Canto C, Houtkooper RH. Mitochondrial response to nutrient availability and its role in metabolic disease. EMBO Mol Med. (2014) 6:580–9. doi: 10.1002/emmm.201303782
96. Akar FG, Aon MA, Tomaselli GF, O'Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. (2005) 115:3527–35. doi: 10.1172/JCI25371
97. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
98. Cai Y, Arikkath J, Yang L, Guo ML, Periyasamy P, Buch S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy (2016) 12:225–44. doi: 10.1080/15548627.2015.1121360
99. Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. (2010) 56:130–40. doi: 10.1097/FJC.0b013e3181e74a14
100. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. (2012) 298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7
101. Roul D, Recchia FA. Metabolic alterations induce oxidative stress in diabetic and failing hearts different pathways, same outcome. Antioxid Redox Signal. (2015) 22:1502–14. doi: 10.1089/ars.2015.6311
102. Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, et al. Origins and evolution of the Western diet health implications for the 21st century. Am J Clin Nutr. (2005) 81:341–54. doi: 10.1093/ajcn.81.2.341
103. Kien CL, Bunn JY, Stevens R, Bain J, Ikayeva O, Crain K, et al. Dietary intake of palmitate and oleate has broad impact on systemic and tissue lipid profiles in humans. Am J Clin Nutr. (2014) 99:436–45. doi: 10.3945/ajcn.113.070557
104. Fenech M, El-Sohemy A, Cahill L, Ferguson LR, French TA, Tai ES, et al. Nutrigenetics and nutrigenomics: viewpoints on the current status and applications in nutrition research and practice. J Nutrigenet Nutrigenomics (2011) 4:69–89. doi: 10.1159/000327772
105. Kolwicz SC Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. (2013) 113:603–16. doi: 10.1161/CIRCRESAHA.113.302095
106. Zaragoza C, Gomez-Guerrero C, Martin-Ventura JL, Blanco-Colio L, Lavin B, Mallavia B, et al. Animal models of cardiovascular diseases. J Biomed Biotechnol. (2011) 2011:497841. doi: 10.1155/2011/497841
107. Mohlke KL, Boehnke M, Abecasis GR. Metabolic and cardiovascular traits an abundance of recently identified common genetic variants. Hum Mol Genet. (2008) 17:R102–8. doi: 10.1093/hmg/ddn275
108. Wende AR, Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta (2010) 1801:311–9. doi: 10.1016/j.bbalip.2009.09.023
109. Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. (2012) 15:805–12. doi: 10.1016/j.cmet.2012.04.006
110. Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, et al. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. (2008) 118:3588–97. doi: 10.1172/JCI35847
111. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. (2015) 21:805–21. doi: 10.1016/j.cmet.2015.05.014
112. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. (2010) 40:280–93. doi: 10.1016/j.molcel.2010.09.023
113. Van Linthout S, Miteva K, Tschope C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res. (2014) 102:258–69. doi: 10.1093/cvr/cvu062
114. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. (2014) 11:255–65. doi: 10.1038/nrcardio.2014.28
115. Yang L, Gao L, Nickel T, Yang J, Zhou J, Gilbertsen A, et al. Lactate promotes synthetic phenotype in vascular smooth muscle cells. Circ Res. (2017) 121:1251–62. doi: 10.1161/CIRCRESAHA.117.311819
116. Lee D, Oka T, Hunter B, Robinson A, Papp S, Nakamura K, et al. Calreticulin induces dilated cardiomyopathy. PLoS ONE (2013) 8:e56387. doi: 10.1371/journal.pone.0056387
117. Kurtenbach S, Kurtenbach S, Zoidl G. Gap junction modulation and its implications for heart function. Front Physiol. (2014) 5:82. doi: 10.3389/fphys.2014.00082
118. Hu YF, Chen YJ, Lin YJ, Chen SA. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol. (2015) 12:230–43. doi: 10.1038/nrcardio.2015.2
119. Staerk L, Sherer JA, Ko D, Benjamin EJ, Helm RH. Atrial fibrillation: epidemiology, pathophysiology, and clinical outcomes. Circ Res. (2017) 120:1501–17. doi: 10.1161/CIRCRESAHA.117.309732
120. Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, et al. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation (2014) 129:145–56. doi: 10.1161/CIRCULATIONAHA.113.006641
121. Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci USA. (2015) 112:11389–94. doi: 10.1073/pnas.1513047112
122. Landstrom AP, Dobrev D, Wehrens XHT. Calcium signaling and cardiac arrhythmias. Circ Res. (2017) 120:1969–93. doi: 10.1161/CIRCRESAHA.117.310083
123. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. (2006) 99:172–82. doi: 10.1161/01.RES.0000232546.92777.05
124. Kohlhaas M, Nickel AG, Maack C. Mitochondrial energetics and calcium coupling in the heart. J Physiol. (2017) 595:3753–63. doi: 10.1113/JP273609
125. Li Q, Su D, O'Rourke B, Pogwizd SM, Zhou L. Mitochondria-derived ROS bursts disturb Ca(2)(+) cycling and induce abnormal automaticity in guinea pig cardiomyocytes: a theoretical study. Am J Physiol Heart Circ Physiol. (2015) 308:H623–36. doi: 10.1152/ajpheart.00493.2014
126. Dietl A, Maack C. Targeting mitochondrial calcium handling and reactive oxygen species in heart failure. Curr Heart Fail Rep. (2017) 14:338–49. doi: 10.1007/s11897-017-0347-7
127. Gao G, Xie A, Zhang J, Herman AM, Jeong EM, Gu L, et al. Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ Arrhythm Electrophysiol. (2013) 6:1018–24. doi: 10.1161/CIRCEP.113.000274
128. Liu M, Dudley SC Jr. Role for the unfolded protein response in heart disease and cardiac arrhythmias. Int J Mol Sci. (2015) 17:52. doi: 10.3390/ijms17010052
129. Mercier A, Clement R, Harnois T, Bourmeyster N, Bois P, Chatelier A. Nav1.5 channels can reach the plasma membrane through distinct N-glycosylation states. Biochim Biophys Acta (2015) 1850:1215–23. doi: 10.1016/j.bbagen.2015.02.009
130. Mayourian J, Cashman TJ, Ceholski DK, Johnson BV, Sachs D, Kaji DA, et al. Experimental and computational insight into human mesenchymal stem cell paracrine signaling and heterocellular coupling effects on cardiac contractility and arrhythmogenicity. Circ Res. (2017) 121:411–23. doi: 10.1161/CIRCRESAHA.117.310796
131. Mueller EE, Momen A, Masse S, Zhou YQ, Liu J, Backx PH, et al. Electrical remodelling precedes heart failure in an endothelin-1-induced model of cardiomyopathy. Cardiovasc Res. (2011) 89:623–33. doi: 10.1093/cvr/cvq351
132. Kohl P, Gourdie RG. Fibroblast-myocyte electrotonic coupling: does it occur in native cardiac tissue? J Mol Cell Cardiol. (2014) 70:37–46. doi: 10.1016/j.yjmcc.2013.12.024
133. Vasquez C, Benamer N, Morley GE. The cardiac fibroblast: functional and electrophysiological considerations in healthy and diseased hearts. J Cardiovasc Pharmacol. (2011) 57:380–8. doi: 10.1097/FJC.0b013e31820cda19
134. Yue L, Xie J, Nattel S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc Res. (2011) 89:744–53. doi: 10.1093/cvr/cvq329
135. Nitsan I, Drori S, Lewis YE, Cohen S, Tzlil S. Mechanical communication in cardiac cell synchronized beating. Nat Phys. (2016) 12:472. doi: 10.1038/nphys3619
136. Hong J, Kim K, Kim JH, Park Y. The role of endoplasmic reticulum stress in cardiovascular disease and exercise. Int J Vasc Med. (2017) 2017:2049217. doi: 10.1155/2017/2049217
137. George I, Sabbah HN, Xu K, Wang N, Wang J. beta-adrenergic receptor blockade reduces endoplasmic reticulum stress and normalizes calcium handling in a coronary embolization model of heart failure in canines. Cardiovasc Res. (2011) 91:447–55. doi: 10.1093/cvr/cvr106
138. Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation (2004) 110:705–12. doi: 10.1161/01.CIR.0000137836.95625.D4
139. Groenendyk J, Lee D, Jung J, Dyck JR, Lopaschuk GD, Agellon LB, et al. Inhibition of the unfolded protein response mechanism prevents cardiac fibrosis. PLoS ONE (2016) 11:e0159682. doi: 10.1371/journal.pone.0159682
140. Jensen BC, Bultman SJ, Holley D, Tang W, de Ridder G, Pizzo S, et al. Upregulation of autophagy genes and the unfolded protein response in human heart failure. Int J Clin Exp Med. (2017) 10:1051–8.
141. Castillero E, Akashi H, Pendrak K, Yerebakan H, Najjar M, Wang C, et al. Attenuation of the unfolded protein response and endoplasmic reticulum stress after mechanical unloading in dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. (2015) 309:H459–70. doi: 10.1152/ajpheart.00056.2015
142. Liu X, Kwak D, Lu Z, Xu X, Fassett J, Wang H, et al. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension (2014) 64:738–44. doi: 10.1161/HYPERTENSIONAHA.114.03811
143. Cheng WP, Wang BW, Lo HM, Shyu KG. Mechanical stretch induces apoptosis regulator TRB3 in cultured cardiomyocytes and volume-overloaded heart. PLoS ONE (2015) 10:e0123235. doi: 10.1371/journal.pone.0123235
144. Doroudgar S, Volkers M, Thuerauf DJ, Khan M, Mohsin S, Respress JL, et al. Hrd1 and ER-associated protein degradation, ERAD, are critical elements of the adaptive ER stress response in cardiac myocytes. Circ Res. (2015) 117:536–46. doi: 10.1161/CIRCRESAHA.115.306993
145. Jia S, Qiao X, Ye J, Fang X, Xu C, Cao Y, et al. Nogo-C regulates cardiomyocyte apoptosis during mouse myocardial infarction. Cell Death Dis. (2016) 7:e2432. doi: 10.1038/cddis.2016.331
146. Zhang S, Lin X, Li G, Shen X, Niu D, Lu G, et al. Knockout of Eva1a leads to rapid development of heart failure by impairing autophagy. Cell Death Dis. (2017) 8:e2586. doi: 10.1038/cddis.2017.17
147. Azevedo PS, Polegato BF, Minicucci MF, Paiva SA, Zornoff LA. Cardiac remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol. (2016) 106:62–9. doi: 10.5935/abc.20160005
148. Schirone L, Forte M, Palmerio S, Yee D, Nocella C, Angelini F, et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid Med Cell Longev. (2017) 2017:3920195. doi: 10.1155/2017/3920195
149. Leonard BL, Smaill BH, LeGrice IJ. Structural remodeling and mechanical function in heart failure. Microsc Microanal. (2012) 18:50–67. doi: 10.1017/S1431927611012438
150. Fountoulaki K, Dagres N, Iliodromitis EK. Cellular communications in the heart. Card Fail Rev. (2015) 1:64–8. doi: 10.15420/cfr.2015.1.2.64
151. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. (2016) 97:245–62. doi: 10.1016/j.yjmcc.2016.06.001
152. Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell (2003) 114:763–76. doi: 10.1016/S0092-8674(03)00687-1
153. Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, et al. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. (2004) 95:911–21. doi: 10.1161/01.RES.0000147315.71699.51
154. Li RK. Intrinsic cardiac stem cells are essential for regeneration. J Thorac Cardiovasc Surg. (2016) 152:583–4. doi: 10.1016/j.jtcvs.2016.03.044
155. Terzic A, Behfar A. Stem cell therapy for heart failure: ensuring regenerative proficiency. Trends Cardiovasc Med. (2016) 26:395–404. doi: 10.1016/j.tcm.2016.01.003
156. MacKenna D, Summerour SR, Villarreal FJ. Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc Res. (2000) 46:257–63. doi: 10.1016/S0008-6363(00)00030-4
157. Tilley DG. G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ Res. (2011) 109:217–30. doi: 10.1161/CIRCRESAHA.110.231225
158. Vega H, Agellon LB, Michalak M. The rise of proteostasis promoters. IUBMB Life (2016) 68:943–54. doi: 10.1002/iub.1576
159. Rani S, Sreenivasaiah PK, Kim JO, Lee MY, Kang WS, Kim YS, et al. Tauroursodeoxycholic acid (TUDCA) attenuates pressure overload-induced cardiac remodeling by reducing endoplasmic reticulum stress. PLoS ONE (2017) 12:e0176071. doi: 10.1371/journal.pone.0176071
160. Sung MM, Dyck JR. Therapeutic potential of resveratrol in heart failure. Ann N Y Acad Sci. (2015) 1348:32–45. doi: 10.1111/nyas.12839
161. Lin Y, Zhu J, Zhang X, Wang J, Xiao W, Li B, et al. Inhibition of cardiomyocytes hypertrophy by resveratrol is associated with amelioration of endoplasmic reticulum stress. Cell Physiol Biochem. (2016) 39:780–9. doi: 10.1159/000447788
162. Takeda N, Manabe I. Cellular interplay between cardiomyocytes and nonmyocytes in cardiac remodeling. Int J Inflam. (2011) 2011:535241. doi: 10.4061/2011/535241
163. Appari M, Breitbart A, Brandes F, Szaroszyk M, Froese N, Korf-Klingebiel M, et al. C1q-TNF-related protein-9 promotes cardiac hypertrophy and failure. Circ Res. (2017) 120:66–77. doi: 10.1161/CIRCRESAHA.116.309398
164. Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension (2007) 49:241–8. doi: 10.1161/01.HYP.0000254415.31362.a7
165. Sun L, Zang WJ, Wang H, Zhao M, Yu XJ, He X, et al. Acetylcholine promotes ROS detoxification against hypoxia/reoxygenation-induced oxidative stress through FoxO3a/PGC-1alpha dependent superoxide dismutase. Cell Physiol Biochem. (2014) 34:1614–25. doi: 10.1159/000366364
166. Luchetti F, Crinelli R, Cesarini E, Canonico B, Guidi L, Zerbinati C, et al. Endothelial cells, endoplasmic reticulum stress and oxysterols. Redox Biol. (2017) 13:581–7. doi: 10.1016/j.redox.2017.07.014
167. Jia LX, Zhang WM, Li TT, Liu Y, Piao CM, Ma YC, et al. ER stress dependent microparticles derived from smooth muscle cells promote endothelial dysfunction during thoracic aortic aneurysm and dissection. Clin Sci. (2017) 131:1287–99. doi: 10.1042/CS20170252
168. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. (2007) 13:952–61. doi: 10.1038/nm1613
169. Weinberger T, Schulz C. Myocardial infarction: a critical role of macrophages in cardiac remodeling. Front Physiol. (2015) 6:107. doi: 10.3389/fphys.2015.00107
170. Chess DJ, Stanley WC. Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc Res. (2008) 79:269–78. doi: 10.1093/cvr/cvn074
171. McKeag NA, McKinley MC, Woodside JV, Harbinson MT, McKeown PP. The role of micronutrients in heart failure. J Acad Nutr Diet. (2012) 112:870–86. doi: 10.1016/j.jand.2012.01.016
172. Sciatti E, Lombardi C, Ravera A, Vizzardi E, Bonadei I, Carubelli V, et al. Nutritional deficiency in patients with heart failure. Nutrients (2016) 8:442. doi: 10.3390/nu8070442
173. Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. (2008) 58:88–111. doi: 10.1016/j.phrs.2008.06.007
174. Wang Y, Hill JA. Electrophysiological remodeling in heart failure. J Mol Cell Cardiol. (2010) 48:619–32. doi: 10.1016/j.yjmcc.2010.01.009
175. Tse G, Yeo JM. Conduction abnormalities and ventricular arrhythmogenesis: the roles of sodium channels and gap junctions. Int J Cardiol Heart Vasc. (2015) 9:75–82. doi: 10.1016/j.ijcha.2015.10.003
176. Hearse DJ, Sutherland FJ. Experimental models for the study of cardiovascular function and disease. Pharmacol Res. (2000) 41:597–603. doi: 10.1006/phrs.1999.0651
177. Camacho P, Fan H, Liu Z, He JQ. Small mammalian animal models of heart disease. Am J Cardiovasc Dis. (2016) 6:70–80. doi: 10.3390/jcdd3040030
178. Piro M, Della Bona R, Abbate A, Biasucci LM, Crea F. Sex-related differences in myocardial remodeling. J Am Coll Cardiol. (2010) 55:1057–65. doi: 10.1016/j.jacc.2009.09.065
179. Salama G, Bett GC. Sex differences in the mechanisms underlying long QT syndrome. Am J Physiol Heart Circ Physiol. (2014) 307:H640–8. doi: 10.1152/ajpheart.00864.2013
180. Keller KM, Howlett SE. Sex differences in the biology and pathology of the aging heart. Can J Cardiol. (2016) 32:1065–73. doi: 10.1016/j.cjca.2016.03.017
181. Ko D, Rahman F, Martins MA, Hylek EM, Ellinor PT, Schnabel RB, et al. Atrial fibrillation in women: treatment. Nat Rev Cardiol. (2017) 14:113–24. doi: 10.1038/nrcardio.2016.171
182. InanlooRahatloo K, Liang G, Vo D, Ebert A, Nguyen I, Nguyen PK. Sex-based differences in myocardial gene expression in recently deceased organ donors with no prior cardiovascular disease. PLoS ONE (2017) 12:e0183874. doi: 10.1371/journal.pone.0183874
183. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
184. Lu J, Wang Q, Huang L, Dong H, Lin L, Lin N, et al. Palmitate causes endoplasmic reticulum stress and apoptosis in human mesenchymal stem cells: prevention by AMPK activator. Endocrinology (2012) 153:5275–84. doi: 10.1210/en.2012-1418
185. Rosca MG, Tandler B, Hoppel CL. Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. (2013) 55:31–41. doi: 10.1016/j.yjmcc.2012.09.002
186. Egnatchik RA, Leamy AK, Noguchi Y, Shiota M, Young JD. Palmitate-induced activation of mitochondrial metabolism promotes oxidative stress and apoptosis in H4IIEC3 rat hepatocytes. Metabolism (2014) 63:283–95. doi: 10.1016/j.metabol.2013.10.009
187. Xu S, Nam SM, Kim JH, Das R, Choi SK, Nguyen TT, et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. (2015) 6:e1976. doi: 10.1038/cddis.2015.331
188. Yin J, Wang Y, Gu L, Fan N, Ma Y, Peng Y. Palmitate induces endoplasmic reticulum stress and autophagy in mature adipocytes: implications for apoptosis and inflammation. Int J Mol Med. (2015) 35:932–40. doi: 10.3892/ijmm.2015.2085
189. Akoumi A, Haffar T, Mousterji M, Kiss RS, Bousette N. Palmitate mediated diacylglycerol accumulation causes endoplasmic reticulum stress, Plin2 degradation, and cell death in H9C2 cardiomyoblasts. Exp Cell Res. (2017) 354:85–94. doi: 10.1016/j.yexcr.2017.03.032
Keywords: endoplasmic reticulum stress, proteostasis, cardiac cells, stress responses, membrane contact sites
Citation: Michalak M and Agellon LB (2018) Stress Coping Strategies in the Heart: An Integrated View. Front. Cardiovasc. Med. 5:168. doi: 10.3389/fcvm.2018.00168
Received: 15 August 2018; Accepted: 02 November 2018;
Published: 21 November 2018.
Edited by:
Georges Nemer, American University of Beirut, LebanonReviewed by:
Fouad Zouein, American University of Beirut, LebanonMiguel A. Aon, National Institute on Aging (NIA), United States
Copyright © 2018 Michalak and Agellon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marek Michalak, marek.michalak@ualberta.ca
Luis B. Agellon, luis.agellon@mcgill.ca